
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiologically potent organotin(IV) complexes of N-acetylated β-amino acids with spectroscopic, X-ray powder diffraction and molecular docking studies†
Nagina Naveed Riazab,
Muhammad Mahboob Ahmed *a,
Muhammad Kashifc,
Muhammad Sajida,
Muhammad Alid and
Khalid Mahmooda
*a,
Muhammad Kashifc,
Muhammad Sajida,
Muhammad Alid and
Khalid Mahmooda
aInstitute of Chemical Sciences, Bahauddin Zakariya University, Multan, Pakistan. E-mail: mahboobahmed@bzu.edu.pk
bDepartment of Chemistry, Division of Science & Technology, University of Education, Lahore, Pakistan
cDepartment of Chemistry, Emerson University, Multan, Pakistan
dSchool of Materials Science and Engineering, University of Science and Technology of China, 96 Jinzhai Road, Baohe District, Hefei, 230026, PR China
First published on 5th April 2023
Abstract
Twelve novel organotin(IV) complexes (1–12) of N-acetylated β-amino acids (L1–L8) were synthesized and characterized by elemental analysis, FTIR, multinuclear (1H, 13C, 119Sn) NMR, EI-MS and powder XRD techniques. The XRD results determined lattice parameters, average particle size, and intrinsic strain and confirmed the crystalline nature of complexes as face centered cubic phases. Molecular docking analysis using a catalytic pocket of the α-glucosidase enzyme indicated that most of the compounds displayed a well-fitted orientation and occupied important amino acids in the enzyme's catalytic pocket. Furthermore, in vitro α-glucosidase inhibitory activity results revealed that L1 and complexes 4, 6 and 10 showed the highest activity with IC50 values of 21.54 ± 0.45, 37.96 ± 0.81 and 35.20 ± 1.02, respectively, compared to standard acarbose with an IC50 value of 42.51 ± 0.21. In addition, in vivo antidiabetic activity of selected compounds using alloxan induced diabetic rabbits showed that L4 and complexes 4, 6, 10, 12 showed significant activities like standard metformin. Anti-bacterial activity against the selected Gram-positive and Gram-negative bacterial strains has the following order Escherichia coli > Pseudomonas aeruginosa > Staphylococcus aureus > Bacillus subtilis. Similarly, antioxidant activity by the DPPH scavenging method was also studied with following results: triorganotin > diorganotin > ligands.
Introduction
Organotin compounds, especially organotin(IV) carboxylates, display prime biological activities as biocides such as cytotoxic, anti-microbial, antiproliferative, antitumor, anticancer, antituberculosis, anti-inflammatory and antidiabetic properties.1–10 Organotin(IV) compounds have also gained importance due to a significant contribution as pesticides, bactericides and fungicides, in wood preservation, anti-fouling, polymer, and paint industries.11–15 β-Amino acid derivatives have been reported as essential synthons in the biological systems of living organisms and their organotin(IV) complexes were also reported as biologically active species.7–10,16 Their cytotoxicity depends upon the starting β-amino acid derivatives, carboxylate skeleton, the type of alkyl groups attached to organotin(IV) moieties7–9,17 and the versatility in the geometry of complexes in solution and solid-state.18 Organotin(IV) complexes of β-amino acids have been reported to possess fascinating structural diversity.18–24 As previously reported, organotin(IV) complexes with amino acids and N-phthalimido β-amino acids exhibited tetrahedral and bipyramidal geometry in solution and solid-state, making these compounds more attractive for biological studies.7,25–27On the other hand, diabetes type-2, also known as diabetes mellitus, is a metabolic disorder characterized mainly by a high level of blood glucose (hyperglycemia) caused by insufficient insulin production by the pancreas or the inability of the body's cell to respond inappropriately to insulin or simply both. This hyperglycemia can be controlled temporarily using α-glucosidase inhibitors (AGIs) such as acarbose, sulfonylureas, miglitol, voglibose, etc.7–11,28 Diabetes is one of the top 10 causes of death globally. Together with other non-communicable diseases (NCDs) such as cardiovascular, cancer, and respiratory diseases, diabetes account for over 80% of all premature deaths.29 Resistant bacterial infection in diabetic patients is also common. Diabetic patients have more exposure to antibiotics and have significant problems with the healing of infections because of reduced blood supply, which affects the body to resist against disease.30 It has also been investigated that hyperglycemia-induced free radicals that cause oxidative stress, leads to the development and progression of diabetes and its complications. Cell and tissue damage can occur with the simultaneous decline of antioxidant defense mechanisms and abnormally high levels of free radicals can cause membrane damage due to peroxidation of membrane lipids and protein glycation.31–34 In literature antidiabetic, antimicrobial, and antioxidant activities of various metal complexes have been reported and have shown effective results compared to standard.35–37 Thus, as a part of our ongoing research work on organotin(IV) chemistry with the functionalized N-phthalimido β-amino acid ligands,7 we are interested in exploring further into the chemistry of organotin(IV) complexes with N-acetylated β-amino acids and their di- or triorganotin(IV) complexes. Here in this work, we have chosen the two-step reaction to synthesize N-acetylated β-amino acids derivatives (L1–L8) via the reported procedure in literature,38,39 as shown in Scheme 1. Spectroscopic techniques confirmed all synthesized ligands (L1–L8), and molecular docking analysis was performed into the catalytic pocket of the α-glucosidase enzyme (PDB ID: 3WY1).40 After inspecting the best binding interaction and poses in the catalytic pocket of the enzyme, two ligands L1 and L4 were selected to synthesized the series of di- or triorganotin(IV) complexes by the reported procedure40,42 (R = butyl, phenyl, cyclohexyl), as presented in Scheme 2. To the best of our knowledge and literature review, in the present contribution, it is being reported for the first time, molecular docking analysis of organotin(IV) complexes (1–12) into the catalytic pocket of the α-glucosidase enzyme (PDB ID: 3WY1) using the AutoDock tools.41,43 These complexes were characterized with the help of elemental analysis, FTIR, multinuclear (1H, 13C, 119Sn) NMR, EI-MS, and powder XRD. Antidiabetic activity of ligands L1, L4, and metal complexes (1–12) were evaluated using the α-glucosidase enzyme and compared with standard drug acarbose. Ligands and complexes with best results for in vitro antidiabetic activity were subjected to in vivo study using the alloxan induced rabbits, and results were compared with standard drug metformin. In addition to this, antibacterial and antioxidant activities were also evaluated to explore the best organotin(IV) candidates as a promising antidiabetic agents.
 | ||
| Scheme 1 Structure and numbering scheme of N-acetylated β-amino acids. | ||
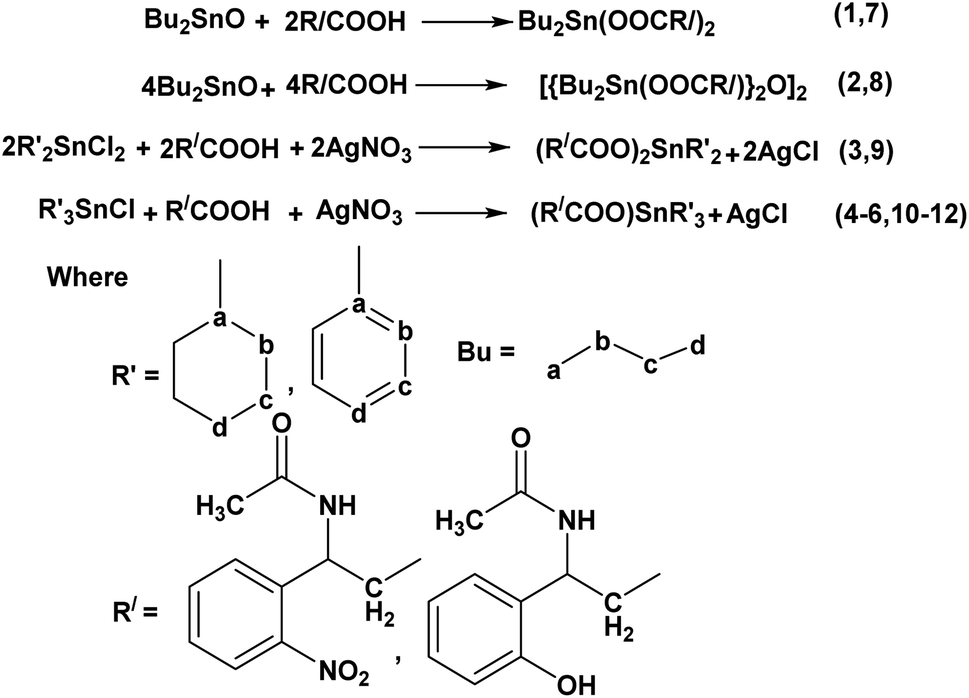 | ||
| Scheme 2 General reactions for the synthesis of complexes (1–12). | ||
Results and discussion
FTIR spectroscopy
The absorption range 4000–400 cm−1 was used to record the solid-state FTIR spectra and principal infrared absorptions of the ligands (L1–L8) and complexes (1–12). The synthesized ligands have medium stretching frequencies at 3495–3285 cm−1 due to ν(–NH) and strong absorbance at 1687–1626 cm−1 belonging to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the amide.44 The carbonyl group ν(CO) of –COOH showed asymmetric and symmetric frequencies at 1790–1682 cm−1 and 1642–1569 cm−1 respectively. The ν(–OH) stretching broad peak was noticed at 3055–2800 cm−1 indicating the carboxyl group of β-amino acid. The –CH and –CH2 vibrations were marked at 3395–3060 cm−1 and 1448–1408 cm−1.45 The substituted ring of benzene presented absorptions at 1606–1410 cm−1 (Fig. S1–S8 in ESI†). In metal complexes (1–12) the ν(–OH) broad band was completely absent, ν(COO)asym and ν(COO)sym frequencies of N-acetylated β-amino acids were shifted to lower values at 1658–1406 cm−1 respectively, suggesting carboxylate coordination with the tin(IV) metal.46 The value of Δυ[ν(COO)asym − ν(COO)sym] helps to determine the mutual coordination mode of carboxylate and tin(IV) atom i.e. if Δν value is <200 cm−1 then bi-dentate mode and if the value is >200 cm−1 then mono-dentate coordination mode results.
O) of the amide.44 The carbonyl group ν(CO) of –COOH showed asymmetric and symmetric frequencies at 1790–1682 cm−1 and 1642–1569 cm−1 respectively. The ν(–OH) stretching broad peak was noticed at 3055–2800 cm−1 indicating the carboxyl group of β-amino acid. The –CH and –CH2 vibrations were marked at 3395–3060 cm−1 and 1448–1408 cm−1.45 The substituted ring of benzene presented absorptions at 1606–1410 cm−1 (Fig. S1–S8 in ESI†). In metal complexes (1–12) the ν(–OH) broad band was completely absent, ν(COO)asym and ν(COO)sym frequencies of N-acetylated β-amino acids were shifted to lower values at 1658–1406 cm−1 respectively, suggesting carboxylate coordination with the tin(IV) metal.46 The value of Δυ[ν(COO)asym − ν(COO)sym] helps to determine the mutual coordination mode of carboxylate and tin(IV) atom i.e. if Δν value is <200 cm−1 then bi-dentate mode and if the value is >200 cm−1 then mono-dentate coordination mode results.
In the case of monomer complexes (1, 3, 7, 9), the band associated with ν(COO)asym and ν(COO)sym stretched at the range of 1658–1615 cm−1 and 1477–1416 cm−1 and Δυ(COO) values difference was less than 200 cm−1, and 1H and 13C NMR spectra gave peaks of only one –COO– group of ligand which suggested that the monomer's have bidentate coordination with trans octahedral structure (Fig. 1(a)).47 Moreover there is notable difference between the Δν values of ligands and the complexes e.g. Δν value for L1 is 125 cm−1, for 1 (monomer) is 160 cm−1 for 2 (dimer) is 193 cm−1 and 4 (tributyl) is 156 cm−1 which give mode of coordination more elaborately as given in the literature i.e. νasym (complex) < νasym (ligand), νsym (complex) < νsym (ligand), Δν (complex) > Δν (ligand). The coordination mode of endo and exo sites in dimer complexes (2, 8) was conveniently illustrated in FTIR spectra. The ν(COO) absorption bands were observed at ν(COO)asym 1630–1610 cm−1 and ν(COO)sym 1465–1460 cm−1 for both endo and exo centers, confirming the two equivalent tin spheres with bidentate bonding and hexa coordination to the tin(IV).48 Similarly a sharp peak of Sn–O–Sn–O ring at about 650–600 cm−1 and in the 1H and 13C NMR spectra of these compounds there were no distinct signals of butyl group attached to endo- and exo-cyclic tin(IV) centers giving the equal status to endo and exo tin(IV) center. It was due to six coordination sites of each endo and exo tin(IV) atom with chemically equivalent nature and the 119Sn NMR spectra have two peaks at −210 ± 8, −215 ± 15 ppm for endo and exo tin(IV) atoms which confirmed the linkage of O-atom of carboxylate of one unit to Sn(IV) atom of other unit which also confirmed dimeric complexes (Fig. 1(b)).49 Triorganotin(IV) compounds (4–6, 10–12) with characteristic ν(COO)asym 1650–1620 cm−l and ν(COO)sym 1440–1410 cm−l have the Δυ(COO) values which suggested that SnR3 groups were coordinated in a bidentate manner with carboxylate group with trigonal bipyramidal geometry (Fig. 1(c)).50,51 In all the complexes (1–12), the vibrations of ν(Sn–O) and ν(Sn–C) bonds were found at 690–650 cm−1 and 575–520 cm−1 (Fig. S9–S20 in ESI†).52
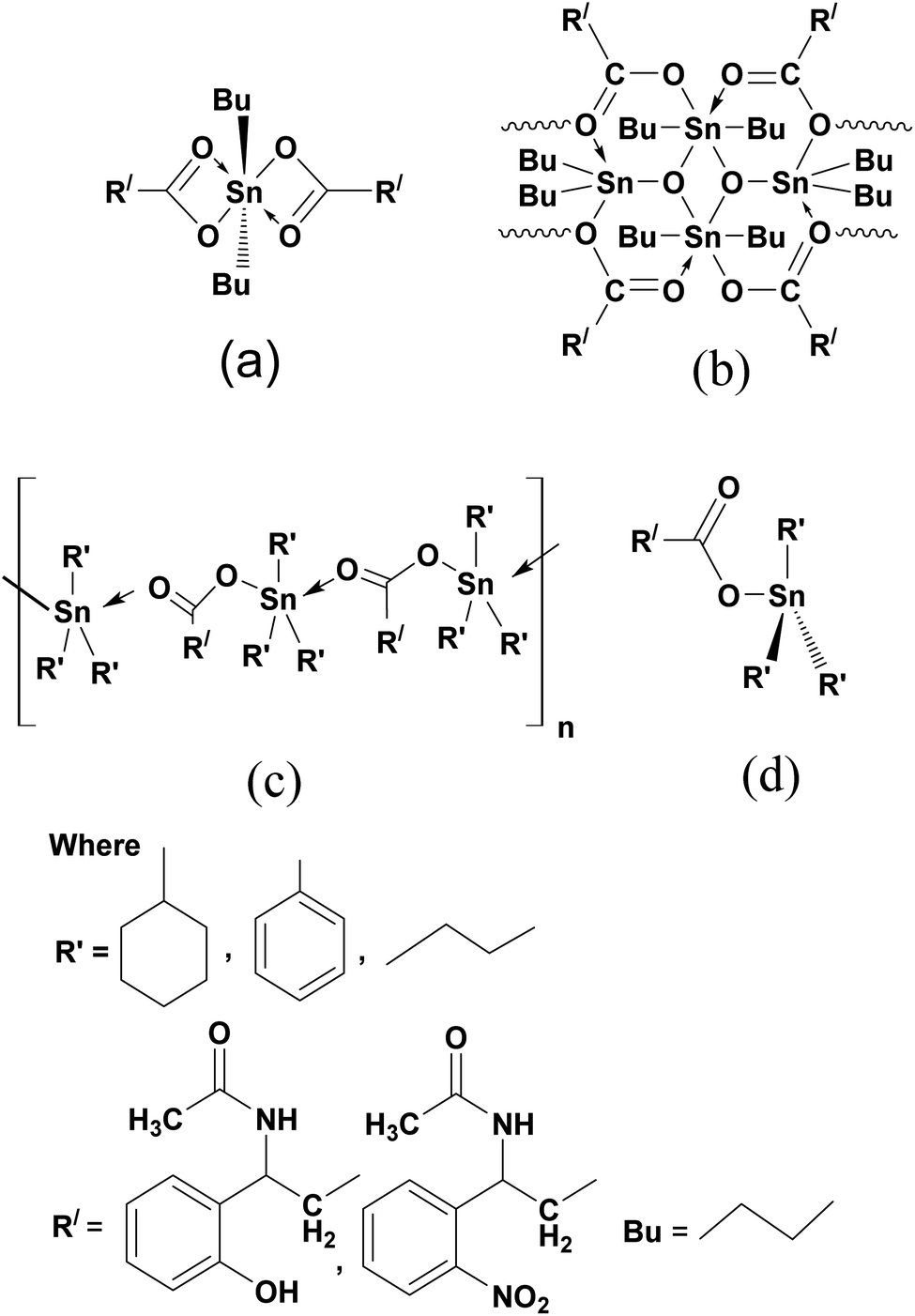 | ||
| Fig. 1 Structure of diorganotin(IV) monomer complexes (1, 3, 7, 9) (a), structure of diorganotin(IV) dimer complexes (2, 8) (b), polymeric structure of triorganotin(IV) complexes (4–6, 10–12) (c), structure of triorganotin(IV) complexes (4–6, 10–12) (d). | ||
Multinuclear (1H, 13C, 119Sn) NMR spectroscopy
The 1H, 13C, 119Sn NMR values of N-acetylated β-amino acids (L1–L8) and its complexes (1–12) in DMSO were also confirmed their synthesis. In 1H NMR, the signals at 12.95–10.59 ppm were due to –OH protons of acids and the distinguished signals at 8.98–8.31 ppm were attributed to –NH protons of –CONH moiety in N-acetylated β-amino acids. The absence of signals for –OH proton at 12.95–10.95 ppm mentioning the deprotonation of –COOH group and coordination to tin(IV) atom.53 The proton signals of –CH2 at 1.38–1.19 ppm, –CH at 1.55–1.40 ppm and aromatic protons at 8.15–7.41 ppm appeared at the same value in the both for ligands and complexes. The –OH group on the aromatic ring exhibited a signal at 4.75–4.35 ppm in the ligand (L1–L3) and complexes (1–6) (Fig. S21–S40 in ESI†). The butyl protons –CH2–CH2–CH2– in monomer complexes (1, 3, 7, 9) linked to metallic center tin(IV) observed as multiplets at 1.59–0.90 ppm and gave a broad triplet signal at 1.93–1.45 ppm due to the terminal –CH3 protons. The literature revealed that such complexes behaved as monomeric diorganotin(IV) dicarboxylates following a hexa-coordination of tin(IV) with trans-octahedral geometry (Fig. 1 (a)).54In dimeric complexes (2, 8), the butyl protons were resolved at proper positions. Only one set of signals for the butyl group indicative of the same tin(IV) environments around the butyl group. There was also one triplet of methylenic protons, which were probably because of the equivalent surroundings of similar protons to the endo and exo tin(IV) atom. Hence, it was challenging to identify the butyl of endo and exo Sn(IV) centers which verified the FTIR results.55 Therefore, it could be suggested that dimer existed in a hexa-coordinated geometry having a unique view linked from one unit to the other by O-atom of carboxylate of one unit to Sn(IV) atom of another unit as shown in Fig. 1(b).56
The 1H NMR spectra for triorganotin(IV) compounds (4–6, 10–12) verified the tetrahedral structure and exhibited that the carboxylate oxygen was coordinated to Sn(IV) core in this arrangement (Fig. 1(d)).57 All the butyl protons (4, 10) at 1.56–0.85 ppm, phenyl protons (5, 11) in multiplets at 7.45–6.79 ppm and cyclohexyl protons (6, 12) at 1.47–1.15 ppm were well resolved.58
In 13C NMR spectra of organotin(IV) complexes, the carboxylate group values were observed in the downfield region (179.45–167.93 ppm) in contrast to the free acid ligands (173.70–164.38 ppm). This was the indication that the carboxylate anions were in coordination with Sn(IV).59 The –CONH (amide) carbon signals were observed almost in the range of 159.81–155.50 ppm, thus confirming this group did not participate in the coordination, which remained almost at the same values as in ligands. The aromatic carbon ring values were observed at 145.77–116.50 ppm, while –CH and –CH2 carbon signals were at 49.43–43.48 ppm and 36.75–20.15 ppm respectively for both ligands and complexes. In all complexes (1–12) the presumed 13C NMR signals were observable and ultimately agreed with the formation of expected compounds (Fig. S41–S60 in ESI†). The 13C NMR shift values of butyl in monomers (1, 3, 7, 9) were observed in the range at 29.45–13.15 ppm with octahedral geometry (Fig. 1 (a)),54 while for dimmers (2, 8) butyl groups of endo and exo centers had no different values indicating equal tin sites. The solution state studies of such complexes suggested that in non-coordinating solvents, there were two different environments for butyl groups linked by endo and exo-cyclic tin(IV) cores,56 but in this study the same environment for the butyl group was observed, which recommended a unique view with hexa coordinated tin(IV) and verified the 1H NMR and FTIR results (Fig. 1(b)). Triorganotin(IV) complexes (4–6, 10–12), 13C NMR values of butyl, phenyl and cyclohexyl have been observed at 29.45–13.15 ppm, 145.65–137.39 ppm, and 29.19–20.44 ppm, respectively, which supported four coordinated tetrahedral geometry (Fig. 1(d)).57,58
The chemical shifts values for 119Sn NMR proposed a coordination number pattern around the tin(IV) atom. The 119Sn shift values increased by increasing in coordination number of tin(IV) atom and usually up field shift was resulted.57 In complexes (4–6, 10–12), the shift values of tributyl, triphenyl and tricyclohexyltin(IV) carboxylates were at −135.74 ppm,-120.72 ppm and 9.18 ppm with four coordination number of tin(IV) atom and supported a tetrahedral arrangement of tin(IV) in DMSO (Fig. 1(d)).60,61 While dibutyltin(IV) monomer complexes (1, 3, 7, 9) exhibited chemical shift value at −140.56 ppm describing more than four coordination numbers, thus favored the trans-octahedral presentation (Fig. 1(a)).55,61 The dimer compounds (2, 8) have a pair of 119Sn resonance frequencies with similar intensities at −210.4 ppm and −216.2 ppm confirmed the endo and exo-cyclic tin(IV) atoms with hexa-coordinated geometry respectively (Fig. 1(b)).56,60
Mass spectrometry
The synthesis of compounds was confirmed by molecular ion peaks, the base peaks and the fragmentation pattern. Mass spectra of ligands (L1–L8) showed prominent molecular ion peaks in the range of 253–223 m/z as well as diorganotin(IV) complexes (1, 3, 7, 9) and triorganotin(IV) derivatives (4–6, 10–12) also showed distinct molecular ion peaks and fragmentation patterns (Fig. S61–S68 in ESI†) in contrast to literature, the molecular ion peaks were observed clearly in dimer complexes (2, 8).7,62 The significant fragmentation was noticed, probably because of losing the ligand moiety. During fragmentation, the R groups (butyl, phenyl, cyclohexyl) were lost successively until the Sn4+ ion was obtained. Further, the remaining substituents of the ligands were lost in successive steps with similar pattern (Fig. S69–S80 in ESI†).62Powder XRD analysis
X-ray diffractometry
X-ray diffraction patterns of ligand (L1) and complexes (1–5) were shown in Fig. 2, indicating that all the samples crystallized as face-centered cubic phases. It is worth noticing that the reflection with the Laue indices HKL = 100 is not a systematic extinction except in complexes 1 and 2, providing the evidence of the uniform filling of tin(IV) atoms in these complexes.63 All the compounds have almost the same lattice parameter (ao), as given in Table 1. Average particle size (D) and intrinsic strain (ε) were calculated based on these diffraction data using different models.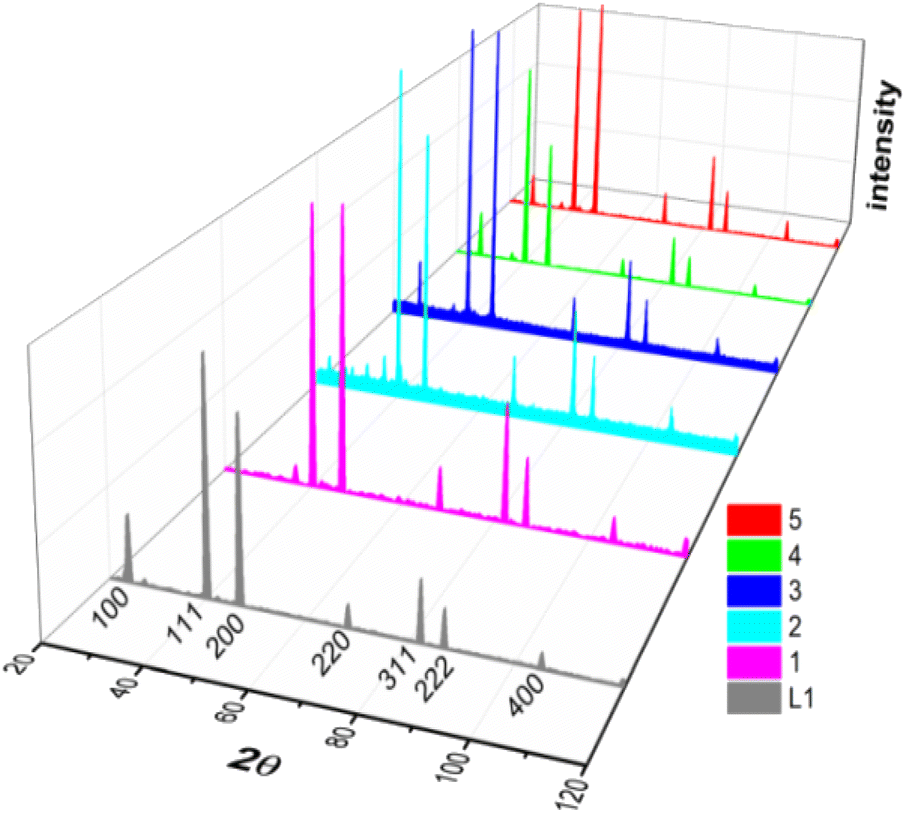 | ||
| Fig. 2 XRD peak profiles of ligand L1 and complexes 1–5 labelled with respective Laue indices (notice that the systematic extinction of Laue indices HKL = 100 occurred only complexes 1 and 2). | ||
| Methods | Sample (ao = nm) | ||||||
|---|---|---|---|---|---|---|---|
| L1 (0.3894) | 1 (0.3896) | 2 (0.3895) | 3 (0.3896) | 4 (0.3896) | 5 (0.3892) | ||
| a UDM = uniform deformation model. | |||||||
| Scherrer method | D (nm) | 28.73 | 34.30 | 33.93 | 41.46 | 35.30 | 34.37 |
| Hall method | D (nm) | 45.70 | 39.57 | 45.86 | 48.32 | 48.73 | 25.46 |
| ε | 0.0016 | −0.0024 | −0.0027 | −0.0019 | −0.0028 | −0.0032 | |
| Williamson–Hall (UDMa) | D (nm) | 45.50 | 37.72 | 43.21 | 32.71 | 29.17 | 22.90 |
| ε | 0.0008 | −0.0023 | −0.0018 | −0.0024 | −0.0013 | −0.0009 | |
| Size–strain plot | D (nm) | 35.50 | 43.13 | 36.78 | 39.80 | 39.33 | 26.68 |
| ε | 0.0006 | −0.0003 | −0.0006 | −0.0006 | −0.0003 | −0.0002 | |
| Halder–Wagner method | D (nm) | 16.31 | 31.48 | 29.44 | 21.14 | 22.43 | 19.92 |
| ε | −0.0630 | −0.0313 | −0.0333 | −0.0411 | −0.0328 | −0.0347 | |
Size and strain analysis
Different approaches such as Scherrer method, Hall method, Williamson–Hall or uniform deformation model (UDM) method, size–strain plot method and Halder–Wagner method were applied to calculate the average particle size and intrinsic strain arising from microstructural defects. Details on distinct features associated with each technique have been described,64 for complex 1 only. The same procedure was followed for other samples and the resulting values are listed in Table 1. The compressive nature (negative value) of strain can be explained as Sn(IV) atoms in the organic framework tended to elongate the lattice and introduced compressive strain in the complexes. Interestingly, in ligand L1 all the methods indicated a small tensile strain except the Halder–Wagner approach yielded a negative value of strain.
D = (kλ)/(β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ) cosθ)
| (1) |
|
βcosθ = (λ)/(D + 2εsinθ)
| (2) |
Taking sinθ as abscissa and βcosθ as ordinate (Fig. 3(a)), the slope and intercept of the resulting straight line resulted in ε = −0.0024 and D = 39.57 nm, for complex 1.66
 | ||
| Fig. 3 Size and strain analysis for complex by (a) Hall plot, (b) Williamson–Hall plot (UDM model), (c) size–strain plot, (d) Halder–Wagner plot. | ||
|
βcosθ = (kλ)/D + 4εsinθ
| (3) |
D and ε values determined for complex 1 are 37.72 nm and −0.0023 respectively (Fig. 3(b)).
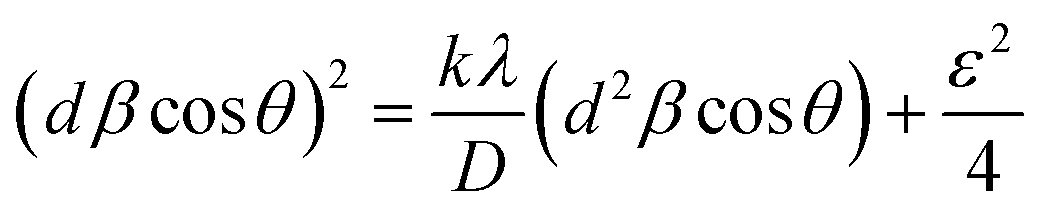 | (4) |
 | (5) |
cosθ/λ; and d* = 2dsinθ/λ. For complex 1, the Halder–Wagner plot is graphically represented in (Fig. 3(d)). D and ε values calculated by this method were found to be 31.48 nm and −0.0313 respectively.69Molecular docking analysis
Docking studies of ligands
In silico analysis was performed to explore the interaction of proposed ligands (L1–L8) into the catalytic pocket of the α-glucosidase enzyme (PDB ID: 3WY1) by employing the MGL tools (Auto Dock) and vina.40,43 The chain A of α-glucosidase co-crystallized with ligand polyacrylic acid (PAA) was selected and unwanted non-standard atoms, ligands and water molecules were removed to prepare the enzyme for docking analysis. The grid box coordinates were adjusted after viewing the size of ligands and essential amino acid residues of the enzyme, i.e. Asp62, Tyr65, Ile146, Phe166, Arg200, Asp202, Thr203, Phe206, Gly228, Tyr235, His332, Asp333 and Arg400 (Fig. 4).70–72 The binding affinity value (−6.7 kcal mol−1) of polyacrylic acid was used as a reference (Fig. 5, 2D and 3D, Table 2). The conformation which has the highest binding affinity value was further used for analysis. The first six compounds (L1–L6) of the series showed docking scores ranging from −7.1 to −7.5 kcal mol−1. Compounds L7 and L8 led to the least predicted binding affinity value of −6.0 and −5.8 kcal mol−1 respectively compared to the reference PAA value of −6.7 kcal mol−1 (Table 2).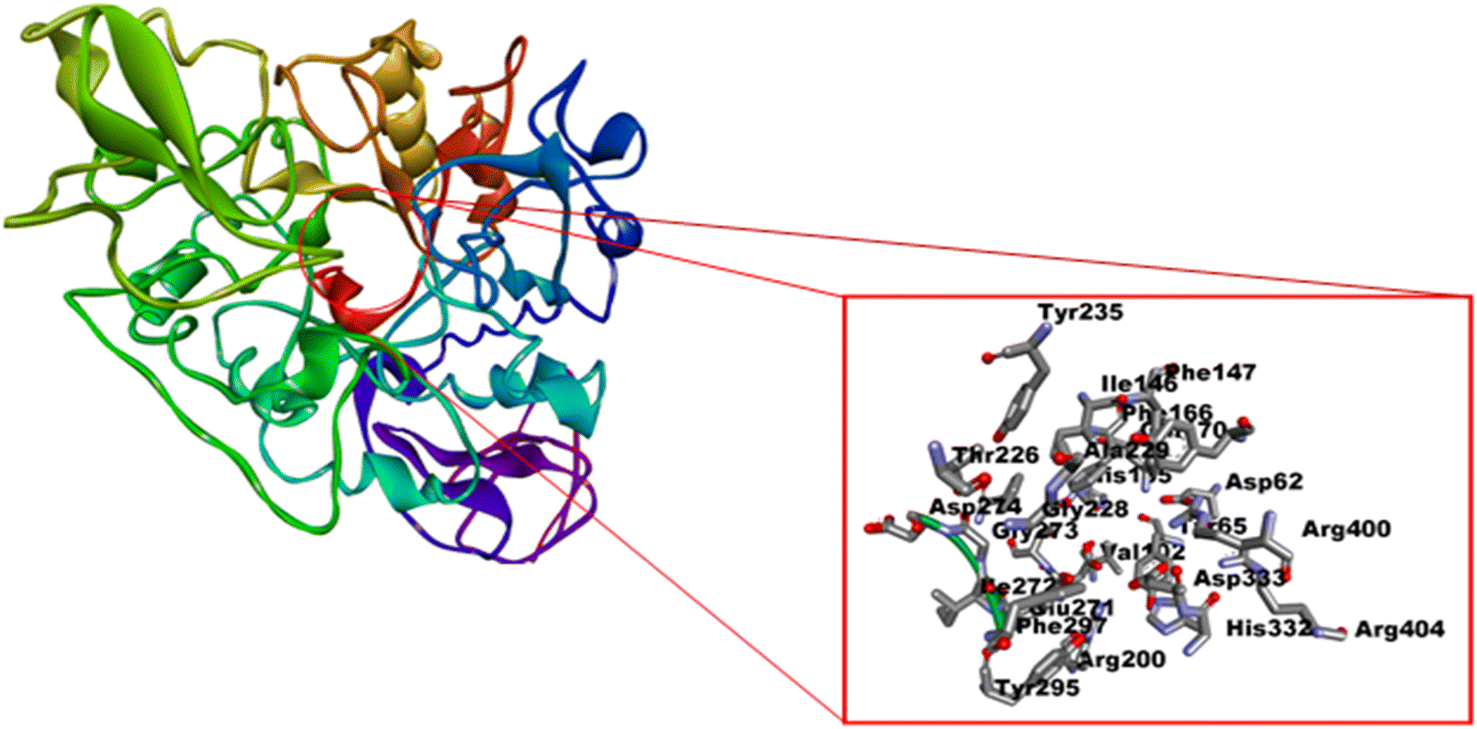 | ||
| Fig. 4 Active amino acid residues on the catalytic pocket of enzyme α-glucosidase enzyme (PDB ID: 3WY1). | ||
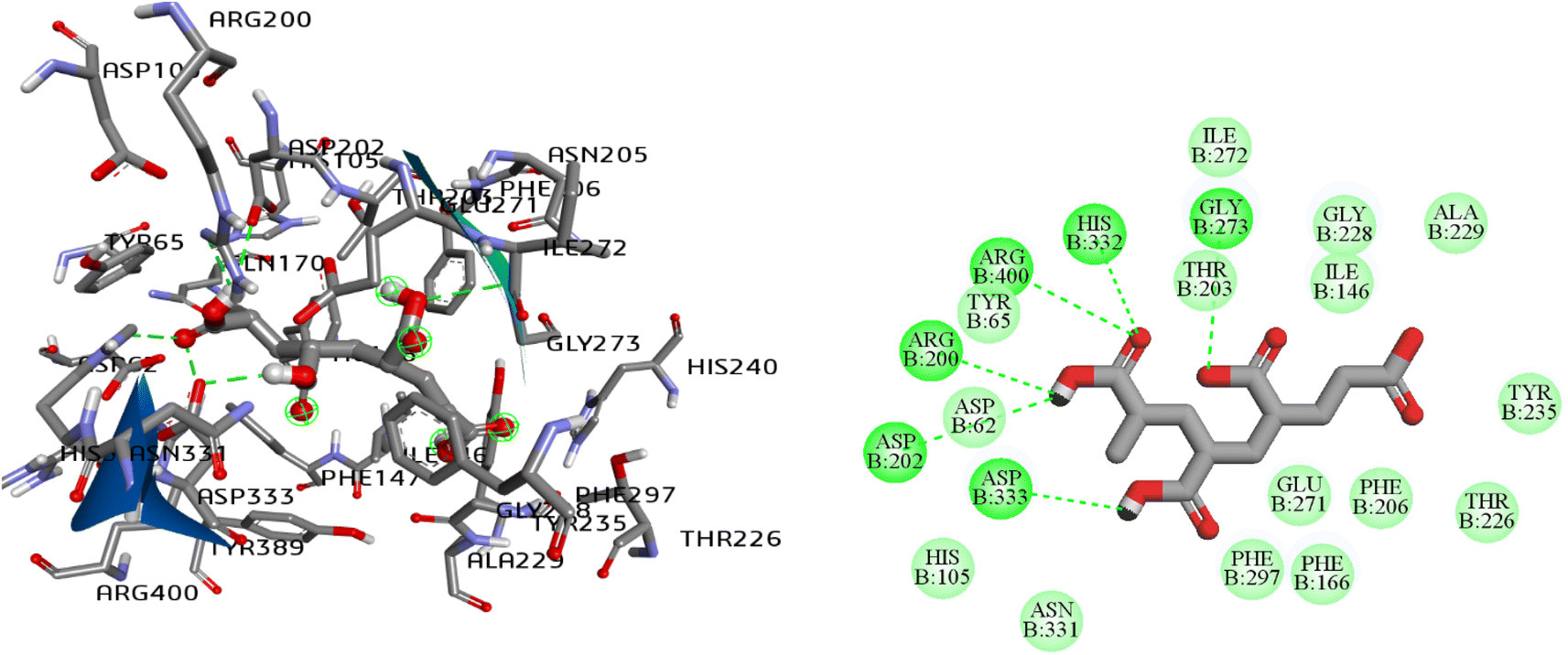 | ||
| Fig. 5 2D and 3D interaction pattern of polyacrylic acid (PAA), natural ligand of α-glucosidase enzyme (PDB ID: 3WY1). | ||
| Ligands/complex | Binding affinity (kcal mol−1) | H-Bonds (Å) | Π-Interactions (stacking, anion and alkyl) | Hydrophobic/van der Waals |
|---|---|---|---|---|
| a PAA = polyacrylic acid natural ligand of α-glucosidase enzyme. | ||||
| L1 | −7.5 | 4. Asp202, Gly228, Glu271, Arg400 | 2. Tyr65, Asp33 (Π–anion) | 8. Asp62, Ile146, Phe166, Arg200, Thr203, Phe206, Gly228, Tyr235, His332 |
| L2 | −7.2 | 3. Asp202, Glu271, His332, Arg400 | 1. Tyr65 | 5. Asp33, Thr203, Phe206, Gly288, Tyr235 |
| L3 | −6.9 | 3. Asp62, His105, His332 | — | Asp62, Tyr65, Ile146, Phe166, Arg200, Thr203, Phe206, Gly228, Tyr235, His332 |
| L4 | −7.5 | 4. Tyr65, Arg200, His332, Arg400 | Asp333 (Π–anion) | Phe166, Arg200, Thr203, Phe206, Gly228, Tyr235 |
| L5 | −7.1 | 4. Asp62, Gln170, His105, His332 | — | Ile146, Phe166, Arg200, Thr203, Phe206, Gly228, Tyr235 |
| L6 | −7.0 | 2. 2× Gly228 | — | Ile146, Phe166, Arg200, Thr203, Phe206 |
| L7 | −6.0 | 2. Glu271, Arg400 | — | Phe206, Gly228, Tyr235, His332 |
| L8 | −5.8 | — | — | Ile146, Phe166, Arg200, Thr203, Phe206 |
| 1 | — | |||
| 2 | — | |||
| 3 | 1.56 | — | Tyr65, Phe147, Phe166, Pro230 | |
| 4 | −6.04 | 1. lle272 | lle146, Phe147, Phe166, Pro230, Tyr389 (Π–alkyl) | Phe166, Arg200, Thr203, Phe206 |
| 5 | −0.71 | — | Lle146, Ala229, Val334 (Π–alkyl) | Phe166, Arg200, Thr203 |
| 6 | −1.70 | 1. Gly228 | Ala229, Tyr235 (Π–anion) | Arg200, Thr203, Phe206 |
| 7 | — | |||
| 8 | — | |||
| 9 | 13.59 | Tyr65, Arg200, Thr226, Asp274 | Gly228, Asp333, Phe297, Tyr389 | lle146, Phe147, Phe166, Pro230, Tyr389 |
| 10 | −5.64 | — | lle146, Phe147, Phe166, Pro230, Tyr389 (Π–alkyl) | |
| 11 | −0.61 | 2. Gln170, Asp274 | Tyr65 | |
| 12 | −1.60 | — | Phe147, Phe166, Pro230, Tyr389 (Π–alkyl) | |
| PAAa | −6.70 | 6. Arg200, Asp202, Gly273, His332, Asp333, Arg400 | — | Asp62, His105, Phe166, Thr226, Gly228 |
The compound L1 with the highest predicted affinity value of −7.5 kcal mol−1 showed an excellent binding pattern in the enzyme's catalytic cavity and exhibited the four hydrogen bond interactions. The –OH group at the second carbon of phenyl ring occupied the Asp202 and Glu271 via hydrogen bond. Moreover, the oxygen atom of the carboxylic acid engaged the Arg400 and the hydrogen atom of the same carboxylic acid moiety were found in interaction with Gly228 via hydrogen bond. The phenyl ring of compound L1 was found in classical π–π interaction with one of the important amino acid residues, Tyr65 and displayed the π–anion interaction with Asp333. The rest of the amino acid residues Asp62, Ile146, Phe166, Arg200, Asp202, Thr203, Phe206, Gly228, Tyr235, His332 was found in hydrophobic and van der Waals interaction with ligand L1 (Fig. 6(a) and (b), 2D and 3D). The ligands L2 and L3 with the same chemical skeleton like compound L1, except the position of –OH group at phenyl ring, showed a predicted binding affinity value of −7.2 and −6.9 kcal mol−1. The compound L2 possessing the –OH group at the third carbon of phenyl ring exhibited similar binding patterns compared to ligand L1. The –OH group at the third carbon of the phenyl ring engaged the Asp202 and His332 via hydrogen bond interaction. The important side chain residue Arg400 showed the hydrogen bond interaction with an oxygen atom of acetamide carbonyl group. The amino acid residue Tyr65, Asp33 and the rest of amino acid residues Thr203, Phe206, Gly228, and Tyr235 showed similar interaction as for compound L1. The unfavorable interaction was observed among –OH hydrogen atoms at the phenyl ring and amino acid residue Arg200. The ligand L3 bearing the –OH group at the fourth carbon of phenyl ring showed completely different conformation in the enzyme's catalytic pocket than ligands L1 and L2. Compound L3 exhibited the three hydrogen bond interactions first between the –OH group of carboxylic acid and Asp62, second between the oxygen atom of the carbonyl group of carboxylic acid and His105. At the same time, third hydrogen bond interaction was found with His332 and acetamide oxygen atom of the carbonyl group. The rest of the amino acid residues showed hydrophobic interactions. Ligands L4, L5 and L6 possessing the –NO2 moiety at the second, third and fourth carbon atom of the phenyl ring displayed the binding affinity value of −7.5, −7.1, −7.0 kcal mol−1 respectively. Compound L4 bearing the –NO2 moiety at the second carbon of phenyl ring showed the four-hydrogen bond interaction in the enzyme's catalytic pocket. A hydrogen bond interaction was formed between the oxygen atom of carbonyl of acetamide and side-chain residue Arg400. Three hydrogen bond interaction between carboxyl oxygen atom and side-chain residues Tyr65, Arg200 and His332. The –NO2 group at the phenyl ring only showed the π–anion interaction with Asp333 residue (Fig. 6(c) and (d), 2D and 3D). Compound L5 was found to mediate the four-hydrogen bond interaction with the target enzyme's active sites of side-chain residue. The hydroxyl group of carboxylic acid involved the Asp62 and Gln170 via hydrogen bond interaction.
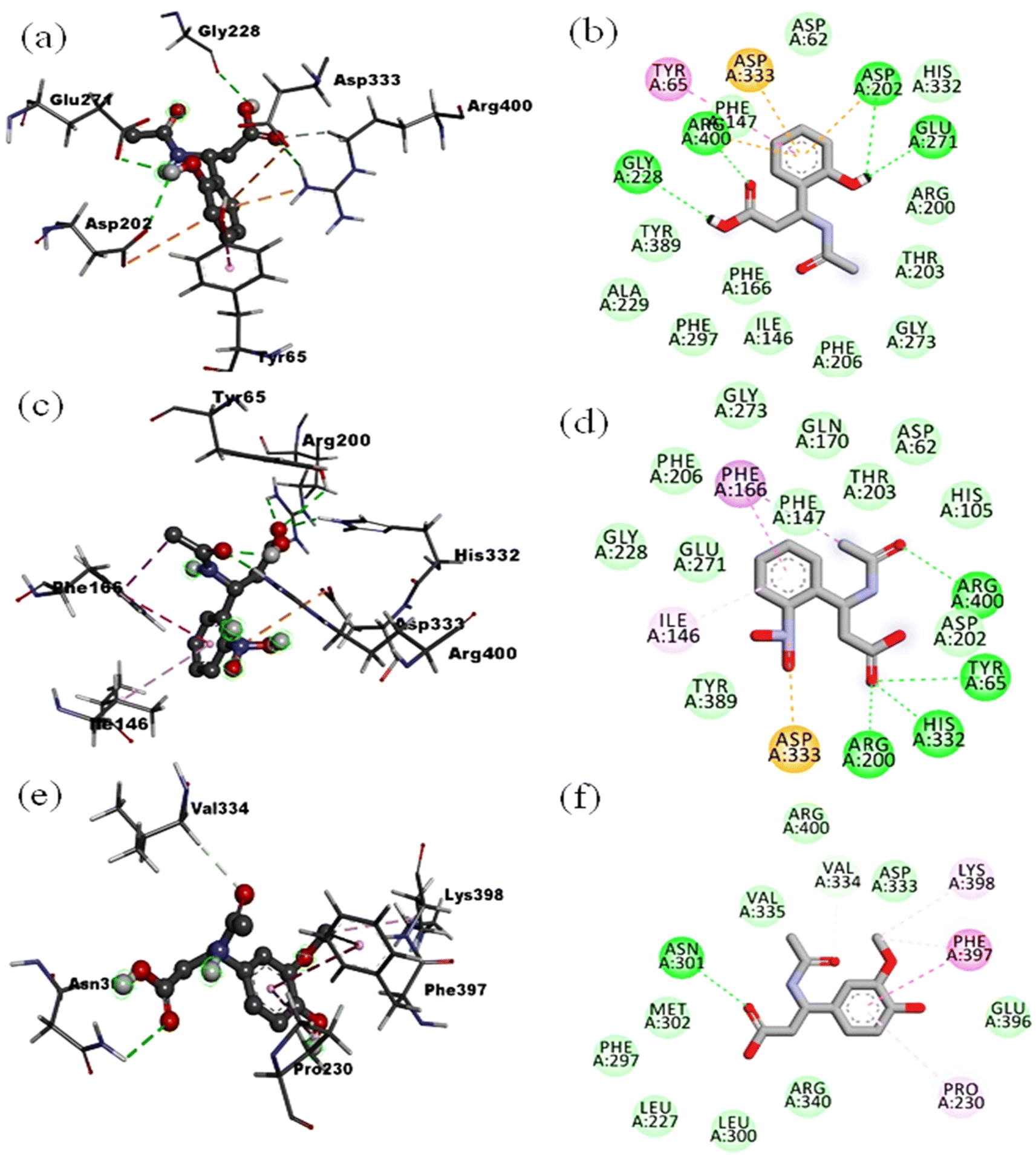 | ||
| Fig. 6 2D and 3D interactions: for ligand L1 (a and b), for ligand L4 (c and d), for ligand L5 (e and f) in catalytic pocket of enzyme (PDB ID: 3WY). | ||
At the same time, the oxygen atom of the carbonyl group of carboxylic acid occupies the His105 with hydrogen bond interactions. The fourth hydrogen bond interaction was found between His332 and the oxygen atom of acetamide carbonyl group. The rest of the amino acid residues in the pocket showed the salt bridge and hydrophobic type interactions. Ligand L6 with a predicted binding affinity value of −7.0 kcal mol−1 exhibited the four-hydrogen bond interaction. The –NO2 group at the fourth carbon of the phenyl group was found to make two hydrogen bond interactions with Gly228. In contrast, for compounds L4 and L5 the –NO2 group at the second and third carbon of the phenyl ring did not show any significant interaction. The oxygen atom of carbonyl and hydroxyl group of carboxylic acid showed the hydrogen bond interaction with His105 and Asp62 respectively.
Compounds L7 possessing the –OH and –OCH3 at the second and third carbon of phenyl ring and compound L8 keeping the –OH and –OCH3 at second and fourth carbon of phenyl ring respectively showed the least predicted binding affinity values of −6.2 and −5.8 kcal mol−1 compared to the reference. Analysis of least confirmation of compound L7 in the targeted enzyme's binding cavity showed two hydrogen bond interactions with Glu271 and Arg400. The Arg400 was found in establishing the two-hydrogen bond interaction with the carbonyl group of acetamide and the hydroxyl group of carboxylic acid. The hydrogen atom of –NH acetamide showed the hydrogen bond interaction with amino acid Glu271. The confirmation of compound L8 in the enzyme's catalytic pocket only showed the hydrophobic interactions (Fig. 6(e) and (f), 2D and 3D with the least value). From the molecular docking poses and binding interaction analysis of proposed ligands (L1–L8) in the catalytic pocket of the α-glucosidase enzyme, it was concluded that such type of derivatives could fit well in catalytic pocket of selected enzyme. The position of substituent as –OH and –NO2 at the second carbon of phenyl ring (for compound L1 and L4) is mainly resulted in minimizing the compound and enzyme complex energy. It is due to the compounds orientation that established the strong bonding with Tyr65 and Arg400 compared to the position of identical substituents at third and fourth carbon of phenyl ring. In comparing chemical moieties –OH and –NO2 we found that the –OH moiety has an advantage over the –NO2 due to its small molecular size and less polarity.
Docking studies of metal complexes
Molecular docking simulation was performed to explore organotin(IV) metal complexes effect and their binding pattern interaction in the catalytic pocket of α-glucosidase enzyme. For this purpose, L1 and L4 ligands has been selected to design the eight organotin(IV) metal complexes (3–6, 9–12). The ligand selection was made due to the higher predicted binding affinity value of −7.5 kcal mol−1, best orientation and excellent binding pattern in the enzyme's pockets. These metal complexes were named diorganotin(IV) and triorganotin(IV) complexes according to the chemistry of organic moieties (n-butyl, phenyl, and cyclohexyl) attached to tin(IV) center. These metal complexes showed the predicted binding affinity value of −6.04 to −0.61 kcal mol−1 (Table 2) and displayed the catalytic pocket's well-fitted orientation (Fig. 7). The insertion of metal ion causes the subtle enhancement of the structural framework ligand and organic moieties on the tin(IV) center, which helped to establish hydrogen bonding, π–π stacking and van der Waals interaction with amino acid residues. Triorganotin(IV) complex 4 showed the highest predicting binding affinity value of −6.04 kcal mol−1 among all the organotin(IV) complexes. The metallic center tin(IV) ion's tributyl group showed the π–alkyl interaction with Ile146, Phe147, Phe166, Pro230, and Tyr389. A hydrogen bond was also found between –OH of the ligand L1 and amino acid residue Ile272. At the same time, the oxygen atom of the carbonyl group, bonded to tin(IV) metal ion displayed the π-lone pair interaction with amino acid Phe297. The rest of the amino acid residues showed the van der Waals interactions (Fig. 8(a) and (b)). Complex 10 exhibited the second highest predicted binding affinity value of −5.64 kcal mol−1, indicating stable conformations in the targeted enzyme's catalytic pocket, did not display high-energy and unfavorable interaction (Fig. 8(c) and (d), 2D and 3D). The butyl chain exhibited the same interaction pattern, as explained for metal complex 4. | ||
| Fig. 7 Cluster of di- and triorganotin(IV) complexes inside the catalytic pocket of enzyme (PDB ID: 3WY1). | ||
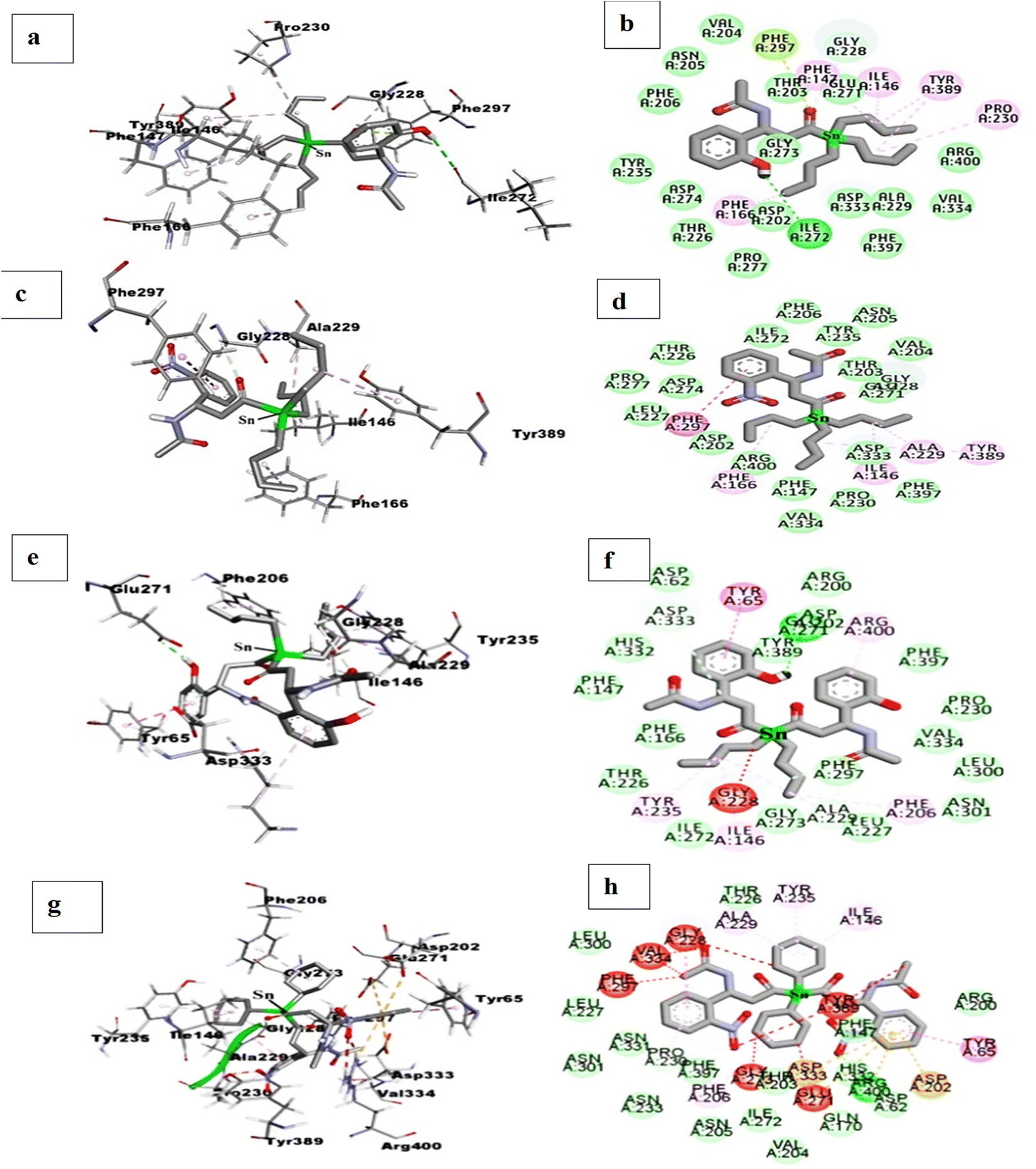 | ||
| Fig. 8 2D and 3D interactions: for triorganotin(IV) complexes 4 (a and b), for triorganotin(IV) complex 10 (c and d), for diorganotin(IV) complex 3 (e and f), for triorganotin(IV) complex 9 (g and h), in catalytic pocket of enzyme (PDB ID: 3WY1). | ||
The third best predicted binding affinity was displayed by complex 6 of −1.70 kcal mol−1 and showed excellent conformation in the catalytic pocket. The –OH group at the second carbon of the ligand's phenyl ring was found hydrogen bond interaction with Gly228. The rest of the amino acid residues in the catalytic pocket only displayed the hydrophobic interaction. At the same time, the tricyclohexyl group showed the π–anion interactions with amino acid residues Ala229 and Tyr235. Complex 5, where the tin(IV) center is coordinated with three phenyl rings, displaying the predicted binding affinity values of −0.71 kcal mol−1. The phenyl rings coordinated with tin(IV) metal ion only participated in the π–alkyl interactions with amino acid residues Ile146, Ala229, and Val334. The organotin(IV) complex 12 showed the docking affinity value of −1.60 kcal mol−1 and fitted well in the target enzyme's catalytic pocket but did not show the hydrogen bond interaction. Complex 11 displayed a binding affinity value of −0.61 kcal mol−1 and engaged the amino acid residues Gln170 and Asp274 via hydrogen bond interactions. The phenyl ring coordinated to tin(IV) metal ion did not show any significant interaction in the pocket, on the other hand the phenyl ring of coordinated compound L4 showed the π–π stacking with amino acid residue Tyr65.
The diorganotin(IV) complex 3 derived from ligand L1 showed the predicted binding affinity value of 1.56 kcal mol−1. The conformation of complex 3 occupied the extra space of the enzyme's pocket due to the more extensive framework than complex 4. The butyl group displayed the same interacting behavior as discussed for complex 4 and 8. But the phenyl ring of ligand showed the classical π–π stacking with one of the important amino acid residues, Tyr65 (Fig. 8(e) and (f), 2D and 3D). While the diorganotin(IV) complex 9 derived from ligand L4 showed a predicted affinity value of 13.59 kcal mol−1, which was found lower than complex 3. The conformation of complex 9 in the catalytic pocket occupied the significant and insignificant amino acid residues. The –NO2 group at both phenyl rings exhibited the four hydrogen bond interactions with two important amino acid residues Tyr65, Arg200 and two non-significant amino acid residues, Thr226 and Asp274. But an unfavorable interaction was also observed for amino acid residue Arg400 which was reported as essential amino acid residue to stabilize the enzyme and ligand in the catalytic pocket. The rest of the amino acid residues exhibited the van der Waals and hydrophobic interactions (Fig. 8(g) and (h), 2D and 3D). Concluding from computational studies that the tin(IV) metal complexes can fit well in the catalytic pocket of enzyme and could occupy the amino acid of vital importance via strong force of attraction as hydrogen bond, van der Waals and hydrophobic interactions.
In a comparison of compound L1 (–OH group at second carbon of phenyl ring) and L4 (–NO2 group at second carbon of phenyl ring), the tin(IV) metal ion complex of compound L1 is more favorable, which may be due to the small size of –OH and can create the hydrogen bond interactions. But in the case of L4 metal complexes, the –NO2 substituent causes them to maximize the enzyme and tin(IV) metal complex energy and resulted in a decrease predicted binding affinity, which may be due to high electron-withdrawing effect and wider electronic environment of –NO2 moiety. In comparing triorganotin(IV) and diorganotin(IV) metal complexes, triorganotin(IV) displayed the highest predicted binding affinity values responsible for energetically stable enzyme and tin(IV) metal complexes. Diorganotin(IV) metal complexes showed the least predicted binding affinity values due to the more extensive framework of tin(IV) metal ion complexes, which caused unfavorable interactions with essential amino acid residues. In comparing of organic moieties coordinated to tin(IV) metal ion, it is observed that the butyl chain has the advantage over phenyl and cyclohexyl group, due to single bond rotation and flexibility, so the butyl chain adjusted fit well in the catalytic pocket. The rigidity of the phenyl ring and torsion of the cyclohexyl ring may be another factor. Finally it can conclude that di- and triorganotin(IV) metal complexes can bind the important amino acid residues in target enzyme pockets and could be used as potential candidates to develop anti-diabetic drugs.
Biological studies
α-Glucosidase inhibitory activity
Antidiabetic potential of ligands L1, L4 and derived organotin(IV) complexes were quantified using α-glucosidase enzyme at various concentration of 25 μg mL−1, 50 μg mL−1, 100 μg mL−1, and 200 μg mL−1 to determine the IC50 values and results were compared with standard acarbose, shown in (Fig. 9). Antidiabetic potential of ligands L1 and L4 and derived organotin(IV) complexes were quantified using α-glucosidase enzyme, and results were compared with the standard acarbose. Among the proposed ligands and complexes, the complexes 4, 6, and 10 showed potent inhibitory potential with IC50 values of 21.54 ± 0.45, 35.20 ± 1.02 and 37.96 ± 0.81 respectively compared to standard acarbose with IC50 values of 42.51 ± 0.21. The metal complex 12 displayed IC50 values of 44.00 ± 0.41 which was found near the standard. While complexes 5 and 11 showed IC50 values of 73.61 ± 0.71 and 87.91 ± 1 which were found about one-fold high than the acarbose. On the other hand an abrupt decrease in inhibition value was observed for complexes 1, 2, 3, 7 and 8 (Table 3). In comparing α-glucosidase inhibition of selected ligands L1 and L4, L1 displayed the IC50 values of 40.96 ± 1.33 which was found equal to standard. In contrast, L4 exhibited the IC50 values of 44.00 ± 1.43, which can be considered better than standard (Fig. 9).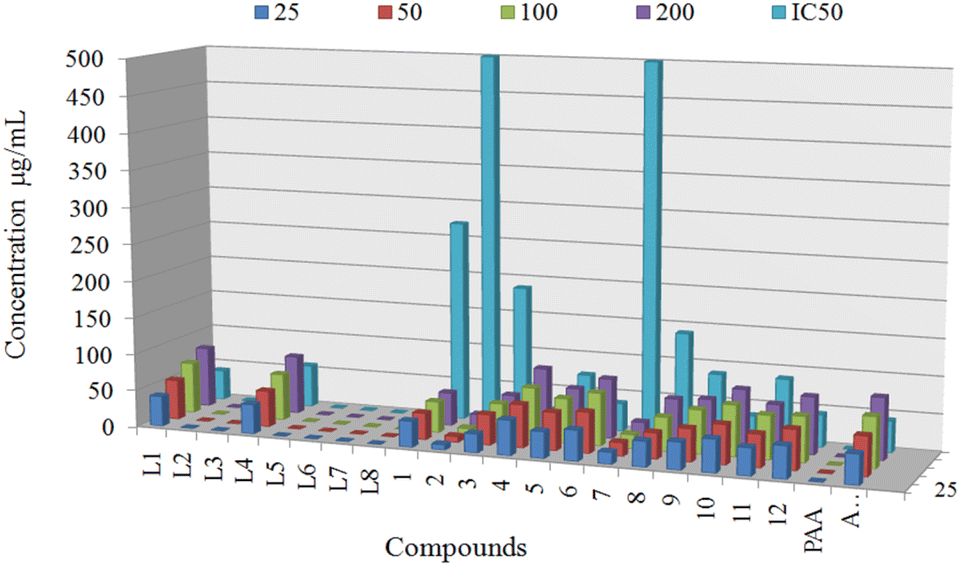 | ||
| Fig. 9 In vitro α-glucosidase inhibitory activity of compounds at different concentrations (25 μg mL−1, 50 μg mL−1, 100 μg mL−1, and 200 μg mL−1) with IC50 values. | ||
| Ligands/complexes | IC50 (μg mL−1) | |
|---|---|---|
| In vitro antioxidant activity | In vitro α-glucosidase inhibitory activity | |
| Std** = quercetin = standard used for antioxidant DPPH assay, Std*** = acarbose = standard used for α-glucosidase inhibition activity. | ||
| L1 | 89.04 ± 2.11 | 40.96 ± 0.96 |
| L2 | 84.56 ± 1.91 | — |
| L3 | 109.57 ± 2.4 | — |
| L4 | 49.48 ± 1.01 | 57.59 ± 0.7 |
| L5 | 78.24 ± 1.0 | — |
| L6 | 61.70 ± 1.4 | — |
| L7 | 96.76 ± 2.01 | — |
| L8 | 87.4 ± 3.02 | — |
| 1 | 33.20 ± 2.11 | 271.22 |
| 2 | 26.33 ± 2.96 | <500 |
| 3 | 31.90 ± 1.04 | 187.44 |
| 4 | 11.74 ± 1.12 | 21.54 ± 0.45 |
| 5 | 16.51 ± 1.31 | 73.61 ± 0.71 |
| 6 | 19.39 ± 1.01 | 37.96 ± 0.81 |
| 7 | 32.01 ± 1.73 | 239.88 |
| 8 | 30.24 ± 2.01 | <500 |
| 9 | 27.9 ± 1.63 | 87.91 ± 0.54 |
| 10 | 14.45 ± 1.02 | 35.20 ± 1.02 |
| 11 | 17.80 ± 1.40 | 87.91 ± 1.0 |
| 12 | 13.10 ± 1.11 | 44.00 ± 0.41 |
| Std** | 9.548 ± 0.23 | — |
| Std*** | — | 42.5 ± 0.21 |
The structure–activity relationship of these compounds suggested that the ligand L1 possessing the –OH at the second carbon of phenyl ring has an advantage over the ligand L4 bearing the –NO2 at the second carbon of phenyl. If we compare these ligand's binding and interaction mode in the target enzyme's catalytic pocket, the –OH group participated in establishing hydrogen bond interaction while the –NO2 group remained inert.
Triorganotin(IV) complexes (4, 10) possessing the butyl group coordinated to tin(IV) center showed the best inhibitory value of α-glucosidase. It is noteworthy that complexes 4 and 10 also have the highest predicted binding affinities value of −6.4 and −5.64 kcal mol−1. Both complexes showed the well fitted orientation in the enzyme's catalytic pocket and butyl moieties established the π–alkyl interactions. On the other hand triorganotin(IV) complexes 6 and 12 possessing the cyclohexyl moieties attached to the metallic center showed enzyme inhibition with IC50 37.96 ± 0.81 and 44.01 ± 1.4 respectively. Comparing the inhibitory activity with the molecular docking interaction pattern of both complexes 6 and 12 in the enzyme catalytic pocket displayed that the cyclohexyl moieties established the hydrophobic bonding with amino acid residues and did not form any unfavorable bump. A hydrogen bond interaction was also observed between –OH of ligand and amino acid residue Gly228 for complex 6. The cyclohexyl moieties fitted well inside the catalytic pocket due to flexibility in the ring. In contrast, complexes 5 and 11 having the phenyl group coordinated to metallic center tin(IV), showed inhibition values of IC50 73.61 ± 0.71 and 87.91 ± 0.54 for the α-glucosidase enzyme. The rise in inhibitory values may be due to rigidity and the bigger size of the phenyl group by occupying more space in the cavity of the enzyme. The interaction pattern of these complexes in the catalytic pocket of enzymes displayed that the phenyl ring caused unfavorable bumps with insignificant amino acid residues outside the catalytic pocket. Diorganotin(IV) metal complexes 1, 2, 3, 7, and 8 displayed an abrupt increase in values of IC50 139 to <500 (Table 3). This behavior showed that the smaller metal framework (tri-organotin(IV)) has an advantage over the broader metal framework (diorganotin(IV)). Overall, the inhibition trend of organotin(IV) metal complexes depending on the organic moieties can be summarized as butyl > cyclohexyl > phenyl and triorganotin(IV) > diorganotin(IV).
Antioxidant activity
The 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging assay was employed to determine the antioxidant activities of synthesized ligands (L1–L8) and proposed organotin(IV) complexes (1–12) at four different concentration 25, 50, 75 and 100 μg mL−1. The quercetin was used as a standard to compare the results at the same concentrations. The DPPH method has advantages over the other methods due to its simplicity and short time required for the analysis. DPPH is a stable free radical with an odd number of electrons.7,73 The DPPH can be reduced by the hydrogen donating ability of antioxidants or electron transfer mechanism that involve the transfer of an electron from a metal or ligand. The reduction in absorbance of the solution of freshly prepared DPPH compared with DPPH solution and antioxidant solution. The absorbance was measured at 517 nm spectroscopically.73,74Antioxidant activity results of ligands and organotin(IV) complexes were calculated in terms of IC50 and shown in Table 4. All organotin(IV) complexes (1–12) displayed the best antiradical activity with the value of IC50 11.74 ± 1.12 to 32.01 ± 1.73 μg mL−1, compared to standard quercetin IC50 value of 9.45 ± 0.23 μg mL−1. Whereas corresponding ligands (L1–L8) displayed the lowest antioxidant activity with the value of IC50 49.48 ± 1.01 to 109.57 ± 2.40 μg mL−1. The antioxidant IC50 values are evaluated on the following scale, less than the 20 μg mL−1 were considered the best, above 20 μg mL−1 to 30 μg mL−1 are moderate and antioxidant values higher than 30 μg mL−1 are inactive. Antioxidant activity of compounds at different concentrations with IC50 values is shown in Fig. 10. Three triorganotin(IV) complexes 4, 10 and 12 displayed the most significant antioxidant activity value of 11.74 ± 1.12, 13.10 ± 1.11, and 14.45 ± 1.02 μg mL−1 respectively which were found near to the standard quercetin IC50 value of 9.45 ± 0.23 μg mL−1. While the complexes 5, 6 and 11 displayed the best antiradical activity with values of 16.51 ± 1.31, 19.39 ± 1.01 and 17.80 ± 1.40 μg mL−1 respectively. On the other hand, diorganotin(IV) complexes 2, 3, 7 and 8 showed the weakest antioxidant values.
| Ligands/complexes (500 ppm) | Gram negative | Gram positive | ||||||
|---|---|---|---|---|---|---|---|---|
| E. coli | P. aeruginosa | S. aureus | B. subtilis | |||||
| Inhibition/mm | Results | Inhibition/mm | Results | Inhibition/mm | Results | Inhibition/mm | Results | |
| a Where ++++ = most significant, +++ = more significant, ++ = significant, Std* = ciprofloxacin = 1000 ppm doze. | ||||||||
| L1 | 19 | + | 16 | + | 22 | +++ | 21 | +++ |
| L2 | 17 | + | 18 | + | 20 | ++ | 18 | ++ |
| L3 | 20 | + | 18 | + | 21 | ++ | 20 | ++ |
| L4 | 19 | + | 17 | + | 20 | ++ | 22 | ++ |
| L5 | 21 | + | 20 | + | 23 | +++ | 25 | +++ |
| L6 | 18 | + | 16 | + | 26 | +++ | 25 | +++ |
| L7 | 17 | + | 19 | + | 17 | ++ | 20 | ++ |
| L8 | 19 | + | 16 | + | 22 | +++ | 23 | +++ |
| 1 | 22 | ++ | 25 | +++ | 23 | +++ | 24 | +++ |
| 2 | 20 | ++ | 17 | ++ | 18 | ++ | 21 | ++ |
| 3 | 23 | +++ | 21 | ++ | 26 | +++ | 25 | +++ |
| 4 | 25 | +++ | 28 | +++ | 29 | ++++ | 30 | ++++ |
| 5 | 28 | +++ | 25 | +++ | 29 | ++++ | 27 | +++ |
| 6 | 26 | +++ | 27 | +++ | 20 | ++ | 26 | +++ |
| 7 | 24 | +++ | 21 | ++ | 20 | ++ | 21 | ++ |
| 8 | 20 | ++ | 18 | ++ | 20 | ++ | 18 | ++ |
| 9 | 28 | +++ | 27 | +++ | 24 | ++++ | 19 | ++ |
| 10 | 32 | ++++ | 29 | ++++ | 32 | ++++ | 31 | ++++ |
| 11 | 28 | +++ | 27 | +++ | 29 | ++++ | 30 | ++++ |
| 12 | 28 | +++ | 28 | +++ | 32 | ++++ | 30 | ++++ |
| Std* | 30 | ++++ | 30 | ++++ | 31 | +++++ | 32 | ++++ |
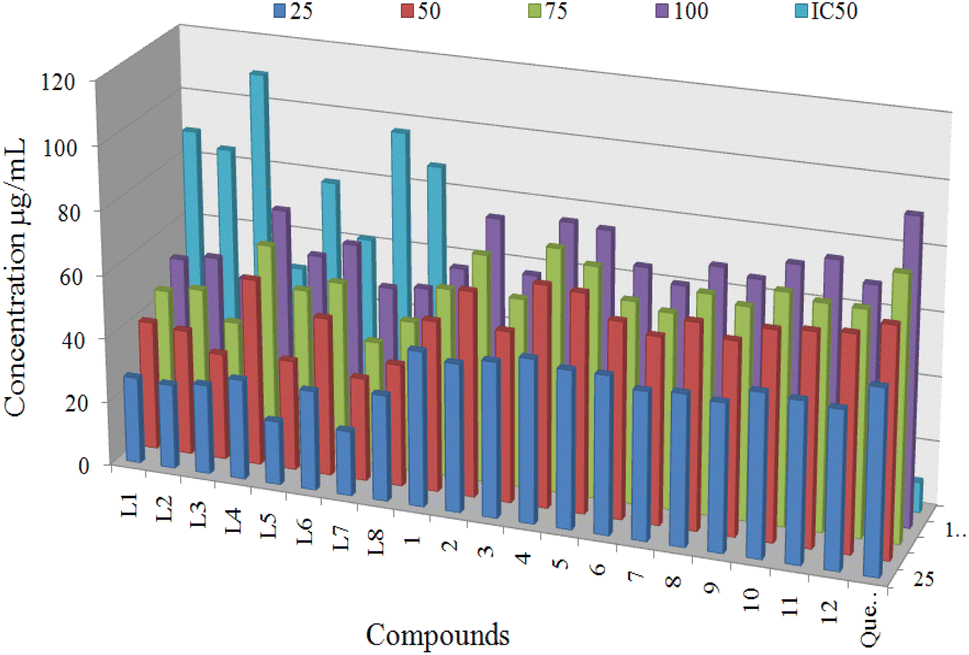 | ||
| Fig. 10 Antioxidant activity of compounds at different concentrations (25 μg mL−1, 50 μg mL−1, 75 μg mL−1, and 100 μg mL−1) with IC50 values. | ||
This behaviour showed that the DPPH scavenging activity of complexes is higher than the corresponding ligands. Thus it can conclude that ligand's activity become enhanced upon coordination with di- or triorganotin(IV) moiety. The presence of organic substituents butyl, phenyl and cyclohexyl attached to the metallic center played a significant role in enhancing the antioxidant activity. As results described that n-butyl chain bearing complexes 4 and 10 displayed the best antioxidant activity, while complex 12 possessing the cyclohexyl moiety exhibited moderate antioxidant activity. In addition, complexes having the phenyl moiety attached to the metallic center exhibited the lowest antiradical activity. In general antioxidant activity in terms of organic moieties attached to the metallic center can be shown as n-butyl > cyclohexyl > phenyl. Overall antioxidant activity can be arranged as: triorganotin > diorganotin > ligands.
Antibacterial activity
The ligands (L1–L8) and proposed organotin(IV) complexes (1–12) were screened in vitro for their antimicrobial activity at the concentration of 500 ppm and ciprofloxacin was used as standard.Significant inhibitory activity (inhibitory zone > 15 mm) was observed at this single concentration, and results (Table 3) were considered as an antimicrobial potential and minimum inhibitory concentration (MIC) of the organotin(IV) compounds. The obtained results are graphically summarized in Fig. 11. Antimicrobial results were evaluated as; inhibition zone of 15–21 mm, inactive; 22–28 mm, moderately active; 29–32 mm, highly active. Overall the organotin(IV) complexes displayed excellent activity against the bacterial strains than the free ligands. The highest activity of organotin(IV) complexes can be related to chelate formation, structure diversity, nature of attached ligand, molecular weight and lipophilicity.75 Generally in tin(IV) complexes, the antimicrobial mechanism of action involved the cellular enzyme, cellular impairment due to denaturing of proteins and alteration of the standard cell process due to the various interactions as hydrogen bonding and van der Waals forces.76
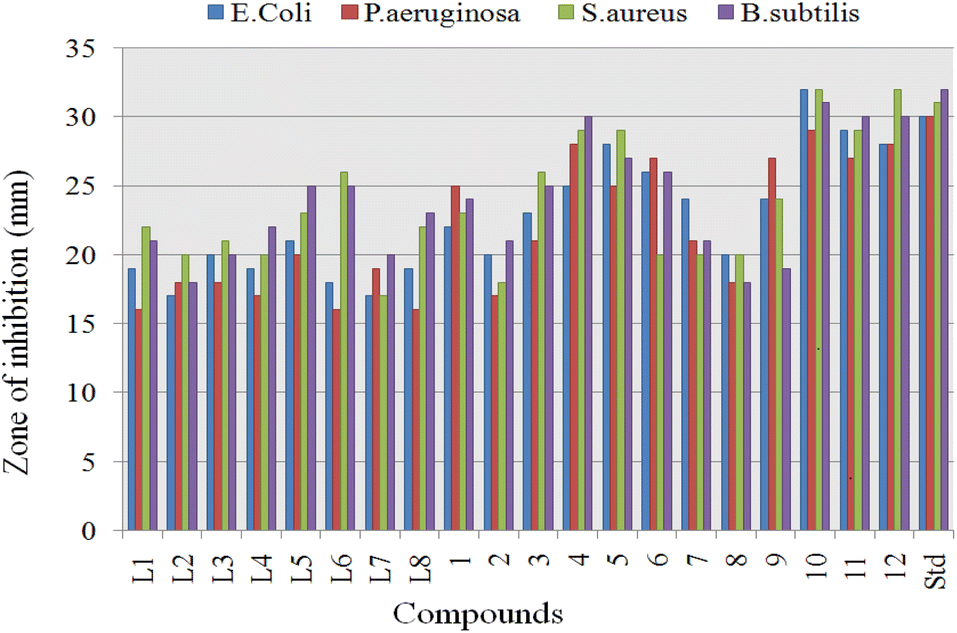 | ||
| Fig. 11 Antibacterial bioassay of ligands (L1–L8) and their organotin(IV) complexes (1–12). | ||
Complex formation and unique structure of organotin(IV) complexes cause an increased antimicrobial activity, which may be associated with the delocalization of electrons and alter the polar nature of metal. This electron delocalization causes an increase in the plasma membrane permeability. Comparing the cell wall structure of Gram-positive and Gram-negative, Gram-positive bacteria possess the simpler cell wall, thus, were more induced by complexes to give higher activity. In contrast, Gram-negative bacteria have an outer lipid membrane formed from lipopolysaccharide, which involved antigenic specificity of Gram-negative bacteria.77,78 Triorganotin(IV) complexes 4, 10 (R = butyl), 5, 11 (R = phenyl), and 6, 12 (R = cyclohexyl), displayed the highest to moderate antibacterial activity with inhibitory zone 25–32 mm.
Complex 10 showed the highest inhibition value for Gram-negative and Gram-positive with inhibition zone 30–32 mm. These results can be associated with flexibility and polarity of butyl chain length, which may increase lipophilicity and penetrate via the bacterial wall. L1 and triorganotin(IV) complex 4 displayed the highest activity against Gram-positive but moderate for Gram-negative bacteria. On the other hand, triorganotin(IV) complexes 5 and 11 possessing the phenyl moiety displayed moderate activity for Gram-positive and negative strains with similar inhibition zone. The lower activity of phenyl bearing organotin(IV) complexes may be due to the more electron-withdrawing effect of phenyl ring and rigidity. Triorganotin(IV) complexes 6 and 12 bearing the cyclohexyl moiety showed excellent inhibition for Gram-positive bacterial strains but poor inhibitory values for Gram-negative. Diorganotin(IV) complexes 1–3 and 7–9 exhibited moderate to minimum inhibitory activity for Gram-positive and Gram-negative strains. Overall antimicrobial activity of ligands and complexes could be arranged in the following order: triorganotins (alkyl) > triorganotins (aryl) > triorganotins (cyclohexyl) > diorganotin > ligands.
Generally, it can conclude that the bacterial organisms are more susceptible to the organotin(IV) complexes than the free ligands. This observed enhanced antimicrobial activity of complexes compared to ligands can be justified based on Overtons' concepts and Tweedy's chelation theories.79–81 The coordination of ligands with metal increases the lipophilicity of complex, while chelate formation reduces the polarity of the central metallic atom. The increase in lipophilicity of the metal ions favors permeability through the lipid layer membrane of the cell.
| No. of rabbit groups | Compounds | Average blood glucose level (mg dL−1) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Before alloxan | After alloxan (24 h) | Dose (mg kg−1) | Mean blood glucose level after treatment | ||||||
| 3 h | 6 h | 9 h | 12 h | 24 h | |||||
| a All alloxan induced rabbits were randomly divided into seven groups (n = 7), blood glucose was measured in triplicate and average values are reported, M* = metformin which was used as standard drug to decrease the blood glucose level and served as a positive control. | |||||||||
| GI | L1 | 75 | 295 | 0.5 | 298 | 295 | 215 | 205 | 198 |
| 1.0 | 332 | 310 | 215 | 215 | 175 | ||||
| 1.5 | 345 | 314 | 189 | 189 | 170 | ||||
| GII | L4 | 96 | 320 | 0.5 | 240 | 219 | 175 | 260 | 264 |
| 1.0 | 278 | 220 | 145 | 245 | 275 | ||||
| 1.5 | 208 | 194 | 130 | 190 | 240 | ||||
| GIII | 4 | 86 | 310 | 0.5 | 210 | 169 | 105 | 140 | 254 |
| 1.0 | 198 | 142 | 115 | 190 | 290 | ||||
| 1.5 | 170 | 134 | 109 | 240 | 300 | ||||
| GIV | 6 | 97 | 296 | 0.5 | 265 | 230 | 165 | 240 | 294 |
| 1.0 | 300 | 247 | 145 | 195 | 268 | ||||
| 1.5 | 295 | 244 | 154 | 205 | 242 | ||||
| GV | 10 | 106 | 303 | 0.5 | 250 | 229 | 165 | 240 | 264 |
| 1.0 | 248 | 212 | 165 | 195 | 245 | ||||
| 1.5 | 198 | 164 | 135 | 230 | 270 | ||||
| GVI | 12 | 88 | 305 | 0.5 | 210 | 169 | 105 | 140 | 254 |
| 1.0 | 198 | 142 | 115 | 190 | 290 | ||||
| 1.5 | 170 | 134 | 109 | 240 | 300 | ||||
| GVII | M* | 89 | 305 | 0.5 | 265 | 235 | 104 | 190 | 250 |
| 1.0 | 245 | 225 | 135 | 165 | 215 | ||||
| 1.5 | 169 | 140 | 105 | 140 | 190 | ||||
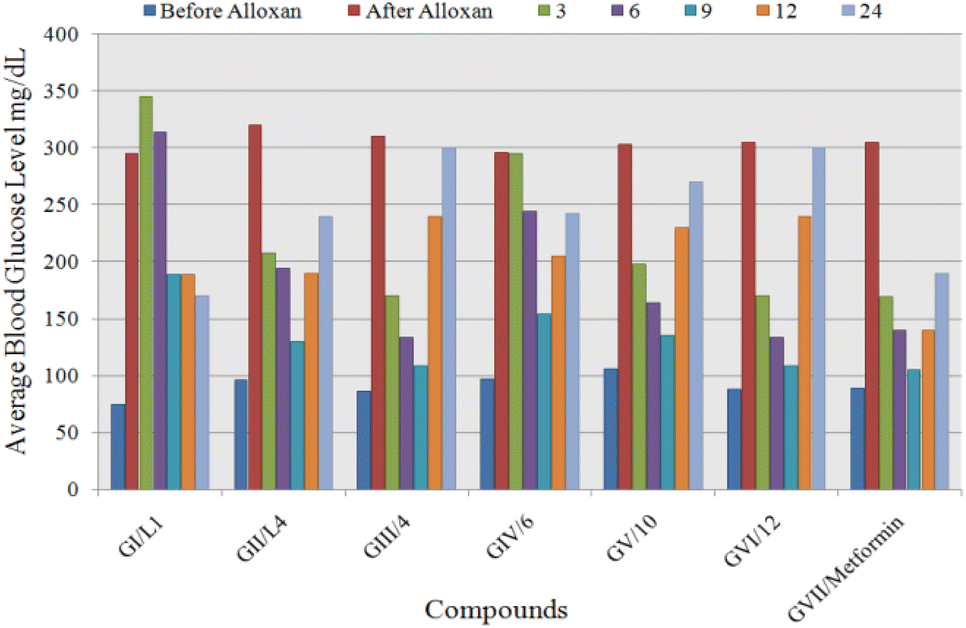 | ||
| Fig. 12 Comparison of the average blood glucose level of seven tested groups at different time interval. | ||
Experimental
Materials and instruments
2-Hydroxy benzaldehyde, 3-hydroxy benzaldehyde, 4-hydroxy benzaldehyde, 2-nitro benzaldehyde, 3-nitro benzaldehyde, 4-nitro benzaldehyde, 2-hydroxy, 3-methoxy benzaldehyde, 4-hydroxy, 3-methoxy benzaldehyde, malonic acid, ammonium acetate, acetyl chloride, dibutyltin(IV) oxide, dibutyltin(IV) dichloride, tributyltin(IV) chloride, triphenyltin(IV) chloride and tricyclohexyltin(IV) chloride were purchased from Merck Company (Germany). Melting points were determined by Fisher-Johns melting point apparatus (USA) and were found uncorrected. An Eager 300 mass analyzer (USA) was used for elemental analyses. A Bruker FTIR (USA) spectrophotometer TENSOR27 (ZnSe) was used to record the FTIR spectra of the pure solid samples covering 4000–400 cm−1. EI-MS spectra were observed in terms of % m/z on a Finnigan MAT 312 spectrometer (USA). 1H, 13C and 119Sn NMR spectra were calculated with a Bruker AM 400 NMR spectrometer (USA).Synthesis
The ligands were synthesized by the following procedure.3-Acetylamino-3(2-hydroxyphenyl) propanoic acid (L1)
2-Hydroxy benzaldehyde (1.21 g, 10 mmol), malonic acid (1.04 g, 10 mmol) and ammonium acetate (0.778 g, 10 mmol) were refluxed in 1-butanol for 4 hours to obtain 3-amino-3(2-hydroxy phenyl) propanoic acid by Radionov Johnson method.38,39 In the second step the corresponding β-amino acid 2 g (11 mmol) and acetyl chloride 0.78 mL (11 mmol) were poured in 50 mL dioxane/water (2:1, v/v) and the mixture was refluxed for about 6 hours. The solvent separation was done under vacuum and the product obtained (L1) was washed with n-hexane and collected. The above method was followed for the synthesis of rest of the ligands (L2–L8) with corresponding benzaldehyde and respective β-amino acid (Scheme 1). Yield: 60%. Mp: 180 °C. Anal. calc. for C13H11NO5: C, 46.4, H, 5.8, N, 8.9, found: C, 46.2, H, 5.7, N, 8.8%. FTIR νmax/cm−1: 3375 ν(N–H), 2900–2800 ν(O–H), 1737 ν(COO)asym, 1672 ν(CO), 1612 ν(COO)sym. 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 12.26 (s, 1H, OH), 8.68 (s, 1H, CONH), 7.86 (d, J = 7.3 Hz, 1H, H-10), 7.70 (d, J = 2.5 Hz, 1H, H-9), 7.58 (d, J = 0.5 Hz, 1H, H-8), 7.41 (d, J = 2.3 Hz, 1H, H-7), 4.75 (s, 1H, OH), 1.40 (t, J = 7.1 Hz, 3H, H-3), 1.25 (d, J = 7.5 Hz, 2H, H-2). 13C NMR (100 MHz, DMSO) δC: 164.38 (C-1), 157.15 (C-4), 134.66 (C-5), 130.59 (C-6), 125.20 (C-7), 118.70 (C-8), 118.36 (C-9), 116.50 (C-10), 45.19 (C-3), 20.15 (C-2). EI-MS m/z: M+ 223 [C11H13NO4]+, 206 [C11H12NO3]+, 178 [C10H12NO2]+, 135 [C8H9NO]+, 118 [C8H8N]+, 103 [C8H7]+, 76 [C6H4]+.
3-Acetyl amino-3(3-hydroxyphenyl) propanoic acid (L2)
Yield: 74%. Mp: 153 °C. Anal. calc. for C11H13NO4: C, 46.1, H, 5.9, N, 8.6, found: C, 46.2, H, 5.7, N, 8.8%. FTIR νmax/cm−1: 3420 ν(N–H), 3000–2921 ν(O–H), 1715 ν(COO)asym, 1690 ν(CO), 1590 ν(COO)sym. 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 12.40 (s, 1H, OH), 8.98 (s, 1H, CONH), 7.74 (d, J = 7.5 Hz, 1H, H-10), 7.66 (d, J = 2.3 Hz, 1H, H-9), 7.59 (d, J = 0.3 Hz, 1H, H-8), 7.43 (d, J = 7.4 Hz, 1H, H-6), 4.48 (s, 1H, OH), 1.43 (t, J = 7.0 Hz, 3H, H-3), 1.29 (d, J = 7.1 Hz, 2H, H-2), 13C NMR (100 MHz, DMSO) δC: 169.85 (C-1), 155.50 (C-4), 139.73 (C-5), 135.70 (C-6), 129.20 (C-7), 123.35 (C-8), 121.45 (C-9), 119.75 (C-10), 49.39 (C-3), 25.45 (C-2), EI-MS m/z: M+ 223 [C11H13NO4]+, 178 [C10H12NO2]+, 161 [C10H11NO]+, 146 [C9H8NO]+, 118 [C8H8N]+, 103 [C8H7]+, 76 [C6H4]+.
3-Acetyl amino-3(4-hydroxyphenyl) propanoic acid (L3)
Yield: 70%. Mp: 135 °C. Anal. calc. for C11H13NO4: C, 46.5, H, 5.9, N, 8.7, found: C, 46.2, H, 5.7, N, 8.8%. FTIR νmax/cm−1: 3495 ν(N–H), 3023–2950 ν(O–H), 1735 ν(COO)asym, 1667 ν(CO), 1600 ν(COO)sym. 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 12.95 (s, 1H, OH), 8.31 (s, 1H, CONH), 7.63 (d, J = 7.1 Hz, 1H, H-10) 7.55 (d, J = 2.2 Hz, 1H, H-9), 7.43 (d, J = 2.4 Hz, 1H, H-7),7.35 (d, J = 7.5 Hz, 1H, H-6), 4.63 (s, 1H, OH), 1.51 (t, J = 7.1 Hz, 3H, H-3), 1.23 (d, J = 7.3 Hz, 2H, H-2), 13C NMR (100 MHz, DMSO) δC: 173.70 (C-1), 157.79 (C-4), 140.45 (C-5), 138.50 (C-6), 136.73 (C-7), 133.53 (C-8), 132.39 (C-9), 138.50 (C-10), 48.45 (C-3), 31.95 (C-2). EI-MS m/z: M+ 223 [C11H13NO4]+, 180 [C9H8NO3]+, 163 [C9H9NO2]+, 146 [C9H8NO]+, 118 [C8H8N]+, 103 [C8H7]+, 76 [C6H4]+.
3-Acetylamino-3(2-nitrophenyl) propanoic acid (L4)
Yield: 68%. Mp: 195 °C. Anal. calc. for C11H12N2O5: C, 48.5, H, 5.7, N, 8.7, found: C, 48.6, H, 5.9, N, 8.8%. FTIR νmax/cm−1: 3357 ν(N–H), 2966–2850 ν(O–H), 1790 ν(COO)asym, 1672 ν(CO), 1642ν(COO)sym, 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 12.61 (s, 1H, OH), 8.39 (s, 1H, CONH), 8.15 (d, J = 7.3 Hz, 1H, H-10), 8.08 (d, J = 2.5 Hz, 1H, H-9), 7.66 (d, J = 0.7 Hz, 1H, H-8), 7.59 (d, J = 2.5 Hz, 1H, H-7), 1.54 (t, J = 7.1 Hz, 3H, H-3), 1.29 (d, J = 7.3 Hz, 2H, H-2). 13C NMR (100 MHz, DMSO) δC: 167.56 (C-1), 158.58 (C-4), 141.77 (C-5), 136.49 (C-6), 134.34 (C-7), 130.63 (C-8), 124.69 (C-9), 123.08 (C-10), 49.35 (C-3), 36.75 (C-2). EI-MS m/z: M+ 252 [C11H12N2O5]+, 207 [C10H11N2O3]+, 164 [C8 H8N2O2]+, 149 [C8H7NO2]+, 103 [C8H7]+, 76 [C6H4]+.
3-Acetyl amino-3(3-nitrophenyl) propanoic acid (L5)
Yield: 74%. Mp: 220 °C. Anal. calc. for C11H12N2O5: C, 47.5, H, 5.6, N, 8.9, found: C, 48.6, H, 5.9, N, 8.8%. FTIR νmax/cm−1: 3395 ν(N–H), 3000–2958 ν(O–H), 1769 ν(COO)asym, 1687 ν(CO), 1629ν(COO)sym. 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 12.45 (s, 1H, OH), 8.56 (s, 1H, CONH), 7.74 (d, J = 7.7 Hz, 1H, H-10), 7.69 (d, J = 2.9 Hz, 1H, H-9), 7.53 (d, J = 0.3 Hz, 1H, H-8), 7.49 (d, J = 7.5 Hz, 1H, H-6), 1.43 (t, J = 7.5 Hz, 3H, H-3), 1.19 (d, J = 7.3 Hz, 2H, H-2). 13C NMR (100 MHz, DMSO) δC: 171.45 (C-1), 156.85 (C-4), 143.30 (C-5), 139.95 (C-6), 137.43 (C-7), 134.29 (C-8), 133.63 (C-9), 131.91 (C-10), 49.43 (C-3), 27.75 (C-2). EI-MS m/z: M+ 252 [C11H12N2O5]+, 235 [C11H11N2O4]+, 192 [C9H8N2O3]+, 164 [C8H8N2O2]+, 118 [C8H8N]+, 103 [C8H7]+, 76 [C6H4]+.
3-Acetyl amino-3(4-nitrophenyl) propanoic acid (L6)
Yield: 63%. Mp: 195 °C. Anal. calc. for C12H15NO5: C, 48.7, H, 5.9, N, 8.6, found: C, 48.6, H, 5.9, N, 8.8%. FTIR νmax/cm−1: 3290 ν(N–H), 3000–2800 ν(O–H), 1682 ν(COO)asym, 1626 ν(CO), 1569 ν(COO)sym. 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 12.51 (s, 1H, OH), 8.69 (s, 1H, CONH), 7.98 (d, J = 7.7 Hz, 1H, H-10), 7.75 (d, J = 2.2 Hz, 1H, H-9), 7.69 (d, J = 2.5 Hz, 1H, H-7), 7.53 (d, J = 7.8 Hz, 1H, H-6), 1.53 (t, J = 7.5 Hz, 3H, H-3), 1.31 (d, J = 7.5 Hz, 2H, H-2). 13C NMR (100 MHz, DMSO) δC: 172.75 (C-1), 159.81 (C-4), 145.51 (C-5), 143.43 (C-6), 138.74 (C-7), 135.41 (C-8), 133.52 (C-9), 131.65 (C-10), 47.54 (C-3), 28.94 (C-2). EI-MS m/z: M+ 252 [C11H12N2O5]+, 235 [C11H11N2O4]+, 207 [C10H11N2O3]+, 161 [C10H11NO]+, 146 [C9H8NO]+, 118 [C8H8N]+, 103 [C8H7]+, 76 [C6H4]+.
3-Acetylamino-3(2-hydroxy-3-methoxyphenyl) propanoic acid (L7)
Yield: 69%. Mp: 205 °C. Anal. calc. for C12H15NO5: C, 45.7, H, 5.9, N, 8.6, found: C, 45.5, H, 5.7, N, 8.5%, FTIR νmax/cm−1: 3345 ν(N–H), 3055–2845 ν(O–H), 1730 ν(COO)asym, 1680 ν(CO), 1590 ν(COO)sym. 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 10.85 (s, 1H, OH), 8.78 (s, 1H, CONH), 7.78 (d, J = 7.5 Hz, 1H, H-10), 7.63 (d, J = 2.3 Hz, 1H, H-9), 7.59 (d, J = 0.5 Hz, 1H, H-8), 4.85 (s, 1H, OH), 3.75 (t, 1H, OCH3), 1.59 (t, J = 7.0 Hz, 3H. H-3), 1.34 (d, J = 7.2 Hz, 2H, H-2). 13C NMR (100 MHz, DMSO) δC: 169.59 (C-1), 159.73 (C-4), 139.54 (C-5), 137.71 (C-6), 135.45 (C-7), 134.91 (C-8), 131.73 (C-9), 129.57 (C-10), 43.48 (C-3), 27.43 (C-2). EI-MS m/z: M+ 253 [C12H15NO5]+, 210 [C10H12NO4]+, 193 [C10H11NO3]+, 176 [C10H10NO2]+, 148 [C9H10NO]+, 117 [C8H7N]+, 102 [C8H6]+, 75 [C6H3]+.
3-Acetylamino-3(4-hydroxy-3-methoxyphenyl) propanoic acid (L8)
Yield: 70%. Mp: 250 °C. Anal. calc. for C12H15NO5: C, 45.2, H, 5.9, N, 8.7, found: C, 45.5, H, 5.7, N, 8.5%. FTIR νmax/cm−1: 3495 ν(N–H), 3000–2850 ν(O–H), 1728 ν(COO)asym, 1664 ν(CO), 1585 ν(COO)sym. 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 10.59 (s, 1H, OH), 8.63 (s, 1H, CONH), 7.63 (d, J = 7.5 Hz, 1H, H-10), 7.59 (d, J = 2.6 Hz, 1H, H-9), 7.43 (d, J = 7.3 Hz, 1H, H-6), 4.45 (s, 1H, OH), 3.93 (t, 1H, OCH3), 1.55 (t, J = 7.2 Hz, 3H, H-3), 1.38 (d, J = 7.0 Hz, 2H, H-2). 13C NMR (100 MHz, DMSO) δC: 170.25 (C-1), 155.53 (C-4), 145.37 (C-5), 145.02 (C-6), 129.02 (C-7), 126.13 (C-8), 124.92 (C-9), 120.83 (C-10), 46.53 (C-3), 28.95 (C-2). EI-MS m/z: M+ 253 [C12H15NO5]+, 236 [C12H14NO4]+, 208 [C11H14NO3]+, 165 [C9H11NO2]+, 148 [C9H10NO]+, 133 [C9H9O]+, 102 [C8H6]+, 75 [C6H3]+.
Organotin(IV) complexes have been synthesized by the procedures given in Scheme 2 and literature.41,42
Dibutyltin(IV)-di-3-acetylamino-3(2-hydroxyphenyl) propanoate (1)
3-Acetylamino-3(2-hydroxyphenyl) propanoic acid 1 g (6.29 mmol) was refluxed for about 6 hours with dibutyltin(IV) oxide 0.55 g (3.14 mmol) in a ratio of 2:1 with (3:1, v/v) 66.0 mL ethanol and 33.0 mL toluene with the azeotropical removal of water using Dean Stark Apparatus. The solvent was separated out under vacuum. The synthesized white crystalline product was obtained which was recrystallized in 1:2 ratio chloroform and ethanol. Yield: 78%. Mp: 170 °C. Anal. calc. for (C4H9)2Sn(O2CC10H12NO2)2: C, 41.2, H, 6.3, N, 5.9, found: C, 41.3, H, 6.2, N, 5.10%. FTIR νmax/cm−1: 3359 ν(N–H), 1650 ν(CO), 1620ν(COO)asym, 1460 ν(COO)sym, 653ν(Sn–O), 526 ν(Sn–C). 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 7.78 (s, 1H, CONH), 7.65 (d, J = 2.3 Hz, 1H, H-7), 7.59 (d, J = 0.5 Hz, 1H, H-8), 7.54 (d, J = 2.5 Hz, 1H, H-9), 7.51 (d, J = 7.3 Hz, 1H, H-10), 4.58 (s, 1H, OH), 1.49 (t, J = 7.1 Hz, 3H, H-3), 1.22 (d, J = 7.5 Hz, 2H, H-2), Sn–nBu skeleton: 1.64 (t, 2H, H-d), 1.59 (m, 2H, H-c), 1.26 (m, 2H, H-b), 0.90 (m, 3H, H-a). 13C NMR (100 MHz, DMSO) δC: 175.25 (C-1), 168.95 (C-4), 136.76 (C-5), 134.33 (C-6), 125.43 (C-7), 120.25 (C-8), 118.40 (C-9), 116.85 (C-10), 45.19 (C-3), 29.25 (C-2), Sn–nBu skeleton: 23.70 (C-a), 22.13 (C-b), 20.48 (C-c), 13.45 (C-d). 119Sn NMR ((CH3)4Sn): −140.56; EI-MS m/z: M+ 676 [(C4H9)2Sn {O2CC10H12NO2}2]+, 632 [(C4H9)2SnO2C{C10H12–NO2}2]+, 619 [(C4H9)Sn{O2CC10H12NO2}2]+, 575 [(C4H9)SnO2C{C10H12N–O2}2]+, 531 [(C4H9)Sn{C10H12NO2}2]+, 454 [(C4H9)2Sn{O2CC10H12NO2}]+, 410 [(C4H9)2Sn{C10H12NO2}]+, 353 [(C4H9)Sn{C10H12NO2}]+, 296 [Sn{C10–H12NO2}]+, 178 [C10H12NO2]+, 119 [Sn]+, 57 [CH3CH2CH2CH2]+, 43 [CH3–CH2CH2]+, 29 [CH3CH2]+, 15 [CH3]+.
Dibutyltin(IV)-di-stannoxane-di-3-acetylamino-3(2-hydroxyphenyl) propanoate (2)
3-Acetylamino-3(2-hydroxy phenyl) propanoic acid 1 g (6.29 mmol) and dibutyltin(IV) oxide 1.1 g (6.29 mmol) were reacted in 1:1 ratio with (3:1, v/v) 66.0 mL ethanol and 33.0 mL toluene as a solvent. Recrystallization was done with chloroform and n-hexane 1:2 ratio mixture. Yield: 72%. Mp: 180 °C. Anal. calc. for (C4H9)Sn(O2CC10H12NO2)2: C, 41.5, H, 6.6, N, 3.4. Found: C, 41.3, H, 6.4, N, 3.4%. FTIR νmax/cm−1: 3459 ν(N–H), 1690 ν(CO), 1670 ν(COO)asym, 1487 ν(COO)sym, 687ν(Sn–O), 530 ν(Sn–C). 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 8.56 (s, 1H, CONH), 7.43 (d, J = 2.3 Hz, 1H, H-7), 7.39 (d, J = 0.5 Hz, 1H, H-8), 7.35 (d, J = 2.5 Hz, 1H, H-9), 7.31 (d, J = 7.3 Hz, 1H, H-10), 4.42 (s, 1H, OH), 1.55 (t, J = 7.1 Hz, 3H, H-3), 1.51 (m, 2H, H-2), Sn–nBu skeleton: 1.43 (t, 2H, H-d), 1.32 (d, J = 7.5 Hz, 2H, H-c), 1.25 (m, 2H, H-b), 0.84 (m, 3H, H-a). 13C NMR (100 MHz, DMSO) δC: 170.42 (C-1), 159.95 (C-4), 138.75 (C-5), 132.65 (C-6), 128.43 (C-7), 125.98 (C-8), 120.35 (C-9), 118.34 (C-10), 43.19 (C-3), 27.45 (C-2), Sn–nBu skeleton: 25.50 (C-a), 22.59 (C-b), 20.63 (C-c), 13.72 (C-d). 119Sn NMR ((CH3)4Sn): −210.4, −216.2; EI-MS m/z: M+ 619 [(C4H9)Sn{O2CC10H12NO2}2]+, 575 [(C4H9)SnO2C {C10H12NO2}2]+, 531 [(C4H9)Sn{C10H12NO2}2]+, 454 [(C4H9)2 Sn{O2CC10H12NO2}]+, 410 [(C4H9)2Sn{C10H12NO2}]+, 353 [(C4H9)Sn {C10H12NO2}]+, 296 [Sn{C10H12–NO2}]+, 178 [C10H12NO2]+, 119 [Sn]+, 57 [CH3CH2CH2–CH2]+.
Dibutyltin(IV)-di-3-acetylamino-3(2-hydroxyphenyl) propanoate (3)
3-Acetylamino 3(2-hydroxyphenyl) propanoic acid 1 g (6.29 mmol) and AgNO3 1.06 g (6.29 mmol) were reacted with dibutyltin(IV) chloride 0.68 g (3.14 mmol) in 2:1 ratio in 66.0 mL toluene and 33.0 mL ethanol (3:1, v/v). The mixture went for reflux of about 6 hours. The solvent was separated under vacuum and a mixture of chloroform and benzene in 1:2 ratio was used for recrystallization. Yield: 79%. Mp: 175 °C. Anal. calc. for (C4H9)2Sn(O2CC10H12NO2)2: C, 41.3, H, 6.4, N, 5.8, found: C, 41.3, H, 6.2, N, 5.10%. FTIR νmax/cm−1: 3455 ν(N–H), 1670ν(CO), 1606 ν(COO)asym, 1440 ν(COO)sym, 664 ν(Sn–O), 544 ν(Sn–C). 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 7.98 (s, 1H, CONH), 7.79 (d, J = 2.3 Hz, 1H, H-7), 7.62 (d, J = 0.5 Hz, 1H, H-8), 7.51 (d, J = 2.5 Hz, 1H, H-9), 7.48 (d, J = 7.3 Hz, 1H, H-10), 4.55 (s, 1H, OH), 1.62 (t, J = 7.1 Hz, 3H, H-3), 1.27 (d, J = 7.5 Hz, 2H, H-2), Sn–nBu skeleton: 1.93 (t, 2H, H-d), 1.32 (m, 2H, H-c), 1.24 (m, 2H, H-b), 0.95 (m, 3H, H-a). 13C NMR (100 MHz, DMSO) δC: 173.65 (C-1), 165.48 (C-4), 137.39 (C-5), 133.73 (C-6), 129.95 (C-7), 125.20 (C-8), 118.74 (C-9), 116.45 (C-10), 41.95 (C-3), 24.39 (C-2), Sn–nBu skeleton: 23.76 (C-a), 21.35 (C-b), 20.45 (C-c), 13.85 (C-d). 119Sn NMR ((CH3)4Sn): −149.59. EI-MS m/z: M+ 676 [(C4H9)2Sn{O2CC10H12NO2}2]+, 619 [(C4H9)Sn{O2CC10H12NO2}2]+, 575 [(C4H9)SnO2C{C10H12NO2}2]+, 531 [(C4H9)Sn{C10H12NO2}2]+, 454 [(C4–H9)2Sn{O2CC10H12NO2}]+, 410 [(C4H9)2Sn{C10H12NO2}]+, 353 [(C4H9)Sn- {C10H12NO2}]+, 232 [(C4H9)2Sn]+, 175 [(C4H9)Sn]+, 119 [Sn]+, 57 [CH3CH2 CH2CH2]+, 43 [CH3CH2CH2]+, 29 [CH3CH2]+, 15 [CH3]+.
Tributyltin(IV)-3-acetylamino-3(2-hydroxyphenyl) propanoate (4)
3-Acetylamino-3(2-hydroxyphenyl) propanoic acid 1 g (6.29 mmol) and AgNO3 1.06 g (6.29 mmol) in 66.0 mL toluene and 33.0 mL ethanol (3:1, v/v) were made to react with tributyltin(IV) chloride 1.45 g (6.29 mmol) in 1:1 ratio. The whole mixture was subjected to reflux for 6 hours. The solvent was separated under vacuum. Recrystallization was performed in 1:2 ratio chloroform and ethanol. Yield: 85%. Mp: 185 °C. Anal. calc. for (C4H9)3Sn(O2CC10–H12NO2): C, 48.2, H, 7.9, N, 3.3, found: C, 48.2, H, 7.8, N, 3.1%. FTIR νmax/cm−1: 3396 ν(N–H)asym, 1636 ν(CO), 1612 ν(COO)asym, 1456 ν(COO)sym, 654 ν(Sn–O), 564 ν(Sn–C). 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 8.25 (s, 1H, CONH), 7.65 (d, J = 2.3 Hz, 1H, H-7), 7.59 (d, J = 0.5 Hz, 1H, H-8), 7.53 (d, J = 2.5 Hz, 1H, H-9), 7.50 (d, J = 7.3 Hz, 1H, H-10), 4.79 (s, 1H, OH), 1.45 (t, J = 7.1 Hz, 3H, H-3),1.42 (m, 2H, H-2), Sn–nBu skeleton: 1.39 (t, 2H, H-d), 1.23 (d, J = 7.5 Hz, 2H, H-c), 1.19 (m, 2H, H-b), 0.89 (m, 3H, H-a). 13C NMR (100 MHz, DMSO) δC: 167.93 (C-1), 155.49 (C-4), 138.39 (C-5), 137.45 (C-6), 132.39 (C-7), 129.73 (C-8), 124.79 (C-9), 120.20 (C-10), 47.33 (C-3), 29.91 (C-2), Sn–nBu skeleton: 28.50 (C-a), 27.12 (C-b), 25.30 (C-c), 15.65 (C-d). 119Sn NMR ((CH3)4Sn): −135.74. EI-MS m/z: M+ 511 [(C4H9)3Sn{O2CC10H12NO2}]+, 454 [(C4H9)2Sn{O2CC10H12NO2}]+, 410 [(C4H9)2Sn{C10H12NO2}]+, 353 [(C4H9)Sn{C10H12NO2}]+, 289 [(C4H9)3–Sn]+, 232 [(C4H9)2Sn]+, 178 [C10H12NO2]+, 175 [(C4H9)Sn]+, 119 [Sn]+, 57 [CH3CH2CH2CH2]+.
Other triorganotin(IV) complexes (5, 6, 10, 11, 12) were synthesized with corresponding triorganotin(IV) chlorides by the procedure as illustrated above.
Triphenyltin(IV)-3-acetylamino-3(2-hydroxyphenyl) propanoate (5)
Yield: 78%. Mp: 180 °C. Anal. calc. for (C6H5)3Sn(O2CC10H12NO2): C, 56.8, H, 4.6, N, 2.8. Found: C, 56.7, H, 4.5, N, 2.7%. FTIR νmax/cm−1: 3212 ν(N–H), 1677 ν(CO), 1650 ν(COO)asym, 1495 ν(COO)sym, 684 ν(Sn–O), 545 ν(Sn–C), 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 8.48 (s, 1H, CONH), 7.95 (d, J = 2.3 Hz, 1H, H-7), 7.79 (d, J = 0.5 Hz, 1H, H-8), 7.65 (d, J = 2.5 Hz, 1H, H-9), 7.61 (d, J = 7.3 Hz, 1H, H-10), 4.69 (s, 1H, OH), 1.49 (t, J = 7.1 Hz, 3H, H-3), 1.29 (d, J = 7.5 Hz, 2H, H-2), Sn–Ph skeleton: 7.51 (d, 1H, H-d), 7.49 (d, 1H, H-c), 7.45 (d, 1H, H-b). 13C NMR (100 MHz, DMSO) δC: 165.92 (C-1), 163.65 (C-4), 145.61 (C-5), 143.54 (C-6), 139.18 (C-7), 137.39 (C-8), 135.43 (C-9), 133.78 (C-10), 48.23 (C-3), 26.75 (C-2), Sn–Ph skeleton: 127.34 (C-a), 124.80 (C-b), 120.90 (C-c), 118.74 (C-d), 119Sn NMR ((CH3)4Sn): −120.72. EI-MS m/z: M+ 571 [(C6H5)3Sn{O2CC10H12NO2}]+, 494 [(C6H5)2–Sn{O2CC10H12NO2}]+, 450 [(C6H5)2Sn{C10H12NO2}]+, 373 [(C6H5)Sn{C10–H12NO2}]+, 349 [(C6H5)3Sn]+, 272 [(C6H5)2Sn]+, 195 [(C6H5)Sn]+, 178 [C10H12NO2]+, 119 [Sn]+, 77 [C6H5]+.
Tricyclohexyltin(IV)-3-acetylamino-3(2-hydroxyphenyl) propanoate (6)
Yield: 76%. Mp: 192 °C. Anal. calc. (C6H11)3Sn(O2CC10H12NO2): C, 54.8, H, 7.9, N, 2.6, found: C, 54.7, H, 7.8, N, 2.6%. FTIR νmax/cm−1: 3470 ν(N–H), 1670 ν(CO), 1650 ν(COO)asym, 1470 ν(COO)sym, 615 ν(Sn–O), 535 ν(Sn–C). 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 8.35 (s, 1H, CONH), 8.15 (d, J = 2.3 Hz, 1H, H-7), 8.08 (d, J = 0.5 Hz, 1H, H-8), 7.66 (d, J = 2.5 Hz, 1H, H-9), 7.59 (d, J = 7.3 Hz, 1H, H-10), 4.85 (s, 1H, OH), 1.50 (t, J = 7.1 Hz, 3H, H-3), 1.24 (d, J = 7.5 Hz, 2H, H-2), Sn–cyclohex skeleton: 1.47 (m, 1H, H-d), 1.34 (m, 2H, H-c), 1.31 (m, 2H, H-b), 1.15 (m, 2H, H-a), 13C NMR (100 MHz, DMSO) δC: 174.52 (C-1), 158.85 (C-4), 139.80 (C-5), 135.65 (C-6), 133.47 (C-7), 128.29 (C-8), 125.75 (C-9), 122.23 (C-10), 47.43 (C-3), 24.15 (C-2), Sn–cyclohex skeleton: 23.75 (C-a), 22.69 (C-b), 21.89 (C-c), 20.44 (C-d). 119Sn NMR ((CH3)4Sn): 9.18. EI-MS m/z: M+ 589 [(C6H11)3Sn{O2CC10H12NO2}]+, 456 [(C6H11)2Sn{O2CC10–H12NO2}]+, 412 [(C6H11)2Sn{C10H12NO2}]+, 379 [(C6H11)Sn{C10H12–NO2}]+, 367 [(C6H11)3Sn]+, 284 [(C6H11)2Sn]+, 201 [(C6H11)Sn]+, 178 [C10H12NO2]+, 119 [Sn]+, 83 [C6H11]+.
Dibutyltin(IV)-di-3-acetylamino-3(2-nitrophenyl) propanoate (7)
3-Amino-3(2-nitro phenyl) propanoic acid 1 g (6.29 mmol) was made to react with dibutyltin(IV) oxide 0.49 g (3.14 mmol) in 2:1 ratio with (3:1, v/v) ethanol and toluene as a solvent. Recrystallization was performed in 1:2 ratio chloroform and ethanol mixture. Yield: 78%. Mp: 204 °C. Anal. calc. (C4H9)2Sn(O2CC10H11–N2O3)2: C, 43.2, H, 6.4, N, 5.9, found (): C, 43.3, H, 6.2, N, 5.10%. FTIR νmax/cm−1: 3446 ν(N–H), 3146, 1682ν(CO), 1628 ν(COO)asym, 1445ν(COO)sym, 658 ν(Sn–O), 540 ν(Sn–C). 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 8.15 (s, 1H, CONH), 7.67 (d, J = 2.5 Hz, 1H, H-7), 7.61 (d, J = 0.7 Hz, 1H, H-8), 7.53 (d, J = 2.5 Hz, 1H, H-9), 7.45 (d, J = 7.3 Hz, 1H, H-10), 1.50 (t, J = 7.1 Hz, 3H, H-3), 1.23 (d, J = 7.3 Hz, 2H, H-2), Sn–nBu skeleton: 1.46 (t, 2H, H-d), 1.34 (m, 2H, H-c), 1.20 (m, 2H, H-b), 0.91 (m, 3H, H-a). 13C NMR (100 MHz, DMSO) δC: 170.35 (C-1), 167.34 (C-4), 137.72 (C-5), 133.73 (C-6), 129.75 (C-7), 125.45 (C-8), 122.92 (C-9), 120.73 (C-10), 48.45 (C-3), 27.75 (C-2), Sn–nBu skeleton: 25.45 (C-a), 24.35 (C-b), 22.45 (C-c), 13.25 (C-d). 119Sn NMR ((CH3)4Sn): −126.13. EI-MS m/z: M+ 734 [(C4H9)2Sn{O2CC10H11N2O3}2]+, 690 [(C4H9)2SnO2C{C10H11N2–O3}2]+, 677 [(C4H9) Sn{O2CC10H11N2O3}2]+, 633 [(C4H9)SnO2C{C10H11N2–O3}2]+, 589 [(C4H9) Sn{C10H11N2O3}2]+, 483 [(C4H9)2Sn{O2CC10H11N2–O3}]+, 439 [(C4H9)2Sn {C10H11N2O3}]+, 382 [(C4H9) Sn{C10H11N2O3}]+, 325 [Sn{C10H11N2O3}]+, 207 [C10H11N2O3]+, 119 [Sn]+, 57 [CH3CH2CH2–CH2]+, 43 [CH3CH2CH2]+, 29 [CH3CH2]+, 15 [CH3]+.
Dibutyltin(IV)-di-stannoxane-di-3-acetylamino-3(2-nitrophenyl) propanoate (8)
The compound was synthesized by adopting the procedure similar to compound (7) with the 1:1 ligand to metal salt ratio. Yield: 79%. Mp: 240 °C. Anal. calc. for (C4H9)2SnO2C(C10H11–N2O3)2: C, 42.3, H, 6.5, N, 3.6, found: C, 42.1, H, 6.4, N, 3.4%. FTIR νmax/cm−1: 3423 ν(N–H), 1697 ν(CO), 1647 ν(COO)asym, 1490 ν(COO)sym, 685 ν(Sn–C), 544 ν(Sn–O). 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 8.09 (s, 1H, CONH), 7.59 (d, J = 2.5 Hz, 1H, H-7), 7.53 (d, J = 0.7 Hz, 1H, H-8), 7.49 (d, J = 2.5 Hz, 1H, H-9), 7.45 (t, J = 7.3 Hz, 1H, H-10), 1.54 (t, J = 7.1 Hz, 3H, H-3), 1.33 (d, J = 7.3 Hz, 2H, H-2), Sn–nBu skeleton: 1.73 (t, 2H, H-d), 1.62 (m, 2H, H-c), 1.24 (m, 2H, H-b), 0.99 (m, 3H, H-a). 13C NMR (100 MHz, DMSO) δC: 179.45 (C-1), 169.79 (C-4), 138.38 (C-5), 135.45 (C-6), 133.34 (C-7), 130.21 (C-8), 129.29 (C-9), 126.37 (C-10), 44.73 (C-3), 29.25 (C-2), Sn–nBu skeleton: 27.65 (C-a), 26.67 (C-b), 24.96 (C-c), 14.19 (C-d). 119Sn NMR ((CH3)4Sn): −212.45,-215.67. EI-MS m/z: M+ 690 [(C4H9)2SnO2C{C10H11 N2O3}2]+, 677 [(C4H9)Sn{O2CC10H11N2O3}2]+, 633 [(C4H9)Sn O2C{C10H11–N2O3}2]+, 589 [(C4H9)Sn{C10H11N2O3}2]+, 483 [(C4H9)2Sn {O2CC10H11N2–O3}]+, 439 [(C4H9)2Sn{C10H11N2O3}]+, 382 [(C4H9)Sn{ C10H11N2O3}]+, 325 [Sn{C10H11N2O3}]+, 207 [C10H11N2O3]+, 119 [Sn]+, 57 [CH3CH2CH2CH2]+.
Dibutyltin(IV)-di-3-acetylamino-3(2-nitrophenyl) propanoate (9)
3-Acetylamino-3(2-nitrophenyl) propanoic acid 1 g (6.29 mmol) and AgNO3 1.06 g (6.29 mmol) were subjected to react with dibutyltin(IV) chloride 0.60 g (3.14 mmol) in 2:1 ratio in toluene and ethanol (3:1, v/v). Further it went to reflux for about 6 hours. The solvent was removed out and the synthesized compound (9) undergoes recrystallization in 1:2 ratio chloroform and benzene mixture and then collected. Yield: 75%. Mp: 175 °C. Anal. calc. for (C4H9)2Sn(O2CC10H11 N2O3)2: C, 41.3, H, 6.3, N, 5.9, found: C, 41.3, H, 6.3, N, 5.10%. FTIR νmax/cm−1: 3359 ν(N–H), 1697 ν(CO), 1628 ν(COO)asym, 1470 ν(COO)sym, 609 ν(Sn–O), 539 ν(Sn–C). 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 8.35 (s, 1H, CONH), 7.79 (d, J = 2.5 Hz, 1H, H-7), 7.65 (d, J = 0.7 Hz, 1H, H-8), 7.53 (d, J = 2.5 Hz, 1H, H-9), 7.43 (d, J = 7.3 Hz, 1H, H-10), 1.54 (t, J = 7.1 Hz, 3H, H-3), 1.25 (d, J = 7.3 Hz, 2H, H-2), Sn–nBu skeleton: 1.45 (t, 2H, H-d), 1.33 (m, 2H, H-c), 1.22 (m, 2H, H-b), 0.95 (m, 3H, H-a). 13C NMR (100 MHz, DMSO) δC: 175.43 (C-1), 163.35 (C-4), 139.54 (C-5), 134.47 (C-6), 131.25 (C-7), 128.65 (C-8), 126.91 (C-9), 124.99 (C-10), 48.15 (C-3), 26.75 (C-2), Sn–nBu skeleton: 25.65 (C-a), 24.50 (C-b), 22.93 (C-c), 13.15 (C-d). 119Sn NMR ((CH3)4Sn): −130.29. EI-MS m/z: M+ 734 [(C4H9)2Sn{O2CC10–H11N2O3}2]+, 677 [(C4H9)Sn{O2CC10H11N2O3}2]+, 620 [SnO2C{C10H11N2–O3}2]+, 483 [(C4H9)2Sn{O2CC10H11N2O3}]+, 439 [(C4H9)2Sn{C10H11N2–O3}]+, 369 [Sn{O2CC10H11N2O3}]+, 325 [Sn{C10 H11N2O3}]+,232 [(C4H9)2–Sn]+, 207 [C10H11N2O3]+, 119 [Sn]+, 43 [CH3–CH2CH2]+, 29 [CH3CH2]+, 15 [CH3]+.
Tributyltin(IV)-3-acetylamino-3(2-nitrophenyl) propanoate (10)
Yield: 75%. Mp: 110 °C. Anal. calc. for (C4H9)3Sn(O2CC06H11N2O3): C, 48.3, H, 7.9, N, 3.2. Found: C, 48.2, H, 7.8, N, 3.1%. FTIR νmax/cm−1: 3299 ν(N–H), 1680 ν(CO), 1629 ν(COO)asym, 1464 ν(COO)sym, 608 ν(Sn–O)asym, 570 ν(Sn–C). 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 8.43 (s, 1H, CONH), 7.98 (d, J = 2.5 Hz, 1H, H-7), 7.89 (d, J = 0.7 Hz, 1H, H-8), 7.63 (d, J = 2.5 Hz, 1H, H-9), 7.46 (d, J = 7.3 Hz, 1H, H-10), 1.48 (t, J = 7.1 Hz, 3H, H-3), 1.27 (d, J = 7.3 Hz, 2H, H-2), Sn–nBu skeleton: 1.56 (t, 2H, H-d), 1.36 (m, 2H, H-c), 1.21 (m, 2H, H-b), 0.85 (m, 3H, H-a). 13C NMR (100 MHz, DMSO) δC: 170.93 (C-1), 164.47 (C-4), 136.37 (C-5), 134.49 (C-6), 132.91 (C-7), 130.73 (C-8), 128.81 (C-9), 125.93 (C-10), 49.73 (C-3), 31.34 (C-2), Sn–nBu skeleton: 29.45 (C-a), 27.53 (C-b), 26.35 (C-c), 14.85 (C-d). 119Sn NMR ((CH3)4Sn): −137.97. EI-MS m/z: M+ 540 [(C4H9)3Sn{O2CC06H11N2O3}]+, 483 [(C4H9)2–Sn{O2CC10H11N2O3}]+, 439 [(C4H9)2Sn{C10H11N2O3}]+, 382 [(C4H9)Sn{C10H11N2O3}]+, 289 [(C4H9)3 Sn]+, 232 [(C4H9)2Sn]+, 207 [C10H11N2O3]+, 175 [(C4H9)Sn]+, 119 [Sn]+, 57 [CH3CH2CH2CH2]+, 43 [CH3CH2CH2]+, 29 [CH3CH2]+, 15 [CH3]+.
Triphenyltin(IV)-3-acetylamino-3(2-nitrophenyl) propanoate (11)
Yield: 78%. Mp: 135 °C. Anal. calc. for (C6H5)3Sn(O2CC10H11N2O3): C, 56.9, H, 4.7, N, 2.8, found: C, 56.7, H, 4.5, N, 2.7%. FTIR νmax/cm−1: 3368 ν(N–H), 1691 ν(CO), 1649 ν(COO)asym, 1474 ν(COO)sym, 658 ν(Sn–O), 548 ν(Sn–C). 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 8.30 (s, 1H, CONH), 7.91 (d, J = 2.5 Hz, 1H, H-7), 7.79 (d, J = 0.7 Hz, 1H, H-8), 7.64 (d, J = 2.5 Hz, 1H, H-9), 7.54 (d, J = 7.3 Hz, 1H, H-10), 1.52 (t, J = 7.1 Hz, 3H, H-3), 1.31 (d, J = 7.3 Hz, 2H, H-2), Sn–Ph skeleton: 7.41 (d, 1H, H-d), 7.29 (d, 1H, H-c), 6.79 (d, 1H, H-b). 13C NMR (100 MHz, DMSO) δC: 176.52 (C-1), 167.39 (C-4), 145.65 (C-5), 144.67 (C-6), 141.58 (C-7), 139.87 (C-8), 135.93 (C-9), 133.82 (C-10), 43.43 (C-3), 33.85 (C-2), Sn–Ph skeleton: 131.38 (C-a), 129.28 (C-b), 127.93 (C-c), 125.21 (C-d), 119Sn NMR ((CH3)4Sn): −119.81. EI-MS m/z: M+ 600 [(C6H5)3Sn{O2CC10H11N2O3}]+, 523 [(C6H5)2 Sn{O2CC10H11N2 O3}]+, 479 [(C6H5)2Sn{C10H11N2O3}]+, 402 [(C6H5)Sn {C10H11N2O3}]+, 349 [(C6H5)3Sn]+, 272 [(C6H5)2Sn]+, 207 [C10H11N2O3]+, 195 [(C6H5)Sn]+, 119 [Sn]+, 77 [C6H5]+.
Tricyclohexyltin(IV)-3-acetylamino-3(2-nitrophenyl) propanoate (12)
Yield: 79%. Mp: 125 °C. Anal. calc. for (C6H11)3Sn(O2C–C10H11N2O3): C, 54.9, H, 7.9, N, 2.7, found: C, 54.7, H, 7.8, N, 2.6%. FTIR νmax/cm−1: 3461 ν(N–H), 1690 ν(CO), 1650 ν(COO)asym, 1495 ν(COO)sym, 660 ν(Sn–O), 570 ν(Sn–C). 1H NMR (400 MHz, DMSO) δH, ligand skeleton: 7.99 (s, 1H, CONH), 7.53 (d, J = 2.5 Hz, 1H, H-7), 7.50 (d, J = 0.7 Hz, 1H, H-8), 7.47 (d, J = 2.5 Hz, 1H, H-9), 7.45 (d, J = 7.3 Hz, 1H, H-0), 1.56 (t, J = 7.1 Hz, 3H, H-3), 1.38 (d, J = 7.3 Hz, 2H, H-2), Sn–cyclohex skeleton: 1.50 (m, 1H, H-d), 1.32 (m, 2H, H-c), 1.25 (m, 2H, H-b), 1.18 (m, 2H, H-a). 13C NMR (100 MHz, DMSO) δC: 175.37 (C-1), 163.38 (C-4), 137.43 (C-5), 135.39 (C-6), 134.68 (C-7), 132.35 (C-8), 129.76 (C-9), 127.64 (C-10), 46.73 (C-3), 32.45 (C-2), Sn–cyclohex skeleton: 29.19 (C-a), 27.86 (C-b), 26.54 (C-c), 23.95 (C-d). 119Sn NMR ((CH3)4Sn): 9.74. EI-MS m/z: M+ 618 [(C6H11)3Sn{O2CC10H11N2O3}]+, 535 [(C6H11)2Sn{O2CC10H11N2O3}]+, 491 [(C6H11)2Sn{C10H11N2O3}]+, 408 [(C6H11)Sn{C10H11N2O3}]+, 367 [(C6H11)3Sn]+, 284 [(C6H11)2Sn]+, 203 [C10H11N2O3]+, 195 [(C6H11)Sn]+, 119 [Sn]+, 83 [C6H11]+.
Powder XRD analysis
X-ray diffractometry (powder method) of ligand (L1) and complexes (1–5) were carried out in the Bruker D2 diffractometer over a 2θ range of 20° to 120° with a step size of 0.02°.63 Powder samples were irradiated by CuKα1 radiation having the wavelength of 0.1542 nm. A standard crystalline Si sample was exploited to calibrate the position and instrumental broadening of peaks. Instrumental broadening is a key parameter to find out the physical broadening of peaks related to the size and strain in the crystallites; each measured as the full width at half maximum (FWHM).64Molecular docking studies
The molecular docking simulation was performed to explore the predicting binding affinity value of ligands (L1–L8) and their organotin(IV) derivatives, using Autodock vina and Autodock tools 4.2.43 The target protein was retrieved from the protein data bank (https://www.rcsb.org). The natural ligand of α-glucosidase (3WY1), polyacrylic acid (ID: PRU), was used as a reference. The AutoDock4.2 and Discovery studio client 2020 software was used to prepare the target enzyme removing non-standard molecules or hetero-atoms or ions (glycerol, polyacrylic acid, water, and Mg2+) and polar hydrogen atoms were also added. Kollaman charges were considered for each amino acid residues in the target enzyme. To determine the interaction and best confirmation of proposed compounds in the catalytic pocket of target enzyme the final space dimension for grid construction, x = y = z = 35 Å and −10, −8.120, −17.33, 19.010 (x, y, and z) on the geometric center was considered.Ligands and organotin(IV) complexes structures were sketched, and energy was minimized using Marvin Sketch software,82 and for metal complexes, the energy was further minimized by applying MMFF94x field by using Avogadro software.83 The Gasteiger charges were added to each ligand by AutoDock4.2 and saved in PDBQT file format. For organotin(IV) metal complexes, parameters were set as Rii = 4.38 Å and van der Waals well depth (epsii) 0.567 in kcal mol−1. For docking simulation, Lamarckian Genetic Algorthim4.2 scoring function with default parameters were applied. Discovery studio visualizer client was employed to create the 2D and 3D interaction of ligands and metal complexes in the catalytic pocket of enzyme 3WY1.
Bioactivity studies
In vitro α-glucosidase inhibitory activity
The α-glucosidase inhibitory assay of selected ligands and tin(IV) metal complexes were evaluated by reported procedure with slight modifications.84 In brief, the ligands L1 and L4 and tin(IV) metal complexes (1–12) were dissolved in 100% dimethyl sulfoxide (DMSO) to prepare the 200 μg mL−1 solution. A serial dilution in 5% DMSO was then performed to obtain the varying concentrations of 25 μg mL−1, 50 μg mL−1, and 100 μg mL−1. α-Glucosidase (0.1 U mL−1) in phosphate buffer pH 6.8 was mixed with varying concentration of ligands and tin(IV) metal complexes in 96 microwell plates and mixture were incubated at room temperature for 35 minutes. The reaction was then started by adding substrate 1.25 mM p-nitrophenyl-α-D-glucopyranoside (pNPG) in each well, and enzymatic reaction was carried out for 30 minutes at optimum temperature. Subsequently, the enzymatic reaction was quenched by adding the 170 μL 0.1 M Na2CO3. A Thermo Fisher microplate reader (ELISA) was used to measure the solution absorbance at 405 nm. Acarbose was used as a positive control at the varying concentration of 25 μg mL−1, 50 μg mL−1, 100 μg mL−1 and 200 μg mL−1 and for control, the sample solution was replaced by DMSO. The following formula calculated enzymatic inhibition.| % Inhibition = [Acontrol − Asample]/[Acontrol] × 100 |
Each experiment was repeated three times. The data was processed by Bio-Stat (version 5) software and results were reported as ±deviation. The Origin Pro 2018 was used for graphical analysis and IC50 was calculated using regression equation by plotting the concentration along X-axis and % age inhibition along Y-axis.
Antioxidant activity (free radical scavenging)
The antioxidant screening of the ligands (L1–L8) and complexes (1–12) was accomplished by DPPH (2,2-diphenyl-1-picrylhydrazyl) method.80,85 Methanol was used to prepare DPPH solution (0.1 mM in MeOH). Various concentrations of the test compounds about 25, 50, 75, 100 μg mL−1 in different solvents were mixed with 2 mL DPPH in separate test tubes and were exposed for 30 minutes to incubation at 37 °C. The absorbance was measured at 517 nm using Synergy HT BioTek® USA microplate reader. The reference standard used in this experiment was quercetin. The solution of methanolic DPPH was taken as a negative control. IC50 values were also calculated using regression equation. This equation measured the scavenging activity (%).| Scavenging activity (%) = [(Ac − As)/Ac × 100] |
Antibacterial activity
All the ligands (L1–L8) and complexes (1–12) were investigated using the disc diffusion method against four bacterial strains, i.e., Staphylococcus aureus, Bacillus subtilis (Gram-positive) and Escherichia coli, Pseudomonas aeruginosa (Gram-negative). Before use, the glassware was sterilized at 150 °C. The microbial specimens as swabs of pus, blood, urine, sputum, etc., were collected from different wards of Nishter Hospital Multan and accumulated for further use. MacConkey agar (10.0 g) and C.L.E.D mediums (10.0 g) in 250.0 mL distilled water were firstly autoclaved and then used further for the composition of Petri plates. The ligands and complexes (1–12) of about 500 ppm in DMSO were prepared. The composed discs, after soaking in test solutions, were dried and then autoclaved. The assembled Petri plates were kept in an incubator for 24 hours at 37 °C. The standard used was ciprofloxacin, and DMSO served as a negative control. This procedure was repeated in triplicate for each organism.7,86 In this activity, complete inhibition (mm) was determined by measuring the diameter of zones.In vivo antidiabetic activity
In this study, adult male healthy rabbits weighing about 1 to 2 kg were used. These rabbits were classified into eight groups, and every group has three rabbits each. One group acted as a control group, and the other is experimental groups. Alloxan was injected into the rabbits (150 mg kg−1 of body weight) to made them diabetic. The average blood glucose level of all rabbits was determined before alloxan injection. All the rabbits were observed as diabetic after 24 hours because all the rabbit's blood glucose level was raised to 293–320 mg dL−1 and was employed to study further.87,88 Metformin was used as a standard drug and was given to the control group. The test samples were composed by dissolving different concentrations i.e. 0.5 mg, 1.0 mg, 1.5 mg of the compounds in DMSO. After administration of the test solutions to all the corresponding rabbit groups, the blood was collected for every 3 hours interval i.e. at 3, 6, 9, 12, and 24 hours by using a Glucometer (SD Biosensor, Inc.) by the glucose oxidase method.7,88 All experiments were performed in compliance with the relevant laws and followed the institutional guidelines. Furthermore, all animal procedures were performed in accordance with the Guidelines of Bahauddin Zakariya University, Multan and approved by the ethical committee of Bahauddin Zakariya University, Multan.Conclusions
The synthesis of biological useful di- and triorganotin(IV) complexes from N-acetylated β-amino acids has been accomplished. Complete characterization of the complexes was achieved by elemental analysis, FTIR, multinuclear (1H, 13C, 119Sn) NMR spectroscopy, and mass spectrometry. The results verified monodentate coordination of the triorganotin(IV) complexes with tetrahedral geometry in solid form and bidentate coordination with trigonal bipyramidal geometry in solution form. Diorganotin(IV) complexes exhibited bidentate coordination with trans-octahedral geometry as monomers and a hexa-coordinated geometry as dimers. Powder XRD analysis of selected complexes revealed that complexes crystallized as face-centered cubic phases and negative value of strain explained tin(IV) insertion in the organic framework, which resulted in elongation of the lattice. Molecular Docking analysis of ligands, di- and triorganotin(IV) complexes (monomer) on the catalytic pocket of the α-glucosidase enzyme (PDB ID: 3WY1) revealed that ligands and triorganotin(IV) complexes (4, 6 and 10) showed the highest values of predicted binding affinity. The poses analysis of complexes in the catalytic pocket of enzyme displayed that triorganotin(IV) complexes can occupy the amino acid residues and have well fitted in conformation in the catalytic pocket compared to diorganotin(IV) complexes. In vitro antidiabetic activity of compounds was evaluated against the α-glucosidase enzyme and compared with standard acarbose. The results of activity reveal that triorganotin(IV) complexes 4, 6, and 10 exhibited the potent enzyme inhibition activity with values of IC50 21.54 ± 0.45, 37.96 ± 0.81 and 35.20 ± 1.02 μg mL−1 respectively compared to standard acarbose with IC50 values of 42.51 ± 0.21 μg mL−1 and diorganotin(IV) complexes. A good correlation among molecular docking of metal complexes and in vitro antidiabetic activity was also found. In vitro, antioxidant and antibacterial studies were also evaluated to determine the biocidal potential of synthesized ligands and complexes. Triorganotin(IV) complexes 4, 5, 6, 10, 11, and 12 displayed significant antiradical activity near to standard. Complexes displayed the highest to moderate antibacterial activities influenced by the nature of alkyl groups (butyl, phenyl, and cyclohexyl) attached to the central tin(IV) atom. Moreover, lipophilicity, polarity, and geometry of complexes may be another factor. Furthermore, in vivo antidiabetic assay of triorganotin(IV) complexes 4, 6, 10, 12, and ligands L1, L4 were performed to determine the hypoglycemic effect. This study suggests that triorganotin(IV) complexes may be a good choice in treating diabetes after further clinical screenings and determining a specific mechanism of action in vivo.Author contributions
The specification of individual contribution of each author is as follow:(a) Nagina Naveed Riaz: data correction, Formal analysis, investigation, software, visualization, writing original draft, writing – review & editing.
(b) Muhammad Mahboob Ahmed: conceptualization, data correction, formal analysis, investigation, project administration, resources, software, validation, visualization, writing original draft, writing – review & editing.
(c) Muhammad Kashif: conceptualization, data correction, investigation, software, visualization, writing – review & editing.
(d) Muhammad Ali: investigation, software, investigation, software.
(e). Muhammad Sajid: data correction, software, validation, visualization, writing – review & editing.
(f) Khalid Mahmood: investigation, software, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are very thankful to Institute of Chemical Sciences (Research Project No. DR & EL/D-740 and Research project No. DR & EL/D-454), Bahauddin Zakariya University, Multan, Pakistan for providing research facilities. The Department of Chemistry, Quid-e-Azam University, Islamabad, Pakistan is also acknowledged for spectroscopic analysis.References
- X. Y. Zhang, H. B. Song, Q. S. Lee, X. F. Liu and L. F. Tang, Polyhedron, 2007, 26, 3743–3749 CrossRef CAS.
- Metallotherapeutic drugs and metal-based diagnostic agents: the use of metals in medicine, ed. M. Gielen and E. R. Tiekink, John Wiley & Sons, 2005 Search PubMed.
- M. Gielen, M. Biesemans, D. de Vos and R. Willem, J. Inorg. Biochem., 2000, 79, 139–145 CrossRef CAS PubMed.
- D. K. Dimertzi, V. Dokouro, A. Primikiri, R. Vergas, C. Silvestru, U. Russo and M. A. Dimertzis, J. Inorg. Biochem., 2009, 103, 738–744 CrossRef PubMed.
- V. Dokouro, A. Primikiri and D. K. Dimertzis, J. Inorg. Biochem., 2011, 105, 195–201 CrossRef PubMed.
- D. Tzimopoulos, I. Sanidas, A. C. Varvogli, A. Czapik, M. Gdaniec, E. Nikolakaki and P. D. Akrivos, J. Inorg. Biochem., 2010, 104, 423–430 CrossRef CAS PubMed.
- M. M. Ahmed, N. N. Riaz, M. Kashif, M. Ashfaq, M. N. Arshad and M. Sajid, J. Braz. Chem. Soc., 2021, 32, 1082–1099 Search PubMed.
- P. Debnath, K. S. Singh, T. S. Devi, S. S. Singh, R. J. Butcher, L. Sieron and W. Maniukiewicz, Inorg. Chim. Acta, 2020, 510, 119736 CrossRef CAS.
- M. Roy, S. Roy, K. S. Singh, J. Kalita and S. S. Singh, New J. Chem., 2016, 40, 1471–1484 RSC.
- P. Debnath, K. S. Singh, K. K. Singh, S. S. Singh, L. Sieron and W. Maniukiewicz, New J. Chem., 2020, 44, 5862–5872 RSC.
- G. Duaa, R. Zahraa and Y. Emad, Arc. Org. Inorg. Chem. Sci., 2018, 3, 344–352 Search PubMed.
- S. K. Hadjikakou and N. Hadjiliadis, Coord. Chem. Rev., 2009, 253, 235–249 CrossRef CAS.
- S. Shoaib Ahmad Shah, M. Ashfaq, A. Waseem, M. Mehboob Ahmed, T. Najam, S. Shaheen and G. Rivera, Mini-Rev. Med. Chem., 2015, 15, 406–426 CrossRef PubMed.
- G. Zaloga, Nutr. Clin. Pract., 1990, 5, 231–237 CrossRef CAS PubMed.
- I. Omae, Appl. Organomet. Chem., 2003, 17, 81–105 CrossRef CAS.
- N. N. Riaz, F. Rehman and M. M. Ahmad, Med. Chem., 2017, 7, 302–307 CAS.
- M. Nath, S. Pokharia and R. Yadav, Coord. Chem. Rev., 2001, 215, 99–149 CrossRef CAS.
- N. Rabiee, M. Safarkhani and M. M. Amini, Rev. Inorg. Chem., 2019, 39, 13–45 CrossRef CAS.
- S. M. Abbas, S. Ali, S. T. Hussain and S. Shahzadi, J. Coord. Chem., 2013, 66, 2217–2234 CrossRef CAS.
- G. Prabusankar and R. Murugavel, Organometallics, 2004, 23, 5644–5647 CrossRef CAS.
- S. Shahzadi and S. Ali, J. Iran. Chem. Soc., 2008, 5, 16–28 CrossRef CAS.
- H. Ullah, V. Previtali, H. B. Mihigo, B. Twamley, M. K. Rauf, F. Javed, A. Waseem, R. J. Baker and I. Rozas, Eur. J. Med. Chem., 2019, 181, 111544 CrossRef CAS PubMed.
- C. Ma, Q. Jian, R. Zhang and D. Wang, Chem.–Eur. J., 2006, 12, 420–428 CrossRef PubMed.
- R. Garcia-Zarracino and H. Hopfl, J. Am. Chem. Soc., 2005, 127, 3120–3130 CrossRef CAS PubMed.
- H. Yin, C. Yue, M. Hong, J. Cui, Q. Wu and X. Zhang, Eur. J. Med. Chem., 2012, 58, 533–542 CrossRef CAS PubMed.
- M. Ashfaq, M. K. Baloch, A. Majeed, A. W. K Khanzada and W. U. Shah, J. Chin. Chem. Soc., 2003, 50, 361–368 CrossRef CAS.
- M. Nath and P. K. Saini, Dalton Trans., 2011, 40, 7077–7121 RSC.
- H. Sakurai, A. Katoh, T. Kiss, T. Jakusch and M. Hattori, Metallomics, 2010, 2, 670–682 CrossRef CAS PubMed.
- M. H. Forouzanfar, P. Liu, G. A. Roth, M. Ng, S. Biryukov, L. Marczak, L. Alexander, K. Estep, K. H. Abate, T. F. Akinyemiju and R. Ali, JAMA, J. Am. Med. Assoc., 2017, 317, 165–182 CrossRef PubMed.
- X. Lin, Y. Xu, X. Pan, J. Xu, Y. Ding, X. Sun, X. Song, Y. Ren and P. F. Shan, Sci. Rep., 2020, 10, 1–11 CrossRef PubMed.
- A. Ceriello, Diabetes Care, 2003, 26, 1589–1596 CrossRef CAS PubMed.
- R. Rahimi, S. Nikfar, B. Larijani and M. Abdollahi, Biomed. Pharmacother., 2005, 59, 365–373 CrossRef CAS PubMed.
- L. Tang, W. Wei, L. Chen and S. Liu, J. Ethnopharmacol., 2006, 108, 109–115 CrossRef CAS PubMed.
- A. C. Maritim, R. A. Sanders and J. B. Watkins, J. Biochem. Mol. Toxicol., 2003, 17, 24–38 CrossRef CAS PubMed.
- A. F. Santos, D. F. Brotto, L. R. Favarin, N. A. Cabeza, G. R. Andrade, M. Batistote, A. A. Cavalheiro, A. Neves, D. Rodrigues and A. D. Anjos, Rev. Bras. Farmacogn., 2014, 24, 309–315 CrossRef CAS.
- M. A. Riswan Ahamed, R. S. Azarudeen and N. M. Kani, Bioinorg. Chem. Appl., 2014, 2014, 764085 Search PubMed.
- T. Bal-Demirci, M. Sahin, E. Kondakci, M. Ozyurek, B. Ulkuseven and R. Apak, Spectrochim. Acta, Part A, 2015, 138, 866–872 CrossRef CAS PubMed.
- A. V. Lebedev, A. B. Lebedeva, V. D. Sheludyakov, E. A. Kovaleva, O. L. Ustinova and I. B. Kozhevnikov, Russ. J. Gen. Chem., 2005, 75, 1113–1124 CrossRef CAS.
- W. M. Rodionow and E. A. Postovskaja, J. Am. Chem. Soc., 1929, 51, 841–847 CrossRef.
- X. Shen, W. Saburi, Z. Gai, K. Kato, T. Ojima-Kato, J. Yu, K. Komoda, Y. Kido, H. Matsui, H. Mori and M. Yao, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 1382–1391 CrossRef CAS PubMed.
- M. Ashfaq, M. M. Ahmed, S. Shaheen, H. Oku, K. Mehmood, A. Khan, S. B. Niazi, T. M. Ansari, A. Jabbar and M. I. Khan, Inorg. Chem. Commun., 2011, 14, 5–12 CrossRef CAS.
- M. Ashfaq, M. M. Ahmed, S. Shaheen, R. Tabussam and G. Rivera, Quim. Nova, 2016, 39, 19–25 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 16, 2785–2791 CrossRef PubMed.
- R. Alizadeh, M. Afzal and F. Arjmand, Spectrochim. Acta, Part A, 2014, 131, 625–635 CrossRef CAS PubMed.
- S. Packianathan and N. Raman, Inorg. Chem. Commun., 2014, 45, 55–60 CrossRef CAS.
- C. Pettinari, F. Marchetti, R. Pettinari, D. Martini, A. Drozdov and S. Troyanov, Inorg. Chim. Acta, 2001, 325, 103–114 CrossRef CAS.
- M. Ashfaq, M. I. Khan, M. K. Baloch and A. Malik, J. Organomet. Chem., 2004, 689, 238–245 CrossRef CAS.
- M. Pervez, S. Anwar, A. Badshah, B. Ahmad, A. Majeed and M. Ashfaq, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 159–160 CrossRef PubMed.
- K. Hans, M. Pervez, M. Ashfaq, M. Saeed, A. B. Amin and A. Majeed, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, m466–m468 CrossRef CAS.
- X. Song, Q. Xie and X. Fang, Heteroat. Chem., 2002, 13, 592–598 CrossRef CAS.
- T. S. BasuBaul, K. S. Singh, X. Song, A. Zapata, G. Eng, A. Lycka and A. Linden, J. Organomet. Chem., 2004, 689, 4702–4711 CrossRef CAS.
- M. Kemmer, L. Ghys, M. Gielen, M. Biesemans, E. R. T. Tiekink and R. Willem, J. Organomet. Chem., 1999, 582, 195–203 CrossRef CAS.
- T. S. B. Baul, C. Masharing, G. Ruisi, R. Jirasko, M. Holcapek, D. de Vos, D. Wolstenholme and A. Linden, J. Organomet. Chem., 2007, 692, 4849–4862 CrossRef.
- A. Azadmeher, M. M. Amini, N. Hadipour, H. R. Khavasi, H. K. Fun and C. Chen, Appl. Organomet. Chem., 2008, 22, 19–24 CrossRef CAS.
- M. M. Amini, A. Azadmeher, H. R. Khavasi and S. W. Ng, J. Organomet. Chem., 2007, 692, 3922–3930 CrossRef CAS.
- M. Gielen, A. Bouhdid, R. Willem, V. I. Bregadze, L. V. Ermanson and E. R. T. Tiekink, J. Organomet. Chem., 1995, 501, 277–281 CrossRef CAS.
- J. Li, G. Zhao, C. Xiong and Y. Ma, Synth. React. Inorg. Met.-Org. Chem., 2001, 31, 85–93 CrossRef CAS.
- M. Roy, S. S. Devi, S. Roy, C. B. Singh and K. S. Singh, Inorg. Chim. Acta, 2015, 426, 89–98 CrossRef CAS.
- M. T. Masood, S. Ali, M. Danish and M. Mazhar, Synth. React. Inorg. Met.-Org. Chem., 2002, 32, 9–23 CrossRef CAS.
- W. Rehman, M. K. Baloch and A. Badshah, Eur. J. Med. Chem., 2008, 43, 2380–2385 CrossRef CAS PubMed.
- M. Gielen, A. El Khloufi, M. Biesemans, F. Kayser, R. Willem, B. Mahieu, D. Maes, J. N. Lisgarten, L. Wyns, A. Moreira, T. K. Chattopadhay and R. A. Palmer, Organometallics, 1994, 13, 2849–2854 CrossRef CAS.
- M. X. Li, D. Zhang, L. Z. Zhang, J. Y. Niu and B. S. Ji, J. Organomet. Chem., 2011, 696, 852–858 CrossRef CAS.
- B. D. Cullity and S. R. Stock, Elements of X-ray Diffraction, Harlow, 2014 Search PubMed.
- D. Nath, F. Singh and R. Das, Mater. Chem. Phys., 2020, 239, 122021 CrossRef CAS.
- P. Bindu and S. Thomas, J. Theor. Appl. Phys., 2014, 8, 123–134 CrossRef.
- R. Jacob and J. Isac, J. Chem. Stud., 2015, 2, 12–21 Search PubMed.
- W. H. Hall, Proc. Phys. Soc., London, Sect. A, 1949, 62, 741 CrossRef.
- D. Balzar and H. Ledbetter, J. Appl. Crystallogr., 1993, 26, 97–103 CrossRef.
- L. Motivalezadeh, Z. Heidary and M. Abrishami, Bull. Mater. Sci., 2014, 37, 397–405 CrossRef.
- H. A. Abuelizz, R. E. Dib, M. Marzouk, E. H. Anouar, Y. A. Maklad, H. N. Attia and R. Al-Salahi, Molecules, 2017, 22, 1094 CrossRef PubMed.
- A. Bhatia, B. Singh, R. Arora and S. Arora, BMC Complementary Altern. Med., 2019, 19, 1–9 CrossRef PubMed.
- R. Al-Salahi, R. Ahmad, E. Anouar, N. I. Iwana Nor Azman, M. Marzouk and H. A. Abuelizz, Future Med. Chem., 2018, 10, 1889–1905 CrossRef CAS PubMed.
- A. Corona-Bustamante, J. M. Viveros-Paredes, A. Flores-Parra, A. L. Peraza-Campos, F. J. Martinez-Martinez, M. T. Sumaya-Martínez and A. Ramos-Organillo, Molecules, 2010, 15, 5445–5459 CrossRef CAS PubMed.
- M. Jabeen, S. Ali, S. Shahzadi, M. Shahid, Q. M. Khan, S. K. Sharma and K. Qanungo, J. Iran. Chem. Soc., 2012, 9, 307–320 CrossRef CAS.
- H. Zafarian, T. Sedaghat, H. Motamedi, D. Trzybinski and K. Wozniak, J. Organomet. Chem., 2017, 853, 184–192 CrossRef CAS.
- T. Sedaghat, L. Tahmasbi, H. Motamedi, R. Reyes-Martinez and D. Morales-Morales, J. Coord. Chem., 2013, 66, 712–724 CrossRef CAS.
- R. Prabhakaran, A. Geetha, M. Thilagavathi, R. Karvembu, V. Krishnan, H. Bertagnolli and K. Natarajan, J. Inorg. Biochem., 2004, 98, 2131–2140 CrossRef CAS PubMed.
- A. Regiel-Futyra, J. M. Dąbrowski, O. Mazuryk, K. Spiewak, A. Kyzioł, B. Pucelik, M. Brindell and G. Stochel, Coord. Chem. Rev., 2017, 351, 76–117 CrossRef CAS.
- O. S. Jung, J. H. Jeong and Y. S. Sohn, Polyhedron, 1989, 8, 1413–1417 CrossRef CAS.
- R. Yadav, M. Trivedi, R. Chauhan, R. Prasad, G. Kociok-Köhn and A. Kumar, Inorg. Chim. Acta, 2016, 450, 57–68 CrossRef CAS.
- A. N. Gupta, V. Kumar, V. Singh, A. Rajput, L. B. Prasad, M. G. B. Drew and N. Singh, J. Organomet. Chem., 2015, 787, 65–72 CrossRef CAS.
- The Marvin Sketch, 20.12 software; Chem Axon, Budapest, Hungary, 2020 CAS; O. E. Trott and A. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- G. C. Wang, Y. P. Peng, Z. Z. Xie, J. Wang and M. Chen, MedChemComm, 2017, 8, 1477–1484 RSC.
- W. B. Williams, M. E. Cuvelier and C. Berset, LWT--Food Sci. Technol., 1995, 28, 25–30 CrossRef.
- N. Awang, N. Mokhtar, N. M. Zin and N. F. Kamaludin, J. Chem. Pharm. Res., 2015, 7, 379–383 CAS.
- A. Wadood, N. Wadood and S. A. Shah, J. Pak. Med. Assoc., 1989, 398, 208–212 Search PubMed.
- R. V. R. Reddy, T. A. R. Raja and K. Uma Maheshwara Rao, World J. Pharm. Res., 2016, 3, 4739–4746 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR Fig. S1–S20, 1H NMR Fig. S21–S40, 13C NMR Fig. S41–S60 and EI-MS Fig. S61–S80 of the synthesized compounds. See DOI: https://doi.org/10.1039/d2ra06718h |
| This journal is © The Royal Society of Chemistry 2023 |