
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regio- and stereoselective divergent cross-coupling of alkynes and disubstituted alkenes via photoredox cobalt dual catalysis†
Shi-Qi
Zhang
ae,
Yan-Lin
Li
a,
Kun
Cui
a,
Chen
Chen
ae,
Zheng-Yang
Gu
 c,
Hu
He
*d and
Ji-Bao
Xia
*ab
c,
Hu
He
*d and
Ji-Bao
Xia
*ab
aState Key Laboratory for Oxo Synthesis and Selective Oxidation, Suzhou Research Institute of LICP, Lanzhou Institute of Chemical Physics (LICP), University of Chinese Academy of Sciences, Chinese Academy of Sciences, Lanzhou 730000, China
bCollege of Material Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, Hangzhou 311121, Zhejiang, China
cCollege of Textiles and Clothing, Key Laboratory for Advanced Technology in Environmental Protection of Jiangsu Province, Yancheng Institute of Technology, Jiangsu, China
dHangzhou SynRx Therapeutics Biomedical Technology Co., Hangzhou, 311121, China. E-mail: hehu@synrx.cn
eUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 28th October 2023
Abstract
Achieving divergent synthesis from the same substrates quickly to generate different products remains an attractive objective in synthetic chemistry and the medicinal industry. Here, we report a discovery of highly selective divergent cross-coupling of alkynes and disubstituted alkenes by merging visible light photoredox and cobalt catalysis. Under otherwise identical conditions, the use of either a hemilabile P,N-ligand or a strong bidentate diphosphine ligand leads to ene-type coupling or reductive coupling of alkynes and Tulipalin A, producing stereodefined 1,4-diene or trisubstituted alkene products, respectively. The approaches feature considerable advantages for the straightforward synthesis of stereodefined multiple substituted alkenes, such as easily available substrates, low catalyst loading of an organophotocatalyst, excellent regio- and stereoselectivity, good functional group tolerance, and mild reaction conditions. Reasonable catalytic reaction pathways from the same cobaltacyclopentene intermediate have been proposed.
Introduction
As essential constituents, 2-furanones (butyrolactones) have been found in over 5800 natural compounds and drug molecules and possess diverse biological activities such as anthelmintic, antiviral, antifungal, and anticancer activities (Scheme 1A).1 Many methods have been developed to access butyrolactones, including the widely used intramolecular esterification or selective oxidation of furan derivatives.2 Tulipalin A, α-methylene-γ-butyrolactone (MBL), is a structurally simple and commercially available sesquiterpene lactone present in bulb scales of tulips with antibacterial and antitumor properties.3 Bearing an exo-methylene group at the α-position of lactone, Tulipalin A is considered as a cyclic analog of methyl methacrylate (MMA). Thus, it has been broadly used as a naturally originating vinyl monomer for the synthesis of biocompatible and biodegradable polymers.4 In addition, Tulipalin A has been used as a reactant in organic synthesis, such as a Michael acceptor or an electron-deficient olefin.5 However, it is rarely used as an efficient coupling partner in transition-metal-catalyzed cross-coupling reactions. It is suspected that the use of Tulipalin A as a simple synthon to couple with other easily accessed materials would be a straightforward and modular approach to access complex targets containing 2-furanone frameworks.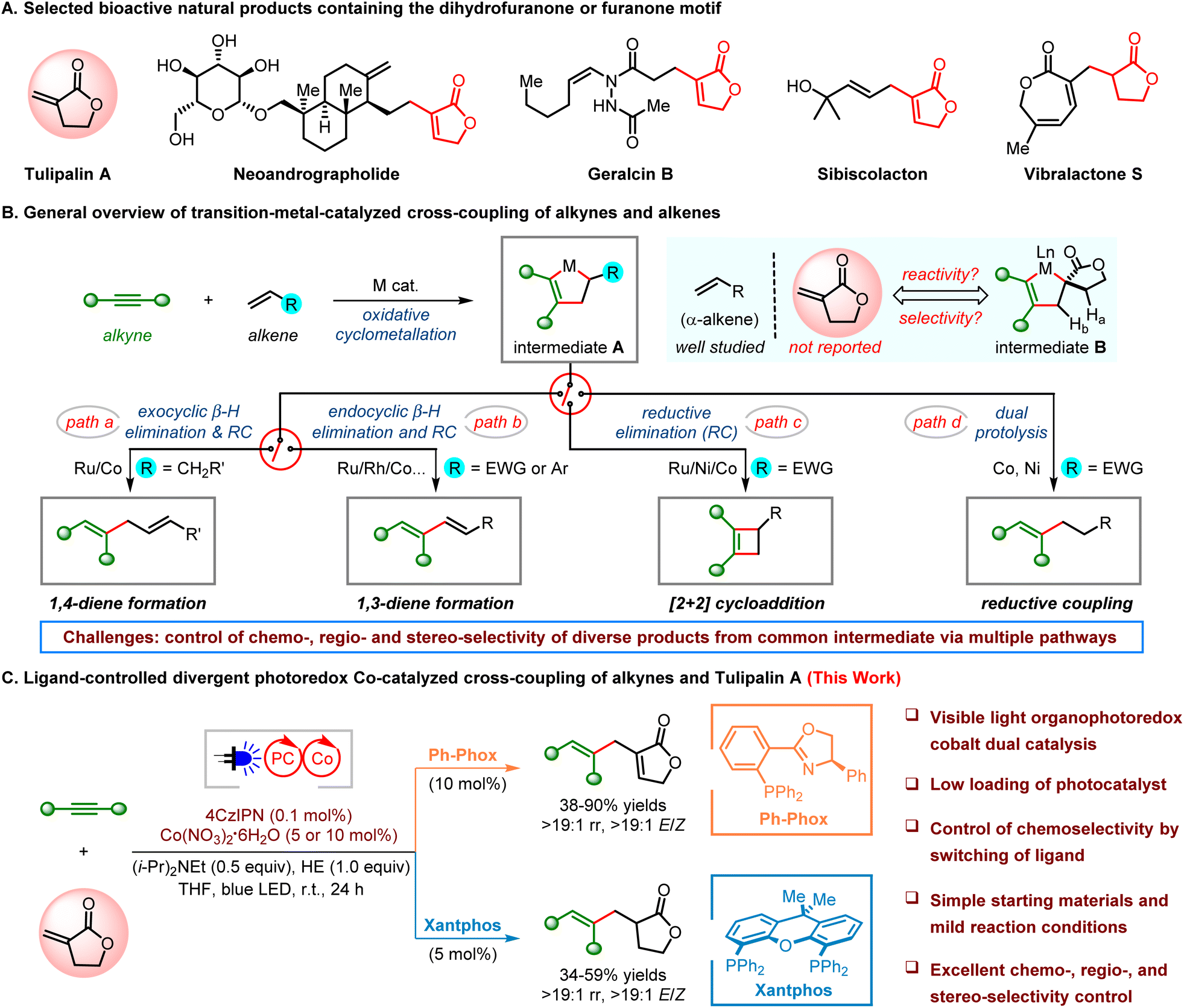 | ||
| Scheme 1 Transition-metal-catalyzed selective cross-coupling of alkynes and alkenes. | ||
Transition-metal-catalyzed intermolecular cross-coupling of alkynes and alkenes provides a highly atom- and step-economical route to form C–C bonds with easily accessed materials in organic synthesis.6 The main challenge of this reaction is the control of its chemo-, regio- and stereoselectivities because diverse products can be obtained via a common intermediate A of metallacyclopentene generated from oxidative cyclometallation of alkynes and alkenes (Scheme 1B). To minimize the steric strain and reduce the regioisomeric products, less hindered monosubstituted alkenes (α-alkenes) have been mainly used as the substrates. For instance, Ru7- or Co-catalyzed8 ene-type coupling of alkynes and monosubstituted alkyl alkenes produced 1,4-dienes as the favored products via exocyclic β-hydride elimination followed by reductive elimination (path a). Through endocyclic β-hydride elimination followed by reductive elimination, 1,3-dienes were obtained using styrenes9 (Co catalysis) or electron-deficient alkenes (Ni,10 Rh,11 Ru12 or Pd13 catalysis) as the coupling partners since exocyclic β-hydride does not exist in intermediate A (path b). Interestingly, cyclobutenes were favored by coupling monosubstituted electron-deficient alkenes under cobalt catalysis via [2 + 2]-cyclization through direct C–C bond reductive elimination of A (path c).14 In addition, Co- or Ni-catalyzed cross-coupling of alkynes and electron-deficient alkenes easily produces multi-substituted alkenes as the reductive coupling products under reducing conditions (path d).15 Recently, a few elegant examples have been reported other than monosubstituted alkenes. Well-designed multi-substituted alkenes containing a directing group were found to be efficient coupling partners in Ru-catalyzed ene-type coupling with silyl alkynes, or propargyl or homopropargyl alcohols.16 Symmetrical cyclic alkenes have also been used in Rh- or Co-catalyzed ene-type alkyne/alkene couplings.17 On the other hand, few examples of Co-catalyzed reductive coupling of alkynes and simple electron-deficient internal alkenes have been reported.18 We questioned whether simple Tulipalin A could be used as a versatile gem-disubstituted alkene to couple with alkynes, providing a direct and concise route to 2-furanone-containing stereodefined alkene targets. Two challenges have been considered in this reaction. First, a crowded spirocyclic metallacyclopentene B should be generated, which may cause a strong steric effect. Second, selective control of the coupling product will be attractive but difficult because intermediate B could possibly undergo either of the following reaction pathways, exocyclic β-Ha or endocyclic β-Hb elimination, direct reductive elimination, or reductive coupling. Photocatalysis has captured the imagination of both synthetic and medicinal chemists and has become an attractive tool in green synthesis.19 In particular, photoredox metal dual catalysis has been proved to be a powerful strategy in the synthesis of complex targets under mild reaction conditions.20,21 Herein, we reported for the first time a photoredox Co-catalyzed regio- and stereoselective ene-type coupling of alkynes and Tulipalin A, a gem-disubstituted alkene, producing 2-furanone-containing 1,4-dienes with excellent control of geometry (Scheme 1C). Moreover, the reductive coupling of alkynes and gem-disubstituted alkenes has also be achieved, affording stereodefined trisubstituted alkenes using a different ligand under otherwise identical conditions.
Results and discussion
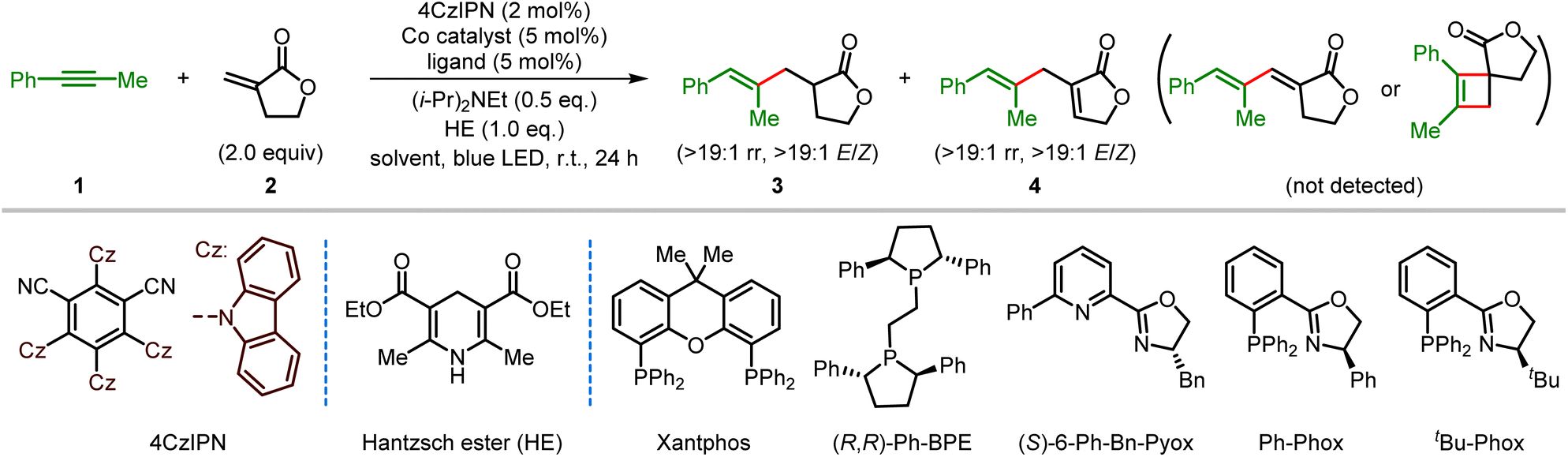
We started our research by investigating the cross-coupling of unsymmetrical 1-phenyl-1-propyne (1) and Tulipalin A (2) via visible light photoredox cobalt dual catalysis (Table 1). Initially, using the commercially available CoCl2 as the catalyst, Xantphos as the ligand, and 2,4,5,6-tetrakis(carbazol-9-yl)-1,3-dicyanobenzene (4CzIPN) as the organic photocatalyst,22 the anticipated reductive coupling reaction occurred smoothly, affording the trisubstituted alkene product 3 in 65% yield with >19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioselectivity (rr) and >19:1 E/Z stereoselectivity using Hantzsch ester (HE) as an organic reducing reagent in tetrahydrofuran (THF) under 5 W blue LED irradiation (Table 1, entry 1). After extensive screening of a variety of cobalt catalysts, an improved yield was obtained with Co(NO3)2·6H2O as the metal catalyst (Table 1, entry 2, also see Table S1 in the ESI†). To our surprise, the ene-type coupling product 1,4-diene 4 was also obtained in 5% yield under these typical reductive coupling conditions. Interestingly, the cross-coupling reaction afforded skipped diene 4 as the major product when N,N-dimethylformamide (DMF) was used as the solvent, indicating that the reductive coupling or ene-type coupling reaction might be possibly controlled by changing the reaction conditions (Table 1, entry 3). Since one chiral center exist in product 3, various chiral ligands were then tested to achieve an enantioselective reductive coupling reaction. Unfortunately, low enantioselectivity control was observed with a wide range of chiral phosphine or nitrogen ligands (Table 1, entries 4 and 5; for details see Table S2 in the ESI†). To our delight, the 1,4-diene product 4 was obtained as the major product in 66% yield with >19:1 regioselectivity and >19:1 E/Z stereoselectivity when Ph-Phox was used as the ligand under otherwise identical conditions (Table 1, entry 6). The ligand plays an important role in the reaction. No reaction occurred with the structurally similar tBu-Phox as the ligand, but dppp gave a similar yield of 4 (Table 1, entries 7 and 8). The yield of product 4 was further increased to 86% after detailed optimizations of the ratio of substrates, the catalyst loadings of the cobalt catalyst and photocatalyst and the amount of HE (Table 1, entries 9–12). Notably, an excellent yield was still maintained when the catalyst loading of 4CzIPN was decreased to 0.1%. It should be noted that the 1,3-diene or cyclobutene product was not observed under the tested conditions. Finally, control experiments demonstrate that no reductive coupling or ene-type coupling product was observed in the absence of a cobalt catalyst, ligand, photocatalyst, or light, which means that they are all required for the progression of the current cross-coupling reactions (Table 1, entries 13 and 14, also see Table S8 in the ESI†).
:1 regioselectivity (rr) and >19:1 E/Z stereoselectivity using Hantzsch ester (HE) as an organic reducing reagent in tetrahydrofuran (THF) under 5 W blue LED irradiation (Table 1, entry 1). After extensive screening of a variety of cobalt catalysts, an improved yield was obtained with Co(NO3)2·6H2O as the metal catalyst (Table 1, entry 2, also see Table S1 in the ESI†). To our surprise, the ene-type coupling product 1,4-diene 4 was also obtained in 5% yield under these typical reductive coupling conditions. Interestingly, the cross-coupling reaction afforded skipped diene 4 as the major product when N,N-dimethylformamide (DMF) was used as the solvent, indicating that the reductive coupling or ene-type coupling reaction might be possibly controlled by changing the reaction conditions (Table 1, entry 3). Since one chiral center exist in product 3, various chiral ligands were then tested to achieve an enantioselective reductive coupling reaction. Unfortunately, low enantioselectivity control was observed with a wide range of chiral phosphine or nitrogen ligands (Table 1, entries 4 and 5; for details see Table S2 in the ESI†). To our delight, the 1,4-diene product 4 was obtained as the major product in 66% yield with >19:1 regioselectivity and >19:1 E/Z stereoselectivity when Ph-Phox was used as the ligand under otherwise identical conditions (Table 1, entry 6). The ligand plays an important role in the reaction. No reaction occurred with the structurally similar tBu-Phox as the ligand, but dppp gave a similar yield of 4 (Table 1, entries 7 and 8). The yield of product 4 was further increased to 86% after detailed optimizations of the ratio of substrates, the catalyst loadings of the cobalt catalyst and photocatalyst and the amount of HE (Table 1, entries 9–12). Notably, an excellent yield was still maintained when the catalyst loading of 4CzIPN was decreased to 0.1%. It should be noted that the 1,3-diene or cyclobutene product was not observed under the tested conditions. Finally, control experiments demonstrate that no reductive coupling or ene-type coupling product was observed in the absence of a cobalt catalyst, ligand, photocatalyst, or light, which means that they are all required for the progression of the current cross-coupling reactions (Table 1, entries 13 and 14, also see Table S8 in the ESI†).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Co catalyst | Ligand | Solvent | Yield of 3b (%) |
ee of 3c (%) |
Yield of 4b (%) |
| a Reaction conditions: 1 (0.2 mmol), 2 (2.0 equiv.), Co catalyst (5 mol%), ligand (5 mol%), 4CzIPN (2 mol%), (i-Pr)2NEt (0.5 equiv.), HE (1.0 equiv.), solvent (3.0 mL), 5 W blue LED, r.t., 24 h, unless otherwise noted. b Determined by GC with dodecane as an internal standard, and isolated yield is shown in parentheses. c Determined by chiral HPLC analysis. d With 2 (3.0 equiv.). e With 4CzIPN (0.1 mol%). f With Co(NO3)2·6H2O (10 mol%) and Ph-Phox (10 mol%). g With HE (0.5 equiv.). h Without light. | ||||||
| 1 | CoCl2 | Xantphos | THF | 65 | — | Trace |
| 2 | Co(NO3)2·6H2O | Xantphos | THF | 68 (59) | — | 5 |
| 3 | Co(NO3)2·6H2O | Xantphos | DMF | 29 | — | 46 |
| 4 | Co(NO3)2·6H2O | (R,R)-Ph-BPE | THF | 17 | 5 | <5 |
| 5 | Co(NO3)2·6H2O | (S)-6-Ph-Bn-Pyox | THF | 11 | 12 | Trace |
| 6 | Co(NO3)2·6H2O | Ph-Phox | THF | 5 | — | 66 |
| 7 | Co(NO3)2·6H2O | t Bu-Phox | THF | 0 | — | 0 |
| 8 | Co(NO3)2·6H2O | dppp | THF | Trace | — | 62 |
| 9d | Co(NO3)2·6H2O | Ph-Phox | THF | 5 | — | 82 |
| 10d,e | Co(NO3)2·6H2O | Ph-Phox | THF | <5 | — | 79 |
| 11d,e,f | Co(NO3)2·6H2O | Ph-Phox | THF | <5 | — | 86 (81) |
| 12d,e,f,g | Co(NO3)2·6H2O | Ph-Phox | THF | <5 | — | 38 |
| 13d,e | No Co catalyst | No ligand | THF | 0 | — | 0 |
| 14d,e,f,h | Co(NO3)2·6H2O | Ph-Phox | THF | 0 | — | 0 |
With the optimized reaction conditions in hand, we first examined the generality of the ene-type coupling of alkynes and gem-disubstituted alkenes leading to 1,4-dienes via visible light organophotoredox cobalt dual catalysis (Fig. 1). In general, excellent regio- (>19:1 rr) and stereoselectivities (>19:1 E/Z or Z/E) were obtained. Coupling of aryl alkyl alkynes and Tulipalin A produced the desired products 5–7 in 52–90% yields, tolerating electron-donating (OMe) and electron-withdrawing (CO2Me, Ac) groups at the meta- or para-position of the aryl ring. Halide (F, Cl, Br) groups are also tolerated to give the corresponding 1,4-dienes in good yields (8–10). Aryl methyl alkynes bearing a bulky ortho-methyl group on the aryl ring (11) or a multi-substituted aryl substituent reacted smoothly, indicating good steric compatibility in this transformation (12 and 13). The alkyne derived from a natural steroid was a suitable coupling partner (14), demonstrating the utility of this reaction for the late-stage functionalization of complex molecules. In addition, an internal alkyne containing a naphthalene or pyridine group was also compatible, generating the corresponding product 15 or 16 in a good yield with excellent regio- and stereoselectivity.
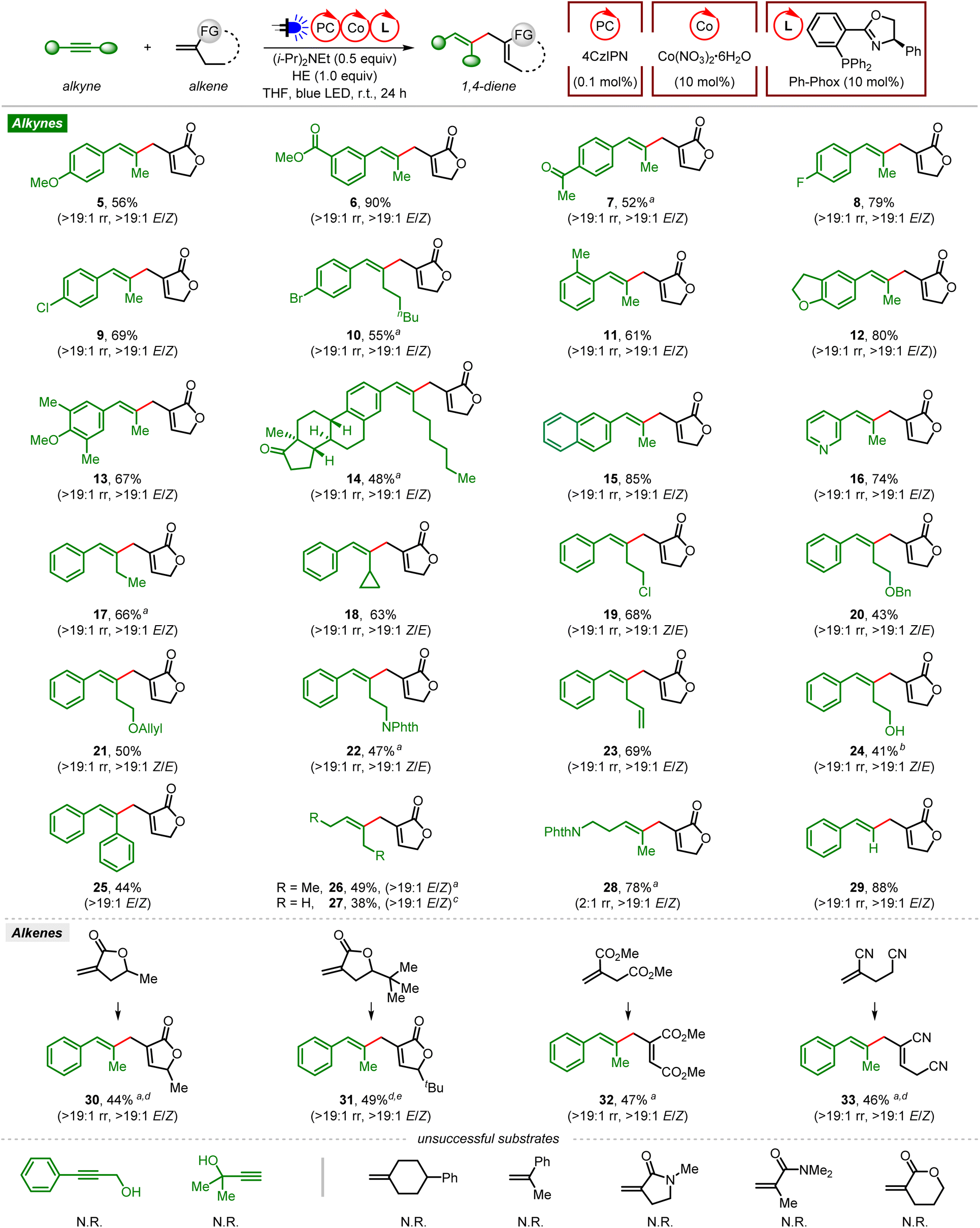 | ||
| Fig. 1 Reaction scope of organophotoredox Co-catalyzed ene-type coupling of alkynes and alkenes. Reaction conditions: alkyne (0.2 mmol), alkene (3.0 equiv.), 4CzIPN (0.1 mol%), Co(NO3)2·6H2O (10 mol%), Ph-Phox (10 mol%), (i-Pr)2NEt (0.5 equiv.), HE (1.0 equiv.), THF (3.0 mL), 5 W blue LED, r.t., 24 h, isolated yield, unless otherwise noted. aWith 4CzIPN (2 mol%). bWith TMS-protected homopropargylic alcohol as the substrate. cWith 2-butyne (50 equiv., 1 mL, 10 M solution in THF). dWith DMF as the solvent. eWith 4CzIPN-Br (2 mol%) as the photocatalyst. | ||
Besides, ene-type coupling of Tulipalin A and terminal alkyl group (ethyl or cyclopropyl) substituted phenylacetylene also occurred smoothly, giving the desired products 17 and 18. A variety of functional groups on the alkyl chain of the phenyl alkyl alkynes, such as halogen (Cl), ether (OBn, OAllyl), imide (Phth, phthalimide), and terminal alkene, were well tolerated, leading to the corresponding products in moderate to good yields (19–23). Notably, the free alcohol product 24 was obtained when trimethylsilyl (TMS)-protected homopropargylic alcohol was used as the substrate, with the removal of the TMS protecting group during isolation using flash column chromatography on silica gel.
Next, we tested symmetrical internal alkynes, such as diphenylethyne, 3-hexyne and 2-butyne, in this cross-coupling reaction. They reacted smoothly with Tulipalin A, affording the corresponding products in moderate yields (25–27). When the unsymmetrical dialkyl alkyne PhthN-substituted 2-pentyne was used as the substrate, 1,4-diene 28 was obtained in 78% yield albeit with low regioselectivity (2:1 rr). Notably, the terminal alkyne phenylacetylene was also proved to be a suitable coupling partner in this reaction, producing 29 in a good yield with excellent regio- and stereoselectivity. However, no reaction occurred with internal or terminal propargylic alcohol under the standard conditions. Then the scope of gem-disubstituted alkenes was explored. The O-alpha alkyl (methyl and tert-butyl) substituted Tulipalin A analogs were successfully employed as the substrates, affording the desired products in moderate yields with excellent regio- and stereoselectivities (30 and 31). Linear alpha-alkyl substituted acrylate and acrylonitrile were also suitable coupling partners, leading to functionalized 1,4-dienes with excellent regio- and stereoselectivities (32 and 33). A variety of other gem-disubstituted alkenes have also been investigated in this ene-type coupling, such as 1,1-dialkyl substituted ethene, α-methylstyrene, cyclic or linear acrylamide, and 2-methylene-5-pentanolide. Unfortunately, no desired product was observed with these substrates.
We then examined the scope of organophotoredox Co-catalyzed reductive coupling of alkynes and gem-disubstituted alkenes (Fig. 2). Generally, stereodefined trisubstituted alkenes were obtained with excellent regio- (>19:1 rr) and stereoselectivities (>19:1 E/Z). A variety of unsymmetrical aryl alkyl alkynes bearing diverse functional groups (p-OMe, m-COOMe, p-Cl, and o-Me) on different sites of the phenyl ring were found to be efficient coupling partners, affording the desired reductive coupling products in moderate yields (34–37). Similar to the ene-type coupling reaction, reductive coupling of a cyclopropane-containing alkyne and Tulipalin A produced the corresponding product 38 without ring-opening of cyclopropane. Moreover, a terminal alkene on the side alkyl chain of a phenyl alkyl alkyne was also tolerated in this reductive coupling reaction (39).
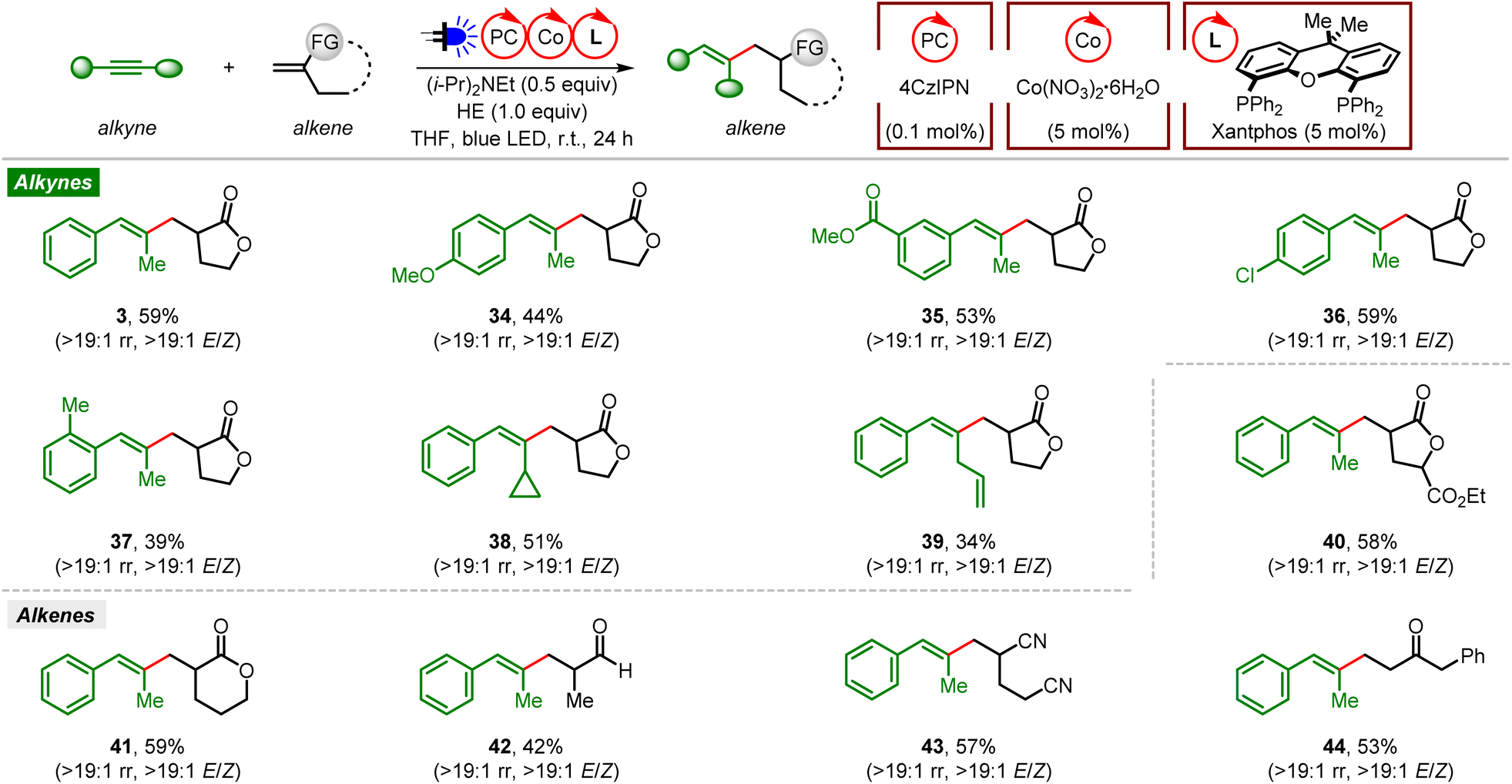 | ||
| Fig. 2 Reaction scope of organophotoredox Co-catalyzed reductive coupling of alkynes and alkenes. Reaction conditions: alkyne (0.2 mmol), alkene (3.0 equiv.), 4CzIPN (0.1 mol%), Co(NO3)2·6H2O (5 mol%), Ph-Phox (5 mol%), (i-Pr)2NEt (0.5 equiv.), HE (1.0 equiv.), THF (3 mL), 5 W blue LED, r.t., 24 h, isolated yield. | ||
On the other hand, various gem-disubstituted alkenes were tested with 1-phenylpropyne under these conditions. Reductive coupling of alkynes with analogs of Tulipalin A or 2-methylene-5-pentanolide proved to be successful, affording the corresponding products in good yields (40 and 41). Methacrolein and linear alpha-alkyl substituted acrylonitrile were also compatible in this reaction (42 and 43). Furthermore, reductive coupling of 1-phenylpropyne and a simple vinyl ketone also occurred smoothly, leading to the desired product 44 in a moderate yield. Unfortunately, no reductive coupling occurred with 1,1-dialkyl substituted ethene, α-methylstyrene, or cyclic or linear acrylamide either.
A variety of synthetic transformations have been carried out to demonstrate the synthetic uses of the two organophotoredox Co-catalyzed cross-coupling reactions (Scheme 2). First, scale-up reactions of both cross-couplings were performed for the synthesis of 3 and 4 (Scheme 2A). The scale-up reductive coupling reaction produced the stereodefined trisubstituted alkene 3 in 51% yield with >19:1 regioselectivity and >19:1 E/Z stereoselectivity under the standard conditions. The scale-up ene-type coupling reaction also occurred smoothly, affording the desired product 4 in 75% yield without any loss of regio- and enantioselectivity control using 2 mol% Co(NO3)2·6H2O/Ph-Phox as the catalyst with a slight increase of the catalyst loading of 4CzIPN (0.4 mol%).
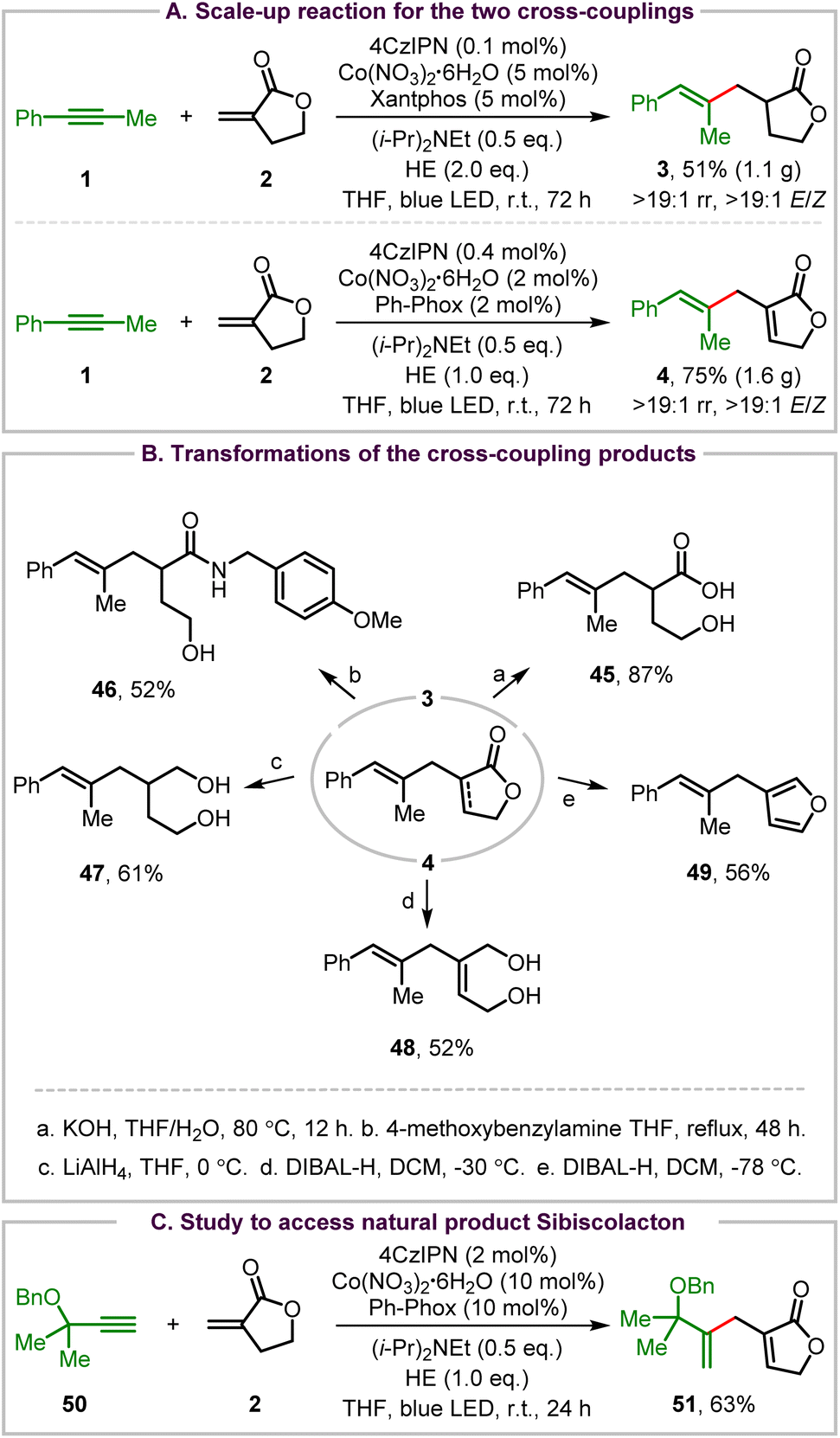 | ||
| Scheme 2 Synthetic applications. | ||
Then, selective transformations of the reductive coupling and ene-type coupling products were investigated. Hydrolysis of the reductive coupling product 3 provided γ-hydroxy carboxylic acid 45 in an excellent yield (Scheme 2Ba). And aminolysis of 3 with 4-methoxybenzylamine afforded γ-hydroxy amide 46 in a moderate yield with incomplete conversion of 3 (Scheme 2Bb). Next, reduction of the 2-furanone group of the ene-type coupling product 4 with LiAlH4 afforded alkene-containing 1,4-diol 47 in 61% yield (Scheme 2Bc). However, reduction of 2-furanone of 4 with DIBAL-H at −30 °C generated allylic diol 48 in 52% yield (Scheme 2Bd). Interestingly, a reductive isomerization of 4 took place, leading to furan-containing trisubstituted alkene 49 in 56% yield when the reaction was performed with DIBAL-H at −78 °C (Scheme 2Be).
Although 2-methyl-3-butyn-2-ol failed to produce the ene-type coupling product, its benzyl-protected derivative 50 was further tested for coupling with Tulipalin A to access the natural product sibiscolacton (Scheme 1A).23 Interestingly but unfortunately, selective ene-type coupling at the internal sp carbon of this terminal alkyne occurred smoothly, leading to product 51 in 63% yield (Scheme 2C). This result also indicates that the regioselectivity control of an alkyne is determined by both the electronic and steric effect of the substituents.
To explore the reaction mechanism of this visible light organophotoredox Co-catalyzed cross-coupling reaction, a range of control experiments were conducted (Scheme 3). First, deuterium labeling experiments were carried out using d3-HE with CoCl2 as the catalyst to exclude the influence of water in Co(NO3)2·6H2O under the standard conditions. The 1,4-diene 4 was obtained without any deuterium atom incorporation in the presence of d3-HE, indicating that HE does not provide the hydrogen atom in the ene-type coupling reaction (Scheme 3A). When d2-Tulipalin A (d2-2) was used as the coupling partner, there was no deuterium atom scrambling, with deuterium totally maintained at the methylene carbon of the Tulipalin A motif (Scheme 3B). These results demonstrate that the transfer of the allylic C–H of Tulipalin A to the alkyne triple bond might take place in this reaction. Then a radical inhibition experiment was also carried out with 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) as the radical trapping reagent. Although the radical trapping product was not observed, the ene-type reaction was indeed suppressed, revealing that some free radical intermediates might exist in this reaction (Scheme 3C). Finally, Co-catalyzed cross-coupling of 1 and 2 was carried out using excess amounts of manganese powder as the reductant. A trace amount of 4 was observed, which indicates the high efficiency of the photoredox cobalt dual catalytic system (Scheme 3D).
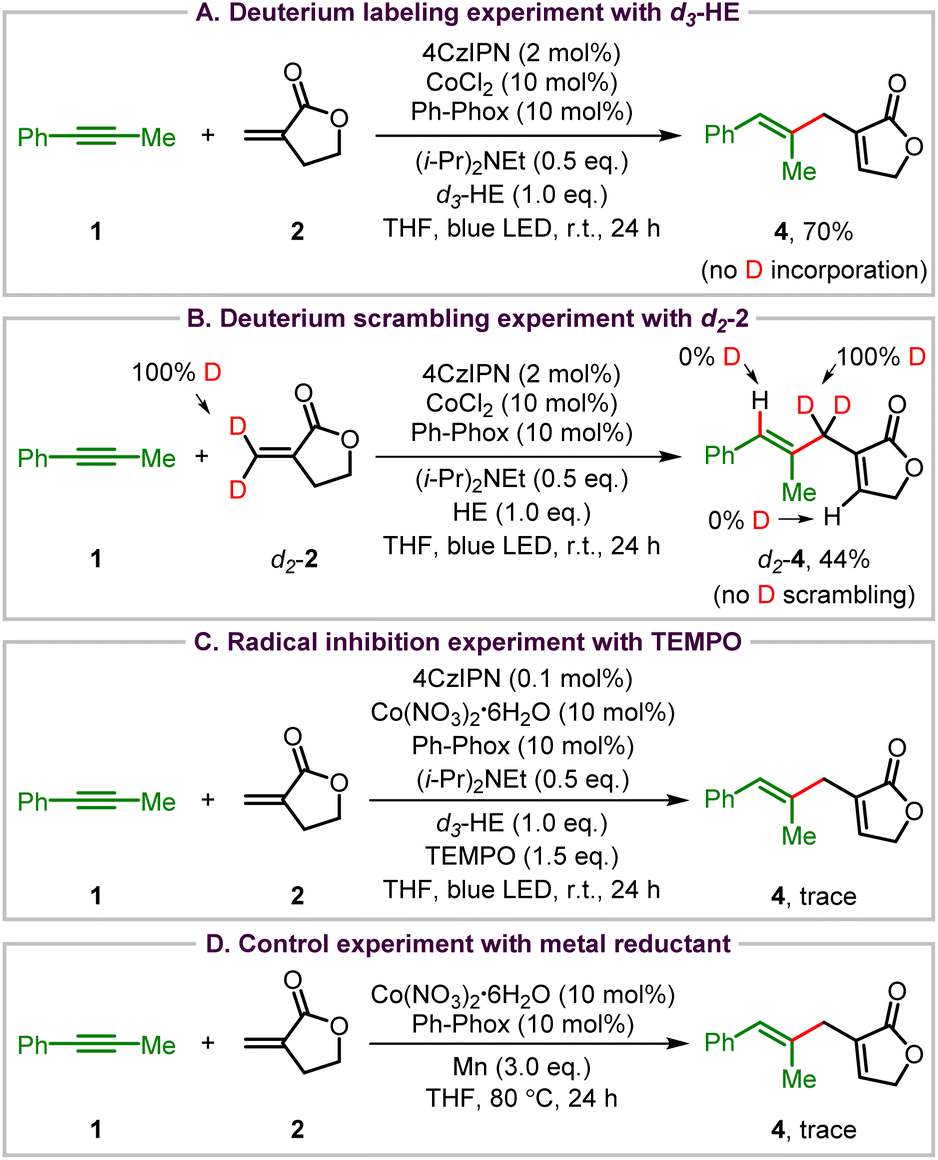 | ||
| Scheme 3 Control experiments and mechanistic studies. | ||
Based on the control experiments and previous reports, we have proposed the possible catalytic cycles of the two coupling reactions (Scheme 4). With coupling of Tulipalin A as the example, upon irradiation with blue LED light, single electron oxidation of HE [Eox1/2 (HE/HE˙+) = +0.51 V vs. Fc+/Fc in MeCN]24 with photoexcited 4CzIPN* produces HE˙+ and 4CzIPN˙−[Ered1/2(4CzIPN*/4CzIPN˙−) = +1.43 V vs. SCE in MeCN].22 Next, reduction of the ligand coordinated Co(II) complex [Ered1/2 (CoII/CoI) = −0.75 V vs. SCE in MeCN]21c with 4CzIPN˙− [Ered1/2 (4CzIPN/4CzIPN˙−) = −1.24 V vs. SCE in MeCN]25 affords the low valent Co(I) species and regenerates 4CzIPN. Then coordination of the alkyne and Tulipalin A to the Co(I) species followed by oxidative cyclization gives the crowded spirocyclic cobaltacyclopentene INT 1, which is the common intermediate of the two cross-couplings. The selective control of the ene-type coupling or reductive coupling is achieved by using different ligands. Using hemilabile Ph-Phox as the ligand, the alkenyl CoIII–H species INT 2 is formed by exocyclic β-Hb elimination (Path a). The following reductive elimination affords the 1,4-diene product and regenerates the CoI catalyst. The 1,3-diene product from endocyclic β-hydride elimination is not observed, probably due to cobalt and β-Ha is not in a syn coplanar arrangement. It is supposed that β-Hb elimination is much faster than β-Ha elimination in the spirocyclic cobaltacyclopentene INT 1. On the other hand, when Xantphos is used as the ligand, protolysis of INT 1 with HE˙+ takes place, generating HEH˙21c and the α-carbonyl alkyl CoIII species INT 3, which can be stabilized by the isomerized cobalt enolate (Path b). Next, the strong electron donor HEH˙ (Eox = −1.13 V vs. SCE) can also donate a radical to 4CzIPN*, leading to 4CzIPN˙− and the pyridinium ion PyH+.26 Furthermore, single electron reduction of CoIII species INT 3 with 4CzIPN˙− affords CoII species INT 4 [Ered1/2 (CoIII/CoII) = −0.51 V vs. Fc+/Fc] and 4CzIPN.27 Finally, protonation of the alkyl Co(II) species INT 4 with PyH+ produces a stereodefined alkene as the reductive coupling product and HP as the by-product. It is surprising and interesting that the β-hydride elimination occurs efficiently under this photoredox reductive conditions in the presence of a suitable ligand.
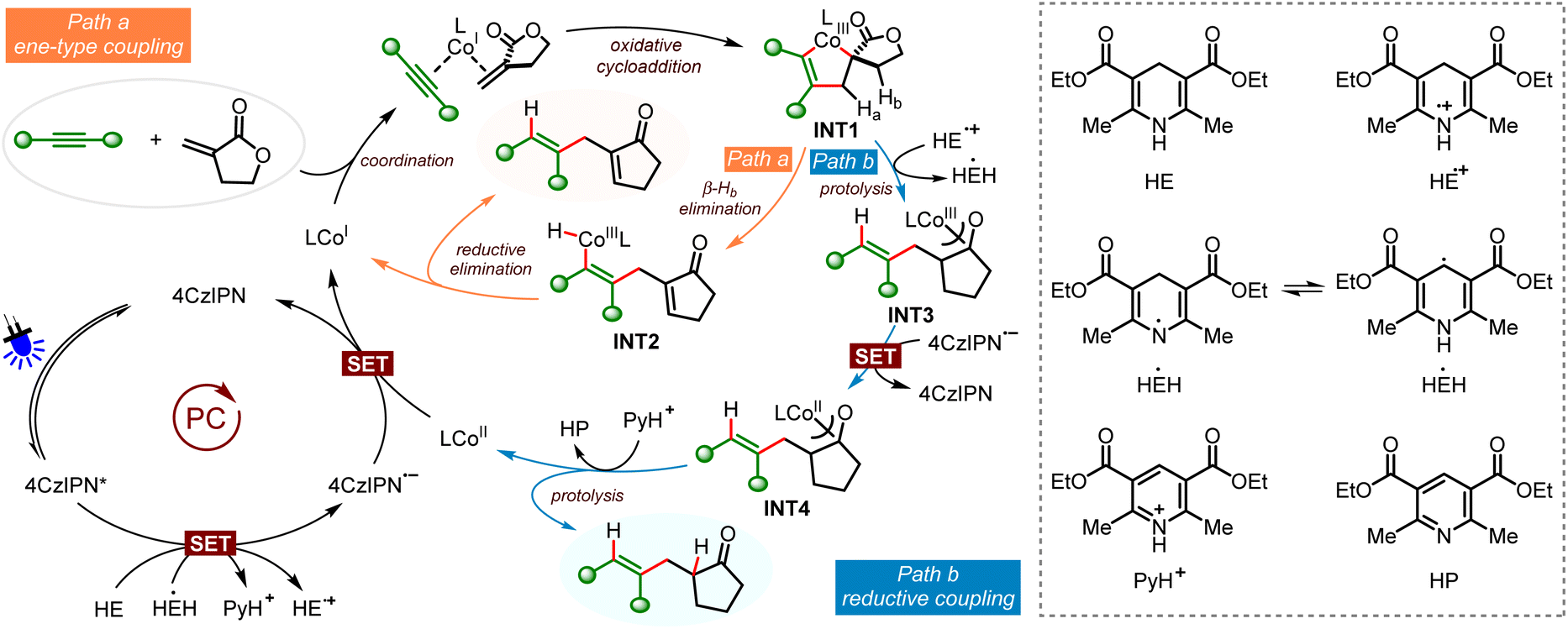 | ||
| Scheme 4 The proposed reaction pathway. | ||
Conclusions
In summary, we reported here the first general method of ligand-controlled ene-type or reductive coupling of alkynes and gem-disubstituted alkenes via visible light organophotoredox cobalt dual catalysis. Using the natural product Tulipalin A and other gem-disubstituted alkenes to couple with internal or terminal alkynes, functionalized 1,4-dienes have been obtained via ene-type coupling in moderate to good yields with excellent chemo-, regio-, and stereoselectivities using a PHOX ligand. Similarly, stereodefined trisubstituted alkenes were synthesized via reductive coupling with Xantphos as the ligand under otherwise similar conditions. Notably, as low as a thousandth of a commercially available organic photocatalyst has been used to efficiently catalyze the two coupling reactions. In general, this study shows that coupling of alkynes with multisubstituted alkenes is much difficult compared to well-studied monosubstituted alkenes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (22271296 and 21772208), the Major Science and Technology Project of Gansu Province (22ZD6GA003), the Natural Science Foundation of Jiangsu Province (BK20201193), and the Major Program of the Lanzhou Institute of Chemical Physics (No. ZYFZFX-1).References
- (a) B. H. B. Kwok, B. Koh, M. I. Ndubuisi, M. Elofsson and C. M. Crews, The Anti-Inflammatory Natural Product Parthenolide from the Medicinal Herb Feverfew Directly Binds to and Inhibits Iκb Kinase, Chem. Biol., 2001, 8, 759–766 CrossRef CAS PubMed; (b) R. R. A. Kitson, A. Millemaggi and R. J. K. Taylor, The Renaissance of α-Methylene-γ-butyrolactones: New Synthetic Approaches, Angew. Chem., Int. Ed., 2009, 48, 9426–9451 CrossRef CAS PubMed; (c) R. Lagoutte, C. Serba, D. Abegg, D. G. Hoch, A. Adibekian and N. Winssinger, Divergent Synthesis and Identification of the Cellular Targets of Deoxyelephantopins, Nat. Commun., 2016, 7, 12470–12480 CrossRef CAS PubMed; (d) J. Hur, J. Jang and J. Sim, Review of the Pharmacological Activities and Recent Synthetic Advances of γ-Butyrolactones, Int. J. Mol. Sci., 2021, 22, 2769–2817 CrossRef CAS PubMed.
- (a) A. Hashem and E. Kleinpeter, The Chemistry of 2(5H)-furanones, Adv. Heterocycl. Chem., 2001, 81, 107–165 CrossRef CAS; (b) T. G. Elford and D. G. Hall, Advances in 2-(Alkoxycarbonyl)allylboration of Carbonyl Compounds and Other Direct Methods for the Preparation of α-Exo-Alkylidene γ-Lactones, Synthesis, 2010, 893–907 CAS; (c) B. Mao, M. Fananas-Mastral and B. L. Feringa, Catalytic Asymmetric Synthesis of Butenolides and Butyrolactones, Chem. Rev., 2017, 117, 10502–10566 CrossRef CAS PubMed; (d) K. J. R. Murauski, A. A. Jaworski and K. A. Scheidt, A Continuing Challenge: N-Heterocyclic Carbene-Catalyzed Syntheses of γ-Butyrolactones, Chem. Soc. Rev., 2018, 47, 1773–1782 RSC; (e) W. Liu and N. Winssinger, Synthesis of α-Exo-Methylene-γ-Butyrolactones: Recent Developments and Applications in Natural Product Synthesis, Synthesis, 2021, 53, 3977–3990 CrossRef CAS.
- (a) J.-P. Lepoittevin, V. Berl and E. Giménez-Arnau, α-Methylene-γ-Butyrolactones: Versatile Skin Bioactive Natural Products, Chem. Rec., 2009, 9, 258–270 CrossRef CAS PubMed; (b) J. McCluskey, M. Bourgeois and R. Harbison, Tulipalin A Induced Phytotoxicity, Int. J. Crit. Illn. Inj. Sci., 2014, 4, 181–183 CrossRef PubMed.
- (a) S. Agarwal, Q. Jin and S. Maji, in Biobased Monomers, Polymers, and Materials, American Chemical Society, 2012, vol. 1105, ch. 13, pp. 197–212 Search PubMed; (b) M. Mousa, H. Bergenudd, A. L. Kron and E. Malmström, Biobased Lactones-Exploring Their Free-Radical Polymerization and Polymer Properties, Macromolecules, 2021, 54, 6127–6134 CrossRef CAS; (c) K. Onita, M. Onishi, T. Omura, T. Wakiya, T. Suzuki and H. Minami, Preparation of Monodisperse Bio-Based Polymer Particles Via Dispersion Polymerization, Langmuir, 2022, 38, 7341–7345 CrossRef CAS PubMed.
- (a) A. Arcadi, M. Chiarini, F. Marinelli, Z. Berente and L. Kollàr, Palladium-Catalyzed Arylation of α-Methylene-γ-Butyrolactones: 3-Benzylfuran-2(5H)-ones Vs (Z)-Benzylidene-γ-Butyrolactones and Their Reduction to 3-Benzyl-γ-Butyrolactones, Org. Lett., 2000, 2, 69–72 CrossRef CAS PubMed; (b) K. Tsuchikama, Y. Kuwata and T. Shibata, Highly Enantioselective Construction of a Chiral Spirocyclic Structure by the [2+2+2] Cycloaddition of Diynes and Exo-Methylene Cyclic Compounds, J. Am. Chem. Soc., 2006, 128, 13686–13687 CrossRef CAS PubMed; (c) R. J. Mayer, P. W. A. Allihn, N. Hampel, P. Mayer, S. A. Sieber and A. R. Ofial, Electrophilic Reactivities of Cyclic Enones and α,β-Unsaturated Lactones, Chem. Sci., 2021, 12, 4850–4865 RSC.
- (a) M. Jeganmohan and C.-H. Cheng, Cobalt- and Nickel-Catalyzed Regio- and Stereoselective Reductive Coupling of Alkynes, Allenes, and Alkenes with Alkenes, Chem. – Eur. J., 2008, 14, 10876–10886 CrossRef CAS PubMed; (b) G. Hilt, Hydrovinylation Reactions–Atom-Economic Transformations with Steadily Increasing Synthetic Potential, Eur. J. Org. Chem., 2012, 4441–4451 CrossRef CAS; (c) P. Gandeepan and C.-H. Cheng, Cobalt Catalysis Involving π Components in Organic Synthesis, Acc. Chem. Res., 2015, 48, 1194–1206 CrossRef CAS PubMed; (d) M. Hirano, Recent Advances in the Catalytic Linear Cross-Dimerizations, ACS Catal., 2019, 9, 1408–1430 CrossRef CAS.
- (a) B. M. Trost, On Inventing Reactions for Atom Economy, Acc. Chem. Res., 2002, 35, 695–705 CrossRef CAS PubMed; (b) B. M. Trost and A. Indolese, Ruthenium-Catalyzed Addition of Alkenes to Acetylenes, J. Am. Chem. Soc., 1993, 115, 4361 CrossRef CAS; (c) H. Butenschön, Construction of Carbon Frameworks with the Help of Ruthenium Complexes: 1,5-Cyclooctadiene as a Reagent in Transition Metal Catalyzed Reactions, Angew. Chem., Int. Ed. Engl., 1994, 33, 636–638 CrossRef; (d) B. M. Trost, A. B. Pinkerton, F. D. Toste and M. Sperrle, Synthesis of 1,1-Disubstituted Alkenes Via a Ru-Catalyzed Addition, J. Am. Chem. Soc., 2001, 123, 12504–12509 CrossRef CAS PubMed; (e) B. M. Trost and J.-P. Surivet, An Atom-Economic Three-Carbon Chain Extension to Give Enamides, Angew. Chem., Int. Ed., 2001, 40, 1468–1471 CrossRef CAS; (f) A. C. Gutierrez and T. F. Jamison, Continuous Photochemical Generation of Catalytically Active [CpRu]+ Complexes from CpRu(η6-C6H6)PF6, Org. Lett., 2011, 13, 6414–6417 CrossRef CAS PubMed; (g) B. M. Trost, D. C. Koester and A. N. Herron, Stereocontrolled Synthesis of Vinyl Boronates and Vinyl Silanes Via Atom-Economical Ruthenium-Catalyzed Alkene–Alkyne Coupling, Angew. Chem., Int. Ed., 2015, 54, 15863–15866 CrossRef CAS PubMed.
- (a) G. Hilt and J. Treutwein, Cobalt-Catalyzed Alder–Ene Reaction, Angew. Chem., Int. Ed., 2007, 46, 8500–8502 CrossRef CAS PubMed; (b) G. Hilt, F. Erver and K. Harms, Regioselective Cobalt-Catalyzed Alder-Ene Reaction toward Silicon- and Boron-Functionalized Building Blocks, Org. Lett., 2011, 13, 304–307 CrossRef CAS PubMed; (c) F. Erver and G. Hilt, Cobalt- Versus Ruthenium-Catalyzed Alder–Ene Reaction for the Synthesis of Credneramide A and B, J. Org. Chem., 2012, 77, 5215–5219 CrossRef CAS PubMed.
- S. Mannathan and C.-H. Cheng, Cobalt-catalyzed Regio- and Stereoselective Intermolecular Enyne Coupling: An Efficient Route to 1,3-Diene Derivatives, Chem. Commun., 2010, 46, 1923–1925 RSC.
- H. Horie, I. Koyama, T. Kurahashi and S. Matsubara, Nickel-Catalyzed Intermolecular Codimerization of Acrylates and Alkynes, Chem. Commun., 2011, 47, 2658–2660 RSC.
- Y. Shibata, M. Hirano and K. Tanaka, Rhodium-catalyzed Regio- and Stereoselective Codimerization of Alkenes and Electron-Deficient Internal Alkynes Leading to 1,3-Dienes, Org. Lett., 2008, 10, 2829–2831 CrossRef CAS PubMed.
- (a) T. Nishimura, Y. Washitake and S. Uemura, Ruthenium/Halide Catalytic System for C-C Bond Forming Reaction between Alkynes and Unsaturated Carbonyl Compounds, Adv. Synth. Catal., 2007, 349, 2563–2571 CrossRef CAS; (b) N. M. Neisius and B. Plietker, The Ruthenium-Catalyzed Hydrovinylation of Internal Alkynes by Acrylates: An Atom Economic Approach to Highly Substituted 1,3-Dienes, Angew. Chem., Int. Ed., 2009, 48, 5752 CrossRef CAS PubMed; (c) J. Zhang, A. Ugrinov, Y. Zhang and P. Zhao, Exploring Bis(Cyclometalated) Ruthenium(II) Complexes as Active Catalyst Precursors: Room-Temperature Alkene–Alkyne Coupling for 1,3-Diene Synthesis, Angew. Chem., Int. Ed., 2014, 53, 8437–8440 CrossRef CAS PubMed.
- A. T. Lindhardt, M. L. H. Mantel and T. Skrydstrup, Palladium-Catalyzed Intermolecular Ene–Yne Coupling: Development of an Atom-Efficient Mizoroki–Heck-Type Reaction, Angew. Chem., Int. Ed., 2008, 47, 2668–2672 CrossRef CAS PubMed.
- M. M. Parsutkar, V. V. Pagar and T. V. RajanBabu, Catalytic Enantioselective Synthesis of Cyclobutenes from Alkynes and Alkenyl Derivatives, J. Am. Chem. Soc., 2019, 141, 15367–15377 CrossRef CAS PubMed.
- (a) A. Herath, B. B. Thompson and J. Montgomery, Catalytic Intermolecular Reductive Coupling of Enones and Alkynes, J. Am. Chem. Soc., 2007, 129, 8712–8713 CrossRef CAS PubMed; (b) S. Mannathan and C.-H. Cheng, Synthesis of Trans-Disubstituted Alkenes by Cobalt-Catalyzed Reductive Coupling of Terminal Alkynes with Activated Alkenes, Chem. – Eur. J., 2012, 18, 11771–11777 CrossRef CAS PubMed; (c) M. J. González and B. Breit, Visible-Light-Driven Intermolecular Reductive Ene–Yne Coupling by Iridium/Cobalt Dual Catalysis for C(Sp3)−C(Sp2) Bond Formation, Chem. – Eur. J., 2019, 25, 15746–15750 CrossRef PubMed; (d) P. Rai, K. Maji and B. Maji, Photoredox/Cobalt Dual Catalysis for Visible-light-mediated Alkene–Alkyne Coupling, Org. Lett., 2019, 21, 3755–3759 CrossRef CAS PubMed; (e) S. Gao, C. Liu, J. Yang and J. Zhang, Cobalt-Catalyzed Electrochemical Reductive Coupling of Alkynes and Alkenes, Chin. J. Org. Chem., 2023, 43, 1559–1565 Search PubMed.
- (a) B. M. Trost and J. J. Cregg, Ruthenium-Catalyzed Alkene–Alkyne Coupling of Disubstituted Olefins: Application to the Stereoselective Synthesis of Trisubstituted Enecarbamates, J. Am. Chem. Soc., 2015, 137, 620–623 CrossRef CAS PubMed; (b) S. M. Rummelt, G.-J. Cheng, P. Gupta, W. Thiel and A. Fürstner, Hydroxy-Directed Ruthenium-catalyzed Alkene/Alkyne Coupling: Increased Scope, Stereochemical Implications, and Mechanistic Rationale, Angew. Chem., Int. Ed., 2017, 56, 3599–3604 CrossRef CAS PubMed; (c) S. Kiyota, S. In, N. Komine and M. Hirano, Regioselectivity Control by Added MeCN in Ru(0)-Catalyzed Cross-Dimerization of Internal Alkynes with Methyl Methacrylate, Chem. Lett., 2017, 46, 1040–1043 CrossRef CAS.
- (a) D. Zhang, M. Li, J. Li, A. Lin and H. Yao, Rhodium-catalyzed Intermolecular Enantioselective Alder–Ene Type Reaction of Cyclopentenes with Silylacetylenes, Nat. Commun., 2021, 12, 6627–6634 CrossRef CAS PubMed; (b) G. Hilt, A. Paul and J. Treutwein, Cobalt Catalysis at the Crossroads: Cobalt-Catalyzed Alder−Ene Reaction Versus [2+2] Cycloaddition, Org. Lett., 2010, 12, 1536–1539 CrossRef CAS PubMed.
- (a) C.-H. Wei, S. Mannathan and C.-H. Cheng, Enantioselective Synthesis of β-Substituted Cyclic Ketones Via Cobalt-Catalyzed Asymmetric Reductive Coupling of Alkynes with Alkenes, J. Am. Chem. Soc., 2011, 133, 6942–6944 CrossRef CAS PubMed; (b) K. Cui, Y.-L. Li, G. Li and J.-B. Xia, Regio- and Stereoselective Reductive Coupling of Alkynes and Crotononitrile, J. Am. Chem. Soc., 2022, 144, 23001–23009 CrossRef CAS PubMed.
- (a) J. Xuan and W.-J. Xiao, Visible-Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (b) N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Photocatalysis in Organic and Polymer Synthesis, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC; (c) M. D. Kärkäs, J. A. Porco Jr. and C. R. J. Stephenson, Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed; (d) B. Chen, L.-Z. Wu and C.-H. Tung, Photocatalytic Activation of Less Reactive Bonds and Their Functionalization Via Hydrogen-Evolution Cross-Couplings, Acc. Chem. Res., 2018, 51, 2512–2523 CrossRef CAS PubMed; (e) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Alkoxy Radicals See the Light: New Paradigms of Photochemical Synthesis, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed; (f) N. Holmberg-Douglas and D. A. Nicewicz, Photoredox-Catalyzed C–H Functionalization Reactions, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed; (g) S. P. Pitre and L. E. Overman, Strategic Use of Visible-Light Photoredox Catalysis in Natural Product Synthesis, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed.
- (a) K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (b) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Alkyl Carbon–Carbon Bond Formation by Nickel/Photoredox Cross-Coupling, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed; (c) H.-H. Zhang, H. Chen, C. Zhu and S. Yu, A Review of Enantioselective Dual Transition Metal/Photoredox Catalysis, Sci. China: Chem., 2020, 63, 637–647 CrossRef CAS; (d) F.-D. Lu, J. Chen, X. Jiang, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Recent Advances in Transition-Metal-Catalysed Asymmetric Coupling Reactions with Light Intervention, Chem. Soc. Rev., 2021, 50, 12808–12827 RSC; (e) K. Kwon, R. T. Simons, M. Nandakumar and J. L. Roizen, Strategies to Generate Nitrogen-Centered Radicals That May Rely on Photoredox Catalysis: Development in Reaction Methodology and Applications in Organic Synthesis, Chem. Rev., 2022, 122, 2353–2428 CrossRef CAS PubMed.
- (a) K.-H. He, F.-F. Tan, C.-Z. Zhou, G.-J. Zhou, X.-L. Yang and Y. Li, Acceptorless Dehydrogenation of N-Heterocycles by Merging Visible-Light Photoredox Catalysis and Cobalt Catalysis, Angew. Chem., Int. Ed., 2017, 56, 3080–3084 CrossRef CAS PubMed; (b) S. M. Thullen and T. Rovis, A Mild Hydroaminoalkylation of Conjugated Dienes Using a Unified Cobalt and Photoredox Catalytic System, J. Am. Chem. Soc., 2017, 139, 15504–15508 CrossRef CAS PubMed; (c) J. Hou, A. Ee, W. Feng, J.-H. Xu, Y. Zhao and J. Wu, Visible-Light-Driven Alkyne Hydro-/Carbocarboxylation Using CO2 Via Iridium/Cobalt Dual Catalysis for Divergent Heterocycle Synthesis, J. Am. Chem. Soc., 2018, 140, 5257–5263 CrossRef CAS PubMed; (d) Q.-Y. Meng, T. E. Schirmer, K. Katou and B. König, Controllable Isomerization of Alkenes by Dual Visible-Light-Cobalt Catalysis, Angew. Chem., Int. Ed., 2019, 58, 5723–5728 CrossRef CAS PubMed; (e) M. Kojima and S. Matsunaga, The Merger of Photoredox and Cobalt Catalysis, Trends Chem., 2020, 2, 410–426 CrossRef CAS; (f) À. Cristòfol, B. Limburg and A. W. Kleij, Expedient Dual Co/Organophotoredox Catalyzed Stereoselective Synthesis of All-Carbon Quaternary Centers, Angew. Chem., Int. Ed., 2021, 60, 15266–15270 CrossRef PubMed; (g) M.-J. Zhou, L. Zhang, G. Liu, C. Xu and Z. Huang, Site-Selective Acceptorless Dehydrogenation of Aliphatics Enabled by Organophotoredox/Cobalt Dual Catalysis, J. Am. Chem. Soc., 2021, 143, 16470–16485 CrossRef CAS PubMed; (h) W.-L. Yu, Z.-G. Ren, K.-X. Ma, H.-Q. Yang, J.-J. Yang, H. Zheng, W. Wu and P.-F. Xu, Cobalt-Catalyzed Chemoselective Dehydrogenation through Radical Translocation under Visible Light, Chem. Sci., 2022, 13, 7947–7954 RSC; (i) H. Jiang, X.-K. He, X. Jiang, W. Zhao, L.-Q. Lu, Y. Cheng and W.-J. Xiao, Photoinduced Cobalt-Catalyzed Desymmetrization of Dialdehydes to Access Axial Chirality, J. Am. Chem. Soc., 2023, 145, 6944–6952 CrossRef CAS PubMed; (j) A. Suzuki, Y. Kamei, M. Yamashita, Y. Seino, Y. Yamaguchi, T. Yoshino, M. Kojima and S. Matsunaga, Photocatalytic Deuterium Atom Transfer Deuteration of Electron-Deficient Alkenes with High Functional Group Tolerance, Angew. Chem., Int. Ed., 2023, 62, e202214433 CrossRef CAS PubMed; (k) H. Yan, Q. Liao, Y. Chen, G. G. Gurzadyan, B. Lu, C. Wu and L. Shi, Photocatalytic Metal Hydride Hydrogen Atom Transfer Mediated Allene Functionalization by Cobalt and Titanium Dual Catalysis, Angew. Chem., Int. Ed., 2023, 62, e202302483 CrossRef CAS PubMed; (l) Z. Jia and S. Luo, Visible Light Promoted Direct Deuteration of Alkenes Via Co(III)-H Mediated H/D Exchange, CCS Chem., 2023, 5, 1069–1076 CrossRef CAS; (m) Y.-L. Li, S.-Q. Zhang, J. Chen and J.-B. Xia, Highly Regio- and Enantioselective Reductive Coupling of Alkynes and Aldehydes Via Photoredox Cobalt Dual Catalysis, J. Am. Chem. Soc., 2021, 143, 7306–7313 CrossRef CAS PubMed.
- (a) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence, Nature, 2012, 492, 234–238 CrossRef CAS PubMed; (b) J. Luo and J. Zhang, Donor–Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(Sp3)–C(Sp2) Cross-Coupling, ACS Catal., 2016, 6, 873–877 CrossRef CAS.
- (a) B. Li, J.-B. Lee, K. Hayashi, Y. Ito, W.-J. Sun, X. Wang, Y. Kano and T. Hayashi, Two New Monoterpenes from Sibiraea Angustata, J. Nat. Med., 2010, 64, 89–92 CrossRef CAS PubMed; (b) Y. Deng, J.-Q. Zhao, L.-J. Mei and Y.-D. Tao, Two New Monoterpenes from Sibiraea Laevigata, J. Asian Nat. Prod. Res., 2017, 19, 877–883 CrossRef CAS PubMed.
- (a) L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, Enantioselective Photoredox Catalysis Enabled by Proton-Coupled Electron Transfer: Development of an Asymmetric Aza-Pinacol Cyclization, J. Am. Chem. Soc., 2013, 135, 17735–17738 CrossRef CAS PubMed; (b) R. A. Aycock, D. B. Vogt and N. T. Jui, A Practical and Scalable System for Heteroaryl Amino Acid Synthesis, Chem. Sci., 2017, 8, 7998–8003 RSC; (c) P.-Z. Wang, J.-R. Chen and W.-J. Xiao, Hantzsch Esters: An Emerging Versatile Class of Reagents in Photoredox Catalyzed Organic Synthesis, Org. Biomol. Chem., 2019, 17, 6936–6951 RSC; (d) J. A. Leitch, T. Rossolini, T. Rogova and D. J. Dixon, α-Tertiary Dialkyl Ether Synthesis Via Reductive Photocatalytic α-Functionalization of Alkyl Enol Ethers, ACS Catal., 2020, 10, 11430–11437 CrossRef CAS.
- E. Speckmeier, T. G. Fischer and K. Zeitler, A Toolbox Approach to Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor–Acceptor Cyanoarenes, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed.
- G. Park, S. Y. Yi, J. Jung, E. J. Cho and Y. You, Mechanism and Applications of the Photoredox Catalytic Coupling of Benzyl Bromides, Chem. – Eur. J., 2016, 22, 17790–17799 CrossRef CAS PubMed.
- N. Elgrishi, D. A. Kurtz and J. L. Dempsey, Reaction Parameters Influencing Cobalt Hydride Formation Kinetics: Implications for Benchmarking H2-Evolution Catalysts, J. Am. Chem. Soc., 2017, 139, 239–244 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qo01281f |
| This journal is © the Partner Organisations 2023 |