
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile preparation of covalently functionalized graphene with 2,4-dinitrophenylhydrazine and investigation of its characteristics
S. Alipour *,
M. Hassani,
S. M. H. Hosseini and
S. M. Mousavi-Khoshdel
*,
M. Hassani,
S. M. H. Hosseini and
S. M. Mousavi-Khoshdel
Department of Chemistry, Iran University of Science and Technology (IUST), Narmak, Tehran, Iran. E-mail: sae.alipour@gmail.com; Fax: +982177240480; Tel: +982177240480
First published on 22nd December 2022
Abstract
This article reports a fast and easy method for simultaneously in situ reducing and functionalizing graphene oxide. 2,4-Dinitrophenylhydrazine hydrate salt molecules are reduced by graphene oxide by reacting with oxide groups on the surface and removing these groups, and 2,4-dinitrophenylhydrazone groups are replaced with oxide groups. The synthesized materials have been investigated using Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy, X-ray diffraction (XRD), thermogravimetric analysis (TGA), X-ray photoelectron spectroscopy (XPS), and UV absorption. Also, the morphology has been examined with a scanning electron microscope (SEM) and Brunauer–Emmett–Teller (BET) analysis. The result of the photocurrent response and electrochemical behavior of the samples through cyclic voltammetry, galvanostatic charge/discharge, and electrochemical impedance spectroscopy (EIS) have been analyzed to investigate the effect of physical and chemical changes compared to graphene.
1. Introduction
Graphene is a two-dimensional monolayer of carbon atoms with a hexagonal honeycomb-like structure in which the carbon bonds have sp2 hybridization, and the out-of-plane π bond creates a network of decentralized electrons that is responsible for electron conduction and also causes a weak interaction between graphene layers or between graphene and a substrate. Due to its unique structural features, graphene has shown exceptional physical properties, which has attracted the attention of many researchers in various sciences.1–4Various methods have been used to synthesize graphene and its derivatives. Due to weak van der Waals forces among the layers in graphite, pristine graphene can be obtained from the mechanical exfoliation of graphite.5,6 The growth of graphene sheets using bottom-up techniques such as chemical vapor deposition (CVD) has been used to grow single-layer and multi-layer graphene sheets on metal surfaces.7–9 However, these methods are not suitable for large-scale production. Therefore, oxidation and exfoliation of graphite oxide, followed by a reduction process, have been widely used and considered to prepare reduced graphene oxide (R-GO).10–13 However, the precursor of R-GO, graphite needs to be exfoliated through the reaction with a mixture of oxidants to create graphene oxide (GO) sheets.14 Therefore, GO sheets are strongly oxidized and characterized by oxygen-containing groups.15,16 To remove these groups and prepare R-GO, various reducing agents; such as hydrazine,17,18 strong alkaline environments,19 vitamin C,20,21 and bovine serum albumin (BSA);22,23 and various methods; such as electrochemical,24,25 photochemical,26,27 and thermal reduction28,29 procedures; have been used.
Reduced graphene oxide has a wide range of applications, including its use in energy storage devices,30,31 electronic devices,32 biomedical applications,33 sensors,34 membranes,35 catalysts,36,37 water purification,30 etc. However, the presence of these functional groups in GO and R-GO compared to pristine graphene allows the ability to adjust the electrical and optical properties through chemical reactions. However, unfortunately, due to the incomplete reduction and the presence of multiple defects, which disrupt the sp2 lattice, R-GO exhibits lower electrical conductivity than pristine graphene.38–40 In addition, the solubility of R-GO in water is minimal, so the direct dispersion of graphene sheets in water is impossible without the help of dispersing agents.41 To improve these properties, various functional groups have been attached to the graphene structure by chemical modification,42,43 covalent,44–48 or non-covalent49–52 functionalization.
The functional groups covalently attached to the carbon atoms on the graphene convert the triangular sp2 orbital to a tetragonal sp3 orbital. Such transformations drastically change the electronic properties of pristine graphene.53–56 In like manner, by functionalizing graphene, the specific capacity increases by increasing the quantum capacity of these sheets due to the formation of new levels in the vicinity of the Fermi level.57,58 Also, due to the creation of steric repulsion caused by the presence of functional groups, the graphene sheets' recombination rate decreases, which leads to an increase in the available surface in functionalized graphene.59,60 Recent experimental studies have shown that the covalent functionalization of graphene using a nitrophenyl group directly affects the magnetic properties of the graphene surface.61–64 During the functionalization process, converting the carbon centers from sp2 to sp3 saturates the carbon atoms. It opens a barrier band gap for electrons, which enables the production of insulating and semiconducting regions in graphene wafers.65,66
This article presents a method for preparing graphene functionalized with nitrophenyl groups, in which graphene oxide is in situ and simultaneously reduced and functionalized with 2,4-dinitrophenylhydrazone groups. Also, this method was performed at a pH close to neutral and without dispersing agents, while for the synthesis of reduced graphene oxide (R-GO), to avoid re-adhesion of the layers and obtain the lowest number of layers as well as the lowest amount of defects, the pH is usually set at about 10, and a scattering agent is used.67 The structural and chemical differences of the material synthesized with graphene were studied. Also, the electrochemical behavior of this material with graphene was investigated to check the effectiveness of the changes made, due to the high sensitivity of electrochemical techniques to changes in the structure.
2. Materials and procedures
2.1. Materials
Graphite powder (<45 μm), sulfuric acid (H2SO4, 98%), potassium persulfate (K2S2O8, 99%), sodium nitrate (NaNO2), potassium permanganate (KMnO4, 98%), hydrochloric acid (HCl, 37%) and hydrogen peroxide (H2O2, 30 wt%) were used to synthesize GO. To prepare graphene, hydrazine was used to reduce graphene oxide. Likewise, 2,4-dinitrophenylhydrazine was used to reduce and functionalize graphene oxide. Sodium sulfate was used to make electrolytes. Graphite powder, hydrazine, and 2,4-dinitrophenyl hydrazine were bought from Merck, and the rest of the materials from Sigma-Aldrich.2.2. Preparing graphene oxide (GO)
The Hummers method was used to synthesize the graphene oxide. Concisely, K2S2O8 (5 g), P2O5 (5 g), and concentrated H2SO4 (50 mL) were mixed at 90 °C, and then the temperature was lowered to 80 °C. Then, graphite powder (10 g) was added to this mixture and stirred for 5 hours. After that, distilled water (1000 mL) was added to the mixture and after 24 hours, the suspension was filtered and dried at ambient temperature. Afterward, using an ice bath, concentrated H2SO4 (200 mL) was cooled to 0 °C and the obtained deposit was added. KMnO4 (20 g) was slowly added while the temperature was kept below 10 °C. The suspension was stirred for 2 hours at a temperature of 35 °C, and then distilled water (460 mL) was added. After 2 hours, distilled water (140 mL) and 30% H2O2 (25 mL) were added. Then the suspension was left at rest for 24 hours, and the clear liquid was drained. The remaining sediment was washed using a centrifuge with 10% HCl solution and distilled water.2.3. Reduction and functionalization of graphene oxide with DNPH (NP-GO)
200 mg of the graphene oxide obtained from the previous procedure was dispersed in distilled water (200 mL) using an ultrasonic bath model ELEMA-P30H for 1 h. The pH was adjusted to 8 by 1 M NaOH (using Istek pH meter model 240L). Then 2,4-dinitrophenylhydrazine salt (2 g) was added to the suspension and stirred for 2 hours at 80 °C. Afterward, the suspension was filtered using PTFE filter paper (0.22 μm), and the obtained product was washed with ethanol, acetone, as well as a mixture of acetonitrile/water and, finally, dried for 24 hours at 70 °C.2.4. Synthesize of graphene (R-GO)
200 mL of GO suspension (1 mg/1 mL) was prepared to prepare graphene, and 1 w% sodium dodecyl sulfate (SDS) as a dispersing agent was added. Subsequently, the suspension was put in the ultrasonic bath for 1 h. Then, the pH of the solution was adjusted to 10 via 1 M NaOH. Afterward, hydrazine hydrate 60% (2 mL) was added and the suspension was stirred for 2 h at 90 °C.3. Investigation of structure and properties
3.1. Characterization of the samples
FTIR (SHIMADZU company, model 8400S), Raman spectroscopy (TEKSAN company model Takram), and thermogravimetric analysis (Bahr device model STA 540) were used to investigate the effect of reducing and functionalizing procedures. XRD (Bruker D8 advance) at 40 kV using Cu Kα radiation (λ = 1.54 Å) was used to identify phases presented in the samples. UV absorption (spectral absorption measurements) was measured with Shimadzu model UV-160 A. X-ray photoelectron spectroscopy (XPS) was carried out via an ESCA2000 under the vacuum pressure of ∼10−10 torr with an X-ray source of Al Ka (1486.6 eV). The surface features of powders were studied according to the Brunauer–Emmett–Teller (BET) method (Micrometric company model ASAP 2020). The morphology of synthesized materials was also studied by SEM (Scanning electron microscopy, TESCAN model VEGA 2).3.2. Photoelectrochemical measurements
A homemade system was used to measure the photoelectrochemical behavior. As a source of radiation, a Xe lamp (500 W) with a cut-off filter (λ > 325 nm) was employed. The powder of the samples (20 mg) was dissolved in acetone and ultrasonicated for 30 minutes, then a grain of iodine was added to the suspension. The ultrasonication continued for another 30 minutes. Afterward, the prepared samples were electroplated (15 volts for 10 minutes) on the surface of ITO glass with an area of 1 cm2. The photocurrent response was measured in a 0.5 M Na2SO4 (sodium sulfate solution) in a three-electrode configuration system containing indium tin oxide (ITO) as the working electrode, platinum wire as the counter electrode, and saturated Ag/AgCl as the reference electrode. The interval between turning on and off the light was 4 seconds. Electrochemical impedance spectroscopy (EIS) was conducted at 100 kHz to 100 mHz in an open circuit potential (OCP).3.3. Electrochemical measurements
The electrochemical behavior of the samples was investigated by cyclic voltammetry and galvanostatic charge/discharge methods, KIMIA STAT device model 126. Electrochemical impedance spectroscopy (EIS) was studied by the Autolab device model PGSTAT204. A three-electrode system including prepared samples (working electrode), a platinum electrode (counter electrode), Ag/AgCl (reference electrode), and sodium sulfate solution (0.5 M) as the electrolyte was used.To make electrode materials for electrochemical analyses, prepared material, black acetylene and polytetrafluoroethylene (PTFE) were mixed with a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:5, and by adding ethanol and then 15 min ultrasonication, a grout was prepared with an appropriate concentration. The prepared material for the electrode was deposited on nickel foam with an area of 1 cm2 by the painting method. In the end, the prepared electrodes were dried for 24 h at 70 °C. EIS measurement was conducted in OCP at a frequency range between 100 kHz to 10 mHz. Capacities of the materials were obtained from the galvanostatic charge/discharge method by the following formula:
:15:5, and by adding ethanol and then 15 min ultrasonication, a grout was prepared with an appropriate concentration. The prepared material for the electrode was deposited on nickel foam with an area of 1 cm2 by the painting method. In the end, the prepared electrodes were dried for 24 h at 70 °C. EIS measurement was conducted in OCP at a frequency range between 100 kHz to 10 mHz. Capacities of the materials were obtained from the galvanostatic charge/discharge method by the following formula:
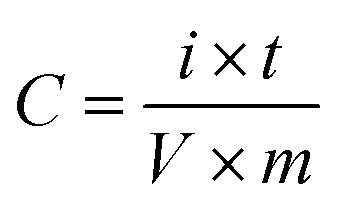
4. Result and discussions
4.1. Structural investigation
Fig. 1a illustrates the results of FTIR spectra corresponding to GO, R-GO, and NP-GO. The existence of oxygen-containing groups; C–O groups (at 1050 cm−1), ketone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (at 1620 cm−1), carboxyl CO (at 1720 cm−1), O–H and CO–OH groups, as well as water molecules in GO layers (at 3500 cm−1); can be verified in the spectrum of GO. In the graphene (R-GO) sample, carboxyl, ketone, ether, and O–H peaks disappeared, indicating that most of the oxygen groups were removed due to the reaction with hydrazine. In addition, 2850 to 2920 cm−1 peaks are associated with asymmetric C–H stretching. After reducing and functionalizing graphene oxide with 2,4-dinitrophenylhydrazine, peaks that appeared at 1345 and 1515 cm−1 are connected to the asymmetric and symmetric stretching vibration related to NO2 groups. The peak appearing at 1590 cm−1 assumes to be associated with the aromatic stretch. Likewise, the peak corresponding to stretching vibrations due to the presence of C–N nitrobenzene groups appears at 850 cm−1. The broad peak in the 3300–3500 cm−1 region seems to correspond to the N–H group of 2,4-dinitrophenylhydrazone.
O (at 1620 cm−1), carboxyl CO (at 1720 cm−1), O–H and CO–OH groups, as well as water molecules in GO layers (at 3500 cm−1); can be verified in the spectrum of GO. In the graphene (R-GO) sample, carboxyl, ketone, ether, and O–H peaks disappeared, indicating that most of the oxygen groups were removed due to the reaction with hydrazine. In addition, 2850 to 2920 cm−1 peaks are associated with asymmetric C–H stretching. After reducing and functionalizing graphene oxide with 2,4-dinitrophenylhydrazine, peaks that appeared at 1345 and 1515 cm−1 are connected to the asymmetric and symmetric stretching vibration related to NO2 groups. The peak appearing at 1590 cm−1 assumes to be associated with the aromatic stretch. Likewise, the peak corresponding to stretching vibrations due to the presence of C–N nitrobenzene groups appears at 850 cm−1. The broad peak in the 3300–3500 cm−1 region seems to correspond to the N–H group of 2,4-dinitrophenylhydrazone.
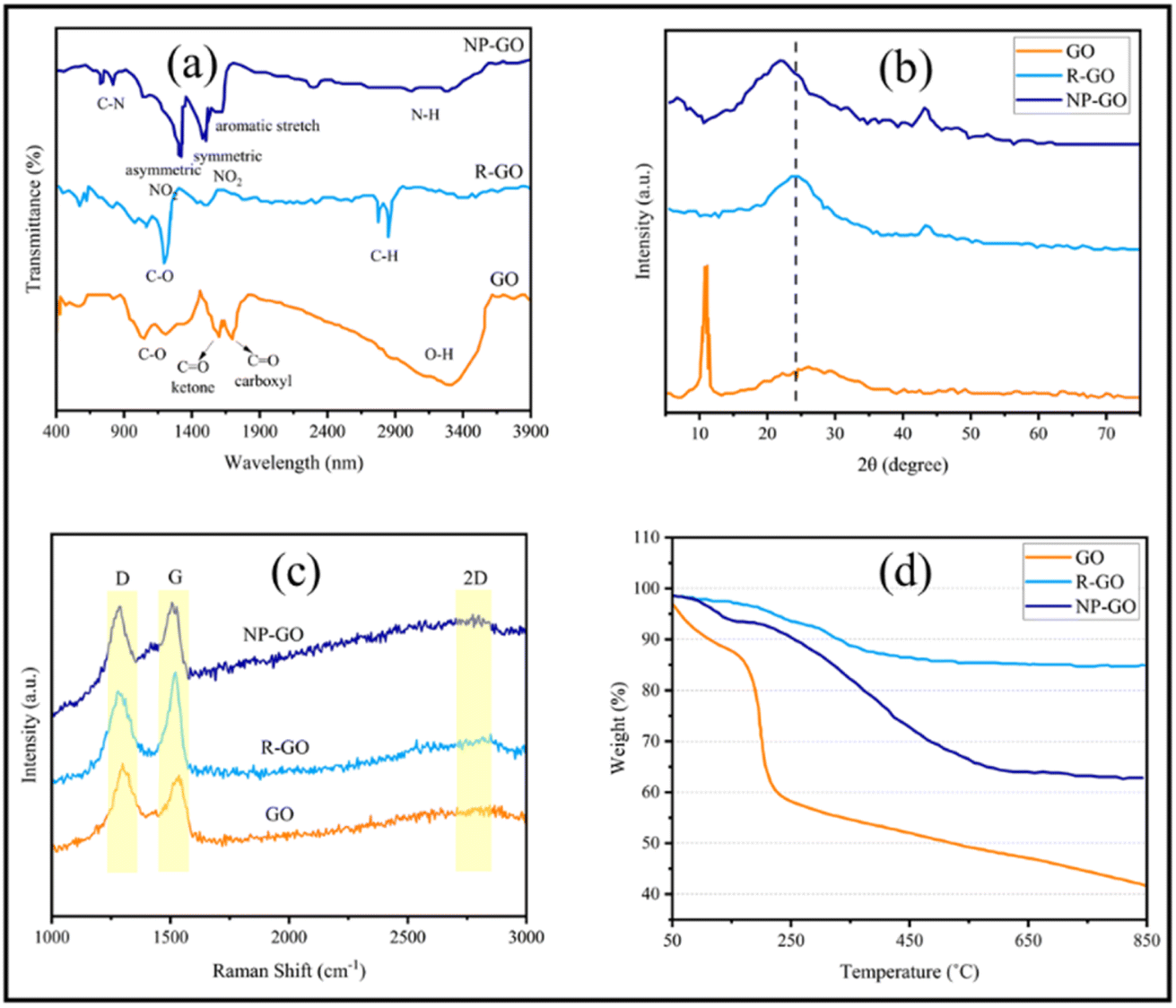 | ||
| Fig. 1 (a) FTIR, (b) XRD, (c) Raman spectra, and (d) TGA plots of GO, R-GO, and NP-GO. | ||
Fig. 1b displays the results of the X-ray diffraction (XRD) analysis of the samples. In GO, bonding with groups that contain oxygen increases the spacing between the layers of graphite. Therefore, the position of the peak indicates the oxidation level of graphite.68 Accordingly, the peak at 11.6° denotes the 7.57 Å d-spacing in graphene oxide. After GO reduction, R-GO sheets approach together and result in a peak at 23.8° (3.73 Å d-spacing) with low intensity. In NP-GO, the reaction of GO with DNPH leads to the removal of oxygen groups in GO and the elimination of the sharp, strong peak related to GO. Furthermore, it yields a broad peak at 22.7° (3.89 Å d-spacing). Based on the results of the FTIR analysis that show the functional groups' existence and by comparing the d-spacing value for R-GO and NP-GO, it seems that the difference in the distance of the layer is caused by the reduction and functionalizing of graphene oxide.
Fig. 1c shows the results of the Raman spectra of samples. The Raman device was equipped with a 532 nm laser and a thermoelectrically cooled CCD detector. A 60×-objective microscope lens was used to focus on the surface, and the exposure time was 5 s. As it is apparent, there are two bands. The first one relates to band D which signifies the graphene flakes' defect and irregularity, and the second corresponds to the G band that represents the first scattering of E2g phonon associated with carbon–carbon bonds with the hybridization of sp2. The 2D band is derived from the second-order double-resonant process between the K and K′ points in the BZ involving two iTO phonons. The 2D band is highly sensitive to the Fermi velocity and iTO phonon band slope due to the unique electronic band structure of graphene.69 The increased intensity of the D and 2D bands after functionalization indicates the attachment of functional groups.70 The level of reduction and defect is determined through the ratio between ID and IG which in GO, R-GO, and NP-GO are 0.89, 1.32, and 1.02, respectively. After the reduction of graphene oxide, the band D intensity increases due to the formation of defects in graphene sheets due to the reaction with hydrazine.71 It has been shown that the I2D/IG ratio of the Raman spectra can indicate the number of layers in graphene materials.72 This ratio for GO, R-GO, and NP-GO are 0.81, 0.75, and 0.94, respectively, which indicates that these materials are multilayered. Considering these increases in the ratio of I2D/IG and ID/IG after the reaction of GO with DNPH, it can be concluded that these changes are attributable to the reduction and replacing oxygen-containing groups in GO with 2,4-dinitrophenylhydrazone groups. In addition, the lower ratio of ID/IG compared to the graphene suggests that there are fewer irregularities and defects in the NP-GO structure.
Fig. 1d exhibits the thermogravimetric analysis (TGA) of the samples. During this process, the samples were heated to a temperature of 850 °C at a rate of 10 °C min−1 in an Ar atmosphere, and the weight changes of the samples were for the temperature. The initial weight loss of graphene oxide (GO) at 100 to 200 °C is associated with water and residual decomposition. The gradual weight loss from 200 to 650 °C is due to the decomposition of oxygen-containing groups on the surface and edge of GO sheets. Graphene (R-GO) shows a much lower reduction than GO, which may be related to removing a high percentage of oxidizing groups during reduction by hydrazine. In the sample of NP-GO, reducing 5% of the original weight under 200 °C indicates that there are no more than 5% unstable oxide groups in this sample. The lower weight loss of NP-GO compared to GO and R-GO can be a confirmation of reducing and functionalizing of graphene oxide with 2,4-dinitrophenylhydrazone.
Using XPS, the chemical composition of the samples was investigated more closely. Fig. 2a represents the XPS spectra of GO, R-GO, and NP-GO. In addition, the binding energy and percentage of compounds from C 1s, O 1s, and N 1s are listed in Table 1. The results showed carbon, oxygen, and nitrogen, which shows that the nitrophenyl groups are present on the surface of the NP-GO. In Fig. 2b it is obvious that the C 1s spectrum of graphene oxide consists of peaks at 284.6 eV (sp2 carbon), 286.6 eV (epoxide), and 288.5 eV (carboxyl groups).73 From the O 1s spectrum of GO (Fig. 2c), the oxygen-containing groups are evident as C–O, CO, C–O–C, O–H, and H2O groups. The C 1s spectrum of R-GO (Fig. 2d) shows a band at 284.6. The intensity of epoxide, carboxyl, or other bands is greatly reduced compared to graphene oxide, which means that the CC bonds relative percentage increased and it decreased for C–O bonds. The XPS spectrum of R-GO results shows that this sample only contains C and O, which designates that there are no impurities in the sample. The spectrum of C 1s related to NP-GO (Fig. 2e) shows the presence of the CC peak related to graphene, the CC bond corresponds to nitrophenyl groups (284.8 eV), the graphene C–C defect (at the site of bonding, 285 eV), and the peak related to C–NO2 (285.6 eV). The ratio of O/C in GO, R-GO, and NP-GO samples is 1.8, 0.04, and 0.2, respectively, showing that most oxygen-containing groups are removed and GO is reduced. The N 1s spectrum of the NP-GO (Fig. 2f) indicates that some nitro groups were reduced to other groups such as amines, and protonated amines due to the reduction by the irradiation of X-ray.74 The O 1s spectrum of NP-GO (Fig. 2g) shows three peaks at 531.3 eV (carboxyl groups), 532.7 (nitro groups), and 533.1 eV (epoxy groups) in the NP-GO sample. The nitro groups come from 2,4 dinitrophenylhydrazien, while the carboxyl and epoxy groups come from non-reduced groups in GO.
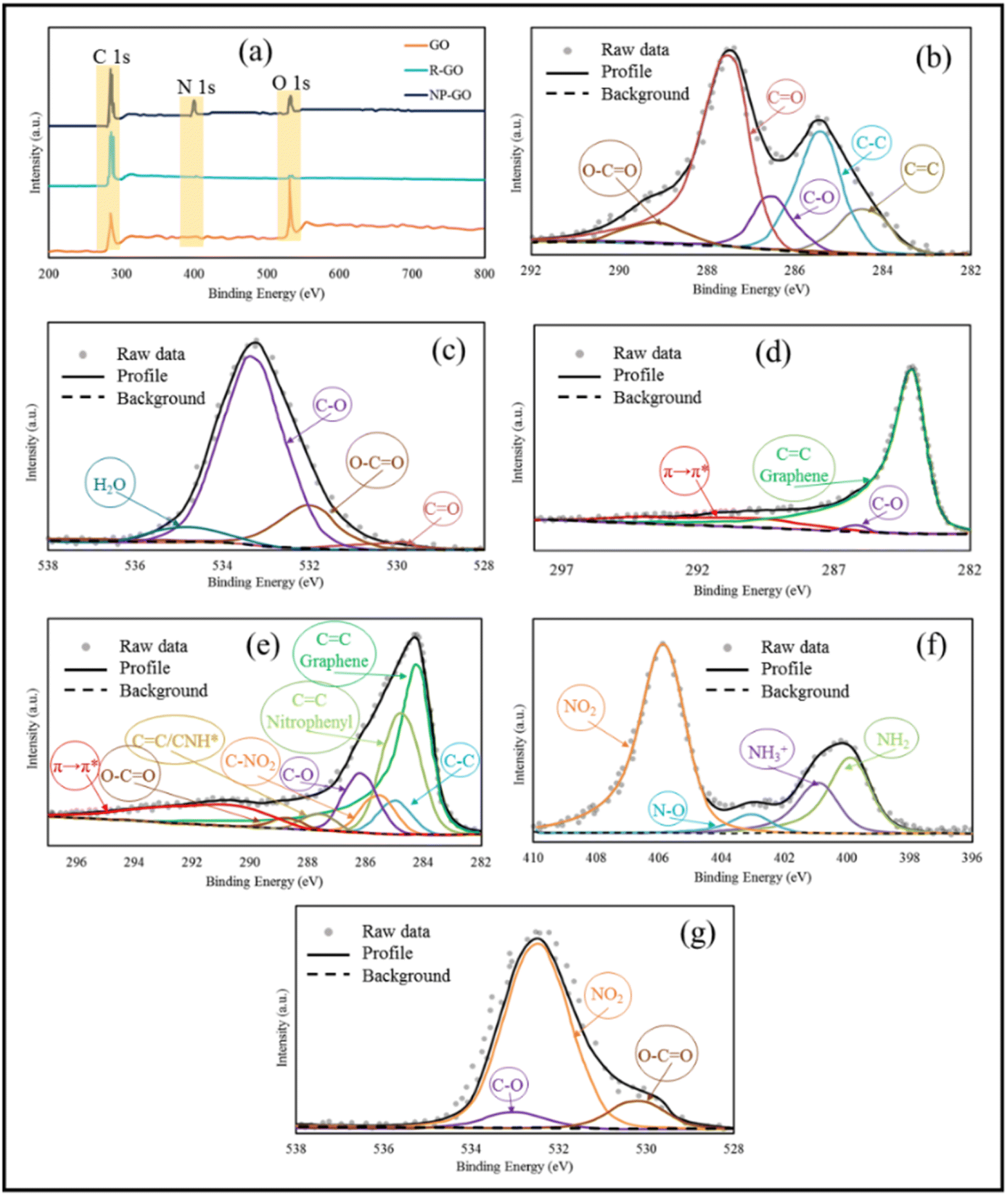 | ||
| Fig. 2 (a) XPS spectra of GO, R-GO, and NP-GO, (b) high-resolution C 1s spectra for GO, (c) high-resolution O 1s spectra for GO, (d) high-resolution C 1s spectra for R-GO, (e) high-resolution C 1s spectra for NP-GO, (f) high-resolution N 1s spectra for NP-GO, and (g) high-resolution O 1s spectra for NP-GO. | ||
| Sample | Peak | Assignment | Binding energy (eV) | Atomic (%) |
|---|---|---|---|---|
| GO | C 1s | CC |
284.5 | 11.41 |
| C–C | 285.4 | 24.58 | ||
| C–O | 286.6 | 9.64 | ||
| CO |
287.9 | 48.48 | ||
| COOH | 289.2 | 5.89 | ||
| O 1s | CO |
530.1 | 4.34 | |
| CO, O–H, C–O–C |
532.3 | 13.87 | ||
| C–O | 533.8 | 74.14 | ||
| H2O | 535.2 | 7.65 | ||
| R-GO | C 1s | CC graphene |
284.5 | 90.62 |
| C–O | 286.4 | 3.75 | ||
| π → π* | 291.3 | 5.63 | ||
| NP-GO | C 1s | CC graphene |
284.1 | 42.84 |
| CC nitrophenyl |
284.7 | 25.89 | ||
| C–C | 285.3 | 5.26 | ||
| C–NO2 | 285.8 | 4.76 | ||
| C–O | 286.4 | 8.55 | ||
| C=O, C–NH3+ | 287.8 | 1.87 | ||
| O–CO |
288.7 | 1.25 | ||
| π → π* | 290.7 | 9.94 | ||
| N 1s | NH2 | 399.9 | 19.54 | |
| NH3+ | 401.3 | 12.13 | ||
| NO | 403.5 | 4.92 | ||
| NO2 | 406.1 | 63.41 | ||
| O 1s | O–CO |
531.4 | 14.28 | |
| NO2 | 532.8 | 74.35 | ||
| C–O | 533.4 | 11.37 |
UV-vis spectroscopy was used to probe the interactions on the surface of the samples. As can be seen in Fig. 3, graphene oxide shows a peak at 235 nm, corresponding to π–π* transitions related to the presence of the remaining CC bonds with sp2 hybridization,75 which were shifted to 265 nm (longer wavelengths) after reduction, rebuilding the π-bond network.76 With the increase in π-bonding, the absorption peak shifts to a longer wavelength because less energy is needed for transition. The UV-visible spectrum of 2,4-dinitrophenylhydrazine showed two peaks at 222 nm related to the π–π* and 360 nm corresponding to n–π* transitions related to the aromatic compound.77 The absorption band of NP-GO species corresponding to the π–π* transition becomes broad and also shows a redshift from 222 to 229 nm, while the n–π* transition peak shifts from 360 to 354 nm. Also, the peak of GO had a redshift to 247 nm, indicating the recovery of the π bond network as a result of the reduction. However, due to the presence of functional groups, this displacement is lower than that of the R-GO sample. These changes and shifts in absorption peaks indicate the existence of π–π* interactions, electronic connections among the functional groups, and reduced graphene oxide sheets.
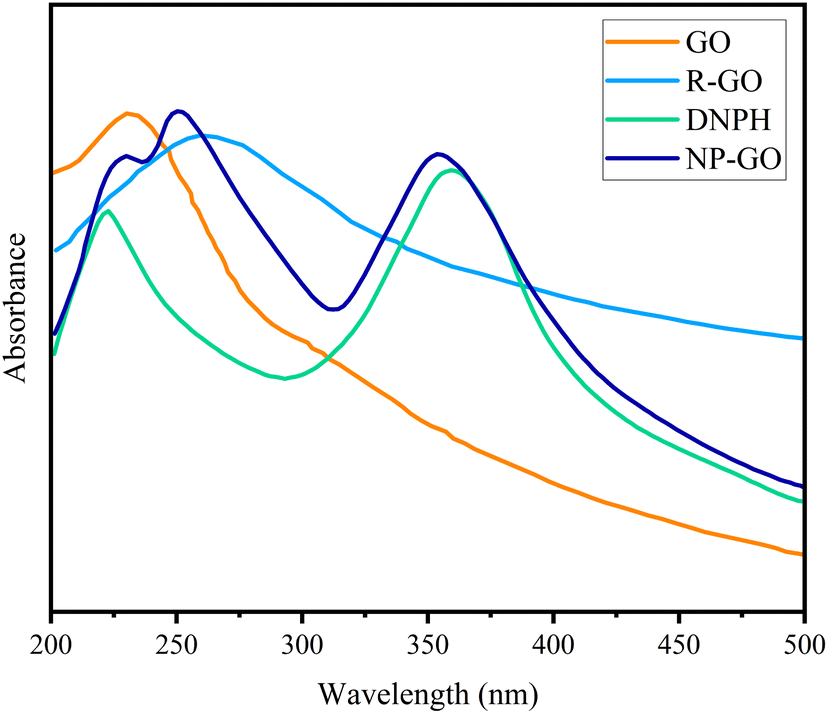 | ||
| Fig. 3 UV spectra of GO, R-GO, DNPH, and NP-GO. | ||
4.2. Morphological characterizations
Fig. 4 shows the SEM images of R-GO and NP-GO. It is obvious from Fig. 4a and b that the morphology of R-GO is wrinkled and crumpled, due to agglomeration through van der Waals forces and π–π interactions among sheets. As can be seen, the flakes of R-GO are in layers. Small R-GO flakes aggregate adjacent to each other and seem to have a bigger mass. The obtained layer structure seems to have formed during the filtering of the suspension. | ||
| Fig. 4 SEM images related to the morphology and structure of (a and b) R-GO and (c and d) NP-GO in different magnifications. | ||
Fig. 4c and d shows the morphology of NP-GO. Due to the presence of 2,4-dinitrophenylhydrazine groups and, as a result, reducing van der Waals forces and forming spatial repulse among functionalized sheets, aggregation is reduced in comparison to the R-GO, thus there is a more accessible active surface in this sample.
The binding effect of organic molecules on the surface of NP-GO can be measured and compared with graphene through the specific surface area (Fig. 5a) and the distribution of pore size (Fig. 5b). Fig. 5a show that the specific surface area of R-GO is near to 500 m2 g−1. It contains small mesoporous (2–50 nm), indicating that the reduction of graphene oxide by hydrazine with the loss of oxygen functional groups causes porosity. However, the value lower than the theoretical specific surface area for a single graphene sheet (2600 m2 g−1) is due to the sheets' accumulation and overlying after the drying process.78 After the grafting of 2,4-dinitrophenylhydrazine groups, the surface area for NP-GO increased to 650 m2 g−1. This increase indirectly suggests the binding 2,4-dinitrophenylhydrazine on the surface of NP-GO. The removal of oxygen-containing groups and the replacement of functional groups causes the creation of pores in the NP-GO sheets and as a result, causes a significant increase in the surface area and a significant difference in the distribution of pore size. Interestingly, the attachment of groups in NP-GO, which may be caused by the decarboxylation of groups in graphene oxide,79 leads to smaller pores than graphene.
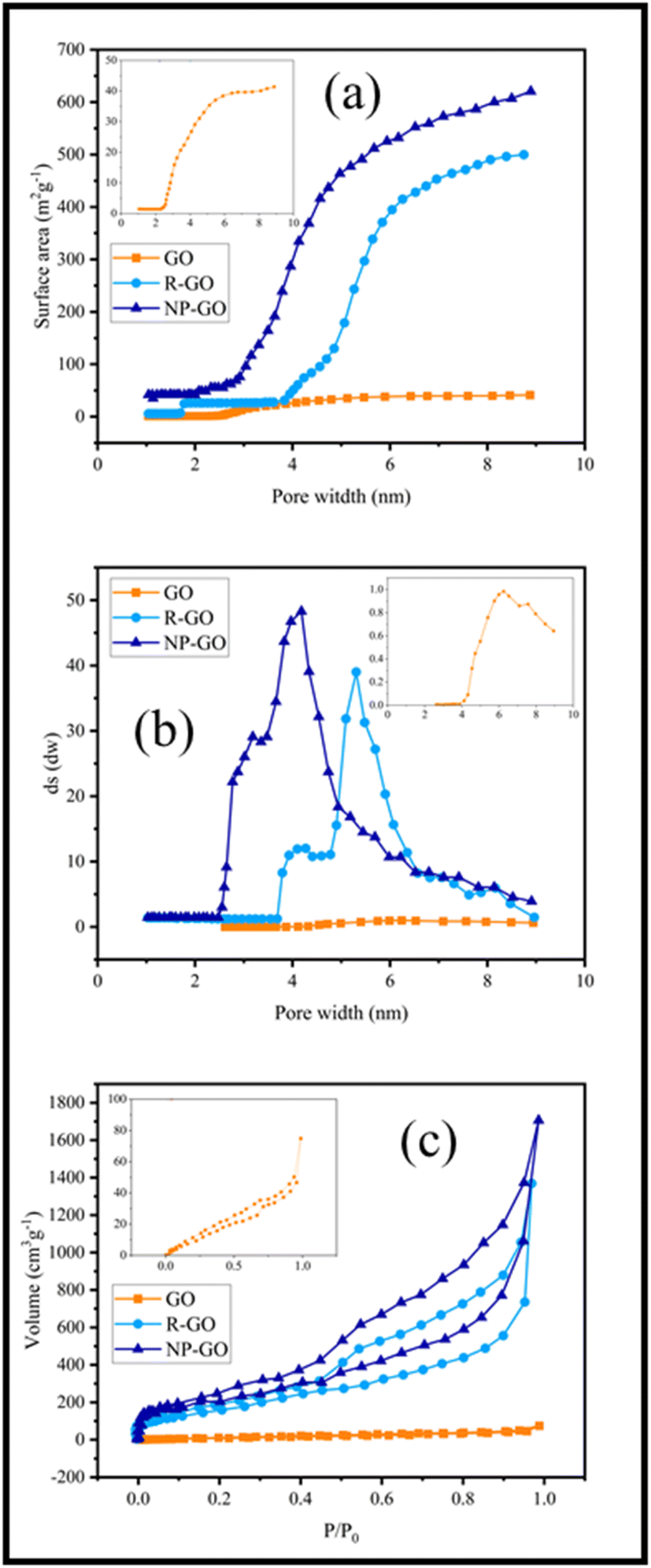 | ||
| Fig. 5 (a) Surface area versus pore width in R-GO and NP-GO, (b) the distribution of pore size of R-GO and NP-GO, and (c) N2 adsorption isotherms in R-GO and NP-GO. | ||
Fig. 5c displays the results of the N2 adsorption and desorption isotherm for R-GO and NP-GO at 77 K. Both samples show type I and II mixing isotherms of relative pressure (P/P0) in low and high regions.80 Low absorption volume at low P/P0 (0–0.5) for samples is representative of mesoporous materials, confirmed by an H3 hysteresis loop and a plateau.81 As it is apparent, absorb volume increases after the reduction and functionalization of graphene oxide with DNPH.
4.3. Photoelectrochemical descriptions
The efficiency of charge transfer/separation is very important in optical nonlinearity. Therefore, Photoelectrochemical investigations were conducted to study the electronic interactions on the samples. Fig. 6a shows the photocurrent performances of GO, R-GO, and NP-GO versus the time of the virtual on-and-off cycles of sunlight. For each on/off cycle, increases and decreases in photocurrent are obvious. When simulated sunlight irradiation is employed, photocurrent is produced via transferring and separating light charges to the working electrodes.82 At off cycle, the working electrode's responses are weak and keep their dark current state. However, when the switch is on, the photocurrent's intensity increases due to the rapid separation of charge carriers that are sensitive to light.83 Obviously, GO and R-GO exhibit weak photocurrent responses under simulated radiation of sunlight. At the same time, NP-GO, owing to the creation of paths for direct conduction through covalent bonds originating between 2,4-dinitrophenylhydrazine and reduced graphene oxide sheets, shows better performance. As a result, the separation and transportation of light-generated carriers were enhanced by removing oxygen-containing groups and introducing 2,4-dinitrophenylhydrazine groups on the GO surface.84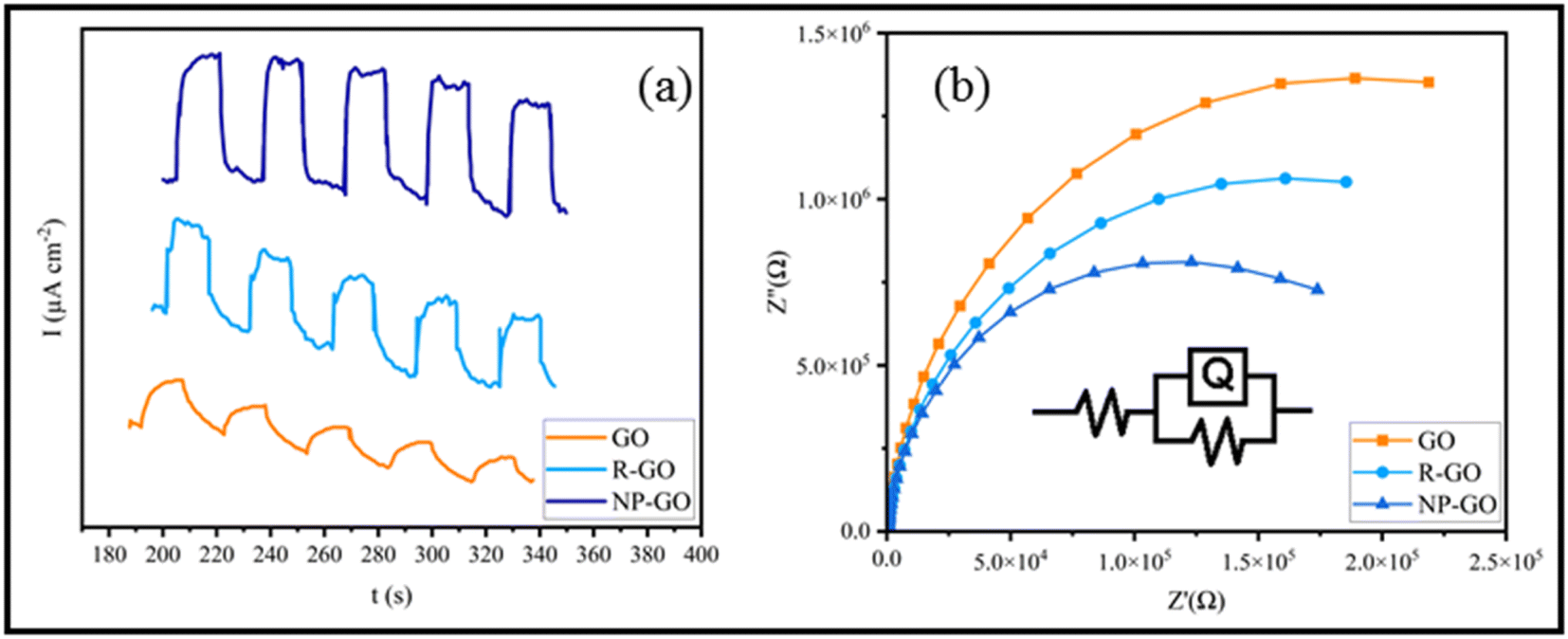 | ||
| Fig. 6 (a) The photocurrent performances of GO, R-GO, and NP-GO and (b) EIS spectra of GO, R-GO, and NP-GO under simulated sunlight. | ||
The Nyquist diagram of electrochemical impedance spectroscopy (EIS) was studied to further describe the charge carrier migration of the prepared samples. The results are shown in Fig. 6b. Subsequently, the proposed Rs(RctQct) model is used to study the resistance behavior of the oxide layer in terms of applied potential at a constant frequency range. In the resultant equivalent model, Rs, Rct, and Qct are the solution resistance, charge transfer resistance, and constant phase element (CPE), correspondingly. Since the EIS capacitor element does not designate steady and regular behavior owing to the concurrent complex corrosion procedure at the border, the constant phase element (CPE) as an expressive deviation from the ideal capacitor-based circuit was used.85 The best-fitted parameters for the reached equivalent circuit are listed in Table 2. As can be seen, NP-GO shows a smaller charge transfer resistance than GO and R-GO, which indicates the reduction of the resistance of the solid-state interface layer and charge transfer through the electrode/electrolyte boundary by forming covalent bonds among 2,4-dinitrophenylhydrazine groups and reduced graphene oxide sheets. These results are in good agreement with the measurements of photocurrent. They indicate that photo-induced carriers become separate and more effective, and charges transfer more rapidly on the surface of NP-GO.
| Samples | Rs (Ω cm2) | Rct (KΩ cm2) | Qct | |
|---|---|---|---|---|
| Y0 (μF cm−2) | n | |||
| GO | 82.54 | 56.43 | 0.132 | 0.8952 |
| R-GO | 108.51 | 49.28 | 0.205 | 0.9601 |
| NP-GO | 89.68 | 39.78 | 0.263 | 0.9522 |
4.4. Electrochemical measurements
Fig. 7a displays the cyclic voltammetry of the electrodes made of R-GO and NP-GO at a scan rate of 5 mV s−1. For R-GO, an almost rectangular curve which is a characteristic of carbon materials, and a very small peak at −400 mVAg/AgCl related to the reduction of unreduced oxide groups, can be seen.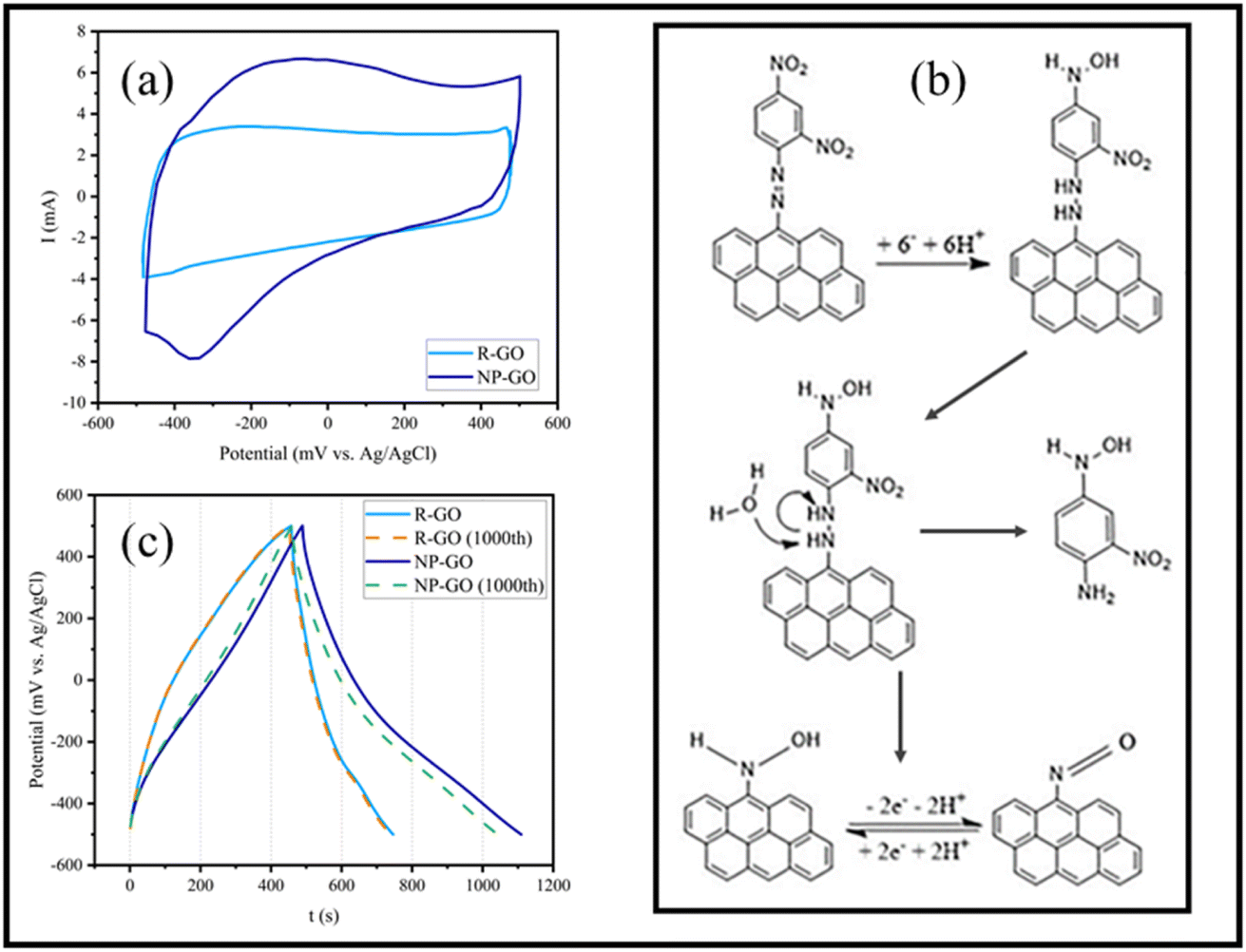 | ||
| Fig. 7 (a) Cyclic voltammetry of electrodes made of R-GO and NP-GO at the scan rate of 5 mV s−1, (b) the mechanism of the reduction of 2,4-dinitrophenylhydrazone presented on the surface of NP-GO in an aqueous environment and the redox reaction of the nitroso group, (c) First and 1000th galvanostatic charge/discharge of R-GO and NP-GO at 0.5 A g−1. | ||
In the curve related to NP-GO, there is an anodic peak in the range of −100 mVAg/AgCl and a cathodic one in −300 mVAg/AgCl. At the potential close to the initial reduction of NO2 groups from the 4-position of the 2,4-dinitrophenylhydrazine and before the reduction at the 2-position, reduction of the azo bond occurs, separating 1-amino-2-nitro-4-nitrophenyl and forming arylnitroso group on NP-GO86 (Fig. 7b). The redox peak observed in the cyclic voltammetry of the NP-GO sample is related to the arylnitroso groups' redox reaction.
Fig. 7c shows the results of the galvanostatic charge and discharge test related to R-GO and NP-GO electrodes in the initial cycle and after 1000 cycles at a current density of 0.5 A g−1. The discharging time of the electrodes made of R-GO and NP-GO are 290 and 620 seconds, respectively, and their respective specific capacities are 145 and 310 F g−1. Table 3 presents the reported capacities of some graphene-based electrodes and those made of functionalized graphene by nitrophenyl in neutral electrolytes. It can be seen that the prepared electrode materials show suitable performances compared to the literature. The results of this test show that R-GO has better stability than NP-GO so that after 1000 charge and discharge cycles, its capacity remains almost constant and equals 144 F g−1, while in the functionalized sample, the capacity has decreased by about 8% and reaches 291 F g−1. It should be noted that the high stability of carbon materials is one of the most important reasons for their use as an electrode material. For this reason, the higher stability of the R-GO electrode is not unconventional. However, the reduction in the capacity of the NP-GO electrode can be attributed to the reduction of the active surface or electrical conductivity due to the loss of layers or the breaking of the bond of the functional groups during the charge and discharge cycles.87
| Electrode materials | Electrolyte | Current density or scan rate | Capacitance | Reference |
|---|---|---|---|---|
| R-GO, CNT, MnO2 | Na2SO4 | 50 mV s−1 | 275 F g−1 | 88 |
| R-GO, CNT, PVDF | Na2SO4 | 0.1 A g−1 | 129 F g−1 | 89 |
| R-GO, CNT, MnO2, PTFE, acetylene black | Na2SO4 | 1 A g−1 | 120 F g−1 | 90 |
| R-GO, CNT, MnO2, PTFE, carbon black | Na2SO4 | o.5 A g−1 | 202 F g−1 | 91 |
| Graphene foil produced by exfoliating graphite | Na2SO4 | 0.1 A g−1 | 106 F g−1 | 92 |
| R-GO, PTFE, carbon black | Na2SO4 | 0.5 A g−1 | 144 F g−1 | This work |
| NP-R-GO (diazonium salt) NP-GO | K2SO4 | 0.1 A g−1 | 315 F g−1 | 93 |
| 0.1 A g−1 | 290 F g−1 | |||
| NP-GO | Na2SO4 | 0.5 A g−1 | 310 F g−1 | This work |
Fig. 8a shows the Nyquist diagram of R-GO and NP-GO electrodes. The obtained data were fitted using the Randles model as an equivalent electric circuit and their values are presented in Table 4. In this equivalent circuit, RS represents the electrolyte resistance, Rdl is the charge transfer resistance, Cdl is the electric double-layer capacitance, ZW is the Warburg impedance, and Cct is the electrode's pseud-capacitance. It should be noted that lower Rct and ZW indicate lower charge transfer resistance and higher ion transfer rate.94 The electrolyte resistance (Rct) in R-GO and NP-GO is 2.23, and 1.07 Ω, respectively, which suggests that the functionalization process leads to improved electrical conductivity and easy electron transfer. Establishing internal connections between the plain and functional groups in NP-GO can be the reason for that. In addition, the presence of ZW with a higher slope in NP-GO than R-GO specifies better ion diffusion, and it can be the result of the existence of more active are in the morphology in this sample. It can also be seen in the Bod-phase diagrams (Fig. 8b) that the maximum value for phase changes is −73 and −84 for R-GO and NP-GO, respectively. Therefore, it can be concluded that NP-GO shows better capacitive features than R-GO due to its morphology and the presence of functional groups. The lower resistance of NP-GO compared to R-GO can be seen in the Bode-magnetite diagrams (Fig. 8c).
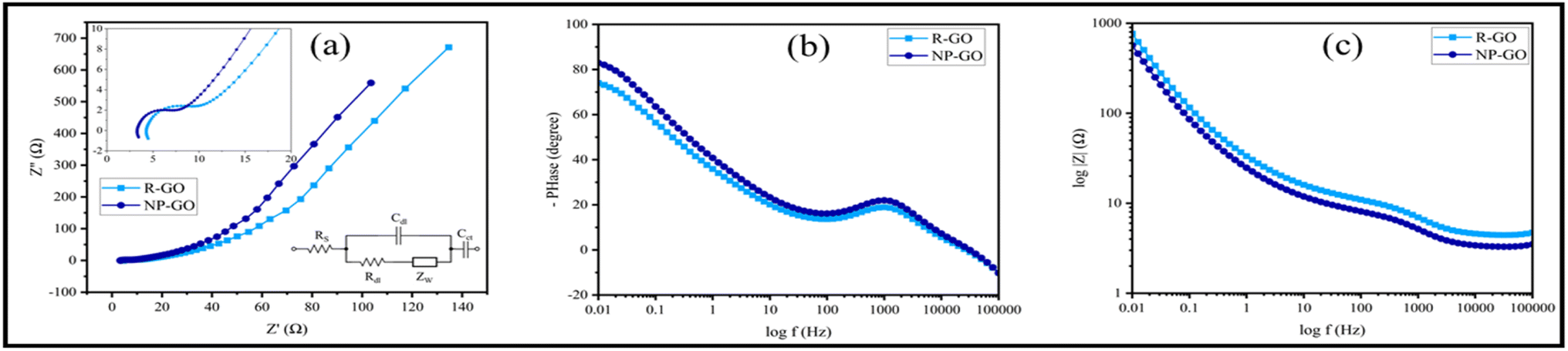 | ||
| Fig. 8 (a) Nyquist plots of R-GO, NP-GO, and the electrical equivalent circuit, (b) Bode-phase diagrams, and (c) Bode-magnetite diagrams. | ||
| Sample | RS (Ω) | Rdl (Ω) | Cdl (F g−1) | ZW | Cct (F g−1) |
|---|---|---|---|---|---|
| R-GO | 4.46 | 2.46 | 3.27 × 10−4 | 0.016 | 0.0167 |
| NP-GO | 3.43 | 1.84 | 6.53 × 10−4 | 0.021 | 0.0639 |
5. Conclusion
Graphene oxide was synthesized by the hummer method and in situ and simultaneously reduced and functionalized with 2,4-dinitrophenylhydrazine. Also, graphene is synthesized through the reaction of GO with hydrazine for comparing it with the functionalized sample. FTIR, Raman, TGA, and XRD of the samples were conducted to investigate the presence and effect of functional groups in samples. In addition, for further study, XPS of the samples was employed. According to the obtained results, due to the reaction with 2,4-dinitrophenylhydrazine, groups containing oxygen in GO are reduced and replaced with 2,4-dinitrophenylhydrazone groups. SEM and BET results display that the morphology of NP-GO has less agglomeration and, as a result, a more accessible active surface compared to graphene. The results of UV absorption show that shifts in absorption peaks of the NP-GO sample indicate the existence of electronic connections and π–π* interactions among the functional groups and reduced graphene oxide sheets. The photocurrent response of the sample exhibits that the separation/transportation of light-induced carriers was enhanced by replacing oxygen-containing groups with 2,4-dinitrophenylhydrazine groups. Galvanostatic charge and discharge show that the electrode made of NP-GO has a much higher capacity but lower stability compared to the electrode made of reduced graphene oxide. Based on the EIS results, the NP-GO electrode has a lower charge transfer resistance and better electrochemical performance than R-GO.Conflicts of interest
There are no conflicts of interest to declare.References
- W. Yu, L. Sisi, Y. Haiyan and L. Jie, RSC Adv., 2020, 10, 15328–15345 RSC.
- G. Yang, L. Li, W. B. Lee and M. C. Ng, Sci. Technol. Adv. Mater., 2018, 19, 613–648 CrossRef CAS PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung and E. Tutuc, Science, 2009, 324, 1312–1314 CrossRef CAS PubMed.
- J. Hass, W. De Heer and E. Conrad, J. Phys.: Condens. Matter, 2008, 20, 323202 CrossRef.
- P. Avouris, M. Freitag and V. Perebeinos, Nat. Photonics, 2008, 2, 341–350 CrossRef CAS.
- P. Blake, E. Hill, A. Castro Neto, K. Novoselov, D. Jiang, R. Yang, T. Booth and A. Geim, Appl. Phys. Lett., 2007, 91, 063124 CrossRef.
- Z. Chen, Y. Qi, X. Chen, Y. Zhang and Z. Liu, Adv. Mater., 2019, 31, 1803639 CrossRef.
- K. Xia, W. Wu, M. Zhu, X. Shen, Z. Yin, H. Wang, S. Li, M. Zhang, H. Wang and H. Lu, Sci. Bull., 2020, 65, 343–349 CrossRef CAS.
- Š. Meškinis, A. Vasiliauskas, A. Guobienė, M. Talaikis, G. Niaura and R. Gudaitis, RSC Adv., 2022, 12, 18759–18772 RSC.
- J.-Y. Moon, M. Kim, S.-I. Kim, S. Xu, J.-H. Choi, D. Whang, K. Watanabe, T. Taniguchi, D. S. Park and J. Seo, Sci. Adv., 2020, 6, eabc6601 CrossRef CAS.
- Q. He, S. Wu, S. Gao, X. Cao, Z. Yin, H. Li, P. Chen and H. Zhang, ACS Nano, 2011, 5, 5038–5044 CrossRef CAS PubMed.
- X. Qi, K. Y. Pu, H. Li, X. Zhou, S. Wu, Q. L. Fan, B. Liu, F. Boey, W. Huang and H. Zhang, Angew. Chem., Int. Ed., 2010, 49, 9426–9429 CrossRef CAS PubMed.
- H. G. Sudibya, Q. He, H. Zhang and P. Chen, ACS Nano, 2011, 5, 1990–1994 CrossRef CAS PubMed.
- X. Cao, Q. He, W. Shi, B. Li, Z. Zeng, Y. Shi, Q. Yan and H. Zhang, Small, 2011, 7, 1199–1202 CrossRef CAS PubMed.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- X. Lv, Y. Huang, Z. Liu, J. Tian, Y. Wang, Y. Ma, J. Liang, S. Fu, X. Wan and Y. Chen, Small, 2009, 5, 1682–1687 CrossRef CAS.
- L. Zhang, X. Li, Y. Huang, Y. Ma, X. Wan and Y. Chen, Carbon, 2010, 48, 2367–2371 CrossRef CAS.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- V. Dua, S. P. Surwade, S. Ammu, S. R. Agnihotra, S. Jain, K. E. Roberts, S. Park, R. S. Ruoff and S. K. Manohar, Angew. Chem., Int. Ed., 2010, 49, 2154–2157 CrossRef CAS PubMed.
- S. Sravya, D. RamaDevi, N. Belachew, K. E. Rao and K. Basavaiah, RSC Adv., 2021, 11, 12030–12035 RSC.
- J. Liu, S. Fu, B. Yuan, Y. Li and Z. Deng, J. Am. Chem. Soc., 2010, 132, 7279–7281 CrossRef CAS PubMed.
- A. Baral, L. Satish, D. P. Das, H. Sahoo and M. K. Ghosh, J. Biomol. Struct. Dyn., 2020, 38, 2038–2046 CrossRef CAS PubMed.
- H.-L. Guo, X.-F. Wang, Q.-Y. Qian, F.-B. Wang and X.-H. Xia, ACS Nano, 2009, 3, 2653–2659 CrossRef CAS PubMed.
- Z. Wang, X. Zhou, J. Zhang, F. Boey and H. Zhang, J. Phys. Chem. C, 2009, 113, 14071–14075 CrossRef CAS.
- X. Huang, X. Zhou, S. Wu, Y. Wei, X. Qi, J. Zhang, F. Boey and H. Zhang, Small, 2010, 6, 513–516 CrossRef CAS.
- X. H. Tai, S. W. Chook, C. W. Lai, K. M. Lee, T. C. K. Yang, S. Chong and J. C. Juan, RSC Adv., 2019, 9, 18076–18086 RSC.
- W. Chen and L. Yan, Nanoscale, 2010, 2, 559–563 RSC.
- C. Mattevi, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- A. Amiri, M. Baghayeri, F. Karimabadi, F. Ghaemi and B. Maleki, Microchim. Acta, 2020, 187, 1–8 CrossRef.
- H. Kumar, R. Sharma, A. Yadav and R. Kumari, J. Energy Storage, 2021, 33, 102032 CrossRef.
- R. Tarcan, O. Todor-Boer, I. Petrovai, C. Leordean, S. Astilean and I. Botiz, J. Mater. Chem. C, 2020, 8, 1198–1224 RSC.
- P. Sandhya, J. Jose, M. Sreekala, M. Padmanabhan, N. Kalarikkal and S. Thomas, Ceram. Int., 2018, 44, 15092–15098 CrossRef CAS.
- S. M. Majhi, A. Mirzaei, H. W. Kim and S. S. Kim, Sensors, 2021, 21, 1352 CrossRef CAS.
- A. P. Straub, D. S. Bergsman, B. A. Getachew, L. M. Leahy, J. J. Patil, N. Ferralis and J. C. Grossman, Nano Lett., 2021, 21, 2429–2435 CrossRef CAS.
- M. Tarahomi, H. Alinezhad and B. Maleki, Appl. Organomet. Chem., 2019, 33, e5203 CrossRef CAS.
- H. Boroumand, H. Alinezhad, B. Maleki and S. Peiman, Polycyclic Aromat. Compd., 2022, 1–17 Search PubMed.
- G. Eda, Y. Y. Lin, C. Mattevi, H. Yamaguchi, H. A. Chen, I. S. Chen, C. W. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505–509 CrossRef CAS PubMed.
- A. Hashimoto, K. Suenaga, A. Gloter, K. Urita and S. Iijima, nature, 2004, 430, 870–873 CrossRef CAS PubMed.
- S. Stankovich, R. D. Piner, X. Chen, N. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155–158 RSC.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- S. Park, J. An, I. Jung, R. D. Piner, S. J. An, X. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593–1597 CrossRef CAS PubMed.
- S. Park, J. An, R. D. Piner, I. Jung, D. Yang, A. Velamakanni, S. T. Nguyen and R. S. Ruoff, Chem. Mater., 2008, 20, 6592–6594 CrossRef CAS.
- T. A. Strom, E. P. Dillon, C. E. Hamilton and A. R. Barron, Chem. Commun., 2010, 46, 4097–4099 RSC.
- X. Wu, H. Cao, B. Li and G. Yin, Nanotechnology, 2011, 22, 075202 CrossRef PubMed.
- H.-P. Cong, X.-C. Ren, P. Wang and S.-H. Yu, Energy Environ. Sci., 2013, 6, 1185–1191 RSC.
- G. Pognon, C. Cougnon, D. Mayilukila and D. Bélanger, ACS Appl. Mater. Interfaces, 2012, 4, 3788–3796 CrossRef CAS PubMed.
- W. Ai, W. Zhou, Z. Du, Y. Du, H. Zhang, X. Jia, L. Xie, M. Yi, T. Yu and W. Huang, J. Mater. Chem., 2012, 22, 23439–23446 RSC.
- H. Bai, C. Li, X. Wang and G. Shi, Chem. Commun., 2010, 46, 2376–2378 RSC.
- S. Liu, J. Tian, L. Wang, H. Li, Y. Zhang and X. Sun, Macromolecules, 2010, 43, 10078–10083 CrossRef CAS.
- Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191–3195 CrossRef CAS.
- W. Tu, J. Lei, S. Zhang and H. Ju, Chem. - Eur. J., 2010, 16, 10771–10777 CrossRef CAS PubMed.
- D. K. Samarakoon, Z. Chen, C. Nicolas and X. Q. Wang, Small, 2011, 7, 965–969 CrossRef CAS PubMed.
- D. K. Samarakoon and X.-Q. Wang, ACS Nano, 2009, 3, 4017–4022 CrossRef CAS PubMed.
- D. K. Samarakoon and X.-Q. Wang, ACS Nano, 2010, 4, 4126–4130 CrossRef CAS.
- U. K. Wijewardena, S. E. Brown and X.-Q. Wang, J. Phys. Chem. C, 2016, 120, 22739–22743 CrossRef CAS.
- F. Chen, Q. Qing, J. Xia, J. Li and N. Tao, J. Am. Chem. Soc., 2009, 131, 9908–9909 CrossRef CAS PubMed.
- E. Paek, A. J. Pak, K. E. Kweon and G. S. Hwang, J. Phys. Chem. C, 2013, 117, 5610–5616 CrossRef CAS.
- M. Mousavi-Khoshdel, E. Targholi and M. J. Momeni, J. Phys. Chem. C, 2015, 119, 26290–26295 CrossRef CAS.
- S. M. Mousavi-Khoshdel and E. Targholi, Carbon, 2015, 89, 148–160 CrossRef CAS.
- R. N. Gunasinghe, D. G. Reuven, K. Suggs and X.-Q. Wang, J. Phys. Chem. Lett., 2012, 3, 3048–3052 CrossRef CAS PubMed.
- T. R. Nanayakkara, U. K. Wijewardena, A. B. Arampath, K. Suggs, N. Ravi and X.-Q. Wang, in Theoretical and Computational Chemistry, Elsevier, 2022, vol. 21, pp. 131–155 Search PubMed.
- X. Tian, J. Gu and J.-b. Xu, J. Chem. Phys., 2014, 140, 044712 CrossRef PubMed.
- Y. Feng, H. Liu, W. Luo, E. Liu, N. Zhao, K. Yoshino and W. Feng, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- E. Bekyarova, S. Sarkar, S. Niyogi, M. Itkis and R. Haddon, J. Phys. D: Appl. Phys., 2012, 45, 154009 CrossRef.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W.-F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS PubMed.
- C. Bosch-Navarro, E. Coronado, C. Martí-Gastaldo, J. Sánchez-Royo and M. G. Gómez, Nanoscale, 2012, 4, 3977–3982 RSC.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S.-J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS.
- Y. Liu, Y. Shi, W. Zhou, W. Shi, W. Dang, X. Li and B. Liang, Opt. Laser Technol., 2021, 139, 106960 CrossRef CAS.
- B. Vinayan, R. Nagar, V. Raman, N. Rajalakshmi, K. Dhathathreyan and S. Ramaprabhu, J. Mater. Chem., 2012, 22, 9949–9956 RSC.
- B. Rajagopalan and J. S. Chung, Nanoscale Res. Lett., 2014, 9, 1–10 CrossRef CAS PubMed.
- H. D. Le, T. T. T. Ngo, D. Q. Le, X. N. Nguyen and N. M. Phan, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2013, 4, 035012 Search PubMed.
- F. T. Johra, J.-W. Lee and W.-G. Jung, J. Ind. Eng. Chem., 2014, 20, 2883–2887 CrossRef CAS.
- K. Roodenko, M. Gensch, J. Rappich, K. Hinrichs, N. Esser and R. Hunger, J. Phys. Chem. B, 2007, 111, 7541–7549 CrossRef CAS PubMed.
- V. Kumar, R. K. Gupta, R. K. Gundampati, D. K. Singh, S. Mohan, S. H. Hasan and M. Malviya, RSC Adv., 2018, 8, 619–631 RSC.
- Y. Zhou, Q. Bao, L. A. L. Tang, Y. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950–2956 CrossRef CAS.
- K. Bijudas, J. Appl. Chem., 2019, 8, 1887–1892 CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- A. Le Comte, D. Chhin, A. Gagnon, R. Retoux, T. Brousse and D. Bélanger, J. Mater. Chem. A, 2015, 3, 6146–6156 RSC.
- A. Gambou-Bosca and D. Bélanger, J. Electrochem. Soc., 2015, 162, A5115 CrossRef CAS.
- T. Ohkubo, J. Miyawaki, K. Kaneko, R. Ryoo and N. A. Seaton, J. Phys. Chem. B, 2002, 106, 6523–6528 CrossRef CAS.
- W. Zhao, C. Li, A. Wang, C. Lv, W. Zhu, S. Dou, Q. Wang and Q. Zhong, Phys. Chem. Chem. Phys., 2017, 19, 28696–28709 RSC.
- W. Zhao, A. Wang, Y. Wang, C. Lv, W. Zhu, S. Dou, Q. Wang and Q. Zhong, J. Alloys Compd., 2017, 726, 164–172 CrossRef CAS.
- J. Zhang, A. Wang, W. Zhao, C. Li, X. Chen, Y. Wang, W. Zhu and Q. Zhong, Dyes Pigm., 2018, 153, 241–247 CrossRef CAS.
- J. Yang, J. Wu, C. Zhang, S. Zhang, B. Yang, W. Emori and J. Wang, J. Alloys Compd., 2020, 819, 152943 CrossRef CAS.
- C. A. Thorogood, G. G. Wildgoose, J. H. Jones and R. G. Compton, New J. Chem., 2007, 31, 958–965 RSC.
- L. Zhang, K. Hui, K.-S. Hui, X. Chen, R. Chen and H. Lee, Int. J. Hydrogen Energy, 2016, 41, 9443–9453 CrossRef CAS.
- Y. Cheng, S. Lu, H. Zhang, C. V. Varanasi and J. Liu, Nano Lett., 2012, 12, 4206–4211 CrossRef CAS PubMed.
- O. Okhay, A. Tkach, P. Staiti and F. Lufrano, Electrochim. Acta, 2020, 353, 136540 CrossRef CAS.
- L. Deng, Z. Hao, J. Wang, G. Zhu, L. Kang, Z.-H. Liu, Z. Yang and Z. Wang, Electrochim. Acta, 2013, 89, 191–198 CrossRef CAS.
- B. Ding, D. Guo, Y. Wang, X. Wu and Z. Fan, J. Power Sources, 2018, 398, 113–119 CrossRef CAS.
- A. Ambrosi and M. Pumera, Chem. - Eur. J., 2016, 22, 153–159 CrossRef CAS PubMed.
- S. Alipour and S. M. Mousavi-Khoshdel, Electrochim. Acta, 2019, 317, 301–311 CrossRef CAS.
- Y. Zhu, W. Chu, N. Wang, T. Lin, W. Yang, J. Wen and X. Zhao, RSC Adv., 2015, 5, 77958–77964 RSC.
| This journal is © The Royal Society of Chemistry 2023 |