
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, spectroscopic, SC-XRD/DFT and non-linear optical (NLO) properties of chromene derivatives†
Nadia Arifa,
Zahid Shafiq *a,
Sajida Noureenb,
Muhammad Khalid*cd,
Abida Ashrafa,
Muhammad Yaquba,
Shabana Irshadcd,
Muhammad Usman Khane,
Muhammad Nadeem Arshadf,
Ataualpa Albert Carmo Bragag,
Ahmed H. Ragabh and
Saedah R. Al-Mhyawii
*a,
Sajida Noureenb,
Muhammad Khalid*cd,
Abida Ashrafa,
Muhammad Yaquba,
Shabana Irshadcd,
Muhammad Usman Khane,
Muhammad Nadeem Arshadf,
Ataualpa Albert Carmo Bragag,
Ahmed H. Ragabh and
Saedah R. Al-Mhyawii
aInstitute of Chemical Sciences, Organic Chemistry Division, Bahauddin Zakariya University, Multan-60800, Pakistan. E-mail: zahidshafiq25@hotmail.com
bMaterials Chemistry Laboratory, Institute of Chemistry, The Islamia University of Bahawalpur, 63100, Pakistan
cInstitute of Chemistry, Khwaja Fareed University of Engineering & Information Technology, Rahim Yar Khan, 64200, Pakistan. E-mail: khalid@iq.usp.br; muhammad.khalid@kfueit.edu.pk
dCentre for Theoretical and Computational Research, Khwaja Fareed University of Engineering & Information Technology, Rahim Yar Khan, 64200, Pakistan
eDepartment of Chemistry, University of Okara, Okara-56300, Pakistan
fCenter of Excellence for Advanced Materials Research (CEAMR), Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
gDepartamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes 748, São Paulo, 05508-000, Brazil
hDepartment of Chemistry, Faculty of Science, King Khalid University, Abha 62224, Saudi Arabia
iDepartment of Chemistry, College of Science, University of Jeddah, Jeddah 21419, Saudi Arabia
First published on 21st December 2022
Abstract
In the present study, we reported the efficient synthesis of novel, heterocyclic, coumarin-based pyrano-chromene derivatives, 2-amino-8-methyl-5-oxo-4-[2-(2-oxo-2H-chromen-3-ylmethoxy)-phenyl]-4H,5H-pyrano[3,2-c]chromene-3-carbonitrile (4a) and 2-amino-8-methyl-5-oxo-4-[2-(2-oxo-2H-chromen-3-ylmethoxy)-phenyl]-4H,5H-pyrano[3,2-c]chromene-3-carboxylic acid methyl ester (4b). The chemical structures of synthesized compounds were resolved by employing various spectroscopic techniques like UV-Vis, FT-IR, 1H & 13C NMR, and single crystal X-ray diffraction (SC-XRD) analysis. The compounds; 4a and 4b, with appealing π-bonded skeleton were further analyzed in terms of their electronic and structural aspects using an integral approach of density functional theory (DFT) and time-dependent DFT (TD/DFT). The methodology: M06-2X/6-31G(d,p) level of theory was applied to compare their experimental data with theoretical outcomes using quantum chemical analysis. The frontier molecular orbitals (FMOs) study revealed that, 4a possesses a low band gap (5.168 eV) as compared to 4b (6.308 eV). Global reactivity parameters were associated with Egap values as 4a, with the lowest band gap showed the smaller value of hardness (0.094 eV) and a larger value of softness (5.266 eV). The non-linear optical (NLO) insight exhibited that, the average polarizability 〈α〉 and second hyperpolarizability (γtot) were observed in 4a as 6.77005 × 10−23 and 0.145 × 104 esu, respectively. Overall, the computational studies suggest that the investigated compounds have distinct NLO properties.
1. Introduction
Diversity-oriented synthesis of heterocyclic frameworks has become indispensable in the construction of novel fused ring heterocyclic compounds and thus aims to design natural product ring systems with useful pharmacophores for drug development.1 The structural and activity profiles of various coumarin derivatives have inspired pharmaceutical chemists to develop new drugs with safe activity and selectivity in their action for a variety of therapeutic disorders.2–5 Coumarin derivatives are well known for their critical role in the prevention and treatment of pathogenic and infectious diseases.6,7 Coumarin-based analogues are intended to show discrete valuable biological and clinical activities vulnerable to substitution patterns exhibited by the parent benzopyran core.8,9 Amongst the highly studied pharmaceutical activities, it is noteworthy for antibacterial,10,11 antiviral,12 antifungal,13 anticancer,14 anti-inflammatory,15 antitubercular,16 antioxidant,17 antimutagenic,18 anticoagulant,19 scavenging of reactive oxygen species (ROS),20 cyclooxygenase,21 lipoxygenase,22 cholinesterase (ChE), and monoamine oxidase (MAO) inhibitory activities, vasodilator,23 CNS stimulant,24 and cytotoxic25 potential.The analysis of supramolecular architecture has greatly revolutionized research in the field of crystal engineering, which accommodates non-covalent interactions where molecules accumulate in self-assembly, possessing various inter and intramolecular interactions.26,27 The strong interactions like hydrogen bonding (inter and intramolecular) also play a pivotal role in molecular biology, supramolecular frameworks and crystal engineering.28,29 There are various structural diversities found in the heterocyclic frameworks, like the presence of electronegative groups (such as –F), which influence the molecular stability as well as the molecular level interactions with the biological receptors. The various privileged biological compounds that undergo structural diversities are found with enhanced efficiencies. Moreover, the rapid synthesis of wide-ranged heterocyclic compounds with the help of multi-component reaction techniques has attracted the attention of researchers.30,31
The heterocyclic chromene-based ring compounds are an important class of benzopyran family compounds that are efficiently being used in several pharmacological industries as anti-bacterial,32–34 antiviral,35 anti-malaria,36 anti-inflammatory,37 anti-cancer,38 anti-HIV,39 antifungal,40 anti-anaphylactic, and anti-proliferative.41 Computational analysis, also known as density functional (DFT) analysis, is an important method for predicting structural parameters such as non-covalent interactions, electronic properties, stability, and chemical reactivity.42 The DFT and time-dependent DFT approaches envisage the bond distances, bond angles, FMOs, global reactivity descriptors (GRDs), natural population analysis (NPA), NBOs, molecular electrostatic potential (MEP), statistical average of molecular dipoles moment, linear and non-linear polarizabilities for various crystalline and non-crystalline compounds, including hydrazones, organic co-crystals,43,44 pyrimidine rings45 etc.
Non-linear optics (NLO) is one of the significant research areas which are being focused nowadays. In the present investigation, the chromene-based synthesized compounds are deliberated for their NLO parameters, i.e., 〈α〉, βtot, and γtot, which were not evaluated so far, as evident from the literature review. Such organic compounds having a π-conjugated framework are efficiently utilized in wide bandwidth optical switching devices. The unique properties of such compounds as intramolecular charge transfer (ICT), electron delocalization, polarizability, and hyperpolarizability have paved the way for new research into efficient NLO devices.46
The chromene-based synthesized crystalline compounds, i.e., 4a and 4b, were studied via the DFT approach. The NLO study of the entitled compounds might not be reported yet. Hence, to overcome the research gap, computational analysis is carried out to examine NLO properties. This research paper would be a new addition to the advancement of NLO compounds and surely serve as a synthesized non-fullerene (NF) based crystalline organic NLO materials in the research discipline for the future.
2. Experimental section
2.1 Synthesis of coumarin-based pyrano-chromene derivatives (4a and 4b)
An equimolar mixture of coumarin-based aldehyde (1) (1 mmol) and appropriate nitrile (2a–2b) (1 mmol) in EtOAc was initially stirred for half an hour at room temperature in the presence of 2–3 drops of Et3N as a catalyst, and then 7-methyl-4-hydroxy coumarin (3) (1 mmol) was added. The course of the reaction was monitored by TLC. After completion of the reaction, the precipitates were formed, which were filtered and washed twice with hot ethyl acetate. Further purification was done by recrystallization using a mixture of CHCl3 (4 parts) and MeOH (1 part) to furnish pure, newly synthesized heterocyclic coumarin-based pyrano-chromene compounds (4a and 4b) with excellent yield. Physical and spectroscopic data for compounds (4a and 4b) are shown below, while NMR and FT-IR spectra are shown in Fig. S1–S6.†2.2 2-Amino-8-methyl-5-oxo-4-[2-(2-oxo-2H-chromen-3-ylmethoxy)-phenyl]-4H,5H-pyrano[3,2-c]chromene-3-carbonitrile (4a)
Colorless crystalline solid; m.p. 268–270 °C; yield: 97%; solubility: DMSO; FT-IR; KBr (cm−1); 1681, 1718 (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 2180 (–CN), 3311, 3387 (NH2-str.); 1H NMR; (400 MHz, DMSO-d6, δ, ppm) 2.37 (s, 3H, CH3–H), 4.67 (s, 1H, CH, C4–H), 4.80 (d, 1H, J = 10.2 Hz, OCH2–Ha), 4.94 (d, 1H, J = 10.2 Hz, OCH2–Hb), 6.89 (d, 1H, J = 6.4 Hz, Ar–H), 6.93 (t, 1H, J = 5.8 Hz, Ar–H), 7.07 (d, 1H, J = 6.5 Hz, Ar–H), 7.18–7.26 (m, 5H, NH2–H, Ar–H), 7.34–7.37 (m, 3H, Ar–H), 7.51 (d, 1H, J = 6.2 Hz, Ar–H), 7.62 (td, 1H, J = 1.2 Hz, 7.3 Hz, Ar–H), 7.90 (s, 1H, CH,
O), 2180 (–CN), 3311, 3387 (NH2-str.); 1H NMR; (400 MHz, DMSO-d6, δ, ppm) 2.37 (s, 3H, CH3–H), 4.67 (s, 1H, CH, C4–H), 4.80 (d, 1H, J = 10.2 Hz, OCH2–Ha), 4.94 (d, 1H, J = 10.2 Hz, OCH2–Hb), 6.89 (d, 1H, J = 6.4 Hz, Ar–H), 6.93 (t, 1H, J = 5.8 Hz, Ar–H), 7.07 (d, 1H, J = 6.5 Hz, Ar–H), 7.18–7.26 (m, 5H, NH2–H, Ar–H), 7.34–7.37 (m, 3H, Ar–H), 7.51 (d, 1H, J = 6.2 Hz, Ar–H), 7.62 (td, 1H, J = 1.2 Hz, 7.3 Hz, Ar–H), 7.90 (s, 1H, CH,  ); 13C NMR; (100 MHz, DMSO-d6, δ, ppm) 21.71, 33.98, 49.06, 57.12, 65.32, 102.68, 110.69, 113.03, 116.60, 116.79, 119.07, 120.11, 121.42, 121.95, 123.98, 124.94, 125.77, 128.90, 129.07, 131.06, 132.27, 141.43, 143.85, 152.47, 153.46, 154.40, 156.44, 158.80, 159.90, 160.33; anal. calcd. for C30H20O6N2 (504): C = 71.42, H = 4.00, N = 5.55; found (%): C = 71.35, H = 4.03, N = 5.58.
); 13C NMR; (100 MHz, DMSO-d6, δ, ppm) 21.71, 33.98, 49.06, 57.12, 65.32, 102.68, 110.69, 113.03, 116.60, 116.79, 119.07, 120.11, 121.42, 121.95, 123.98, 124.94, 125.77, 128.90, 129.07, 131.06, 132.27, 141.43, 143.85, 152.47, 153.46, 154.40, 156.44, 158.80, 159.90, 160.33; anal. calcd. for C30H20O6N2 (504): C = 71.42, H = 4.00, N = 5.55; found (%): C = 71.35, H = 4.03, N = 5.58.
2.3 2-Amino-8-methyl-5-oxo-4-[2-(2-oxo-2H-chromen-3-ylmethoxy)-phenyl]-4H,5H-pyrano[3,2-c]chromene-3-carboxylic acid methyl ester (4b)
Colorless crystalline solid; m.p. 230–232 °C; yield: 92%; solubility (DMSO, CHCl3); FT-IR; KBr (cm−1); 1687, 1717 (CO), 3324, 3420 (NH2-str.); 1H NMR; (300 MHz, DMSO-d6, δ, ppm) 2.13 (s, 3H, CH3–H), 3.52 (s, 3H, COOMe–CH3–H), 4.73 (d, 1H, J = 12.6 Hz, OCH2–Ha), 4.76 (s, 1H, CH, C4–H), 4.82 (d, 1H, J = 12.8 Hz, OCH2–Hb), 6.90 (t, 1H, J = 7.2 Hz, Ar–H), 6.99 (d, 1H, J = 8.1 Hz, Ar–H), 7.16 (t, 1H, J = 7.5 Hz, Ar–H), 7.24–7.30 (m, 3H, Ar–H), 7.34–7.41 (m, 3H, Ar–H), 7.48 (d, 1H, J = 7.5 Hz, Ar–H), 7.62–7.67 (m, 3H, NH2–H, Ar–H), 7.81 (s, 1H, CH,  ); 13C NMR, (75 MHz, DMSO-d6, δ, ppm) 20.92, 34.02, 50.95, 65.50, 75.86, 105.11, 113.17, 113.29, 116.52, 116.64, 119.15, 120.78, 122.04, 124.17, 125.04, 128.43, 128.92, 131.81, 132.29, 132.59, 133.54, 134.02, 141.55, 150.60, 153.48, 153.90, 156.98, 159.36, 159.91, 160.52, 168.75; anal. calcd for C31H23O8N (537.14): C = 69.27, H = 4.31, N = 2.61; found (%): C = 69.24, H = 4.33, N = 2.68.
); 13C NMR, (75 MHz, DMSO-d6, δ, ppm) 20.92, 34.02, 50.95, 65.50, 75.86, 105.11, 113.17, 113.29, 116.52, 116.64, 119.15, 120.78, 122.04, 124.17, 125.04, 128.43, 128.92, 131.81, 132.29, 132.59, 133.54, 134.02, 141.55, 150.60, 153.48, 153.90, 156.98, 159.36, 159.91, 160.52, 168.75; anal. calcd for C31H23O8N (537.14): C = 69.27, H = 4.31, N = 2.61; found (%): C = 69.24, H = 4.33, N = 2.68.
2.4 Chemistry
In order to obtain o-ring heterocyclic (pyrano-chromenes), a multicomponent approach was designed to synthesize novel fused ring coumarin-based dihydropyranochromenes (4a and 4b) (Scheme 1). Coumarin-based aldehyde (1) was synthesized by utilizing the Baylis–Hillman reaction between salicylaldehyde and methyl acrylate.47,48 The multi-component reaction of coumarin-based aromatic aldehyde (1), nitriles (2a–2b), and 7-methyl-4-hydroxy coumarin (3) in the presence of Et3N through the Knoevenagel–Michael-cyclization path was utilized for the synthesis of these heterocyclic compounds with excellent yield. The optimization of various parameters like temperature, solvent, and catalyst, during the reaction was obtained to enhance the regioselectivity of the reaction as well as the yield of the product. Thus, ethyl acetate as solvent, Et3N or Me3N as a base, and 60 °C temperature were found to be the suitable parameters of choice for this reaction. The structures of synthesized compounds (4a and 4b) were elaborated via spectroscopic techniques like FT-IR, NMR, and CHN analysis. In FTIR spectroscopic analysis, the CO functional group appeared in the range of 1671–1718 cm−1, while the cyano group appeared at 2179 cm−1, and NH2 groups in the relevant products were identified by absorption bands in the range of 3387–3420 cm−1.
 | ||
| Scheme 1 Synthesis of coumarin-based pyrano-chromenes (4a and 4b). | ||
The 1H NMR spectra of (4a and 4b) gave evidence for cyclized products as there is no downfield signal of CHO proton at δ 10.48–10.53 ppm. Instead, a singlet of two protons for the NH2 group was observed at δ 7.18–7.68 ppm and this peak is merged into the aromatic range by shielding or deshielding. Furthermore, the disappearance of the signal for the singlet of the OH group in 4-hydroxylated coumarins confirmed the cyclization. The appearance of one signal for the singlet of 3H at δ 3.34 ppm illustrated the presence of methyl-ester in compounds (4b). Total proton counts in the aromatic region also fulfill the structural requirements of the compound. Their chemical shifts were allocated on behalf of spin-multiplicity as well as coupling constant.
The 13C NMR spectra of (4a and 4b) unveiled the accordance of chemical shifts of aliphatic, aromatic, and carbonyl moieties with the desired structures. Further confirmation of the representative compounds (4a and 4b) came from the absorption peaks of aliphatic carbons at 21.71, 33.98, and 49.06 ppm, aromatic carbons from 120 to 158 ppm, and carbonyl at 159.90 and 160.33 ppm. CHN analyses also favored the desired structures of pyrano-chromenes, as experimentally collected values were in accordance with calculated % ages. By utilizing the animation of Avogadro software, we calculated the results of vibrational analysis of fore-said compounds and compared with experimental results. The simulated stretching symmetrical and asymmetrical vibration band for –NH2 groups in 4a and 4b are observed in the range of 3742–3509 and 3709–3538 cm−1, respectively, which are in good harmony with experimental absorption bands (3466 and 3576 cm−1, accordingly). Similarly, the experimentally determined stretching vibrational frequencies for –CN group in 4a are found as 2361 cm−1 which showed good agreement with their DFT results (2376 cm−1). The symmetrical stretching absorption bands for –CO are noted as 1886 and 1815 cm−1 while their experimental bands are determined at 1719 and 1716 cm−1, respectively. Many other kind of simulated vibrations frequencies in 4a and 4b are also studied which showed good agreement with their experimental absorption bands (see in Tables S10, S11 and Fig. S5, S6†). In this research paper, the non-fullerene-natured synthesized crystals, i.e., 4a and 4b, are evaluated in terms of their NLO characteristics. For this purpose, SC-XRD characterization is done and inspection of molecular configuration is carried out thoroughly via different parameters like bond lengths, bond angles, etc. The experimental data are compared with the theoretical results (DFT) to verify the chemical nature of the studied compounds.
2.5 Crystallization, data collection and structure solution
The crystals of compounds 4a and 4b were grown by slow evaporation in a methanol/chloroform solvent system in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Colorless crystals appeared after a few days. Good quality single crystals were chosen for the diffraction experiment. The crystal was mounted on a goniometer head using grease. The data sets were collected on a Bruker D8 Venture with a PHOTON II detector and Mo microfocus source (λ = 0.71073 Å) (Bruker 2016) under ambient conditions using omega and phi scan methods. The diffracted intensities were processed using APEX3 tools. An analytical absorption correction was applied using the face indices of the crystal. Table 1 summarizes the crystallographic and refinement details.
:1. Colorless crystals appeared after a few days. Good quality single crystals were chosen for the diffraction experiment. The crystal was mounted on a goniometer head using grease. The data sets were collected on a Bruker D8 Venture with a PHOTON II detector and Mo microfocus source (λ = 0.71073 Å) (Bruker 2016) under ambient conditions using omega and phi scan methods. The diffracted intensities were processed using APEX3 tools. An analytical absorption correction was applied using the face indices of the crystal. Table 1 summarizes the crystallographic and refinement details.
| Comp. | 4a | 4b |
|---|---|---|
| Crystal data | ||
| Chemical formula | C30H20N2O6·CH4O | C31H23NO8 |
| Mr | 536.55 | 537.50 |
| Crystal system, space group | Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Triclinic, P |
| Temperature (K) | 297(1) | 297(1) |
| a, b, c (Å) | 9.1201(7), 11.5487(7), 12.5810(8) | 8.6139(15), 10.6283(16), 14.384(2) |
| α, β, γ (°) | 93.498(2), 96.303(3), 100.317(3) | 80.026(6), 80.436(7), 81.990(6) |
| V (Å3) | 1291.38(15) | 1270.7(4) |
| Z | 2 | 2 |
| Radiation type | Mo Kα | Mo Kα |
| μ (mm−1) | 0.10 | 0.10 |
| Crystal size (mm) | 0.22 × 0.18 × 0.17 | 0.32 × 0.22 × 0.13 |
|
||
| Data collection | ||
| Diffractometer | Bruker D8 Venture Photon II detector | |
| Absorption correction | Analytical SADABS2016/2 (Bruker, 2016/2) was used for absorption correction. wR2(int) was 0.1247 before and 0.0684 after correction. The ratio of minimum to maximum transmission is 0.7828. The λ/2 correction factor is not present | |
| Tmin, Tmax | 0.667, 0.746 | 0.584, 0.745 |
| No. of measured, independent and observed [I ≥ 2u(I)] reflections | 35003, 5884, 3085 |
33043, 5205, 2618 |
| Rint | 0.064 | 0.083 |
| (Sinθ/λ)max (Å−1) |
0.649 | 0.625 |
|
||
| Refinement | ||
| R[F2 > 2σ(F2)], wR(F2), S | 0.064, 0.248, 1.22 | 0.056, 0.194, 1.07 |
| No. of reflections | 5884 | 5205 |
| No. of parameters | 365 | 371 |
| H-atom treatment | H-atom parameters constrained | H atoms are treated by a mixture of independent and constrained refinement |
| Δ〉max, Δ〉min (e Å−3) | 0.35, −0.35 | 0.29, −0.25 |
Both the crystal structures were solved by direct methods in the triclinic (P) space group using Olex2 (ref. 49) (Fig. 1). All the hydrogen atoms could be located in different Fourier maps. However, they are fixed using a riding model.
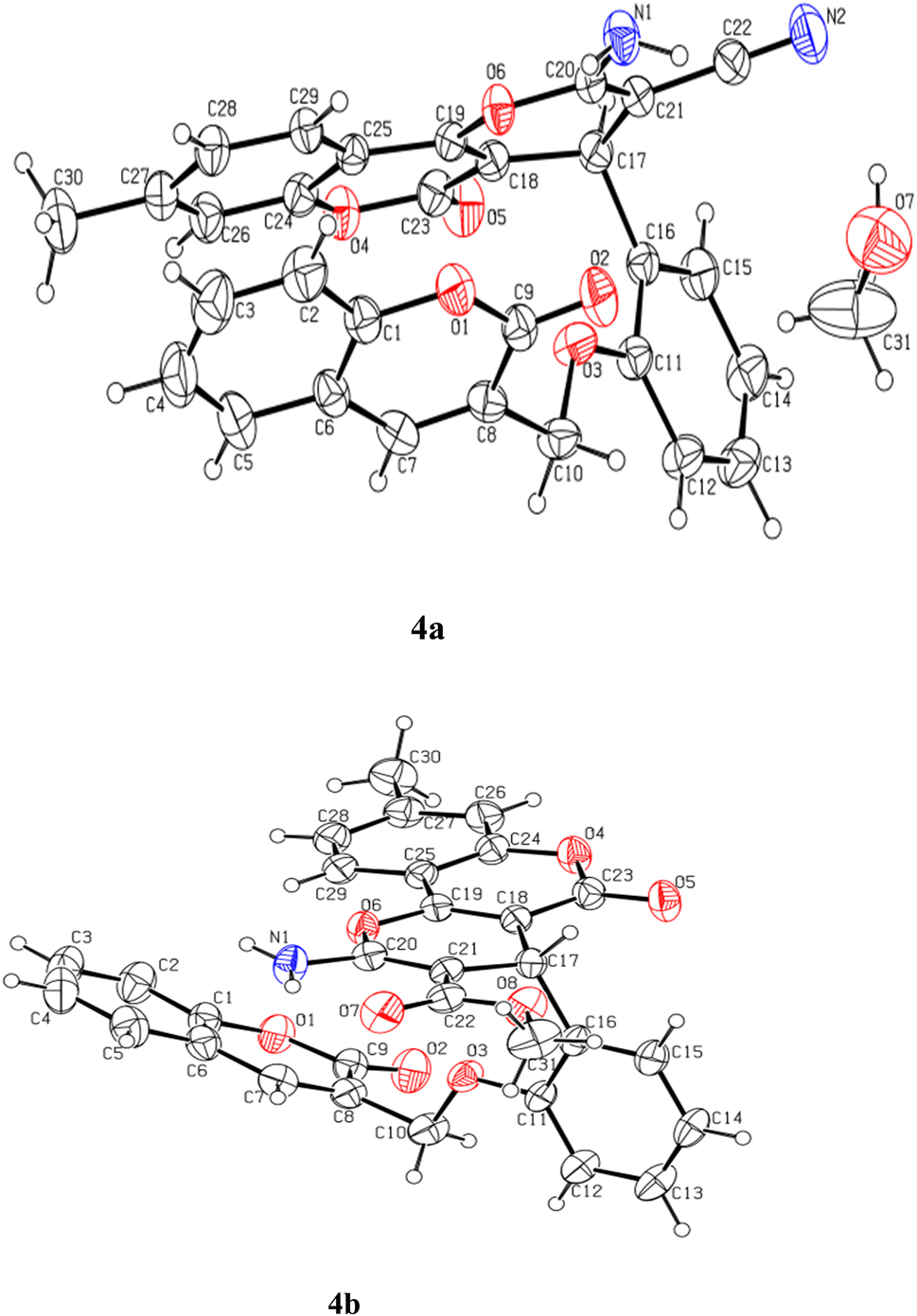 | ||
| Fig. 1 View of the atom-labeled structures of compounds 4a and 4b (thermal ellipsoids were drawn at 30% probability level for both molecules). | ||
Both molecules crystallized with the P space group. The asymmetric unit of 4a contains one molecule of the organic compound along-with one molecule of methanol as solvent while the asymmetric unit of 4b contains only one molecule of the organic moiety Fig. 1. The angles between the planes of the fitted atoms of pyrano-chromene ring system (C17–C29/O4/O6) and the central phenyl ring (C11–C16) are 88° and 88.35° in 4a and 4b, respectively, while the angles between the phenyl (C11–C16) and the coumarin ring system(C1–C9/O1) are 78.38° and 72.35° in 4a and 4b, respectively. The molecular structures (ORTEP diagrams) of coumarin-based pyrano-chromenes 4a and 4b with their crystallographic numbering are shown in Fig. 1.
In the case of 4a, the molecular assembly is primarily built based on strong hydrogen bonds like N1–H1B⋯O2 and C14–H14⋯O5. Both interactions result in molecular dimerization. The interaction N1–H1B⋯O2, at a distance of 2.20(3) Å and N–H⋯O angle of 135.07(3)°, connects the molecules in dimers and generate twenty-four (24) membered ring motifs R22(24). Similarly, C14–H14⋯O5, connects the molecules to produce sixteen membered ring motifs R22(16), these two interactions go in a zigzag fashion at right angle. All these further extended into three-dimensional spaces through a large network of van der Waals's interactions. The amino group is involved in the formation of two hydrogen bonds. One we have discussed above while the other forms a hydrogen bond with N1–H1a⋯O7 at a distance of 2.171(3) Å and N–H⋯O angle of 155.1(3)° with the solvent molecule (methanol). The methanol molecule also forms another strong hydrogen bond with the reference molecule through the O7–H7A⋯O1 hydrogen bond (Fig. 2a). The hydroxyl group of methanol is bridging between the amino group and O1 of coumarin Fig. 2a. The interaction C3–H3⋯N2 connects the molecules along the diagonal of ab-plane. Other weak interactions from C5–H5⋯O5 and C3–H3⋯N2 generate fourteen membered ring motifs R32(14) Fig. 2a and Table 2.
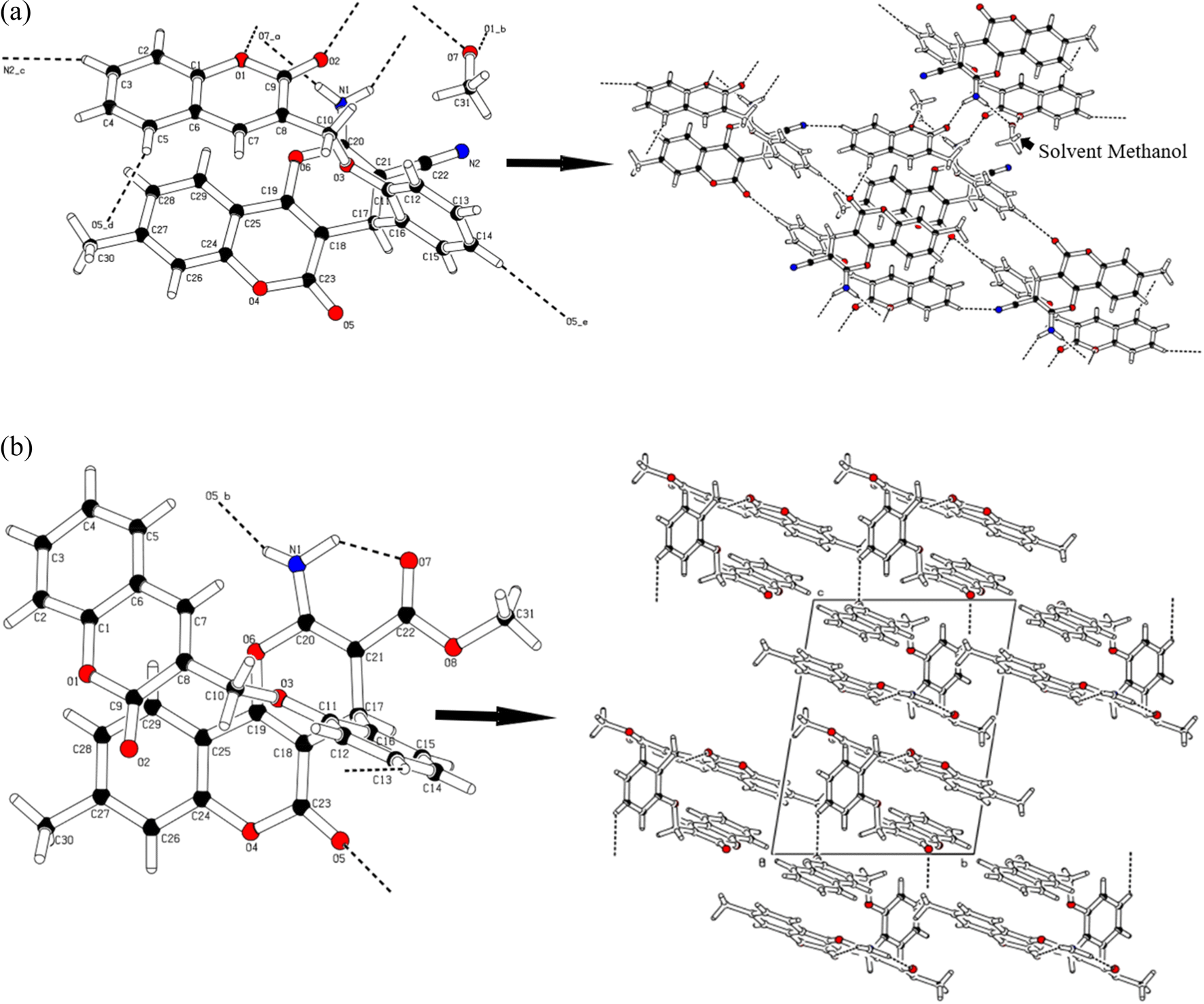 | ||
| Fig. 2 (a) A diagram showing the hydrogen bonding interactions. Formation of dimer in through a centrosymmetric network of reciprocal strong N–H⋯O and bridging via O–H⋯O hydrogen bonds of methanol. (b) A diagram showing the hydrogen bonding interactions. Chain formation running along a-axis. | ||
| D | H | A | d(D–H)/Å | d(H–A)/Å | d(D–A)/Å | D–H–A/° |
|---|---|---|---|---|---|---|
| a 1 − X, 1 − Y, −Z.b 1 − X, 2 − Y, 1 − Z.c −1 + X, −1 + Y, +Z.d −X, 1 − Y, 1 − Z.e 1 + X, +Y, +Z.f −X, 1 − Y, 2 − Z. | ||||||
| 4a | ||||||
| N1 | H1A | O7 | 0.86 | 2.17 | 2.973(4) | 154.6 |
| O7 | H7A | O1 | 0.82 | 2.40 | 2.997(4) | 130.7 |
| N1 | H1B | O2a | 0.86 | 2.20 | 2.874(3) | 135.4 |
| C14 | H14 | O5b | 0.93 | 2.55 | 3.470(4) | 168.8 |
| C3 | H3 | N2c | 0.93 | 2.61 | 3.483(4) | 156.3 |
| C5 | H5 | O5d | 0.93 | 2.60 | 3.433(4) | 150.0 |
|
||||||
| 4b | ||||||
| N1 | H1A | O7 | 1.05(4) | 1.84(4) | 2.674(4) | 133(3) |
| N1 | H1B | O5e | 0.84(3) | 2.32(3) | 3.114(4) | 158(3) |
| C13 | H13 | O2f | 0.93 | 2.55 | 3.221(4) | 129.6 |
Although the 4b has no solvent of crystallization but it is affording three different type of hydrogen bonding interactions. There is one classical hydrogen bond between N1–H1b⋯O5 at a distance of 2.321(3) Å holding the successive molecules together in Fig. 2b. In addition, there is a nonclassical weak interaction from C13–H13⋯O2 interaction that help in the expansion of the molecular assembly along a-axis, base vector (1 0 0). There is an intramolecular hydrogen bonding interaction via N1–H1⋯O7 (Table 2) forming the stable six membered (H1A/N1/C20/C21/C22/O7) ring motif S(6) with root mean square deviation of 0.0110(3) Å and this ring is fused with another ring (O6/C20/C21/C17/C18/C19). The presence of more hydrogen bonds in 4a than in 4b lends higher thermal stability to the former, which is evident from the relatively higher m.p. of 4a (268 °C) than in 4b (230 °C). The root mean square deviation for the ring (O6/C20/C21/C17/C18/C19) is 0.0588(16) Å and puckering parameters are Q = 0.144(3) Å, θ = 72.0(12)° and φ = 176.9(11)° which are indicating that the ring is slightly deviating from planer to chair shape conformation. Both the rings (H1A/N1/C20/C21/C22/O7) and (O6/C20/C21/C17/C18/C19) are twisted by 4.232(4)°.
2.6 Hirshfeld surface and fingerprint plot analysis
Hirshfeld surface analysis allows us to quantify the various intermolecular interactions contributing to the packing of a whole molecule in a crystal.50,51 The Hirshfeld surface and fingerprint plot of 4a and 4b molecules were mapped with dnorm (Fig. 3 and 4) using Crystal Explorer.58 The white colour shows the contacts, which are close to Vander Waal's radii; the blue colour indicates the longer contacts; and the red colour represents the stronger interactions present in the crystal of HA. In Fig. 3, the surface map for 4a represents the dark red region in the vicinity of O1, O2, and N1, showing the presence of strong O7–H7A⋯O1, N1–H1b⋯O2, and N1–H1a⋯O7, intermolecular interactions in the crystal. The rest of the surface does not show the presence of any strong interaction and appears neutral. These colour indications are very useful for the identification and relative strength of possible intermolecular interactions. | ||
| Fig. 3 A Hirshfeld surface of the 4a showing the regions of strong interactions along with fingerprint plots showing the contributions of various interactions. | ||
 | ||
| Fig. 4 A Hirshfeld surface of the 4b showing the regions of strong interactions along with fingerprint plots showing the contributions of various interactions. | ||
Similarly, in Fig. 4, the surface map for 4b represents a single dark red region only in the vicinity of N1–H1b, while the rest of the surface appears neutral. These colour indications are very useful for the identification and relative strength of possible intermolecular interactions.
Fingerprint plots analyse the intermolecular interactions and molecular packing in a crystal on the basis of the Hirshfeld surface.56–58 The fingerprint plots of 4a and 4b molecules are shown in Fig. 3 and 4. In both molecules, the H⋯H interactions predominate on the surface, with 39% and 40.2% contributions, respectively. This is followed by the contributions of O⋯H contacts, which account for 28.1% and 21.3%, respectively. Contributions by C⋯H interactions are next in the hierarchy, with values of 20.8% and 25.3%, respectively. The N⋯H contribution in 4a is 11.6%, while in 4b this contribution is very negligible (0.3%). The Hirshfeld surface map enables us to understand the intermolecular interactions and the strength of above-mentioned strong and weak interactions that contribute to the surface of the molecules.
2.7 Computational procedure
For the computational analysis of synthesized crystalline compounds 4a and 4b, the Gaussian 09 program package52 was used. Primarily, the geometries of compounds were attained through SC-XRD by utilizing the crystallographic information file (CIF). Subsequently, DFT calculations for the entitled compounds were accomplished at M06-2X/6-31G(d,p) level of theory. Furthermore, NLO and FMOs analyses were performed for 4a and 4b at the aforementioned level of DFT, as well as NLO responses were calculated by using eqn (1)–(4).53–55 However, natural bonding orbitals (NBOs) were investigated using the M06-2X/6-31G(d,p) level of theory,56 because non-covalent interactions can be effectively studied at this level of theory. For interpretation of results from output files, Gauss View,57 Avogadro,58 Chemcraft,59 PyMOlyze 2.0,60 Origin 8.0,61 and Multiwfn 3.7 (ref. 62) were employed.| μ = (μx2 + μy2 + μz2)1/2 | (1) |
| 〈α〉 = (axx + ayy + azz)/3 | (2) |
| βtot = (βx2 + βy2 + βz2)1/2 | (3) |
 | (4) |

2.8 Result and discussions
In this research paper, the non-fullerene natured synthesized crystals, i.e., 4a and 4b are evaluated in terms of their NLO characteristics. For this purpose, SC-XRD characterization is done and inspection of molecular configuration is carried out thoroughly via different parameters like bond lengths, bond angles etc. The experimental data are compared with the theoretical results (DFT) in order to verify the chemical nature of the analyzed compounds. Moreover, the structural and optimized representation of entitled compounds is shown in Fig. 5. | ||
| Fig. 5 The optimized structures of 4a and 4b. | ||
2.9 Geometrical parameters
The geometrical parameters of entitled compounds were optimized at the M06-2X/6-31G(d,p) level of theory, and their data is organized in ESI (Tables S1 and S2†). The results of SC-XRD and DFT are found in excellent agreement with a few variations.The simulated C–C bond lengths in the benzene ring of compound 4a are found to be in the range of 1.162–1.507 Å, while in the range of 1.149–1.523 Å, as determined by XRD as shown in Table S1.† The bond lengths between carbon and oxygen atoms calculated through DFT are found to be 1.355, 1.366, 1.368, 1.364, 1.369, 1.415, 1.36, 1.385, 1.202, and 1.211 Å for O1–C10, O1–C15, O3–C20, O3–C22, O4–C9, O4–C31, O5–C17, O5–C18, O6–C18, and O7–C20, respectively, while through XRD the values were 1.366, 1.368, 1.373,1.375, 1.372, 1.431, 1.381, 1.387, 1.202, and 1.204 Å, respectively. Furthermore, the bond length for N2–C15 obtained by XRD is 1.342 Å, while the DFT calculated value for the above-mentioned bond length is examined to be 1.344 Å as given in Table S1.†
Similarly, for compound 4b, the aforesaid bond lengths are observed in the range of 1.345–1.526 Å, which have good agreement with experimental data as 1.336–1.534 Å (see Table S2†). Moreover, for O1–C10, O1–C32, O2–C15, O2–C16, O3–C19, O3–C27, O4–C22, O4–C24, O5–C26, O5–C40, O6–C24, O7–C19, and O8–C26 bond lengths in compound 4b, DFT values are noted to be 1.381, 1.418, 1.361, 1.364, 1.374, 1.362, 1.361, 1.38, 1.345, 1.425, 1.202, 1.205 and 1.224 Å, which are correspondingly matched with SC-XRD data at 1.381, 1.421, 1.364, 1.377, 1.376, 1.378, 1.376, 1.387, 1.338, 1.436, 1.215, 1.202, and 1.222 Å, respectively. For N9–C16 the bond length by both XRD and DFT calculation is found to be 1.355 Å and 1.336 Å, respectively.
In 4a, bond angle values in C10–O1–C15, O1–C15–N2, N2–C15–C14, C20–O3–C22, O3–C20–C26, C9–O4–C31, O4–C9–C8, O4–C31–C26, C17–O5–C18, O5–C17–C27, O5–C18–C11, and O7–C20–C26 were observed through DFT as 118.3°, 110.4°, 117.6°, 122.8°,117.6°, 117.2°, 116°, 108.8°, 122.6°, 117.6°, 117.2°, and 124.8°, respectively. Similarly, through XRD, prominent values are 118.5°, 109.8°, 128°, 122.6°,117.8°, 118.3°, 115°, 106.2°, 121.9°, 117.7°, 118.1°, and 125.9°, respectively, as shown in Table S1.†
Similarly, in 4b, the calculated bond angles between C10–O1–C32, O1–C10–C30, O1–C10–C35, O1–C32–C18, C15–O2–C16, O2–C15–C11, O2–C16–N9, O2–C16–C13, C19–O3–C27, O3–C19–O7, O3–C19–C18, O4–C22–C14, C26–O5–C40, O5–C26–O8, and N9–C16–C13 are observed through DFT as 114.7°, 120°, 118.9°, 107.9°,118.8°, 123.2°, 109.6°, 123°, 122.3°, 118.4°, 117°,114.7°, 121.6°, 115.1°, 121.6°, and 127.5 as well as 118.6°, 123.3°, 115.6°, 107.1°, 118.7°, 123.6°, 109.4°, 122.1°, 122.1°, 116.5°, 117.7°,114.7°, 121.2°, 117.6°, 121.7°, and 128.5° through XRD, respectively as shown in Table S2.† The torsion angles experimentally determined in the solid state are shown in Table S3.† The above-discussion explored that a good agreement is noted in computed and experimental findings, which confirms that M06-2X/6-31G(d,p) is suitable level of theory for DFT study. The geometrical structures are shown in Fig. S7.†
2.10 Frontier molecular orbitals (FMOs) analysis
The electronic characteristics of the studied molecules are investigated using the energies of HOMO/LUMO and their frontier molecular orbitals (FMOs) diagrams to calculate their chemical reactivity and stability.63 The Lewis donor is commonly known HOMO (highest occupied molecular orbital) and the Lewis acceptor is called as LUMO (lowest unoccupied molecular orbital).64,65 The electrons are transferred from the donor to the acceptor (HOMO to LUMO), which provides information about the intramolecular charge transfer (ICT) and NLO behavior.66 An inverse relationship exists between the ΔE and ICT of a compound. Molecules with a greater ability to transfer charge between orbitals exhibit significant NLO properties with a lower ΔE value, and vice versa.67 Herein, we calculated the orbital energies and band gaps of our entitled compounds as shown in Table 3.| MOs | 4a | 4b | ||
|---|---|---|---|---|
| E (eV) | ΔE (eV) | E (eV) | ΔE (eV) | |
| LUMO | −1.540 | 5.168 | −0.932 | 6.308 |
| HOMO | −6.708 | −7.240 | ||
| LUMO+1 | −0.700 | 6.669 | −0.693 | 6.767 |
| HOMO−1 | −7.369 | −7.460 | ||
| LUMO+2 | −0.055 | 7.445 | −0.436 | 7.330 |
| HOMO−2 | −7.500 | −7.766 | ||
The data of energies obtained for 4a are found to be −6.708 eV for HOMO and −1.540 eV for LUMO, while those for 4b are noted to be −7.240 eV and −0.932 eV, respectively. The ΔE of 4a is examined to be 5.168 eV, which is abridged to 6.308 eV in 4b. Interestingly, a significant reduction in orbital energies and Egap is observed for 4a as compared to 4b. This reduction of energies in 4a is due to the presence of the nitro (–NO2) group, as it is known that the nitro group has a stronger electron-withdrawing effect and inductive effect than the –CN group.64 In addition to the energies of orbitals, ICT has also been studied in the entitled compounds, as shown in Fig. S9.† The electronic charge density for HOMO is concentrated over the entire molecule in both compounds, while for LUMO the electronic cloud is distributed significantly all over the molecular region except the nitro and cyano groups in 4a and 4b, respectively.
2.11 Global reactivity descriptors
The stability and reactivity of 4a and 4b molecules can be excellently elaborated with various global reactivity parameters (GRPs) calculated by utilizing the LUMO–HOMO energy gap.68 The results of these parameters, including electron affinity (EA), ionization potential (I),69 global hardness (η),70 global electrophilicity (ω),71 electronegativity (X),72 chemical potential (μ)73 and global softness (σ), are estimated with the help of eqn (S5)–(S11)† and their outcomes are tabulated in Table S4.†Ionization potential (IP) is the tendency to donate electrons, and electronegativity represents the ability to attract electrons.68 Table 3 reveals that compound 4a showed a higher value of electron affinity, electronegativity, and electrophilicity than compound 4b. The reason for this enhanced value for 4a might be the presence of highly electron-withdrawing nitro groups in the 4a molecule. The parameters like chemical potential (μ) and hardness (η) are directly correlated with the energy gap. The compounds with the higher values of these parameters are considered as kinetically more stable and less reactive.74 In our synthesized compounds, 4a had a lower global hardness (η = 0.094 eV) and chemical potential (μ = −0.151 eV) with a higher softness (σ = 5.266 eV) than 4b, indicating that 4a is more kinetically stable and less reactive than 4b. This GRPs investigation indicated that our prepared compounds are kinetically more stable and show excellent concurrence with NBO and SC-XRD data (Table 4).
| GRPs | 4a | 4b |
|---|---|---|
| a Units are in eV. | ||
| IP | 0.246 | 0.266 |
| EA | 0.056 | 0.034 |
| X | 0.151 | 0.150 |
| η | 0.094 | 0.116 |
| μ | −0.151 | −0.150 |
| ω | 0.121 | 0.097 |
| σ | 5.266 | 4.314 |
2.12 Natural bond orbitals (NBOs) analysis
The NBOs are important for the investigation of bond interactions, the distribution of charges, and their delocalization among filled and vacant orbitals in a compound.73 Moreover, the NBO analysis also facilitates the study of charge densities among various parts of a donor–π–acceptor molecule. It has been explored from the literature that most commonly the donor moiety holds a positive charge, while the acceptors are found with negatively charged values. Whereas, the π-spacer may have positive or negative charges due to its role as a facilitator in the studied donor–π–acceptor framework. Assuming the above facts, the second-order perturbation theory of the said molecules is accomplished, and the stabilization energy for these molecules is calculated using eqn (5), which is as follows:
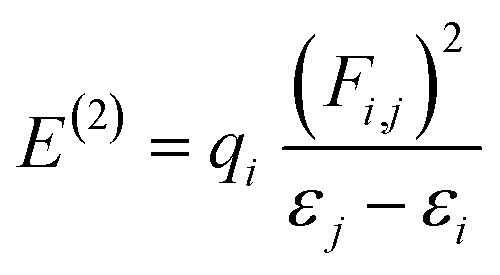 | (5) |
| Comp. | Donor (i) | Type | Acceptor (j) | Type | E(2)a [kcal mol−1] | E(j) − E(i)b (a.u.) | F(i, j)c (a.u.) |
|---|---|---|---|---|---|---|---|
| a E(2) represents the energy of hyper conjugative interaction (stabilization energy in kcal mol−1).b Energy difference between D and A NBO orbitals.c F(i, j) is the Fock matrix element between i and j NBO orbitals. | |||||||
| 4a | C27–C33 | π | C12–C17 | π* | 34.85 | 0.34 | 0.100 |
| C8–C24 | π | C14–C15 | π* | 0.56 | 0.34 | 0.013 | |
| C11–C18 | σ | O1–C10 | σ* | 6.80 | 1.20 | 0.081 | |
| C26–C31 | σ | C31–H54 | σ* | 0.51 | 1.23 | 0.022 | |
| N2 | LP(1) | C14–C15 | π* | 77.62 | 0.36 | 0.153 | |
| O4 | LP(1) | C31–H55 | σ* | 0.66 | 1.15 | 0.025 | |
| 4b | C16–C19 | π | O8–C33 | π* | 34.95 | 0.37 | 0.106 |
| O7–C23 | π | O7–C23 | π* | 0.78 | 0.51 | 0.019 | |
| C13–C30 | σ | O2–C18 | σ* | 6.82 | 1.19 | 0.080 | |
| C22–C28 | σ | C42–H43 | σ* | 0.51 | 1.34 | 0.023 | |
| O5 | LP(2) | O8–C33 | π* | 59.26 | 0.43 | 0.148 | |
| O8 | LP(1) | O5–C60 | σ* | 0.50 | 1.14 | 0.021 | |
Four different forms of electronic transitions are frequently observed: π → π*, σ → σ*, LP → π*, and LP → σ*. The most dominant electronic transitions are found as π → π* among the above-mentioned transitions, while σ → σ* is examined as the least prominent, and LP → π* and LP → σ* are noticed as slightly dominant transitions.
In the case of compound 4a, the most prominent π → π* transitions are π(C27–C33) → π*(C12–C17) and π(C8–C24) → π*(C14–C15), which showed the highest and the least energies of stabilization as 34.85 and 0.56 kcal mol−1, respectively. The weaker σ → σ* transitions are also examined with some prominent values as 6.80 and 0.51 kcal mol−1 observed in transitions σ(C11–C18) → σ*(O1–C10) and σ(C26–C31) → σ*(C31–H54), respectively. Some resonance transitions that are also important for discussion include LP1(N2) → π*(C14–C15), which determined the highest stabilization energy of 77.62 kcal mol−1, and LP1(O8) → σ*(C31–H55), which showed the lowest stabilization energy of 0.66 kcal mol−1 (see Table 5).
In compound 4b, the highest stabilization energy obtained is 34.95 kcal mol−1, which is exhibited during π(C16–C19) → π*(O8–C33) transitions, while, π(O7–C23) → π*(O7–C23) is noted with the smallest stabilization energy value as 0.78 kcal mol−1. The other feeble electronic transitions that are significant to discuss for compound 4a are: σ(C13–C30) → σ*(O2–C18), σ(C22–C28) → σ*(C42–H43), LP2(O5) → π*(O8–C33), and LP1(O8) → σ*(O5–C60), which showed energies of 6.82, 0.51, 59.26, and 0.50 kcal mol−1, respectively.
Overall, the results obtained from the above discussion validate the formation of charge separation states in the investigated molecules. Whereas, the stabilization energies also revealed comparable stability in both compounds, for which the ICT, conjugation, and hyper conjugative interactions might be the main reasons for their stabilities.
2.13 UV-vis analysis
The UV-Vis absorption spectra of the coumarin-based pyrano-chromene derivatives 4a and 4b in chloroform, dimethyl sulfoxide (DMSO), and 1,4-dioxane were recorded at room temperature. TDDFT UV-Vis absorption data were calculated in the gas phase. This study provides a reasonable apprehension of optoelectronic properties, probability of charge transfer, and molecular orbital configurations of electronic transitions for the studied compounds.75–77 The TD-DFT methodology was employed using the above-mentioned level of theory to execute the UV-Vis absorption spectra. Table 6 presents the data obtained for 4a and 4b, including the maximum wavelength (λmax), vertical excitation energies (eV), oscillator strength (fos), and orbital transitions. The detailed results are recorded in Table S7,† and graphs of absorption maxima for both compounds are displayed in Fig. S8.†The data obtained from the analysis show the λmax of 4a and 4b are 268.579 and 269.513 nm, respectively. Interestingly, the experimentally calculated values (in solvents i.e., chloroform, 1,4-dioxane, and DMSO) of λmax are found bathochromically shifted as compared to the theoretical values (in the gas phase) of compounds (4a and 4b), which may occur due to change in environmental conditions as expressed in Table 6. 4a displayed a value of the absorbance at 268.579 nm with corresponding oscillation strength of 0.337 and 4.616 eV transition energy, which are associated with H−1 → L (68%) as a major contribution. Similarly, λmax calculated in the case of 4b was 269.513 nm with two major transitions as H → L+1 (36%), and H−1 → L+2 (30%), possessing oscillation strengths of 0.374 and 4.600 eV transition energy.
2.14 Non-linear optical (NLO) properties
Non-linear optical materials have immense potential in photonics with striking applications in the optoelectronics and telecommunication sectors.74,75 The NLO response varies with the variation of the position of diverse substituents on the compounds. Here, we compute the statistical average of molecular dipoles moment (u), linear and non-linear responses of 4a and 4b at the aforementioned level of theory, and the major results are tabulated in Table 7, whereas the detailed calculations are present in the ESI (Tables S8 and S9†).| Comp. | μtot | 〈α〉 ×10−23 | γtot × 104 |
|---|---|---|---|
| 4a | 6.236 | 6.770 | 0.145 |
| 4b | 3.256 | 6.699 | 0.145 |
Table S8† holds the linear polarizability with major contributing tensors along the x, y, and z-axes. The polarizability tensor along the x-direction (αxx) shows the highest value and thus has more contribution towards linear polarizability in both 4a and 4b compounds. The total values of statistical average of molecular dipoles moment (μtot), average polarizability 〈α〉, and second order hyperpolarizability (γtot) are also shown in Table 7. The μtot is found to be 6.236 and 3.256 D, for compounds 4a and 4b, respectively. The highest computed value of statistical average of molecular dipoles moment exists for compound 4a as 6.236 D, while the smallest amplitude of 3.256 D is examined for compound 4b. All the entitled molecules showed significant statistical average of molecular dipoles moment values when compared with the standard urea molecule, which showed a value of 1.3732 D (ref. 78) (Table S8†). The average linear polarizability 〈α〉 values for compounds 4a and 4b are observed to be 6.770 × 10−23 and 6.699 × 10−23 esu, respectively. Hence, the greater 〈α〉 value is noticed in 4a as compared to 4b.
The second-order polarizability (γtot) values are observed in both the studied compounds as 0.145 × 104 esu. It is also observed that the highest contributing value of γtot for 4a is shown along the z-axis (γzz = 2.324 × 104 esu) and for 4b along the x-axis (8.907 × 104 esu), which indicates the higher polarization along the z and x-directions for titled compounds (Table S8†). Interestingly, in our studied crystals, remarkable NLO results were obtained as their various parameters such as μtot, 〈α〉, and γtot when compared with urea molecule.79,80 This comparative analysis of 4a and 4b with urea found to be many folds higher than urea. This urea-related examination implied that all considered molecules were appropriate to be NLO entities. This showed their effective structural tailoring using efficient donor and acceptor moieties to obtain significant NLO materials. The orientation of the 4a and 4b molecule along x, y and z axis are shown in Fig. S10 and S11.†
3. Conclusion
The current work indicates the quantum chemical investigation of two novel coumarin-based pyrano-chromene crystal chromophores, 4a and 4b. The compounds were synthesized in a good yield through the Knoevenagel–Michael-cyclization path using a one-pot multicomponent reaction. The structures of these o-ring heterocyclic compounds were ascertained by spectroscopic techniques like UV-Vis, FT-IR, 1H & 13C NMR, and single crystal X-ray diffraction analysis. The SC-XRD analysis of 4a and 4b revealed the triclinic crystal lattice with space group P. The spectroscopic results were found to be in good agreement with the SC-XRD data. The DFT and SC-XRD data were also perceived in good accordance, as shown by their computed bond lengths and bond angles. The NBO probe also confirmed the presence of charge separation, which showed that the entitled compounds are involved in the process of ICT, hence, endorsing their higher molecular stability. The higher energy band gap of 4b (6.308 eV) as compared to 4a (5.168 eV) was obtained due to the presence of highly electronegative cyano group in 4b. The GRPs were derived utilizing energies of HOMOs and LUMOs of said compounds. Fantastically, large 〈α〉 and γtot responses are computed for 4a than that of standard urea molecule. We infer that the present DFT-based study might be regarded as a remarkable achievement and may have prospective applications in developing new NLO materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank King Khalid University's Deanship of Scientific Research for funding this study under grant number (R.G.P.1/28/43). Dr Muhammad Khalid gratefully acknowledges the financial support of HEC Pakistan (project no. 20-14703/NRPU/R&D/HEC/2021). Authors are also thankful for cooperation and collaboration of A. A. C. B. from IQ-USP, Brazil especially for his continuous support and providing computational lab facilities.References
- J. D. Sunderhaus and S. F. Martin, Applications of multicomponent reactions to the synthesis of diverse heterocyclic scaffolds, Chem.–Eur. J., 2009, 15, 1300–1308 CrossRef CAS PubMed.
- K. C. Nicolaou, J. A. Pfefferkorn, H. J. Mitchell, A. J. Roecker, S. Barluenga, G.-Q. Cao, R. L. Affleck and J. E. Lillig, Natural product-like combinatorial libraries based on privileged structures. 2. Construction of a 10000-membered benzopyran library by directed split-and-pool chemistry using NanoKans and optical encoding, J. Am. Chem. Soc., 2000, 122, 9954–9967 CrossRef CAS.
- K. C. Nicolaou, J. A. Pfefferkorn, S. Barluenga, H. J. Mitchell, A. J. Roecker and G.-Q. Cao, Natural product-like combinatorial libraries based on privileged structures. 3. The “libraries from libraries” principle for diversity enhancement of benzopyran libraries, J. Am. Chem. Soc., 2000, 122, 9968–9976 CrossRef CAS.
- G. M. Cragg and D. J. Newman, Natural products: a continuing source of novel drug leads, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- A. Jashari, F. Imeri, L. Ballazhi, A. Shabani, B. Mikhova, G. Dräger, E. Popovski and A. Huwiler, Synthesis and cellular characterization of novel isoxazolo-and thiazolohydrazinylidene-chroman-2,4-diones on cancer and non-cancer cell growth and death, Bioorg. Med. Chem., 2014, 22, 2655–2661 CrossRef CAS PubMed.
- X.-M. Peng, G. L. V. Damu and H. Zhou, Current developments of coumarin compounds in medicinal chemistry, Curr. Pharm. Des., 2013, 19, 3884–3930 CrossRef CAS PubMed.
- L. Ma, Y. Xiao, C. Li, Z.-L. Xie, D.-D. Li, Y.-T. Wang, H.-T. Ma, H.-L. Zhu, M.-H. Wang and Y.-H. Ye, Synthesis and antioxidant activity of novel Mannich base of 1,3,4-oxadiazole derivatives possessing 1,4-benzodioxan, Bioorg. Med. Chem., 2013, 21, 6763–6770 CrossRef CAS PubMed.
- M. A. Musa, V. L. Badisa, L. M. Latinwo, J. Cooperwood, A. Sinclair and A. Abdullah, Cytotoxic activity of new acetoxycoumarin derivatives in cancer cell lines, Anticancer Res., 2011, 31, 2017–2022 CAS.
- P. Borah, P. S. Naidu and P. J. Bhuyan, Synthesis of some tetrazole fused pyrido[2,3-c]coumarin derivatives from a one-pot three-component reaction via intramolecular 1,3-dipolar cycloaddition reaction of azide to nitriles, Tetrahedron Lett., 2012, 53, 5034–5037 CrossRef CAS.
- M. Ninomiya, K. Shirabe, H. Kayashima, T. Ikegami, A. Nishie, N. Harimoto, Y. Yamashita, T. Yoshizumi, H. Uchiyama and Y. Maehara, Functional assessment of the liver with gadolinium–ethoxybenzyl-diethylenetriamine penta-acetate-enhanced MRI in living-donor liver transplantation, Br. J. Surg., 2015, 102, 944–951 CrossRef CAS PubMed.
- G. J. Keating, J. G. Quinn and R. O'Kennedy, Immunoassay for the determination of 7-hydroxycoumarin in serum using ‘real-time’ biosensor analysis, 1999 Search PubMed.
- J. Neyts, E. D. Clercq, R. Singha, Y. H. Chang, A. R. Das, S. K. Chakraborty, S. C. Hong, S.-C. Tsay, M.-H. Hsu and J. R. Hwu, Structure-activity relationship of new anti-Hepatitis C virus agents: heterobicycle-coumarin conjugates, J. Med. Chem., 2009, 52, 1486–1490 CrossRef CAS PubMed.
- U. Salar, K. M. Khan, A. Jabeen, S. Hussain, A. Faheem, F. Naqvi and S. Perveen, Diversified Thiazole Substituted Coumarins and Chromones as Non-Cytotoxic ROS and NO Inhibitors, Lett. Drug Des. Discovery, 2020, 17, 547–555 CrossRef CAS.
- K. V. Sashidhara, A. Kumar, M. Kumar, J. Sarkar and S. Sinha, Synthesis and in vitro evaluation of novel coumarin–chalcone hybrids as potential anticancer agents, Bioorg. Med. Chem. Lett., 2010, 20, 7205–7211 CrossRef CAS PubMed.
- C. A. Kontogiorgis and D. J. Hadjipavlou-Litina, Synthesis and antiinflammatory activity of coumarin derivatives, J. Med. Chem., 2005, 48, 6400–6408 CrossRef CAS PubMed.
- R. S. Keri, B. S. Sasidhar, B. M. Nagaraja and M. A. Santos, Recent progress in the drug development of coumarin derivatives as potent antituberculosis agents, Eur. J. Med. Chem., 2015, 100, 257–269 CrossRef CAS PubMed.
- G. Borges Bubols, D. da Rocha Vianna, A. Medina-Remon, G. von Poser, R. M. Lamuela-Raventos, V. Lucia Eifler-Lima and S. Cristina Garcia, The antioxidant activity of coumarins and flavonoids, Mini-Rev. Med. Chem., 2013, 13, 318–334 Search PubMed.
- T. Matsumoto, K. Takahashi, S. Kanayama, Y. Nakano, H. Imai, M. Kibi, D. Imahori, T. Hasei and T. Watanabe, Structures of antimutagenic constituents in the peels of Citrus limon, J. Nat. Med., 2017, 71, 735–744 CrossRef CAS PubMed.
- O. M. Abdelhafez, K. M. Amin, R. Z. Batran, T. J. Maher, S. A. Nada and S. Sethumadhavan, Synthesis, anticoagulant and PIVKA-II induced by new 4-hydroxycoumarin derivatives, Bioorg. Med. Chem., 2010, 18, 3371–3378 CrossRef CAS PubMed.
- Y. K. Al-Majedy, A. A. Al-Amiery, A. A. H. Kadhum and A. B. Mohamad, Antioxidant activities of 4-methylumbelliferone derivatives, PLoS One, 2016, 11, e0156625 CrossRef PubMed.
- A. Nargotra, S. Sharma, M. I. Alam, Z. Ahmed, A. Bhagat, S. C. Taneja, G. N. Qazi and S. Koul, In silico identification of viper phospholipaseA2 inhibitors: validation by in vitro, in vivo studies, J. Mol. Model., 2011, 17, 3063–3073 CrossRef CAS PubMed.
- O. S. Kwon, J. S. Choi, M. Islam, Y. S. Kim and H. P. Kim, Inhibition of 5-lipoxygenase and skin inflammation by the aerial parts of Artemisia capillaris and its constituents, Arch. Pharmacal Res., 2011, 34, 1561–1569 CrossRef CAS PubMed.
- T. Thomas, Monoamine oxidase-B inhibitors in the treatment of Alzheimers disease, Neurobiol. Aging, 2000, 21, 343–348 CrossRef CAS PubMed.
- P. O. Patil, S. B. Bari, S. D. Firke, P. K. Deshmukh, S. T. Donda and D. A. Patil, Bioorg. Med. Chem., 2013, 21, 2434–2450 CrossRef CAS PubMed.
- I. Kostova, Synthetic and natural coumarins as cytotoxic agents, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 29–46 CrossRef CAS PubMed.
- K. Biradha, Crystal engineering: from weak hydrogen bonds to co-ordination bonds, CrystEngComm, 2003, 5, 374–384 RSC.
- R. Jawaria, M. Hussain, Z. Shafiq, H. B. Ahmad, M. N. Tahir, H. A. Shad and M. M. Naseer, Robustness of thioamide dimer synthon, carbon bonding and thioamide–thioamide stacking in ferrocene-based thiosemicarbazones, CrystEngComm, 2015, 17(12), 2553–2561 RSC.
- S. K. Seth, D. Sarkar and T. Kar, Use of π–π forces to steer the assembly of chromone derivatives into hydrogen bonded supramolecular layers: crystal structures and Hirshfeld surface analyses, CrystEngComm, 2011, 13, 4528–4535 RSC.
- M. Małecka, L. Chęcińska, A. Rybarczyk-Pirek, W. Morgenroth and C. Paulmann, Electron density studies on hydrogen bonding in two chromone derivatives, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 687–695 CrossRef PubMed.
- A. Poursattar Marjani, B. Ebrahimi Saatluo and F. Nouri, An efficient synthesis of 4H-chromene derivatives by a one-pot, three-component reaction, Iran, Iran. J. Chem. Chem. Eng., 2018, 37, 149–157 CAS.
- M. R. Khumalo, S. N. Maddila, S. Maddila and S. B. Jonnalagadda, A facile and one-pot synthesis of new tetrahydrobenzo[b]pyrans in water under microwave irradiation, BMC Chem., 2019, 13, 1–7 CrossRef PubMed.
- D. Kumar, V. B. Reddy, S. Sharad, U. Dube and S. Kapur, A facile one-pot green synthesis and antibacterial activity of 2-amino-4H-pyrans and 2-amino-5-oxo-5,6,7,8-tetrahydro-4H-chromenes, Eur. J. Med. Chem., 2009, 44, 3805–3809 CrossRef CAS PubMed.
- A. M. El-Saghier, M. B. Naili, B. K. Rammash, N. A. Saleh and K. M. Kreddan, Synthesis and antibacterial activity of some new fused chromenes, Arkivoc, 2007, 16, 83–91 Search PubMed.
- Ž. B. Milanović, Z. S. Marković, D. S. Dimić, O. R. Klisurić, I. D. Radojević, D. S. Šeklić, M. N. Živanović, J. D. Marković, M. Radulović and E. H. Avdović, Synthesis, structural characterization, biological activity and molecular docking study of 4,7-dihydroxycoumarin modified by aminophenol derivatives, C. R. Chim., 2021, 24, 215–232 CrossRef.
- P. M. Ronad, M. N. Noolvi, S. Sapkal, S. Dharbhamulla and V. S. Maddi, Synthesis and antimicrobial activity of 7-(2-substituted phenylthiazolidinyl)-benzopyran-2-one derivatives, Eur. J. Med. Chem., 2010, 45, 85–89 CrossRef CAS PubMed.
- R. Devakaram, D. S. Black, K. T. Andrews, G. M. Fisher, R. A. Davis and N. Kumar, Synthesis and antimalarial evaluation of novel benzopyrano[4,3-b]benzopyran derivatives, Bioorg. Med. Chem., 2011, 19, 5199–5206 CrossRef CAS PubMed.
- S. M. Hasan, M. M. Alam, A. Husain, S. Khanna, M. Akhtar and M. S. Zaman, Synthesis of 6-aminomethyl derivatives of benzopyran-4-one with dual biological properties: anti-inflammatory-analgesic and antimicrobial, Eur. J. Med. Chem., 2009, 44, 4896–4903 CrossRef CAS PubMed.
- A. E.-F. G. Hammam, Novel fluoro substituted benzo[b]pyran with anti-lung cancer activity, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2005, 44B, 1887–1893 Search PubMed.
- I. P. Singh and H. S. Bodiwala, Recent advances in anti-HIV natural products, Nat. Prod. Rep., 2010, 27, 1781–1800 RSC.
- M. Nawaz, M. W. Abbasi and S. Hisaindee, Synthesis, characterization, anti-bacterial, anti-fungal and nematicidal activities of 2-amino-3-cyanochromenes, J. Photochem. Photobiol., B, 2016, 164, 160–163 CrossRef CAS PubMed.
- I. Fatima, R. Saxena, G. Kharkwal, M. K. Hussain, N. Yadav, K. Hajela, P. L. Sankhwar and A. Dwivedi, The anti-proliferative effect of 2-[piperidinoethoxyphenyl]-3-[4-hydroxyphenyl]-2H-benzo(b)pyran is potentiated via induction of estrogen receptor beta and p21 in human endometrial adenocarcinoma cells, J. Steroid Biochem. Mol. Biol., 2013, 138, 123–131 CrossRef CAS PubMed.
- C. Corminboeuf, F. Tran and J. Weber, The role of density functional theory in chemistry: some historical landmarks and applications to zeolites, J. Mol. Struct.: THEOCHEM, 2006, 762, 1–7 CrossRef CAS.
- M. Ashfaq, A. Ali, A. Kuznetsov, M. N. Tahir and M. Khalid, DFT and single-crystal investigation of the pyrimethamine-based novel co-crystal salt: 2,4-diamino-5-(4-chlorophenyl)-6-ethylpyrimidin-1-ium-4-methylbenzoate hydrate (1:1:1) (DEMH), J. Mol. Struct., 2021, 1228, 129445, DOI:10.1016/j.molstruc.2020.129445.
- E. H. Avdović, D. S. Dimić, M. Fronc, J. Kožišek, E. Klein, Ž. B. Milanović, A. Kesić and Z. S. Marković, Structural and theoretical analysis, molecular docking/dynamics investigation of 3-(1-m-chloridoethylidene)-chromane-2,4-dione: the role of chlorine atom, J. Mol. Struct., 2021, 1231, 129962 CrossRef.
- A. Ali, M. Khalid, M. F. U. Rehman, S. Haq, A. Ali, M. N. Tahir, M. Ashfaq, F. Rasool and A. A. C. Braga, Efficient synthesis, SC-XRD, and theoretical studies of O-benzenesulfonylated pyrimidines: role of noncovalent interaction influence in their supramolecular network, ACS Omega, 2020, 5, 15115–15128 CrossRef CAS PubMed.
- M. Akram, M. Adeel, M. Khalid, M. N. Tahir, M. U. Khan, M. A. Asghar, M. A. Ullah and M. Iqbal, A combined experimental and computational study of 3-bromo-5-(2,5-difluorophenyl)pyridine and 3,5-bis(naphthalen-1-yl)pyridine: insight into the synthesis, spectroscopic, single crystal XRD, electronic, nonlinear optical and biological properties, J. Mol. Struct., 2018, 1160, 129–141 CrossRef.
- P. T. Kaye, M. A. Musa, X. W. Nocanda and R. S. Robinson, Does the DABCO-catalysed reaction of 2-hydroxybenzaldehydes with methyl acrylate follow a Baylis–Hillman pathway?, Org. Biomol. Chem., 2003, 1, 1133–1138 RSC.
- A. Hameed, Z. Shafiq, M. Yaqub, M. Hussain, H. B. Ahmad, M. N. Tahir and M. M. Naseer, Robustness of a thioamide {⋯H–N–CS}2 synthon: synthesis and the effect of substituents on the formation of layered to cage-like supramolecular networks in coumarin–thiosemicarbazone hybrids, New J. Chem., 2015, 39, 6052–6061 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- D. Milenković, E. Avdović, D. Dimić, S. Sudha, D. Ramarajan, Ž. Milanović, S. Trifunović, Z. Marković and N. B. O. Vibrational, Hirshfeld surface analyses and molecular docking study of m-toluidine-coumarin derivative and its corresponding palladium(II) complex, J. Mol. Struct., 2020, 1209, 127935 CrossRef.
- Ž. B. Milanović, D. S. Dimić, E. H. Avdović, D. A. Milenković, J. D. Marković, O. R. Klisurić, S. R. Trifunović and Z. S. Marković, Synthesis and comprehensive spectroscopic (X-ray, NMR, FTIR, UV-Vis), quantum chemical and molecular docking investigation of 3-acetyl-4-hydroxy-2-oxo-2H-chromen-7-yl acetate, J. Mol. Struct., 2021, 1225, 129256 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. Alparone, Linear and nonlinear optical properties of nucleic acid bases, Chem. Phys., 2013, 410, 90–98 CrossRef CAS.
- A. Plaquet, M. Guillaume, B. Champagne, F. Castet, L. Ducasse, J.-L. Pozzo and V. Rodriguez, In silico optimization of merocyanine-spiropyran compounds as second-order nonlinear optical molecular switches, Phys. Chem. Chem. Phys., 2008, 10, 6223–6232 RSC.
- K. B. Lipkowitz and D. B. Boyd, Reviews in Computational Chemistry, John Wiley & Sons, 2009, vol. 12 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- R. D. Dennington, T. A. Keith and J. M. Millam, GaussView 5.0, Gaussian, Inc., Wallingford, 2008 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- G. A. Zhurko and D. A. Zhurko, ChemCraft, Version 1.6, 2009, https://www.chemcraftprog.com Search PubMed.
- N. M. O'boyle, A. L. Tenderholt and K. M. Langner, Cclib: a library for package-independent computational chemistry algorithms, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- L. A. Deschenes, D. A. Vanden Bout and University of Texas, Origin 6.0: Scientific Data Analysis and Graphing Software, Origin Lab Corporation (Formerly Microcal Software, Inc.), 2000, commercial price: 595, academic price: 446, https://www.originlab.com Search PubMed.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- M. N. Arshad, A.-A. M. Al-Dies, A. M. Asiri, M. Khalid, A. S. Birinji, K. A. Al-Amry and A. A. Braga, Synthesis, crystal structures, spectroscopic and nonlinear optical properties of chalcone derivatives: a combined experimental and theoretical study, J. Mol. Struct., 2017, 1141, 142–156 CrossRef CAS.
- M. S. Ahmad, M. Khalid, M. A. Shaheen, M. N. Tahir, M. U. Khan, A. A. C. Braga and H. A. Shad, Synthesis and XRD, FT-IR vibrational, UV-vis, and nonlinear optical exploration of novel tetra substituted imidazole derivatives: a synergistic experimental-computational analysis, J. Phys. Chem. Solids, 2018, 115, 265–276 CrossRef CAS.
- M. Shahid, M. Salim, M. Khalid, M. N. Tahir, M. U. Khan and A. A. C. Braga, Synthetic, XRD, non-covalent interactions and solvent dependent nonlinear optical studies of Sulfadiazine-Ortho-Vanillin Schiff base: (E)-4-((2-hydroxy-3-methoxy-benzylidene)amino)-N-(pyrimidin-2-yl)benzene-sulfonamide, J. Mol. Struct., 2018, 1161, 66–75 CrossRef CAS.
- K. Colladet, M. Nicolas, L. Goris, L. Lutsen and D. Vanderzande, Low-band gap polymers for photovoltaic applications, Thin Solid Films, 2004, 451, 7–11 CrossRef.
- A. Saeed, S. Muhammad, S. Rehman, S. Bibi, A. G. Al-Sehemi and M. Khalid, Exploring the impact of central core modifications among several push-pull configurations to enhance nonlinear optical response, J. Mol. Graphics Modell., 2020, 100, 107665 CrossRef CAS PubMed.
- M. Miar, A. Shiroudi, K. Pourshamsian, A. R. Oliaey and F. Hatamjafari, Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo[d]thiazole-2(3H)-imine and its para-substituted derivatives: solvent and substituent effects, J. Chem. Res., 2021, 45, 147–158 CrossRef CAS.
- C.-G. Zhan, J. A. Nichols and D. A. Dixon, Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: molecular properties from density functional theory orbital energies, J. Phys. Chem. A, 2003, 107, 4184–4195 CrossRef CAS.
- R. G. Parr and R. G. Pearson, Absolute hardness: companion parameter to absolute electronegativity, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- R. Parthasarathi, V. Subramanian, D. R. Roy and P. K. Chattaraj, Electrophilicity index as a possible descriptor of biological activity, Bioorg. Med. Chem., 2004, 12, 5533–5543 CrossRef CAS PubMed.
- R. G. Parr, R. A. Donnelly, M. Levy and W. E. Palke, Electronegativity: the density functional viewpoint, J. Chem. Phys., 1978, 68, 3801–3807 CrossRef CAS.
- P. Politzer and D. G. Truhlar, Chemical applications of atomic and molecular electrostatic potentials: reactivity, structure, scattering, and energetics of organic, inorganic, and biological systems, Springer Science & Business Media, 2013 Search PubMed.
- A. Ali, M. Khalid, K. P. Marrugo, G. M. Kamal, M. Saleem, M. U. Khan, O. Concepción and F. Alexander, Spectroscopic and DFT/TDDFT insights of the novel phosphonate imine compounds, J. Mol. Struct., 2020, 1207, 127838 CrossRef CAS.
- M. Khalid, M. U. Khan, R. Hussain, S. Irshad, B. Ali, A. A. C. Braga, M. Imran and A. Hussain, Exploration of second and third order nonlinear optical properties for theoretical framework of organic D–π–D–π–A type compounds, Opt. Quantum Electron., 2021, 53, 1–19 CrossRef.
- L. Dalton, M. Lauermann and C. Koos, NLO: Electro-Optic Applications, in The WSPC Reference on Organic Electronics: Organic Semiconductors. Volume 2: Fundamental Aspects of Materials and Applications, World Scientific, 2016, pp. 369–396 Search PubMed.
- C. Qin and A. E. Clark, DFT characterization of the optical and redox properties of natural pigments relevant to dye-sensitized solar cells, Chem. Phys. Lett., 2007, 438, 26–30 CrossRef CAS.
- P. N. Prasad and D. J. Williams, Introduction to nonlinear optical effects in molecules and polymers, Wiley, New York, 1991 Search PubMed.
- M. Khalid, R. Jawaria, M. U. Khan, A. A. C. Braga, Z. Shafiq, M. Imran, H. M. A. Zafar and A. Irfan, An efficient synthesis, spectroscopic characterization, and optical nonlinearity response of novel salicylaldehyde thiosemicarbazone derivatives, ACS Omega, 2021, 6, 16058–16065 CrossRef CAS PubMed.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Design and construction of molecular assemblies with large second-order optical nonlinearities. Quantum chemical aspects, Chem. Rev., 1994, 94, 195–242 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2115636–2115638. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra07134g |
| This journal is © The Royal Society of Chemistry 2023 |