
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of potential polycyclic energetic materials using bicyclic triazole and azetidine structures as building blocks†
Xin-bo Yang‡
 ab,
Chen-hui Jia‡c,
Xiang-yan Miaoa,
Yu-chuan Li*a and
Si-ping Pang*a
ab,
Chen-hui Jia‡c,
Xiang-yan Miaoa,
Yu-chuan Li*a and
Si-ping Pang*a
aSchool of Materials Science & Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: liyuchuan@bit.edu.cn; pangsp@bit.edu.cn
bSchool of Mechatronical Engineering, Beijing Institute of Technology, Beijing 100081, China
cBeijing Composite Materials Co., Ltd, No. 261 Kangxi Road, Badaling Economic Development Zone, Yanqing District, Beijing, 102101, China
First published on 17th January 2023
Abstract
Exploring the design strategy of new energetic materials is crucial to promote the development of energetic materials. In this study, a method for designing polycyclic energetic materials is proposed by combining the azetidine structure with azobis-1,2,4-triazole or bi-1,2,4-triazole. A series of typical triazolyl polycyclic compounds were designed and synthesized by simple nucleophilic reaction, which included 5,5′-dichloro-3,3′-bis(3,3′-difluoroazetidine)-4,4′-azobis-1,2,4-triazole (1), 5,5′-dichloro-3,3′-bis(3,3′-difluoroazetidine)-4,4′-bi-1,2,4-triazole (2), 5,5′-dichloro-3-(N,N-dimethyl)-3′-(3,3′-difluoroazetidine)-4,4′-bi-1,2,4-triazole (3) 5,5′-dichloro-3,3′-bis(3,3′-dinitroazetidine)-4,4′-bi-1,2,4-triazole (4), 5,5′-dichloro-3-(N,N-dimethyl)-3′-(3,3′-dinitroazetidine)-4,4′-bi-1,2,4-triazole (5), and 5,5′-diazido-3,3′-bis(3,3′-difluoroazetidine)-4,4′-azo-1,2,4-triazole (6). These designed and synthesized polycyclic compounds (1, 2, 3) have high decomposition temperatures (>200 °C). The molecular van der Waals surface electrostatic potentials suggested the reactivity of compounds 1, 2, and 3 when attacked by nucleophiles. The natural bond orbital and Hirshfeld surface analysis proved the essential reason for the stability of these compounds in theory. The formula design example suggests that some triazolyl polycyclic compounds (4, 5, and 6) are potentially explosives, suggesting that this strategy is feasible for constructing the triazolyl polycyclic energetic compounds.
1 Introduction
Energetic materials have important applications in modern national production and national defense construction. It is generally believed that the crystal density and energy level of energetic materials with cyclic molecular skeletons are often higher than those of chain-like molecular skeleton materials. In the ring skeleton energetic compounds, the double-ring or multi-ring skeleton structures have higher crystal density and energy than the single-ring skeleton structures. For example, 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20), octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX), and 1,3,5-trinitro-1,3,5-triazinane (RDX) all belong to non-aromatic cyclic nitramine compounds.1,2 The molecular structure of CL-20 can be considered a cage of multiple rings, which determines that CL-20 has a higher energy potential among the three from the molecular skeleton. Furthermore, the cage structure increases the stacking efficiency, which increases the density of CL-20 higher.In addition, there is a similar phenomenon for aromatic ring energetic materials. For example, the crystal density and enthalpy of a solid formation of bi-1,2,4-triazole and azobis-1,2,4-triazole are higher than those of the energetic compounds with the mono-triazole ring skeleton.3–5 Therefore, the study of polycyclic skeleton energetic compounds is one of the development trends of energetic materials in future. The selection of a suitable building block is crucial for the design of polycyclic energetic materials. 1,3,3-Trinitroazetidine (TNAZ)6,7 is a well-known azetidine structure that is a highly energetic material more powerful than RDX. However, TNAZ has the characteristics of relatively high vapour pressure, large volume shrinkage, forming pores in the process of melt solidification, and expensive price, which limits its practical applications. Another important azetidine structure is 1H-3,3-dinitroazetidine (DNAZ),8,9 which is also a precursor for TNAZ preparation.10,11 Compared with TNAZ because the nitrogen atom in azetidine is easily protonated, DNAZ tends to exist in the cationic form, making it easy to form ionic salts with other anions.12,13 In addition, DNAZ is prone to nucleophilic addition reaction with electron deficient reagents,14 N-nitrosation,15 and even the Mannich condensation reaction under some conditions.16 For example (see Scheme 1), DNAZ–trifluoride methane sulfonate reacts with activated halogenated benzene (1-fluoro-2,4,6-trinitrobenzene) to produce 1-dinitroazetidine-2,4,6-trinitrobenzene.17 When DNAZ–trifluoride methane sulfonate reacts with cyanuric chloride, 2,4,6-tris(3,3-dinitroazetidin-1-yl)-1,3,5-triazine could be synthesized. Ma et al.18 obtained 1-(2,4-dinitrophenyl)-3,3-dinitroazetidine by the reaction between DNAZ and 2,4-dinitrochlorobenzene. Hiskey et al.16 also investigated the nucleophilic aromatic substitution of DNAZ with 3,6-dichloro-1,2,4,5-tetrazine. The main product and subproduct of this reaction are 3-chloro-6-(3,3-dinitroazetidineyl)-1,2,4,5-tetrazine and 3,6-di(3,3-dinitroazetidineyl)-1,2,4,5-tetrazine, respectively. Other studies have shown that DNAZ reacts with paraformaldehyde to produce 1,1′-bis-(3,3-dinitrosacyclobutane-1-yl)methane (DNAZ–CH2–DNAZ).16,19 These compounds containing azetidine structures encouraged us to further explore other energetic materials containing such structures. With the development of nitrogenous heterocycles, triazolyl energetic materials have gradually attracted the attention of researchers because of their high nitrogen content and stability. The common construction idea of triazolyl polycyclic compounds is to combine triazole rings with other nitrogen-rich heterocyclic groups by bridging20–22 or fused-ring23,24 connections. However, the process of synthesizing these fused-ring and bridged polycyclic structures are relatively cumbersome. In the multi-membered ring molecular structure, with the increase in the number of nitrogen atoms in the ring, the molecular stability decreases gradually. It is extremely attractive to develop polycyclic energetic compounds with simple synthetic route and good stability. Therefore, we attempted to construct new polycyclic energetic compounds by nucleophilic substitution mechanism using bicyclic triazolyl structure (azobis-1,2,4-triazole3,25–27 and bi-1,2,4-triazole5) and azetidine structure (DNAZ or 3,3′-difluoroazetidine (DFAZ)28), which is a different strategy from fused rings and bridges. The azetidine structures not only can increase the nitrogen content of the molecule, but also has higher ring strain, which can improve the energetic performance from the additional energy release upon opening of the strained ring system during decomposition.18,29 Furthermore, it is generally believed that nitro can improve the molecular density, enthalpy of formation, oxygen balance and nitrogen content, which promotes the application of DNAZ in energetic materials; fluorine atoms can increase density and release a large amount of heat by reacting with metal aluminum or aluminum oxide, which suggests that DFAZ derivatives may be used as fluorine-containing oxidants in propellants28 and melt cast explosives.30
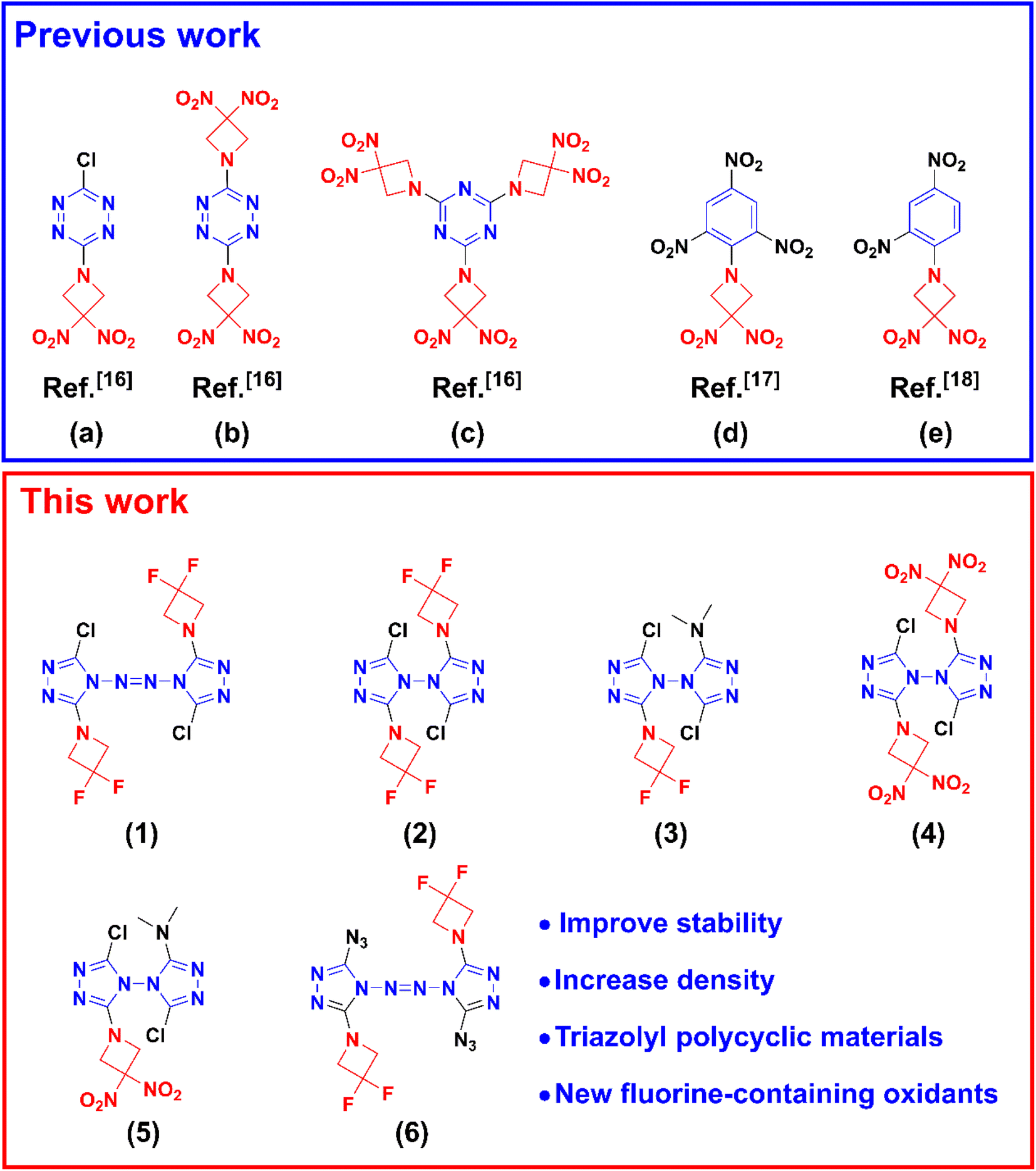 | ||
| Scheme 1 Some energetic materials (a–e) with azetidine structures involved in previous works and triazolyl polycyclic compounds designed in this work (1–6). | ||
The purpose of this work was to explore and verify a design method for developing new polycyclic energetic compounds. Some new triazolyl polycyclic intermediates were prepared successfully by nucleophilic substitution reactions between chlorinated azobis-1,2,4-triazole/chlorinated bi-1,2,4-triazole with DNAZ or DFAZ under alkaline conditions. Owing to the steric hindrance of the azetidine structures, all chlorine atoms in azobis-1,2,4-triazole/bi-1,2,4-triazole cannot be substituted. The strategy of introducing high ring strain azetidine structure into nitrogen-rich triazole structure could be used as a reference for the development of other energetic materials.
2 Experiment
A series of chlorinated triazolyl polycyclic intermediate compounds were obtained through the synthetic process described in Scheme 2. In the presence of K2CO3 and N,N-dimethylformamide (DMF), the nucleophilic substitution reaction between chlorinated bicyclotriazole and DFAZ/DNAZ was carried out to introduce the azetidine into the framework of bicyclotriazole. The reason that compounds 3 and 5 contained dimethylamino structure may be that DMF contains some degraded samples of dimethylamine upon the long-term storage process.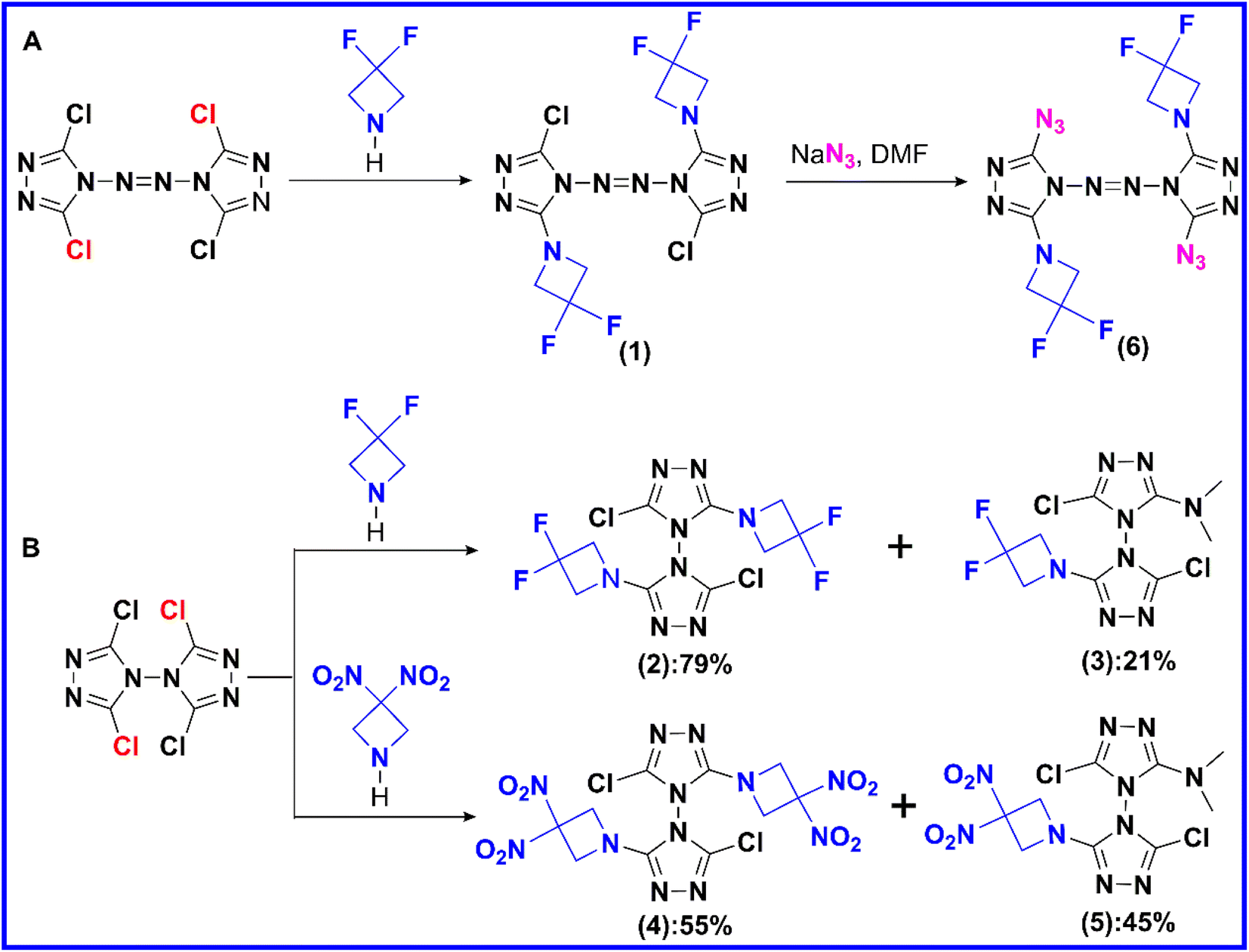 | ||
| Scheme 2 Brief synthetic processes (A and B) of compounds 1–6. | ||
Caution! The compounds reported in this work are potentially energetic materials that could explode under certain conditions, such as impact, friction, or electric discharge. Therefore, experiments should be performed on a small scale. Appropriate safety precautions, including the use of safety shields and personal protections (safety glasses, ear plugs, and gloves) is suggested all the time when handling these compounds.
2.1 Synthesis of 5,5′-dichloro-3,3′-bis (3,3′-difluoroazetidine)-4,4′-azo-1,2,4-triazole (1)
Compound 1 was synthesized according to the following procedure: 30 ml of N,N-dimethylformamide (DMF), 4.5 mmol of DFAZ hydrochloride and 0.5 g (4.7 mmol) anhydrous sodium carbonate were added to a 100 ml three-necked flack. The mixture was stirred for 30 minutes. Then, 1 mmol 3,3′,5,5′-tetrachloro-4,4′-azo-1,2,4-triazole (TCAT) was added to the above solution with stirring for 10 minutes. 0.5 g (3.6 mmol) anhydrous potassium carbonate was added to the reaction solution at 30 °C under stirring for 24 hours. The reaction solution was filtered for the next step. The filtrate was extracted with ethyl acetate (30 ml × 3). Then, the extracted organic phase was dried with anhydrous magnesium sulfate. The solution was filtered and the organic phase was removed by vacuum evaporation to obtain the 0.30 g yellow solid with a yield of 73.2%; 1H NMR (DMSO-d6) δ:4.58 (dd, 4H), 4.51 (dd, 4H); 13C NMR (DMSO-d6) δ: 64.00, 116.88, 135.32, 153.16 ppm; Fourier-transform infrared spectroscopy (FTIR) (KBr) ν: 2957, 2872, 1578, 1484, 1461, 1442, 1395, 1353, 1250, 1209, 1160, 910, 884, 725, 650 cm−1. Elemental analysis (%) calcd for (13): C, 25.91; H, 1.93; N, 36.94; found: C, 25.64; H, 1.86; N, 36.76. The structure of (1) was supported by single crystal X-ray analysis.2.2 Synthesis of 5,5′-dichloro-3,3′-bis(3,3′-difluoroazetidine)-4,4′-bi-1,2,4-triazole (2) and 5,5′-dichloro-3-(N,N′-dimethyl)-3′-(3,3′-difluoroazetidine)-4,4′-bi-1,2,4-triazole (3)
Compounds 2 and 3 were synthesized according to the following procedure: 30 ml DMF, 4.5 mmol DFAZ hydrochloride, and 0.5 g (4.7 mmol) anhydrous sodium carbonate were added to a 100 ml three-necked flack with stirring for 30 minutes. 1 mmol 3,3′,5,5′-tetrachloro-4,4′-bi-1,2,4-triazole (TCBT) was added to the above solution with stirring for 10 minutes. Then, 0.5 g (3.6 mmol) anhydrous potassium carbonate was added into the reaction solution at 30 °C for stirring 24 hours. The solution was filtered and the filtrate was extracted with ethyl acetate (30 ml × 3). Anhydrous magnesium sulfate was used to dry the extracted organic phase and filter it. The organic phase was removed by vacuum evaporation to obtain two white solids 2 (0.2844 g, yield 79%) and 3 (0.066 g, 21%), the total yield is 93%. For 2, m.p.: 194.0 °C; 1H NMR (acetone-d6) δ: 4.61 (dd, 4H), 4.49 (dd, 4H): 13C NMR (acetone-d6) δ: 63.69, 116.05, 137.20, 154.89 ppm; FTIR (KBr) ν: 2956, 1577, 1496, 1463, 1453, 1360, 1298, 1249, 1228, 1214, 918, 716 cm−1; For 3, m.p.: 169.2 °C; 1H NMR (acetone-d6) δ: 4.54 (dd, 2H), 4.47 (dd, 2H), 2.91 (s, 6H); 13C NMR (acetone-d6) δ: 38.85, 63.29 ppm; FTIR (KBr) ν: 3009–2814, 1597, 1577, 1512, 1495, 1464, 1445, 1443, 1431, 1416, 1359, 1278, 1245, 1220, 1056, 990, 914, 877, 706 cm−1. Elemental analysis (%) calcd for 2: C, 31.03; H, 2.08; N, 28.95; found: C, 31.26; H, 2.16; N, 28.75; elemental analysis (%) calcd for 3: C, 31.88; H, 2.97; N, 33.04; found: C, 31.61; H, 3.04; N, 33.16. The structure of 2 and 3 were supported by single-crystal X-ray analysis.2.3 Synthesis of 5,5′-dichloro-3,3′-bis(3,3′-dinitroazetidine)-4,4′-bi-1,2,4-triazole (4) and 5,5′-dichloro-3-(N,N′-dimethyl)-3′-(3,3′-dinitroazetidine)-4,4′-bi-1,2,4-triazole (5)
Compounds 4 and 5 were synthesized according to the following procedure: 30 ml DMF, 4.5 mmol DNAZ hydrochloride, and 0.5 g (4.7 mmol) anhydrous sodium carbonate were added to a 100 ml three-necked flack under stirring for 30 minutes. 1 mmol TCBT was added into the above solution with stirring for 10 minutes. Then, 0.5 g (3.6 mmol) anhydrous potassium carbonate was added to the reaction solution at 40 °C and stirred for 24 hours. The solution was filtered and the filtrate was extracted with ethyl acetate (30 ml × 3). Anhydrous magnesium sulfate was used to dry the extracted organic phase and was filtered. The organic phase was separated by vacuum evaporation. Two types of yellow oils were obtained by silica gel column chromatography: Compound 4-0.204 g (55%) and compound 5-0.133 g (45%). The total yield was 75%. For 4, FTIR (KBr) ν: 3012–2960, 1729, 1676, 1576, 1495, 1440, 1368, 1330, 1311, 1220, 1049, 858, 835 cm−1; For 5, FTIR (KBr) ν: 3011–2853, 1679, 1573, 1467, 1358, 1333, 1311, 1211, 866, 843 cm−1. Elemental analysis (%) calcd for 4: C, 24.26; H, 1.63; N, 33.95; found: C, 24.45; H, 1.70; N, 34.02; elemental analysis (%) calcd for 5: C, 27.50; H, 2.56; N, 35.63; found: C, 27.63; H, 2.54; N, 35.31.2.4 Synthesis of 5,5′-diazido-3,3′-bis(3,3′-difluoroazetidine)-4,4′-azo-1,2,4-triazole (6)
Compound 6 was synthesized according to the following procedure: 15 ml of DMF and 0.042 g (0.1 mmol) compound 1 were added to a 100 ml three-necked flack. The mixture was stirred for 10 minutes. 0.016 g (0.25 mmol) sodium azide was added to the above solution and it was kept in a water bath temperature at 30 °C. The reaction was monitored every 2 hours. After the reaction, the DMF solution was diluted 4–5 times with distilled water and then extracted with ethyl acetate (30 ml × 3). The organic phase was dried with anhydrous magnesium sulfate, filtered, and evaporated to obtain 0.024 g of yellow solid in 56% yield. FTIR (KBr) ν: 3080–2767, 2153, 1721, 1607, 1536, 1500, 1460, 1407, 1356, 1292, 1247, 1227, 1117, 977, 915, 802 cm−1. Elemental analysis (%) calcd for 6: C, 25.12; H, 1.87; N, 55.34; found: C, 25.46; H, 1.66; N, 55.60.3 Computational methods
The initial geometries in this work were calculated using the M06-2X exchange–correlation functional31 in conjunction with 6-311G (d, p) basis set32 in the gas phase. All optimized structures discussed were characterized to be local minima without imaginary frequencies. Gaussian 09 (D.01) program33 was used for the quantum chemistry calculations for initial geometries.The NBOs isosurface and molecular electrostatic potential analyses were performed using the Multiwfn 3.8 (dev) code34 based on the formatted checkpoint file of Gaussian 09. The isosurface maps of NBO orbitals and ESP were rendered by means of Visual Molecular Dynamics (VMD) software35 based on the files exported from Multiwfn.
The calculation of enthalpy of formation consisted of the following parts: the double hybrid PWPB95 level of theory functional in the ORCA program36 was used to calculate the final single point energy of the stable molecular structure and the input files of ORCA program was prepared with the help of Multiwfn code. The DFT single-point energy calculations were conducted with ORCA applying TightSCF and Grid4 options. The quadruple-ζ def2-QZVP37 basis set was used for DFT calculations. The D3 London dispersion correction was generally applied to DFT calculations. The resolution-of-identity (RI) approximation38 for coulomb (RIJ) and exchange (RIJK) integrals were used in combination with matching auxiliary basis sets as implemented in ORCA (def2/J, def2/JK options, def2-QZVPP/C39 for the MP2 correlation part) to speed up the DFT calculations.40 The vibration analysis files of the initial structure were used to calculate various thermodynamic constants by the Shermo program.41 For M06-2X functional, the resonant frequency correction factor of zero-point energy (ZPE) was 0.97. Finally, according to the definition of enthalpy of formation, the enthalpy of formation in the gas phase was calculated. See ESI† for a detailed calculation method for the enthalpy of formation.
The two-dimensional fingerprint and Hirshfeld surface42 plot spectra of the compounds were calculated by using CrystalExplorer 17 code43 based on the crystallographic information file (cif) of the corresponding compounds.
4 Experimental results and analysis
4.1 Crystal structures
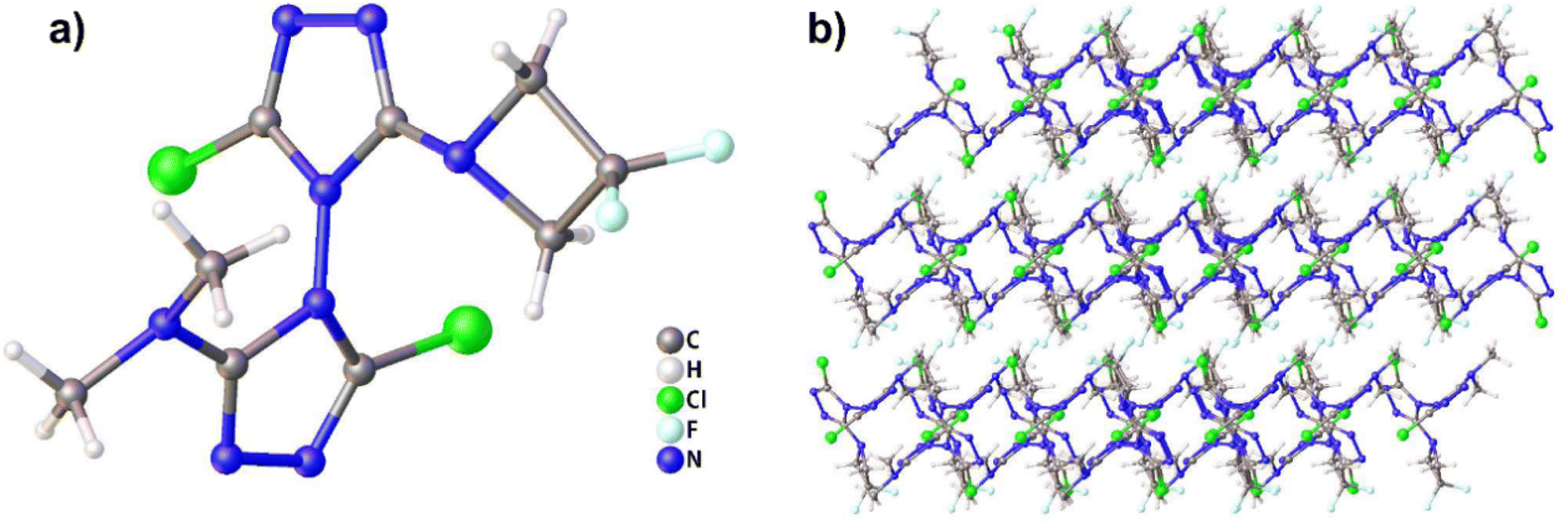
Compound 1, which possesses disordered fragments, crystallizes in the monoclinic space group P21/c with two molecules per unit cell (Z = 2). The molecular structure is shown in Fig. 1(a). The measured crystal density is 1.813 g cm−3 at 153 K. It can be seen from the crystal structure that the atoms in a skeleton and the chlorine atoms connected with them are in the same plane. Along the a axis, the layers are stacked by π–π interactions and separated by distances of 3.515 Å (see in Fig. 1(b)), which is helpful to transform the external mechanical energy into intermolecular interaction energy to elude the accumulation of hot spot formation, molecular vibration during explosive decomposition and subsequent final detonation.44 The detailed information is given in Table S1 in the ESI.†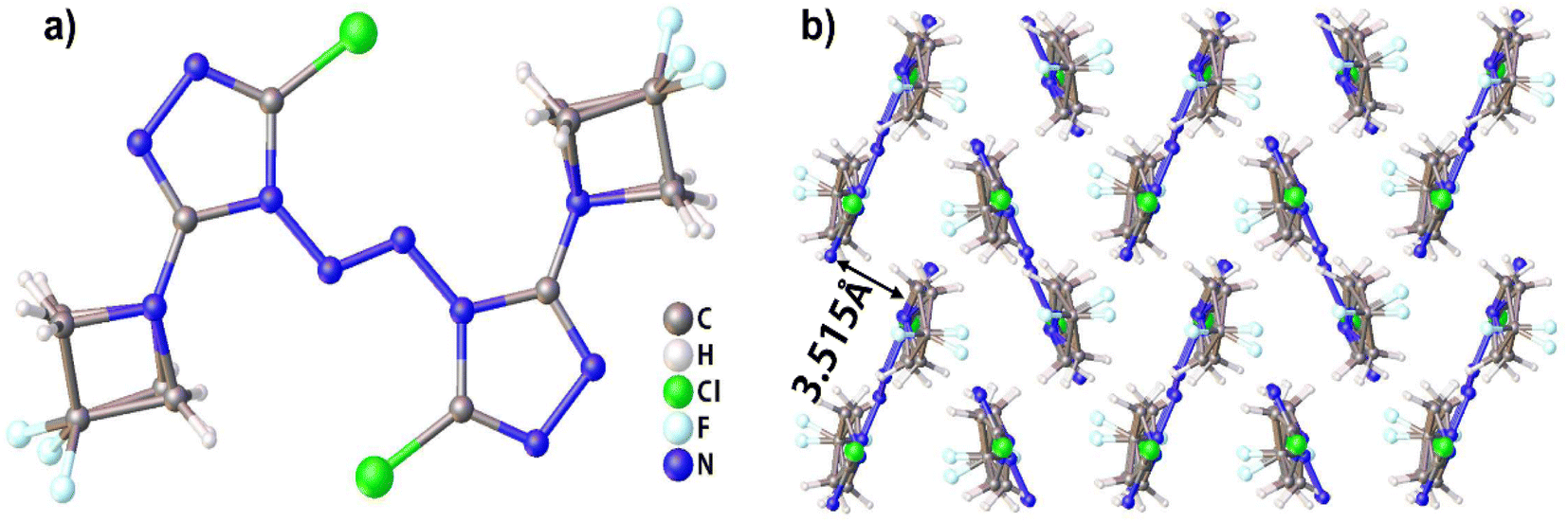 | ||
| Fig. 1 (a) Molecular structure of compound 1. (b) Packing diagram of compound 1. | ||
Compound 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two molecules per unit cell (Z = 2). The molecular structure is shown in Fig. 2(a). The measured crystal density was 1.726 g cm−3 at 296 K. The angle between the two triazole rings in compound 2 is 85.68°, which is not different from that in TCBT (84.53°). The results show that the introduction of two DFAZ rings has little effect on the molecular structure of BTR. In addition, the two DFAZ rings have a certain torsion angle, which relieves the ring strain of azetidine itself and helps stabilize the four-membered ring structure. Other relevant crystallographic parameters and diffraction data are shown in Table S1 in ESI.†
with two molecules per unit cell (Z = 2). The molecular structure is shown in Fig. 2(a). The measured crystal density was 1.726 g cm−3 at 296 K. The angle between the two triazole rings in compound 2 is 85.68°, which is not different from that in TCBT (84.53°). The results show that the introduction of two DFAZ rings has little effect on the molecular structure of BTR. In addition, the two DFAZ rings have a certain torsion angle, which relieves the ring strain of azetidine itself and helps stabilize the four-membered ring structure. Other relevant crystallographic parameters and diffraction data are shown in Table S1 in ESI.†
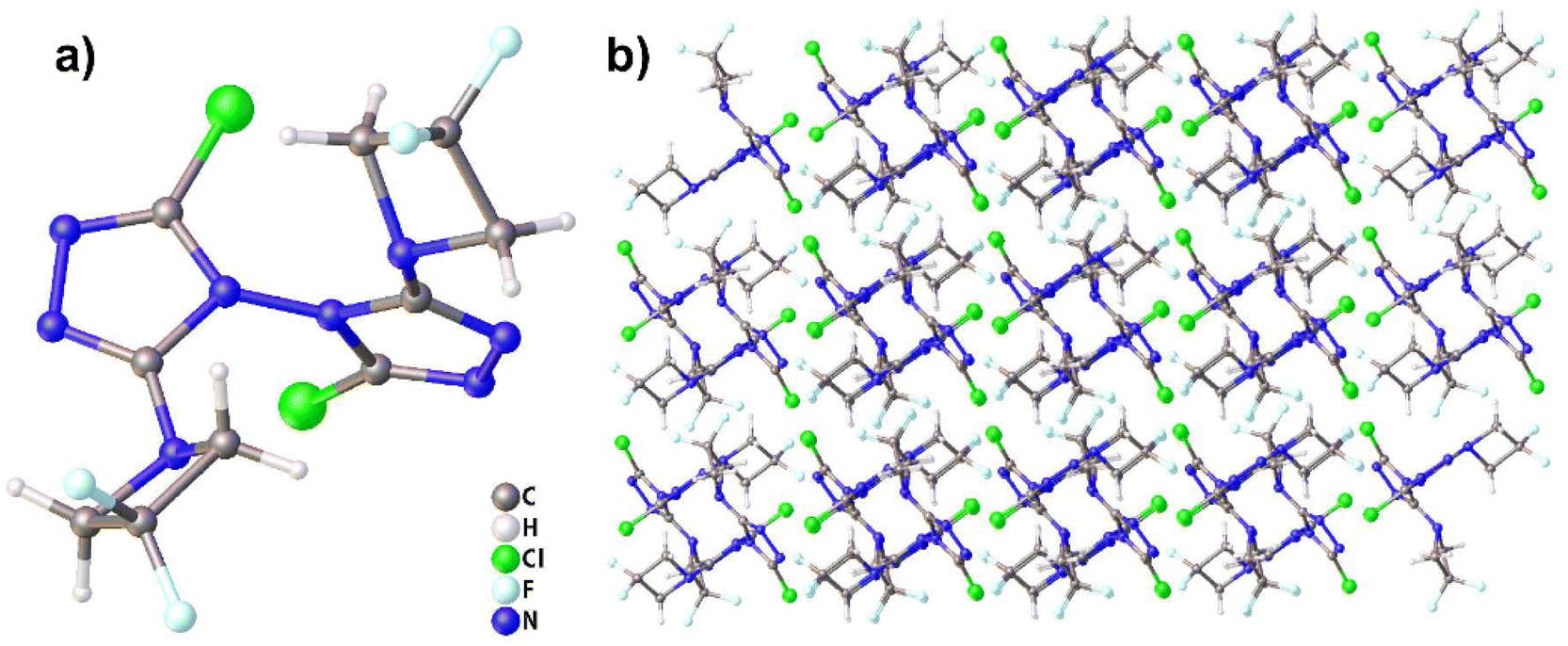 | ||
| Fig. 2 (a) Molecular structure of compound 2. (b) Packing diagram of compound 2. | ||
Compound 3 crystallizes in the triclinic space group P with two molecules per unit cell (Z = 2). The molecular structure is shown in Fig. 3(a). The measured crystal density was 1.601 g cm−3 at 296 K. The angle between the two triazole rings in compound 3 is 89.88°, which is almost a right angle. The DFAZ rings have a certain torsion angle, which is similar to that of compound 2, that is, the existence of a twist angle further stabilizes the strain of the four-membered rings and promotes its stable existence in the molecule. The detailed information is given in Table S1 in ESI.†
 | ||
| Fig. 3 (a) Molecular structure of compound 3. (b) Packing diagram of compound 3. | ||
4.2 Physicochemical properties
In order to preliminarily understand the thermal stability of the compounds in this paper, differential scanning calorimetry was used for analysis. The thermal behaviour of the DFAZ, compound 1, compound 2, and compound 3 was determined using the TA-DSC Q2000 differential scanning calorimeter (New Castle, DE, USA) at a heating rate of 5 °C min−1. In Fig. 4(a), DFAZ has an obvious exothermic peak at 147.05 °C, which indicates that decomposition occurred at this temperature. However, in (b), (c), and (d), the thermal decomposition temperatures are 210.1 °C, 267.2 °C, and 254.4 °C respectively, which are higher than 147.05 °C in (a). It is not difficult to find that azetidine in polycyclic 1,2,4-triazole has higher stability than DFAZ. In addition, the higher decomposition temperature also shows that the triazolyl ring skeleton increases the thermal stability of the DFAZ ring. The reason for this phenomenon may be the p–π conjugation effect between the lone pair electrons of the N atom in azetidine and the π electrons in the 1,2,4-triazole ring.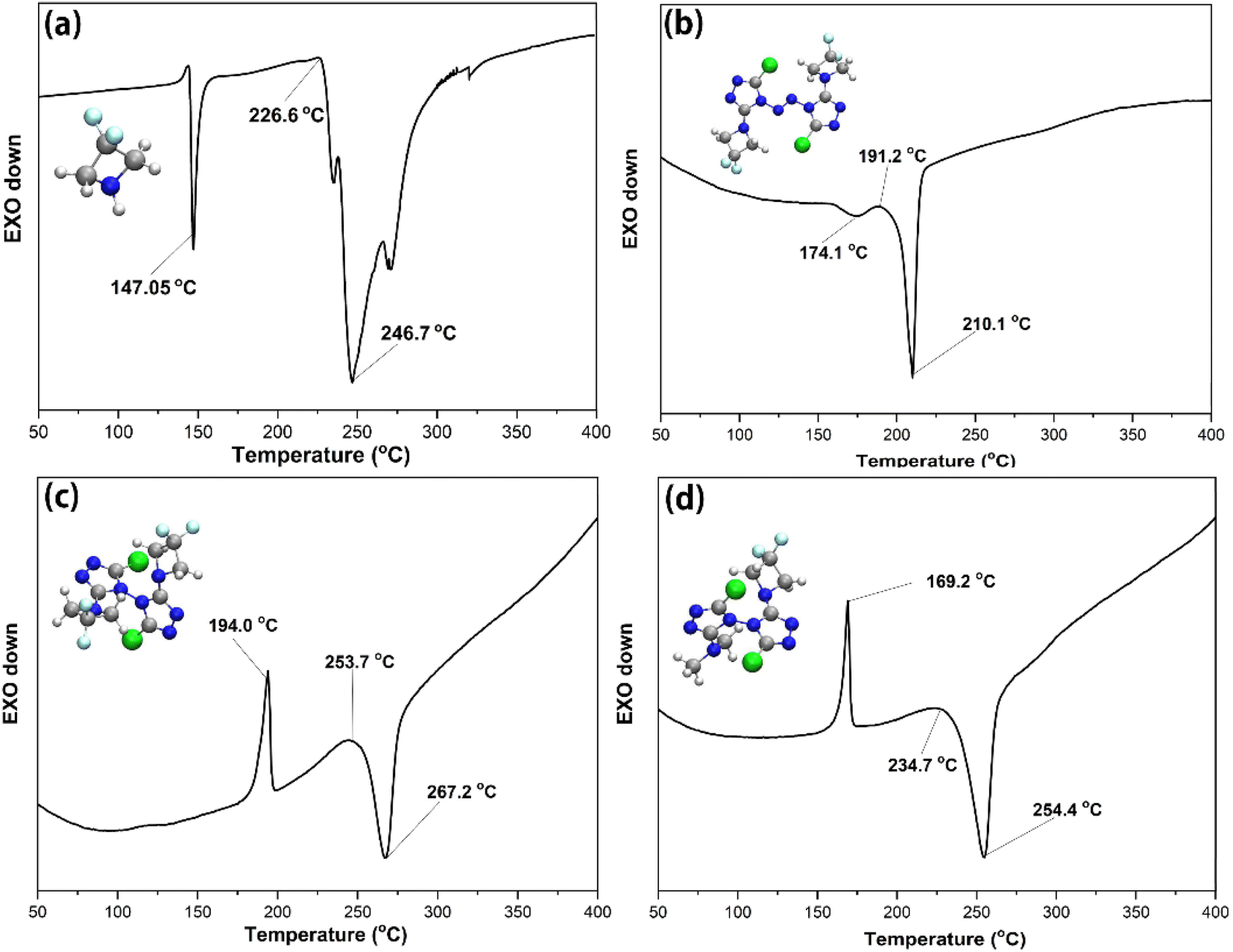 | ||
| Fig. 4 Differential scanning calorimetry curves of DFAZ (a), compound 1 (b), compound 2 (c), and compound 3 (d). The cyan, grey, blue, white, and green spheres correspond to the F, C, N, H, and Cl atoms, respectively. | ||
In order to further illustrate the stability mechanism of triazolyl polycyclic compounds, the p–π conjugation effect in the molecular structures was analyzed. The natural bond orbital (NBO) theory45 is a powerful tool to study the p–π conjugation effect. The conjugation effect from the p lone electron pair of the nitrogen atom in the azetidine group with the π–electron of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond in the 1,2,4-triazole ring is presented by the orbital overlap. As can be seen from Fig. 5, there is a conjugation effect between p–π in the two parts of the polycyclic triazole energetic compounds (azetidine and 1,2,4-triazole). The second-order perturbation theory analysis of the Fock matrix in the NBO confirms strong intramolecular conjugative interactions. The second-order perturbation energies (2nd-perturbation energy, E(2)) of compounds 1, 2, and 3 are 25.13 kcal mol−1 (for NBO91–NBO510 and NBO92–NBO493 in compound 1), 50.04 kcal mol−1 (for NBO85–NBO460 in compound 2), 38.61 kcal mol−1 (for NBO84–NBO470 in compound 2) and 42.20 kcal mol−1 (for NBO79–NBO430 in compound 3), respectively. These interactions are formed by the orbital overlap between two orbitals (p with π orbital), which results in the increase of the system's stabilization energy. The expanded conjugated region is helpful to stabilize the molecular structure. This intramolecular conjugative interaction dominates the contributions of all delocalizations for the stabilization energies, which further explains the experimental results of DSC.
N double bond in the 1,2,4-triazole ring is presented by the orbital overlap. As can be seen from Fig. 5, there is a conjugation effect between p–π in the two parts of the polycyclic triazole energetic compounds (azetidine and 1,2,4-triazole). The second-order perturbation theory analysis of the Fock matrix in the NBO confirms strong intramolecular conjugative interactions. The second-order perturbation energies (2nd-perturbation energy, E(2)) of compounds 1, 2, and 3 are 25.13 kcal mol−1 (for NBO91–NBO510 and NBO92–NBO493 in compound 1), 50.04 kcal mol−1 (for NBO85–NBO460 in compound 2), 38.61 kcal mol−1 (for NBO84–NBO470 in compound 2) and 42.20 kcal mol−1 (for NBO79–NBO430 in compound 3), respectively. These interactions are formed by the orbital overlap between two orbitals (p with π orbital), which results in the increase of the system's stabilization energy. The expanded conjugated region is helpful to stabilize the molecular structure. This intramolecular conjugative interaction dominates the contributions of all delocalizations for the stabilization energies, which further explains the experimental results of DSC.
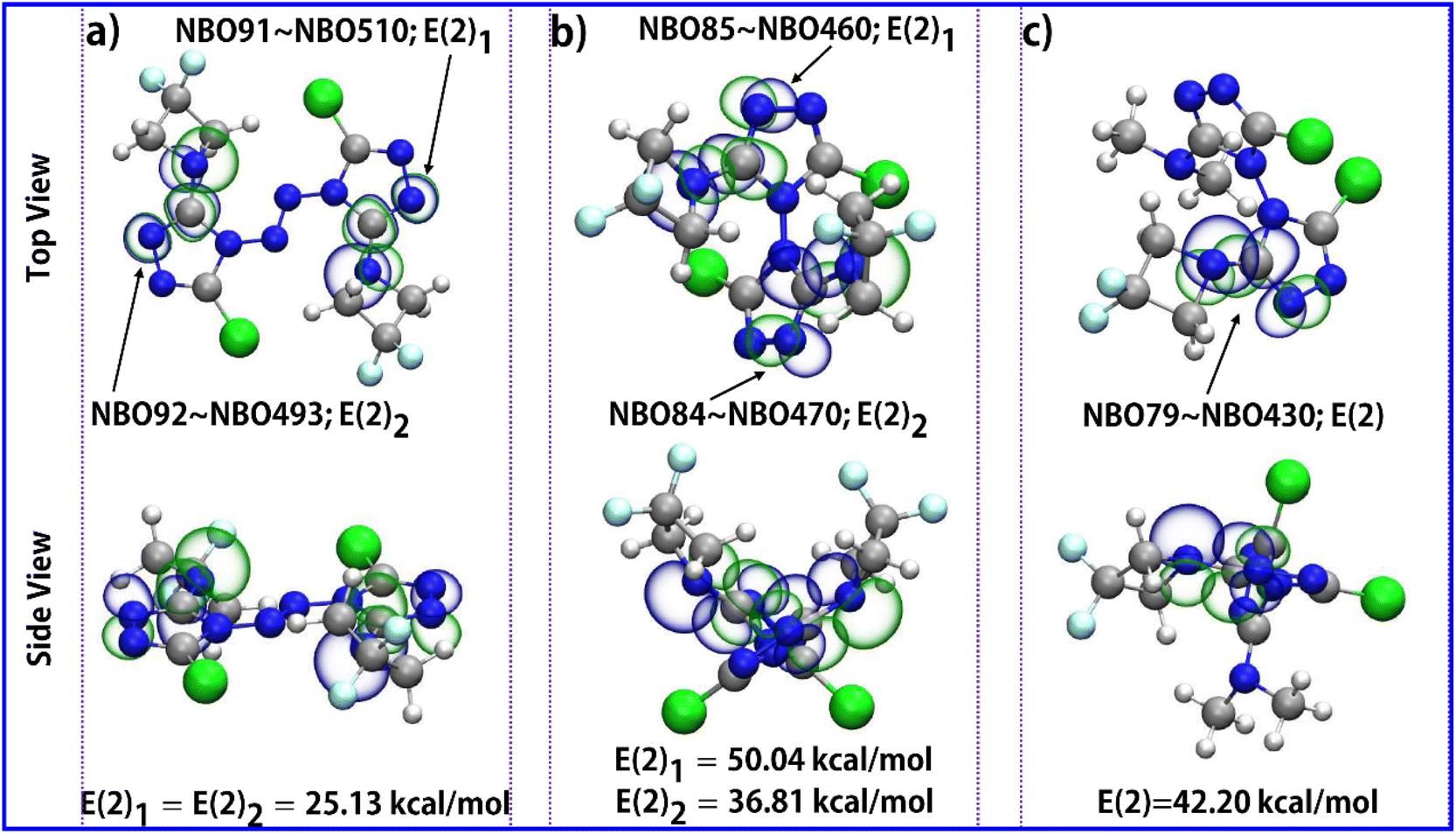 | ||
| Fig. 5 The p–π conjugative NBOs isosurface of compound 1 (a), compound 2 (b), and compound 3 (c) with isovalue of +0.05 and −0.05. The cyan, grey, blue, white, and green spheres correspond to F, C, N, H, and Cl atoms, respectively. The theoretical calculation level is M06-2X/6-311G (d, p). | ||
The molecular van der Waals surface electrostatic potential (ESP)46,47 is a very useful descriptor in understanding noncovalent interactions and chemical reactions. The negative (blue) regions are related to electrophilic reactivity and the positive (red) ones to nucleophilic reactivity. The surface local minima and maxima of ESP are represented as cyan and orange spheres (in kcal mol−1). As shown in Fig. 6, among compound 1 (a), compound 2 (b), and compound 3 (c), the surface minima of ESP basically appear near the nitrogen atoms, while the surface maximum prefers to be mainly distributed near the hydrogen atoms and carbon atoms. The global maxima of ESP on compound 1, compound 2 and compound 3 are 32.98, 32.50, and 29.69 kcal mol−1, respectively, which implies that the order of the reactive activities of these four compounds is 1 > 2 > 3 when attacked by nucleophiles.
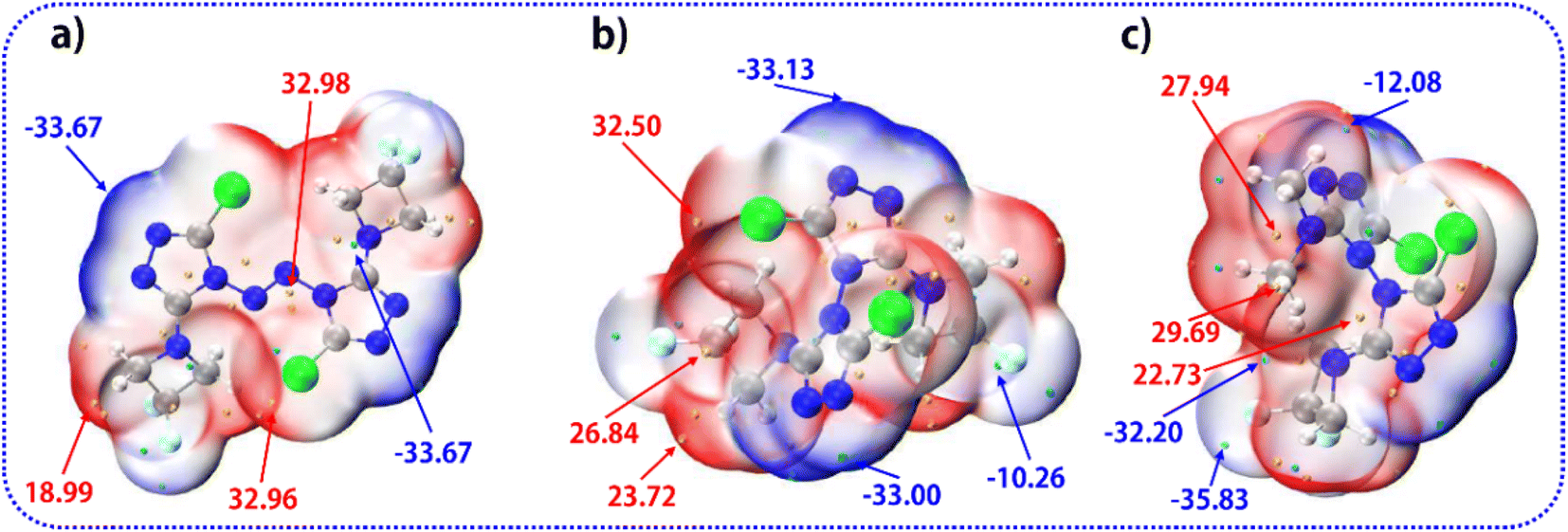 | ||
| Fig. 6 Isosurface maps of ESP of compound 1 (a), compound 2 (b), and compound 3 (c) with the isovalue of 0.001 a.u. (electrons per bohr3). The cyan, grey, blue, white, and green spheres correspond to F, C, N, H, and Cl atoms, respectively. The theoretical calculation level is M06-2X/6-311G (d, p). | ||
To better understand the interactions within crystals, the two-dimensional fingerprint plot and Hirshfeld surface plot analysis of TCBT, compound 1, compound 2, and compound 3 in the crystal structures were investigated and the data are shown in Fig. 7. The red dot region mainly appears near the hydrogen atom in the DFAZ group and the nitrogen atom in the 1,2,4-triazole ring in compounds 1 (b), 2 (c), and 3 (d), indicating that the intermolecular interaction mainly occurs between the atoms of the external molecule and these H atoms or nitrogen atoms. From the relative contribution of the contacts, the major interactions are N⋯Cl (22.3%), Cl⋯Cl (29.3%), Cl⋯N (26.6%) for TCBT (e), N⋯N (12.8%), F⋯H (12.8%) for compound 1 (f); and N⋯H (10.6%), Cl⋯H (10.1%) for compound 2 (g) and N⋯H (12.9%), Cl⋯H (13.9%), and H⋯H (14.6%) for compound 3 (h). To sum up, the close intermolecular contacts for hydrogen bonds in molecules are 47.3% (f), 59.8% (g), and 61.1% (h) for compound 1, compound 2, and compound 3, respectively. The remarkably abundant hydrogen bonds may give rise to the lowest sensitivity of compound 3 (61.1%) among them.48
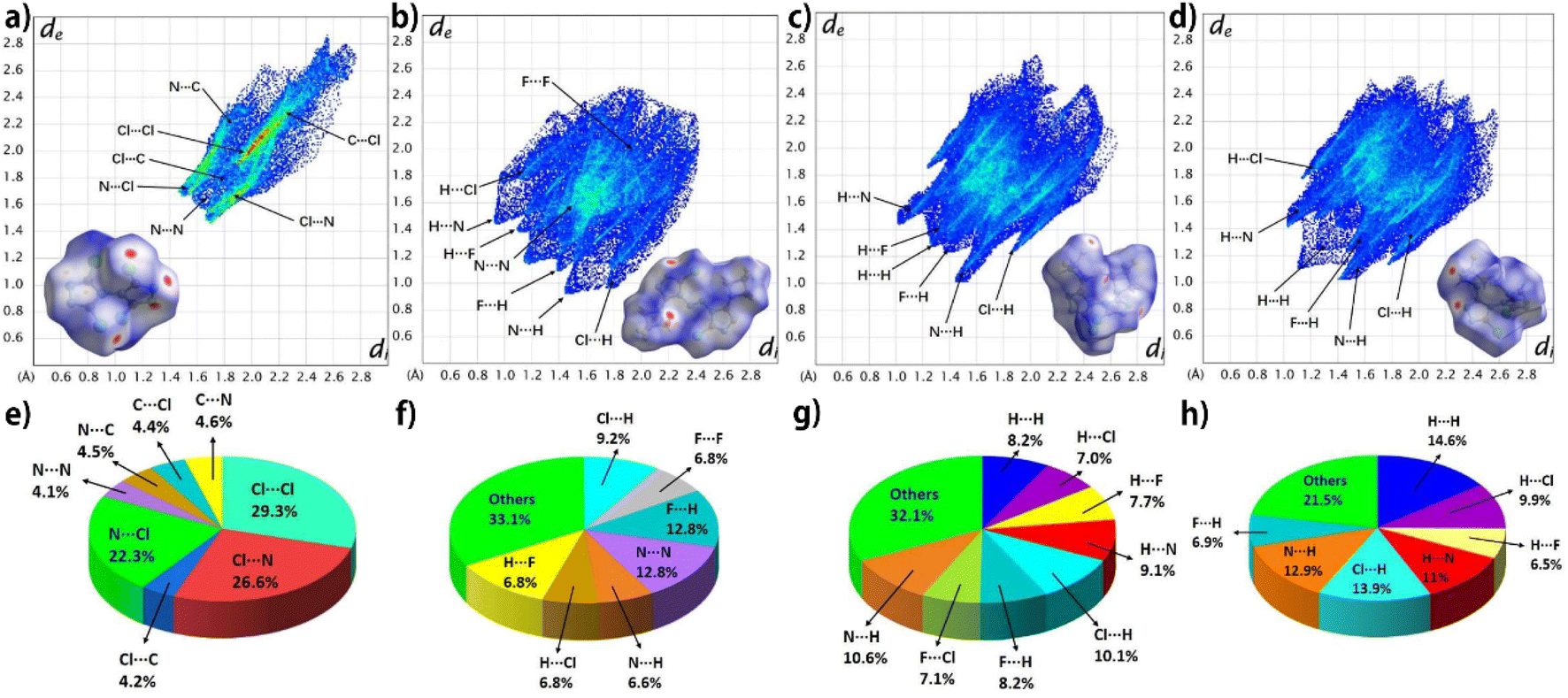 | ||
| Fig. 7 Fingerprint plots and Hirshfeld surfaces (inside) in crystal stacking for TCBT, compound 1, compound 2, and compound 3 (a–d), respectively; the pie graphs for TCBT, compound 1, compound 2, and compound 3 (e–h) show the percentage contributions of the individual atomic contacts to the Hirshfeld surface, respectively. | ||
4.3 Detonation and safety performance
The detonation performance of triazolyl polycyclic compounds was calculated, as described in this section. The impact and friction sensitivity measurements were performed using a BAM fall hammer apparatus (BFH-10) and a BAM friction apparatus (FSKM-10) (San Diego, CA, USA). Densities were measured at 25 °C using a Micromeritics Accupyc II 1340 gas pycnometer (Norcross, GA, USA) for 1, 2, 3, and were calculated for others.49 The gas-phase formation enthalpy of all compounds was calculated by quantum chemistry. The solid-phase formation enthalpy was obtained by subtracting the enthalpy sublimation from the gas-phase formation enthalpy.50 The enthalpy of sublimation was predicted from the method in the literature.51Various physical quantities of related compounds are listed in Table 1. The results showed that: (1) the higher densities of all triazolyl polycyclic compounds than those of bi-1,2,4-triazole, azobis-1,2,4-triazole, DFAZ and DNAZ·HCl shows that the introduction of azetidine derivative group strategy can effectively improve the molecular density with a simple way than positional isomerism;52 (2) when nitro and azido groups were introduced into the molecule, the enthalpy of formation of these triazolyl polycyclic compounds increased significantly (1039.2 kJ mol−1 for 4, 1104.6 kJ mol−1 for 6); (3) because of the four-membered ring distorted spatial structure of azetidine and the larger space volume of chlorine atoms are not conducive to the close packing of molecules, the detonation performance of chlorinated intermediates (1–3) are not good as a whole; (4) the comprehensive detonation performance of 4, 5, 6 are better than that of TNT; (5) chlorinated intermediates (compound 1 to compound 5) containing difluoroazetidine substituents maybe provide new ideas for the development of new fluorine-containing oxidants.53 However, the conclusion that the azetidine structure increases the density of the compounds is questioned among compounds 1–5 for chlorine atoms. Therefore, we theoretically designed four compounds, calculated their densities, and compared them with those of azobis-1,2,4-triazole or bi-1,2,4-triazole (see in ESI†). The results show that the azetidine structure can indeed increase the density of the system. In brief, the introduction of azetidine structure into molecules is an effective strategy for the design of polycyclic energetic compounds.
| Compound | ρa | Db | Pc | ISd | FSe | ΔHff | Tdg |
|---|---|---|---|---|---|---|---|
| a Measured densities-gas pycnometer at room temperature (g cm−3) for 1, 2, 3.b Detonation velocity (m s−1).c Detonation pressure (GPa).d Impact sensitivity (J).e Friction sensitivity (N).f Enthalpy of formation (kJ mol−1).g Thermal decomposition temperature (onset) under nitrogen gas (DSC, 5 °C min−1, (°C)). All the detonation performance of triazolyl polycyclic compounds were predicted by empirical Kamlet–Jacobs56 in EXPLO5 v6.05.57 | |||||||
| 1 | 1.813 | 6652 | 16.8 | 14 | 230 | 370.3 | 210.1 |
| 2 | 1.726 | 6307 | 15.3 | 17 | 270 | 75.6 | 267.2 |
| 3 | 1.601 | 6285 | 14.6 | 16 | 280 | 361.9 | 254.4 |
| 4 | 1.825 | 7932 | 26.9 | 13 | 220 | 1039.2 | 196.2 |
| 5 | 1.716 | 7054 | 19.4 | 14 | 230 | 837.9 | 215.5 |
| 6 | 1.750 | 7727 | 22.4 | 12 | 220 | 1104.6 | 230.4 |
| Azobis-1,2,4-triazole | 1.58 | 7833 | 21.5 | 14 | 240 | 896.8 | 315.8 |
| Bi-1,2,4-triazole | 1.57 | 7379 | 18.4 | >40 | >360 | 634.0 | 268.0 |
| DFAZ28 | 1.52 | 5931 | 11.9 | — | — | −321.0 | 147.1 |
| DNAZ·HCl54 | 1.63 | 6881 | 20.9 | — | — | 145.3 | 194.7 |
| TNT55 | 1.65 | 6809 | 18.7 | 15 | 353 | −67.0 | 295 |
4.4 Application potential
Compared with TNT, compounds 4, 5, and 6 have higher density and formation enthalpy and have the potential to replace TNT in mixed explosives. In addition, more oxidizing groups (fluorine atoms and nitro groups) can further improve energy performance.Compounds 4, 5, and 6 can replace TNT in classical B Explosive (40% TNT and 60% RDX, F0 in Table 2) to obtain formulations F1–F3. These formulations have detonation velocities exceeding that of F0. These results show that the prepared triazolyl polycyclic compounds have good application potential.
5 X-ray crystallography
Data collection for 1, 2, and 3 were performed using the Mercury CCD and the Saturn 724 CCD diffractometer with graphite monochromatic MoKα radiation. The raw data were refined using SHELXL-97 and SHELXS-97 program.58 CCDC-1589971, 1589973, and 1589972† contain the details of the structures of compounds 1, 2, and 3.6 Conclusions
In this work, a strategy method for designing new triazolyl polycyclic energetic materials was established by combining the structure of azetidine with bi-1,2,4-triazole or azobis-1,2,4-triazole. This strategy can effectively improve the density and decomposition temperature (>200 °C) of the system. The strong p–π conjugation effect and abundant intra-crystal interactions explain the stability of these compounds. X-ray single crystal diffraction analysis intuitively showed that the introduction of azetidine structure does not affect the molecular skeleton configuration of bi-1,2,4-triazole or azobis-1,2,4-triazole. The calculation of the energetic formula design shows that some triazolyl polycyclic compounds designed in this work are potential energetic materials. Furthermore, chlorinated intermediate compounds encourage us to further develop research on triazolyl polycyclic derivatives with various energetic groups replacing chlorine atoms in future work. We hope that this well-proven design strategy will help the researchers develop other novel polycyclic energetic materials.Author contributions
Xin-bo Yang: investigation, data curation, visualization, theoretical calculation, editing, writing-original draft. Chen-hui Jia: data curation, experimental synthesis. Xiang-yan Miao: editing. Yu-chuan Li and Si-ping Pang: writing-review & editing, funding acquisition, methodology, conceptualization. The manuscript was written through the contributions of all authors.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this study.Acknowledgements
The authors would like to acknowledge the National Natural Science Foundation of China (22135003 and 21975023) for providing funds for conducting experiments.References
- P. W. Cooper, Explosives Engineering, Wiley-VCH, New York, 1996 Search PubMed.
- J. Y. Huang, A. Li and J. R. Li, An efficient approach for the synthesis of oligosaccharides using ionic liquid supported glycosylation, Carbohydr. Polym., 2011, 83(1), 297–302 CrossRef.
- C. Qi, S. H. Li, Y. C. Li, Y. Wang, X. K. Chen and S. P. Pang, A novel stable high-nitrogen energetic material: 4,4'-azobis(1,2,4-triazole), J. Mater. Chem., 2011, 21(9), 3221–3225 RSC.
- R. R. Gupta and M. V. Gupta, Heterocyclic Chemistry: Volume II: Five-Membered Heterocycles, Springer Science & Business Media, 2013 Search PubMed.
- J. G. Haasnoot and W. L. Groeneveld, Complexes of 1,2,4-triazoles, VII [1]. Preparation and vibrational spectra of 4,4’-bi-1,2,4-triazole and some of its complexes with transition metal(II) thiocyanates, Z. Naturforsch., B: J. Chem. Sci., 1979, 34(11), 1500–1506 CrossRef.
- T. G. Archibald, R. Gilardi, K. Baum and C. George, Synthesis and X-ray crystal structure of 1,3,3-trinitroazetidine, J. Org. Chem., 1990, 55(9), 2920–2924 CrossRef.
- Z. Jalový, S. Zeman, M. Sućeska, P. Vávra, K. Dudek and M. Rajić, 1,3,3-trinitroazetidine (TNAZ). Part I. Syntheses and properties, J. Energ. Mater., 2001, 19(2–3), 219–239 CrossRef.
- M. A. Hiskey, M. D. Coburn, M. A. Mitchell and B. C. Benicewicz, Synthesis of 3, 3-dinitroazetidine from 1-t-butyl-3, 3-dinitroazetidine, J. Heterocycl. Chem., 1992, 29(7), 1855–1856 CrossRef.
- M. A. Hiskey, Synthesis of 3, 3-dinitroazetidine, US Pat., US5395945 A, 1995 Search PubMed.
- M. A. Hiskey and M. D. Coburn, Synthesis of 1, 3, 3-trinitroazetidine, Los Alamos National Lab.(LANL), Los Alamos, NM, United States, 1994 Search PubMed.
- K. G. Herring, L. E. Toombs, G. F. Wright and W. J. Chute, Catalyzed nitration of amines: I. dinitroxydiethylnitramine, Can. J. Res., 1948, 26b(1), 89–103 CrossRef.
- M. A. Hiskey, M. M. Stinecipher and J. E. Brown, Synthesis and initial characterization of some energetic salts of 3, 3-dinitroazetidine, J. Energ. Mater., 1993, 11(3), 157–165 CrossRef CAS.
- R. Gao, H. X. Ma, B. Yan, J. R. Song and Y. H. Wang, Structure-property investigation on TDNAZ · HNO3 and DNAZ · HCl, Chem. J. Chin. Univ., 2009, 30(3), 577–582 CAS.
- L. R. Fedor, T. C. Bruice, K. L. Kirk and J. Meinwald, Aminolysis of Phenyl Acetates in Aqueous Solutions. V.1 Hypernucleophilicity Associated with Constraint of Bond Angles, J. Am. Chem. Soc., 1966, 88(1), 108–111 CrossRef CAS.
- L. W. Deady, G. J. Leary, R. D. Topsom and J. Vaughan, N-Arylazetidines, J. Org. Chem., 1963, 28(2), 511–514 CrossRef CAS.
- M. A. Hiskey, M. C. Johnson and D. E. Chavez, Preparation of 1-substituted-3, 3-dinitroazetidines, J. Energ. Mater., 1999, 17(2–3), 233–252 CrossRef CAS.
- A. Fischer, R. E. J. Hutchinson, R. D. Topsom and G. J. Wright, Rates of reaction of cyclic imines with p-fluoronitrobenzene, J. Chem. Soc. B, 1969, 544 RSC.
- B. Yan, H. X. Ma, Y. Hu, Y. L. Guan and J. R. Song, 1-(2,4-Dinitro-phen-yl)-3,3-dinitro-azetidine, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65(Pt 12), o3215 CrossRef CAS PubMed.
- H. Li, Y. J. Shu, Y. Huang, S. J. Liu and Q. Jiang, Studt on the synthesis of 3,3-dinitroazetidine and 1,1′-bis-(3,3-dinitroazetidin-1-yl)methane, Chin. J. Org. Chem., 2004, 24(7), 775–777 CAS.
- P. Yin, Q. H. Zhang and J. M. Shreeve, Dancing with energetic nitrogen atoms: versatile N-functionalization strategies for N-heterocyclic frameworks in high energy density materials, Acc. Chem. Res., 2016, 49(1), 4–16 CrossRef CAS.
- Y. Qu and S. P. Babailov, Azo-linked high-nitrogen energetic materials, J. Mater. Chem. A, 2018, 6(5), 1915–1940 RSC.
- X. Li, Q. Sun, Q. Lin and M. Lu, [N-N=N-N]-linked fused triazoles with π-π stacking and hydrogen bonds: Towards thermally stable, Insensitive, and highly energetic materials, Chem. Eng. J., 2021, 406, 126817–126823 CrossRef CAS.
- P. Yin, C. He and J. M. Shreeve, Fused heterocycle-based energetic salts: alliance of pyrazole and 1,2,3-triazole, J. Mater. Chem. A, 2016, 4(4), 1514–1519 RSC.
- H. Gao, Q. Zhang and J. M. Shreeve, Fused heterocycle-based energetic materials (2012–2019), J. Mater. Chem. A, 2020, 8(8), 4193–4216 RSC.
- Y. C. Li, C. Qi, S. H. Li, H. J. Zhang, C. H. Sun, Y. Z. Yu and S. P. Pang, 1,1'-Azobis-1,2,3-triazole: a high-nitrogen compound with stable N8 structure and photochromism, J. Am. Chem. Soc., 2010, 132(35), 12172–12173 CrossRef CAS PubMed.
- C. Qi, S. H. Li, Y. C. Li, Y. Wang, X. X. Zhao and S. P. Pang, Synthesis and promising properties of a new family of high-nitrogen compounds: polyazido- and polyamino-substituted N,N'-azo-1,2,4-triazoles, Chem.–Eur. J., 2012, 18(51), 16562–16570 CrossRef CAS PubMed.
- S. H. Li, S. P. Pang, X. T. Li, Y. Z. Yu and X. Q. Zhao, Synthesis of new tetrazene(N–N=N–N)-linked bi(1,2,4-triazole), Chin. Chem. Lett., 2007, 18(10), 1176–1178 CrossRef.
- J. Chen, Y. Yu, S. Zhang, Y. Li and S. Pang, Energetic materials with fluorinated four-membered heterocyclic ring:3,3'-difluoroazetidine (DFAZ) salts, New J. Chem., 2019, 43(38), 15115–15119 RSC.
- A. E. Frumkin, A. M. Churakov, Y. A. Strelenko, V. V. Kachala and V. A. Tartakovsky, Synthesis of 1,2,3,4-tetrazino[5,6-f]benzo-1,2,3,4-tetrazine 1,3,7,9-tetra-N-oxides, Org. Lett., 1999, 1(5), 721–724 CrossRef PubMed.
- Y. Li, P. Chen, Y. Liu, P. Yin, C. He and S. Pang, Synthesis and characterization of fluorodinitrobenzenes with tunable melting point: potential low sensitive energetic plasticizer and melt-cast carrier, Chin. J. Chem., 2020, 38(12), 1619–1624 CrossRef.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2007, 120(1–3), 215–241 Search PubMed.
- M. W. Wong, P. M. W. Gill, R. H. Nobes and L. Radom, 6-311G(MC)(d,p): a second-row analogue of the 6-311G(d,p) basis set: calculated heats of formation for second-row hydrides, J. Phys. Chem., 2002, 92(17), 4875–4880 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peratta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09; Revision; D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graph., 1996, 14(1), 33–38 CrossRef PubMed.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2(1), 73–78 Search PubMed.
- O. Vahtras, J. Almlöf and M. W. Feyereisen, Integral approximations for LCAO-SCF calculations, Chem. Phys. Lett., 1993, 213(5–6), 514–518 CrossRef.
- A. Hellweg, C. Hättig, S. Höfener and W. Klopper, Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn, Theor. Chem. Acc., 2007, 117(4), 587–597 Search PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- M. Bursch, A. Hansen, P. Pracht, J. T. Kohn and S. Grimme, Theoretical study on conformational energies of transition metal complexes, Phys. Chem. Chem. Phys., 2021, 23(1), 287–299 RSC.
- T. Lu and Q. Chen, Shermo: A general code for calculating molecular thermochemistry properties, Comput. Theor. Chem., 2021, 1200, 113249 CrossRef.
- M. A. Spackman and D. Jayatilaka, Hirshfeld surface analysis, CrystEngComm, 2009, 11(1), 19–32 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef PubMed.
- B. M. Abraham, V. D. Ghule and G. Vaitheeswaran, A comparative study of the structure, stability and energetic performance of 5,5'-bitetrazole-1,1'-diolate based energetic ionic salts: future high energy density materials, Phys. Chem. Chem. Phys., 2018, 20(47), 29693–29707 RSC.
- E. D. Glendening, C. R. Landis and F. Weinhold, Natural bond orbital methods, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2(1), 1–42 CAS.
- T. Lu and F. Chen, Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS PubMed.
- T. Lu and S. Manzetti, Wavefunction and reactivity study of benzo[a]pyrene diol epoxide and its enantiomeric forms, Struct. Chem., 2014, 25(5), 1521–1533 CrossRef CAS.
- B. M. Abraham and G. Vaitheeswaran, From van der Waals interactions to structures and properties of 3,3′-dinitro-5,5′-bis-1,2,4-triazole-1,1′-diolate based energetic materials, Mater. Chem. Phys., 2020, 240, 122175 CrossRef CAS.
- B. M. Rice and E. F. Byrd, Evaluation of electrostatic descriptors for predicting crystalline density, J. Comput. Chem., 2013, 34(25), 2146–2151 CAS.
- P. Atkins and J. d. Paula, Physical chemistry thermodynamics, structure, and change, WH Freeman and Company, New York, 2014 Search PubMed.
- E. F. Byrd and B. M. Rice, Improved prediction of heats of formation of energetic materials using quantum mechanical calculations, J. Phys. Chem. A, 2006, 110(3), 1005–1013 CrossRef CAS PubMed.
- Q. Sun, N. Ding, C. Zhao, J. Ji, S. Li and S. Pang, Positional isomerism for strengthening intermolecular interactions: Toward monocyclic nitramino oxadiazoles with enhanced densities and energies, Chem. Eng. J., 2022, 427, 130912 CrossRef CAS.
- S. K. Valluri, M. Schoenitz and E. Dreizin, Fluorine-containing oxidizers for metal fuels in energetic formulations, Def. Technol., 2019, 15(1), 1–22 CrossRef.
- M. Coburn, M. Hiskey, T. Archibald, An Alternative Synthesis of 1, 3, 3-Trinitroazetidine, Los Alamos National Laboratory Report LA-CP-95-145, USA, Los Alamos, 1995 Search PubMed.
- W. Cao, W. Dong, Z. Lu, Y. Bi, Y. Hu, T. Wang, C. Zhang, Z. Li, Q. Yu and J. Zhang, Construction of Coplanar Bicyclic Backbones for 1,2,4-Triazole-1,2,4-Oxadiazole-Derived Energetic Materials, Chemistry, 2021, 27(55), 13807–13818 CrossRef CAS.
- M. J. Kamlet and S. J. Jacobs, Chemistry of detonations. I. A simple method for calculating detonation properties of C-H-N-O explosives, J. Chem. Phys., 1968, 48(1), 23–35 CrossRef CAS.
- M. Sućeska, EXPLO5, version 6.05.02, Brodarski Institute, Zagreb, Croatia, 2018 Search PubMed.
- M. Sheldrick, SHELXS-97, Program for crystal structure solution, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1589971–1589973. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra06646g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |