
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom temperature synthesis of flower-like hollow covalent organic framework for efficient enrichment of microcystins†
Zhenli Yua,
Hui Chena,
Wenming Zhangb,
Qingqing Dinga,
Qidong Yua,
Min Fangb and
Lan Zhang *a
*a
aMinistry of Education Key Laboratory for Analytical Science of Food Safety and Biology, Fujian Provincial Key Laboratory of Analysis and Detection Technology for Food Safety, College of Chemistry, Fuzhou University, Fuzhou, Fujian 350002, China. E-mail: zlan@fzu.edu.cn; Fax: +86-591-22866135; Tel: +86-591-22866135
bSchool of Chemical and Biological Technology, Minjiang Teachers College, Fuzhou, Fujian 350108, China
First published on 1st February 2023
Abstract
The morphology of nanomaterials is one of essential factors for their unique properties. Herein, a hollow covalent organic framework with a flower-like structure (HFH-COF) was synthesized at room temperature. The synthesized HFH-COF has a very large specific surface area, mesoporous structure, excellent chemical stability, and good crystallinity. The special morphology endowed HFH-COF with high specific surface area utilization and rapid mass transfer rate, resulting in faster equilibration time and better extraction performance than spherical COF (S-COF). Subsequently, combined with high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS), an efficient and sensitive method was established for microcystins (MCs) detection. The developed method has low detection limits (0.6–0.8 pg mL−1), wide linear ranges (1.5–1000.0 pg mL−1, R ≥ 0.9993), and acceptable reproducibility (RSD ≤ 7.6%, n = 6). Real biological samples were analyzed by the developed method, and trace levels of MC-YR, MC-RR and MC-LR were detected. The results indicate that the synthesized HFH-COF is an ideal sorbent for efficient extraction of MCs from complex biological samples.
1. Introduction
Sample pretreatment is an essential step in chromatographic analysis, accounting for about two-thirds of total analysis time.1 Among various pretreatment methods, dispersive-solid phase extraction (d-SPE) combines extraction and concentration into one, which has the advantages of time saving, low solvent consumption, simple operation and large contact area with targets.2 Notably, the performance of d-SPE is highly dependent on the properties of adsorbents, affecting the sensitivity and selectivity of final analytical results.3It is well known that the morphology of adsorbents is crucial for their available specific surface area, chemical kinetics and stability.4 In recent years, biomimetic materials (plant fibers, pearl layers, spider webs etc.) have attracted the attention of researchers because of their unique layered structure and excellent mechanical/chemical properties.4–6 For example, in 2020, our group demonstrated adsorbents with fish-scale morphology that made its adsorption capacity against targets much higher than that of granular adsorbents because of the increased contact area of adsorbents with targets.7 Asefa's group investigated the adsorption behaviours of dendritic silica and cylindrical pore mesoporous silica on DNA. The results showed that the adsorption capacity of dendritic silica is higher than that of cylindrical pore mesoporous silica because the dendritic shape has a large pore, wide orifice, and fibrous structure that makes it easier for DNA molecules to pass and diffuse.8 Wang's group prepared a bamboo-like nanotube for removal of Cr(VI) ions from wastewater, which was demonstrated that the adsorption capacity of bamboo-like nanotubes for Cr(VI) ions was much higher than that of conventional nanoparticles.9 Up to now, there is a great demand to develop materials with different morphologies.
Covalent organic frameworks (COFs), a new generation of porous crystalline materials, is formed by connecting molecular building blocks through covalent bonds based on reticulation chemistry.10,11 Since 2005, COFs has been widely used in various fields such as gas adsorption/separation,12,13 catalysis,14 and biomedicine due to their large specific surface area,15 tunable pore size, good chemical selectivity and thermal stability. Since the properties of COFs are highly dependent on their morphology, controlling morphology is very effective in improving the performances of COFs.16 In this regard, various shapes of COFs, including tubular COFs,17 core–shell structured COFs,18 hollow structured COFs,19 and COF nanosheets20 were prepared and showed excellent performance in catalysis, gas storage/separation and pollutant detection. In recent years, researchers have developed a number of methods for the synthesis of COFs with different morphologies, including microwave-assisted, ionothermal, mechanochemical and room-temperature synthesis methods, in addition to the traditional solvothermal method. Among them, room-temperature synthesis is of great interest due to its simplicity and easy control. For example, S. P. Mukherjee synthesized bromine ion covalent organic backbone (BE-COF:Br) by a simple mechanochemical method and used it for the preparation of CsPbBr3/Cs4PbBr6 perovskite@COF nanocomposites.21 Gu's group recently reported a multi-component strategy for the synthesis of highly crystalline buta-1,3-diene-linked COF.22 The COF-LZU1 nanorods were synthesized at room temperature by Zhang's group using CO2/H2O as a solvent, which is a fast and simple preparation method, but the combined COF-LZUI nanoparticles will adhere to each other and have poor dispersion.23 O. Lyutakov applied readily available copper nanoparticles (CuNPs) as a cheap plasmonic template, functionalized with the “anchor layer” of CTF-1 building block, for the preparation of the COF on the surface of NPs under LSPR excitation. Although this method is simple, the crystallinity of the combined CTF-1 is low.24 So, although synthetic methods to control the physical and chemical functions of COFs at the molecular level by using different structural units and linking chemicals have become increasingly sophisticated,25 harsh reaction conditions or too fast reaction rates have led to low crystallinity and inhomogeneous morphology of the synthesized COFs. Therefore, the precise synthesis of COFs with high crystallinity, homogeneous and well-dispersed specific morphology remains a considerable challenge.
In this work, a hollow flower-like covalent organic backbone (HFH-COF) was synthesized by a simple room-temperature method and used as a d-SPE adsorbent to extract microcystins (MCs) from biological samples. MCs is a group of monocyclic polypeptide toxins secreted by cyanobacteria, which is of great concern because of their acute toxicity to humans as well as carcinogenicity after long-term exposure (Fig S1†).26–28 However, low levels of MCs in biological samples and complexity of sample matrices urgently require an efficient and reproducible pretreatment method for MCs extraction. Therefore, the obtained HFH-COF attempt to solve the above issues. The special flower-like morphology of HFH-COF could provide more adsorption sites that greatly increases the adsorption capacity of HFH-MOF against MCs. In addition, the hollow structure was beneficial to improve the extraction efficiency by reducing the mass transfer resistance. For comparison, spherical COF (S-COF) was also synthesized to explore the adsorption behaviour of different morphological COFs. This work provides a reference for the application of flower-shaped hollow COFs in sample pretreatment. Finally, combined with high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS), an efficient and sensitive method was successfully established for MCs detection in biological samples.
2. Experimental
2.1. Synthesis of HFH-COF
The HFH-COF was synthesized by a facile room temperature method.29 Typically, 0.04 mmol TPB and 0.06 mmol DVA were dispersed in a 5 mL of ACN with 10% H2O and sonicated for 1 min until the building blocks were fully dissolved. After that, 0.8 mL of AcOH (glacial acetic acid, 17.4 M) was added and sonicated for 1 min, sealed and allowed to stand at room temperature for 72 h. The yellow precipitate was washed three times with THF and ethanol, respectively, and dried under vacuum at 60 °C for 24 h, yield: 92%.As a comparison, we prepared spherical COF (S-COF). According to the literature,29 the morphology of COF can be adjusted by changing the H2O content in ACN. Therefore, 5 mL of aqueous solution containing 10% H2O was changed to 5 mL of ACN, and other conditions remained unchanged to synthesize S-COF, yield: 90%.
2.2. Process of d-SPE
The pH of the aqueous standard solution or aqueous sample was adjusted to 3.0 with hydrochloric acid prior to the extraction of MCs. Then, 4.0 mg of HFH-COF powder was added to a centrifuge tube containing 20 mL of the prepared solution. The mixture was placed in an oscillator for sorption. After 8 min of extraction, the supernatant was removed by centrifugation. Subsequently, 0.5 mL of methanol solution containing 2% FA was added, vortexed for 10 min to allow sufficient resolution, and filtered through a 0.22 μm membrane prior to HPLC-MS/MS analysis.2.3. Adsorption experiments
For investigating the adsorption kinetic behaviors of HFH-COF and S-COF for MC-RR and MC-YR, we conducted an adsorption experiment of 0.1 mg of HFH-COF and S-COF were used to extract MC-RR and MC-YR (50 ng mL−1) in 0.5 mL of several single standard solutions for the time ranging from 1 to 60 min, respectively. After centrifugation, the supernatant was collected and measured by HPLC-MS/MS.As for isothermal adsorption experiment, 0.1 mg of HFH-COF or S-COF were put into 0.5 mL of MC-RR or MC-YR several single standard solutions that concentrations ranging from 0.05 to 10 μg mL−1 and shaken for 10 min, respectively. After centrifugation, the supernatant was determined through HPLC-MS/MS. The equilibrium adsorption capacities (qe) were measured by the formula:
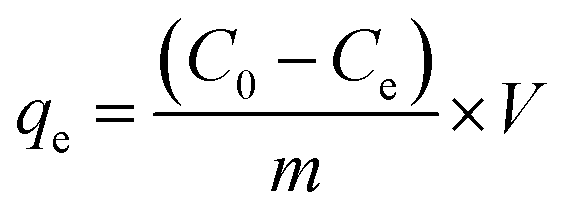 | (1) |
2.4. Sample pretreatment
Crucian, prawns, and pond snails were purchased from the local market (Fuzhou, China). The samples were processed in the same way as the previous ones, with slight modifications.30 The samples were first washed and removed from the shells, then placed on a metal grid with an aperture of about 2 mm to drain the water. After the water was drained, the samples were placed in a homogenizer and stirred at high speed. 5.0 g of the above homogenizer pulp was weighed into a 50 mL centrifuge tube, to which 25 mL of methanol solution was added. The suspended samples were vortexed for 5 min and sonicated for 20 min. The samples were then centrifuged at 8000 rpm for 10 minutes and the supernatant was collected. The remaining residue was re-extracted twice with 10 mL of MeOH. Thereafter, the sample dissolved in MeOH was spun and diluted with ultrapure water into a 250 mL volumetric flask. Finally, each solution was filtered twice through a 0.22 μm filter and stored in a sealed environment at 4 °C.3. Results and discussion
3.1. Characterization of HFH-COF
The surface morphology of the HFH-COF was characterized by TEM and SEM. From TEM and SEM, it is clear that HFH-COF has a unique flower-like morphology and presents a hollow structure (Fig. 1d–f), which is considered to be beneficial to rapid mass transfer.31,32 It can be noted that the flower-like morphology facilitates better access to the MCs. S-COF has a uniform solid spherical morphology (Fig. 1a–c). The above morphological structures proved that S-COF and HFH-COF had been prepared successfully. | ||
| Fig. 1 TEM images of (a and b) S-COF and (d and e) HFH-COF; SEM images of (c) S-COF and (f) HFH-COF. | ||
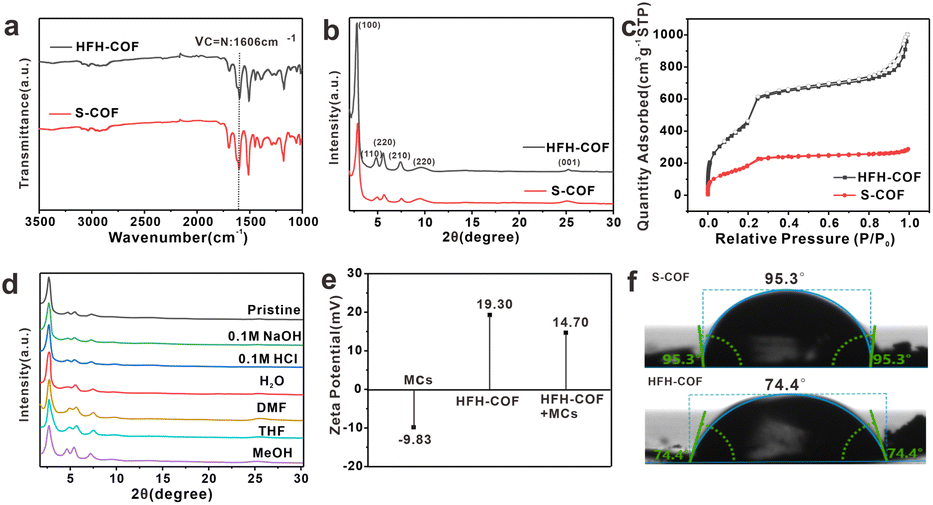
The FT-IR (Fig. 2a) peak of the HFH-COF at 1606 cm−1 was related to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching, along with the disappearance of the aldehyde groups –C–H (2872 and 2777 cm−1) and –CO (1676 cm−1) of DVA and –N–H (3432 and 3352 cm−1) of TPB, indicating a successful Schiff base condensation reaction between the amino group of TPB and the aldehyde group of DVA to form HFH-COF.33 It is noteworthy that a clear strong band at around 1680 cm−1 is present in both FTIR spectra, noticeably smaller in case of HFH-COF, which we think it may be due to the presence of an incomplete reaction where some of the aldehyde groups of the aldehyde ligands are not fully reacted.34 And the extent of the reaction for HFH-COF is a little more complete than that for S-COF.
N stretching, along with the disappearance of the aldehyde groups –C–H (2872 and 2777 cm−1) and –CO (1676 cm−1) of DVA and –N–H (3432 and 3352 cm−1) of TPB, indicating a successful Schiff base condensation reaction between the amino group of TPB and the aldehyde group of DVA to form HFH-COF.33 It is noteworthy that a clear strong band at around 1680 cm−1 is present in both FTIR spectra, noticeably smaller in case of HFH-COF, which we think it may be due to the presence of an incomplete reaction where some of the aldehyde groups of the aldehyde ligands are not fully reacted.34 And the extent of the reaction for HFH-COF is a little more complete than that for S-COF.
 | ||
| Fig. 2 (a) FT-IR spectra of HFH-COF and S-COF; (b) XRD patterns of HFH-COF and S-COF; (c) N2 adsorption–desorption isotherm of HFH-COF and S-COF; (d) PXRD patterns of HFH-COF after treatment with different solvents for 48 h; (e) zeta potential of MCs, HFH-COF and HFH-COF after adsorbing MCs; (f) the water contact angle of S-COF and HFH-COF. | ||
PXRD confirms the crystallinity of HFH-COF. It was observed from Fig. 2b, that the diffraction pattern of the HFH-COF shows a strong peak at 2.80°, along with other relatively weak peaks at 4.76, 5.54, 7.40, 9.68, and 25.06°, which is in good agreement with previous work,35 and demonstrates the good crystallinity of the prepared HFH-COF. It is noteworthy that the peak intensity of HFH-COF is higher than that of S-COF, indicating that the crystallinity of HFH-COF is better than that of S-COF. We performed XPS characterization of HFH-COF (Fig. S2†), and the C1s spectrum of XPS showed peaks associated with the CC bond at 284.6 eV, the C–C bond at 283.3 eV, and the CN at 288.5 eV, further verifying that the HFH-COF was successfully synthesized.
The porous structure of HFH-COF and S-COF were confirmed by N2 sorption–desorption isotherms (Fig. 2c). It was found that BET surface areas of HFH-COF were calculated to be 2134.5 m2 g−1, and the specific surface area of the S-COF was 784.8 m2 g−1. We list the BET comparison of HFH-COF with the materials in your proposed literature (Table S2†). The results revealed that the BET of HFH-COF is much larger than most of the COFs materials that would provide more adsorption sites for analytes. The pore-size distribution of HFH-COF and S-COF were calculated by NLDFT and the results demonstrated that regular pores with narrow size distribution at 3.11 nm of HFH-COF (Fig. S4†) and 2.54 nm of S-COF were obtained (Fig. S3†). It is worth noting that the average pore size of HFH-COF is about 3.11 nm, while the molecular length of MCs are all less than 2.0 nm.28 Obviously, the pore structure of HFH-COF can effectively accommodate MCs, which is beneficial and necessary for the efficient enrichment of MCs.
In addition, the chemical stability of HFH-COF was investigated by PXRD after immersing HFH-COF in different solvent treatments for 48 h. The results verified that the PXRD pattern of HFH-COF was almost unchanged (Fig. 2d), which verified the high chemical stability of HFH-COF.
Fig. 2e reveals the ζ potential information of HFH-COF as well as MCs. From the Fig. 2e, it is known that MCs are negatively charged and HFH-COF dissolved in water is positively charged, which facilitates the enrichment between the two by electrostatic interaction.
Furthermore, the hydrophilicity of S-COF and HFH-COF were determined by water contact angle analysis. The water contact angles of S-COF and HFH-COF were 95.3° and 74.4°, respectively (Fig. 2f). The result indicated that HFH-COF was much more hydrophilic than S-COF and has better dispersibility in water, which is more conducive to the efficient enrichment of MCs.
3.2. Adsorption characteristics
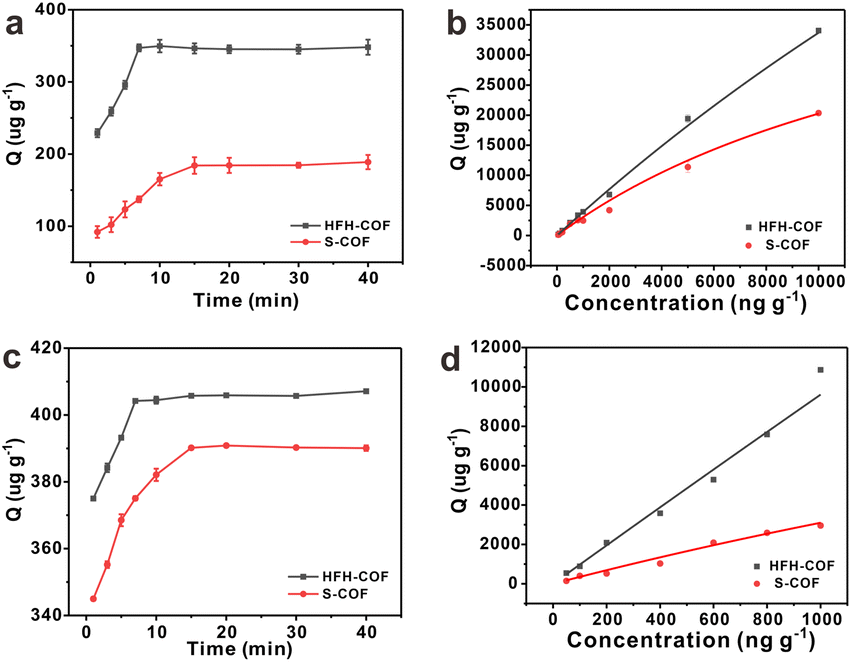
Adsorption kinetics is an important characteristic that provides insights into possible adsorption mechanisms. We investigated the adsorption kinetic behaviors of HFH-COF and S-COF on MC-RR and MC-YR, respectively. (Both HFH-COF and S-COF were 0.1 mg, and the concentrations of MC-RR and MC-YR were 50 ng mL−1). As shown in Fig. 3a and c, the adsorption of HFH-COF on MC-RR and MC-YR reached equilibrium within 7 min, while S-COF took 15 min under the same conditions, indicating that the adsorption of HFH-COF on MC-RR and MC YR is faster than that of S-COF, which is also evident from the proposed second-order constants (Table S3†). This may be due to the hollow flower-like structure of HFH-COF which facilitates the entry of target molecules into the center for accelerated mass transfer and the abundant active sites for adsorption.31 As can be seen from Fig. S5–S12,† the pseudo-second-order model (higher R) generally fitted the kinetic data well for the studied adsorption of MC-RR on HFH-COF compared with the pseudo-first-order model, which indicates that the adsorption process was mainly based on chemical adsorption involving multiple interactions through hydrophobic acting force, π–π interaction, and the hydrogen bond.36,37 The linear forms of the pseudo-first-order and pseudo-second-order models are presented as eqn (2) and (3), respectively.
ln(qe − qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − k1t qe − k1t
| (2) |
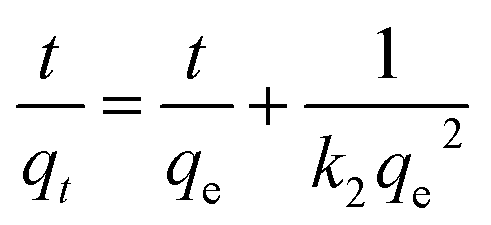 | (3) |
 | ||
| Fig. 3 (a) Extraction time curves of HFH-COF and S-COF for MC-RR. (b) The characteristic adsorption isotherms of HFH-COF and S-COF for MC-RR. (c) Extraction time curves of HFH-COF and S-COF for MC-YR. (d) The characteristic adsorption isotherms of HFH-COF and S-COF MC-YR. | ||
The adsorption isotherms of HFH-COF and S-COF on MC-RR and MC-YR were also determined. As can be seen from the Fig. 3b and d, the adsorption increased with the increase of concentration, but the slopes of both were different. The results indicate that HFH-COF has high adsorption capacity for MC-RR and MC-YR. After Langmuir fitting, the Qmax of HFH-COF for MC-RR and MC-YR were 218816 μg g−1, 570591 μg g−1, respectively, while the Qmax of S-COF for MC-RR and MC-YR were 53910 μg g−1, 24396 μg g−1. It is obvious that the equilibrium time of MC-RR adsorption by HFH-COF was shortened to half of that of S-COF, and the maximum adsorption amount was about 4 times that of S-COF. In summary, the remarkable extraction performance of HFH-COF can be attributed to the strong electrostatic interaction, hydrogen bonding and π–π superposition between MCs and HFH-COF. In addition, the large specific surface area, large mesopores, and hollow flower-like structure of HFH-COF promoted the accessibility of target molecules and accelerated the diffusion, and the high exposed active sites promoted the adsorption performance, which greatly shortened the extraction time and improved the extraction efficiency.
3.3. Optimization of d-SPE conditions
The optimal conditions for the extraction of MCs by HFH-COF were explored by optimizing the adsorption and desorption parameters. The optimization was performed with 20.0 mL of standard solution containing 250.0 pg mL−1. In order to make the results as close as possible to the optimal sorption and desorption conditions, this optimization experiment was performed in multiple rounds using a univariate approach.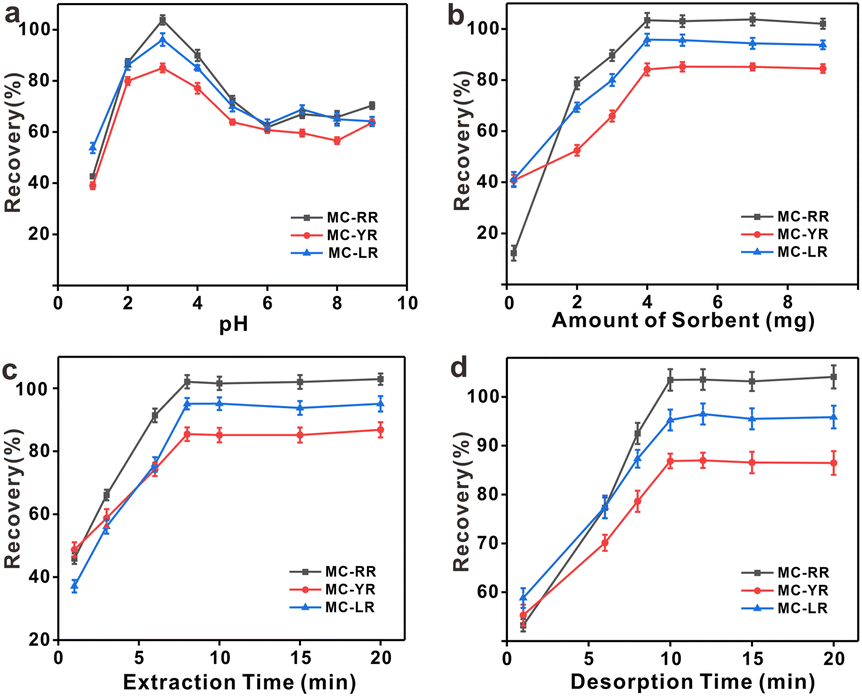 | ||
| Fig. 4 Optimization of (a) pH, (b) adsorbent dosage, (c) extraction time, and (d) desorption time. Error bars show the standard deviations for three replicate extractions. | ||
3.4. Stability test of HFH-COF
The stability test of COF after eight times cycles of MCs extraction were carefully investigated. Under optimal d-SPE conditions, 20 mL 250 ng L−1 MCs extracted with 4 mg HFH-COF, eluted with 2% formic acid in methanol for 10 min, washed and dried, and then the above operation was repeated. The results are shown in Fig. S13.† The adsorption efficiency of more than 80% was maintained after 8 cycles, indicating that HFH-COF has good stability and reusability which can be used as a potential adsorption material.3.5. Interference of ionic species
Considering the presence of salts in the actual sample can affect the pretreatment results, the ions not only affect the surface properties of the adsorbent but also introduce adsorption competition with the target analytes,40 further reducing the extraction performance. Cations (Na+, K+, NH4+, Ca2+, Mg2+, Fe3+), as well as anions (Cl−, CO32−, NO3−, SO42−) that may be present in the actual sample, were chosen for this experiment to examine the salt tolerance of the material, and if the standard deviation (SD) of the recoveries was less than 10%, the material was judged to be undisturbed. The results of the examination are shown in the Table S4† and show that the HFH-COF material has a good tolerance to salts.3.6. Method evaluation
Based on the above optimal optimized conditions, the analytical methods were established for the three MCs by HPLC-MS/MS, standard curves were plotted, the parameters of linear range, LOD, and reproducibility of the analytical methods were investigated (Table 1). The method has wide linear ranges of 1.5–1000.0 pg mL−1 for MC-RR and 2.0–1000.0 pg mL−1 for MC-YR and MC-LR, and good linearity with correlation coefficients (R) between 0.9993 and 0.9996. The LODs (defined as the lowest spiked concentration which generates a chromatographic peak with S/N ≥ 3) and limits of quantification (LOQ, defined as the lowest spiked concentration at which the accuracy is within 20%) values were in the range of 0.6–0.8 pg mL−1 and 1.5–2.0 pg mL−1, respectively. The intra-day and inter-day relative standard deviations (RSDs, n = 6) were 3.5–6.8% and 4.2–6.5% for the three MCs (10.0, 50.0, 250.0 pg mL−1), respectively, demonstrating good reproducibility of the method. The intra-day and inter-day relative standard deviations (RSDs, n = 6) were 3.5–6.8% and 4.2–6.5%, respectively, reflecting the good reproducibility of the method.| Compounds | Linear equation | Linearity range (pg mL−1) | R | LOD (pg mL−1) | RSD (%, n = 6) | |
|---|---|---|---|---|---|---|
| Intra-day | Inter-day | |||||
| a Spiked at 10 pg mL−1.b Spiked at 50 pg mL−1.c Spiked at 250 pg mL−1. | ||||||
| MC-RR | Y = 1214.80x + 939.27 | 1.5–1000.0 | 0.9994 | 0.6 | 4.3a, 4.9b, 5.6c | 4.2a, 5.1b, 5.3c |
| MC-YR | Y = 610.44x − 234.37 | 2.0–1000.0 | 0.9996 | 0.8 | 5.2a, 4.9b, 6.8c | 5.6a, 5.3b, 6.5c |
| MC-LR | Y = 643.72x + 77.38 | 2.0–1000.0 | 0.9993 | 0.8 | 3.5a, 4.8b, 5.2c | 4.5a, 5.2b, 6.3c |
3.7. Method application
The practicality of the HPLC-MS/MS method was verified by detecting three MCs in three biological samples. As shown in Table 2, MC-YR (113.75 pg g−1) was detected in crucian carp, MC-RR (67.50 pg g−1) was detected in prawns, and MC-RR (60.00 pg g−1) and MC-LR (556.25 pg g−1) were detected in pond snails. In order to investigate the interference degree of the actual sample matrix to the analytical method, the spiked experiments were carried out at 10.0, 50.0, and 250.0 pg mL−1, respectively. The spiked recoveries of this method were 85.7–108.7%, with RSDs ≤ 7.6% (n = 6). The typical chromatograms of pond snails samples and their spiked samples are shown in the Fig. 5. The above results show that the method has high precision, practicability, and reproducibility in actual samples.| Sample | Compounds | Found (pg g−1) | Spiked level (pg mL−1) | |||||
|---|---|---|---|---|---|---|---|---|
| 10.0 | 50.0 | 250.0 | ||||||
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | |||
| Crucian | MC-RR | N. D. | 86.4 | 4.7 | 89.7 | 1.2 | 89.0 | 1.1 |
| MC-YR | 113.7 ± 1.2 | 87.6 | 1.4 | 98.8 | 0.7 | 103.8 | 6.1 | |
| MC-LR | N. D. | 92.3 | 0.7 | 97.2 | 6.1 | 94.8 | 2.1 | |
| Prawn | MC-RR | 67.5 ± 5.0 | 85.7 | 1.9 | 87.3 | 1.2 | 87.3 | 3.8 |
| MC-YR | N. D. | 91.4 | 2.4 | 106.7 | 4.5 | 104.0 | 0.6 | |
| MC-LR | N. D | 92.3 | 7.6 | 89.8 | 1.8 | 99.2 | 0.6 | |
| Pond snails | MC-RR | 60.0 ± 7.5 | 88.7 | 1.5 | 87.8 | 1.4 | 87.9 | 1.4 |
| MC-YR | N. D. | 92.6 | 2.8 | 86.6 | 1.2 | 93.7 | 6.4 | |
| MC-LR | 556.2 ± 31.2 | 107.6 | 1.1 | 108.7 | 6.2 | 98.7 | 5.2 | |
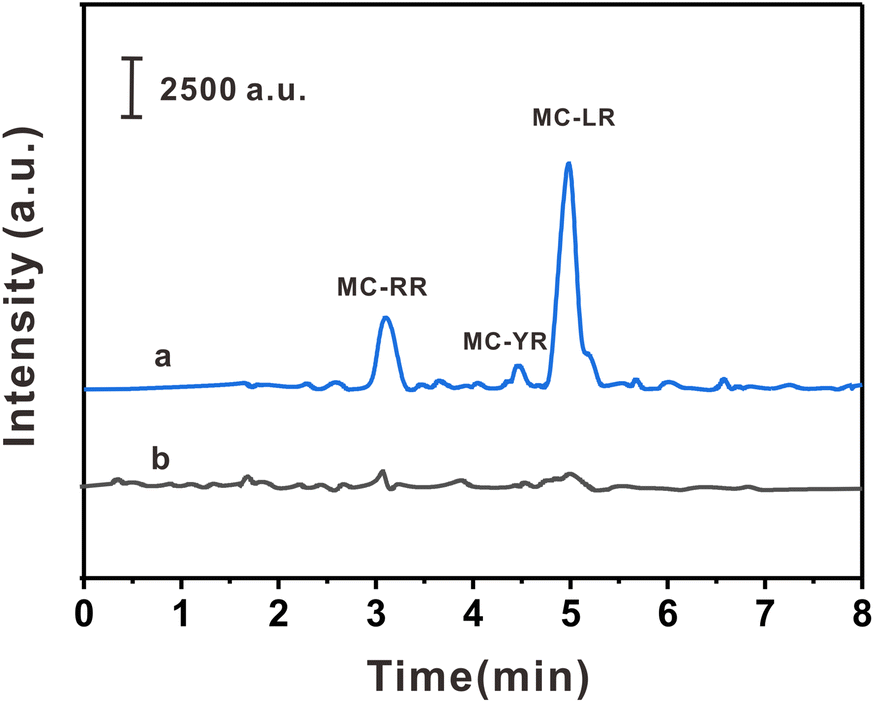 | ||
| Fig. 5 HPLC chromatograms of the escargots sample (a) after and (b) without the developed method. | ||
3.8. Comparisons of the proposed method with previously reported results
The established analytical method was compared with the reported analytical methods (Table 3). It can be seen that the proposed method required less sorbent (only 4.0 mg) to achieve a lower detection limit.41 In addition, the pretreatment time of the developed method (extraction and desorption, 18.0 min) was shorter than most other methods, especially than our group's reported study.42 Although Pan's method has a lower lod and shorter pretreatment time, it requires a large amount of solvent and adsorbent.43 At the same time, the LOD of the methods using HFH-COF as d-SPE adsorbent combined with HPLC-MS/MS for the detection of MCs are lower than those reported in most literature.37,44 Due to the large specific surface area and the unique hollow cavity of the prepared flower-like nanomaterials, it can contact the target faster and therefore requires less adsorbent and a shorter time to reach adsorption equilibrium.| Pretreatment method | Absorbent | Amounts of sorbents (mg) | Sample volume (mL) | Technique | LOD (pg mL−1) | Pretreatment time (min) | Reference |
|---|---|---|---|---|---|---|---|
| SPE | Octadecyl cartridge | 500.0 | 30.0 | LC-MS/MS | 30.0–40.0 | >14 | 41 |
| MSPE | Magnetic porous β-CD-containing polyme | 4.0 | 20.0 | HPLC-MS/MS | 1.0–2.0 | 97 | 42 |
| MSPE | DS-MMIP@GO | 10.0 | 1000.0 | HPLC-MS/MS | 0.03–0.6 | 10 | 43 |
| MSPE | MCNTs@TpPa-1 | 4.0 | 20.0 | HPLC-MS/MS | 0.8–1.5 | 20 | 44 |
| MSPE | Fe3O4@TabTfa-F4 | 9.0 | 50.0 | HPLC-UV | 41.0 | 50 | 37 |
| d-SPE | HFH-COF | 4.0 | 20.0 | HPLC-MS/MS | 0.6–0.8 | 18 | This work |
4. Conclusions
In this work, a hollow flower-like COF was synthesized by a simple room-temperature method. The synthesized HFH-COF as a d-SPE adsorbent has good chemical stability, high specific surface area, and mesoporous pore size. The unique flower-like morphology and hollow cavity structure can improve the specific surface area utilization of adsorbents, accelerate the mass transfer of MCs to adsorbents, and have a faster equilibration time and better extraction performance than that of S-COFs. Based on the above characteristics, an efficient and sensitive method for the detection of three MCs was established by combining with HPLC-MS/MS. Subsequently, the developed method was successful in detecting trace of MCs in biological samples. This work showed COFs with the hollow flower-like structure is an ideal adsorbent for efficient extraction of MCs from complex biological samples, indicating the importance of material morphology in sample pretreatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the special fund for Key Program of Science and Technology of Fujian Province, China (2020YZ019007), Fuzhou Science and Technology Plan Project (No. 2019-S-66).References
- T. Hyotylainen, Anal. Bioanal. Chem., 2009, 394, 743–758 CrossRef CAS PubMed.
- C. Lou, C. Wu, K. Zhang, D. Guo, L. Jiang, Y. Lu and Y. Zhu, J. Chromatogr. A, 2018, 1550, 45–56 CrossRef CAS PubMed.
- Q. Lu, S. Lin, Q. Ding, H. Zhang, P. Tong, M. Fang, W. Zhang and L. Zhang, J. Chromatogr. A, 2022, 1683, 463524 CrossRef CAS PubMed.
- Y. He, N. An, C. Meng, K. Xie, X. Wang, X. Dong, D. Sun, Y. Yang and Z. Hu, J. Mater. Chem. A, 2022, 10, 11030–11038 RSC.
- S. Di, Y. Qian, L. Wang and Z. Li, J. Mater. Sci., 2022, 57, 3085–3113 CrossRef CAS.
- N. Suresh Kumar, R. Padma Suvarna, K. Chandra Babu Naidu, P. Banerjee, A. Ratnamala and H. Manjunatha, Appl. Phys. A, 2020, 126, 445 CrossRef CAS.
- Q. Ding, H. Chen, C. Huang, Q. Lu, P. Tong, W. Zhang and L. Zhang, Analyst, 2020, 145, 5925–5932 RSC.
- X. Huang, Z. Tao, J. C. Praskavich, Jr., A. Goswami, J. F. Al-Sharab, T. Minko, V. Polshettiwar and T. Asefa, Langmuir, 2014, 30, 10886–10898 CrossRef CAS PubMed.
- S. Li, X. Lu, X. Li, Y. Xue, C. Zhang, J. Lei and C. Wang, J. Colloid Interface Sci., 2012, 378, 30–35 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard Iii, J. Am. Chem. Soc., 2008, 130, 11580–11581 CrossRef CAS PubMed.
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W. H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed.
- Z. Xiong, B. Sun, H. Zou, R. Wang, Q. Fang, Z. Zhang and S. Qiu, J. Am. Chem. Soc., 2022, 144, 6583–6593 CrossRef CAS PubMed.
- B. Gole, V. Stepanenko, S. Rager, M. Grune, D. D. Medina, T. Bein, F. Wurthner and F. Beuerle, Angew. Chem., Int. Ed. Engl., 2018, 57, 846–850 CrossRef CAS PubMed.
- G. Lin, C. Gao, Q. Zheng, Z. Lei, H. Geng, Z. Lin, H. Yang and Z. Cai, Chem. Commun., 2017, 53, 3649–3652 RSC.
- Z. J. Yin, S. Q. Xu, T. G. Zhan, Q. Y. Qi, Z. Q. Wu and X. Zhao, Chem. Commun., 2017, 53, 7266–7269 RSC.
- D. Rodriguez-San-Miguel, C. Montoro and F. Zamora, Chem. Soc. Rev., 2020, 49, 2291–2302 RSC.
- P. Kour and S. P. Mukherjee, J. Mater. Chem. A, 2021, 9, 6819–6826 RSC.
- Y. Su, B. Li, H. Xu, C. Lu, S. Wang, B. Chen, Z. Wang, W. Wang, K.-i. Otake, S. Kitagawa, L. Huang and C. Gu, J. Am. Chem. Soc., 2022, 144, 18218–18222 CrossRef CAS PubMed.
- F. Zhang, J. Zhang, B. Zhang, X. Tan, D. Shao, J. Shi, D. Tan, L. Liu, J. Feng, B. Han, G. Yang, L. Zheng and J. Zhang, ChemSusChem, 2018, 11, 3576–3580 CrossRef CAS PubMed.
- O. Guselnikova, Y. Kalachyova, R. Elashnikov, M. Cieslar, Z. Kolska, P. Sajdl, P. Postnikov, V. Svorcik and O. Lyutakov, Microporous Mesoporous Mater., 2020, 309, 110577 CrossRef CAS.
- J. L. Segura, S. Royuela and M. Mar Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- R. Dawson, Toxicon, 1998, 36, 953–962 CrossRef CAS PubMed.
- N. L. McLellan and R. A. Manderville, Toxicol. Res., 2017, 6, 391–405 CrossRef CAS PubMed.
- Z. Svircev, D. Drobac, N. Tokodi, B. Mijovic, G. A. Codd and J. Meriluoto, Arch. Toxicol., 2017, 91, 621–650 CrossRef CAS PubMed.
- W. Ma, G. Li, C. Zhong, Y. Yang, Q. Sun, D. Ouyang, W. Tong, W. Tian, L. Zhang and Z. Lin, Chem. Commun., 2021, 57, 7362–7365 RSC.
- H. Chen, C. Huang, W. Zhang, Q. Ding, J. Gao and L. Zhang, J. Chromatogr. A, 2019, 1608, 460404 CrossRef CAS PubMed.
- Y. Guo, X. He, C. Huang, H. Chen, Q. Lu and L. Zhang, Anal. Chim. Acta, 2020, 1095, 99–108 CrossRef CAS PubMed.
- S. Wang, L. Shao, Y. Sang and J. Huang, J. Chem. Eng. Data, 2019, 64, 1662–1670 CrossRef CAS.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS PubMed.
- G. Hu and J. Lindt, Polym. Bull., 1992, 29, 357–363 CrossRef CAS.
- W. Ma, Q. Zheng, Y. He, G. Li, W. Guo, Z. Lin and L. Zhang, J. Am. Chem. Soc., 2019, 141, 18271–18277 CrossRef CAS PubMed.
- M. Afshari and M. Dinari, J. Hazard. Mater., 2020, 385, 121514 CrossRef CAS PubMed.
- J. Lu, J. Zhou, H. Guo, Y. Li, X. He, L. Chen and Y. Zhang, J. Chromatogr. A, 2022, 1676, 463290 CrossRef CAS PubMed.
- P. G.-J. de Maagd, A. J. Hendriks, W. Seinen and D. T. Sijm, Water Res., 1999, 33, 677–680 CrossRef CAS.
- L. Wang, C. Shi, L. Wang, L. Pan, X. Zhang and J. J. Zou, Nanoscale, 2020, 12, 4790–4815 RSC.
- Y. Huang, J. Peng and X. Huang, J. Chromatogr. A, 2018, 1546, 28–35 CrossRef CAS PubMed.
- W. M. Draper, D. Xu, P. Behniwal, M. J. McKinney, P. Jayalath, J. S. Dhoot and D. Wijekoon, Anal. Methods, 2013, 5, 6796–6806 RSC.
- W. Zhang, M. Lin, M. Wang, P. Tong, Q. Lu and L. Zhang, J. Chromatogr. A, 2017, 1503, 1–11 CrossRef CAS PubMed.
- S. D. Pan, X. H. Chen, X. P. Li, M. Q. Cai, H. Y. Shen, Y. G. Zhao and M. C. Jin, J. Chromatogr. A, 2015, 1422, 1–12 CrossRef CAS PubMed.
- G. Liu, H. Chen, W. Zhang, Q. Ding, J. Wang and L. Zhang, Anal. Chim. Acta, 2021, 1166, 338539 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06901f |
| This journal is © The Royal Society of Chemistry 2023 |