
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafast degradation of SMX and TC by CoSiOx activated peroxymonosulfate: efficiency and mechanism
Xiaowei Liu ab,
Chen Chenb,
Peng Chenb and
Lili Wang*ac
ab,
Chen Chenb,
Peng Chenb and
Lili Wang*ac
aZhejiang Key Laboratory of Drinking Water Safety and Distribution Technology, Zhejiang University, Hangzhou 310058, China. E-mail: liliwang@zafu.edu.cn
bOcean College, Zhejiang University, Hangzhou 310058, China
cEnvironmental Engineering, Jiyang College of Zhejiang A & F University, Zhuji 311800, China
First published on 20th January 2023
Abstract
To address the concern about residual antibiotics in effluent of sewage treatment plants, cobalt silicate (CoSiOx) was prepared by hydrothermal method and employed as an activator of peroxymonosulfate (PMS) for the rapid degradation of antibiotics. Taking sulfamethoxazole (SMX) and tetracycline (TC) as representatives of antibiotics, the effects of operation parameters (CoSiOx and PMS dosage) and water quality parameters (temperature, solution pH, bicarbonate, chloride, and natural organic matter) on degradation of target pollutants by a CoSiOx activated PMS process (CoSiOx/PMS) were investigated. The mechanism involved in the interaction of CoSiOx and PMS was also elucidated. The results indicated that CoSiOx/PMS can degrade SMX and TC at fast pseudo-first-order rate constants (0.47 and 0.56 min−1 respectively) under optimal conditions. Increasing the dosage of PMS and CoSiOx appropriately was beneficial to the degradation of antibiotics. Chloride, bicarbonate, and HA showed negative effects on the degradation process due to their free radical-scavenging ability and were ranked as chloride < bicarbonate < HA. Abundant ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Co–OHs and oxygen vacancies on the surface of CoSiOx contributed to its excellent activation capability towards PMS. The radical scavenging experiments indicated that target pollutant degradation mainly resulted from the attack of sulfate radicals (43.0% contribution) and hydroxyl radicals (52.9% contribution). The practicality of CoSiOx/PMS was verified by continuous flow test. This study provides a cheap, highly efficient, and feasible advanced depollution method based on CoSiOx.
Co–OHs and oxygen vacancies on the surface of CoSiOx contributed to its excellent activation capability towards PMS. The radical scavenging experiments indicated that target pollutant degradation mainly resulted from the attack of sulfate radicals (43.0% contribution) and hydroxyl radicals (52.9% contribution). The practicality of CoSiOx/PMS was verified by continuous flow test. This study provides a cheap, highly efficient, and feasible advanced depollution method based on CoSiOx.
Introduction
Antibiotic pollution of the natural water environment has become a serious problem due to the abuse of antibiotics in the medical and animal industries.1 Residual antibiotics in the groundwater and surface water are usually found at a trace level of concentration (ng L−1 to μg L−1), which does not immediately cause toxicity or adverse effects in humans and aquatic organisms. However, antibiotics can be enriched and accumulated in the human body via the food chain, which may induce physiological disorders and even organ diseases.2 Moreover, bacteria will develop resistance from continuous exposure to the low concentration of antibiotics, which will pose a serious threat to human health and the ecological environment.1 Given that effluent of sewage treatment plants is one of the important routes of antibiotic exposure, it is imperative to exploit effective techniques to remove the remaining antibiotics.Advanced oxidation processes (AOPs), characterized by the generation of reactive species (hydroxyl radical (HO˙), sulfate radical (SO4˙−), superoxide radical (O2˙−), etc.), are quite attractive for degrading refractory organics. Recently, there has been a surge of interest in SO4˙−-based AOPs thanks to the characteristics of SO4˙− such as high redox potential, long viable period, wide range of pH applicability, and high selectivity to react with aromatic organic compounds.3 Among the precursors of SO4˙−, peroxymonosulfate (PMS) was favoured due to its special asymmetric structure and short O–O bond. When PMS was used to produce radicals, Co2+ was considered to be the most efficient PMS activator.4 Unfortunately, the recovery of Co2+ and Co2+-related secondary pollution hindered the application of Co2+ activated PMS process (PMS/Co2+). Thus, great efforts have been made to develop heterogeneous Co-based activators to overcome the shortcomings of PMS/Co2+.
Silicon is widely distributed in the earth's crust and occurs naturally in the form of silica and complex silicates. Metal silicates are a significant category of fundamental substances with stable chemical quality, numerous structures, and abundant surface functional groups.5 The excellent inherent properties, such as good plasticity and resistance to high temperature, corrosion and wear, make metal silicates innately a kind of functional material for water treatment.5,6 Hao et al.7 found that cobalt silicate showed the best performance for PMS activation among a series of prepared metal silicates. Zhu et al.5 fabricated low-crystalline cobalt silicate hydroxide with abundant active Co–OH+ on the surface, exhibiting high PMS activation ability. Obviously, embedding Co2+ into the silicate framework can clear the obstacles plaguing PMS/Co2+. It should be mentioned that, although previous works successfully fabricated cobalt silicate or metal-dopped cobalt silicate and obtained excellent activation performance towards PMS, the complex preparation processes discounted their advantages. From the viewpoint of practical application, a simple preparation method of cobalt silicate with good PMS activation ability is needed. Moreover, little attention was paid to untraditional active sites such as oxygen vacancies on cobalt silicate surfaces.8
Thus, cobalt silicate was synthesized by a simple hydrothermal method. Sulfamethoxazole (SMX) and tetracycline (TC), two typical antibiotics frequently detected in effluent of sewage treatment plant, were selected as target pollutants to evaluate the degradation performance of the PMS/cobalt silicate process. The effects of key factors on the degradation efficiency of antibiotics were investigated in detail, and the catalytic mechanism was systematically investigated. Finally, the depollution performance of PMS/cobalt silicate was further verified through a continuous flow test using real water. This work contributed to promoting the practical application of PMS-based AOPs and antibiotic pollution control in effluent of sewage treatment plant.
Material and methods
Materials and reagents
Cobalt nitrate hexahydrate, sodium metasilicate pentahydrate, sodium thiosulfate pentahydrate, disodium hydrogen phosphate, potassium dihydrogen phosphate, methanol, and tert-butanol were obtained from Aladdin Reagent Co. Ltd. Sodium chloride, sodium bicarbonate, sodium hydroxide, hydrochloric acid, and phosphoric acid were ordered from Sinopharm Chemical Reagent Co. Ltd. Sulfamethoxazole (SMX), tetracycline (TC), ciprofloxacin (CIP), PMS (Oxone®, KHSO5·0.5KHSO4·0.5K2SO4), acetonitrile, methanol (MeOH), tert-butyl alcohol (TBA), formic acid, oxalic acid, and humic acid (HA) were purchased from Sigma-Aldrich. Alumina (AAO) membrane was provided by Nanjing Xian Feng Nano-Material Technology Co., Ltd. Solutions were prepared with ultrapure water (18.2 MΩ cm).Synthesis of cobalt silicate
Working solutions of Na2SiO3 (250 mL) and Co(NO3)2 (250 mL) at a concentration of 0.1 M were firstly prepared. Na2SiO3 solution was then titrated with Co(NO3)2 through a constant pressure funnel at 1 mL min−1 and stirred with a magnetic stirrer at 300 rpm simultaneously. The pH of the mixture obtained after titration was adjusted to 8–9 and subsequently aged at 40 °C for 12 h. The sediment at the bottom was collected and washed repeatedly until the washing solution reached a neutral pH. The cleaned materials were finally dried under a vacuum at 60 °C for 48 h and denoted as CoSiOx.Catalytic degradation measurements
Batch experiments were conducted in 4 L reactor equipped with thermal jacket and magnetic stirrer. The SMX or TC solution at designed concentration was prepared and transferred to the reactor. Then, the reaction solution was adjusted using phosphate buffer solution. Subsequently, a certain amount of CoSiOx was added to the solution. Finally, the degradation process was considered to have started once PMS solution was added. During the reaction process, samples were drawn and immediately injected into the vial containing quencher, followed by filtration with 0.22 μm PES membrane to intercept particles.Fig. 1 shows the experimental set-up for the continuous flow test. Designed amount of CoSiOx was loaded onto the alumina (AAO) membrane and then installed the membrane into the filter. Raw secondary effluent from wastewater treatment plant was pumped to mix with the PMS dosing stream. Immediately, the mixture flowed through the membrane module through the peristaltic pump. Water samples were taken periodically.
 | ||
| Fig. 1 Experimental setup of the continuous flow test. | ||
Analytical methods
The concentrations of antibiotics were detected by high-performance liquid chromatography (HPLC, Agilent) equipped with a C-18 column (5 μm, 4.6 × 150 mm) and a UV detector. For SMX, the mobile phase was a mixture of formic acid (0.1%) and acetonitrile (HPLC grade, 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, v/v). The mobile phase flow rate was 0.8 mL min−1, and the detection wavelength was 264 nm. For TC, the mobile phase was a mixture of oxalic acid (0.01 M), methanol and acetonitrile (HPLC grade, 67:11:22, v/v). The mobile phase flow rate was 0.7 mL min−1, and the detection wavelength was 355 nm. For CIP, the mobile phase was a mixture of phosphoric acid (0.1%) and acetonitrile (HPLC grade, 85:15, v/v). The mobile phase flow rate was 0.7 mL min−1, and the detection wavelength was 278 nm. The degradation products were analyzed by an Agilent 6460 triple-quad HPLC-MS. Water samples, collected at different reaction time (0–15 min), were concentrated 50 times using solid phase extraction before analysis. A Microtox Model 500 toxicity analyzer coupled with luminescent bacteria Vibrio fisheri was used to test the comprehensive toxicity of water samples (indicated with luminescence inhibition rate).
:30, v/v). The mobile phase flow rate was 0.8 mL min−1, and the detection wavelength was 264 nm. For TC, the mobile phase was a mixture of oxalic acid (0.01 M), methanol and acetonitrile (HPLC grade, 67:11:22, v/v). The mobile phase flow rate was 0.7 mL min−1, and the detection wavelength was 355 nm. For CIP, the mobile phase was a mixture of phosphoric acid (0.1%) and acetonitrile (HPLC grade, 85:15, v/v). The mobile phase flow rate was 0.7 mL min−1, and the detection wavelength was 278 nm. The degradation products were analyzed by an Agilent 6460 triple-quad HPLC-MS. Water samples, collected at different reaction time (0–15 min), were concentrated 50 times using solid phase extraction before analysis. A Microtox Model 500 toxicity analyzer coupled with luminescent bacteria Vibrio fisheri was used to test the comprehensive toxicity of water samples (indicated with luminescence inhibition rate).
Energy dispersive spectrometry (EDS, FEI, FEG650) measurement was employed to investigate the elementary composition. The surface functional groups and chemical bonds were determined by Fourier transform infrared spectroscopy with a resolution ratio of 4 cm−1 and an accumulation of 32 scans (FTIR, Thermo Scientific, NICOLET iS50). The crystal structures were characterized by X-ray diffraction (XRD, PANalytical, X-pert Powder) using a Cu Kα source, with a scanning step of 0.026°. X-ray photoelectron spectroscopy (XPS, V.G ESCALAB) was used to study the element valence distribution of cobalt silicate, and the obtained curve was fitted with Xpspeak software. High performance liquid chromatography-mass spectrometry (HPLC-MS, AB Sciex, Triple TOF 5600+) was used to determine the intermediate products.
Results and discussion
Characterization of CoSiOx
The wide-angle XRD pattern of cobalt silicate powder revealed the structure of amorphous phase (Fig. 2a). There were no typical peaks at 19°, 31°, 37°, 45°, 55°, and 65°, ascribed to the reflecting planes (111), (220), (311), (400), (422), and (440) of cobalt spinel,9 suggesting that cobalt species do not exist in the form of Co3O4. Instead, all the weak diffraction peaks correspond to a pure cobalt talc phase (Co3(Si2O5)2(OH)2, PDF no. 21-0871).10 The broad peak at the 2θ range of 21 to 30° is associated with the (003) plane. And the other two peaks located at around 34.3 and 59.5° correspond to the (210) and (330) planes, respectively. However, the characteristic peak at a low angle (6–9°), corresponding to the basal plane (001) of the Co talc layered structure,11 was not detected. EDS measurement was performed to gain further insight into the chemical composition. Three elements, Co, Si and O at an atom ratio of 1:1:1.9, were observed (Fig. 2b). XPS measurement further confirmed the presence of Co, Si and O elements (Fig. 2c). The coordinate unsaturation of O with Co and Si implied that there were oxygen vacancies (OVs) on the surface. These results indicated that the prepared powder was a pure phase material. Therefore, for the convenience of subsequent discussion, the chemical structure of prepared cobalt silicate powder was represented as CoSiOx according to the analysis results of EDS and XPS.
 | ||
| Fig. 2 Characterization of CoSiOx: (a) XRD pattern, (b) EDS spectra, (c) XPS spectra, (d) FT-IR spectra. | ||
FT-IR spectra provide detailed information on the surface functional groups of CoSiOx, and the results are shown in Fig. 2d. Significantly, there was a band assigned to the O–H stretching of characteristic cobalt hydroxyl groups (3640 cm−1), which played an important role in the interaction of metal oxide and PMS.5 By comparing with the FT-IR spectrum of Na2SiO3, one can find that the typical Si–O–Si stretching band shows a blue shift from 993 to 1015 cm−1 due to the formation of the Co–O–Si bond formed by the combination of cobalt ion and the O atom of [SiO4]4−.5,12 The bands at 661 and 461 cm−1 are attributed to Co–O and Si–O vibrations, respectively.5 It should be mentioned that the characteristic band at around 578 cm−1, correlated with the Co–O stretching modes of CoO and Co3O4,4 was not detected, proving that the prepared material did not contain component of CoO or Co3O4.
Depollution capability evaluation of CoSiOx/PMS
To figure out depollution capability of CoSiOx/PMS and involved contributors for pollutant degradation or removal, CoSiOx/PMS system was compared with systems including PMS oxidation, CoSiOx adsorption, Co3O4 adsorption, and Co3O4/PMS, and the results are showed in Fig. 3a and b. Direct PMS oxidation degrade 10.2% SMX and ∼80% TC within 15 min, and adsorption using CoSiOx removed less than 3% SMX and TC. Once CoSiOx was combined with PMS, both of the two pollutants completely degraded within 10 min. The results demonstrated that there was synergistic effect formed between CoSiOx and PMS. Such synergistic effect is reasonably considered to result from the formation of oxidative radicals such as SO4˙− due to the activation of PMS by CoSiOx. Obviously, CoSiOx was far superior to commercial Co3O4 in terms of combining with PMS. Moreover, CoSiOx/PMS can be competent for degradation of TC and SMX at concentration ranging from 30 μM to 100 μM (Fig. 3c and d).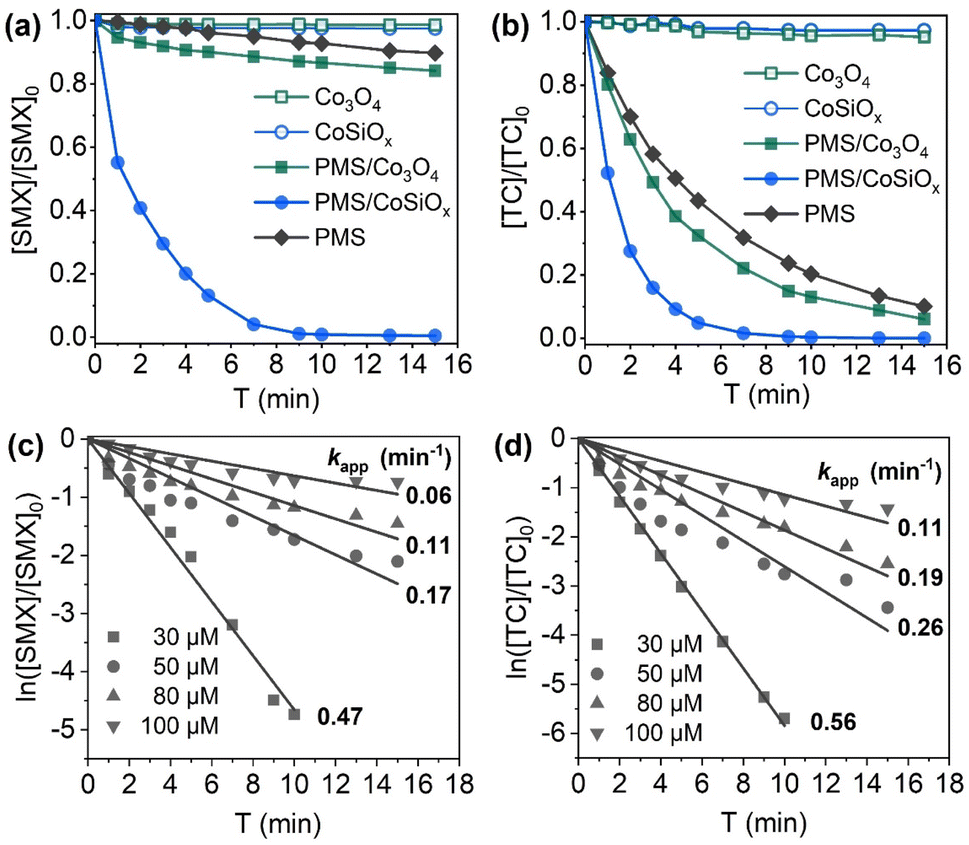 | ||
| Fig. 3 (a) SMX degradation in different systems ([SMX]0 = 30 μM, [CoSiOx]0 = [Co3O4]0 = 40 mg L−1); (b) TC degradation in different systems ([TC]0 = 30 μM, [CoSiOx]0 = [Co3O4]0 = 5 mg L−1); (c) degradation rate constants for SMX at different concentrations ([CoSiOx]0 = 40 mg L−1); (d) degradation rate constants for TC at different concentrations ([CoSiOx]0 = 5 mg L−1). Other experimental conditions: [PMS]0 = 0.2 mM, pH = 7.0 ± 0.2, T = 25 °C. | ||
To rank the PMS activation ability of CoSiOx, rough comparison was also made between CoSiOx and other reported heterogeneous Co-based materials (Table 1). The comparison results demonstrated that activation capacity of CoSiOx was superior to these reported materials even at much lower dosage.
| System | [Catalyst]0 (mg L−1) | Antibiotic | [Antibiotic]0 (μM) | [PMS]0 (mM) | pH | kapp (min−1) | Ref. |
|---|---|---|---|---|---|---|---|
| PMS/CoSiOx | 40 | SMX | 30 | 0.2 | 7.0 ± 0.2 | 0.47 | This work |
| PMS/Co–N–C@SiO2 | 50 | SMX | 39.5 | 2.3 | 7.0 | 0.12 | 13 |
| PMS/NiCo2O4-EG | 60 | SMX | 19.8 | 0.3 | 6.3 ± 0.3 | 0.13 | 14 |
| PMS/CoSiOx | 5 | TC | 30 | 0.2 | 7.0 ± 0.2 | 0.57 | This work |
| PMS/Co–CN | 500 | TC | 22.5 | 0.8 | 6.9 | 0.20 | 15 |
| PMS/CoFe2O4@MoS2 | 200 | TC | 22.5 | 0.5 | 7.0 | 0.06 | 16 |
Effect of operational parameters
Fig. 4 shows the degradation performance of SMX and TC by PMS/CoSiOx under different CoSiOx dosage. A positive correlation was observed between the degradation performance and CoSiOx dosage. Although SMX and TC exhibited obvious difference in treatability, both of them can be completely degraded within 10 min at a low CoSiOx dosage (40 mg L−1 for SMX and 5 mg L−1 for TC). Notably, the addition of 1 mg L−1 of CoSiOx yielded kapp-SMX of 0.04 min−1 and kapp-TC of 0.26 min−1. Based on the fitting equation of kapp Vs CoSiOx dosage, one can infer that 90% degradation of SMX and TC will realize within 3 min at a CoSiOx dosage over 70 mg L−1, which means only a small reactor is needed or loading CoSiOx onto membrane-like carrier for filtration purification is feasible. | ||
| Fig. 4 Degradation rate constant of SMX and TC at different CoSiOx dosages. Experimental conditions: [SMX]0 = [TC]0 = 30 μM, [PMS]0 = 0.2 mM, pH = 7.0 ± 0.2, T = 25 °C. | ||
The effects of PMS dosage are shown in Fig. 5. The degradation efficiency of SMX and TC increased remarkably with PMS dosage ranging from 0.02 to 0.2 mM, and a linear correlation was found. The slopes of kapp Vs PMS dosage for SMX and TC was much higher than those of kapp Vs CoSiOx dosage, implying that depollution efficiency of PMS/CoSiOx was more responsive to PMS dosage. It should be mentioned that, when PMS dosage increased from 0.2 to 0.25 mM, there was only a subtle improvement in TC degradation, and the SMX degradation efficiency was decreased. Thus, the optimal dosing ratios of PMS to CoSiOx for SMX and TC were 0.005 and 0.04 molar per g-CoSiOx, respectively. That is, the number of active sites on CoSiOx surface was limiting factor. Considering efficiency and cost, 0.2 mM was selected as the PMS concentration parameter for the subsequent SMX and TC degradation experiments.
 | ||
| Fig. 5 Degradation rate constants of SMX and TC at different PMS dosages. Experimental conditions: [SMX]0 = [TC]0 = 30 μM, [CoSiOx]0 = 40 mg L−1 (SMX), [CoSiOx]0 = 5 mg L−1 (TC), pH = 7.0 ± 0.2, T = 25 °C. | ||
Effect of water quality parameters
Fig. 6a present the effect of solution pH on SMX and TC degradation. The best degradation performance of SMX and TC were received at pH 7.0 and pH 9.0, respectively. There are two reasons to explain this phenomenon: (1) the pH value will affect the form of existence of PMS (pKa2 = 9.4 for H2SO5), thus affect the formation and transformation of free radicals. In a strong alkaline environment, PMS is largely transformed into SO52− due to deprotonation, resulting in a dramatic reduction in the SO4˙− yield and degradation efficiency; (2) the pH value will affect the morphology and distribution of SMX and TC (Fig. 6b and c). | ||
| Fig. 6 (a) Degradation rate constants of SMX and TC at different pH values; (b) distribution coefficient of SMX; (c) distribution coefficient of TC. Experimental conditions: [SMX]0 = [TC]0 = 30 μM, [PMS]0 = 0.2 mM, [CoSiOx]0 = 40 mg L−1 (SMX), [CoSiOx]0 = 5 mg L−1 (TC), T = 25 °C. | ||
The solid–liquid interface reaction is greatly affected by mass transfer. Temperature is one of the key factors affecting mass transfer efficiency. Thus, degradation of SMX and TC by PMS/CoSiOx was investigated under different temperature. As shown in Fig. 7a, degradation of SMX and TC was improved with the increase of solution temperature ranging from 10–40 °C. Because TC shows a lower reaction activation energy (Ea) than SMX (Fig. 7b), SMX degradation was more sensitive to temperature change. Given that the Ea values for of SMX and TC degradation were much higher than the Ea of the diffusion-controlled reaction (10–13 kJ mol−1),17 it can be concluded that the degradation process is controlled by the chemical reaction rate on the solid–liquid interface.
 | ||
| Fig. 7 (a) Degradation rate constants of SMX and TC at different temperatures; (b) calculated activation energy for degradation of SMX and TC. Experimental conditions: [SMX]0 = [TC]0 = 30 μM, [PMS]0 = 0.2 mM, [CoSiOx]0 = 40 mg L−1 (SMX), [CoSiOx]0 = 5 mg L−1 (TC), pH = 7.0 ± 0.2. | ||
Anions such as Cl− and HCO3− were widely reported to scavenge radicals,18 thus the effects of them on degradation process were evaluated. As presented in Fig. 8a, Cl− acted as an inhibitor for the degradation of SMX and TC herein. When Cl− concentration increased from 0 to 5 mM, the degradation rate constant declined by about 47.2% for SMX and decreased by 27.1% for TC. The inhibition effect of Cl− resulted from its scavenging of SO4˙− and HO˙, which yielded chlorine radicals such as Cl˙ and Cl2˙−.19 The chlorine radicals showed a lower oxidation potential (2.0–2.4 eV) than SO4˙− and HO˙, which means the loss in oxidation power. Similar to Cl−, HCO3− was also verified to be an inhibitor to the degradation process (Fig. 8b). When Cl− concentration was 2 mM, kapp-SMX and kapp-TC decreased to 0.03 and 0.23 min−1, respectively. Obviously, the inhibition effect of HCO3− was stronger than that of Cl−. HCO3− can scavenge SO4˙− and HO˙ to produce CO3˙− and  with lower oxidation potential (1.7 eV), which accounts for the stronger negative influence on the degradation.18
with lower oxidation potential (1.7 eV), which accounts for the stronger negative influence on the degradation.18
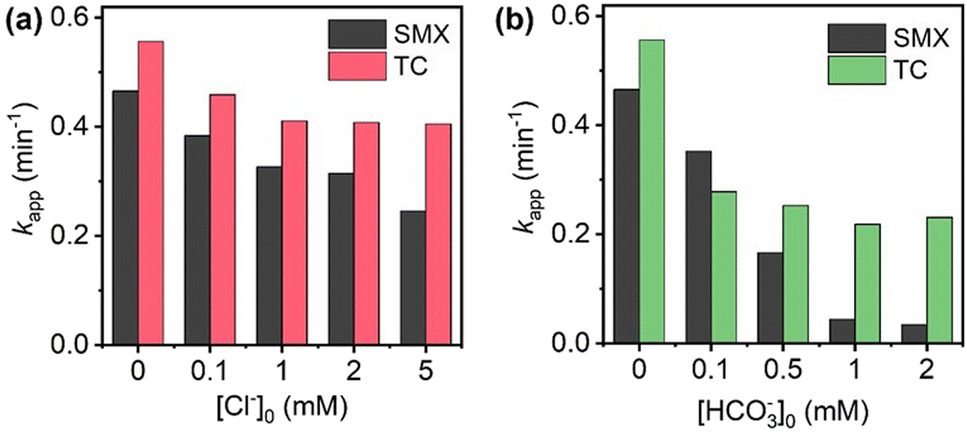 | ||
| Fig. 8 Degradation rate constants of SMX and TC in the presence of different concentrations of (a) Cl− and (b) HCO3−. Experimental conditions: [SMX]0 = [TC]0 = 30 μM, [PMS]0 = 0.2 mM, [CoSiOx]0 = 40 mg L−1 (SMX), [CoSiOx]0 = 5 mg L−1 (TC), pH = 7.0 ± 0.2, T = 25 °C. | ||
As organic scavenger of radicals, natural organic matters (NOM) usually played a significant negative effect on SO4˙−-based AOPs. Fig. 9 shows the effect of HA (representative of NOM) on SMX and TC degradation. Even at a low concentration (0.1 mg L−1), HA decreased kapp-SMX and kapp-TC by 19.7% and 13.2% respectively. When HA reached a common concentration of 1.0 mg L−1, kapp-SMX and kapp-TC were decreased by over 30%. Obviously, the inhibition effect of HA was much stronger than Cl− and HCO3−. The abundant electron-rich and ligand moieties bearing in the chemical structure of HA, acting as radical scavenger and interaction blocker of PMS and CoSiOx, contributed to this strong inhibition effect.20
 | ||
| Fig. 9 Degradation rate constants of SMX and TC in the presence different concentrations of HA. Experimental conditions: [SMX]0 = [TC]0 = 30 μM, [PMS]0 = 0.2 mM, [CoSiOx]0 = 40 mg L−1 (SMX), [CoSiOx]0 = 5 mg L−1 (TC), pH = 7.0 ± 0.2, T = 25 °C. | ||
Radical identification and activation mechanism
It is widely accepted that SO4˙− and HO˙ are the main radicals in Co(II)-activated PMS process. The classical quenching experiments were performed to identify radical involved in the PMS/CoSiOx system. MeOH (scavenger for ˙OH and SO4˙−) and TBA (˙OH scavenger) were employed to distinguish the contribution of SO4˙− and HO˙.21 As shown in Fig. 10, the addition of TBA made the degradation rate constant decreased from 0.47 to 0.22 min−1, validating the participation of HO˙. The presence of MeOH could decrease the degradation rate constant of SMX to 0.02 min−1, which further verified the involvement of SO4˙−. It is worth noting that neither TBA nor MeOH could completely inhibit the degradation of SMX. This part of contribution resulted from the PMS direct oxidation. Moreover, the addition of NaN3 did not affect the SMX degradation, indicating the negligible role of 1O2 in the PMS/CoSiOx system. The contributions of HO˙ and SO4˙− for SMX degradation were calculated to be 52.9% and 43.0%, respectively. | ||
| Fig. 10 SMX degradation by PMS/CoSiOx in the presence of different scavengers. | ||
To figure out how PMS and CoSiOx interact with each other, the valence changes of element (Si, O, and Co) were determined using XPS. In the Si 2p spectrum (Fig. 11a), the peaks locating at 103.8 and 102.9 eV correspond to the Si–O–Si and Si–O–Co bonds, respectively.22 The types and relative contents of Si did not change before and after the reaction, indicating that Si did not participate in the reaction. As shown in Fig. 11b, the O 1s spectrum could be deconvoluted into three characteristic peaks located at 532.9, 531.9, and 530.9 eV, corresponding to lattice oxygen (Olat), hydroxyl groups (O–OH), and adsorbed oxygen species (Oads, O22−, O2− and O−), respectively.23,24 To some extent, the Oads species could represent oxygen vacancies.8 Surface hydroxyl can be used as active sites, producing a profound impact on the PMS activation and the production of reactive species.25 The relative content of Oads decreased, which may be attributed to the neutralization of OVs during the catalytic reaction, confirmed the involvement of OVs.23
 | ||
| Fig. 11 XPS spectra of fresh and used CoSiOx: (a) Si 2p, (b) O 1 s, and (c) Co 2p. | ||
Fig. 11c displays the spectrum of the Co 2p. The two prominent peaks at 782.4 and 798.2 eV with satellite peaks locating at 786.1 and 804 eV can be assigned to Co 2p3/2 and Co 2p1/2.11 The locations and separation of these peaks are related to Co(II) in Co–O–Si bonds. Moreover, the massive high-spin Co(II) occupying octahedral sites is consistent with the high intensity of satellite peaks, corresponding to the Co talc structure.11 Two small peaks corresponding to Co(III) at 780.6 and 796.6 eV could be observed after the interaction of CoSiOx with PMS. The results confirmed a conversion of the valence states of Co(II)/Co(III) during the catalytic reaction, inferring that cobalt plays an essential role as an active site for PMS activation.
Based on the above results and previous reports, the activation mechanism of PMS by CoSiOx includes the following aspects: (1) water molecules firstly adsorbed on the surface of CoSiOx and formed hydroxyl radical (Co–OHs); (2) then, Co–OHs served as active sites and reacted with PMS (HSO5−) to form the [Co(II)–(O)OSO3−] coordination compound precursor; (3) the internal electron transfer occurred in the generated precursor (from Co(II) to HSO5−), followed by the break of the O–O bonds with the aid of active parallel-spin electrons released from the unstable eg-orbitals in high-spin Co(II) (t3↑2↓2ge2↑g) and generation of SO4˙−. During this process, Co(II) was oxidized to Co(III). In addition, the OVs on CoSiOx surface, which could effectively promote the PMS adsorption process, lower the reaction energy barrier of PMS decomposition, and improve the electron transfer efficiency, enhanced the O–O bonds activation; (4) Co(III) was reduced to Co(II) by HSO5− to complete the redox cycle of Co(III)/Co(II). It is important to emphasize that the conversion of Co3+/Co2+ is difficult to occur in the traditional homogeneous catalytic system. However, OVs sites on the CoSiOx surface can increase the charge carrier density and then improve the electrical conductivity, promoting the regeneration of Co(II).23
Degradation pathways of SMX
To further verify the involvement of HO˙ and SO4˙− during the pollutant degradation and figure out whether there is accumulation of intermediate product, SMX was selected as representative and oxidized with PMS/CoSiOx. The degradation products and pathways are presented in Fig. 12. There are three possible degradation pathways. In pathway I, the amino groups on the benzene ring of SMX were oxidized to nitroso, and then the nitroso was further oxidized to form the product NO2-SMX.26 In pathway II, SMX-OH formed through the hydroxylation of heterocycle, and the addition reaction at isoxazole ring produced SMX-2OH.27 The electron density of olefin double bond is enhanced by nitrogen atom. In pathway III, the aniline group was attacked by SO4˙− to produce unstable SMX˙+, and then OH-SMX was formed by hydrolysis. Considering the electron donor effect of –NH2 and the electron withdrawing effect of –SO2–NH–, the hydroxylation of benzene ring would occur at the ortho-position of –NH2. Then, the cleavage of S–N bond produced OH-SAA and AMI. Notably, these degradation products were detected within the first 5 min once reaction started. It is reasonable to speculate that these degradation intermediates, which keep complete or partial functional groups of the parent molecular, are broken into small molecules or mineralized with the extension of reaction time. | ||
| Fig. 12 Pathways of SMX degradation by PMS/CoSiOx. Experimental conditions: [SMX]0 = 30 μM, [PMS]0 = 0.2 mM, [CoSiOx]0 = 40 mg L−1, pH = 7.0 ± 0.2, T = 25 °C, detected intermediates were qualified based on HPLC-MS analysis results of water samples collected at reaction time of 0–5 min. | ||
Practical application scenario test
The continuous flow experiments were performed to investigate the degradation performance of antibiotics in real sewage treatment plant effluent by CoSiOx/PMS. CoSiOx was loaded onto the AAO membrane (CoSiOx@AAO) and the removal performance for three antibiotics was monitored. Although the raw water contained high concentration of radical scavengers including COD and Cl− (Table 2), the three target antibiotics were removed above 82% during the 6-d test period (Fig. 13a). Specifically, the degradation efficiency of the three antibiotics was in the order of TC > CIP > SMX. The results denoted that CoSiOx can stably perform its activation ability during long-time reaction. The results show that CoSiOx has a stable activation capacity towards PMS and that it is feasible to load CoSiOx on antioxidation membrane for water treatment. There were no significant differences between the XRD of the used and fresh CoSiOx (Fig. 13b), demonstrating the good structural stability of the prepared catalyst. Moreover, a significant decrease in toxicity (indicated with luminescence inhibition rate) was detected for the water samples treated by CoSiOx/PMS (Fig. 13c).| Parameter | Value | Parameter | Value |
|---|---|---|---|
| COD (mg O2 L−1) | 16.2 | SMX (μg L−1) | 0.21 |
| Chloride (mg Cl− L−1) | 167 | TC (μg L−1) | 0.047 |
| Sulfate (mg SO42− L−1) | 72 | CIP (μg L−1) | 0.12 |
| pH | 7.80 | Triclosan (μg L−1) | 0.016 |
| Turbidity (NTU)a | 0.56 | Oxytetracycline (μg L−1) | 0.103 |
| Nitrite (mg NO2−-N L−1) | <0.015 | Norfloxacin (μg L−1) | 0.032 |
| Alkalinity (mg L−1 as CaCO3) | 218.6 | Minocycline (μg L−1) | 0.011 |
| Bicarbonate (mg L−1) | 180.3 | UV254 (cm−1) | 0.012 |
 | ||
| Fig. 13 (a) Degradation of antibiotics by CoSiOx@AAO/PMS; (b) XRD pattern of unused and used CoSiOx; (c) toxicity of water samples before and after treatment (indicated with luminescence inhibition rate). Experimental conditions: [PMS]0 = 0.15 mM, membrane area is 4.9 cm2, 40.8 mg CoSiOx per cm2 AAO membrane, flow rate = 3.5 mL min−1, empty bed contact time = 18 s. | ||
Conclusions
The CoSiOx/PMS system realized ultrafast degradation of SMX and TC (100%) within 15 min at a very low dosage of CoSiOx under mild conditions. Although increasing the dosage of CoSiOx and PMS both accelerated the degradation of SMX and TC, the depollution efficiency of PMS/CoSiOx was more responsive to PMS dosage. In addition, excessive PMS addition would cause reduction in depollution efficiency. High solution temperature was beneficial for PMS/CoSiOx to exert the degradation capability, and the calculated activation energy of the SMX (53.4 kJ mol−1) and TC (31.0 kJ mol−1) degradation by CoSiOx/PMS suggested the nature of non-diffusion control. Among the common radical scavengers, HCO3− and HA showed a more pronounced negative effect than Cl−. Competitive kinetics experiments confirmed that SO4˙− and HO˙ acted as main contributors to SMX and TC degradation.Co–OHs and oxygen vacancies on the surface of CoSiOx were found to play an essential role in the reaction of CoSiOx and PMS. The long-time continuous flow test using real water further confirmed that the prepared CoSiOx was efficient to activate PMS and can keep stable during use.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by the Zhejiang Public Welfare Technology Research Program (No. GG21E080021) and the Jiyang 533 Talent Program (No. JY21E0102).References
- M. Qiao, G.-G. Ying, A. C. Singer and Y.-G. Zhu, Environ. Int., 2018, 110, 160–172, DOI:10.1016/j.envint.2017.10.016.
- Q.-Q. Zhang, G.-G. Ying, C.-G. Pan, Y.-S. Liu and J.-L. Zhao, Environ. Sci. Technol., 2015, 49, 6772–6782, DOI:10.1021/acs.est.5b00729.
- Z. Wang, Y. Han, W. Fan, Y. Wang and L. Huang, Sep. Purif. Technol., 2022, 278, 119558, DOI:10.1016/j.seppur.2021.119558.
- P. Shao, J. Tian, X. Duan, Y. Yang, W. Shi, X. Luo, F. Cui, S. Luo and S. Wang, Chem. Eng. J., 2019, 359, 79–87, DOI:10.1016/j.cej.2018.11.121.
- Z.-S. Zhu, X.-J. Yu, J. Qu, Y.-Q. Jing, Y. Abdelkrim and Z.-Z. Yu, Appl. Catal., B, 2020, 261, 118238, DOI:10.1016/j.apcatb.2019.118238.
- J. Qu, W. Li, C.-Y. Cao, X.-J. Yin, L. Zhao, J. Bai, Z. Qin and W.-G. Song, J. Mater. Chem., 2012, 22, 17222–17226, 10.1039/c2jm33178k.
- S.-M. Hao, M.-Y. Yu, Y.-J. Zhang, Y. Abdelkrim and J. Qu, J. Colloid Interface Sci., 2019, 545, 128–137, DOI:10.1016/j.jcis.2019.03.017.
- L.-L. Su, R.-J. Li, N. Huang, T.-T. Zhu, S.-H. Deng, X.-X. Wang, G. Yang, Y.-Z. Zhang and L.-L. Long, J. Colloid Interface Sci., 2022, 628, 955–965, DOI:10.1016/j.jcis.2022.08.022.
- L.-H. Ai and J. Jiang, Powder Technol., 2009, 195, 11–14, DOI:10.1016/j.powtec.2009.05.006.
- G.-Q. Zhang, Y.-Q. Zhao, F. Tao and H.-l. Li, J. Power Sources, 2006, 161, 723–729, DOI:10.1016/j.jpowsour.2006.03.041.
- M. Domínguez, E. Taboada, H. Idriss, E. Molins and J. Llorca, J. Mater. Chem., 2010, 20, 4875–4883 RSC https://pubs.rsc.org/en/content/articlelanding/2010/jm/c0jm00184h.
- R. Al-Oweini and H. El-Rassy, J. Mol. Struct., 2009, 919, 140–145, DOI:10.1016/j.molstruc.2008.08.025.
- Y. Feng, W. Sang, Z. Deng, S. Zhang and C. Lia, Sep. Purif. Technol., 2022, 280, 119783, DOI:10.1016/j.seppur.2021.119783.
- M. Xu, H. Zhou, Z. Wu, N. Li, Z. Xiong, G. Yao and B. Lai, J. Hazard. Mater., 2020, 399, 123103, DOI:10.1016/j.jhazmat.2020.123103.
- X. Zhang, B. Xu, S. Wang, X. Li, C. Wang, B. Liu, F. Han, Y. Xu, P. Yu and Y. Sun, Chem. Eng. J., 2022, 431, 133477, DOI:10.1016/j.cej.2021.133477.
- X. Peng, Z. Yang, F. Hu, C. Tan, Q. Pan and H. Dai, Sep. Purif. Technol., 2022, 287, 120525, DOI:10.1016/j.seppur.2022.120525.
- Y. Yao, Y. Cai, F. Lu, F. Wei, X. Wang and S. Wang, J. Hazard. Mater., 2014, 270, 61–70, DOI:10.1016/j.jhazmat.2014.01.027.
- X. Liu, T. Zhang, Y. Zhou, L. Fang and Y. Shao, Chemosphere, 2013, 93, 2717–2724, DOI:10.1016/j.chemosphere.2013.08.090.
- L. Wang and X. Liu, Processes, 2019, 7, 95, DOI:10.3390/pr7020095.
- J. Sharma, I. M. Mishra, D. D. Dionysiou and V. Kumar, Chem. Eng. J., 2015, 276, 193–204, DOI:10.1016/j.cej.2015.04.021.
- W.-D. Oh, Z. Dong and T.-T. Lim, Appl. Catal., B, 2016, 194, 169–201, DOI:10.1016/j.apcatb.2016.04.003.
- X. Zhang, J. Liang, Y. Sun, F. Zhang, C. Li, C. Hu and L. Lyu, J. Colloid Interface Sci., 2020, 576, 59–67, DOI:10.1016/j.jcis.2020.05.007.
- Q. Lian, A. Roy, O. Kizilkaya, D. D. Gang, W. Holmes, M. E. Zappi, X. Zhang and H. Yao, ACS Appl. Mater. Interfaces, 2020, 12, 57190–57206, DOI:10.1021/acsami.0c20341.
- C. Zhang, L. Zhang, G. Xu, X. Ma, J. Xu, L. Zhang, C. Qi, Y. Xie, Z. Sun and D. Jia, ACS Appl. Nano Mater., 2018, 1, 800–806, DOI:10.1021/acsanm.7b00246.
- Y. Zhao, H. An, G. Dong, J. Feng, T. Wei, Y. Ren and J. Ma, Chem. Eng. J., 2020, 388, 124371, DOI:10.1016/j.cej.2020.124371.
- M. M. Ahmed, S. Barbati, P. Doumenq and S. Chiron, Chem. Eng. J., 2012, 197, 440–447, DOI:10.1016/j.cej.2012.05.040.
- Y. Ji, Y. Fan, K. Liu, D. Kong and J. Lu, Water Res., 2015, 87, 1–9, DOI:10.1016/j.watres.2015.09.005.
| This journal is © The Royal Society of Chemistry 2023 |