
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompositional dependence of Co- and Mo-supported beta zeolite for selective one-step hydrotreatment of methyl palmitate to produce bio jet fuel range hydrocarbons†
Su-Un Lee ,
Tae-Wan Kim,
Kwang-Eun Jeong,
Sungjune Lee,
Min Cheol Shin and
Chul-Ung Kim*
,
Tae-Wan Kim,
Kwang-Eun Jeong,
Sungjune Lee,
Min Cheol Shin and
Chul-Ung Kim*
Chemical & Process Technology Division, Korea Research Institute of Chemical Technology, 141, Gajeong-ro, Yuseong-gu, Daejeon 34114, South Korea. E-mail: cukim@krict.re.kr; Fax: +82-42-860-7590; Tel: +82-42-860-7504
First published on 17th January 2023
Abstract
For producing a drop-in bio jet fuel, one-step hydrotreatment, which includes deoxygenation, isomerization and cracking in one step, is essential to overcome the typical biofuel drawbacks due to high oxygen content, out of jet fuel range hydrocarbons, and low isomerization degree. Herein, Co- or/and Mo-supported Beta(25) zeolites with various Co/Mo ratios were prepared as transition metal-supported zeolite catalysts without the need for sulfidation of conventional transition metal catalysts. Based on the catalyst characterization, the Co/Mo ratio alters the metal phase with the appearance of CoMoO4 and the altered Co metal phase strongly influences the acidic properties of Beta(25) by the formation of Lewis (L) acid sites with different strengths as Co3O4 and CoMoO4 for strong and weak L acid sites, respectively. The catalytic activities were investigated for hydrotreatment of methyl palmitate as a biofuel model compound of fatty acid methyl esters. Primarily, Co is required for deoxygenation and Mo suppresses overcracking to enhance the yield of jet fuel range hydrocarbons. The Co/Mo ratio plays an important role to improve the C8–C16 selectivity by modifying the acidic properties to inhibit excessive cracking. Co5Mo10/Beta(25) achieved the best catalytic performance with the conversion of 94.2%, C8–C16 selectivity of 89.7 wt%, and high isomer ratio of 83.8% in organic liquid product. This unique modification of acidic properties will find use in the design of optimal transition metal-supported zeolite catalysts for selective one-step hydrotreatment to produce bio jet fuel range hydrocarbons.
1. Introduction
The increasing consumption of transportation fuels, the depletion of fossil fuels, and the corresponding environmental concerns have provoked interest in renewable energy sources. By utilizing diverse biomass sources consisting of triglycerides and free fatty acids (FFAs), biomass-derived fuel is a promising candidate to substitute conventional fossil fuels with green biofuel.1–3 The common commercialized technology for converting biomass to biofuel is the transesterification of triglycerides or esterification of free fatty acids (FFAs) with methanol, which produces fatty acid methyl esters (FAMEs). However, because of the high oxygen content, out of jet fuel range hydrocarbons, and low isomerization degree, FAMEs have poor combustion properties such as low heating value, thermal instability, and high freezing point.The potential of biofuels is highlighted in terms of being able to replace conventional jet fuels with renewable jet fuels, as the world jet fuel demand is likely to grow rapidly by 38% from 2005 to 2025.4 Ultimately, feasible alternatives for conventional jet fuel is a ‘drop-in’ jet fuel that is completely compatible with conventional jet fuel and existing engines in the absence of a requirement for any modifications. However, it is difficult to replace conventional jet fuels with unrefined biofuels and satisfy stringent international requirements, such as excellent oxidative stability, freezing points below −40 °C, and heat of combustion greater than 42.8 MJ kg−1.5 To achieve a ‘drop-in’ jet fuel from biofuel, one-step hydrotreatment is essential to overcome the typical biofuel disadvantages, caused by higher oxygen content and out of jet fuel range hydrocarbons, and low isomerization degree. In general, the first step is deoxygenation (DO) to produce n-alkanes, and the following step is the cracking and isomerization of n-alkanes to produce iso-alkanes in the jet fuel range from C8 to C16. The integration of DO, isomerization and cracking in one step can raise the industrial applicability with economic gains, but the development of ideal catalysts is still far behind industrial requirements.6
To upgrade the quality of bio jet fuel, various metal-supported metal oxide and zeolite catalysts have been investigated, such as Rh/ZrO2,7 CoMo/Al2O3,8 Mo/ZSM-5,1 Ni/Beta,9,10 and so forth. The use of metal oxide as a support can stabilize the active metal catalysts with high dispersion for high activity and stability such as mesoporous support11 and LDH-derived metal catalysts.12,13 More than that, zeolites are expected to outperform the intrinsic catalytic performance of metal catalysts because they provide a porous structure to reduce the diffusion limitation9 and acid sites for acid-catalyzed reactions1,10,14 and isomerization to change n-alkanes to iso-alkanes.15 These studies have found that a high-performance zeolite catalyst can be achieved by optimizing the porosity, acidity, and composition. Meanwhile, among the conventional metal catalysts for DO, noble metal catalysts, such as Rh and Pt, exhibit high efficiency, but their exorbitant costs restrict large-scale application. In the case of conventional transition metal catalysts, such as CoMo or NiMo, the introduction of sulfur for transition metal sulfides is necessary to achieve activity comparable to that of noble metal catalysts, which cause the serious deactivation and product contamination by sulfur leaching.8,16 Combining the advantages of zeolites and transition metals as bi-functional catalysts, transition metal-supported zeolite catalysts are highly attractive to cover the low activity of transition metal without sulfidation by exploiting the catalytic properties of zeolites. However, only a few reports have dealt with nonsulfided CoMo or NiMo-supported zeolite catalysts.17
In this work, we prepared Co- or/and Mo-supported Beta(25) zeolites to investigate the effects of transition metal-supported zeolite catalysts on the catalytic properties for selective one-step hydrotreatment of methyl palmitate as a model compound of FAMEs in order to produce a drop-in bio jet fuel. As representative examples of the second and third generation of renewable biofuels that do not compete with food crops such as the first generation biofuels, the biofuels derived from such as styrax japonicas and microalgae contain methyl palmitate and unsaturated fatty acids in double bonds. In the meantime, unsaturated materials in double bonds can be not only easily saturated by hydrogenation before subsequent hydrotreatment,18 but also upgraded for biochemicals through metathesis.19 Therefore, methyl palmitate was selected as model compound of FAMEs to investigate selective one-step hydrotreatment. Beta zeolite was chosen because large pore radius and high surface area can provide the diffusion of large molecules of FAMEs and generate small hydrocarbon molecules in the bio-jet fuel range.20,21 According to the change in the composition ratio of Co and Mo, the metal phase was varied with not only each single metal oxides but also CoMoO4 as the third metal phase. Compared with single metal-supported beta zeolite catalysts, the Co- and Mo-supported Beta(25) zeolites enabled a unique modification of acidic properties with CoMoO4 and the optimal condition enhanced the catalytic properties including high methyl palmitate conversion and selective C8–C16 range alkanes with high isomer ratio as desired products for jet fuel range hydrocarbons. This work provides a meaningful reference for Co- and Mo-supported beta zeolite catalysts for selective one-step hydrotreatment of bio jet fuel range hydrocarbons.
2. Experimental section
2.1 Catalyst preparation
2.2 Catalyst characterization
The characterizations of the catalysts were performed using XRD, Raman, XPS, TEM, H2-TPR, N2 sorption, NH3-TPD, and FTIR analysis.X-ray diffraction (XRD) patterns were obtained using a Rigaku Ultima IV Diffractometer operating at 40 kV and 40 mA with Cu-Kα radiation.
Raman spectra were measured with Nanophoton RAMANforce Raman spectrometer using 532 nm laser excitation.
X-ray photoelectron spectroscopy (XPS) spectra were obtained using a KRATOS AXIS NOVA spectrometer with monochromatic Al Kα X-ray (1486.6 eV). All the XPS data was calibrated based on C 1s peak at 284.8 eV.
Transmission electron microscopy (TEM) images, high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and energy-disperse X-ray spectrometry (EDS) mapping images were obtained by a field emission gun transmission and scanning transmission electron microscope (FEG-S/TEM, FEI-Talos F200S) operating at 200 kV.
Hydrogen-temperature programming reduction (H2-TPR) was estimated by BELCAT-B equipped with a thermal conductivity detector (TCD). The sample of 0.05 g was packed in a U-tube quartz cell with quartz wool. As a pretreatment step, the sample was treated in a He flow by heating to 200 °C at 10 °C min−1 and stabilized for 1 h. After cooling down to 40 °C, He flow was changed to a H2 (3.5%)/Ar flow and maintained for 3 h. When the TCD signal was stabilized, the sample was heated to 1000 °C at 10 °C min−1 in a H2 (3.5%)/Ar flow, and TCD signals were recorded by the temperature. The water product was trapped by passing the effluent gas from the reactor through a trap of molecular sieves 13X.
Nitrogen (N2) adsorption–desorption experiments were carried out by a Micrometrics TriStar 3000 instrument. The sample was reduced in a tubular furnace by H2 flow at 500 °C for 4 h for the same pretreatment with the catalytic reaction test. Then, the reduced sample was degassed under vacuum condition at 300 °C for 5 h and N2 adsorption–desorption experiment was conducted at −196 °C. Total surface area and pore volume were calculated by the BET equation and the N2 adsorption method, respectively. Surface area and pore volume of micropore and mesopore were estimated by t-plot method.
The temperature programming desorption of ammonia (NH3-TPD) was measured by BELCAT-B equipped with a TCD. The sample was reduced in a tubular furnace by H2 flow at 500 °C for 4 h for the same pretreatment with the catalytic reaction test. Then, the sample of 0.05 g was packed in a U-tube quartz cell with quartz wool and pretreated at 150 °C for 2 h to remove the moisture. The sample was placed in a NH3 (30 mol%)/He flow for 30 min, and weakly physisorbed ammonia was eliminated by purging in the He flow for 90 min. The sample was heated from 100 °C to 700 °C at 10 °C min−1 in the He flow, and the amount of the desorbed ammonia was recorded by TCD signals.
For Fourier transformation infrared (FTIR) spectra of adsorbed pyridine, the sample was prepared after the reduction in a tubular furnace by H2 flow at 500 °C for 4 h for the same pretreatment with the catalytic reaction test. Then, thin wafer (ca. 13 mg) was produced by pressing the sample in the type of powder and pretreated in an IR cell at 150 °C for 2 h to remove the moisture. After cooling down to 60 °C, pyridine was purged to adsorb on the acidic sites of pretreated samples at 60 °C for 10 min. The physisorbed and weakly bound species were removed under a He flow at 150 °C for 3 h. IR spectrum was obtained from an FT-IR spectrometer (Thermo SCIENTIFIC Nicolet6700) equipped with thermal detector.
2.3 Catalytic performance tests
The catalytic reaction tests of methyl palmitate as a biofuel model component of fatty acid methyl esters were carried out in a semi-batch reaction system at 280 °C under a pressure of 20 bar for 2 h (the catalytic conversions of palmitic acid and hexadecane were also followed the same procedures). Before conducting the catalytic performance, the catalyst was reduced in a tubular furnace by H2/N2 mixed flow at 500 °C for 4 h. Then, 1.0 g of the catalyst and 10 g of methyl palmitate in the absent of solvent were loaded in the reactor (100 ml, Parr Instrument Co.). Prior to the reaction, the atmosphere in the reactor was charged with purging nitrogen and then hydrogen was pressurized at 20 bar. The temperature in the reactor was raised up to 280 °C and held for 2 h. The reaction was initiated by adjusting the atmosphere to 20 bar of hydrogen and proceeded with stirring (500 rpm). After 2 h, the reaction was terminated by rapidly cooling down to room temperature. The liquid and solid products were separated by centrifugation after the reaction.The liquid product was measured on a balance for mass balance by weight change of before and after the reaction. Prior to gas chromatographic analysis, the liquid samples had to be dissolved in pyridine and silylated with N,O-bis(trimethyl)-trifluoroacetamide (BSTFA) including eicosane as an internal standard for the quantitative analysis. The silylation treatment is required to prevent clogging of the GC column due to the reaction between organic acids and GC packing materials because the product can be produced not only as hydrocarbon component but also as free fatty acid, which is an acid component. Then, the silylated samples were analyzed with a gas chromatograph (YoungLin GC 3000) equipped with HP-5 column and flame ionization detector (FID).
| Conversion of methyl palmitate = (moles of converted methyl palmitate/moles of the starting methyl palmitate) × 100% |
| Selectivity of products in organic liquid product = (moles of each product/moles of total products) × 100%. |
3. Results and discussion
3.1 Compositional characterization of catalysts
The catalysts in the oxidized form were obtained by wet-impregnation and subsequent calcination. As bimetallic catalysts, the Co- and Mo-supported Beta(25) zeolites were described as “CoxMoy/Beta(25)” including Co10Mo5/Beta(25), Co10Mo10/Beta(25), Co5Mo10/Beta(25), Co3Mo10/Beta(25), and Co2Mo10/Beta(25), where ‘x’ and ‘y’ are the weight% of Co and Mo, respectively. As monometallic catalysts, Co10/Beta(25) and Mo10/Beta(25) were prepared containing 10 weight% of Co and Mo, respectively. The crystalline phases were revealed by the X-ray powder (XRD) patterns as shown in Fig. 1. The XRD pattern of Co10/Beta(25) demonstrates the characteristic diffraction peaks of Co3O4 at 19.1°, 31.4°, 36.9°, 44.7°, 55.8°, 59.5°, and 65.4°, which are assigned to (111), (220), (311), (400), (422), (511), (440) planes, respectively. On the other hands, the crystalline phase of Mo species is not observed in the XRD pattern of Mo10/Beta(25), so it can be attributed to amorphous phase and/or high dispersion below the size of the XRD detection limit. When Co and Mo were co-impregnated in the Co- and Mo-supported Beta(25), a new diffraction peak was detected at 2θ of 26.4° corresponding to the (002) plane of CoMoO4. It means that co-impregnation of Co and Mo induces the formation of CoMoO4 phase. When the content of Co was decreased below Co3Mo10/Beta(25) and Co2Mo10/Beta(25), the diffraction peaks of Co3O4 disappeared; therefore, it is assumed that Co would form CoMoO4 with Mo rather than Co3O4 until the unconverted Co to CoMoO4 remains in excess of Mo.22 To confirm the Mo effect on the dispersion of Co species,23,24 the mean crystallite size of Co3O4 was calculated from the diffraction peak at 2θ = 36.9° with the Scherrer equation; 18.2 nm for Co10/Beta(25), 18.4 nm for Co10 Mo5/Beta(25), 15.7 nm for Co10 Mo10/Beta(25), and 16.6 nm for Co5 Mo10/Beta(25). The mean crystallite size of Co3O4 seems to decrease from around 18 nm to around 16 nm for sample loaded with more than 10 wt% Mo, but within the range of no significant difference. Regarding the crystallinity of Beta zeolite, two prominent peaks appear at 2θ of 7.6° and 22.6° indexed to the (101) and (302) planes of Beta zeolite. However, the peak intensities of Beta zeolite weaken with metal loading, especially Co, indicating the incorporation of metal species with zeolite framework,25 and it will be considered with H2-TPR results.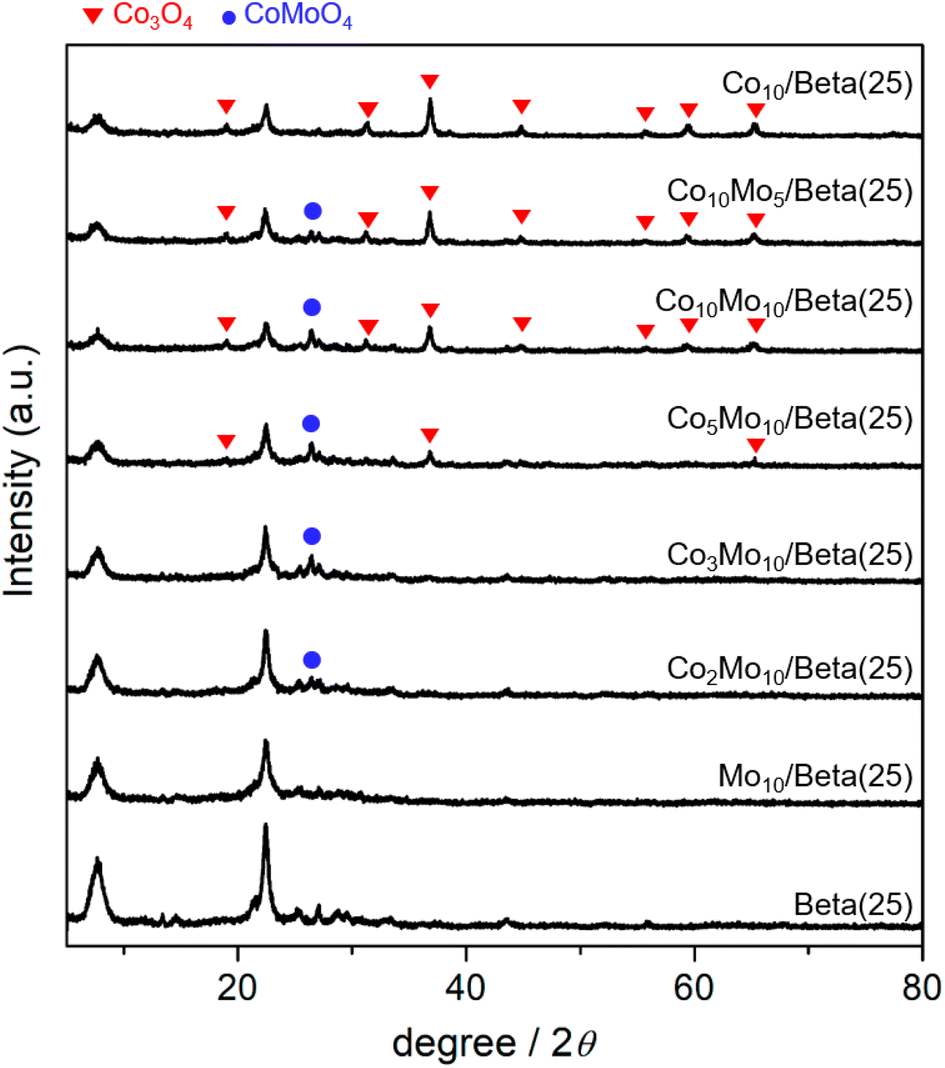 | ||
| Fig. 1 XRD patterns of Co- or/and Mo-supported Beta(25) and Beta(25) obtained by wet-impregnation and subsequent calcination. The positions of characteristic peaks are referenced from the JCPDS database (BEA: PDF# 01-074-8795, Co3O4: PDF# 00-042-1467, CoMoO4: PDF# 00-021-0868). | ||
To further clarify the phases of supported metal oxide on the Beta(25) zeolites, Raman spectra were obtained, as shown in Fig. 2. Co10/Beta(25) shows peaks at 474, 515, 615, and 681 cm−1 in the range of 400–800 cm−1, which correspond to the typical Eg, F22g, F33g, and A1g vibration modes of crystalline Co3O4,26 respectively. Mo10/Beta(25) shows peaks at 952 and 907 cm−1, which are attributed to the symmetric stretching of the Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in the polymeric molybdate species.27 The intense peak at 952 cm−1 is considered to be octahedral molybdate species interacting weakly with the support,28–30 and the weak peak at 907 cm−1 originates from isolated tetrahedral molybdates species interacting strongly with the support.31 Generally, bulk MoO3 crystallites display the Raman peaks around 990 cm−1, 820 cm−1, and 300 cm−1 corresponding to the bending mode of MO bonds, the stretching mode of MO, and the antisymmetric stretching mode of Mo–O–Mo, respectively.32 In our case, the absence of Raman peaks from bulk MoO3 crystallites suggests the small size of (MoO3)n oligomers, which can also explain the absence of X-ray diffraction for MoO3.33 As for Co- and Mo-supported Beta(25), Raman spectra confirm the formation of CoMoO4 with the appearance of new peak at 941 cm−1 and 820 cm−1, which are assigned to the Mo–O–Co stretching vibrations of CoMoO4.34 However, the Raman peaks for crystallite Co3O4 are not detected in Co2Mo10/Beta(25) and Co3Mo10/Beta(25) with low Co/Mo ratio, and start to appear from Co5Mo10/Beta(25). In consistent with the XRD results, it supports the preference for the formation of CoMoO4 than mono metal oxides, and thus Co3O4 is concurrently formed with CoMoO4 above a certain Co/Mo ratio, such as from Co5Mo10/Beta(25), in these findings. The difference in metal phase could be beneficial for better understanding the effect of the Co/Mo ratio on the following physicochemical and catalytic properties.
O bond in the polymeric molybdate species.27 The intense peak at 952 cm−1 is considered to be octahedral molybdate species interacting weakly with the support,28–30 and the weak peak at 907 cm−1 originates from isolated tetrahedral molybdates species interacting strongly with the support.31 Generally, bulk MoO3 crystallites display the Raman peaks around 990 cm−1, 820 cm−1, and 300 cm−1 corresponding to the bending mode of MO bonds, the stretching mode of MO, and the antisymmetric stretching mode of Mo–O–Mo, respectively.32 In our case, the absence of Raman peaks from bulk MoO3 crystallites suggests the small size of (MoO3)n oligomers, which can also explain the absence of X-ray diffraction for MoO3.33 As for Co- and Mo-supported Beta(25), Raman spectra confirm the formation of CoMoO4 with the appearance of new peak at 941 cm−1 and 820 cm−1, which are assigned to the Mo–O–Co stretching vibrations of CoMoO4.34 However, the Raman peaks for crystallite Co3O4 are not detected in Co2Mo10/Beta(25) and Co3Mo10/Beta(25) with low Co/Mo ratio, and start to appear from Co5Mo10/Beta(25). In consistent with the XRD results, it supports the preference for the formation of CoMoO4 than mono metal oxides, and thus Co3O4 is concurrently formed with CoMoO4 above a certain Co/Mo ratio, such as from Co5Mo10/Beta(25), in these findings. The difference in metal phase could be beneficial for better understanding the effect of the Co/Mo ratio on the following physicochemical and catalytic properties.
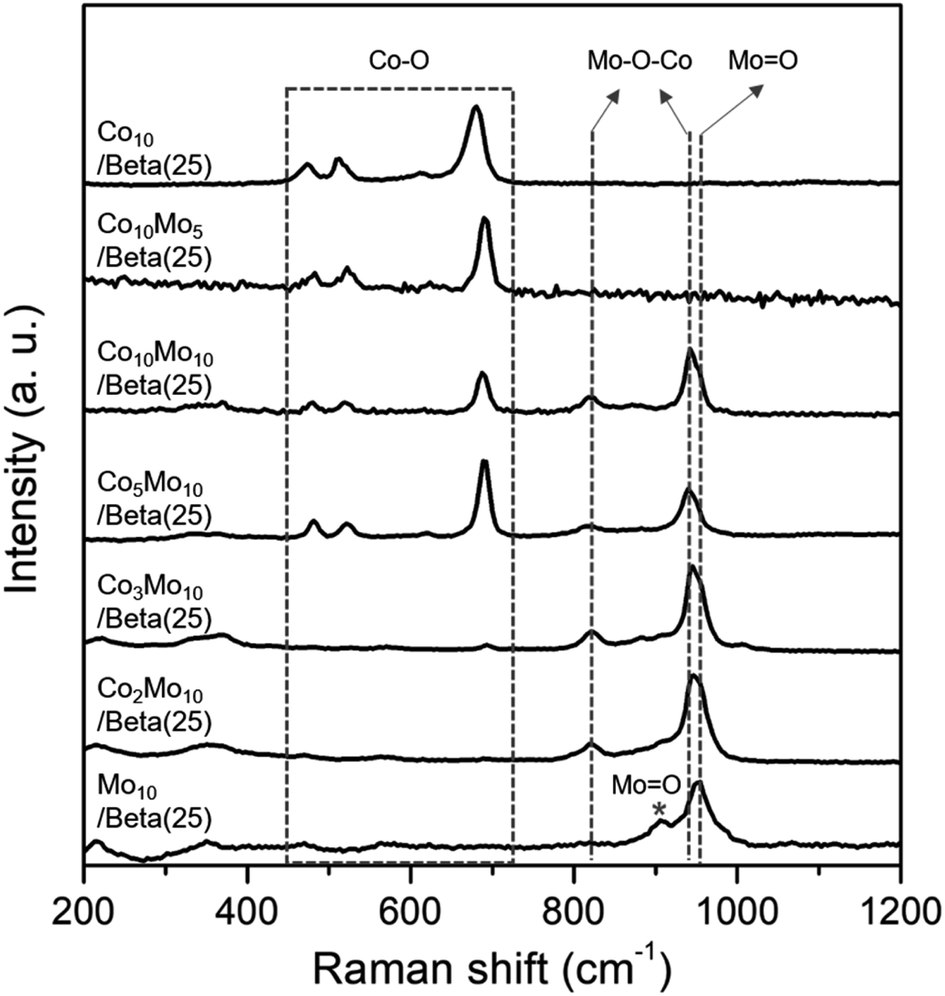 | ||
| Fig. 2 Raman spectra of Co- or/and Mo-supported Beta(25) zeolites after the wet-impregnation and subsequent calcination. | ||
The XPS experiments were conducted in the binding energy (BE) ranges of corresponding to Co 2p and Mo 3d to elucidate the types of surface metal species in the prepared catalysts as shown in Fig. S1.† The Co 2p XPS spectrum of Co10/Beta(25) (Fig. S1a†) presents intense doublet peaks at 797.3 eV (Co 2p1/2) and 781.2 eV (Co 2p3/2), and a satellite accompanying each doublet at higher BEs. Considering that Co10/Beta(25) contains Co3O4 according to the XRD analysis, it has relatively higher BE than the typical BE of Co3O4 observed at 779.9 eV (Co 2p3/2)35 by 1.3 eV. Similarly, the Mo 3d XPS spectrum of Mo10/Beta(25) in Fig. S1c† exhibits the BE of Mo 3d5/2 at 233.5 eV, and it is also higher than the typical BE for Mo6+ (232.8 eV)36 by 0.7 eV. The upward shift in the BEs of Co and Mo are caused by the deficient electron density of the wet-impregnated metal species in Beta(25). It was previously reported that Brønsted acid sites of zeolites act as electron acceptors and induce the electron transfer from metal particles to zeolites.37,38 Therefore, it is worth mentioning that metal–support interaction, especially on Brønsted acid sites. On the other hand, the Co 2p XPS of Co5Mo10/Beta(25) in Fig. S1b† presents the peak of Co 2p3/2 at 783.0 eV, which is attributed to Co3+ in the form of CoMoO4 the typical BE of CoMoO4 observed at 781.2 eV39 with shift to higher BE by 1.8 eV. With regard to the Mo 3d XPS of Co5Mo10/Beta(25) in Fig. S1d,† it can be deconvoluted into two peaks, which can be indexed to Mo6+ at 233.6 eV and CoMoO4 at 232.3 eV. The result confirms the existence of not only CoMoO4 but also MoO3, which cannot be revealed from the XRD analysis. Therefore, the existence of MoO3 cannot be excluded even in the Co- and Mo-supported Beta(25) zeolites with low Co/Mo ratio in spite of that the preference of formation of CoMoO4.
The spatial distribution of elemental composition in association with each metal phase was observed by transmission electron microscopy (TEM), high-angle annular dark-field scanning TEM (HAADF-STEM), and the corresponding energy-dispersive X-ray spectroscopy (EDS) elemental mapping images as shown in Fig. 3. As for Co10/Beta(25), the Co3O4 phase was confirmed as an agglomerated Co domain independent of Beta(25) zeolites by TEM image in Fig. 3a with the dark contrast and the corresponding HAADF-STEM-EDS mapping image in Fig. 3d with the green color. In the case of Mo10/Beta(25), the Mo domain is not distinctly marked with different contrast in the TEM image (Fig. 3b), and the compositional distribution of Mo shows good matching with those of Si and Al in the HADDF-STEM-EDX mapping image (Fig. 3e) due to high dispersion over Beta(25) zeolite. When Co and Mo are co-impregnated in Co5Mo10/Beta(25), the Mo species are observed with an overlap of the Co domain as shown in Fig. 3f, which is also represented by the dark contrast in the TEM image in Fig. 3c. This region simultaneously observed with Co and Mo has the CoMoO4 phase. In contrast, a non-overlapping Co domain with Mo has the additional Co3O4.
 | ||
| Fig. 3 TEM (a–c) and corresponding HADDF-STEM-EDX-mapping (d–f) images of (a and d) Co10/Beta(25), (b and e) Mo10/Beta(25), and (c and f) Co5Mo10/Beta(25) after the wet-impregnation and subsequent calcination. | ||
By analyzing H2-temperature-programmed reduction (TPR) in Fig. 4, the transformation of metal oxide phases was estimated according to reduction temperature. As shown in Fig. 4, Co10/Beta(25) exhibits two intense peaks at 313 °C and 354 °C, which can be attributed to the reduction of Co3O4 through Co3+ to Co2+ and Co2+ to Co0, respectively.40 In the case of Mo10/Beta(25), the H2-TPR profile has two overlapped peaks at 492 °C and 548 °C, and a broad peak over 589 °C. The consecutive reductions are attributed to MoO3 (Mo6+) to MoO2 (Mo4+) and then to Mo (Mo0). Considering that the H2-TPR profiles of Co10/Beta(25) and Mo10/Beta(25) show the overlapped and broad peaks, it is possible that the wet-impregnated metal species loaded on the surface of zeolites establish different degree of metal–support interaction and thus reduced at different temperature. From the H2-TPR profile of the Co and Mo-supported Beta(25) zeolites, the reduction of Co3O4 was detected as the first peak in the temperature range of 285 °C and 450 °C. As decreasing the Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo ratio, the intensity and temperature range of the first peak gradually decreased, which indicates that less Co3O4 was formed with improved reducibility. As the XRD results confirmed that the crystallinity of Beta zeolite was better maintained with low Co content, the improved reducibility at the low Co:Mo ratio compared to Co10/Beta(25) could be attributed to the weak interaction between metal and zeolite framework. Then, H2-consumption from the reduction of Co3O4 was finally disappeared in Co2Mo10/Beta(25) and Co3Mo10/Beta(25). It is in good agreement with that Co combines with Mo to form CoMoO4 rather than independently becoming cobalt oxides when Co and Mo are co-impregnated.22 Instead, the reduction of CoMoO4 phase was observed with new three reduction peaks at higher temperature than that of cobalt oxides. More in detail, the first two peaks between 450–610 °C are attributed to the reduction of CoMoO4 through CoMoO4 to CoMoO3 and CoMoO3 to Co3Mo, respectively. The latter at approximately 780 °C is assigned to a further reduction of Co3Mo to Co2Mo3.41 Especially, the reduction of CoMoO4 to Co3MO accompanies the additional formation of MoO3 and Mo4O11,42 so there is a limitation of separating the reduction from MoO3 due to overlapping H2-consumption. Before conducting catalytic reaction test for hydrotreatment of methyl palmitate at 280 °C under a hydrogen pressure of 20 bar, the catalysts were pretreated under hydrogen atmosphere at 500 °C. From the XRD pattern in Fig. S2,† the phase transformation of metal oxides after hydrogen reduction was identified to define the active phase. The XRD pattern of the reduced Co10/Beta(25) confirms the co-existence of CoO and metallic Co, because the complete reduction of cobalt oxides to metallic cobalt requires to be reduced up to 740 °C according to the TPR analysis. The reduced Co and Mo-supported Beta(25) displayed the typical XRD peaks of Co and Co3Mo except CoO. As the Co:Mo ratio decreases, the peak intensity for Co3Mo increases and that for monometallic Co decreases. Thus, it also demonstrates that the reduced Co and Mo-supported Beta(25) favors to obtain bimetallic Co and Mo oxides/metal rather than single Co metal/oxide because all the Co species would form CoMoO4 with Mo and the excess Co form Co3O4. Therefore, it concludes that all the Co- and Mo-supported Beta(25) contains CoMo alloy metal/oxide and Mo oxides after hydrogen reduction, but the presence of Co metal/oxides are dependent on the Co/Mo ratio. Combined with the above analysis results, the control of the Co/Mo ratio does not simply change the loading amount of each metals/oxides, but diversifies the phase of a single metal/oxide into a mixed metal/oxide with the appearance of CoMoO4 phase. Therefore, it is meaningful to focus on the phase difference in relation with noticeable change in their catalytic properties.
:Mo ratio, the intensity and temperature range of the first peak gradually decreased, which indicates that less Co3O4 was formed with improved reducibility. As the XRD results confirmed that the crystallinity of Beta zeolite was better maintained with low Co content, the improved reducibility at the low Co:Mo ratio compared to Co10/Beta(25) could be attributed to the weak interaction between metal and zeolite framework. Then, H2-consumption from the reduction of Co3O4 was finally disappeared in Co2Mo10/Beta(25) and Co3Mo10/Beta(25). It is in good agreement with that Co combines with Mo to form CoMoO4 rather than independently becoming cobalt oxides when Co and Mo are co-impregnated.22 Instead, the reduction of CoMoO4 phase was observed with new three reduction peaks at higher temperature than that of cobalt oxides. More in detail, the first two peaks between 450–610 °C are attributed to the reduction of CoMoO4 through CoMoO4 to CoMoO3 and CoMoO3 to Co3Mo, respectively. The latter at approximately 780 °C is assigned to a further reduction of Co3Mo to Co2Mo3.41 Especially, the reduction of CoMoO4 to Co3MO accompanies the additional formation of MoO3 and Mo4O11,42 so there is a limitation of separating the reduction from MoO3 due to overlapping H2-consumption. Before conducting catalytic reaction test for hydrotreatment of methyl palmitate at 280 °C under a hydrogen pressure of 20 bar, the catalysts were pretreated under hydrogen atmosphere at 500 °C. From the XRD pattern in Fig. S2,† the phase transformation of metal oxides after hydrogen reduction was identified to define the active phase. The XRD pattern of the reduced Co10/Beta(25) confirms the co-existence of CoO and metallic Co, because the complete reduction of cobalt oxides to metallic cobalt requires to be reduced up to 740 °C according to the TPR analysis. The reduced Co and Mo-supported Beta(25) displayed the typical XRD peaks of Co and Co3Mo except CoO. As the Co:Mo ratio decreases, the peak intensity for Co3Mo increases and that for monometallic Co decreases. Thus, it also demonstrates that the reduced Co and Mo-supported Beta(25) favors to obtain bimetallic Co and Mo oxides/metal rather than single Co metal/oxide because all the Co species would form CoMoO4 with Mo and the excess Co form Co3O4. Therefore, it concludes that all the Co- and Mo-supported Beta(25) contains CoMo alloy metal/oxide and Mo oxides after hydrogen reduction, but the presence of Co metal/oxides are dependent on the Co/Mo ratio. Combined with the above analysis results, the control of the Co/Mo ratio does not simply change the loading amount of each metals/oxides, but diversifies the phase of a single metal/oxide into a mixed metal/oxide with the appearance of CoMoO4 phase. Therefore, it is meaningful to focus on the phase difference in relation with noticeable change in their catalytic properties.
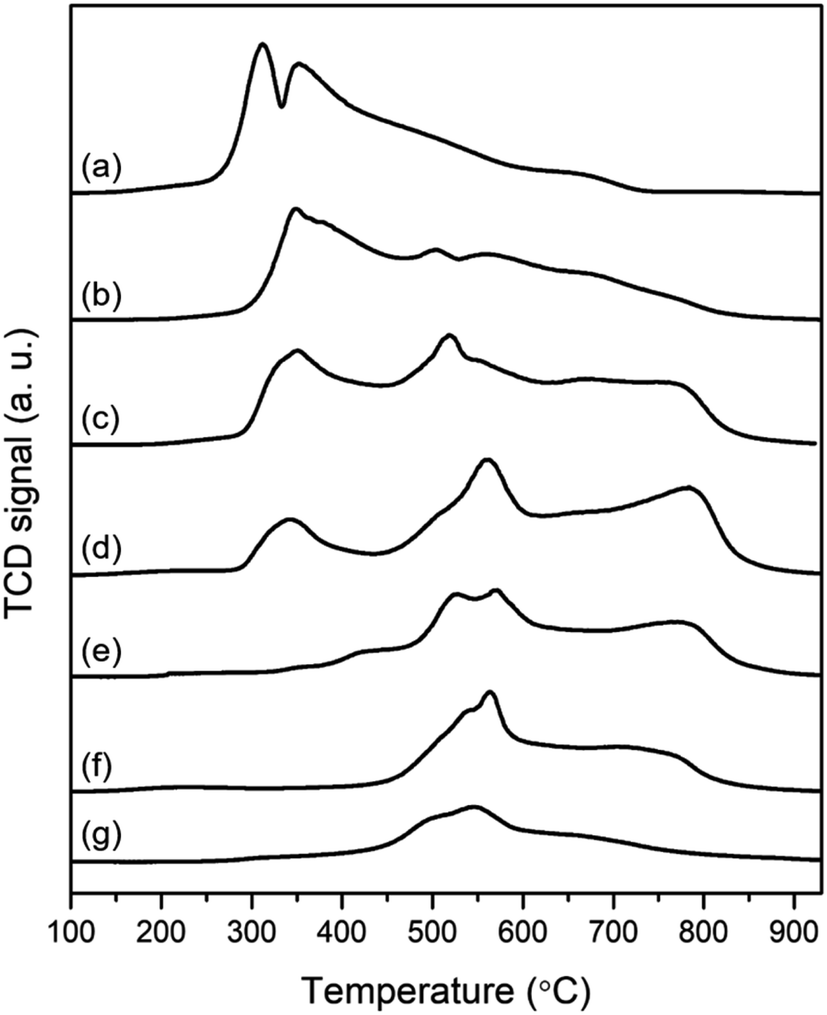 | ||
| Fig. 4 H2-TPR of the catalysts after the wet-impregnation and subsequent calcination; (a) Co10/Beta(25), (b) Co10Mo5/Beta(25), (c) Co10Mo10/Beta(25), (d) Co5Mo10/Beta(25), (e) Co3Mo10/Beta(25), (f) Co2Mo10/Beta(25), and (g) Mo10/Beta(25). | ||
3.2 Modified properties by the Co/Mo ratio with respect to catalytic property: surface area, pore structure and acidity
Typically, the main factors that affect the catalytic activity of a catalyst are surface area, pore structure and acidic property. Among them, surface area and pore volume are basic factors that provide reaction sites and promote diffusion of reactants and products, and thus estimated from the isothermal adsorption and desorption isotherms of N2 at 77 K (Table S1†) with the BET and t-plot method. The catalysts obtained by the wet-impregnation and subsequent calcination were reduced following the same pretreatment conditions of the catalytic reaction test to reflect the same state in the catalytic reaction test. Compared to the pristine Beta(25) zeolite, the surface area and pore volume of the Co- or/and Mo-supported Beta(25) zeolites were reduced, but the difference according to the amount of metal loading was insignificant. Thus, the effects of the surface area and pore volume were excluded to find the factors determining the difference in catalytic activity of individual Co- or/and Mo-supported Beta(25) zeolites.For the analysis of the acidic property, an NH3-temperature-programmed desorption (TPD) was measured in Fig. 5 and the amount of acid sites was estimated in Table 1. Note that all the samples were also reduced prior to NH3-TPD analysis, which is the same pretreatment used for the catalytic reaction test. The supported metal catalyst does not appear to be relevant with respect to the total amount of acid sites. In order to further clarify the difference in acidic properties according to the metal phase difference, the Co- or/and Mo-supported Beta(25) zeolites were classified into four categories based on the catalysts obtained by the wet-impregnation and subsequent calcination for clarity: group I: Co10/Beta(25) with Co3O4; group II: Co10Mo5/Beta(25), Co10Mo10/Beta(25), and Co5Mo10/Beta(25) with Co3O4 and CoMoO4 + MoO3; group III: Co3Mo10/Beta(25) and Co2Mo10/Beta(25) with CoMoO4 and MoO3; group IV: Mo10/Beta(25) with MoO3 as described in Table 1. With respect to the NH3-TPD of the pristine Beta(25) zeolite in Fig. 5a, the NH3 desorption peaks are deconvoluted into three desorption peaks centered in the temperature ranges of 100–200 °C (weak acidity), 200–350 °C (medium acidity), and above 350 °C (strong acidity) using the Gaussian curve fitting method. The detailed deconvolution results are demonstrated in Fig. S3,† and the corresponding peak temperature and density of acid sites are calculated in Table 1. As shown, the NH3-TPD of Beta(25) indicates weak and medium acidity in the main acid sites, but also show a very small amount of strong acidity. When Co is added to Beta(25) zeolite for Co10/Beta(25) of group I, the amounts of weak and medium acid sites decrease and a new intensive desorption peak appears at 371 °C. A similar phenomenon has been reported in the literature, and these results were attributed to the fact that the weak acid sites on the surface of hydroxyl groups are covered by Co species and the strong acid sites are created by the tendency of Co to form a tetrahedral CoO42− arrangement in the zeolite framework.43–45 On the other hand, the addition of Mo induces a slight decrease in the NH3-desorption peak intensity at low temperature and a peak shift of the three NH3-desorption peaks to the lower temperature range in the NH3-TPD when compared with Beta(25) and Mo10/Beta(25) in Fig. 5b and Table 1. In the case of the Co- and Mo-supported Beta(25) zeolites belonging to group II and III, the NH3-desorption peak generally decreased at weak and medium temperature, and the peak appeared at high temperature showed a tendency to increase, which was consistent with Co10/Beta(25). As decreasing the Co/Mo ratio from Co10Mo5/Beta(25) to Co2Mo10/Beta(25), the intensity of NH3-desorption peak shift from high temperature to low temperature while maintaining a similar total amount (see also Table 1). Noticeably, group III showed a dramatic decrease in the NH3-desorption peak at high temperature and the position of the peak also appeared at higher temperature range of 410–430 °C, unlike 380–400 °C in group II. In terms of Co metal phase, group II contains Co3O4 in the same forms as group I and CoMoO4, but group III contains Co only as CoMoO4. Therefore, it can be seen that the Co loading induces different effects on the acidic properties in relation to the metal phase in which Co is present.
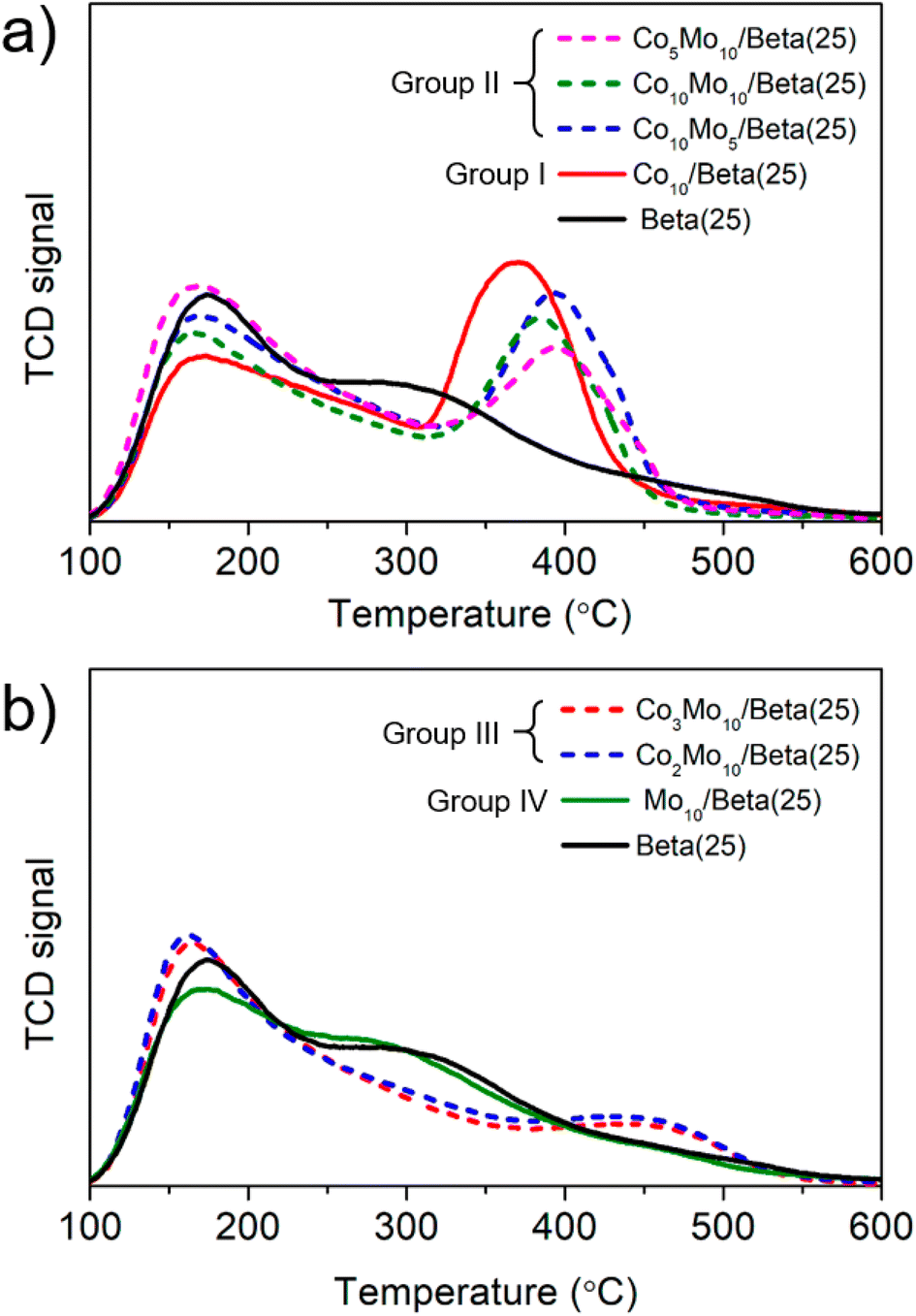 | ||
| Fig. 5 NH3-TPD of catalysts after the reduction under the same pretreatment condition of the catalytic reaction test: (a) Co10/Beta(25), Co10Mo5/Beta(25), Co10Mo10/Beta(25), Co5Mo10/Beta(25), and Beta(25), and (b) Mo10/Beta(25), Co2Mo10/Beta(25), Co3Mo10/Beta(25), and Beta(25), which are separately classified according to the combination of metal phases. | ||
| Group | Sample | Metal phase | NH3-TPD | FTIR of adsorbed pyridine | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Weak acid sitesa | Medium acid sitesa | Strong acid sitesa | Total acid sites (mmol g−1) | Bb (mmol g−1) | Lb (mmol g−1) | B/Lb | ||||||
| T (°C) | Density (mmol g−1) | T (°C) | Density (mmol g−1) | T (°C) | Density (mmol g−1) | |||||||
| a Peak temperature and the corresponding amounts of acid sites calculated from the deconvolution results of NH3-TPD profiles (Fig. S3).b Quantity of Brønsted (B) and Lewis (L) acid sites determined by multiplying the B/L ratio from FTIR spectra of adsorbed pyridine and the total acid sites concentration from NH3-TPD profiles. | ||||||||||||
| I | Co10/Beta(25) | Co3O4 | 164 | 0.24 | 237 | 0.43 | 371 | 0.66 | 1.33 | 0.17 | 1.16 | 0.15 |
| II | Co10Mo5/Beta(25) | Co3O4 + CoMoO4 + MoO3 | 167 | 0.30 | 247 | 0.54 | 396 | 0.53 | 1.37 | 0.19 | 1.18 | 0.16 |
| Co10Mo10/Beta(25) | 164 | 0.26 | 240 | 0.47 | 386 | 0.47 | 1.20 | 0.18 | 1.02 | 0.18 | ||
| Co5Mo10/Beta(25) | 164 | 0.37 | 242 | 0.53 | 393 | 0.47 | 1.37 | 0.20 | 1.17 | 0.17 | ||
| III | Co3Mo10/Beta(25) | CoMoO4 + MoO3 | 164 | 0.30 | 231 | 0.53 | 416 | 0.31 | 1.14 | 0.21 | 0.93 | 0.23 |
| Co2Mo10/Beta(25) | 163 | 0.32 | 238 | 0.59 | 426 | 0.29 | 1.20 | 0.31 | 0.89 | 0.35 | ||
| IV | Mo10/Beta(25) | MoO3 | 167 | 0.31 | 258 | 0.66 | 395 | 0.23 | 1.19 | 0.78 | 0.41 | 1.89 |
| Beta(25) | — | 171 | 0.35 | 276 | 0.79 | 453 | 0.11 | 1.24 | 0.58 | 0.66 | 0.88 | |
To further clarify the types of the acid sites in the catalysts between Brønsted (B) and Lewis (L) acid site, the Fourier transformation infrared (FTIR) spectra of adsorbed pyridine were obtained as shown in Fig. S4.† The quantity of B and L acid sites are reflected on the IR peaks of 1550 cm−1 and 1450 cm−1, respectively, as the stretching bands of pyridine adsorbed to each acid site. As described in Table 1, the B/L ratio was calculated from the ratio of the corresponding peak area, and each quantity is determined by multiplying the B/L ratio from the FTIR spectra of adsorbed pyridine and the total acid site concentration from the NH3-TPD profiles. Relative to Beta(25), Co10/Beta(25) exhibits a significant decrease in the B/L ratio from 0.88 to 0.15 due to the formation of L acid sites. In general, the supported Co component can act as an acceptor of electron pairs as L acid sites.43,46 Along with the NH3-TPD results showing strong acid sites formation on the Co-supported Beta(25) zeolites, new strong acid sites can be interpreted as L acid sites. Meanwhile, Mo10/Beta(25) causes the large increase in the B/L ratio from 0.88 to 1.03, and the quantities of each type of acid site demonstrate the formation of B acid sites and the disappearance of L acid sites. Regarding new B acid sites in the Mo-supported Beta(25) zeolites, a likely cause is the tetrahedral Mo species strongly bound in a monodentate manner to Al in the support or Mo in a polymolybdate layer, according to Rajagopal et al., who made a similar observation over MoO3/silica-aluminas.47,48 The coordination of Mo as tetrahedral molybdates over Beta(25) in Mo10/Beta(25) was confirmed by the Raman spectra shown in Fig. 2. Since the NH3-TPD of Mo10/Beta(25) showed the reduced peak intensity at low temperature and the peak shift to lower temperature, the acid strength of the increased B and decreased L acid sites should result in the decreased acidity than Beta(25). Combined with the characterization of the NH3-TPD and FTIR spectra of adsorbed pyridine, Co results in the formation of strong L acid sites, and Mo generates weak B acid sites in the single metal-supported Beta(25). When Co and Mo were co-supported over the Beta(25) zeolites, the quantity of L acid sites increased by increasing the Co/Mo ratio as observed on the Co10/Beta(25). However, unlike the Mo10/Beta(25), B acid sites decreased over the Co- and Mo-supported Beta(25) zeolites. As previously reported by Kiviat et al., Co–Mo/Al2O3 showed fewer B acid sites than Mo/Al2O3 due to the Co ions interacting with the molybdate (Mo6+) phase.49 More importantly, group III showed a large increase in the formation of L acid sites similar to Co10/Beta(25) from FTIR spectra of adsorbed pyridine, but a slight increase in the amount of strong acid sites the from the NH3-TPD profiles. Therefore, the introduction of Co causes the formation of L acid sites with different strengths, such as strong L acid sites for Co3O4 and weak L acid sites for CoMoO4 depending on Co metal phase. From the above characterization, it was found that each metal species had different modifying effect on the acidic properties depending on single metal or bimetallic phase.
3.3 Catalytic property in one-step hydrotreatment of methyl palmitate
The catalytic performance of the Co- or/and Mo-supported Beta(25) zeolites was evaluated in one-step hydrotreatment of methyl palmitate in a semi-batch reactor at 280 °C and 20 bar of H2 pressure during 120 min and their catalytic activities are shown in Table 2. When it comes to hydrotreatment of methyl palmitate over zeolite catalysts, the acid sites of zeolites are responsible for isomerization and cracking by the generation of carbenium ions,50 so it can be deactivated by coke formation at high temperature during the reaction despite high conversion at initial stage.17 Because of this, the corresponding catalytic reaction condition was chosen to stably monitor the catalytic properties of the prepared catalysts based on the previous related literature performed in the temperature range of 260–300 °C.1,9,17,51 In Scheme 1, the simplified reaction pathway in one-step hydrotreatment of methyl palmitate was based on the earlier proposed reaction pathways and the obtained results. C16 alkanes can be obtained through (i) hydrodeoxygenation without carbon loss in long alkyl chain of methyl palmitate, and C15 alkanes can be produced through (ii) decarbonylation or decarboxylation with removal of carboxyl group by releasing carbon monoxide with water or carbon dioxide, respectively. Then, consecutive catalytic reactions, such as hydrogenation, dehydrogenation, cracking, and isomerization result in the formation of alkanes with carbon numbers lower than 15. In addition, palmitic acid and C32H64O2 are detected as primary intermediate before the above catalytic reactions and esterification of palmitic acid, respectively7,10 proceed. In this regard, the liquid products were classified as alkanes with the carbon numbers of 5–7 (C5–C7), 8–16 (C8–C16), 17–18 (C17–C18), and C32H64O2, and palmitic acid. The gas products included alkanes with low carbon numbers less than 4, dimethyl ether, CO, and CO2. In the production of jet fuel, it is meaningful to focus on the yield of jet fuel range hydrocarbons in C8–C16 range alkanes.| Catalyst | Conv. (%) | Gas product | Organic liquid product | Yield of jet fuel range hydrocarbons (C8–C16, %) | n-C16/(n-C15 + n-C16) | Isomer ratio in C8–C16 (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Yield (%) | Yield (%) | Selectivity (wt%) | |||||||||
| C5–C7 | C8–C16 | C17–C18 | C32H64O2 | Palmitic acid | |||||||
| Beta(25) | 40.1 | 7.9 | 92.1 | 0 | 0 | 0 | 0 | 100 | 0 | — | — |
| Co10/Beta(25) | 100 | 26.1 | 73.9 | 20.2 | 79.1 | 0 | 0 | 0 | 56.5 | 0.69 | 74.9 |
| Co10Mo5/Beta(25) | 99.9 | 24.2 | 75.8 | 14.3 | 84.1 | 1.5 | 0 | 0.1 | 64.7 | 0.66 | 82.0 |
| Co10Mo10/Beta(25) | 99.9 | 23.0 | 77.0 | 13.2 | 85.1 | 1.6 | 0.1 | 0 | 65.5 | 0.68 | 82.3 |
| Co5Mo10/Beta(25) | 94.2 | 10.4 | 89.6 | 6.3 | 89.7 | 2.4 | 1.6 | 0 | 80.4 | 0.65 | 83.8 |
| Co3Mo10/Beta(25) | 61.0 | 11.1 | 88.9 | 0 | 15.6 | 0 | 41.4 | 43.1 | 13.9 | 0.65 | 72.3 |
| Co2Mo10/Beta(25) | 37.8 | 6.2 | 93.8 | 0 | 15.3 | 0 | 7.7 | 77.0 | 14.4 | 1.00 | 78.2 |
| Mo10/Beta(25) | 25.2 | 4.9 | 95.1 | 0 | 0 | 0 | 4.5 | 95.5 | 0 | — | — |
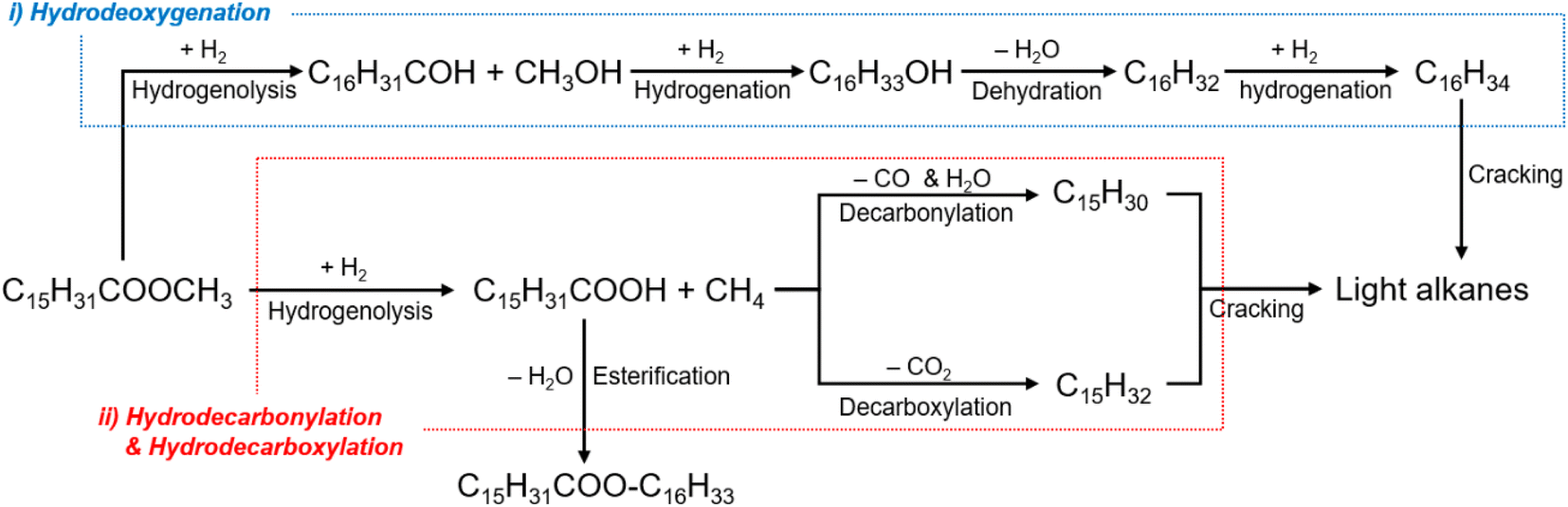 | ||
| Scheme 1 Simplified reaction pathway for catalytic hydrotreatment of methyl palmitate. | ||
As shown in Table 2, Beta(25) zeolite used as support showed a low conversion of 40.1% and palmitic acid selectivity of 100 wt% as a major product in the organic liquid product. It is already known that the transformation of methyl palmitate to palmitic acid is catalyzed by L acid sites.8 Although palmitic acid is straightforwardly formed by L acid sites on Beta(25) zeolites, alkanes with carbon numbers lower than 15 were not observed. It can be deduced that Beta(25) zeolite cannot lead to the next reaction pathways such as deoxygenation, decarboxylation, or decarbonylation, so none of the expected hydrocarbon products were observed.8,14
In the case of the Mo10/Beta(25) catalyst after wet impregnation of Mo, a lower conversion of 25.2% compared to Beta(25) zeolite was obtained. Similar to the product distribution of Beta(25) zeolites, the Mo10/Beta(25) catalyst showed selectivity toward palmitic acid (95.5 wt%) as a major liquid product and C32H64O2 (4.5 wt%) as a minor liquid product. With low acid site concentration on the Mo10/Beta(25), the weak acid sites involved by Mo species seem to be insufficient to facilitate catalytic reactions except hydrogenolysis of methyl palmitate.
To confirm that the bottleneck of further catalytic reaction for jet fuel range hydrocarbon over the Beta(25) and Mo10/Beta(25) catalysts is DO, the catalytic conversion of palmitic acid (PA) and hexadecane (HD) were investigated, as shown in Table 3. For the production of light alkanes, PA should be subjected to DO through decarbonylation or decarboxylation like methyl palmitate, but HD is enough through cracking. In the case of PA, the conversions over the Beta(25) and Mo10/Beta(25) catalysts were very low, 9.5% and 3.9%, respectively, and light alkanes from cracking were not detected. However, HD was completely decomposed to light alkanes with 100% conversion, so the gas product yield also increased to 42.7% and 35.5% over Beta(25) and Mo10/Beta(25), respectively. This proves the possibility of the cracking and isomerization steps with the Beta(25) and Mo10/Beta(25) catalysts, unlike DO. Moreover, wet-impregnated Mo can be used to suppress the degree of cracking according to the decreased yield of cracked light alkanes in comparison between the Beta(25) and Mo10/Beta(25) catalysts, which is consistently observed during the conversion of methyl palmitate.
| Reactant | Catalyst | Conv. (%) | Gas product | Organic liquid product | Yield of jet fuel range hydrocarbons (C8–C16, %) | n-C16/(n-C15 + n-C16) | Isomer ratio in C8–C16 (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Yield (%) | Yield (%) | Selectivity (wt%) | ||||||||||
| C5–C7 | C8–C16 | C17–C18 | C32H64O2 | Palmitic acid | ||||||||
| PA | Beta(25) | 9.5 | 7.0 | 93.0 | 0 | 0 | 22.2 | 77.8 | 0 | 0 | — | — |
| Mo10/Beta(25) | 3.9 | 3.9 | 96.1 | 0 | 0 | 43.5 | 56.5 | 0 | 0 | — | — | |
| HD | Beta(25) | 100 | 42.7 | 57.3 | 41.9 | 58.1 | 0 | 0 | 0 | 33.3 | 0.80 | 78.1 |
| Mo10/Beta(25) | 100 | 35.5 | 64.5 | 31.0 | 69.0 | 0 | 0 | 0 | 44.5 | 0.93 | 63.7 | |
Over the Co10/Beta(25) catalyst, the conversion was significantly raised to 100%, and alkanes with carbon numbers lower than 15 were observed as a result of the subsequent catalytic conversion, as shown in Table 2. Thus, the yield of jet fuel range hydrocarbons reached 56.5%. It is noteworthy that n-C16 and n-C15 alkanes were simultaneously detected with the n-C16/(n-C16 + n-C16) ratio of 0.69. This result indicates that methyl palmitate over the Co10/Beta(25) catalyst converted along with the competing reaction pathways between hydroxygenation for n-C16 and decarboxylation/decarbonylation for n-C15. Therefore, the wet-impregnated Co active for decarbonylation or decarbonylation should be contained for converting methyl palmitate into jet fuel range hydrocarbons (C8–C16). According to the NH3-TPD and FT-IR spectra after pyridine desorption, as shown in Table 1, the introduction of a Co catalyst enables the generation of strong L acid sites, which contribute to the catalysis of not only decarboxylation/decarbonylation but also hydrodeoxygenation with weak L acid sites on the surface of Beta(25) zeolites.14 Then, the n-alkanes generated from the above reaction pathways would be isomerized on the metal sites and the B acid sites of Beta(25) zeolites, thus exhibiting an isomer ratio of 74.9% in the range of C8–C16.10,14 The isomer ratio can reflect the quality of the product as bio jet fuel because the isomerization converts the unsaturated compounds into the branched compounds, which are favorable for combustion quality with high octane number.
As for raising the yield of alkanes in the range of C8–C16 as desirable products for jet fuel range hydrocarbons, Co10/Beta(25) is required to reduce unnecessary cracking including a high gas yield of 26.1% and C5–C7 selectivity of 20.2 wt% in the organic liquid product. In this respect, the Co-supported Beta(25) catalyst was modified by adding Mo, and catalytic testing of samples was conducted to find the optimized composition of Co and Mo for high selectivity of C8–C16 (Fig. 6). In one approach, the loading amount of Mo was increased from Co10Mo5/Beta(25) to Co10Mo10/Beta(25) on the basis of Co10/Beta(25) as shown in Fig. 6a (see also Table 2). Our findings confirmed the deactivation of cracking by Mo in Mo10/Beta(25), and the addition of Mo to Co10/Beta(25) improved the C8–C16 selectivity of 85.1 wt% by decreasing the gas yield and C5–C7 selectivity to 23.0% and 13.2 wt%, respectively. However, there is a limitation to further raising the yield for desirable C8–C16 by increasing the Mo loading; the similar product distributions were observed for Co10Mo5/Beta(25) and Co10Mo10/Beta(25) as 64.7% and 65.5%, respectively. In another approach, the loading amount of Co was increased from Mo10/Beta(25) to Co10Mo10/Beta(25) on the basis of Mo10/Beta(25) as shown in Fig. 6b (see also Table 2). By increasing the amount of added Co, the conversion was improved, and the product distribution shifted to light alkanes. Until Co2Mo10/Beta(25) and Co3Mo10/Beta(25), the increased Co content resulted in the dominant esterification of palmitic acid to C32H64O2. Then, the significant improvement was achieved with Co5Mo10/Beta(25); it exhibited the conversion of 94.2% and the C8–C16 selectivity of 89.7 wt% without palmitic acid remaining in the liquid product. The conversion was consistently improved over Co10Mo10/Beta(25) with 99.9%. These dramatic catalytic improvements from the Co5Mo10/Beta(25) catalyst seem to originate from the different acidic property with the rapidly increase in the quantity of strong L acid sites, which can catalyze not only decarboxylation/decarbonylation but also hydrodeoxygenation as explained above for Co10/Beta(25). However, the higher Co loading amount in Co10Mo10/Beta(25) led to reduced C8–C16 selectivity due to excessive cracking. Thus, Co5Mo10/Beta(25) exhibited the best catalytic activity achieving the yield of fuel range hydrocarbons of 80.4% as desirable products for bio jet fuel production. In addition, the isomer ratio in C8–C16 showed a similar tendency to the C8–C16 selectivity, so Co5Mo10/Beta(25) obtained the highest isomer ratio in C8–C16 as 83.8%. It is also remarkably improved catalytic activity than those of the previous catalysts performed for targeting bio jet fuel range hydrocarbons from FAMEs as shown in Table S2,† such as Ni/desilicated meso Y zeolite (the C8–C16 selectivity of 64.8%, the isomer ratio in C8–C16 of 19.4%).18
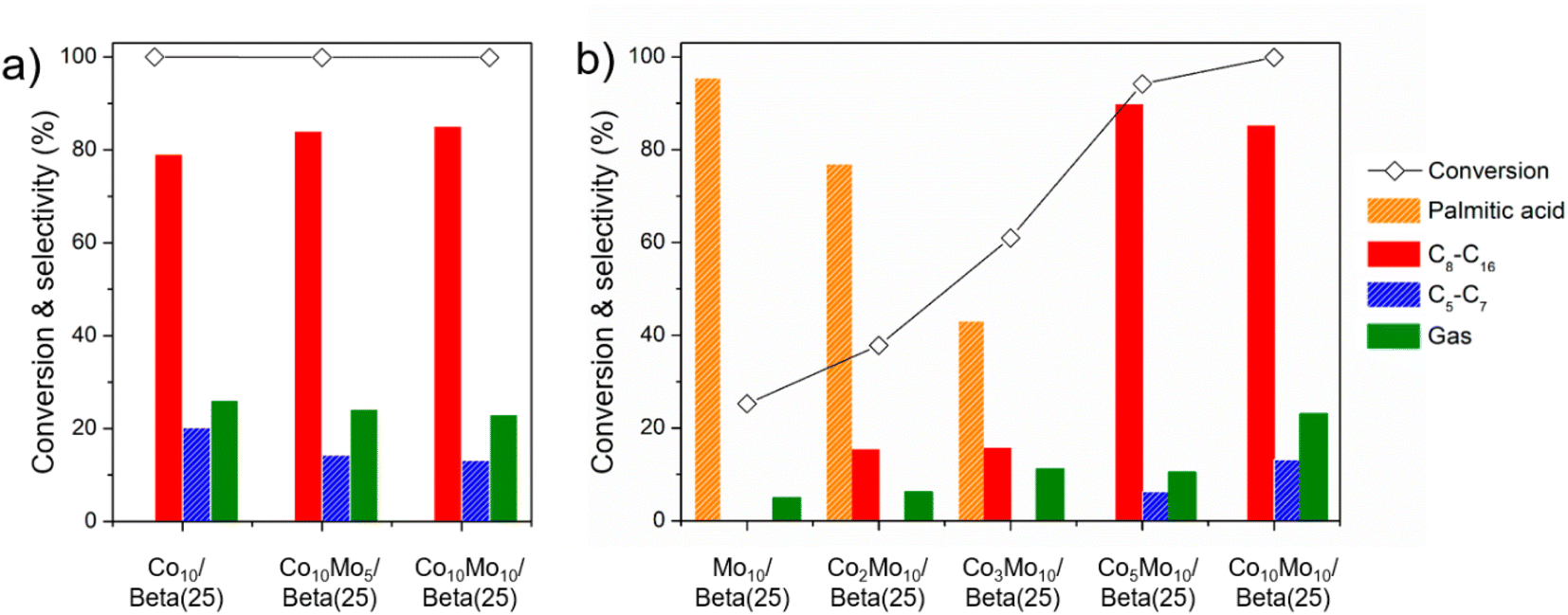 | ||
| Fig. 6 Catalytic activities of methyl palmitate according to the effect of (a) the increased Mo content in the Co10/Beta(25) and (b) the increased Co content in the Mo10/Beta(25). | ||
However, there is still doubt about the requirement of bimetal co-introduced onto the support, so the Co15/Beta(25) catalyst was compared with the same metal loading amount with Co5Mo10/Beta(25). Except the fact that the wet-impregnated Co of Co15/Beta(25) had Co3O4 crystalline phase (Fig. S5†), the quantity of total acid sites and the distribution of B and L acid sites in Co15/Beta(25) were similar to those in Co5Mo10/Beta(25) (Table S3 and Fig. S6†). In addition, as discussed about the strong L acid site formation by Co3O4, the IR peak of 1550 cm−1 representing Brønsted acid sites was hardly detected in Co15/Beta(25) and it can be seen that all acid sites exhibit the characteristics of Lewis acid sites (Fig. S6b†). Over Co15/Beta(25), as seen in Table S4,† the high C8–C16 selectivity of 89.6 wt% was achieved, which is comparable to that of Co5Mo10/Beta(25), but the gas product yield increased by 2.6 times and resulted in lowering the C8–C16 yield to 65.1%. The isomerization also did not proceed well under the promoted cracking, resulting in 77.7% of the isomer ratio in C8–C16. This proves the necessity of co-impregnation of Co and Mo to suppress unnecessary cracking steps and overcome the limitation of single metal-supported zeolites due to not only the metal itself but also the effect of bimetal co-introduced onto the support.
Based on the above findings from hydrotreatment of methyl palmitate over the Co-and Mo-supported Beta(25) zeolites, deoxygenation of methyl palmitate is preceded by Co with the aid of strong L acid sites. In the absence of Co, palmitic acid and C32H64O2 are only obtained, which can be achieved before deoxygenation, as in Mo10/Beta(25) and Beta(25). Then, C16 alkanes and C15 alkanes are simultaneously obtained through not only decarboxylation/decarbonylation but also hydrodeoxygenation, and the subsequent cracking and isomerization result in alkanes with carbon numbers lower than 15 and isomerized hydrocarbons. The introduction of Mo can suppress the cracking and increase the selectivity for light alkanes, and Beta(25) can enhance isomerization with hydrogenation promoted by Co. For better selectivity to jet fuel range hydrocarbons, co-impregnation of Co and Mo is necessary to overcome the limitation of single metal-supported zeolites, which can exploit the catalytic property of metal itself as well as the optimized acidic property according to the Co/Mo ratio with the appearance of CoMoO4 as weak L acid.
4. Conclusion
The Co- or/and Mo-supported Beta(25) zeolites were investigated with different Co/Mo composition ratio for hydrotreatment of methyl palmitate. The variation of the Co/Mo composition ratio led to a distinct change in the metal phase due to the preference for the formation of CoMoO4 than mono metal oxides. Furthermore, the acidic property was strongly dependent on the Co metal phase altered by the Co/Mo ratio because L acid sites with different strength were formed such as Co3O4 for strong L acid sites and CoMoO4 for weak L acid sites. In hydrotreatment of methyl palmitate with the Co- or Mo-supported Beta(25) zeolite as monometallic catalyst, impregnated Co was found to be essential for decarbonylation/decarboxylation to convert methyl palmitate into jet fuel range hydrocarbons (C8–C16), and impregnated Mo was used to suppress the degree of cracking. As bimetallic catalyst, the Co and Mo-supported Beta(25) zeolite was beneficial to improve the C8–C16 selectivity by optimizing the acidic properties to inhibit excessive cracking. Co5Mo10/Beta(25) zeolites achieved the highest C8–C16 selectivity of 89.7 wt% including isomer ratio of 83.8% in organic liquid product with the conversion of 94.2%. This work provides a meaningful reference for Co- and Mo-supported Beta zeolite as transition metal-supported zeolite catalysts with unique modifications of acidic properties and optimal catalytic properties for selective one-step hydrotreatment to produce bio jet fuel range hydrocarbons.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (No. 20212010200100).References
- Y. Shi, Y. Cao, Y. Duan, H. Chen, Y. Chen, M. Yang and Y. Wu, Green Chem., 2016, 18, 4633–4648 RSC.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef PubMed.
- Y. Du, F. Wang, X. Xia, H. Zhu, Z. Zhang, C. You, X. Jiang, J. Jiang and C. Li, Renewable Energy, 2022, 198, 246–253 CrossRef.
- C. Zhang, X. Hui, Y. Lin and C.-J. Sung, Renewable Sustainable Energy Rev., 2016, 54, 120–138 CrossRef CAS.
- F. P. Sousa, L. N. Silva, D. B. de Rezende, L. C. A. de Oliveira and V. M. D. Pasa, Fuel, 2018, 223, 149–156 CrossRef CAS.
- E. S. K. Why, H. C. Ong, H. V. Lee, Y. Y. Gan, W.-H. Chen and C. T. Chong, Energy Convers. Manage., 2019, 199, 112015 CrossRef CAS.
- Y. Bie, J. Lehtonen and J. Kanervo, Appl. Catal., A, 2016, 526, 183–190 CrossRef CAS.
- I. V. Deliy, E. N. Vlasova, A. L. Nuzhdin, E. Y. Gerasimov and G. A. Bukhtiyarova, RSC Adv., 2014, 4, 2242–2250 RSC.
- B. Ma and C. Zhao, Green Chem., 2015, 17, 1692–1701 RSC.
- L. Chen, J. Fu, L. Yang, Z. Chen, Z. Yuan and P. Lv, ChemCatChem, 2014, 6, 3482–3492 CrossRef CAS.
- E. W. Ping, R. Wallace, J. Pierson, T. F. Fuller and C. W. Jones, Microporous Mesoporous Mater., 2010, 132, 174–180 CrossRef CAS.
- Z. Zhang, F. Wang, J. Jiang, H. Zhu, Y. Du, J. Feng, H. Li and X. Jiang, Fuel, 2023, 333, 126341 CrossRef CAS.
- H. Zhu, F. Wang, J. Jiang, Z. Zhang, Y. Du, J. Feng and X. Jiang, Fuel Process. Technol., 2023, 239, 107537 CrossRef CAS.
- C. Wang, Z. Tian, L. Wang, R. Xu, Q. Liu, W. Qu, H. Ma and B. Wang, ChemSusChem, 2012, 5, 1974–1983 CrossRef CAS.
- S. Kovács, T. Kasza, A. Thernesz, I. W. Horváth and J. Hancsók, Chem. Eng. J., 2011, 176–177, 237–243 CrossRef.
- V. A. Yakovlev, S. A. Khromova, O. V. Sherstyuk, V. O. Dundich, D. Y. Ermakov, V. M. Novopashina, M. Y. Lebedev, O. Bulavchenko and V. N. Parmon, Catal. Today, 2009, 144, 362–366 CrossRef.
- H. Imai, M. Abe, K. Terasaka, H. Yamazaki, R. Osuga, J. N. Kondo and T. Yokoi, Fuel Process. Technol., 2020, 197, 106182 CrossRef.
- Z. Zhang, J. Cheng, Y. Qiu, X. Zhang, J. Zhou and K. Cen, Fuel, 2019, 244, 472–478 CrossRef.
- J. Y. Jeon, Y. Han, Y.-W. Kim, Y.-W. Lee, S. Hong and I. T. Hwang, Biofuels, Bioprod. Biorefin., 2019, 13, 690–722 CrossRef.
- P. Chintakanan, T. Vitidsant, P. Reubroycharoen, P. Kuchonthara, T. Kida and N. Hinchiranan, Fuel, 2021, 293, 120472 CrossRef.
- I.-H. Choi, K.-R. Hwang, J.-S. Han, K.-H. Lee, J. S. Yun and J.-S. Lee, Fuel, 2015, 158, 98–104 CrossRef.
- J. E. Herrera, L. Balzano, A. Borgna, W. E. Alvarez and D. E. Resasco, J. Catal., 2001, 204, 129–145 CrossRef CAS.
- Y. Chen, Z. Tian, Q. Liu and B. Bian, Sustainable Energy Fuels, 2020, 4, 3042–3050 RSC.
- K. Shimura, T. Miyazawa, T. Hanaoka and S. Hirata, Appl. Catal., A, 2015, 494, 1–11 CrossRef.
- J. Janas, T. Machej, J. Gurgul, R. P. Socha, M. Che and S. Dzwigaj, Appl. Catal., B, 2007, 75, 239–248 CrossRef.
- B. de Rivas, R. López-Fonseca, C. Jiménez-González and J. I. Gutiérrez-Ortiz, J. Catal., 2011, 281, 88–97 CrossRef.
- Q. Yu, L. Zhang, R. Guo, J. Sun, W. Fu, T. Tang and T. Tang, Fuel Process. Technol., 2017, 159, 76–87 CrossRef.
- L. Zhang, Q. Dai, W. Fu, T. Tang, P. Dong, M. He and Q. Chen, J. Catal., 2018, 359, 130–142 CrossRef.
- P. A. Nikulshin, D. I. Ishutenko, A. A. Mozhaev, K. I. Maslakov and A. A. Pimerzin, J. Catal., 2014, 312, 152–169 CrossRef CAS.
- E. Payen, J. Grimblot and S. Kasztelan, J. Phys. Chem., 1987, 91, 6642–6648 CrossRef CAS.
- W. Fu, L. Zhang, D. Wu, M. Xiang, Q. Zhuo, K. Huang, Z. Tao and T. Tang, J. Catal., 2015, 330, 423–433 CrossRef CAS.
- W. Li, G. D. Meitzner, R. W. Borry and E. Iglesia, J. Catal., 2000, 191, 373–383 CrossRef CAS.
- K. Chen, S. Xie, A. T. Bell and E. Iglesia, J. Catal., 2001, 198, 232–242 CrossRef CAS.
- F. Nti, D. A. Anang and J. I. Han, J. Alloys Compd., 2018, 742, 342–350 CrossRef CAS.
- G. Zhang, D. Wang, P. Feng, S. Shi, C. Wang, A. Zheng, G. Lü and Z. Tian, Chin. J. Catal., 2017, 38, 1207–1215 CrossRef CAS.
- X. Li, W. Zhang, X. Li, S. Liu, H. Huang, X. Han, L. Xu and X. Bao, J. Phys. Chem. C, 2009, 113, 8228–8233 CrossRef CAS.
- A. Y. Stakheev and W. M. H. Sachtler, J. Chem. Soc., Faraday Trans., 1991, 87, 3703–3708 RSC.
- T. M. Tri, J. P. Candy, P. Gallezot, J. Massardier, M. Prlmet, J. C. Védrine and B. Imelik, J. Catal., 1983, 79, 396–409 CrossRef CAS.
- Y. Liu, Y. Xing, S. Xu, Y. Lu, S. Sun and D. Jiang, Chem. Eng. J., 2022, 431, 133240 CrossRef CAS.
- D. P. Upare, S. Park, M. S. Kim, J. Kim, D. Lee, J. Lee, H. Chang, W. Choi, S. Choi, Y. P. Jeon, Y. K. Park and C. W. Lee, J. Ind. Eng. Chem., 2016, 35, 99–107 CrossRef CAS.
- M. de Boer, E. P. F. M. Koch, R. J. Blaauw, E. R. Stobbe, A. N. J. M. Hoffman, L. A. Boot, A. J. van Dillen and J. W. Geus, Solid State Ionics, 1993, 63–65, 736–742 CrossRef CAS.
- J. Chen, Y. Ge, Q. Feng, P. Zhuang, H. Chu, Y. Cao, W. R. Smith, P. Dong, M. Ye and J. Shen, ACS Appl. Mater. Interfaces, 2019, 11, 9002–9010 CrossRef CAS PubMed.
- M. H. M. Ahmed, O. Muraza, A. K. Jamil, E. N. Shafei, Z. H. Yamani and K.-H. Choi, Energy Fuels, 2017, 31, 5482–5490 CrossRef CAS.
- Q. Liu, Y. Bie, S. Qiu, Q. Zhang, J. Sainio, T. Wang, L. Ma and J. Lehtonen, Appl. Catal., B, 2014, 147, 236–245 CrossRef CAS.
- M. Rutkowska, Z. Piwowarska, E. Micek and L. Chmielarz, Microporous Mesoporous Mater., 2015, 209, 54–65 CrossRef CAS.
- D. Kubička, N. Kumar, T. Venäläinen, H. Karhu, I. Kubičková, H. Österholm and D. Y. Murzin, J. Phys. Chem. B, 2006, 110, 4937–4946 CrossRef.
- T. M. Salama, I. Othman, M. Sirag and G. A. El-Shobaky, Microporous Mesoporous Mater., 2006, 95, 312–320 CrossRef CAS.
- S. Rajagopal, J. A. Marzari and R. Miranda, J. Catal., 1995, 151, 192–203 CrossRef CAS.
- F. E. Kiviat and L. Petrakis, J. Phys. Chem., 1973, 77, 1232–1239 CrossRef CAS.
- W. Luo, W. Cao, P. C. A. Bruijnincx, L. Lin, A. Wang and T. Zhang, Green Chem., 2019, 21, 3744–3768 RSC.
- B. Peng, Y. Yao, C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2012, 51, 2072–2075 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06938e |
| This journal is © The Royal Society of Chemistry 2023 |