
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-oxide ligands for selective separations of lanthanides: insights from computation†
Tongyu Liua,
Alexander S. Ivanov b,
Ilja Popovsb,
Santa Jansone-Popovab and
De-en Jiang*cd
b,
Ilja Popovsb,
Santa Jansone-Popovab and
De-en Jiang*cd
aDepartment of Chemistry, University of California, Riverside, CA 92521, USA
bChemical Sciences Division, Oak Ridge National Laboratory, 1 Bethel Valley Road, Oak Ridge, TN 37831, USA
cDepartment of Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, TN 37235, USA. E-mail: de-en.jiang@vanderbilt.edu
dDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235, USA
First published on 3rd January 2023
Abstract
Preorganized ligands such as bis-lactam-1,10-phenanthroline (BLPhen) show unique selectivity trends across the lanthanide series, indicating the synergistic effects of both N and O donors in complexing with lanthanides. We hypothesize that by replacing amide functional groups with an N-oxide functionality would open the door to new ligand architectures with improved selectivities. To test this idea, we computationally examined mixed N,O-donor ligands containing pyridinic N and N-oxide groups and evaluated their relative aqueous La(III)/Ln(III) selectivity by computing free energy changes for the exchange reaction between the designed ligands and a reference ligand. Three novel ligands show promise as excellent extractant agents in selectively separating trivalent lanthanides. The extent of conjugation (and hyperconjugation), the complex geometry, and the electron accumulations on the two O-donors of the N-oxide groups are found to be important factors in dictating the selectivity trends.
Introduction
Rare earth elements (REEs), including fifteen lanthanides (Ln), Sc, and Y, find broad applications in enabling many important technologies and industries.1–6 However, they occur naturally together due to their similar properties and must be separated. Solvent extraction is the primary means to separate different lanthanides on an industrial scale.7 Due to lanthanide contraction, most ligands prefer to bind heavier lanthanides than the lighter ones, because of the decreasing ion size traversing the series. Commercial extractants employ oxygen donors such as tributyl-phosphate (TBP),8 diglycolamide (DGA),9–11 and bis(2-ethylhexyl) phosphoric acid (D2EHPA).12 Ligands with N donors such as alkylated bis-triazinyl pyridines (BTP),13 6,6′-bis-triazinyl-2,2′-bipyridine (BTBP),14 and 2,9-bis-triazinyl-1,10-phenanthroline (BTPhen)15,16 are also used.Recently, ligands combining hard O-donor and soft N-donor atoms have been recognized as efficient extractants. One such example is 2,9-bis-lactam-1,10-phenanthroline (BLPhen)17 that shows unparalleled selectivity for light trivalent lanthanides.18 The rigidity of the BLPhen backbone has been shown via quantum chemical calculations to be an important factor influencing the selectivity of lanthanide ions.18 First principles molecular dynamics simulations suggested a tight binding pocket between BLPhen and Ln(III).19 Although the BLPhen ligand is a relatively new system, first examined for Am(III)/Eu(III) separation17 in 2017 and then for Ln(III) separations18 in 2019, what is known about the system suggests that combining O-donor and N-donor atoms could be a general strategy to design new ligands for separations of lanthanides. An innovation would be to introduce different types of N,O donors on the BLPhen framework.
N-oxide donors are common in chelate complexes of transition metals.20 More interestingly, some pyridine-N-oxide-derived ligands have exhibited abilities to selectively coordinate to Ln(III)21–23 or actinides24 whose separations can provide enlightenment on Ln(III) behaviors. Moreover, computational approaches have been increasingly used to help design new ligands, including the data-driven machine learning approach.25 Our goal here is to computationally examine new mixed N,O-donor ligands based on phenanthroline and N-oxide functionalities for complexation across the Ln(III) series, in order to gain insights into their potential for separating Ln(III)s and to correlate with their molecular structures and electronic structures in terms of key descriptors. Below we explain our computational approach first.
Computational method
It is still difficult to accurately predict the absolute binding free energy (ΔG) between a ligand and a Ln(III) ion computationally, using an implicit solvation model. By computing ΔΔG instead, one can focus on the overall relative trend and benefit from error cancellation. Following a previously established computational strategy,18,26 the relative aqueous selectivity for La(III) over the other Ln(III) ions was evaluated by computing the Gibbs free energy change, ΔΔGaq(La/Ln), of the following ligand-exchange reaction:| [La(Lref)](NO3)3(aq) + [Ln(Ltarget)](NO3)3(aq) ⇌ [La(Ltarget)](NO3)3(aq) + [Ln(Lref)](NO3)3(aq), | (1) |
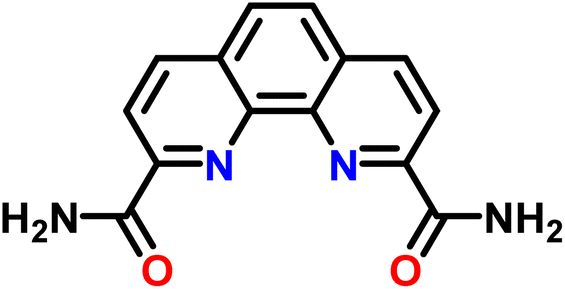 | ||
| Scheme 1 The 2,9-bis-amide-1,10-phenanthroline (BAPhen) ligand, used as the reference ligand to determine free-energy change for the ligand-exchange reaction between La(III) and Ln(III). | ||
The energies of the four 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand–metal complexes in reaction (1) were computed at the B3LYP level of density functional theory (DFT) using the Gaussian 16 (revision C.01) program package.27 6-31+G(d) basis sets were used for the main-group elements and hydrogen. The corresponding large-core (LC) relativistic effective core potentials (RECP) were used for all lanthanides elements.28 Frequency calculations were performed to ensure real vibrational modes for the minimum ground-state structures and to provide zero-point energies (ZPEs). ZPEs and entropy contributions (T = 298.15 K) calculated at the B3LYP/LC/6-31+G(d) level were added to the total energy to obtain the Gibbs free energies. Initial structures for geometry optimizations were generated from the corresponding experimental crystal structures by appropriately modifying donor atoms, substituents, and backbone of the BLPhen ligands. Typical input file and coordinates for converged complex structures are provided in ESI.† All calculations were performed in aqueous environment by employing the IEF-PCM (integral equation formalism of the polarizable continuum model) implicit solvation model to obtain solvation free energies in the aqueous solution.29 To correct the errors on the free energies of low-frequency vibrational modes from the harmonic oscillator model, frequencies lower than 60 cm−1 were set to 60 cm−1 by following the quasiharmonic approximation.30
:1 ligand–metal complexes in reaction (1) were computed at the B3LYP level of density functional theory (DFT) using the Gaussian 16 (revision C.01) program package.27 6-31+G(d) basis sets were used for the main-group elements and hydrogen. The corresponding large-core (LC) relativistic effective core potentials (RECP) were used for all lanthanides elements.28 Frequency calculations were performed to ensure real vibrational modes for the minimum ground-state structures and to provide zero-point energies (ZPEs). ZPEs and entropy contributions (T = 298.15 K) calculated at the B3LYP/LC/6-31+G(d) level were added to the total energy to obtain the Gibbs free energies. Initial structures for geometry optimizations were generated from the corresponding experimental crystal structures by appropriately modifying donor atoms, substituents, and backbone of the BLPhen ligands. Typical input file and coordinates for converged complex structures are provided in ESI.† All calculations were performed in aqueous environment by employing the IEF-PCM (integral equation formalism of the polarizable continuum model) implicit solvation model to obtain solvation free energies in the aqueous solution.29 To correct the errors on the free energies of low-frequency vibrational modes from the harmonic oscillator model, frequencies lower than 60 cm−1 were set to 60 cm−1 by following the quasiharmonic approximation.30
Results and discussion
Changing the amide groups in BLPhen to N-oxides
Our design starts with modification of the O donors on the BLPhen ligand (1a in Fig. 1) by replacing the amide moieties with N-oxides (1b–1d). Fig. 1b summarizes the computed ΔΔG trends across the Ln(III)s for the four ligands. One can see that the La(III)/Ln(III) selectivity becomes worse upon replacing one or both amides in 1a with N-oxide functionality. Fig. 1b also shows an interesting peak at Ce, meaning that the target ligands (1a–1d) prefer to bind Ce(III) over La(III). This non-linear trend at Ce has also been observed in several experimental reports,18,31 suggesting a different chemical nature of Ce(III) from nearby Ln(III)s.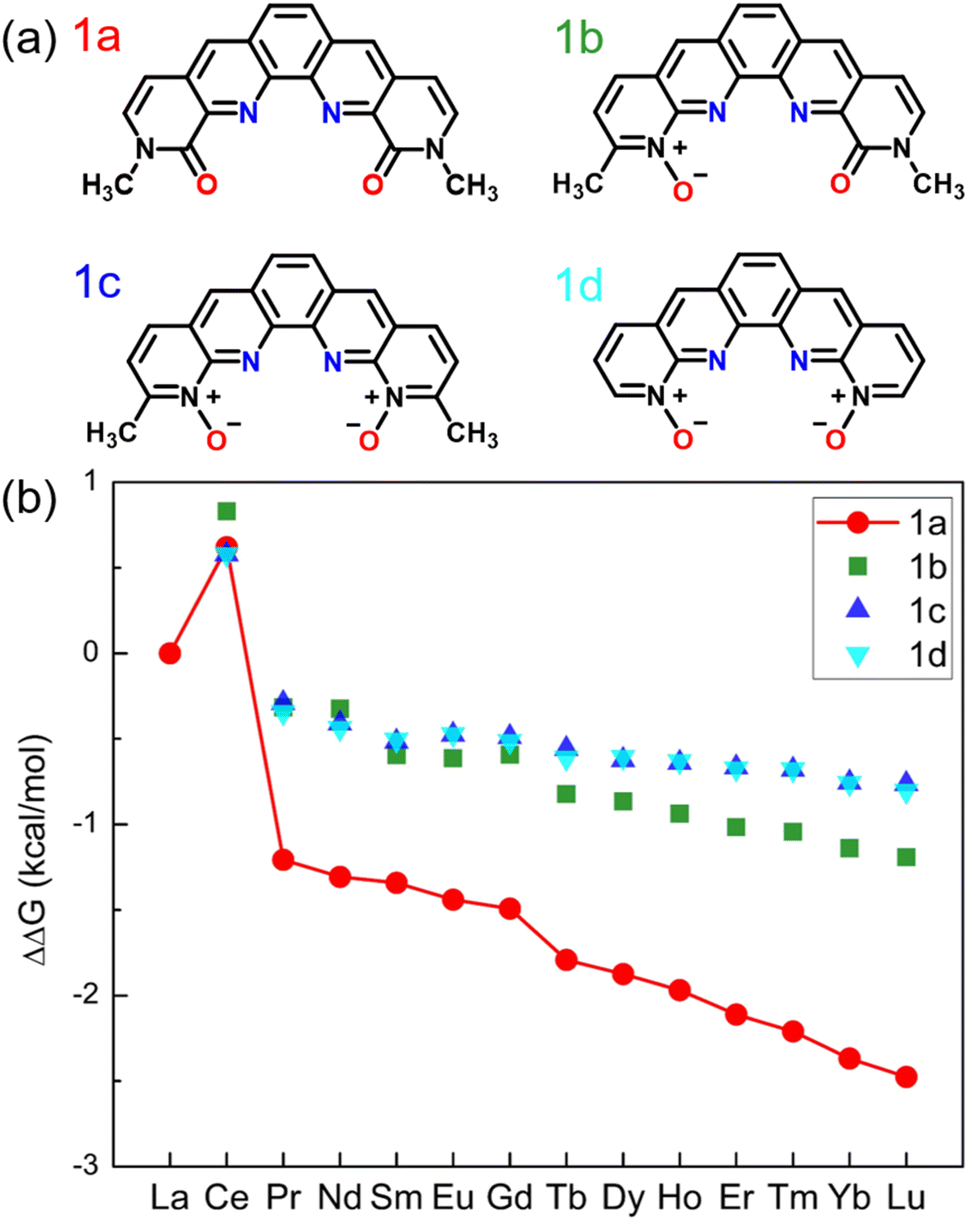 | ||
| Fig. 1 (a) Chemical structures of BLPhen (1a) and BLPhen-derived N-oxide ligands (1b–1d); (b) DFT-calculated relative aqueous-phase selectivity, ΔΔGaq(La/Ln), for the ligands 1a–1d, with respect to the refence ligand (BAPhen; Scheme 1). | ||
To understand the overall performance from the N-oxides (ligands 1b-1d), we determined the partial charges on the O donors by the natural bond orbital (NBO) populations and found the charges of −0.68e for 1a, −0.64e for 1b, and −0.63e for 1c and 1d. In other words, changing amide moieties to N-oxides does not make the O donors more negative, as initially hypothesized. This is likely due to the enhanced π conjugation from the pyridinium group in 1b–1d. Hence our next strategy is to tune the conjugation to see if the performance of N-oxide-based ligands can be improved.
Tuning the conjugation of the N-oxide ligands by varying the center ring
The ligands 1a–1d all have the conjugation throughout the whole molecule. Our idea was to disrupt the conjugation at the middle ring (2a–2d in Fig. 2a). Since 1b, 1c, and 1d show similar performances (Fig. 1b), we selected 1d as a starting ligand. Four new ligands were created based on modifications of 1d: changing the middle top C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond to a single bond (2a); reducing the middle 6-membered ring to a 5-membered ring (2b); enlarging the middle ring to be a 7-membered one (2d) or eliminating the top CC bond (2c). The calculated ΔΔGaq(La/Ln) values for 2a–2d (Fig. 2b) are compared to those of 1a. Interestingly, ligand 2b has much better performance than 1a. On the other hand, the performances of 2a, 2c, and 2d are worse than that of 1a. Therefore, the reduced extent of conjugation in 2b, combined with other factors, makes it a more selective ligand. To reveal those factors, we first compare the optimized geometries after complexation.
C double bond to a single bond (2a); reducing the middle 6-membered ring to a 5-membered ring (2b); enlarging the middle ring to be a 7-membered one (2d) or eliminating the top CC bond (2c). The calculated ΔΔGaq(La/Ln) values for 2a–2d (Fig. 2b) are compared to those of 1a. Interestingly, ligand 2b has much better performance than 1a. On the other hand, the performances of 2a, 2c, and 2d are worse than that of 1a. Therefore, the reduced extent of conjugation in 2b, combined with other factors, makes it a more selective ligand. To reveal those factors, we first compare the optimized geometries after complexation.
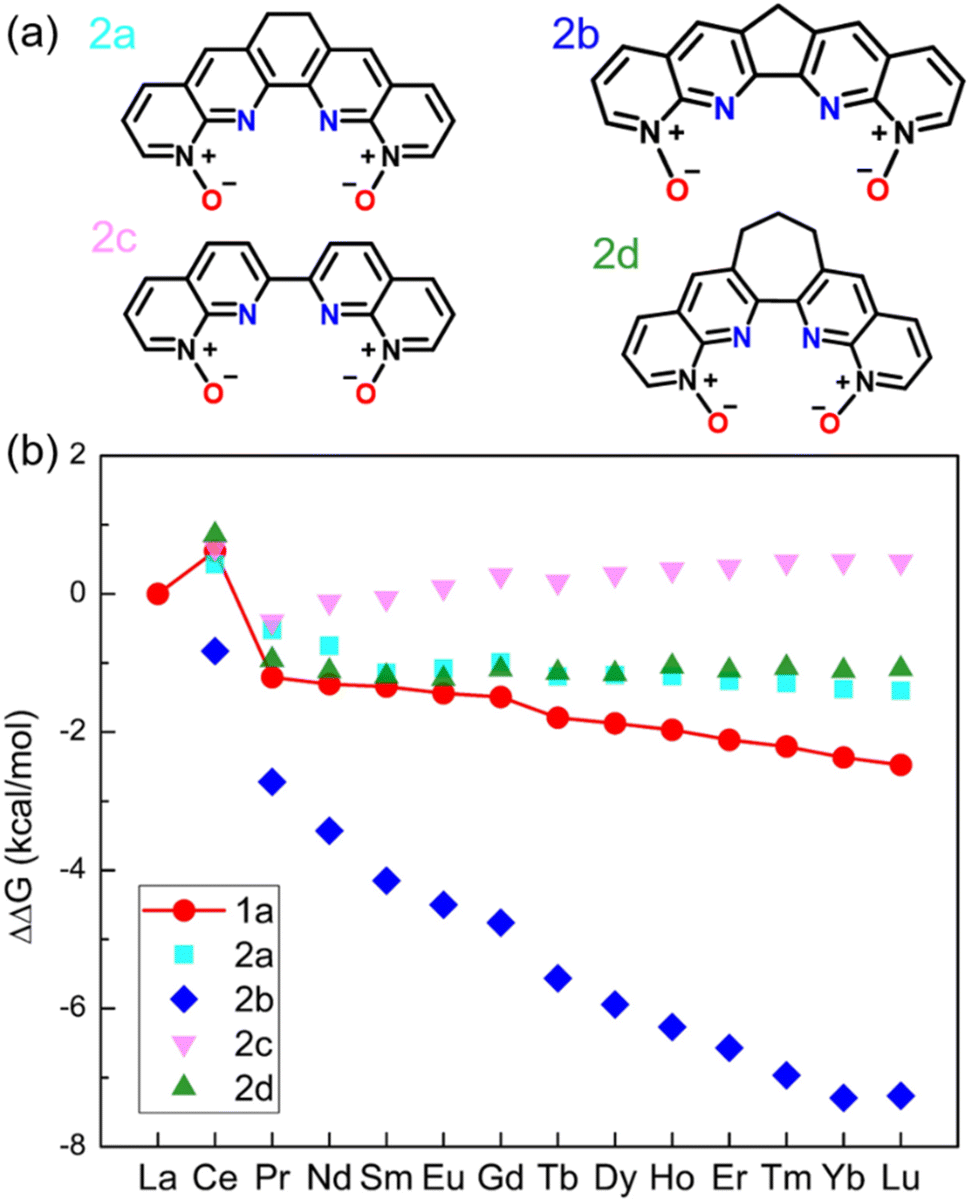 | ||
| Fig. 2 (a) Chemical structures of 1,10-phenanthroline derived N-oxides with reduced conjugation. (b) DFT-calculated relative aqueous phase selectivity (ΔΔGaq(La/Ln)). | ||
Similar to the structure of [La(BLPhen)](NO3)3,17 our optimized [La(2b)](NO3)3 complex (Fig. 3) also has tenfold coordination: four donors from 2b ligand and six from three bidentate nitrates. Both [La(2b)](NO3)3 and [Ln(1a)](NO3)3 complexes are planar, as evidenced by the close-to-zero O1–N1–N2–O2 dihedral angle (Table 1 and Fig. 3). On the other hand, ligands 2a, 2c, and 2d are significantly non-planar, leading to less planar complexes. Therefore, the geometric comparison suggests that the planar geometry combined with the reduced conjugation helps improve the selectivity for 2b, while the non-planarity of ligands 2a, 2c, and 2d indicates that the whole conjugation is completely broken down into two smaller parts which might be detrimental to the complexation. We note that the NO3 coordination in the Ln-complexes varies slightly from ligand to ligand.
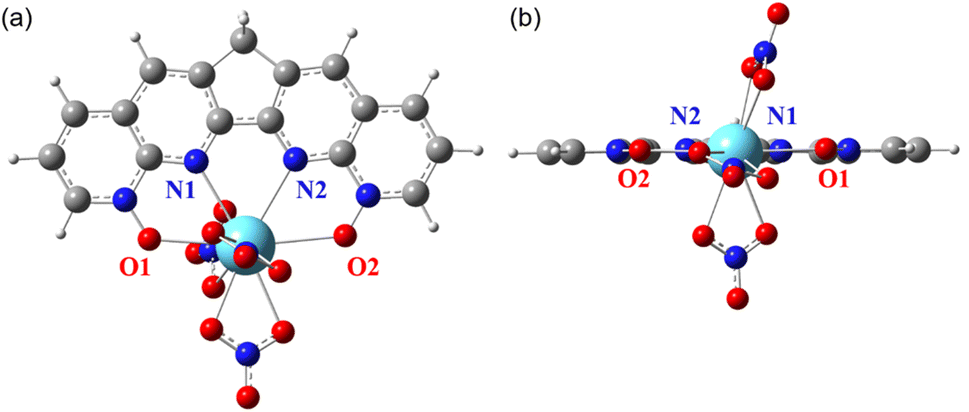 | ||
| Fig. 3 DFT-optimized [La(2b)](NO3)3 complex with four donors on the 2b ligand labelled: (a) top view; (b) side view. | ||
| 1a | 2a | 2b | 2c | 2d | 3a | 3b | 3c | |
|---|---|---|---|---|---|---|---|---|
| Ligand | 0.4° | 9.9° | 0.5° | 18.9° | 26.8° | 0.0° | 0.2° | 0.6° |
| La(ligand)(NO3)3 | 0.8° | 8.3° | 1.3° | 5.0° | 14.3° | 1.4° | 2.0° | 28.0° |
Unlike other ligands considered in this study, 2b possesses a fluorene-like moiety, where the methylene group is known to be involved in hyperconjugation.32 Indeed, our NBO analysis for 2b revealed relatively strong interactions of the electrons in C–H σ-bonds with adjacent CC π* orbitals (Fig. S1†) with estimated second-order stabilization energies E(2) of ∼3.4 kcal mol−1. Such hyperconjugation leads to a better geometric fit for Ln(III) ions, as reflected in the shorter Ln–N bonds in the [Ln(2b)](NO3)3 complexes than in the complexes of other ligands (see Table S1† for some comparisons). On the other hand, NBO charge analyses on ligands 2a–2d do not show significant differences of charges on pyridinic nitrogen atoms or NO oxygen atoms (Table S2†). In other words, we think that the impact of the hyperconjugation from the middle constrained 5-membered ring is more geometric than electronic.
We have further performed NBO charge analysis for [Ln(2a)](NO3)3 and [Ln(2b)](NO3)3 complexes (Fig. S2†) and found that the non-linear trends of Ln partial charges across the Ln series are very similar between 2a and 2b. The charges on Ln are lightly smaller in [Ln(2a)](NO3)3 complexes than in their [Ln(2b)](NO3)3 counterparts; in other words, there are more ligand charge transfers to Ln from 2a than from 2b. On the contrary, ΔΔGaq(La/Ln) values in Fig. 2 show that 2b is more selective than 2a; the selective trend across the Ln series is rather monotonic. So we conclude that the charge transfer is unlikely to be the key factor. This is consistent with our conclusion that the geometric factor is more important.
Further tuning of the conjugation size and the O–O distance of the N-oxides by varying the side rings
Since 2b notably enhances the La(III)/Ln(III) selectivity across the whole lanthanide series, we designed additional ligands based on 2b by modifying the conjugation size via the following approaches as shown in Fig. 4a: changing the two pyridiniums to five-membered rings (3a); changing only one outside ring to a five-membered one (3b); inserting one phenyl ring on each side (3c). In comparison with 2b, the calculated ΔΔG (Fig. 4b) indicates better La(III)/Ln(III) selectivity for 3a and much worse La(III)/Ln(III) selectivity for 3c. Considering the contrast between 3b and 3c in ΔΔGaq(La/Ln) values, we think that the O–O distance is a key factor here: it is too short in 3c (Fig. 4a) which is very detrimental to the La(III)/Ln(III) selectivity. Besides geometric factors such as planarity and O–O distances, orbital interactions may also be important, which we analyze next.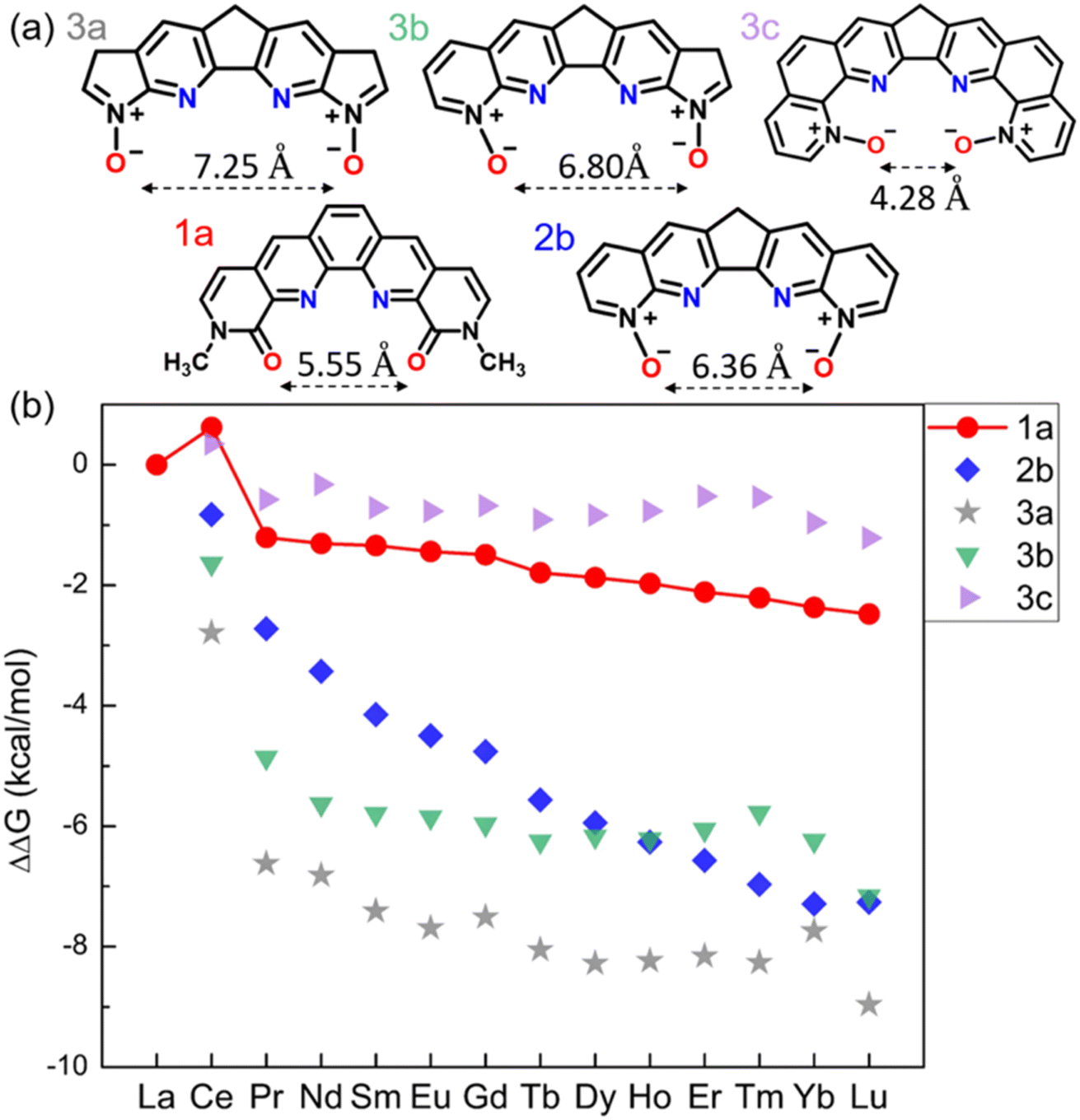 | ||
| Fig. 4 (a) Chemical structures of N-oxide ligands 3a–3c derived from 2b; ligand 1a is also shown for comparison in terms of O–O distance. (b) DFT-calculated relative aqueous phase selectivity, ΔΔGaq(La/Ln), for ligands 3a–3c in comparison with ligands 1a and 2b. | ||
Orbital and charge analyses
Computed ΔΔGaq(La/Ln) for our designed mixed N,O-donor shows that the N-oxide-based ligands 2b, 3a, and 3b have the potential to be more La(III)/Ln(III) selective than the BLPhen ligand 1a. The complexation of these ligands with Ln(III) involves mainly the donation of the lone pairs of electrons from the ligands to the Ln(III) ion. Therefore, it would be interesting to compare the HOMOs of these high-performing ligands. From Fig. 5 one can see more electron accumulation around O donors in 2b, 3a, and 3b than in 1a, which could be a reason for their improved selectivity profile. The other interesting feature is that the conjugation is similar between 1a and 2b, but more fragmented in 3a and 3b. This causes very different electron density distribution at N donors from 1a and 2b to 3a and 3b. We think that this may be the reason for the more non-linear selectivity trends across the Ln series for 3a and 3b (Fig. 4b). | ||
| Fig. 5 (a) HOMO and (b) HOMO−1 of ligands 1a, 2b, 3a and 3b and their energies. | ||
Of course, there could be other important factors such as conjugation sizes and O–O distances discussed above, as the orbitals for all the ligands, when examined together, displays a more complicated picture (Fig. S3†). A machine-learning model that can rank all descriptors in terms of their importance in dictating the selectivity would be highly desirable. Future work is warranted.
Implications for ligand synthesis
Ligand 1a (BLPhen) and its derivatives have been reported recently for Ln(III) separations.17,18 Our results above suggest that the N-oxide-based 2b, 3a, and 3b ligands can potentially surpass the BLPhen-based ligands. Being the simplest among the three, 2b could be the first target and Scheme 2 shows the proposed route. It starts with a commercially available compound, 3-methyl-1,8-naphthyridine-2-carboxylic acid (1), which reacts with methyllithium to yield 2,33 followed by oxidation to 3.34 Equivalent 3 and 2-aminonicotinaldehyde react in potassium hydroxide and ethanol to yield 4,35 which dehydrates in polyphosphoric acid (PPA) to yield 5,36 followed by hydrazine reduction to 6.36 Finally, 6 can be selectively oxidized by reacting with 3.5 equivalent meta-chloroperoxybenzoic acid (mCPBA) in dichloromethane,37 to yield 2b. Similar routes can also be adopted to synthesize 3a and 3b. We hope that the proposed route in Scheme 2 would inspire synthetic chemists to try it or come up with better ones.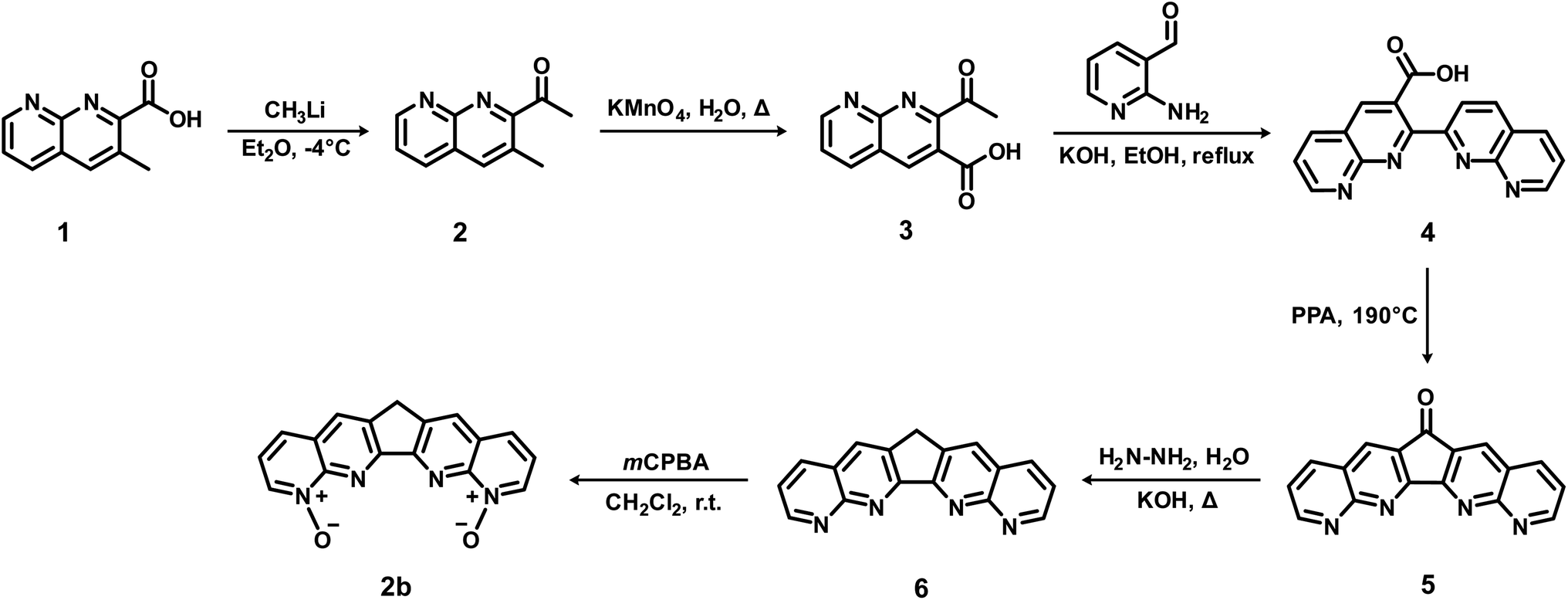 | ||
| Scheme 2 Proposed route to synthesize 2b. | ||
Conclusions
We have computationally evaluated a new family of mixed N,O-donor ligands, derived from BLPhen and incorporating N-oxide functionalities, for their relative aqueous La(III)/Ln(III) selectivity. We found that the conjugation size, the O–O distance, the planarity of the formed complex, and the electron density on the two O atoms are important control knobs that affect ligand's selectivity for lanthanides. Three novel ligands (2b, 3a, and 3b) were identified to be promising and experimentally viable targets in selective separations of trivalent lanthanides. Instead of a center benzene ring as in BLPhen, the three N-oxide ligands share the feature of a center cyclopentadiene ring, which opens up the binding pocket as reflected in the greater O–O distances. The combined effect of the center cyclopentadiene ring and the N-oxide groups leads to higher predicted La(III)/Ln(III) selectivities for the designed ligands 2b, 3a, and 3b than the BLPhen reference ligand. Our computational insights will guide the follow-up efforts towards synthesizing the top candidates and testing them for Ln(III) separations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Separation Science program and Materials Chemistry program under Award Number DE-SC00ERKCG21.References
- T. Cheisson and E. J. Schelter, Science, 2019, 363, 489–493 CrossRef CAS PubMed.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- Z. Zeng, Y. Xu, Z. Zhang, Z. Gao, M. Luo, Z. Yin, C. Zhang, J. Xu, B. Huang and F. Luo, Chem. Soc. Rev., 2020, 49, 1109–1143 RSC.
- F. Saraci, V. Quezada-Novoa, P. R. Donnarumma and A. J. Howarth, Chem. Soc. Rev., 2020, 49, 7949–7977 RSC.
- B. Zheng, J. Fan, B. Chen, X. Qin, J. Wang, F. Wang, R. Deng and X. Liu, Chem. Rev., 2022, 122, 5519–5603 CrossRef CAS PubMed.
- F. Ortu, Chem. Rev., 2022, 122, 6040–6116 CrossRef CAS PubMed.
- F. Xie, T. A. Zhang, D. Dreisinger and F. Doyle, Miner. Eng., 2014, 56, 10–28 CrossRef CAS.
- L. K. Sinclair, J. W. Tester, J. F. H. Thompson and R. V. Fox, Ind. Eng. Chem. Res., 2019, 58, 9199–9211 CrossRef CAS.
- E. A. Mowafy and D. Mohamed, Sep. Purif. Technol., 2014, 128, 18–24 CrossRef CAS.
- R. J. Ellis, D. M. Brigham, L. Delmau, A. S. Ivanov, N. J. Williams, M. N. Vo, B. Reinhart, B. A. Moyer and V. S. Bryantsev, Inorg. Chem., 2017, 56, 1152–1160 CrossRef CAS PubMed.
- X. He, X. Wang, Y. Cui, Z. Su, G. Ye, C. Lu and L. Li, J. Radioanal. Nucl. Chem., 2021, 329, 1019–1026 CrossRef CAS.
- A. Kumari, R. Panda, J. Y. Lee, T. Thriveni, M. K. Jha and D. D. Pathak, Sep. Purif. Technol., 2019, 227, 115680 CrossRef CAS.
- A. Kovács, C. Apostolidis and O. Walter, Inorganics, 2019, 7, 26 CrossRef.
- M. R. Foreman, M. J. Hudson, M. G. Drew, C. Hill and C. Madic, Dalton Trans., 2006, 1645–1653 RSC.
- Y. Yang, J. Liu, L. Yang, K. Li, H. Zhang, S. Luo and L. Rao, Dalton Trans., 2015, 44, 8959–8970 RSC.
- D. M. Whittaker, T. L. Griffiths, M. Helliwell, A. N. Swinburne, L. S. Natrajan, F. W. Lewis, L. M. Harwood, S. A. Parry and C. A. Sharrad, Inorg. Chem., 2013, 52, 3429–3444 CrossRef CAS PubMed.
- S. Jansone-Popova, A. S. Ivanov, V. S. Bryantsev, F. V. Sloop, R. Custelcean, I. Popovs, M. M. Dekarske and B. A. Moyer, Inorg. Chem., 2017, 56, 5911–5917 CrossRef CAS PubMed.
- M. R. Healy, A. S. Ivanov, Y. Karslyan, V. S. Bryantsev, B. A. Moyer and S. Jansone-Popova, Eur. J. Chem., 2019, 25, 6326–6331 CrossRef CAS PubMed.
- T. D. N. Reddy, A. S. Ivanov, D. M. Driscoll, S. Jansone-Popova and D.-e. Jiang, ACS Omega, 2022, 7, 21317–21324 CrossRef CAS PubMed.
- J. Quagliano, J. Fujita, G. Franz, D. Phillips, J. Walmsley and S. Tyree, J. Am. Chem. Soc., 1961, 83, 3770–3773 CrossRef CAS.
- I. Binyamin, S. Pailloux, E. N. Duesler, B. M. Rapko and R. T. Paine, Inorg. Chem., 2006, 45, 5886–5892 CrossRef CAS PubMed.
- M. A. S. Goher and F. A. Mautner, J. Mol. Struct., 2007, 846, 153–156 CrossRef CAS.
- H.-J. Zhang, R.-H. Gou, L. Yan and R.-D. Yang, Spectrochim. Acta, Part A, 2007, 66, 289–294 CrossRef PubMed.
- D. Das, M. Joshi, S. Kannan, M. Kumar, T. K. Ghanty, A. S. Pente, A. Sengupta and C. P. Kaushik, Polyhedron, 2021, 201, 115166 CrossRef CAS.
- T. Liu, K. R. Johnson, S. Jansone-Popova and D.-e. Jiang, JACS Au, 2022, 2, 1428–1434 CrossRef CAS PubMed.
- A. S. Ivanov and V. S. Bryantsev, Eur. J. Inorg. Chem., 2016, 2016, 3474–3479 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian 16 (revision C.01), Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1989, 90, 1730–1734 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2011, 115, 14556–14562 CrossRef CAS PubMed.
- J. Dehaudt, N. J. Williams, I. A. Shkrob, H. Luo and S. Dai, Dalton Trans., 2016, 45, 11624–11627 RSC.
- R. Baker, C. Eaborn and J. Sperry, J. Chem. Soc., 1962, 2382–2385 RSC.
- G. M. Rubottom and C. Kim, J. Org. Chem., 1983, 48, 1550–1552 CrossRef CAS.
- G. K. Wagner, D. Kotschenreuther, W. Zimmermann and S. A. Laufer, J. Org. Chem., 2003, 68, 4527–4530 CrossRef CAS PubMed.
- B. Xiong, S. Zhang, H. Jiang and M. Zhang, Org. Lett., 2016, 18, 724–727 CrossRef CAS PubMed.
- M. E. Hoque, R. Bisht, A. Unnikrishnan, S. Dey, M. M. Mahamudul Hassan, S. Guria, R. N. Rai, R. B. Sunoj and B. Chattopadhyay, Angew. Chem., Int. Ed., 2022, 61, e202203539 CrossRef CAS PubMed.
- R. P. Thummel and Y. Jahng, J. Org. Chem., 1985, 50, 3635–3636 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07029d |
| This journal is © The Royal Society of Chemistry 2023 |