
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light-mediated hydrogen atom transfer and proton transfer for the conversion of (2-vinylaryl)methanol derivatives to aryl aldehydes or aryl ketones†
Jun
Yan
a,
Ziqi
Yu
 a,
Hao-Zhao
Wei
a,
Min
Shi
*ab and
Yin
Wei
*b
a,
Hao-Zhao
Wei
a,
Min
Shi
*ab and
Yin
Wei
*b
aKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: mshi@mail.sioc.ac.cn
bState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Science, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: mshi@mail.sioc.ac.cn; weiyin@sioc.ac.cn
First published on 12th October 2023
Abstract
In this paper, we report a photochemical strategy for the visible light-mediated efficient conversion of (2-vinylaryl)methanol derivatives to the corresponding aryl aldehydes or aryl ketones in moderate to excellent yields with broad substrate scope under mild conditions. This photochemical process takes place from the generation of the triplet state of olefins and involves 1,5-hydrogen atom transfer, enol tautomerization, and a subsequent proton transfer process. The plausible reaction mechanism has been verified by deuterium labeling and control experiments, kinetic and Stern–Volmer analyses, and DFT calculations.
Introduction
Visible light-mediated chemical transformations have become a powerful tool in synthetic chemistry1 and materials science2 for the efficient construction of novel chemical skeletons under mild conditions in the last decade. On the basis of visible light-mediated reaction modes of single electron transfer (SET) and energy transfer (EnT), various robust organic transformations can be achieved under mild conditions, such as C–H bond functionalizations,3 C–C and C–X (X = heteroatom) bond formations as well as their selective cleavage and the subsequent downstream functionalizations and so on.4 The SET pathway relies heavily on a redox process between a photosensitizer and a reaction substrate or a reaction intermediate,5 so the excited-state photosensitizer often performs the role of an oxidant or a reductant. For example, in transition metal-catalyzed reductive coupling reactions,6 the photoredox strategy is often used in place of a stoichiometric metal reductant, and hence the reaction can be performed under more environmentally-benign conditions. It is worth noting that the EnT reaction mode is also an effective way to obtain highly reactive intermediates in the excited state (or the ground state) under mild conditions.7 These highly reactive excited intermediates often undergo pivotal chemical transformations that are not possible with regard to their corresponding ground states.Olefins as unsaturated hydrocarbons are remarkably valuable synthetic scaffolds in organic synthetic chemistry.8 Excited olefins allow for many chemical transformations such as atom transfer reactions,9 [2 + 2] cyclizations,10 and E → Z isomerizations11 that cannot be realized in the ground state. Nevertheless, the direct excitation of most olefinic compounds via visible light irradiation is disfavored, basically due to the comparatively low absorption at longer wavelengths. In general, harsh UV light irradiation is required for the direct excitation of styryl organic molecules, which leads to negative impacts on the selectivity, functional group tolerance and general applicability of the reaction, as well as competitive and uncontrollable side reactions.12,13 For example, the conversion of (2-(1-phenylvinyl)phenyl)methanol to the corresponding 2-(1-phenylethyl)benzaldehyde under UV irradiation was reported by Hornback's group in 1979 (Scheme 1a).14 As shown in Scheme 1a, it must be emphasized here that due to the harsh UV irradiation, the photochemical reaction product was only obtained in 6.3% yield in the absence of a photosensitizer, and even with the addition of xanthone as a photosensitizer, the corresponding benzaldehyde was obtained only in 15.1% yield. The low yield is probably due to the Norrish-type reaction15 of 2-(1-phenylethyl)benzaldehyde under UV light irradiation, which leads to the decomposition of the obtained product. Therefore, it is conceivable that the use of the recently developed visible light-mediated strategy can avoid the further excitation of the formed aldehyde and realize an effective photochemical conversion of (2-vinylaryl)methanol derivatives to aryl aldehydes or aryl ketones.
 | ||
| Scheme 1 Previous work and this work. | ||
Based on the above description and our previous work,9c,d we envisaged that the triplet state of (2-vinylaryl)methanol derivatives would be achieved through an EnT process upon visible light irradiation in corporation with a photocatalyst, in which the unprotected benzyloxy functional group could be used as a hydrogen atom transfer (HAT) partner and the subsequent enol interconversion isomerization as well as proton transfer would take place, resulting in an efficient conversion to aryl aldehydes or aryl ketones (Scheme 1b, this work). In this scenario, we anticipated that the desired product can be obtained in high yield along with broad substrate scope.
To test our working hypothesis, we started our investigation by using (2-(cyclopropylidene(phenyl)methyl)phenyl)methanol 1a (0.2 mmol, 1.0 equiv.) as a substrate, Ir(dF-CF3-ppy)2(dtbbpy)PF6 (2 mol%) as a photosensitizer, and degassed MeCN (2.0 mL) as the solvent under irradiation with 5 W blue LEDs for 12 hours under an argon atmosphere. To our delight, the desired aromatic aldehyde product 2a was obtained in 94% isolated yield (Table 1, entry 1). However, when using photosensitizers such as [Ir(2′,4′-dF-5-CF3-ppy)2(bpy)]PF6 having a slightly lower triplet state energy (Table 1, entry 2) and Ir(dFppy)2pic having a higher triplet state energy (Table 1, entry 3) as the photocatalysts, 2a was obtained in lower NMR yields of 31% and 25%, respectively. The use of fac-Ir(ppy)3, [Ru(phen)3](PF6)2, and [Ru(bpy)3](PF6)2 as the photocatalysts, which have lower triplet state energies, did not give 2a at all (Table 1, entries 4–6). When xanthone was used as a photosensitizer, the expected product 2a was not detected (Table 1, entry 7). This is probably due to that xanthone (λmax = 340 nm) is not absorbed in the visible light region.16 Notably, this photosensitizer was used by Hornback's group in the photochemical synthesis of benzaldehyde. The solvent effects on the reaction outcome were then examined, including dichloromethane (Table 1, entry 8), tetrahydrofuran (Table 1, entry 9), and toluene (Table 1, entry 10), and they afforded 2a in reduced yields compared to MeCN. The substrate concentration did not have a significant impact on the yield of 2a as shown in entries 11 and 12 of Table 1. Control experiments elucidated that the target product was not obtained in the absence of a photosensitizer (Table 1, entry 13) or under dark conditions (Table 1, entry 14). When a higher power lamp was used as the light source, it speeded up the rate of the reaction, but did not increase the yield (Table 1, entry 15).
|
|
|||||
|---|---|---|---|---|---|
| Entry | PC | E T (kcal mol−1) | Solvent | Conc. [M] | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol) and PC (2 mol%) were added in degassed solvent (2.0 mL) under an Ar atmosphere for 12.0 h, in a sealed tube under 5 W blue LED light irradiation. b Yields were determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. c Isolated yield. d No light. e Under 100 W blue LEDs for 6 h. | |||||
| 1 | Ir(dF-CF3-ppy)2(dtbbpy)PF6 | 60.8 | MeCN | 0.1 | 94c |
| 2 | [Ir(2′,4′-dF-5-CF3-ppy)2(bpy)]PF6 | 60.4 | MeCN | 0.1 | 31 |
| 3 | Ir(dFppy)2pic | 61.1 | MeCN | 0.1 | 25 |
| 4 | fac-Ir(ppy)3 | 57.8 | MeCN | 0.1 | N.R |
| 5 | [Ru(phen)3](PF6)2 | 48.4 | MeCN | 0.1 | N.R |
| 6 | [Ru(bpy)3](PF6)2 | 46.5 | MeCN | 0.1 | N.R |
| 7 | Xanthone | 74.3 | MeCN | 0.1 | N.R |
| 8 | Ir(dF-CF3-ppy)2(dtbbpy)PF6 | DCM | 0.1 | 24 | |
| 9 | Ir(dF-CF3-ppy)2(dtbbpy)PF6 | THF | 0.1 | 29 | |
| 10 | Ir(dF-CF3-ppy)2(dtbbpy)PF6 | Toluene | 0.1 | 65 | |
| 11 | Ir(dF-CF3-ppy)2(dtbbpy)PF6 | MeCN | 0.05 | 85 | |
| 12 | Ir(dF-CF3-ppy)2(dtbbpy)PF6 | MeCN | 0.2 | 68 | |
| 13 | None | MeCN | 0.1 | N.R | |
| 14d | Ir(dF-CF3-ppy)2(dtbbpy)PF6 | MeCN | 0.1 | N.R | |
| 15e | Ir(dF-CF3-ppy)2(dtbbpy)PF6 | MeCN | 0.1 | 90 | |

Having determined the optimized conditions, the substrate scope of this newly developed photochemical reaction was investigated with a wide variety of substrates 1 and we found that most of them provided the corresponding arylaldehydes 2 smoothly in good to excellent yields (Table 2). For terminal olefins, substrates 1 with various substituted aryl groups as R1 moieties, regardless of whether they had electron-donating or electron-withdrawing groups, were tolerated in the protocol, delivering the target products 2b–2g in moderate to excellent yields ranging from 65% to 96%. Substrate 1h, in which R1 was a thienyl group, also afforded the corresponding arylaldehyde 2h in 83% yield. We also examined the effects of substituents at the aryl alcohol moiety on the reaction outcome, revealing that substrates 1i–1n having 4-Cl, 5-Cl, 4-Br, 5-Br, 4-Me, and 5-Me substituted aryl alcohols were all compatible in this transformation, providing the desired products 2i–2n in moderate to good yields ranging from 57% to 92% yields. For Br and methyl substituents, changing the position from 5-substitution to 4-substitution induced over a 30% increase in yield (2k and 2l, 2m and 2n). Therefore, we hypothesized that in terms of electronic effects, the Br and methyl substituents at the 4-position probably make the corresponding intermediate IV (shown in Scheme 4) unstable, thus leading to a decrease in yields (2l and 2m). Substrate 1o bearing a naphthalenemethanol unit as a HAT partner was also smoothly converted to the corresponding aldehyde 2o in 70% yield under the standard conditions. When using substrates 1p, 1q and 1r containing a trisubstituted olefinic moiety in this photochemical reaction, the corresponding products 2p–2r were also obtained in moderate yields under 100 W blue light irradiation. For substrates 1p, 1q, and 1r containing trisubstituted olefinic molecules, the E/Z isomerization occurred as a side reaction; thus, 100 W blue LED light was used to ensure that the 1,5-HAT process as a critical step was able to proceed more efficiently to give the desired products in moderate yields. The desired product 2p was obtained in 60% yield when using 1p (E/Z = 4/1) as a substrate. However, it is worth noting that the E/Z ratio of substrate 1q is 6/4, and the Z-trisubstituted olefin 1q′ cannot be excited under the standard conditions due to its higher EnT energy,11a thus leading to a significant decrease in the yield of 2q to 25% yield. Substrate 1r with a free hydroxyl group was also tolerated in this protocol and the target product 2r was obtained in 60% yield.
a Reaction conditions: 1 (0.2 mmol) and PC (2 mol%) were added in solvent (2.0 mL) under an Ar atmosphere, in a sealed tube under 5 W blue LED light irradiation.
b Yield of the isolated products.
c Using 100 W blue light irradiation.
d The E/Z ratio of substrate 1p is 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
e The E/Z ratio of substrate 1q is 6:4.
f A 6:1 mixture of MeCN and H2O was used as the solvent. :1.
e The E/Z ratio of substrate 1q is 6:4.
f A 6:1 mixture of MeCN and H2O was used as the solvent.
|
|---|

|
To further extend the scope of substrates 1, a series of substrates bearing a methylenecyclopropane unit were used in this newly explored visible light-mediated photochemical reaction, and the corresponding products were obtained in good to excellent yields. For substrates 1s–1w, in which R1 was a substituted aryl group, the desired products 2s–2w were also successfully obtained in good to high yields ranging from 60% to 95% regardless of whether the aryl group had an electron-withdrawing group or an electron-donating group. In addition, for substrates 1x and 1y, in which R1 was a 2-thienyl group and a 2-naphthyl group, the reactions proceeded smoothly, delivering the desired products 2x and 2y in 85% and 70% yields, respectively, although a prolonged reaction time was required. This protocol also performed very well for substrate 1z with a methylenecyclobutane unit, providing the desired aldehyde 2z in 59% yield. Moreover, when substrates 1aa (R1 = Me) and 1ab (R1 = H) were used in the reaction, the corresponding aldehydes 2aa and 2ab were obtained in 34% and 24% yields, respectively, despite that the photochemical reactivity decreased dramatically. For the monoaryl substrates 1aa and 1ab, the excitation efficiency was lower than that of the diaryl substrates; thus, 100 W blue LED light is required.
Next, we turned our attention to examine the substrate scope of secondary alcohols. For substrates 1ac and 1ad, in which R3 = Me, the reactions proceeded smoothly, furnishing the corresponding acetophenones 2ac and 2ad in moderate yields of 60% and 81%, respectively. When the methyl group was changed to a sterically hindered group such as an n-butyl, an isobutyl, or an isopropyl group or a 2-ethyl-1,3-dioxolane moiety, the desired products 2ae–2ah were also obtained in moderate yields ranging from 70% to 79%. It is worth noting that in these cases, additional water was necessary to be added into the reaction system as a proton source to accelerate the proton transfer process efficiently.17 Unfortunately, some limitations of this newly developed HAT and proton transfer reaction were observed for several substrates. For example, when non-cyclic tetrasubstituted olefin 1ai was employed as a substrate, no reaction occurred under the standard conditions, probably due to the steric hindrance leading to the non-occurrence of the 1,5-HAT process. Furthermore, none of the desired products 2aj and 2al were detected for substrates 1aj and 1ak containing E-disubstituted olefins, and most of them isomerized to the corresponding Z-olefins 1aj′ and 1ak′ under the standard conditions.11 For the monoaryl disubstituted olefins 1aj and 1ak, the desired reactions did not occur, probably due to that they were more difficult to excite, as well as the presence of E/Z isomerization. When the OH group was changed to the SH group (1al), the reaction system became complex.
To further investigate the reaction mechanism, a series of deuteration experiments and controlled experiments were carried out. First, the kinetic isotope effect of this photochemical reaction was determined using the mono-deuterated substrate [D1]-1a, and kH/kD ≈ 5.7 was obtained for the formation of [D1]-2a, indicating that the C–H bond homolytic cleavage is involved in the rate-determining step (Scheme 2a) (see page S13 in the ESI†). Subsequently, the double-deuterated substrate [D2]-1a was used in the reaction under the standard conditions, providing the target product [D2]-2a in 33% yield along with >99% deuterium incorporation (Scheme 2b). These obtained results are consistent with our working assumption that the olefinic biradical species at the triplet state undergoes a 1,5-HAT process with the C(sp3)–H bond at the benzylic alcohol position to give an open-shell biradical species. To clarify another benzylic proton source in the product, we carried out this reaction using substrate [D1]-1a′, in which the alcoholic hydrogen atom was deuterated, giving the deuterated product in 91% yield along with 87% deuterium incorporation, suggesting that the benzylic proton of [D1]-2a′ indeed comes from the alcoholic hydrogen atom in the substrate (Scheme 2c). As shown in Scheme 2d, when the reaction was carried out using 1ae as a substrate under the standard conditions in anhydrous MeCN, the reaction efficiency dropped down significantly, affording the desired product 2ae in only 33% yield, and benzocyclobutane 3 was detected in 51% yield as a diastereomeric mixture, which was produced by [2 + 2] electrocyclization of the conjugated diene intermediate in the reaction.9d However, when the solvent was replaced with a mixture of MeCN and H2O (6:1), the target product 1ae was obtained in 78% yield as a sole product under otherwise identical conditions (Scheme 2e) (see Table 2 as well). This result suggests that the ambient H2O in the reaction system is likely to accelerate the proton transfer process,17 and as for substrates bearing sterically large groups, the reaction efficiency can be improved.
 | ||
| Scheme 2 Deuterium labeling and control experiments. | ||
To further elucidate the interaction details of substrate 1 and the photocatalyst Ir(dF-CF3-ppy)2(dtbbpy)PF6, a fluorescence quenching experiment was performed using substrate 1a with Ir(dF-CF3-ppy)2(dtbbpy)PF6 (Fig. 1a) (see page S16 in the ESI†). From Stern–Volmer analysis, it can be seen that the emission of Ir(dF-CF3-ppy)2(dtbbpy)PF6 was effectively quenched by substrate 1a, suggesting that substrate 1a may be photoexcited by the photosensitizer via an EnT pathway upon 5 W blue LED irradiation (Fig. 1b). The oxidation potential of substrate 1a was determined by cyclic voltammetry as Eoxp/2 = +1.75 V vs. SCE (Fig. 1c) (see page S17 in the ESI†), showing that it is impossible for substrate 1a to undergo a SET process with the used photosensitizer (Ered (*IrIII/IrII) = 1.21 V vs. SCE). For the terminal olefinic substrate 1l, we also determined its oxidation potential as Eoxp/2 = +1.35 V vs. SCE, suggesting that all these olefinic substrates undergo an EnT process under the standard conditions (see page S17 in the ESI†). Meanwhile, the light–dark interval experiments (Fig. 1d) and the quantum yield of 0.13 for this photochemical reaction (see page S18 in the ESI†) indicated that continuous visible light irradiation is an essential element in the product formation process and the radical chain process might not be involved in the turnover process.
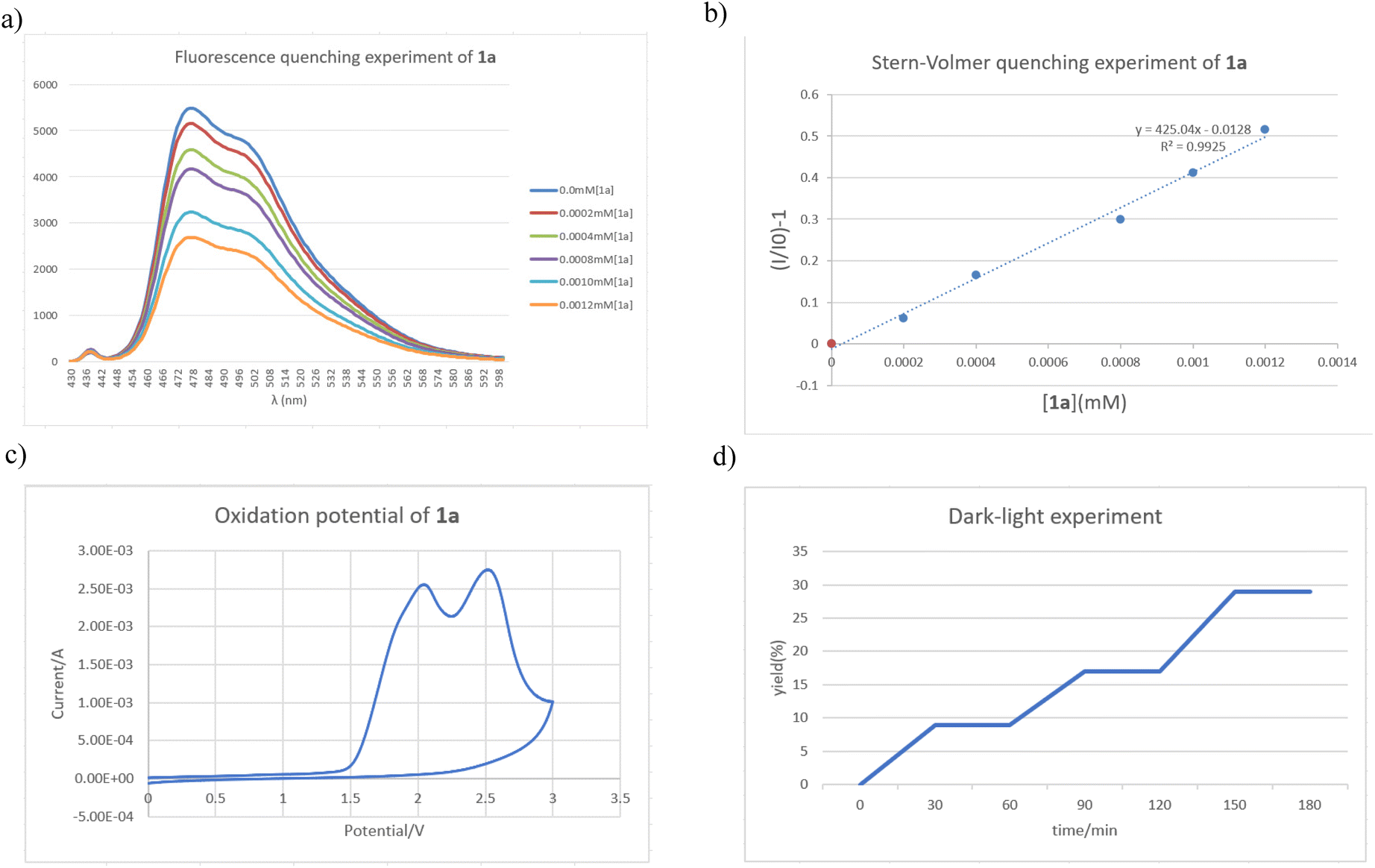 | ||
| Fig. 1 Mechanistic studies. (a) An appropriate amount of 1a was added to a 0.001 M photocatalyst solution in MeCN under an argon atmosphere and the emission from the sample was collected; (b) Stern–Volmer studies; (c) a solution of the substrate 1a in MeCN (0.1 M) was tested with 0.1 M Bu4NPF6 as the supporting electrolyte. Scan rate = 0.1 V s−1; (d) light on/off experiments. | ||
DFT calculations were performed to gain insight into the reaction mechanism. All calculations were performed at the SMD(CH3CN)/M06/def2tzvpp//M06/def2svp level with the Gaussian 16 program.18 The solvation Gibbs free energy profile in acetonitrile for the suggested reaction pathway is shown in Scheme 3 (see page S174 in the ESI†). The first triplet-state of 1a (int-1a) lies higher than the ground state of 1a by 44.5 kcal mol−1, and the Ir(dF-CF3-ppy)2(dtbbpy)PF6 in the excited state (60.8 kcal mol−1) can promote the formation of int-1avia an EnT pathway. Subsequently, int-1a undergoes 1,5-HAT viats-1a to form the open-shell biradical intermediate int-2a (20.8 kcal mol−1) with a reaction energy barrier of 11.8 kcal mol−1. The intersystem crossing of int-2a gives the closed-shell conjugated diene intermediate int-3a (18.9 kcal mol−1). Once int-3a is generated, the subsequent proton transfer takes place to generate the corresponding product 2a (−17.2 kcal mol−1) viats-2a with a reaction barrier of 0.5 kcal mol−1. We also examined the reaction barrier (ts-3a, ΔG‡ = 10.4 kcal mol−1) for int-3a to undergo electrocyclization to form benzocyclobutane 3a (3.0 kcal mol−1). The results show that the pathway to generate benzocyclobutane is unfavourable. For the reaction of the terminal olefin 1b, we also performed DFT calculations; the calculation results are shown in Scheme 3b and they are similar to those of substrate 1a, which undergoes the 1,5-HAT process and the subsequent proton transfer process. DFT calculations for the reaction of the substrate (1ae) having a secondary alcohol moiety were also performed (Scheme 3c). First, the energy transfer of the photosensitiser in the excited state occurs to obtain the excited-state intermediate int-1ae, and the energy of this state is 47.2 kcal mol−1 higher than that of 1a. Subsequently, int-1ae undergoes 1,5-HAT across a low reaction energy barrier (ts-1ae, ΔG‡ = 7.9 kcal mol−1) to form the biradical intermediate int-2ae (27.0 kcal mol−1). Considering that butyl is a sterically hindered group, we calculated the rotational energy barrier for the transformation of int-2ae into int-2ae′, which is 13.5 kcal mol−1 and indicates that both int-2ae and int-2ae′ can exist in the reaction system. DFT calculations show that int-3ae′ cannot be obtained through the direct rotation of int-3ae because the rotation barrier is 38.2 kcal mol−1 which is difficult to overcome under the standard reaction conditions. Finally, int-2ae′ undergoes proton transfer viats-2ae (ΔG‡ = 4.1 kcal mol−1) to produce the corresponding ketone 2ae; the intermediate int-2ae undergoes electrocyclization viats-3ae or ts3–3ae′ to give a pair of diastereomers 3 and 3′, (ΔΔG‡ = 0.2 kcal mol−1) which is basically in line with the dr value obtained in the experiment (dr = 3:2, see Scheme 2d).
 | ||
| Scheme 3 (a) DFT calculations on the reaction of 1a; (b) DFT calculations on the reaction of 1b; (c) DFT calculations on the reaction of 1ae. | ||
Based on the previous literature9 and our own experimental results above, a possible mechanism for this photochemical conversion is depicted in Scheme 4. First, the photosensitizer enters the excited state under visible light irradiation, which is quenched by substrate 1a through an EnT pathway to revert to the ground state, and 1a is converted to its triplet state as a biradical species I. Subsequently, the species I abstracts the hydrogen atom at the benzylic alcohol position via a 1,5-HAT process to produce the open-shell biradical intermediate II, followed by intersystem crossing to give the closed-shell conjugated diene intermediate III. Finally, intermediate IV is formed by an enol intercalative isomerization, and it undergoes a proton transfer process to give the corresponding product 2a.
 | ||
| Scheme 4 A possible mechanism. | ||
To demonstrate the synthetic utility of this newly explored photochemical reaction, a gram-scale reaction of 1a was executed and this reaction worked well, affording 2a in 91% yield (Scheme 5). Subsequently, 2a and 2b were subjected to a series of transformations under different reaction conditions. First, 2a underwent a Witting reaction in THF with the in situ generated phosphonium ylide to produce the corresponding substituted styrene 4 in 90% yield. Considering that product 2a contains an unactivated cyclopropyl group, an attempt was made to achieve ring-opening 1,3-hydroboration of cyclopropane in DCM at room temperature in the presence of phenylsilane and boron tribromide according to the previous literature;19 however, the corresponding benzyl bromide 5 was produced in 31% yield without the formation of the cyclopropane ring-opened product under the standard conditions. Next, substituted benzaldoxime 6 was obtained in 60% yield upon treating 2a with 1.25 equiv. of NH2OH·HCl and 2.50 equiv. of CH3COONa in HCOOH/H2O (6/4) at 80 °C. Moreover, according to the previous literature,20 products 2a and 2b were smoothly transformed into trisubstituted pyridines 7 and 8 under the treatment with acetophenone and an ammonium salt at 130 °C in PhCl in the presence of 3 equiv. of DMSO and molecular oxygen (1.0 atm). In addition, the Knoevenagel reaction of product 2b with dimethyl malonate provided the corresponding condensation product 9 in 84% yield (Scheme 5).
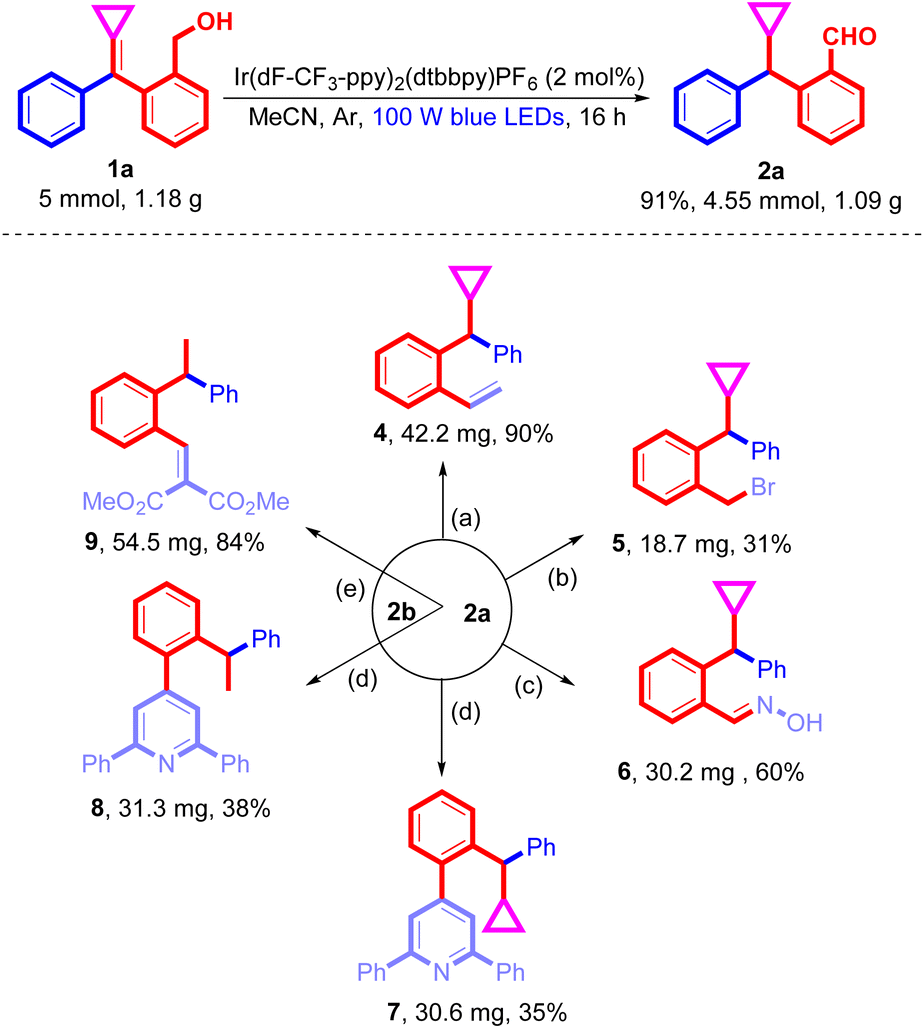 | ||
| Scheme 5 Gram scale and product transformations. (a) 2a (0.2 mmol), t-BuOK (0.40 mmol, 2.0 equiv., 1.0 mol L−1), methyltriphenylphosphonium bromide (0.40 mmol, 2.0 equiv.), DCM, r.t.; (b) 2a (0.20 mmol), phenylsilane (0.24 mmol, 1.2 equiv.), boron tribromide (0.24 mmol, 1.2 equiv., 0.24 mL of 1.0 M solution in DCM), DCM, r.t.; (c) 2a (0.20 mmol), NH2OH·HCl (0.24 mmol, 1.2 equiv.), CH3COONa (0.24 mmol, 1.2 equiv.), HCOOH/H2O (2.0 mL, 3:2), 80 °C; (d) 2a (0.20 mmol, 1.0 equiv.) or 2b (0.20 mmol), phenylacetophenone (0.60 mmol, 3.0 equiv.), NH4I (0.40 mmol, 2 equiv.), DMSO (0.60 mmol, 3.0 equiv.), molecular oxygen (1.0 atm), PhCl, 130 °C; (e) 2b (0.20 mmol), dimethyl malonate (0.40 mmol, 2 equiv.) AcOH (10 mol%), piperazine (10 mol%), toluene, 100 °C. | ||
Conclusions
In conclusion, we have explored a novel approach for the transformation of (2-vinylaryl)methanol derivatives into the corresponding aryl aldehydes or aryl ketones in moderate to excellent yields under mild conditions via visible light-mediated 1,5-HAT, enol interconversion and proton transfer processes with wide substrate scope. Deuterium labeling and kinetic analysis experiments verify that the HAT process takes place at the excited triplet state of olefins and the homolytic cleavage of the C–H bond is the rate-limiting step in this photochemical reaction. Meanwhile, another enol tautomerization-involved proton transfer process is also involved in this photochemical reaction. In addition, the control experiments have demonstrated that the presence of water in the reaction system can accelerate the proton transfer process and the possibility that the photoreaction may involve a radical chain process has also been ruled out by on and off light experiments and the determination of the quantum yield. This novel photochemical strategy further broadens the reaction patterns of visible light-mediated reactions of excited-state olefins. Further investigations based on the visible light-mediated conversion of triplet-state compounds and their applications in materials science and synthetic chemistry such as in natural product total synthesis and medicinal chemistry are currently underway.Author contributions
Yan, J. and Wei, H. Z. contributed to the investigation. Yu, Z. Q., Wei, Y., and Yan, J. contributed to the calculations. Yan, J., Wei, Y., and Shi, M. contributed to conceptualization and writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Key R & D Program of China (2022YFC2303100), the National Natural Science Foundation of China (21372250, 21121062, 21302203, 20732008, 21772037, 21772226, 21861132014, 91956115 and 22171078), a project supported by the Shanghai Municipal Science and Technology Major Project (Grant No. 2018SHZDZX03) and the Fundamental Research Funds for the Central Universities 222201717003.References
- (a) Q. Liu and L.-Z. Wu, Recent Advances in Visible-Light-Driven Organic Reactions, Natl. Sci. Rev., 2017, 4, 359–380 CrossRef CAS; (b) A. K. Bagdi, M. Rahman, D. Bhattacherjee, G. V. Zyryanov, S. Ghosh, O. N. Chupakhin and A. Hajra, Visible Light Promoted Cross-Dehydrogenative Coupling: A Decade Update, Green Chem., 2020, 22, 6632–6681 RSC; (c) S. P. Pitre and L. E. Overman, Strategic Use of Visible-Light Photoredox Catalysis in Natural Product Synthesis, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed; (d) Y.-Z. Cheng, Z. Feng, X. Zhang and S.-L. You, Visible-Light Induced Dearomatization Reactions, Chem. Soc. Rev., 2022, 51, 2145–2170 RSC; (e) H.-Z. Wei, M. Shi and Y. Wei, Visible-Light-Induced Reactions of Methylenecyclopropanes (MCPs), Chem. Commun., 2023, 59, 2726–2738 RSC.
- (a) H. K. Bisoyi and Q. Li, Light-Driven Liquid Crystalline Materials: From Photo-Induced Phase Transitions and Property Modulations to Applications, Chem. Rev., 2016, 116, 15089–15166 CrossRef CAS PubMed; (b) G. Mamba and A. K. Mishra, Graphitic Carbon Nitride (g-C3N4) Nanocomposites: A New and Exciting Generation of Visible Light Driven Photocatalysts for Environmental Pollution Remediation, Appl. Catal., B, 2016, 198, 347–377 CrossRef CAS; (c) J. You, Y. Guo, R. Guo and X. Liu, A Review of Visible Light-Active Photocatalysts for Water Disinfection: Features and Prospects, Chem. Eng. J., 2019, 373, 624–641 CrossRef CAS; (d) R. Weinstain, T. Slanina, D. Kand and P. Klán, Visible-to-NIR-Light Activated Release: From Small Molecules to Nanomaterials, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed; (e) Z. Zhang, J. Jia, Y. Zhi, S. Ma and X. Liu, Porous Organic Polymers for Light-Driven Organic Transformations, Chem. Soc. Rev., 2022, 51, 2444–2490 RSC.
- (a) D. C. Fabry and M. Rueping, Merging Visible Light Photoredox Catalysis with Metal Catalyzed C–H Activations: On the Role of Oxygen and Superoxide Ions as Oxidants, Acc. Chem. Res., 2016, 49, 1969–1979 CrossRef CAS PubMed; (b) Q.-Y. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer and B. König, Photocarboxylation of Benzylic C–H Bonds, J. Am. Chem. Soc., 2019, 141, 11393–11397 CrossRef CAS PubMed; (c) T. Kawasaki, N. Ishida and M. Murakami, Dehydrogenative Coupling of Benzylic and Aldehydic C–H Bonds, J. Am. Chem. Soc., 2020, 142, 3366–3370 CrossRef CAS PubMed; (d) L. Capaldo, D. Ravelli and M. Fagnoni, Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed; (e) E. Le Saux, M. Zanini and P. Melchiorre, Photochemical Organocatalytic Benzylation of Allylic C–H Bonds, J. Am. Chem. Soc., 2022, 144, 1113–1118 CrossRef CAS PubMed; (f) H. Cao, D. Kong, L.-C. Yang, S. Chanmungkalakul, T. Liu, J. L. Piper, Z. Peng, L. Gao, X. Liu, X. Hong and J. Wu, Brønsted Acid-Enhanced Direct Hydrogen Atom Transfer Photocatalysis for Selective Functionalization of Unactivated C(Sp3)–H Bonds, Nat. Synth., 2022, 1, 79803 Search PubMed.
- (a) K. Chen, N. Berg, R. Gschwind and B. König, Selective Single C(Sp3)–F Bond Cleavage in Trifluoromethylarenes: Merging Visible-Light Catalysis with Lewis Acid Activation, J. Am. Chem. Soc., 2017, 139, 18444–18447 CrossRef CAS PubMed; (b) W. Shu, A. Genoux, Z. Li and C. Nevado, γ-Functionalizations of Amines through Visible-Light-Mediated, Redox-Neutral C−C Bond Cleavage, Angew. Chem., Int. Ed., 2017, 56, 10521–10524 CrossRef CAS PubMed; (c) S. T. Nguyen, E. A. McLoughlin, J. H. Cox, B. P. Fors and R. R. Knowles, Depolymerization of Hydroxylated Polymers via Light-Driven C–C Bond Cleavage, J. Am. Chem. Soc., 2021, 143, 12268–12277 CrossRef CAS PubMed; (d) X. Yu, M. Lübbesmeyer and A. Studer, Oligosilanes as Silyl Radical Precursors through Oxidative Si−Si Bond Cleavage Using Redox Catalysis, Angew. Chem., Int. Ed., 2021, 60, 675–679 CrossRef CAS PubMed; (e) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Visible Light-Driven Radical-Mediated C–C Bond Cleavage/Functionalization in Organic Synthesis, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (f) C.-H. Qu, X. Yan, S.-T. Li, J.-B. Liu, Z.-G. Xu, Z.-Z. Chen, D.-Y. Tang, H.-X. Liu and G.-T. Song, Visible Light-Mediated Metal-Free Alkyl Suzuki–Miyaura Coupling of Alkyl Halides and Alkenylboronic Acids/Esters: A Green Method for the Synthesis of Allyl Difluoride Derivatives, Green Chem., 2023, 25, 3453–3461 RSC; (g) X.-S. Zhou, Z. Zhang, W.-Y. Qu, X.-P. Liu, W.-J. Xiao, M. Jiang and J.-R. Chen, Asymmetric [3 + 2] Photocycloaddition of β-Keto Esters and Vinyl Azides by Dual Photoredox/Nickel Catalysis, J. Am. Chem. Soc., 2023, 145, 12233–12243 CrossRef CAS PubMed; (h) X. Jiang, W. Xiong, S. Deng, F.-D. Lu, Y. Jia, Q. Yang, L.-Y. Xue, X. Qi, J. A. Tunge, L.-Q. Lu and W.-J. Xiao, Construction of Axial Chirality via Asymmetric Radical Trapping by Cobalt under Visible Light, Nat. Catal., 2022, 5, 788–797 CrossRef CAS; (i) Q. Li, P. Dai, H. Tang, M. Zhang and J. Wu, Photomediated Reductive Coupling of Nitroarenes with Aldehydes for Amide Synthesis, Chem. Sci., 2022, 13, 9361–9365 RSC.
- (a) A. R. Allen, E. A. Noten and C. R. J. Stephenson, Aryl Transfer Strategies Mediated by Photoinduced Electron Transfer, Chem. Rev., 2022, 122, 2695–2751 CrossRef CAS PubMed; (b) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed; (c) J. Liu, L.-Q. Lu, Y. Luo, W. Zhao, P.-C. Sun, W. Jin, X. Qi, Y. Cheng and W.-J. Xiao, Photoredox-Enabled Chromium-Catalyzed Alkene Diacylations, ACS Catal., 2022, 12, 1879–1885 CrossRef CAS; (d) L.-H. Li, H.-Z. Wei, Y. Wei and M. Shi, The Morita–Baylis–Hillman Reaction for Non-Electron-Deficient Olefins Enabled by Photoredox Catalysis, Chem. Sci., 2022, 13, 1478–1483 RSC; (e) Q. Li, X. Gu, Y. Wei and M. Shi, Visible-Light-Induced Indole Synthesis via Intramolecular C–N Bond Formation: Desulfonylative C(Sp2)–H Functionalization, Chem. Sci., 2022, 13, 11623–11632 RSC.
- (a) Y.-L. Li, W.-D. Li, Z.-Y. Gu, J. Chen and J.-B. Xia, Photoredox Ni-Catalyzed Branch-Selective Reductive Coupling of Aldehydes with 1,3-Dienes, ACS Catal., 2020, 10, 1528–1534 CrossRef CAS; (b) Y.-L. Li, S.-Q. Zhang, J. Chen and J.-B. Xia, Highly Regio- and Enantioselective Reductive Coupling of Alkynes and Aldehydes via Photoredox Cobalt Dual Catalysis, J. Am. Chem. Soc., 2021, 143, 7306–7313 CrossRef CAS PubMed; (c) S. H. Lau, M. A. Borden, T. J. Steiman, L. S. Wang, M. Parasram and A. G. Doyle, Ni/Photoredox-Catalyzed Enantioselective Cross-Electrophile Coupling of Styrene Oxides with Aryl Iodides, J. Am. Chem. Soc., 2021, 143, 15873–15881 CrossRef CAS PubMed; (d) H. Xie and B. Breit, Organophotoredox/Ni-Cocatalyzed Allylation of Allenes: Regio- and Diastereoselective Access to Homoallylic Alcohols, ACS Catal., 2022, 12, 3249–3255 CrossRef CAS.
- (a) F. S. Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Energy Transfer Catalysis Mediated by Visible Light: Principles, Applications, Directions, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC; (b) Q. Zhou, Y. Zou, L. Lu and W. Xiao, Visible–Light–Induced Organic Photochemical Reactions through Energy–Transfer Pathways, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed; (c) F. S. Kalthoff and F. Glorius, Triplet Energy Transfer Photocatalysis: Unlocking the Next Level, Chem, 2020, 6, 1888–1903 CrossRef; (d) H. Jung, H. Keum, J. Kweon and S. Chang, Tuning Triplet Energy Transfer of Hydroxamates as the Nitrene Precursor for Intramolecular C(Sp3)–H Amidation, J. Am. Chem. Soc., 2020, 142, 5811–5818 CrossRef CAS PubMed; (e) M. Zhu, X. Zhang, C. Zheng and S.-L. You, Visible-Light-Induced Dearomatization via [2 + 2] Cycloaddition or 1,5-Hydrogen Atom Transfer: Divergent Reaction Pathways of Transient Diradicals, ACS Catal., 2020, 10, 12618–12626 CrossRef CAS; (f) J. Guo, Y. Chen, C. Wang, Q. He, X. Yang, T. Ding, K. Zhang, R. Ci, B. Chen, C. Tung and L. Wu, Direct Excitation of Aldehyde to Activate the C(Sp2)−H Bond by Cobaloxime Catalysis toward Fluorenones Synthesis with Hydrogen Evolution, Angew. Chem., Int. Ed., 2023, 62, e202214944 CrossRef CAS PubMed; (g) M. J. Oddy, D. A. Kusza and W. F. Petersen, Visible-Light Mediated Metal-Free 6π-Photocyclization of N -Acrylamides: Thioxanthone Triplet Energy Transfer Enables the Synthesis of 3,4-Dihydroquinolin-2-Ones, Org. Lett., 2021, 23, 8963–8967 CrossRef CAS PubMed.
- (a) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Transition-Metal-Catalyzed C–H Alkylation Using Alkenes, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed; (b) L. Massaro, J. Zheng, C. Margarita and P. G. Andersson, Enantioconvergent and Enantiodivergent Catalytic Hydrogenation of Isomeric Olefins, Chem. Soc. Rev., 2020, 49, 2504–2522 RSC; (c) H. Jiang and A. Studer, Intermolecular Radical Carboamination of Alkenes, Chem. Soc. Rev., 2020, 49, 1790–1811 RSC; (d) S. Engl and O. Reiser, Copper-Photocatalyzed ATRA Reactions: Concepts, Applications, and Opportunities, Chem. Soc. Rev., 2022, 51, 5287–5299 RSC; (e) W. Lee, I. Park and S. Hong, Photoinduced Difunctionalization with Bifunctional Reagents Containing N-Heteroaryl Moieties, Sci. China: Chem., 2023, 66, 1688–1700 CrossRef CAS.
- (a) W. Ding, C. C. Ho and N. Yoshikai, Energy-Transfer-Mediated Cyclization of 2-(1-Arylvinyl)Benzaldehydes to Anthracen-9-(10 H)-Ones, Org. Lett., 2019, 21, 1202–1206 CrossRef CAS PubMed; (b) T. Peez, V. Schmalz, K. Harms and U. Koert, Synthesis of Naphthocyclobutenes from α-Naphthyl Acrylates by Visible-Light Energy-Transfer Catalysis, Org. Lett., 2019, 21, 4365–4369 CrossRef CAS PubMed; (c) J. Liu, Y. Wei and M. Shi, Direct Activation of a Remote C(Sp3)–H Bond Enabled by a Visible–Light Photosensitized Allene Moiety, Angew. Chem., Int. Ed., 2021, 60, 12053–12059 CrossRef CAS PubMed; (d) J. Liu, T. Hao, L. Qian, M. Shi and Y. Wei, Construction of Benzocyclobutenes Enabled by Visible–Light–Induced Triplet Biradical Atom Transfer of Olefins, Angew. Chem., Int. Ed., 2022, 61, e202204515 CrossRef CAS PubMed; (e) Y. Xiong, J. Großkopf, C. Jandl and T. Bach, Visible Light–Mediated Dearomative Hydrogen Atom Abstraction/Cyclization Cascade of Indoles, Angew. Chem., Int. Ed., 2022, 61, e202200555 CrossRef CAS PubMed; (f) M. J. Oddy, D. A. Kusza, R. G. Epton, J. M. Lynam, W. P. Unsworth and W. F. Petersen, Visible–Light–Mediated Energy Transfer Enables the Synthesis of B–Lactams via Intramolecular Hydrogen Atom Transfer, Angew. Chem., Int. Ed., 2022, 61, e202213086 CrossRef CAS PubMed.
- (a) P. Franceschi, S. Cuadros, G. Goti and L. Dell'Amico, Mechanisms and Synthetic Strategies in Visible Light–Driven [2 + 2]–Heterocycloadditions, Angew. Chem., Int. Ed., 2023, 62, e202217210 CrossRef CAS PubMed; (b) A. Tröster, R. Alonso, A. Bauer and T. Bach, Enantioselective Intermolecular [2 + 2] Photocycloaddition Reactions of 2(1 H)-Quinolones Induced by Visible Light Irradiation, J. Am. Chem. Soc., 2016, 138, 7808–7811 CrossRef PubMed; (c) J. Zhao, J. L. Brosmer, Q. Tang, Z. Yang, K. N. Houk, P. L. Diaconescu and O. Kwon, Intramolecular Crossed [2 + 2] Photocycloaddition through Visible Light-Induced Energy Transfer, J. Am. Chem. Soc., 2017, 139, 9807–9810 CrossRef CAS PubMed; (d) M. Zhu, C. Zheng, X. Zhang and S.-L. You, Synthesis of Cyclobutane-Fused Angular Tetracyclic Spiroindolines via Visible-Light-Promoted Intramolecular Dearomatization of Indole Derivatives, J. Am. Chem. Soc., 2019, 141, 2636–2644 CrossRef CAS PubMed; (e) M. S. Oderinde, E. Mao, A. Ramirez, J. Pawluczyk, C. Jorge, L. A. M. Cornelius, J. Kempson, M. Vetrichelvan, M. Pitchai, A. Gupta, A. K. Gupta, N. A. Meanwell, A. Mathur and T. G. M. Dhar, Synthesis of Cyclobutane-Fused Tetracyclic Scaffolds via Visible-Light Photocatalysis for Building Molecular Complexity, J. Am. Chem. Soc., 2020, 142, 3094–3103 CrossRef CAS PubMed; (f) R. Kleinmans, T. Pinkert, S. Dutta, T. O. Paulisch, H. Keum, C. G. Daniliuc and F. Glorius, Intermolecular [2π+2σ]-Photocycloaddition Enabled by Triplet Energy Transfer, Nature, 2022, 605, 477–482 CrossRef CAS PubMed.
- (a) K. Singh, S. J. Staig and J. D. Weaver, Facile Synthesis of Z -Alkenes via Uphill Catalysis, J. Am. Chem. Soc., 2014, 136, 5275–5278 CrossRef CAS PubMed; (b) A. Singh, C. J. Fennell and J. D. Weaver, Photocatalyst Size Controls Electron and Energy Transfer: Selectable E/Z Isomer Synthesis via C–F Alkenylation, Chem. Sci., 2016, 7, 6796–6802 RSC; (c) W. Cai, H. Fan, D. Ding, Y. Zhang and W. Wang, Synthesis of Z -Alkenes via Visible Light Promoted Photocatalytic E → Z Isomerization under Metal-Free Conditions, Chem. Commun., 2017, 53, 12918–12921 RSC; (d) H. Li, H. Chen, Y. Zhou, J. Huang, J. Yi, H. Zhao, W. Wang and L. Jing, Selective Synthesis of Z –Cinnamyl Ethers and Cinnamyl Alcohols through Visible Light–Promoted Photocatalytic E to Z Isomerization, Chem. – Asian J., 2020, 15, 555–559 CrossRef CAS PubMed.
- M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Photocatalysis for the Formation of the C−C Bond, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed.
- S. Protti and M. Fagnoni, The Sunny Side of Chemistry: Green Synthesis by Solar Light, Photochem. Photobiol. Sci., 2009, 8, 1499–1516 CrossRef CAS PubMed.
- J. M. Hornback, L. G. Mawhorter, S. E. Carlson and R. A. Bedont, Reactions of O-Xylylenes Produced Photochemically from o-Alkylstyrenes, J. Org. Chem., 1979, 44, 3698–3703 CrossRef CAS.
- (a) R. G. W. Norrish and C. H. Bamford, Photodecomposition of Aldehydes and Ketones, Nature, 1936, 138, 1016 CrossRef CAS; (b) R. G. W. Norrish and C. H. Bamford, Photo-decomposition of Aldehydes and Ketones, Nature, 1937, 140, 195–196 CrossRef CAS; (c) R. G. W. Norrish, On The Principle of Primary Recombination in Relation to The Velocity of Thermal Reactions in Solution, Trans. Faraday Soc., 1937, 33, 1521–1528 RSC; (d) N. C. Yang and D.-D. H. Yang, Photochemical Reactions of Ketones in Solution, J. Am. Chem. Soc., 1958, 80, 2913–2914 CrossRef CAS; (e) P. J. Wagner, Type II Photoelimination and Photocyclization of Ketones, Acc. Chem. Res., 1971, 4, 168–177 CrossRef CAS.
- M. Sayes, G. Benoit and A. B. Charette, Borocyclopropanation of Styrenes Mediated by UV–light Under Continuous Flow Conditions, Angew. Chem., Int. Ed., 2018, 57, 13514–13518 CrossRef CAS PubMed.
- (a) Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z.-X. Yu, An Unexpected Role of a Trace Amount of Water in Catalyzing Proton Transfer in Phosphine-Catalyzed (3 + 2) Cycloaddition of Allenoates and Alkenes, J. Am. Chem. Soc., 2007, 129, 3470–3471 CrossRef CAS PubMed; (b) H. Yuan and J. Zhang, [DBU-H]+ and H2O as Effective Catalyst Form for 2,3-Dihydropyrido[2,3-d]Pyrimidin-4(1 H)-Ones: A DFT Study, J. Comput. Chem., 2015, 36, 1295–1303 CrossRef CAS PubMed; (c) W. M. C. Sameera, M. Hatanaka, T. Kitanosono, S. Kobayashi and K. Morokuma, The Mechanism of Iron(II)-Catalyzed Asymmetric Mukaiyama Aldol Reaction in Aqueous Media: Density Functional Theory and Artificial Force-Induced Reaction Study, J. Am. Chem. Soc., 2015, 137, 11085–11094 CrossRef CAS PubMed; (d) W. Rauf and J. M. Brown, Palladium-Catalysed Directed C–H Activation by Anilides and Ureas; Water Participation in a General Base Mechanism, Org. Biomol. Chem., 2016, 14, 5251–5257 RSC; (e) L. Tao, Y. Wei and M. Shi, Thermally–Induced Intramolecular [4 + 2] Cycloaddition of Allylamino– or Allyloxy–Tethered Alkylidenecyclopropanes, Chem. – Asian J., 2021, 16, 2463–2468 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- D. Wang, X. Xue, K. N. Houk and Z. Shi, Mild Ring-Opening 1,3-Hydroborations of Non-Activated Cyclopropanes, Angew. Chem., Int. Ed., 2018, 57, 16861–16865 CrossRef CAS PubMed.
- J. Chen, H. Meng, F. Zhang, F. Xiao and G.-J. Deng, Transition-Metal-Free Selective Pyrimidines and Pyridines Formation from Aromatic Ketones, Aldehydes and Ammonium Salts, Green Chem., 2019, 21, 5201–5206 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. See DOI: https://doi.org/10.1039/d3qo01266b |
| This journal is © the Partner Organisations 2023 |