DOI:
10.1039/D2RA06693A
(Paper)
RSC Adv., 2023,
13, 4422-4430
Molecular basis of ligand selectivity for melatonin receptors†
Received
22nd October 2022
, Accepted 20th January 2023
First published on 1st February 2023
Abstract
Sleep disorders in adults are related to adverse health effects such as reduced quality of life and increased mortality. About 30–40% of adults are suffering from different sleep disorders. The human melatonin receptors (MT1 and MT2) are family A G protein-coupled receptors that respond to the neurohormone melatonin MEL which regulates circadian rhythm and sleep. Many efforts have been made to develop drugs targeting melatonin receptors to treat insomnia, circadian rhythm disorders, and even cancer. However, designing subtype-selective melatonergic drugs remains challenging due to their high similarities in both sequences and structures. MEL (a function-selective compound with a bulky β-naphthyl group) behaves as an MT2-selective antagonist, whereas it is an agonist of MT1. Here, molecular dynamics simulations were used to investigate the ligand selectivity of MT receptors at the atomic level. We found that the binding conformation of MEL differs in different melatonin receptors. In MT1, the naphthalene ring of MEL forms a structure perpendicular to the membrane surface. In contrast, there is a 130° angle between the naphthalene ring of MEL and the membrane surface in MT2. Because of this conformational difference, the MEL leads to a constant water channel in MT1 which activates the receptor. However, MEL hinders the formation of continuous water channels, resulting in an inactive state of MT2. Furthermore, we found that A1173.29 in MT2 is a crucial amino acid capable of hindering the conformational flip of the MEL molecule. These results, coupled with previous functional data, reveal that although MT1 and MT2 share highly similar orthosteric ligand-binding pockets, they also display distinctive features that could be used to design selective compounds. Our findings provide new insights into functionally selective melatonergic drug development for sleep disorders.
1 Introduction
Melatonin receptors (MTs) are seven transmembrane-spanning proteins belonging to the family A G protein-coupled receptors (GPCRs) superfamily.1,2 They are expressed in the central nervous system (CNS) and peripheral tissues, which mediate melatonin activities in the synchronization of circadian rhythm and sleep regulation.3–5 Melatonin, N-acetyl-5-methoxytryptamine, mainly synthesized in the pineal gland, follows a circadian pattern synchronized to the dark phase of the natural light/dark cycle.6,7 With the development of society, working around the clock, long-distance travel across time zones, and prolonged exposure to light sources have become the norm. The most immediate consequence of these everyday phenomena is disrupting people's circadian rhythms. As a result, sleep disturbances affect large populations in modern society and cause a considerable economic burden.8 Melatonin is one of the most popular supplements in managing sleeping disorders, including insomnia and jet lag.9 In addition to regulating sleep-wake rhythms, melatonin can modulate other physiological processes, including regulating the cardiovascular system10 and buffering the immune system.11 The roles of MTs in cancer protection,12,13 bone formation,14 glucose maintenance,15 and neurodegenerative diseases12 have also been demonstrated recently. Therefore, the design of therapeutic agents targeting MTs has been continuously executed in modern drug discovery.
In humans, the MT1 family consists of two highly conserved members, including MT1 and MT2. They are composed of 350 and 362 amino acids, sharing 55% identity in the overall region and 70% identity within the membrane domains.16 The MT1 receptor is found in the perifornical lateral hypothalamus (PFH), involved in rapid eye movement (REM) phases of the vigilance state in sleep. In contrast, the MT2 receptor is uniquely located in the reticular thalamus, an area involved in non-REM (NREM) triggering.17 Besides that, genetic variants of the MTNR1B locus, encoding the melatonin MT2 receptor, have been considered to be associated with increased type 2 diabetes (T2D) risk.18 Given the different physiological roles of MT1 and MT2, it is highly desirable to obtain selective ligands. However, most drugs on the market or under clinical evaluation, such as ramelteon, tasimelteon, and agomelatine, are nonselective.19,20 Recently, the crystal structures of MT1 and MT2 have been determined by X-ray free-electron laser,21,22 which provides valuable information for studying the agonist binding modes of MT1 and MT2. Therefore, the use of function-selective molecules (MEL23) to accurately delineate the atomic details of ligands in the binding pocket of MTs is of great value for effective drug design. In this study, we used unbiased molecular dynamics (MD) simulations to illustrate the molecular basis of ligand selectivity against MT receptors.
2 Methodology and computational details
2.1 Loop filling and refinement
For the simulations systems of the inactive state used for the unbiased simulations, the crystal structures of MT1 in complex with 2-phenylmelatonin (PDB code: 6ME3) and β2AR in complex with 2-phenylmelatonin (PDB code: 6ME6) were applied.21,22 Since the intracellular loop ICL2 for each receptor was missing because of the insertion of a fusion protein in this region. The missing residues were rebuilt using the Prime package24 included in Schrodinger. A total of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 loops were generated for each receptor in Modeller V9.10.25 A conformation with the lowest Discrete Optimized Protein Energy (DOPE) score was chosen for receptor construction.
000 loops were generated for each receptor in Modeller V9.10.25 A conformation with the lowest Discrete Optimized Protein Energy (DOPE) score was chosen for receptor construction.
2.2 Protein structure preparation
All protein models were prepared with the Protein Preparation Wizard26 included in Maestro under the OPLS_2005 force field:27 hydrogen atoms were added to the repaired structures at physiological pH (7.0) with the PROPKA tool28 to optimize the hydrogen bond network, all water molecules were removed, C- and N-terminal capping were added, disulfide bonds were assigned, and constrained energy minimizations were carried out on the full-atomic models until the RMSD of the heavy atoms converged to 0.3 Å.
2.3 Ligand structure preparation
A functionally selective small-molecule MEL (an agonist in MT1 and an antagonist in MT2 (ref. 23)) was downloaded from Pubchem Database (https://pubchem.ncbi.nlm.nih.gov/). The LigPrep module in the Schrodinger suite was introduced for geometric optimization by using the OPLS_2005 force field. Ionization states of ligands were calculated with the Epik tool29 employing Hammett and Taft methods in conjunction with ionization and tautomerization tools.
2.4 Protein–ligand docking
Glide30 performed the docking procedure in the Schrodinger suite software. The grid files were created around the binding pocket defined by the ligand-bound crystal structure (PDB code: 6ME3 and 6ME6). A cubic box centered on the ligand mass center with a radius of 20 Å for all ligands defined the docking binding regions. SP Glide docking was executed for all structures. The best-scored pose with same position and orientation (functional groups: R1, R2, and R3 (ref. 21 and 22)) for each ligand was chosen as the initial structure for MD simulations (Fig. S1†).
2.5 Molecular dynamics simulation
The OPM webserver (https://opm.phar.umich.edu/ppm_server) was used to align the experimental structures in the lipid bilayer.31 Membrane systems were built using the membrane building tool CHARMMGUI.32 All the simulated systems were assigned for CHARMM36m forcefield,33 and the ligand was assigned for the Charmm CGenFF force field.34 MEL bonded MT1, and MT2 models were embedded in 128 POPC lipids, coupled with TIP3P water molecules and 0.15 M NaCl. The final systems contained 59591 and 59642 atoms, for MT1 and MT2, with a volume of rough 70 × 70 × 110 Å3. The unbiased MD simulations were performed using GROMACS-2018.4.35 The systems were minimized with a 50000-step energy minimization using the steepest descent algorithm. The systems were subjected to temperature equilibrating in the NVT ensemble at 310 K for 200 ps. After that, density equilibrating in the NPT ensemble at 310 K and 1 atm for 10 ns. The heavy atoms were constrained using a harmonic restraint with a force constant of 1000 kJ mol−1 nm−2 in the equilibrating steps. All the production runs were carried out under the NPT ensemble. The production of simulations lasted for 3 μs, and three replicas were used to ensure reproducibility. The details of MD simulations can be found in our previous work.36
2.6 Average water and Na+ density calculations
Water density was calculated in the density module of the MDAnalysis toolkit.37 Based on removing the periodic effect of the simulated box and retaining water molecules, the protein was superimposed in the center. The simulation box was split into lattices with the smallest unit of 1 Å × 1 Å × 1 Å, and the oxygen atoms (OW) above the TIP3P and Na+ were selected as the analysis objects. The average water density was calculated every 500 ns of each long-timescale MD simulation. Final output results were visualized in PyMOL.38
2.7 Angle between membrane plane and MEL, TM area displacement, and amino acids distance calculation
The included angle between MEL and the membrane plane, the displacement of the TM region, and the distance of sandwich structure were conducted using the GROMACS-2018.4 patched with plumed-2.5.1.39 The complete parameters used in PLUMED were listed in Table S1.†
2.8 Interaction fingerprint calculations
The IFP was performed using PLIP software.40 PLIP detects frequent non-covalent protein–ligand interactions, including hydrogen bonds, hydrophobic contacts, pi–stacking, pi–cation interactions, salt bridges, water bridges, and halogen bonds. We used 500 frames from the final 1 μs MD simulations for the IFP analysis. A plot was generated for the combination of all kinds of interactions. Parameters used for IFP calculations were kept as default.
3 Results and discussion
3.1 Statistics of water molecular and Na+ near D2.50 density distribution
Previous studies revealed that water molecules from the cytoplasmic region of rhodopsin (Rho) are involved in the hydrolytic release of retinal upon Rho activation by light.41,42 Crystal structures of Rho and the beta-2 adrenergic receptor (β2AR) revealed further details. Internal, structured water molecules play a critical role in receptor activation; some of these waters connect two TM helices, and some form an H-bond network between receptor and ligand.43 The continuous pathway of water molecules has been observed in previous reporters and is considered essential in GPCR activation.44 Here, the distribution of water molecules in the simulated system is shown in Fig. 1. We found that in the MT1/MEL system, with the prolongation of the calculation time, a continuous water molecule channel was formed, which means that MT1 is in an activated state, indicating that MEL is an agonist of MT1. In the MT2/MEL system, with the calculation time. The prolongation of MT2 does not form continuous water molecule channels, which means that MT2 is in an inactive state, indicating that MEL is an antagonist of MT2. These results are consistent with those reported experimentally.23 Furthermore, the allosteric Na+ next to D2.50 was observed primarily in the structures of inactive GPCR structures but not in their active states.45–47 In our simulations, the same phenomenon was observed (Fig. 1B). In the MT1/MEL system, there is no sodium ion near D2.50 of activated MT1. Conversely, there is a sodium ion (started from approximate 600–900 ns, shown in Fig. 1C) in the inactive MT2 in the MT2/MEL system. Theoretical studies have shown that extracellular Na+ could enter the allosteric D2.50 via the orthosteric site. It has been speculated that this cation stabilizes the inactive states of GPCRs.48
 |
| | Fig. 1 Density distribution. The colored depth was colored according to water density. The figure was rendered in PyMol via the volume tool. The iso level was set to the range of 0.002–0.003, whereas the transparency alpha was sent to 0.24. (A) Distribution diagram of water molecules in MEL-MT1 receptor or MT2 receptor system over simulated time. MEL are displayed as sticks. Blue represents the high-density regions of water molecules. (B) Three-dimensional spatial density distributions of Na+ across MT1 and MT2. Residues D732.50 in MT1 (which is D862.50 in MT2) are displayed as sticks. Blue represents the high-density regions of sodium ion (left). (C) The number of Na+ near the D2.50 over simulated time. | |
3.2 Angle changes of MEL along the membrane plane
According to the results of the above water molecular pathway, it is confirmed that our computational simulation captures the difference in the dynamic interaction between MEL and MT1 and MT2. So, we moved on to a more detailed analysis. The angle of the naphthalene ring in MEL along membrane plane was analyzed over MD simulations. We found that the conformations of the naphthalene rings were different in MT1, and MT2. The naphthalene rings with different conformations conferred different consequences. The conformation of this naphthalene ring can be demonstrated by its relative angle towards the plane of the lipid bilayers (Fig. 2A). As shown in Fig. 2B, the naphthalene ring conformation stabilized at 200 ns and 1500 ns in MT1 and MT2, respectively. The conformations of the naphthalene ring are noticeably different in a stabilized state at the later stage of MD simulations. In MT1, the naphthalene ring forms a conformation perpendicular to the membrane plane. In contrast, naphthalene ring forms an angle of 130° with the membrane plane in MT2 (Fig. 2C). In the previous analysis of the water channel, we found that the conformation of the naphthalene ring is different in MT1 and MT2 (Fig. 1A). We guessed that due to the different conformation, the blocking effect on the water molecular channel would be different, resulting in the different roles of MEL in MT1 and MT2.
 |
| | Fig. 2 The angle between MEL and the membrane plane changes in the MEL-MT1 receptor or MT2 receptor system over simulated time. (A) Definition of the included angle between MEL and the membrane plane; (B) the included angle changes with the simulation time in the MEL-MT1 receptor or MT2 receptor system; (C) comparison of the conformation of the last frame. Magenta and orange represent the conformation of the last frame simulated by MEL in MT1 and MT2, respectively. | |
3.3 Conformational changes in the TM regions of MT1 and MT2
We next analyzed the conformational changes in the TM regions of MT1 and MT2. Generally, the activation of GPCRs is featured by a movement of TM6 helixes, which opens a large space in the cytoplasmic region for G protein bindings.43,44 As shown in Fig. 3 A, at 500 ns, TM6 began to move, and the TM6 region moved outwards with a distance of ∼4 Å at 3000 ns. It is worth noting that when TM6 began to move, a continuous pathway of water molecules is formed in MT1 (Fig. 1A), and the angle between the small molecule and the membrane plane also changed (Fig. 2B). In contrast, no obvious movement of TM6 was observed in the inactive TM2 system. Interestingly, while the TM6 was moving, the TM1 of MT1 was shifted by ∼5 Å relative to TM7 (Fig. 3B). While our manuscript was being prepared, Wang et al. reported the experimental structure of G protein-coupled active MT1 and MT2 in different conformations.49 They found that MT1 coupled to Gi protein adopted an active conformation with the characteristic of TM6 outward displacement, similar to the many other active GPCRs. However, conformational differences between active MT1 and MT2 were also observed in the extracellular region of TM1.49 The results of our simulation were in agreements with their experimental results. We inspected the outward movement of TM1 can increase the cavity space of the GPCR transmembrane region in the MT1 system, which was conducive to the formation of continuous water molecule channels.
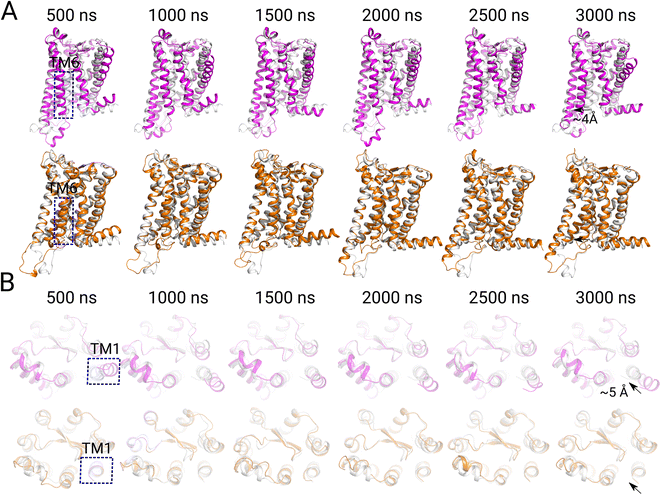 |
| | Fig. 3 Conformational changes in the TM regions of MT1 and MT2. (A) The displacement of TM6 in MT1 and MT2 systems over simulated time. (B) The displacement of TM1 in MT1 and MT2 systems over simulated time. | |
3.4 A sandwich structure
To further analyze why TM1 moves outward upon MT1 activation instead of MT2, we investigated the atomic details of TM1, TM7, and MEL. Although pockets of the inactive MT1 and MT2 show only subtle differences21,22,49 (Fig. S2†), the variants become much larger in the active states observed in our calculated data and confirmed by structural data. Wang et al. reported that the bulky Y2827.40 in MT1, which is linked to the slight differences between the ligand-binding pockets, pushed TM1 more outward from the TM bundles. Packing of Y2827.40 against TM1 moves the two adjacent residues Y2817.39 and Y2857.43 closer to the core of the pocket than the equivalent residues Y2947.39 and Y2987.43 in MT2.49 Here, we found that the differences in the outward movement of the TM1 region between MT1 and MT2 was mainly caused by the interactions among several amino acids including L341.39, Y2857.43 in MT1 and L471.39, Y2987.4 in MT2. In the initial structure, Y2857.43 in MT1 (which is Y2987.43 in MT2) formed a sandwich-like stacking with both the naphthalene ring of MEL and L341.39 (which is L471.39 in MT2) via hydrophobic interactions. Such changes and differences can be well represented by the coordination distance d1–d2 (Fig. 4A). The d1–d2 in the MT1 increased along MD simulations which demonstrated that the sandwich structure was destroying. The final distances of d1 and d2 in MT1 were 9.7 Å and 5.3 Å, respectively (Fig. 4B, up; 4C, left); in MT2, the d1–d2 did not change notably, and the sandwich structure remained. The final distances of d1 and d2 in MT1 were 5.1 Å and 5.5 Å, respectively (Fig. 4B, bottom; 4C, left). During the long-time scaled MD simulations, the conformation of this naphthalene ring in MEL changed: the hydrophobic interaction with Y2857.43 was disrupted because of the fast movement of water molecules (Movie S1†). We assumed that disruption of this sandwich structure opened a gate for the water molecule from the bulk environment to fluctuate into the inner space of MT1, which led to the outward movement of the TM1 region. In MT2, the sandwich structure was stabilized by MEL during the whole MD simulations. MT2 was always in an inactive state.
 |
| | Fig. 4 The distance between crucial amino acids and MEL in TM1 and TM7 regions of MT1 and MT2 varies with the simulation time. (A) Definition of the corresponding position and distance of key amino acids and MEL. D1 is the centroid distance between L341.39/L471.39 and Y2857.43/Y2987.43, whereas d2 is the centroid distance between Y2857.43/Y2987.43 and MEL. (B) The sandwich distances (d1–d2) in MT1 (up) and MT2 (down), respectively, as simulated time changes. (C) The final sandwich structure poses for MT1 and MT2. | |
3.5 Analysis of interaction fingerprint
We analyzed the dynamic binding differences between MEL-MT1 and MEL-MT2 via interaction fingerprint (IFP). As shown in Fig. 5A, the amino acids marked in red are shared by MT1 and MT2 in the sequence alignment, while the amino acids observed in black are unique to MT1 or MT2. There are many shared amino acids in the dynamic process of MT1 and MT2 because their binding pockets are identical. These included residues Y7.43, A7.42, Y7.37, L6.51, W6.48, V5.43, Q181ECL2 (Q194ECL2), F179ECL2 (F192ECL2), L168ECL2 (L181ECL2), V3.36, and M3.32. Some differences were also observed: G2586.55, N2556.52, F2476.44, F1965.47, H1955.46, V1915.42, T178ECL2, I1153.40, and G1083.33 in MT1; A1173.29 and N1754.60 in MT2. Noticeably, the naphthalene ring of MEL stabilized the sandwich structure, blocking the formation of the water channel and MT2 activation. Thus, MEL is an antagonist of MT2 (Fig. 4A). Residue G1043.29 in MT1 (A1173.29 in MT2) in the orthosteric site seems to play an essential role in stabilizing the sandwich interaction as indicated by Fig. 5B. Subsequently, we performed 1 μs unbiased molecular dynamics simulations of amino acid mutations (G104A3.29 in MT1 and A117G3.29 in MT2) to verify our conclusions. The results showed that the damaged sandwich structure is re-established in the mutant MT1 system. In contrast, in the mutant MT2, the stable sandwich structure was destroyed (Fig. 5C). As shown in Fig. 5D, the stabilized sandwich structure blocks the continuous water channel, and TM1 and TM6 were in place in the native MT1. In the MT2, TM1 moved about 5 Å compared to the unmutated system. Moreover, continuous water channels formed in the mutated system due to the destruction of the sandwich stacking. Furthermore, we speculated that if the naphthalene ring of MEL was replaced by other more minor, for example, benzene rings, such molecules would be agonists of MT1 and MT2. This was consistent with previously reported experimental results.23As mentioned above, the unique configuration of the MEL-Y7.43-L2.46 sandwich structure and particular binding sites between MT1 and MT2 could be targeted to design MT subtype-selective drugs.
 |
| | Fig. 5 Interaction diagram of MEL with MT1 and MT2 in a dynamic process. (A) Radar map. The red amino acids represent those common to MT1 and MT2, and the black amino acids represent those unique to MT1 or MT2. (B) The conformation of the critical amino acid A117@MT2 (corresponding to G in MT1) in the binding pocket. Sequence alignment of MT1 and MT2 at key amino acid locations. (C) The final sandwich structure poses for mutated MT1 and MT2. (D) The TM region displacement and continuous water channel changes in mutated MT1 and MT2 systems vary with the simulated time. | |
4 Conclusions
The ligand specificity is an essential question in modern drug discovery. Why a molecule is an agonist for one receptor, whereas is an antagonist for the other, is always fascinating to the pharmaceutical industry. In this work, we investigated the ligand functional selectivity of ligands of melatonin receptors using all-atom long-time scaled MD simulations. MEL an agonist of MT1 receptor whereas it is also an antagonist of MT2 receptor. Our simulations indicated that the model mode as well as IFP of MEL differed between MT1-MEL and MT2-MEL. In MT1, the naphthalene ring forms a structure perpendicular to lipid bilayers, whereas it creates an internal angle of ∼130° towards the same membrane surface in MT2. Moreover, further analysis found that A1173.29 in MT2 was essential in hindering MEL molecules' rotation. It stabilized the hydrophobic MEL-Y2987.43-L341.39 sandwich structure, which blocked the water channel formation keeping MT2 in the inactive state. In contrast, a residue G3.29 in the corresponding position in MT1 provided enough space for the molecular switches of both MEL and Y2857.43. In the long-time scaled MD simulations, it induced a continuous water channel inside MT1 and led to the noticeable movements of TM1. Finally, MT1 was activated. Our findings provide an insightful view into the design of selective ligands targeting melatonin receptors.
Data availability
All data generated or analyzed during this study are included in this article. Also, the data supporting the study's findings are available from the corresponding author upon reasonable request.
Author contributions
Shuguang Yuan & Rongfeng Zou designed and led the project; Wenqiang Cui made the computational simulations and analyzed the data; all authors wrote and revised the articles.
Conflicts of interest
Shuguang Yuan and Horst Vogel are co-founders of Alpha Mol Science Ltd, Shenzhen, China.
Acknowledgements
The work of Shuguang Yuan was supported by funding from Chinese Academy of Sciences, the Shenzhen Institute of Advanced Technology, CAS, Shenzhen government (grant no. JCYJ20200109114818703) as well as that from Guangdong province (grant no. 2019QN01Y306). We would like to thank the support from the following grants: Shenzhen Key Laboratory for Computer Aided Drug Discovery in SIAT (ZDSYS20201230165400001), Advanced Technology, Chinese Academy of Sciences, Shenzhen, Guangdong; Guangdong Retired Expert; The Chinese Academy of Science President's International Fellowship Initiative (PIFI) (2020FSB0003) as well as Shenzhen Pengcheng Scientist; NSFC-SNSF Funding (no. 32161133022); Shenzhen Government Top-talent Working Funding ; Guangdong Province Academician Work Funding.
References
- R. Jockers, P. Delagrange, M. L. Dubocovich, R. P. Markus, N. Renault, G. Tosini, E. Cecon and D. P. Zlotos, Br. J. Pharmacol., 2016, 173, 2702–2725 CrossRef CAS PubMed
 .
. - M. L. Dubocovich, P. Delagrange, D. N. Krause, D. Sugden, D. P. Cardinali and J. Olcese, Pharmacol. Rev., 2010, 62, 343–380 CrossRef CAS PubMed .
- F. Confente, M. Rendon, L. Besseau, J. Falcon and J. A. Munoz-Cueto, Gen. Comp. Endocrinol., 2010, 167, 202–214 CrossRef CAS PubMed .
- S. M. Reppert, D. R. Weaver and T. Ebisawa, Neuron, 1994, 15, 1177–1185 CrossRef PubMed .
- S. M. Reppert, C. Godson, C. D. Mahle, D. R. Weaver, S. A. Slaugenhaupt and J. F. Gusella, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 8734–8738 CrossRef CAS PubMed .
- F. G. D. Amaral and J. Cipolla-Neto, Arch. Endocrinol. Metab., 2018, 62, 472–479 CrossRef PubMed .
- S. Ganguly, S. L. Coon and D. C. Klein, Cell Tissue Res., 2002, 309, 127–137 CrossRef CAS PubMed .
- M. K. Stoller, Clin. Ther., 1994, 16, 873–897 CAS .
- L. A. Erland and P. K. Saxena, J. Clin. Sleep Med., 2017, 13, 275–281 CrossRef PubMed .
- E. Sewerynek, Neuroendocrinol. Lett., 2002, 23(1), 79–83 CAS .
- A. Carrillo-Vico, P. J. Lardone, N. Alvarez-Sanchez, A. Rodriguez-Rodriguez and J. M. Guerrero, Int. J. Mol. Sci., 2013, 14, 8638–8683 CrossRef PubMed .
- J. Liu, S. J. Clough, A. J. Hutchinson, E. B. Adamah-Biassi, M. Popovska-Gorevski and M. L. Dubocovich, Annu. Rev. Pharmacol. Toxicol., 2016, 56, 361–383 CrossRef CAS PubMed .
- S. G. Grant, M. A. Melan, J. J. Latimer and P. A. Witt-Enderby, Expert Rev. Mol. Med., 2009, 11, e5 CrossRef PubMed .
- E. J. Sanchez-Barcelo, M. D. Mediavilla, D. X. Tan and R. J. Reiter, J. Osteoporosis, 2010, 2010, 830231 CAS .
- M. Garaulet, J. Qian, J. C. Florez, J. Arendt, R. Saxena and F. Scheer, Trends Endocrinol. Metab., 2020, 31, 192–204 CrossRef CAS PubMed .
- E. Cecon, A. Oishi and R. Jockers, Br. J. Pharmacol., 2018, 175, 3263–3280 CrossRef CAS PubMed .
- G. Gobbi and S. Comai, Front. Endocrinol., 2019, 10, 87 CrossRef PubMed .
- A. Imam, E. C. Winnebeck, N. Buchholz, P. Froguel, A. Bonnefond, M. Solimena, A. Ivanova, M. Bouvier, B. Plouffe, G. Charpentier, A. Karamitri, R. Jockers, T. Roenneberg and C. Vetter, Front. Physiol., 2020, 11, 564140 CrossRef PubMed .
- D. P. Zlotos, R. Jockers, E. Cecon, S. Rivara and P. A. Witt-Enderby, J. Med. Chem., 2014, 57, 3161–3185 CrossRef CAS PubMed .
- J. A. Boutin, P. A. Witt-Enderby, C. Sotriffer and D. P. Zlotos, J. Pineal Res., 2020, 69, e12672 CrossRef CAS PubMed .
- B. Stauch, L. C. Johansson, J. D. McCorvy, N. Patel, G. W. Han, X. P. Huang, C. Gati, A. Batyuk, S. T. Slocum, A. Ishchenko, W. Brehm, T. A. White, N. Michaelian, C. Madsen, L. Zhu, T. D. Grant, J. M. Grandner, A. Shiriaeva, R. H. J. Olsen, A. R. Tribo, S. Yous, R. C. Stevens, U. Weierstall, V. Katritch, B. L. Roth, W. Liu and V. Cherezov, Nature, 2019, 569, 284–288 CrossRef CAS PubMed .
- L. C. Johansson, B. Stauch, J. D. McCorvy, G. W. Han, N. Patel, X. P. Huang, A. Batyuk, C. Gati, S. T. Slocum, C. Li, J. M. Grandner, S. Hao, R. H. J. Olsen, A. R. Tribo, S. Zaare, L. Zhu, N. A. Zatsepin, U. Weierstall, S. Yous, R. C. Stevens, W. Liu, B. L. Roth, V. Katritch and V. Cherezov, Nature, 2019, 569, 289–292 CrossRef CAS PubMed .
- S. Rivara, A. Lodola, M. Mor, A. Bedini, G. Spadoni, V. Lucini, M. Pannacci, F. Fraschini, F. Scaglione, R. O. Sanchez, G. Gobbi and G. Tarzia, J. Med. Chem., 2007, 50, 6618–6626 CrossRef CAS PubMed .
- M. P. Jacobson, D. L. Pincus, C. S. Rapp, T. J. Day, B. Honig, D. E. Shaw and R. A. Friesner, Proteins, 2004, 55, 351–367 CrossRef CAS PubMed .
- N. Eswar, B. Webb, M. A. Marti-Renom, M. S. Madhusudhan, D. Eramian, M. Y. Shen, U. Pieper and A. Sali, Curr. Protoc. Bioinf., 2006 DOI:10.1002/0471250953.bi0506s15 , chapter 5, unit-5.6.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef PubMed .
- D. Shivakumar, E. Harder, W. Damm, R. A. Friesner and W. Sherman, J. Chem. Theory Comput., 2012, 8, 2553–2558 CrossRef CAS PubMed .
- D. C. Bas, D. M. Rogers and J. H. Jensen, Proteins, 2008, 73, 765–783 CrossRef CAS PubMed .
- J. C. Shelley, A. Cholleti, L. L. Frye, J. R. Greenwood, M. R. Timlin and M. Uchimaya, J. Comput.-Aided Mol. Des., 2007, 21, 681–691 CrossRef CAS PubMed .
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed .
- M. A. Lomize, A. L. Lomize, I. D. Pogozheva and H. I. Mosberg, Bioinformatics, 2006, 22, 623–625 CrossRef CAS PubMed .
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed .
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmuller and A. D. MacKerell Jr, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed .
- K. Vanommeslaeghe, E. P. Raman and A. D. MacKerell Jr, J. Chem. Inf. Model., 2012, 52, 3155–3168 CrossRef CAS PubMed .
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS .
- Y. Xia, R. Zou, M. Escouboue, L. Zhong, C. Zhu, C. Pouzet, X. Wu, Y. Wang, G. Lv, H. Zhou, P. Sun, K. Ding, L. Deslandes, S. Yuan and Z. M. Zhang, Nat. Commun., 2021, 12, 5969 CrossRef CAS PubMed .
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed .
- W. L. Delano, 2002.
- M. Bonomi, D. Branduardi, G. Bussi, C. Camilloni and M. Parrinello, Comput. Phys. Commun., 2009, 180, 1961–1972 CrossRef CAS .
- S. Salentin, S. Schreiber, V. J. Haupt, M. F. Adasme and M. Schroeder, Nucleic Acids Res., 2015, 43, W443–W447 CrossRef CAS PubMed .
- B. Jastrzebska, K. Palczewski and M. Golczak, J. Biol. Chem., 2011, 286, 18930–18937 CrossRef CAS PubMed .
- T. E. Angel, M. R. Chance and K. Palczewski, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8555–8560 CrossRef CAS PubMed .
- J. Li, A. L. Jonsson, T. Beuming, J. C. Shelley and G. A. Voth, J. Am. Chem. Soc., 2013, 135, 8749–8759 CrossRef CAS PubMed .
- S. Yuan, S. Filipek, K. Palczewski and H. Vogel, Nat. Commun., 2014, 5, 4733 CrossRef CAS PubMed .
- E. Segala, D. Guo, R. K. Cheng, A. Bortolato, F. Deflorian, A. S. Dore, J. C. Errey, L. H. Heitman, I. J. AP, F. H. Marshall and R. M. Cooke, J. Med. Chem., 2016, 59, 6470–6479 CrossRef CAS PubMed .
- A. Batyuk, L. Galli, A. Ishchenko, G. W. Han, C. Gati, P. A. Popov, M. Y. Lee, B. Stauch, T. A. White, A. Barty, A. Aquila, M. S. Hunter, M. Liang, S. Boutet, M. Pu, Z. J. Liu, G. Nelson, D. James, C. Li, Y. Zhao, J. C. Spence, W. Liu, P. Fromme, V. Katritch, U. Weierstall, R. C. Stevens and V. Cherezov, Sci. Adv., 2016, 2, e1600292 CrossRef PubMed .
- A. Borodovsky, C. M. Barbon, Y. Wang, M. Ye, L. Prickett, D. Chandra, J. Shaw, N. Deng, K. Sachsenmeier, J. D. Clarke, B. Linghu, G. A. Brown, J. Brown, M. Congreve, R. K. Cheng, A. S. Dore, E. Hurrell, W. Shao, R. Woessner, C. Reimer, L. Drew, S. Fawell, A. G. Schuller and D. A. Mele, J. Immunother. Cancer, 2020, 8, e000417 CrossRef PubMed .
- X. Wang, S. Yuan and H. C. S. Chan, J. Chem. Inf. Model., 2022, 62, 3090–3095 CrossRef CAS PubMed .
- Q. Wang, Q. Lu, Q. Guo, M. Teng, Q. Gong, X. Li, Y. Du, Z. Liu and Y. Tao, Nat. Commun., 2022, 13, 454 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a




