
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure and emission properties of dinuclear copper(I) complexes with pyridyltriazole†
Alexey Gusev *a,
Mikhail Kiskinb,
Elena Bragaa,
Ekaterina Zamniusa,
Mariya Kryukovac,
Nataliya Karaush-Karmazind,
Glib Baryshnikovde,
Boris Minaevd and
Wolfgang Linertf
*a,
Mikhail Kiskinb,
Elena Bragaa,
Ekaterina Zamniusa,
Mariya Kryukovac,
Nataliya Karaush-Karmazind,
Glib Baryshnikovde,
Boris Minaevd and
Wolfgang Linertf
aV.I. Vernadsky Crimean Federal University, Simferopol, 295007, Crimea. E-mail: galex0330@gmail.com
bN.S. Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, Moscow, 119991, Russia
cInstitute of Chemistry, Saint Petersburg State University, Universitetskaya Nab. 7/9, Saint Petersburg, Russia
dDepartment of Chemistry and Nanomaterials Science, Bohdan Khmelnytsky National University, 18031 Cherkasy, Ukraine
eLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, SE-60174 Norrköping, Sweden
fInstitute of Applied Physics, Vienna University of Technology, Wiedner Hauptstraße 8-10, 1040 Vienna, Austria
First published on 27th January 2023
Abstract
A new series of five highly emissive binuclear heteroleptic pyridyltriazole-Cu(I)-phosphine complexes 1–5 was synthesized and examined by different experimental (IR, elemental and thermogravimetric analysis, single crystal X-ray diffraction technique, UV-vis and fluorescence spectroscopy) and quantum chemical aproaches. Complexes 1–5 exhibited excellent stimuli-responsive photoluminescent performance in the solid state at room temperature (quantum yield (QY) = 27.5–52.0%; lifetime (τ) = 8.3–10.7 μs) and when the temperature was lowered to 77 K (QY = 38.3–88.2; τ = 17.8–134.7 μs). The highest QY was examined for complex 3 (52%) that can be explained by the small structural changes between the ground S0 and exited S1 and T1 states leading to the small S1–T1 triplet gap and efficient thermally-activated delayed fluorescence. Moreover, complex 4 demonstrates reversible mechanochromic and excitation dependent luminescence.
Introduction
Luminescent Cu(I) complexes due to their excellent luminescence properties, such as a high quantum yield at room temperature, thermo- and vapochromic luminescence, thermally activated delayed fluorescence (TADF), and stimuli-responsive luminescence, has become of increasing interest recently for their promising prospects in materials science.1,2 Development of new systems based on Cu(I), especially heteroleptic copper complexes, is strongly motivated as they are good alternatives to those based on the more expensive platinum group metals and rare earths.One of the most interesting peculiarities of Cu(I) complexes is the number of different molecular and crystalline structures obtainable by simply varying reaction conditions. Among several systems, copper(I) halide aggregates represent a prominent and well-investigated family which currently lies at the forefront of both coordination chemistry and crystal engineering. The structural diversity of copper(I) halide complexes has enabled them to show a wide range of emission spectra from blue to near-infrared. Generally, such emission originates from several different excited states, such as metal-to-ligand charge transfer (MLCT), halide-to-ligand charge transfer (XLCT), a cluster-centered (CC) state and so on.2
While photochemical and photophysical Cu(I) complexes and clusters have been extensively studied since the early 70s their stimuli responsive photoluminescence has been studied only in the last two decades. Luminescent materials that respond to external stimuli (such as grinding, shearing, stretching or pressing) are of scientific and technological interest owing to their wide potential applications in the fields, including chemosensors, data storage, optoelectronic devices, and biomedical imaging.3 Despite that significant number of publications on mechanoluminescence have been addressed to organic systems,4 there are fewer examples based on Cu(I) complexes.5 It was found that in contrast to the photoluminescence which respond to the stimuli mainly by molecular chemical structural transition, most of the mechanoluminescent materials work on the disruption of an intermolecular metal⋯metal contact or inter-ligand π⋯π stacking and other non-covalent supramolecular interactions. Therefore, the investigation on mechanoluminescent behavior of materials is more challenging, which needs to consider not only the molecule itself but also its complicated surroundings in the condensed solid state.
Cu(I) complexes possessing diimine and phosphine ligands are the most common and photophysical properties of these Cu(I) complexes can be highly tuned by judicious choice of ligands since the lowest unoccupied molecular orbital (LUMO) is basically localized on the diimine ligand.1,6 To explore further the relationship between the luminescence properties of Cu(I) complexes and the structure of the N-heterocyclic chelating ligand, it is necessary to synthesize more Cu(I) complexes with multi-nitrogen-containing heterocyclic ligands.
The 2-pyridyl substituted azoles owing to their simple and economical preparation, good optical characteristics, and rigid structure are of particular interest from this point of view. Nevertheless, most pyridylazole copper complexes have a predominantly mononuclear structure,6 whereas from the point of view of realising mechanoluminescence it is preferable to obtain bi- and/or tetranuclear clusters due to strong impact of external stimuli on metallophilic interaction in the excited state.5b In contrast of above 3/4-pyridyl derivatives of azoles can form polynulcear clusters and polymers in a multidentate (bi- or tridentate) fashion. The different degrees of bending which are caused by the changing position of the pyridyl N atoms could benefit the formation of different topological structures. We expect that the peculiarities of the structure of such ligands will contribute to the formation of di and/or polynuclear complexes, in contrast to the 2-pyridyl analogues. Despite these reasons the use of 3/4-pyridyl derivatives of azoles for the synthesis of photoluminescent copper complexes is quite rare.
Herein, five binuclear heteroleptic 3/4-pyridyltriazole-Cu(I)-phosphine complexes are reported. They have been synthesized and characterized by using IR, elemental and thermogravimetric analysis (TGA), UV-vis and fluorescence spectroscopic methods, as well as single crystal X-ray diffraction analysis and quantum chemical calculations. It has been shown that bidentate complexes are formed in solutions, which in some cases exhibit mechano- and excitation-dependent luminescence.

Results and discussion
Syntheses and solid state characterization
The above five heteroleptic Cu(I) complexes were synthesized by treatment of cuprous iodide with the corresponding triazoles L1–L5 and triphenylphosphine ligand in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 stoichiometric ratio in chloroform/methanol mixture (Scheme 1). The numbering of the complexes corresponds to the numbering of the ligands (see. Exp. section for detail).
:1:1 stoichiometric ratio in chloroform/methanol mixture (Scheme 1). The numbering of the complexes corresponds to the numbering of the ligands (see. Exp. section for detail).
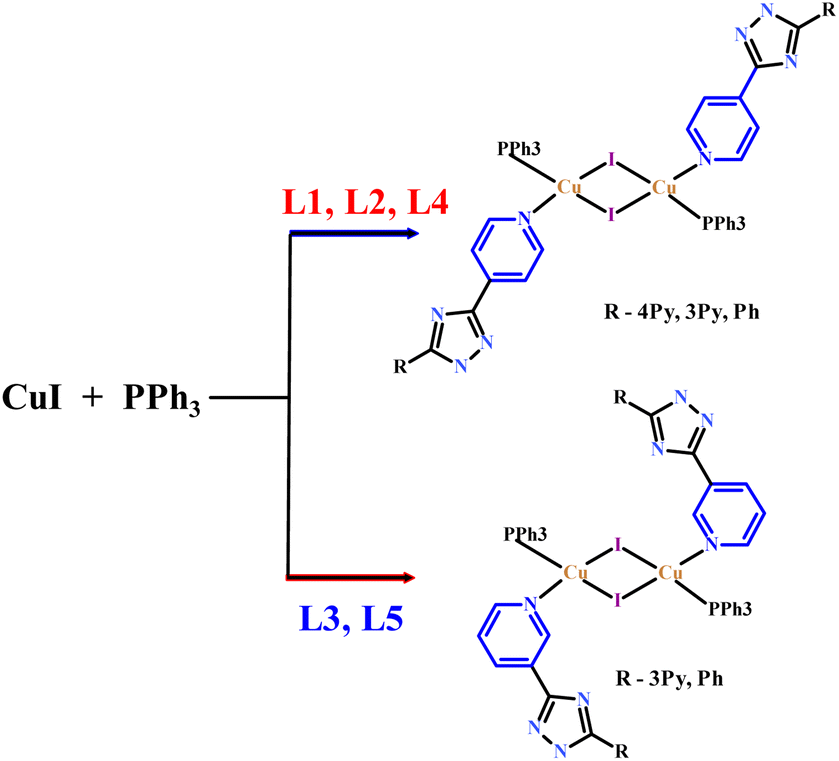 | ||
| Scheme 1 Schematic synthetic route for complexes 1–5. | ||
All the products were air stable and soluble in methanol, acetonitrile and halogenated solvents but insoluble in diethyl ether and petroleum ether. Complexes 1–5 were fully characterized by elemental analysis, infrared and electronic absorption spectroscopy. Phase purities were confirmed by the powder XRD.
The infrared spectrum of 1–5 show all the bands required due to the presence of triazole ligands and PPh3. Characteristic vibration bands of the aromatic heterocyclic appear at 1400–1625 cm−1. Upon P–Cu(I) coordination, the characteristic peak of PPh3 in 1–5 slightly shifted to higher frequency (∼1091–1095 cm−1) relative to that of free PPh3 (1089 cm−1).
The thermal stability of obtained complexes has been investigated by thermogravimetric analysis experiments of mass loss (Fig. S1 in ESI†). The first weight loss for complex 1 (6.20%) occurs in the 100–140 °C range, which is assigned to the release of three methanol solvate molecules (6.65%). The residual framework starts to decompose beyond 233 °C with a series of complicated weight losses and does not stop until heating ends at 650 °C. For the TGA curve of complex 2, there are several mass loss steps, although without a clear plateau. The first observed weight loss of 13.5% in the region of 40–130 °C corresponds to the release of the lattice chloroform guests (calculated 13.52%). The residual framework starts to decompose beyond 215 °C with and does not stop until heating ends at 645 °C. For 4, the weight loss of the lattice methanol (4.5%) occurs in the range of 50–140 °C (calculated 4.52%). The residual framework starts to decompose beyond 242 °C the following step, corresponds to the decomposition of all organic ligands and the iodine-anion. The TGA curves of 3 and 5 are similar. The complexes are stable up to 232 °C for 3 and 224 °C for 5 followed by a series of consecutive steps of weight loss and does not stop until heating ends at 650 °C. Spectroscopic results were further complemented by the analysis of the crystal and molecular structures of complexes 1–5 (Fig. 1). Key bond parameter data are summarized in Table S2.†
 | ||
| Fig. 1 Molecular structures of 1-a, 2-b, 3-c and 4-d, 5-e. Solvate molecules are omitted for clarity. The color map for atom types is as follows: I – pink, Cu – turquoise, P – green, N – blue, C – grey, H – white. | ||
The structures of 1–5 prepared in this study are typical for dimeric complexes with the formula [(μ-I)2Cu2L2(PPh3)2].1b As shown in Fig. 1, the crystal structure studies of the complexes 1–5 establish that they have a similar Cu2I2 rhombic core with trans arrangement of the two PPh3 and the two triazole ligands.
Each Cu(I) center is in a tetrahedral coordination environment consisting of two bridging iodo-ligands, one PPh3 and the nitrogen atom of 4-pyridinylsubstituent of L1, L2, or L5 and 3-pyridinyl substituent of L3 or L4, respectively. Interestingly, in the case of competitive coordination in the L2 case, coordination of the 4-pyridyl ring is preferred. Notably that L1–L3 potentially can act as bridging ligands due to coordination on the N-atoms from the second pyridine ring but here they demonstrate monodentate coordination mode. Each molecule is on a crystallographic center of inversion at the center of the Cu2I2 rhomb so that there is half a molecule in the asymmetric unit.
Within each dimer, the two unique Cu–I distances lie in the range from 2.6386(5) to 2.724(1) Å across the series. These distances were comparable to the other iodo-bridged Cu(I) complexes with N-donor ligands and PPh3. The angles at Cu center vary in the range of 110.08–116.33°, while the angles at the iodine center vary in the range of 63.67–69.92°, indicating about planar geometry of Cu2I2 core.
The Cu–Cu distances, being important for emission properties of the complexes,7 were altered by the triazole-ligands structures from 2.827(2) Å (2) to 3.037(2) Å (3). Complexes with 4-pyridine coordinating group tend to show shorter Cu⋯Cu separation while those with 3 pyridine coordinating derivatives tend to show the longer Cu⋯Cu separation. Obtained Cu⋯Cu distances are longer than the sum of the van der Waals radii of Cu (2.8 Å), and the metallophilic interactions in these dinuclear cores were also negligible.
The Cu–P bond lengths were less affected by the triazole ligands. The average Cu–P distances for the series of dimeric complexes are 2.229–2.248 Å. However, different coordinating pyridine rings (L1-L5) have moderate impact on the Cu–N bond (2.048–2.069 Å) lengths with shorter ones for the L4 and L5 suggesting that the differing electronic properties of the ligands extend to the environment around the Cu(I) centers.
An examination of the crystal structures of the 1–5 shows that they exhibit complicated supramolecular architecture due to the different H-bonding networks and π⋯π interactions (Fig. S2†). In the crystalline lattice of 1, the non-coordinating N atom of 4Py ring of L1 form a H-bond with the H atom of the methanol molecule which in turn binds to the hydrogen atom of the triazole ring, the second solvate molecule of methanol also participates in H⋯I bonds; these interactions lead to the formation of a supramolecular chain structure (Table S3†). π⋯π stacking between phenyl rings of PPh3 ligand generate an supramolecular layers (Fig. S2, Table S4†), which are linked by C–H⋯π interactions into a three-dimensional structure (Table S5†).
The main supramolecular motifs in 2 and 3 is 1D chain structure in which separate complexes are connected through hydrogen bonds (Ntz-H⋯NPy for 2 and 3, and Cpy-H⋯I for 3, Fig. S2, Table S3†). C–H⋯π interactions in 3 result in a three-dimensional structure (Table S5†).
In 4 a supramolecular chain is formed as the result of weak π⋯π stacking interactions between triazole and phenyl rings and C–H⋯π interactions (Tables S4 and S5†). In crystal 5, 1D chains are formed via Ntz-H⋯I bonds (Table S3†). The chains are linked in a layered structure via π⋯π stacking interactions between the triazole and phenyl rings, and then into a three-dimensional supramolecular structure through C–H⋯π interactions (Tables S4 and S5†).
Photophysical properties
The absorption spectra of the complexes 1–5 were measured in solid state (Fig. 2a) and in THF solution (Fig. S3†). Each absorption spectrum consists of two major sections, a strong absorption in high energy UV region from 250 nm to 330 nm and a weak absorption in low energy region from 350 nm to ∼450 nm. According to the literature reports2f the strong absorption peaks corresponds to the ligand-based π–π* transitions, while the low-energy weak absorption band can be assigned to the metal-to-ligand charge-transfer (MLCT) that are responsible for the yellow or orange color of the complexes. This is in agreement with quantum-chemical calculations discussed later in the text. | ||
| Fig. 2 Absorption spectra of solid state complexes (a), normalized emission spectra of THF solution (10−5 M) (b) and solid polycrystalline samples (c) of 1–5 at room temperature (above) and luminescence images of 1–5 under UV lamp (d). | ||
At the first step, we examined luminescent properties of the synthesized complexes in THF solution at 298 K. Exciting the complexes in the energy range of MLCT band (330–380 nm) leads to blue emission with low intensity (QY < 1%). Complexes 1, 2 and 4 display single-band emission maximized at 385, 408 and 382 nm (Fig. 2b) with lifetime of 7.16, 6.82 and 8.60 ns, respectively. The shape of the emission band and its location indicate that the emission originates from the ligand to ligand charge transfer (LLCT) or intra-ligand charge transfer (ILCT) or mixture of them. Luminescent spectra of 3 and 5 are more complicated and contain two vibronic maxima at 393 and 410 nm (3) and 387 and 408 nm (5) respectively. All complexes showed no obvious MLCT emission in air atmosphere due to the quenching by oxygen and the active intramolecular motion.
Further the solid-state emission properties of 1–5 at ambient temperature were studied using polycrystalline samples. Related luminescence data are demonstrated in Table 1.
| Complex | Emission λmax (nm) (RT/77 K) | Quantum yield, % (RT/77 K) | kr(RT) × 10−4/kr(77) × 10−4, s | Lifetime, μs (RT/77 K) |
|---|---|---|---|---|
| a Ground sample. | ||||
| 1 | 606/625 | 35.1/78.1 | 3.5/3.1 | 9.9/25.0 |
| 2 | 591/612 | 27.5/58.2 | 2.9/2.7 | 9.5/21.9 |
| 3 | 543/568 | 52.0/88.2 | 4.9/0.65 | 10.7/134.7 |
| 4 | 507 (551)a/527 | 28.2/55.3 | 3.3/3.1 | 8.6/17.8 |
| 5 | 528/528 | 15.6/38.3 | 1.9/1.6 | 8.3/23.4 |
The purity of each sample was checked by powder X-ray diffraction (PXRD) patterns and elemental analysis. All of the compounds are strongly emissive at room temperature and exhibit green to orange luminescence when they are excited with UV light (Fig. 2d).
As can be seen from Fig. 2b and d solid complexes 1–5 demonstrate bright broad band photoluminescence centered at 606 (ex. at 465 nm), 591 (ex. at 465 nm), 543 (ex. at 400 nm), 551 (ex. at 395 nm), 528 (ex. at 390 nm) nm, respectively. The choice of excitation wavelength was determined by the maximum in the excitation spectra (Fig. S4†). Emission bands were assigned to the MLCT nature. As comparing with the ligands (Fig. S5†), MLCT emission for all the complexes is strongly red-shifted. Lowering the temperature down to 77 K induces for complexes 1–4 a bathochromic shift of the emission profile on 19–25 nm accompanied remarkable increase of the emission intensity (Fig. S6–S10†). On the other hand, decreasing temperature did not affect on emission maxima for complex 5.
It should be noted that varying the substituents in complexes 1, 2 and 4, which differ in the uncoordinated substituent of the ligand (4Py, 3Py and Ph respectively) results in a shift of the maximum to the blue region as a result of different degree of delocalisation of electron density. A similar trend is observed for complexes 3 and 5.
The difference in the photophysical properties of complexes 1–5 in solutions and in the solid state indicates that metal-to-ligand charge transfer (MLCT) states are no emissive in solution but it could be turned on in an aggregated state. Notably, luminescent aggregates of copper iodide clusters obtained through the microemulsion method have been reported very recently and AIE (aggregation induced emission) properties were observed, but not thoroughly investigated.8
We therefore investigated whether these complexes were AIE active by promoting increasing aggregation in THF solutions through the addition of water as an antisolvent. This experiment was conducted by adjusting the ratio of THF/water from 0% to 80% (volume/volume percentages) to monitor the changes in luminescence while maintaining the concentration of the solution at 10−5 M, with an increase in the water content gradually causing aggregation. As shown in Fig. 3 gradual increasing of water amount leads to de-intensification of LC emission at ∼405 nm, close to what was observed for the emission of THF solutions. With the addition of increasing water content over this range, the solubility of both complexes continuously decreased and microsuspensions formed, which was linked to a decrease in the intensity of the high-energy emission and the appearance of a red-shifted, broad emission band that we assigned to a MLCT emission. The luminescence intensities of complexes 1 and 2 in a THF/water mixture with 80 vol% water were 12 and 3.2 times higher than those in solution in THF, respectively. The insets depict the changes in luminescence intensity with different water fractions.
 | ||
| Fig. 3 Emission spectra of 1, measured in THF/water mixtures with different water fractions (0–80% v/v) at room temperature. Insets: plots of the luminescence y of the complexes in THF–H2O mixtures with 0 and 80% water amount. | ||
As we mentioned before, the absence of MLCT emission in the solution phase caused by efficient oxygen quenching. This means that when microsuspension is formed, the oxygen can not efficiently penetrate the microcrystals and quench long-lived triplet excited states, thus MLCT emission appears at 550 nm. However, for the solid state sample of 1, MLCT emission at room temperature is observed at 606 nm, i.e. red-shifted to microsuspension state. This means that photoluminescence of complex 1 is sensitive to the bulk crystal architecture including intermolecular interactions network and coupling with methanol solvate molecules comparing to the microcrystalline form (typical for AIE materials).
To elucidate the emission behavior of compounds 1–5 in details, their emission lifetimes and quantum yields were measured in the solid state at both 298 and 77 K (Fig S4–S8†). The results are summarized in the Table 1. Among the complexes 1–5, sample 3 shows the highest quantum yield both at room temperature (52%) and at 77 K (88%).
The time-resolved luminescence behavior of complexes 1–5 can be described by a mono-exponential decay with emission lifetimes ranging at 8.3–10.7 μs at room temperature. Lowering the temperature down to 77 K yields a prolongation of the excited state lifetimes (17.8–25 μs) for the complexes 1, 2, 4 and 5 preserving the monoexponential character. On the other hand, the emission lifetime for 3 changes drastically and at 77 K it reaches 134 μs.
Using the obtained data and the quantum yields values the radiative rates kr were determined both for 298 and 77 K (Table 1).
The noticeable red-shift of the emission, as well as decreasing of the radiative rate by about 9 times, give reason to assume the TADF manifestation for complex 3 while the other ones seem to show less pronounced TADF features.9 To support this assumption, the temperature dependence of the lifetimes for complex 3 has been recorded (Fig. 4).
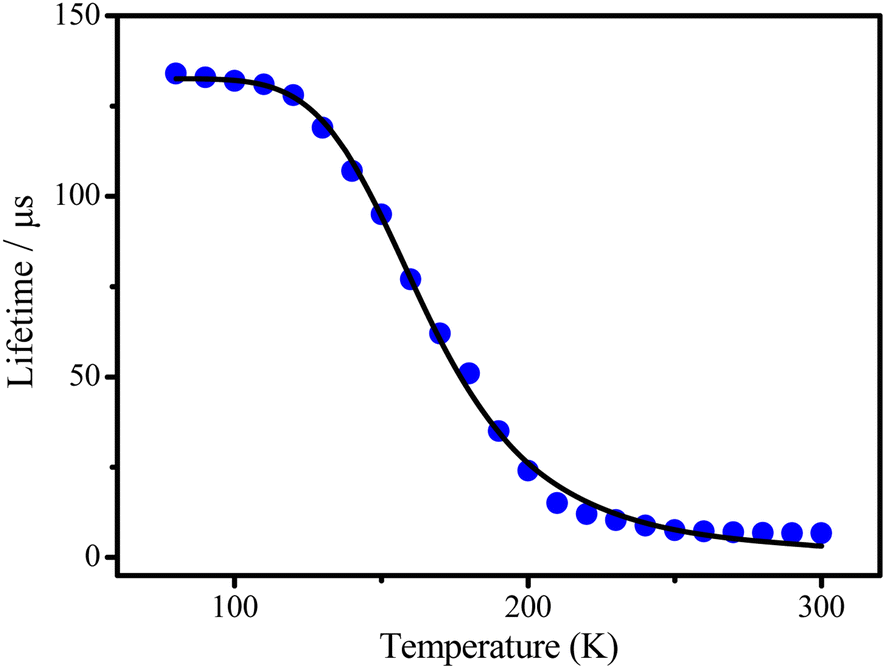 | ||
| Fig. 4 Temperature dependences of the lifetimes observed for 3. | ||
At low temperature, i.e. in the range of the plateau up to T < 130 K only a long-lived phosphorescence (T1–S0) with τ(T1) = 134 μs is observed. With increasing temperature, fast up intersystem crossing to the S1 state takes place and opens the additional radiative TADF process via the decay path from the S1 state. This leads to a drastic decrease of the emission decay time and to the observed blue shift of the emission spectrum, since the emitting S1 state lies higher in energy than the T1 state.
Applying following to the temperature dependences of lifetime allows us to determine important TADF parameters10

Copper-based complexes have recently appeared to be relevant candidates as new stimuli-responsive materials.5 The mechanochromic properties of the investigated dinuclear complexes show rather striking differences within the series. We examined the emission spectra of the samples that were ground in a mortar. Complexes 1–3 and 5 show no luminescence change upon mechanical grinding. In contrast of above complex 4 exhibits mechanochromic luminescent properties. Indeed, the greenish emission (λmax = 508 nm, λex = 395 nm) of crystalline 4 is transformed into a much more intense yellow one (λmax = 551 nm, λex = 395 nm) (Fig. 5). This change in luminescence is reversible by soft annealing of 4 powder at 100 °C for 30 min. To elucidate the mechanism of the mechanochromic luminescence of 4, PXRD patterns of this sample were measured (Fig. S11†). The powder X-ray diffraction patterns of the ground sample are generally match the patterns of crystalline sample indicate that no chemical reaction or phase transition occurred during the grinding process. The diffraction peaks became weaker and broader than those before grinding, indicating that crystal-to-amorphous transition occurred.
 | ||
| Fig. 5 Emission spectra of unground and ground crystalline powders of 4. | ||
It was previously reported that the grinding could alter Cu–Cu distances to affect the energy of 3MLTC excited states.5b Since the remarkable color and luminescence changes and the mechanochromic behavior of 4 suggested a change in the origin of luminescence, the emission lifetime and quantum yield of the ground sample were determined at 298. In contrast of crystalline sample ground sample shows a bi-exponential decay, being τ1 = 14.4 μs (67%), τ2 = 6.3 μs (33%). Considering that mono exponential kinetic was observed in the emission decay of the crystalline sample of 4, different emissive state(s) may be generated in the ground sample. Thus, the mechanochromic properties of complex 4 are most likely associated with a change in the emitting state upon crystal-to-amorphous transition. Lu et al.11 reported that grinding the related dinuclear Cu(I) complexes leads to removal of solvent molecules that shifts the emission spectrum significantly to the red similar to our observations. It seems that crystallized solvent species indeed affect the energy and lifetime of the corresponding 3MLTC states and in the grinding sample we have both microcrystals still containing the coupled solvent molecules but also the complexes that lost methanol molecules and thus emitting at lower energies (Fig. 5).
Another interesting phenomenon was also found for the complex 4 which specifically exhibits excitation-dependent colour emission behaviour. The relevant spectra of the complexes upon excitation are presented in Fig. 6. Upon excitation wavelengths below 385 nm, only minor shifts in the maximum emission wavelength at 508 nm are observed generating bright greenish-yellow emission; then excitation between 405–425 nm leads to the emission maximum shifts to 595 nm with registration by the naked eyes pink emission. As the excitation wavelength exceeds 435 nm, the emission intensity quickly lowered without noticeable shifting. In our opinion, there are two probable reasons of the excitation-dependent properties: (1) it might relate to existence of intermolecular charge-transfer states together with molecular-centered MLCT. These states can be populated and depopulated independently and the non-adiabatic coupling between them (if assume all are triplets) should be close to zero because of different space localization of them. This results in actually forbidden internal conversion between them and allows to distinguish these states independently by different excitation pulses; (2) surface traps and crystal defects in which the solvent molecules are absent or complex geometry is distorted. This is in line with mechanochromic properties of 4 discussed above when the grinding leads to bathochromic shift of emission maximum through appearance of additional emissive states most likely by removal of methanol solvent. As a result, bulk crystal species emit at shorter wavelength, but the complexes located at surface traps and defects demonstrate the red-shifted and less intensive emission.
 | ||
| Fig. 6 Excitation-dependent photoluminescence spectra of 4 in the solid state at 298 K with excitation wavelengths from 385 to 445 nm at 10 nm intervals. | ||
Theoretical studies
To obtain better insight into the origin of the emission properties of complexes 1–5, density functional theory (DFT) and time-dependent DFT (TDDFT) calculations were performed.Quantum chemical calculations of 1–5 in gas phase confirm tetragonal coordination geometry for each Cu(I) centre with the corresponding ligand atom coordination environment and two bridging iodine atoms. For 1–5 series the Cu2I2 core is planar with two Cu–I distances varying within 2.778–2.813 Å (Fig. S12–S16 in ESI†); the angles at each Cu centre are in the range 109.49–111.49°, while angles at each I centre are smaller - 68.51–70.52°. The calculated Cu⋯Cu distances, being longer (3.148–3.252 Å) than the experimental ones, display the trend of shorter Cu⋯Cu distances with 4-pyridine coordinating group and longer Cu⋯Cu distances with 3-pyridine coordinating derivatives. The Cu–P distances were calculated in the range of 2.348–2.352 Å (Fig. S12–S16 in ESI†). The Cu–N bond lengths are 2.150–2.172 Å and show the shortest distances 2.150 and 2.156 Å for the L4 and L5, respectively.
In the first excited singlet (S1) and triplet (T1) states, each Cu(I) center maintains the tetragonal coordination geometry and planar Cu2I2 core in complexes 1–5. For the S1 of 1–5, the main structural peculiarities are associated with two more aligned Cu–I bond lengths (2.723–2.762 Å, Fig. S12–S16† in ESI). The angles at each Cu center in S1 state (109.27–110.48°) are close similar to those in S0 state, while the angles at each I center are slightly larger (70.73–72.40°) than those one in S0 state. In S1 state the calculated Cu⋯Cu distances vary within 3.180–3.229 Å across all the series of 1–5. The Cu–P distances become more elongated (2.360–2.373 Å), while the Cu–N bond lengths become shorter (2.150–2.172 Å) than those calculated in S0 state of 1–5 (Fig. S12–S16 in ESI†).
For the T1 of 1–5 the two Cu–I distances vary in a narrow range of 2.725–2.776 Å (Fig. S19–S14 in ESI†). The corresponding angles at each Cu centre and at each I centre become less obtuse (105.64–107.48°) and less acute (72.52–74.36°), respectively, compared to S0 and S1 states of 1–5. The Cu–Cu distances are longer (3.235–3.350 Å) than those in S0 and S1 states of 1–5. The Cu–P distance values in T1 of 1–5 are placed between those calculated in S0 and S1 states (2.359–2.373 Å, Fig. S12–S16 in ESI†). The Cu–N bond lengths are smaller (2.077–2.140 Å) than those in S0 and S1 states of 1–5.
For the Cu(I) complexes 1–5 the first S0 → S1 transition of medium intensity was calculated at 534, 517, 483, 468 and 425 nm, respectively (Table S3, Fig. S17 in ESI†). For all studied complexes 1–5 this transition correspond to the charge transfer from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) with contribution up to 98%. Notably, the HOMO is similar for all the complexes 1–5 and is placed mainly at the Cu2I2 core (Fig. 7). The LUMO has the same nature only for the 1–3 and is placed at the coordinating pyridine conjugated with triazole fragment. Interestingly, for 4 the LUMO is localized throughout the triazole-ligand, while for 5 the LUMO covers only the triazole fragment and the terminal benzene ring (Fig. S18–S22†).
 | ||
| Fig. 7 Orbital diagram for the complexes 1–5 calculated by using B3LYP functional and 6-31G(d) basis set for the C, H, N and P atoms and the effective core potential Lanl2DZ basis set for the heavy Cu and I atoms. | ||
TD DFT calculations indicate that the observed emission bands at 606, 591, 543, 551 and 528 nm (Table 1) correspond to the vertical S1 → S0 transition calculated at 693, 638, 601, 555 and 502 nm for 1–5 (Table S4†) showing a perfect qualitative correlation between theory and experiment.
As mentioned above, among complexes 1–5 the quantum yield (QY) is the highest for 3 (52%), and the lowest for 5 (15%). This is because the small structural changes between the ground and exited states for 3 compared to other complexes 1, 2, 4, 5. For example, for 3 the angles at each Cu center and at each I center in S0, S1 and T1 change less than 2–3°, while for 5 these changes are more than 5°. For the other complexes 1, 2, 4 the changes in angles are about 4°. The small structural distortion of the S1 and T1 states of 3 reduces S–T gap and results in more efficient TADF, which leads finally to a high QY. Indeed, complex 3 demonstrate the smallest shift between vertical S1–S0 and T1–S0 emission (Tables S4 and S5 in ESI†) as well as smallest calculated adiabatic S1–T1 gap (0.013 eV) comparing to other complexes (0.036, 0.040, 0.037, 0030 for complexes 1, 2, 4, 5, respectively).
Experimental
Materials and methods
All the reagents and solvents were commercially available and used as received without further purification: 4-cyanopyridine, 3-cyanopyridine isoniazid, benzoic and nicotinic acids hydrazide, triphenylphosphine, CuI (Sigma-Aldrich). 3,5-bis(pyridin-4-yl)-1H-1,2,4-triazole (L1), 5-(pyridin-4-yl)-3-(pyridin-3-yl)-1H-1,2,4-triazole (L2), 3,5-bis(pyridin-4-yl)-1H-1,2,4-triazole (L3), 5-phenyl-3-(pyridin-4-yl)-1H-1,2,4-triazole (L4), 5-phenyl-3-(pyridin-3-yl)-1H-1,2,4-triazole (L5), and was prepared according to the literature method.12Elemental analyses of C, H, and N were performed with the EuroEA 3000 analyzer. The IR spectra were measured by the FSM 2202 spectrometer in the range of 4000–400 cm−1. Diffuse reflection spectra were recorded with a Cintra-3000 spectrophotometer for the solid-state samples. Photoluminescence and excitation spectra were recorded on the Fluorolog FL3-22 spectrometer for the complexes in solid state. Luminescence decays were measured using the same spectrometer equipped a xenon flash lamp. The luminescence quantum yields of the solid samples were determined by the absolute method using an integrating sphere.
Synthetic procedures
[Cu2(L1)2I2(PPh3)2] 3CH3OH. A mixture of CuI (0.190 g, 1 mmol), PPh3 (0.262 g, 1 mmol) and 44bpt (0.222 g, 1 mmol) was dissolved in a mixture of CHCl3 (15 ml) and CH3OH (15 ml). Stirring was continued for 6 hours and undissolved solid was filtrated. Pale yellow prismatic crystals suitable for single-crystal X-ray crystallography analysis began to form from the limpid filtrate after two days, which were collected by filtration.The procedures for the synthesis of complexes 2–5 were essentially identical to that described for 1, only different ligands were used.
The single crystal X-ray diffraction data for 1, 2, 4, and 5 were collected using the Bruker APEX II diffractometer equipped with a CCD detector and a graphite-monochromated MoKα radiation source (λ = 0.71073 Å). The single crystal X-ray diffraction data for 3 were collected using the SuperNova diffractometer equipped with a HyPix-3000 detector and a micro-focus CuKα radiation source (λ = 1.54184 Å). The structures of complexes were solved by the direct methods and refined in the full-matrix anisotropic approximation for all non-hydrogen atoms. The hydrogen atoms of water molecule were found in differential Fourier maps and their parameters were refined using the riding model. The hydrogen atoms of the carbon-containing ligand were positioned geometrically and refined by using a riding model. All the calculations were performed by direct methods in SHELX13 using Olex2.14 The crystallographic parameters and the structure refinement statistics for 1–5 are shown in Table S1.†
The thermal behavior of complexes 1–5 was studied using the simultaneous thermal analysis (STA) technique for parallel recording of TG (thermogravimetry) and DSC (differential scanning calorimetry) curves. The study was performed on an STA 6000 instrument Al-crucibles under a lid with a hole to ensure a vapour pressure of 1 atm during the thermal decomposition of the samples. The rate of heating to 700 °C was 10 °C min−1 under an N2 atmosphere. The accuracy of temperature measurements was ±0.7 °C, and that of mass changes was ±1 × 10−2 mg.
Computational details
Geometry optimizations of the complexes 1–5 in the ground singlet (S0) state were carried out at the density functional theory (DFT) computational level in vacuum approximation using the Becke–Lee–Yang–Parr three-parametric exchange-correlation functional (B3LYP)15 and the 6-31G(d)16a–c basis set for the light atoms (C, H, N and P) and the effective core potential Lanl2DZ16d basis set for the heavy Cu and I atoms. Starting from the ground-state (S0) geometries, the first excited singlet (S1) and triplet (T1) states geometries of 1–5 were optimized within the time-dependent (TD) DFT17 approach in gas phase.All the DFT and TD DFT calculations were performed using the Gaussian 16 program package.18
Conclusions
In this paper, five new highly photoluminescent dinuclear Cu(I) complexes with 3- and 4-pyridyl azole derivatives of the general formula [(μ-I)2Cu2L2(PPh3)2] were prepared. These Cu(I) complexes 1–5 exhibit a broad emission spectra in solid state at room temperature with maxima at 606, 591, 543, 551, 528 nm, respectively, shifted to the red region compared to the corresponding ligands. According to the quantum chemical calculations the observed emission is of MLCT nature. All the complexes 1–5 represent potential photoluminescent materials with quantum yields 27.5–52.0% and lifetimes 8.3–10.7 μs at room temperature and quantum yields 38.3–88.2% and lifetimes 17.8–134.7 μs at the temperature lowering to 77 K. Complex 3 shows the highest quantum yield both at room temperature (52%) and at 77 K (88%) because of efficient thermally activated delayed fluorescence behavior. Energy levels of the frontier orbitals that were responsible for photoexcitation were estimated by DFT calculations to qualitatively understand the difference in emission color. According DFT calculation the smallest structural changes between the first singlet and triplet exited states resulting in the smallest singlet-triplet energy gap for complex 3. Complex 4 demonstrates the emission transformed from the moderate greenish into more intense yellow one upon the external mechanic force stimuli (grinding). Additionally, complex 4 demonstrates excitation-dependent emission. Such mechanochromic and excitation-dependent behaviors most likely are associated with the crucial role of solvated methanol molecules, surface traps and crystal defects on the energy of emissive triplet state. The present work has demonstrated that the 3/4-pyridyl derivatives of 1,2,4-triazole can effectively construct functional Cu(I) clusters.Author contributions
Alexey Gusev, Mikhail Kiskin, Glib Baryshnikov: investigation, formal analysis, methodology, writing-original draft; Elena Braga, Mariya Kryukova, Nataliya Karaush-Karmazin: investigation, formal analysis, methodology; Ekaterina Zamnius: data curation, formal analysis, software, writing-original draft, conceptualization, funding acquisition; Boris Minaev, Wolfgang Linert: supervision, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was funded by Russian Science Foundation grant (project no. 22-73-00043). This study was also supported by the Ministry of Education and Science of Ukraine (projects no. 0121U107533 and 0122U000760). M. Kiskin thanks the state assignment of the IGIC RAS in the field of fundamental scientific research for possibility to perform X-ray diffraction analyses. GB acknowledges the financial support of the Swedish Research Council (starting grant 2020-04600).References
- (a) R. Czerwieniec, M. J. Leitl, H. H. H. Homeier and H. Yersin, Coord. Chem. Rev., 2016, 325, 2–28 CrossRef CAS; (b) J. Troyano, F. Zamora and S. Delgado, Chem. Soc. Rev., 2021, 50, 4606–4628 RSC; (c) C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed; (d) Y. Liu, S.-C. Yiu, C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2018, 375, 514–557 CrossRef CAS; (e) P. C. Ford, E. Cariati and J. Bourassa, Chem. Rev., 1999, 99, 3625–3648 CrossRef CAS PubMed; (f) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed; (g) L. P. Ravaro, K. P. S. Zanoni and A. S. S. de Camargo, Energy Rep., 2020, 6, 37–45 CrossRef.
- (a) E. Cariati, E. Lucenti, C. Botta, U. Giovanella, D. Marinotto and S. Righetto, Coord. Chem. Rev., 2016, 306, 566–614 CrossRef CAS; (b) P. C. Ford, E. Cariati and J. Bourassa, Chem. Rev., 1999, 99, 3625–3648 CrossRef CAS PubMed; (c) P. C. Ford, Coord. Chem. Rev., 1994, 132, 129–140 CrossRef CAS; (d) M. Hashimoto, S. Igawa, M. Yashima, I. Kawata, M. Hoshino and M. Osawa, J. Am. Chem. Soc., 2011, 133, 10348–10351 CrossRef CAS PubMed; (e) G. Farias, C. A. M. Salla, J. Toigo, L. G. T. A. Duarte, A. J. Bortoluzzi, E. Girotto, H. Gallardo, T. D. Z. Atvars, B. de Souza and I. H. Bechtold, Dalton Trans., 2022, 51, 1008–1018 RSC; (f) Y. Zhang, M. Schulz, M. Wächtler, M. Karnahl and B. Dietzek, Coord. Chem. Rev., 2018, 356, 127–146 CrossRef CAS.
- (a) F. Ciardelli, G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2013, 42, 857–870 RSC; (b) Z. Qiu, W. Zhao, M. Cao, Y. Wang, J. W. Y. Lam, Z. Zhang, X. Chen and B. Zhong Tang, Adv. Mater., 2018, 30, 1803924 CrossRef PubMed.
- (a) H. Tsujimoto, D.-G. Ha, G. Markopoulos, H. S. Chae, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2017, 139, 4894–4900 CrossRef CAS PubMed; (b) Z. Yang, Z. Chi, Z. Mao, Y. Zhang, S. Liu, J. Zhao, M. P. Aldred and Z. Chi, Mater. Chem. Front., 2018, 2, 861–890 RSC; (c) R. Tan, S. Wang, H. Lan and S. Xiao, Curr. Org. Chem., 2017, 21, 236–248 CrossRef CAS.
- (a) S. Perruchas, X. F. Le Goff, S. Maron, I. Maurin, F. Guillen, A. Garcia, T. Gacoin and J.-P. Boilot, J. Am. Chem. Soc., 2010, 132, 10967–10969 CrossRef CAS PubMed; (b) A. Kobayashi and M. Kato, Chem. Lett., 2017, 46, 154–162 CrossRef CAS; (c) B. Huitorel, R. Utrera-Melero, F. Massuyeau, J.-Y. Mevelec, B. Baptiste, A. Polian, T. Gacoin, C. Martineau-Corcosd and S. Perruchas, Dalton Trans., 2019, 48, 7899–7909 RSC; (d) X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC.
- (a) C. Bizzarri, C. Flechon, O. Fenwick, F. Cacialli, F. Polo, M. D. Galvez-Lopez, C.-H. Yang, S. Scintilla, Y. Sun, R. Frohlich and L. De Cola, ECS J. Solid State Sci. Technol., 2016, 5(6), R83–R90 CrossRef CAS; (b) R. Peng, M. Li and D. Li, Coord. Chem. Rev., 2010, 254, 1–18 CrossRef CAS; (c) J.-P. Zhang, Y.-Y. Lin, X.-C. Huang and X.-M. Chen, J. Am. Chem. Soc., 2005, 127, 5495–5506 CrossRef CAS PubMed; (d) J.-L. Chen, X.-F. Cao, J.-Y. Wang, L.-H. He, Z.-Y. Liu, H.-R. Wen and Z.-N. Chen, Inorg. Chem., 2013, 52, 9727–9740 CrossRef CAS PubMed; (e) J.-L. Chen, X.-Z. Tan, X.-F. Fu, X.-X. Chen, J.-Y. Wang, L.-H. Hea and H.-R. Wen, J. Coord. Chem., 2014, 67, 1186–1197 CrossRef CAS.
- R. Kobayashi, H. Imoto and K. Naka, Eur. J. Inorg. Chem., 2020, 37, 3548–3553 CrossRef , https://chemistry-europe.onlinelibrary.wiley.com/toc/10990682c/2020/2020/37.
- C. Chen, R.-H. Li, B.-S. Zhu, K.-H. Wang, J.-S. Yao, Y.-C. Yin, M.-M. Yao, H.-B. Yao and S.-H. Yu, Angew. Chem., Int. Ed., 2018, 57, 7106–7110 CrossRef CAS PubMed.
- C. Sandoval-Pauker, M. Santander-Nelli and P. Dreyse, RSC Adv., 2022, 12, 10653–10674 RSC.
- A. Schinabeck, M. J. Leitl and H. Yersin, J. Phys. Chem. Lett., 2018, 9, 2848–2856 CrossRef CAS PubMed.
- T. Lu, J.-Y. Wang, D. Tu, Z.-N. Chen, X.-T. Chen and Z.-L. Xue, Inorg. Chem., 2018, 57, 13618 CrossRef CAS PubMed.
- (a) A. Gusev, E. Braga, E. Zamnius, M. Kiskin, M. Kryukova, A. Baryshnikova, B. Minaev, G. Baryshnikov, H. Ågrend and W. Linert, RSC Adv., 2019, 9, 22143–22152 RSC; (b) A. Gusev, E. Braga, Y. Baluda, M. Kiskin, M. Kryukova, N. Karaush-Karmazin, G. Baryshnikov, A. Kuklin, B. Minaev, H. Ågren and W. Linert, Polyhedron, 2020, 191, 114768 CrossRef CAS.
- (a) G. M. Sheldrick, SADABS. Program for Scanning and Correction of Area Detector Data, Guttingen Univ., Guttingen, 1997 Search PubMed; (b) G. M. Sheldrick, SHELX97. Program for the Solution of Crystal Structures, Guttingen Univ., Guttingen, 1997 Search PubMed; (c) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- (a) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS; (b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS; (c) M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS; (d) P. J. Hay and P. J. Wadt, J. Phys. Chem., 1985, 82, 270–283 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, et al.Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2216203–2216207. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra06986e |
| This journal is © The Royal Society of Chemistry 2023 |