
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering the degree of proton-transfer in pyrido-cyclophanes by chiroptical outcomes in non-aqueous solvents†
Jonathan
Álvarez-García
a,
Víctor
Rubio-Pisabarro
a,
Luis
García-Río
 b and
María
Magdalena Cid
*a
b and
María
Magdalena Cid
*a
aDepartamento de Química Orgánica, Edificio Ciencias Experimentais, Campus Lagoas-Marcosende, Vigo E-36310, Spain. E-mail: mcid@uvigo.gal
bDepartamento de Química Física, Facultade de Química, Avda das Ciencias, s/n, Santiago de Compostela, E-15782, Spain
First published on 18th September 2023
Abstract
Proton transfer equilibria are of pivotal importance due to the role they play in a plethora of biological and phamaceutical processes. With the aim of explaining the relative position of the hydrogen to be transferred, we investigated the behavior of chiral pyrido-cyclophanes with different morphologies using circular dichroism in the presence of different acids in acetonitrile. The results showed that all three compounds underwent double protonation and formation of cascade ion-pairs, leading to the appearance of diagnostic signals in their ECD spectra. The presence of water fosters the crystallization of several intermediates that do not correspond to those in solution. By using Brønsted correlations, it was found that proton transfer from the acid to the pyridine occurred regardless of the acid's pKa.
Introduction
Proton transfer and H-bonding are of paramount importance in numerous (bio)chemical processes, such as stabilization of protein structure and catalysis.1–3 It is therefore unsurprising that imbalances in maintaining the correct pH in bodily functions are related to a variety of diseases, such as cancer or Alzheimer's.4,5Total or partial hydrogen transfer between acid and base partners results in the formation of salts or a hydrogen-bonding driven complex, respectively. The degree of proton transfer between two species in the solid state can be predicted using the pKa rule, that suggests the potential formation of a co-crystal when the difference in pKa values of the interacting partners, denoted as ΔpKa, is below 0, whereas if it exceeds 3, the formation of a molecular salt is likely.6,7 Nonetheless, there are always exceptions and for an intermediate value no accurate prediction can be made. On the other hand, in solution, salts are present as separated, contact or aggregated ion-pairs depending on the medium polarity. In particular, ion-pairing, in which understanding the degree of proton transfer is essential, explain unexpected observations in the behaviour of synthetic ion receptors.8–10
Shape-persistent pyridocyclophanes have been the subject of research in Supramolecular Chemistry and Material Science due, in part, to the properties of the pyridine motif.11–14 So, it is not surprising that proton transfer between acids and pyridine nitrogen atoms had drawn considerable attention.15–18
Our research group has exploited pyridines and allenes as construction motifs in the synthesis of chiral shape-persistent cyclophanes with applicability in the field of molecular recognition in non-aqueous solvents.19 We reported the preparation and full characterization of pyrido-cyclophane 1 (hereafter, pyrido-allenophane to evidence the presence of allenyl motifs, Fig. 1), which showed one of the strongest chiroptical responses described for small organic molecules with a g-factor‡ of 0.011.20 However, its rapid racemization under ambient conditions, driven by ring strain, did not allow us to exploit its sensing capabilities. With the aim of overcoming ring strain, we synthesized bipyrido-allenophane 2 and found that, in the presence of TfOH, it not only undergoes a stereoselective double protonation process, which is quite remarkable for a cis-locked bipyridine, but also captures one of the corresponding counteranions forming a contact ion-pair (also referred to as a cascade complex, in which the anion coordinates to the cation center).20 In this respect, pyridine-decorated chiral cyclic peptides have shown simultaneous complexation of cations and anions.21
 | ||
| Fig. 1 Potential pyrido-allenophanes as ion-pair receptors: known 1, 2 and 4, and new [72]-pyridoallenophane 3. | ||
Our aim is to investigate the degree of the proton transfer in acid–base processes involving pyridine derivatives via chiroptical signals in non-aqueous media. For such endeavor, allenes are used to provide axial chirality to pyrido-cyclophanic systems (Fig. 1).
Having laid the foundation with bipyrido-allenophane 2, and taking into account the interest that proton transfer phenomena and pyridine H-bonding have, we decided to expand our study to a greater number of acids. Besides, we now envision the introduction of a pyridine motif into the butadiyne bond of 1 to afford the new pyridoallenophane 3, since, in addition to its role as a stability factor, the second pyridine moiety provides an extra recognition site. In addition, we want to sort out whether allenophane 4,22,23 whose conformational space in solution is defined mainly by two conformers, would undergo a conformation-selective molecular recognition process and thus susceptible to being monitored by circular dichroism (CD).
So, here we present the preparation and complete characterization of the new chiral macrocycle [72]-pyrido-allenophane 3, the behavior of allenophanes 2, 3 and 4 against different acids (oxoacids and HCl) in acetonitrile and their abilities to capture the counteranions and consequently reduce their conformational space via ion-pair recognition. These three chiral allenophanes each contain two pyridine units in three different spatial morphologies, as the aromatic spacers are separated by zero (2), one (3) or two (4) allene units, which could supply crucial information to correlate chirality signals with geometry rigidity and recognition properties.
Results and discussion
Synthesis and characterization of pyrido-allenophanes
Pyridoallenophanes 2 and 4 were prepared following literature procedures.20,24 These compounds showed a dissymmetry factor of 0.007 and 0.006, respectively, which is notably high for small organic molecules. Macrocycle 3 was synthesized according to the procedure shown in Scheme 1. (P)-Diethynylallene (DEA)25,26 was submitted to a Sonogashira coupling with an excess of both 2,6-dibromo- and 2,6-diiodopyridine in order to obtain the mono-haloderivatives 5 and 7, respectively. Then, deprotection of pyridine 5 upon treatment with NaOH in toluene rendered 2-bromopyridine 6, that via a chemoselective Sonogashira cross-coupling with 2-iodopyridine 7 afforded 8 in very high overall yield. The key step in the synthetic pathway involved treating 8 with NaOH in toluene at 110 °C, followed by an intramolecular Sonogashira coupling under high dilution conditions to give the enantiopure target macrocycle 3 in 63% in both steps. When (P2)-3 was allowed to crystallize from a Et3N solution, suitable small crystals for X-ray diffraction analysis were obtained at room temperature.§ The resulting structure exhibited a molecular geometry very similar to the only conformer obtained in DFT computations. Different geometries were explored by DFT calculations at the CAMB3LYP/6-31g+(d,p) level using Gaussian09.27 To ensure the characterization of the structures as energy minima, calculation of vibrational frequencies were performed (Scheme 1). NICS indices have also been computed for 3 using the same level of theory and employing the GIAO approximation (see ESI† for details) revealing its non-aromatic nature. | ||
| Scheme 1 Synthetic sequence followed to obtain macrocycle 3. Bottom: Crystal structure resolved by X-ray diffraction ((P2)-3) and conformation obtained by DFT computations (CAM-B3LYP/6-31g+(d,p)). | ||
Allenophane 3 exhibited a maximum dissymmetry factor‡g = 0.006, which is the same as that of the [142] analogue 4, even though the number of chiral units is reduced by 50%. This result supports the importance of the ratio between the number of chiral units and conformation stability to render outstanding chiral systems. Also, in contrast to compound 1, 3 is configurationally stable under ambient conditions of light and temperature.
Titration experiments
In order to test the sensing capabilities of the enantiopure allenophanes 2, 3 and 4 and study their chiroptical responses when exposed to different Brønsted acids, we chose acetonitrile as an aprotic polar solvent. This choice was based on several factors, including the solubility properties of our macrocycles, acetonitrile's ability to facilitate the dissociation of the acids, and the wide range of pKa values documented in this medium.28,29 In addition, the study of acid–base reactions in acetonitrile is important to obtain information on the degree of ionization of a molecule in more lipophilic media rather than in water.30Previous research by our group showed that both CD and NMR titration experiments gave consistent equilibrium constants (K) values for allenophanes in the presence of TFA in acetonitrile.20,31 In the current study, we therefore focused primarily on CD spectroscopy due to its sensitivity to conformational changes32,33 and the large chiroptical responses of this family of macrocycles. In addition, CD has proven to be a very useful tool for studying protonation of other chiral pyridocyclophanes.34 The details for each titration experiment may be found in the ESI.¶
Titrations with oxoacids
We first extended our previous study of bipyrido-allenophane 2 with TFA and TfOH by including MsOH, TsOH and H2SO4 to ensure coverage of a wide pKa range.For the five oxoacids tested, the CD spectrum (Fig. 2A, red line) evolved until a first saturation point was reached that is characterized by two negative bands centered at 290 and 370 nm (Fig. 2A, black line). Upon further increasing the acid concentration, the band at 370 nm moved to 388 nm and a new band appeared at 330 nm that is diagnostic for fixation of the bipyridine axis in the M configuration (Fig. 2A, blue line), as we previously showed for TFA.20
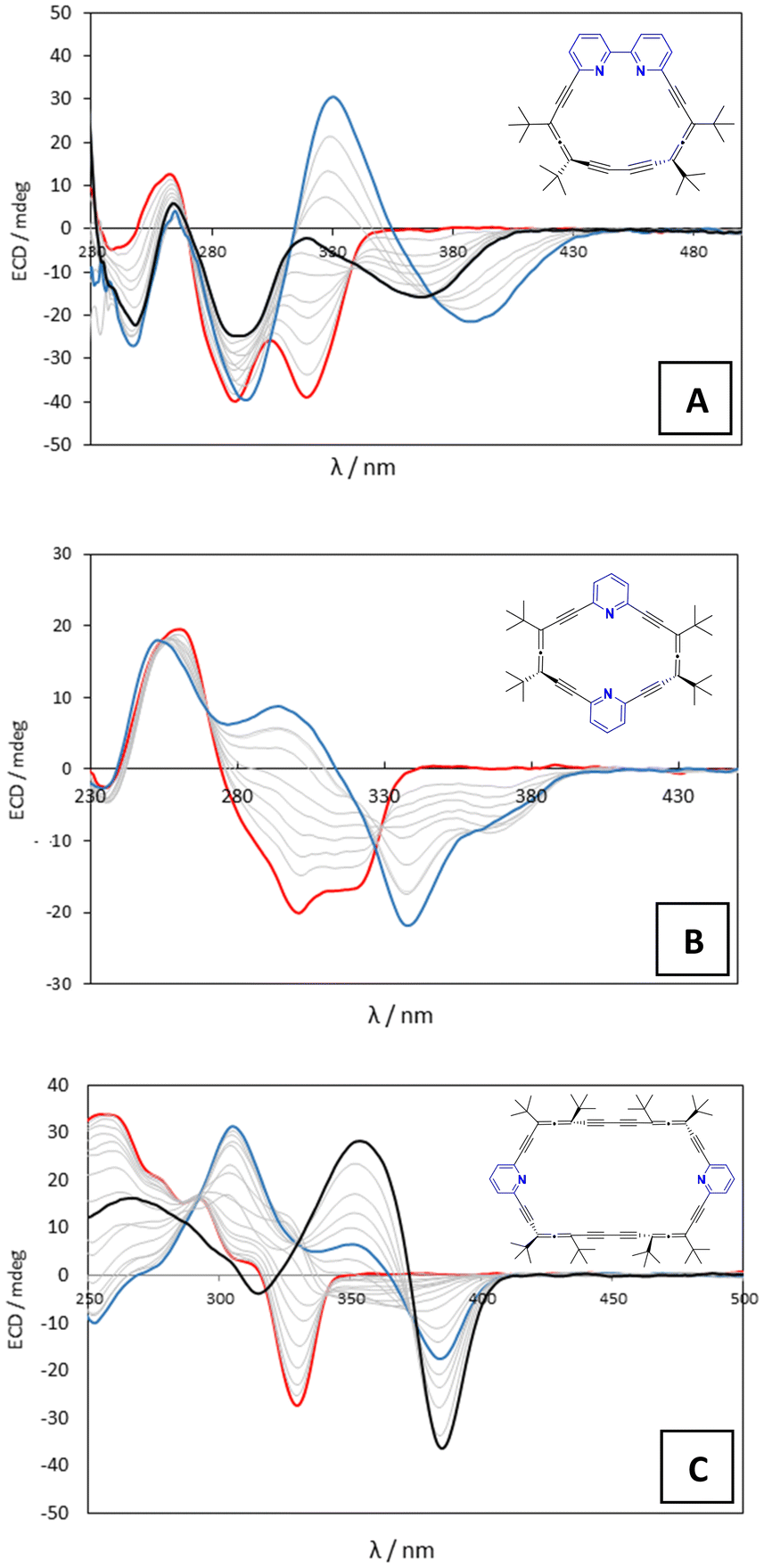 | ||
| Fig. 2 Titration course for TfOH (concentration ranging from 0 to 0.2 M) in acetonitrile for 2 (A; 4 × 10−5 M), 3 (B; 1.5 × 10−5 M) and 4 (C; 6.24 × 10−6 M). The red line represents the initial ECD spectral bands. The blue line represents the end of the titration for oxoacids. The black line represents the end of the titration for HCl in dioxane. In section A, the black line also represents the first saturation point reached with the oxoacids. | ||
Given the results from allenophane 2, pyridoallenophane 3 was treated with TfOH, H2SO4 and TFA as representative acids. Upon increasing amounts of acid in acetonitrile, allenophane 3 showed important changes in its CD spectrum. Initial negative bands at 302 and 320 nm (Fig. 2B, red line) gradually transformed into a new positive band with a maximum centered at 302 nm and two new negative bands appeared with minima at 340 and 375 nm (Fig. 2B, blue line).
We found that the experimental data obtained from the different titrations for 2 and 3 could be fitted mathematically to the same set of equilibria (Scheme 2), although with different association constants. From a general point of view, at low acid concentrations, the free macrocycle begins to disappear in favor of the monoprotonated (AP·H)+ and AP·HA species (AP = allenophane 2 or 3). Later, when the concentration of acid in solution increases, both species disappear in favor of the doubly protonated macrocycle complexed with a counteranion (AP·H2A)+, which turns out to be the final species in all the titrations.
 | ||
| Scheme 2 Titration scheme for allenophanes 2, 3 and 4 with oxoacids that was modeled mathematically to obtain equilibrium constants from the experimental data. The equilibrium in orange is only relevant for compound 4. (AP = Allenophane). | ||
Finally, we evaluated the behavior of allenophane 4 in the presence of TfOH, H2SO4 and TFA. We observed the disappearance of the initial negative band centered at 330 nm (Fig. 2C, red line) followed by the gradual rise of three new diagnostic bands, one negative at 384 nm and two positives at 351 and 305 nm (Fig. 2C, blue line). In this case, the titration data suggest that 4·H2A2 is the final species (Scheme 2, orange), in which the free macrocycle captures two molecules of acid in contrast to macrocycles 2 and 3, whose endpoints correspond to the diprotonated species capturing one counteranion.
Inspired by the ability of macrocycle 4 to form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complexes with several acids, we directed our efforts to achieve a 1:1 host–guest inclusion complex and obtain clues about the size and adaptability of the binding cavity. For this purpose, we thought that 1,2-ethanedisulfonic acid might be a suitable target for two reasons. First, it could function as a bidentate guest, and second, sulfonate counterions are key players in the pharmaceutical salt portfolio because of their unique properties, which include high melting points, fair aqueous solubility and stability, and the absence of hydrates. As a consequence, sulfonates often offer attractive advantages over the hydrochloride salt forms.35 Thus, the use of this bidentate acid as titrant led to the same experimental CD outcome as the other oxoacids but this time forming a 1:1 complex.
:2 complexes with several acids, we directed our efforts to achieve a 1:1 host–guest inclusion complex and obtain clues about the size and adaptability of the binding cavity. For this purpose, we thought that 1,2-ethanedisulfonic acid might be a suitable target for two reasons. First, it could function as a bidentate guest, and second, sulfonate counterions are key players in the pharmaceutical salt portfolio because of their unique properties, which include high melting points, fair aqueous solubility and stability, and the absence of hydrates. As a consequence, sulfonates often offer attractive advantages over the hydrochloride salt forms.35 Thus, the use of this bidentate acid as titrant led to the same experimental CD outcome as the other oxoacids but this time forming a 1:1 complex.
Our attempts to crystallize several of these complexes under anhydrous conditions were unsuccessful. However, using commercial TfOH, where traces of water are to be expected, we managed to crystallize allenophane 3 both in its monoprotonated (3·H)+ form (Fig. 3A) from acetonitrile and the diprotonated (3·H2)+2 (Fig. 3B) form from a 1:1 acetonitrile:ethanol solution at 4 °C.§ Both have an encapsulated water molecule slightly displaced towards one of the pyridine rings. In (3·H)+, a water molecule acts as a bridge between both Npyr and the triflate anion. In (3·H2)+2, the water molecule acts as an H-bond acceptor for both NpyrHs and as a donor for the two counterions that are located on opposite sides of the macrocycle. In neither of the structures was the initial conformation of the starting macrocycle modified.
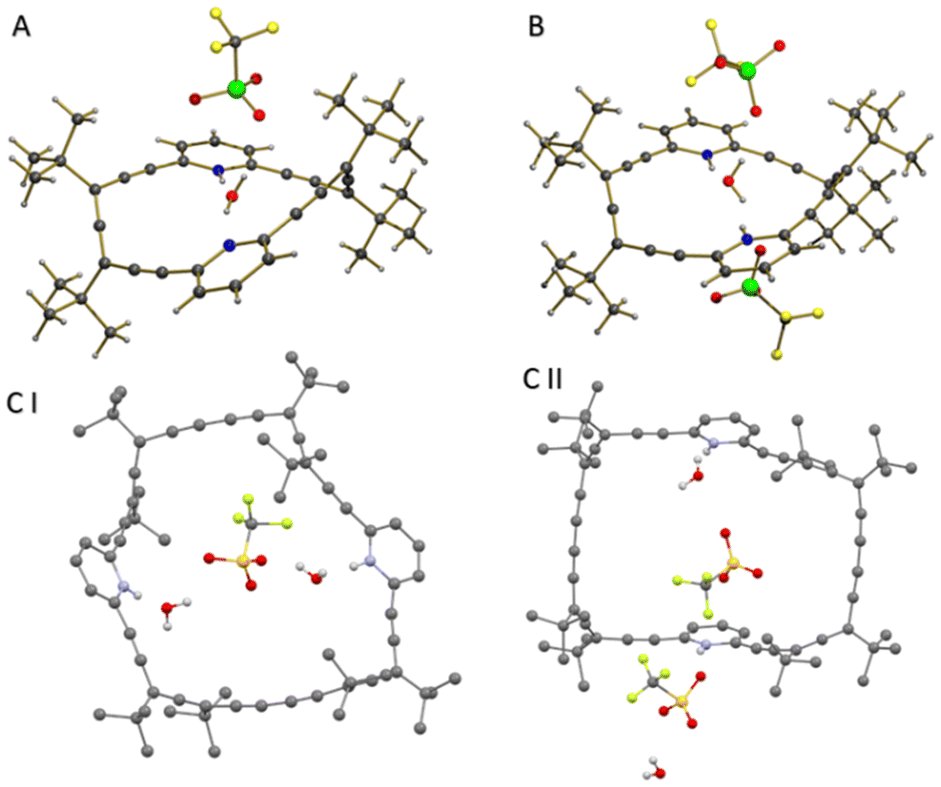 | ||
| Fig. 3 (A) Crystal structure of 3·H2O·OTf. (B) Crystal structure of 3·(H2O·OTf)2. (C) Crystal structure of 4·(H2O·OTf)2: asymmetric units I and II. Hydrogens were omitted for clarity. | ||
Under the same conditions, allenophane 4 crystallized in the form of (4·H2)+2 from a 8:2 dichloromethane:ethanol solution.§ Here, the asymmetric unit contains two slightly different conformations of the allenophane framework: I- each of the two water molecules establishes H-bonding with NpyrHs and with a triflate anion (Fig. 3CI); II- both pyridine rings act independently due to the orientation of respective N–H bonds (Fig. 3CII). In the three crystals, the protons are located on the pyridine rings, in accord with predictions made by the pKa rule for triflic acid6 and the equilibrium constants previously obtained in our mathematical analysis (see Table S3†).
Next, we wanted to address the question “Where is the proton located?” in the final species of each titration. Different techniques have been used for this purpose, including NMR,36 IR,18,37 XRPD7,38 and polarography,39 although mainly in carboxylic acids. Here we apply a simple and intuitive Brønsted correlation, plotting the logK2 obtained from the mathematical fits (Table S3†) against the pKa of the tested oxoacids. As shown in Fig. 4, we observed linear correlations for all three macrocycles, indicating that regardless of the pKa the extent of proton transfer must be the same for all the acids tested.
 | ||
| Fig. 4 Brønsted plots of logK2vs. the pKa of the oxoacid used to titrate macrocycles 2 (blue), 3 (green), and 4 (orange). | ||
For allenophanes 3 and 4, the lines are practically parallel to each other with a slope α ≈ |1|, which indicates complete transfer of the proton from the acid to the pyridine (Fig. 4a). In the case of 2, the slope turns out to be α ≈ |0.43|, which a priori could indicate that the proton is equally shared between the acid and the pyridine.|| However, the pyridines in 2 are not independent systems, so we are inclined to think that the proton has been transferred as in 3 and 4 and that it is shared by the bipyridine moiety (Fig. 4b).
Titration with HCl in dioxane
In view of the interesting behavior of allenophanes 2, 3 and 4 against different oxoacids with pKa values in the range ca. 2–12, we wanted to ascertain whether anion-shape selection effects might be important. With such goal, we decided to use a 4 M solution of HCl in dioxane since allenophanes 2–4 show poor solubility in water. This initial commercial solution was diluted in acetonitrile to soften the effect of dioxane in the medium (ESI†).Surprisingly, when bipyrido-allenophane 2 was titrated with HCl, only the first saturation point was reached (Fig. 2A, black line). The spectrum did not evolve further and diagnostic bands for the stereoselective discrimination of the bipyridine axis never appeared, pointing to a different final species. In contrast, treatment of a solution of pyrido-allenophane 3 in acetonitrile with HCl/dioxane led to the same spectrum changes as those seen for the oxoacids (Fig. 2B, blue line). Completely different behavior was observed in the case of pyrido-allenophane 4. At low acid concentrations, the experiment yielded the same outcome as the oxoacids (Fig. 2C, blue line), but as the concentration of acid increased, the bands at 384 and 351 nm intensified and the positive band at 305 nm became a weak negative band (Fig. 2C, black line).
These observations reveal a pivotal role of the counteranion in selecting the conformation of the final ion-pair complex. When fitting the experimental data to calculate the equilibrium constants, we have to keep in mind that the protonating species can be either H+, HCl or (H·dioxane)+. To make it simpler and since our main interest is to determine which species is the final one, we performed the mathematical analysis by just considering the pKa of HCl in acetonitrile (see the ESI† for details). The mathematical fitting suggested that the final species are 2·H2Cl2/(2·H2·Dioxane2)+2, (3·H2Cl)+/(3·H2·Dioxane)+2 and 4·H2Cl2/(4·H2·Dioxane2)+2, with calculated CD spectra that closely resemble the experimental ones (see below).
Computational exploration
To explain our results and shed light on the experimental outcome of each titration at the molecular level, we carried out a computational exploration using DFT calculations at the CAM-B3LYP/6-31G+(d,p) level using acetonitrile as implicit solvent as included in the SMD package. The structures of the final species obtained by mathematically fitting the experimental data for each of the titrations were optimized and their CD spectra calculated (see ESI† for details).The conformational space of allenophane 2 was described by two possible conformers depending on the configuration adopted by the axis that joins both pyridine rings, namely P or M. When we computed and optimized the structure of the complexes with all five oxoacids (2·H2A)+, we found that the bipyridine nitrogens share the two protons with an oxygen atom of the corresponding oxoanion while the side chain of each of the acids is located towards the outside of the cavity, moving away from the tert-butyl groups (Fig. 5A). As a result of this interaction, the bipyridine axis is fixed in an M configuration. The calculated spectrum of this species (Fig. 5A, dash green line) coincides remarkably well with the experimental one, and when the axis adopts only the M configuration does the diagnostic band at 330 nm appear.
 | ||
| Fig. 5 Blue lines represent the experimental endpoint of every titration. Green dash lines represent the calculated ECD spectra of the corresponding complex: (A) (2·H2TfO)+, (B) (3·H2TfO)+ and (3·H2dioxane)+2, (C) 4·H2(ethanedisulfonate) and (D) 4·H2(H2O)2dioxane. Bottom: Calculated geometry for (2·H2(dioxane)2)+2 and crystal structure of 2 along with two CHCl3 molecules. | ||
In the case of HCl, the computationally optimized structure of both 2·H2A2 complexes (A being Cl− or dioxane) shows the guest molecules located on each of the faces of the macrocycle (Fig. 5, bottom). Since bipyridine nitrogens do not coordinate the same molecule cooperatively, the bipyridine scaffold can continue to adopt both configurations, namely M and P, and therefore the diagnostic band at 330 nm never appeared. Interestingly, the optimized 2·H2A2 structure is very reminiscent of the structure obtained by X-ray diffraction of macrocycle 2 in chloroform20 (Fig. 5, bottom).
For allenophane 3, the calculated geometries of the complexes (3·H2A)+ showed the oxoanions acting as bidentate ligands and consequently forcing the macrocycle to adopt a V-shaped conformation (Fig. 5B). The calculated spectrum of these species (Fig. 5B, dash green line) coincides remarkably well with the final experimental spectrum, and only when the macrocycle acquires the new conformation, in addition to being diprotonated, does the positive diagnostic band at 302 nm appear, supporting the idea that one of the anions must be located inside the macrocycle. To ensure this V-like conformation in the case of HCl, we ascribed a bidentate character to the dioxane molecules. Indeed, the calculated and experimental CD spectra were coincident when a dioxane molecule was introduced as a bridge between the two N–Hs via the oxygen atoms, with an average NH⋯O distance of 1.86 Å.**
On the other hand, the conformational space in solution of allenophane 4 consists of two conformers: chair and twist.23 So, once again, the geometries of the 4·H2A2 complexes for the oxoacids were optimized and their CD spectra calculated. The outcomes showed a new half-chair conformation in which each anion is oriented towards the cavity of the allenophanic ring with an average distance of 10.50 Å between the two nitrogens of the pyridine moieties.** The calculated spectra of these complexes reproduced the experimental spectrum very well (Fig. 5C, dashed green line). In the case of 1,2-ethanedisulfonic acid, simultaneous interactions between the two acidic moieties and the two pyridine rings were observed (Fig. 5C), with the two sulfonic groups adopting an antiperiplanar disposition.
Modeling the obtained complex upon treatment of 4 with HCl proved to be more of a challenge. In order to arrive at a computed geometry whose CD spectrum resembled that seen experimentally, we devised a system in which the two protonated pyridines and two water molecules were bridged by one dioxane molecule (Fig. 5D, dash green line). The optimized geometry here showed the macrocycle adopting a new distorted twist conformation, with bond distances of 1.65 Å and 1.77 Å for the NH⋯O and HO⋯O(CH2)4O interactions, respectively. In addition, the distance between the two nitrogens of the pyridine moieties is larger (12.98 Å) than that in the complexes formed with the other acids (10.50 Å) (Fig. 5C).** The difference between the two new conformations in the final species explains the different outcomes in their ECD spectra.
It should be mentioned that the ECD spectra of the salt geometries obtained by X-ray diffraction were also computed. In none of the cases, the ECDs calculated coincided with the experimental end point of the titrations nor with those of the optimized crystallographic geometries (Fig. S73, S74 and S80†). Therefore, the complexes in solution at the end point must be different from those obtained in the solid state in the presence of water. This supports the idea that the geometries of the complexes shown in Fig. 5 should be responsible for the observed changes in the ECD during the titrations.
Furthermore, in all the optimized structures the protons were located on the pyridine moieties, matching their computed spectra with the experimental results. This agrees with what the Brønsted correlation called for.
Conclusions
New chiral pyrido-allenophane 3 was synthesized in high overall yield and fully characterized. Its conformational space consists of only one conformer that results in a strong g-factor for such small organic molecule. The behavior of this allenophane, together with that of 2 and 4, was studied with different acids in acetonitrile by circular dichroism. It was found that in all cases, in addition to a double protonation, the complexation of one or two counteranions occurred and, consequently, diagnostic signals appear in their ECD spectra. Moreover, ECD proved to be a useful technique to elucidate the role of water molecules in proton transfer processes in acetonitrile, since the conformation of the receptors turned out to be completely different when water molecules were incorporated. Furthermore, Brønsted correlations suggest full proton transfer from the acid to the pyridine, regardless of the pKa of the acid used. Hence, by combining circular dichroism and Brønsted correlations, it is possible to accurately discern whether the investigated species exist as salts or ion-pairs. These investigations pave the way to tailored synthetic receptors that contain strategically placed pyridine moieties for anionic guests and/or salts depending on the sought application.Author contributions
JAG and VRP run the experimental work, preliminary data analysis and wrote the original draft. LGR did the mathematical fit and validated the extracted conclusions. MMC was in charge of the conceptualization, supervision and editing the final version of the manuscript.Conflicts of interest
There is no conflict to declare.Acknowledgements
We thank Dr Inma R. Lahoz for preliminary experiments, Dr Berta Covelo for her commitment with X-ray studies and Prof. Donald Hilvert for his valuable comments. Financial support from Ministerio de Economia y Competitividad of Spain (PID2021-128057NB-I00) and Xunta de Galicia (EC431C 2021/41) is acknowledge. Also, we also thank CESGA for allocation of HPC resources and Universidade de Vigo Structural analysis unit (CACTI).References
- T. P. Silverstein, The Proton in Biochemistry: Impacts on Bioenergetics, Biophysical Chemistry, and Bioorganic Chemistry, Front. Mol. Biosci., 2021, 8, 764099 CrossRef CAS PubMed.
- T. Ke and A. M. Klibanov, Markedly Enhancing Enzymatic Enantioselectivity in Organic Solvents by Forming Substrate Salts, J. Am. Chem. Soc., 1999, 121, 3334–3340 CrossRef CAS.
- S. Hammes-Schiffer, Virtual Issue on Proton Transfer, J. Phys. Chem. B, 2021, 125, 3725–3726 CrossRef CAS PubMed.
- O. A. Saibu, S. O. Hammed, O. O. Oladipo, T. T. Odunitan, T. M. Ajayi, A. J. Adejuyigbe, B. T. Apanisile, O. E. Oyeneyin, A. T. Oluwafemi, T. Ayoola, O. T. Olaoba, A. O. Alausa and D. A. Omoboyowa, Protein-protein interaction and interference of carcinogenesis by supramolecular modifications, Bioorg. Med. Chem., 2023, 81, 117211 CrossRef CAS PubMed.
- L. Schwartz, S. Peres, M. Jolicoeur and J. da Veiga Moreira, Cancer and Alzheimer's disease: intracellular pH scales the metabolic disorders, Biogerontology, 2020, 21, 683–694 CrossRef CAS PubMed.
- A. J. Cruz-Cabeza, M. Lusi, H. P. Wheatcroft and A. D. Bond, The role of solvation in proton transfer reactions: implications for predicting salt/co-crystal formation using the ΔpKa rule, Faraday Discuss., 2022, 235, 446–466 RSC.
- A. Lemmerer, S. Govindraju, M. Johnston, X. Motloung and K. L. Savig, Co-crystals and molecular salts of carboxylic acid/pyridine complexes: Can calculated pKa's predict proton transfer? A case study of nine complexes, CrystEngComm, 2015, 17, 3591–3595 RSC.
- Q. He, G. I. Vargas-Zúñiga, S. H. Kim, S. K. Kim and J. L. Sessler, Macrocycles as Ion Pair Receptors, Chem. Rev., 2019, 119, 9753–9835 CrossRef CAS PubMed.
- S. K. Kim and J. L. Sessler, Ion pair receptors, Chem. Soc. Rev., 2010, 39, 3784 RSC.
- S. Roelens, A. Vacca, O. Francesconi and C. Venturi, Ion-Pair Binding: Is Binding Both Binding Better?, Chem. – Eur. J., 2009, 15, 8296–8302 CrossRef CAS PubMed.
- K. Miki and K. Ohe, π–Conjugated Macrocycles Bearing Angle–Strained Alkynes, Chem. – Eur. J., 2020, 26, 2529–2575 CrossRef CAS PubMed.
- H. Abe, K. Ohtani, D. Suzuki, Y. Chida, Y. Shimada, S. Matsumoto and M. Inouye, Alternating 2,6-/3,5-Substituted Pyridine-Acetylene Macrocycles: π-Stacking Self-Assemblies Enhanced by Intermolecular Dipole–Dipole Interaction, Org. Lett., 2014, 16, 828–831 CrossRef CAS PubMed.
- S. Kobayashi, Y. Yamaguchi, T. Wakamiya, Y. Matsubara, K. Sugimoto and Z. Yoshida, Shape-persistent cyclyne-type azamacrocycles: synthesis, unusual light-emitting characteristics, and specific recognition of the Sb(V) ion, Tetrahedron Lett., 2003, 44, 1469–1472 CrossRef.
- K. Campbell, N. M. Tiemstra, N. S. Prepas-Strobeck, R. McDonald, M. J. Ferguson and R. R. Tykwinski, Synthesis of a Pyridinophane with endo -Annular Donor Sites, Synlett, 2004, 182–186 CAS.
- Y. Yano, T. Ono, S. Hatanaka, D. T. Gryko and Y. Hisaeda, Salt-cocrystal continuum for photofunction modulation: Stimuli-responsive fluorescence color-tuning of pyridine-modified intramolecular charge-transfer dyes and acid complexes, J. Mater. Chem. C, 2019, 7, 8847–8854 RSC.
- V. Balevicius, R. Bariseviciute, K. Aidas, I. Svoboda, H. Ehrenberg and H. Fuess, Proton transfer in hydrogen-bonded pyridine/acid systems: the role of higher aggregation, Phys. Chem. Chem. Phys., 2007, 9, 3181 RSC.
- C. C. Seaton, Proton location in acid…pyridine hydrogen bonds of multi-component crystals, CrystEngComm, 2014, 16, 5878–5886 RSC.
- G. M. Barrow, The Nature of Hydrogen Bonded Ion-Pairs: The Reaction of Pyridine and Carboxylic Acids in Chloroform, J. Am. Chem. Soc., 1956, 78, 5802–5806 CrossRef CAS.
- S. Míguez-Lago and M. M. Cid, Axially Chiral Shape-Persistent Encapsulating Agents, Synthesis, 2017, 4111–4123 Search PubMed.
- S. Castro-Fernández, J. Álvarez-García, L. García-Río, C. Silva-López and M. M. Cid, Double Protonation of a cis-Bipyridoallenophane Detected via Chiral-Sensing Switch: The Role of Ion Pairs, Org. Lett., 2019, 21, 5898–5902 CrossRef PubMed.
- A. Fuertes, M. Amorín and J. R. Granja, Versatile symport transporters based on cyclic peptide dimers, Chem. Commun., 2019, 56, 46–49 RSC.
- I. R. Lahoz, A. Navarro-Vázquez, A. L. Llamas-Saiz, J. L. Alonso-Gómez and M. M. Cid, Rotation-locked 2,6-pyrido-allenophanes: Characterization of all stereoisomers, Chem. – Eur. J., 2012, 18, 13836–13843 CrossRef CAS PubMed.
- D. Padula, I. R. Lahoz, C. Díaz, F. E. Hernández, L. Di Bari, A. Rizzo, F. Santoro and M. M. Cid, A Combined Experimental-Computational Investigation to Uncover the Puzzling (Chiro-)optical Response of Pyridocyclophanes: One- and Two-Photon Spectra, Chem. – Eur. J., 2015, 21, 12136–12147 CrossRef CAS PubMed.
- S. Castro-Fernández, I. R. Lahoz, A. L. Llamas-Saiz, J. L. Alonso-Gómez, M. M. Cid and A. Navarro-Vázquez, Preparation and characterization of a halogen-bonded shape-persistent chiral alleno-acetylenic inclusion complex, Org. Lett., 2014, 16, 1136–1139 CrossRef PubMed.
- S. Odermatt, J. L. Alonso-Gómez, P. Seiler, M. M. Cid and F. Diederich, Shape-persistent chiral alleno-acetylenic macrocycles and cyclophanes by acetylenic scaffolding with 1,3-diethynylallenes, Angew. Chem., Int. Ed., 2005, 44, 5074–5078 CrossRef CAS PubMed.
- R. Livingston, L. R. Cox, S. Odermatt and F. O. Diederich, 1,3-diethynylallenes: Carbon-rich modules for three-dimensional acetylenic scaffolding, Helv. Chim. Acta, 2002, 85, 3052–3077 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.2, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. Kütt, S. Tshepelevitsh, J. Saame, M. Lõkov, I. Kaljurand, S. Selberg and I. Leito, Strengths of Acids in Acetonitrile, Eur. J. Org. Chem., 2021, 1407–1419 CrossRef.
- D. Himmel, S. K. Goll, I. Leito and I. Krossing, A Unified pH Scale for All Phases, Angew. Chem., Int. Ed., 2010, 49, 6885–6888 CrossRef CAS PubMed.
- M. Vallaro, G. Ermondi, J. Saame, I. Leito and G. Caron, Ionization and Lipophilicity in Nonpolar Media Mimicking the Cell Membrane Interior, Bioorg. Med. Chem., 2023, 81, 117203 CrossRef CAS PubMed.
- S. Roelens, A. Vacca and C. Venturi, Binding of Ionic Species: A General Approach to Measuring Binding Constants and Assessing Affinities, Chem. – Eur. J., 2009, 15, 2635–2644 CrossRef CAS PubMed.
- G. Pescitelli, L. Di Bari and N. Berova, Application of electronic circular dichroism in the study of supramolecular systems, Chem. Soc. Rev., 2014, 43, 5211–5233 RSC.
- C. Wolf and K. W. Bentley, Chirality sensing using stereodynamic probes with distinct electronic circular dichroism output, Chem. Soc. Rev., 2013, 42, 5408–5424 RSC.
- A. Shimizu, Y. Inoue and T. Mori, A Combined Experimental and Theoretical Study on the Circular Dichroism of Staggered and Eclipsed Forms of Dimethoxy[2.2]-, [3.2]-, and [3.3]Pyridinophanes and Their Protonated Forms, J. Phys. Chem. A, 2017, 121, 8389–8398 CrossRef CAS PubMed.
- S. S. Bharate, Modulation of biopharmaceutical properties of drugs using sulfonate counterions: A critical analysis of FDA-approved pharmaceutical salts, J. Drug Delivery Sci. Technol., 2021, 66, 102913 CrossRef CAS.
- H. H. Limbach, M. Pietrzak, S. Sharif, P. M. Tolstoy, I. G. Shenderovich, S. N. Smirnov, N. S. Golubev and G. S. Denisov, NMR parameters and geometries of OHN and ODN hydrogen bonds of pyridine-acid complexes, Chem. – Eur. J., 2004, 10, 5195–5204 CrossRef CAS PubMed.
- B. Koeppe, S. A. Pylaeva, C. Allolio, D. Sebastiani, E. T. J. Nibbering, G. S. Denisov, H. H. Limbach and P. M. Tolstoy, Polar solvent fluctuations drive proton transfer in hydrogen bonded complexes of carboxylic acid with pyridines: NMR, IR and ab initio MD study, Phys. Chem. Chem. Phys., 2017, 19, 1010–1028 RSC.
- C. Vladiskovic, S. Mantegazza, G. Razzetti and N. Masciocchi, Salt or Cocrystal? Using XRPD to Infer Proton Transfer in Three Adducts of Iodoxybenzoic Acid by Analyzing Iodine-Oxygen Bond Lengths, Cryst. Growth Des., 2023, 23, 1119–1126 CrossRef CAS.
- K. Tsuji and P. J. Elving, Polarographic Measurement of Relative Strengths of Brønsted Acids in Pyridine Dispersion and Solvation Effects in Acid-Base Equilibria. Analysis of Brønsted Acid Mixtures, Anal. Chem., 1969, 41, 286–294 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic data and experimental details for the preparation of 3. Crystal data of 6, 3, 3·H2O·OTf, 3·(H2O·OTf)2 and 4·(H2O·OTf)2; geometries and spectra for the calculated species. CCDC 2280776, 2280778, 2280777, 2280775 and 2280779. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01180a |
| ‡ Dissymmetry factor (g) quantifies the effective chiroptical activity of a system by correlating the circular dichroism signal intensity with the average absorption signal intensity at a given frequency. |
| § CCDC 2280776 contains the supplementary crystallographic data for (P)-6, 2280778 for (P2)-3, 2280777 for (rac)-3·TfOH, 2280775 for (rac)-3·(TfOH)2 and 2280779 for (P4)-4·(TfOH)2.† |
| ¶ The allenophanes 2, 3 and 4 are configurational stable under the tested conditions and upon addition of Et3N the original circular dichroism spectra were recovered. |
| || The hydrogen coordination to both nitrogens yields a larger N⋯H bond that is compatible with a partial hydrogen transfer. A plausible interpretation of this result implies that the counterion acts as a shuttle for the mobile H+, provided both pyridines behave independently. |
| ** The measurements were made using GaussView 6 software. |
| This journal is © the Partner Organisations 2023 |