
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Celastraceae sesquiterpene pyridyl ligands
Chen
Dai
 and
Alan C.
Spivey
*
and
Alan C.
Spivey
*
Department of Chemistry, Molecular Sciences Research Hub (MSRH), Imperial College London, White City campus, 82 Wood Lane, London W12 0BZ, UK. E-mail: a.c.spivey@imperial.ac.uk
First published on 31st August 2023
Abstract
Celastraceae plant extracts have been widely used as traditional medicines and insecticides in China and South America. More recently, the extracts from Celastraceae plants have been studied at the molecular level and many of the interesting medicinal and agrochemical properties can be attributed to a large class of sesquiterpene alkaloids found in these extracts. These are generally based on highly oxygenated dihydroagarofuran cores with pyridyl diacid macrodilactone bridging ligands. Whereas previous reviews have focused on the dihydroagarofuran cores, in this article, the history, structure, and syntheses of the macrodilactone bridging ligands are reviewed.
 Chen Dai | Chen Dai received an MSci in chemistry in 2022 from Imperial College London and is currently in the first year of research towards a PhD exploring approaches to the total synthesis of bioactive Celastraceae sesquiterpene alkaloids under the direction of Professor Alan Spivey at Imperial College London. |
 Alan C. Spivey | Alan Spivey (https://www.imperial.ac.uk/people/a.c.spivey) has been professor of synthetic chemistry at Imperial College London since 2008. Previously, he was a lecturer then reader in the Department of Chemistry, University of Sheffield. His research encompasses the development of new reactions, synthesis of complex molecules of biomedical interest, including studies towards the synthesis of bioactive Celastraceae sesquiterpenoids, chemical biology and medicinal chemistry. For further details of his groups research activities see: https://www.imperial.ac.uk/spiveygroup. |
Introduction
Plant extracts have a long and venerable history of providing new lead compounds for drug discovery, and many drug candidates and approved therapeutics are derived from natural products.1 The Celastraceae family has approximately 88 genera and 1300 individual species and is found in tropical and subtropical regions of the world, including North Africa, South America, and many parts of East Asia, especially China. In particular, the leaves and roots of the genera Maytenus, Tripterygium and Celastrus have been utilized in traditional medicine and agriculture for centuries and extracts of these have been shown to exhibit e.g., stimulant, appetite suppressive, sedative, emetic, purgative, memory restorative, male contraceptive, anti-tumour, anti-leukemic, anti-bacterial, insecticidal and insect repellent activities (Fig. 1).2–4 | ||
| Fig. 1 (a) The properties and uses of thunder god vine (Tripterygium wilfordii) as traditional Chinese medicine documented in Addendum to Compendium of Materia Medica (Xuemin Zhao, 1765, Qing dynasty of China); (b) a vintage chromolithograph of spindle tree (Euonymus europaeus). | ||
Among all the bioactive natural products extracted from Celastraceae plants, a large set of sesquiterpenene alkaloids have proven to be predominantly responsible for their medicinal properties. These compounds are widely distributed in the Celastraceae and comprise a dihydroagarofuran core adorned with between two and nine esterified alcohol groups (Chart 1).2–4 These peripheral esterifying residues range from acetic and benzoic acids to the stereochemically unique pyridine-containing dicarboxylic acids which are characteristic of these natural products. The diacid ligands form macrodilactone bridges between C-3 and C-13 and/or C-8 and C-14, making these secondary metabolites topologically complex.2
 | ||
| Chart 1 The dihydroagarofuran skeleton. Two different numbering systems are commonly used, and the left system will be used in this review. | ||
The first reported isolation of a Celastraceae alkaloid was in 1887 when Katin was isolated from the root bark of Catha edulis Forskal. However, the sample obtained was impure and it was not until 14 years later that pure Katin was isolated and determined to have the molecular formula C10H18N2O (and a bitter taste!).5 This study was reported in Die Alkaloide in 1931, together with an account of the isolation of a few additional Celastraceae extracts, all of which were categorised as being ‘alkaloide mit unbekannter stammsubstanz’.6 The presence of alkaloids in plants belonging to the Euonymus genus were first reported in 1934,7 and Celastraceae alkaloids have been investigated extensively since then.
Previous reviews of dihydroagarofuran sesquiterpenoids have mainly focused on the tricyclic polyol core, with less attention paid to the macro-dilactone bridging ligands. In this article, the history, structure, and syntheses of the bridging ligands are surveyed (Chart 2).
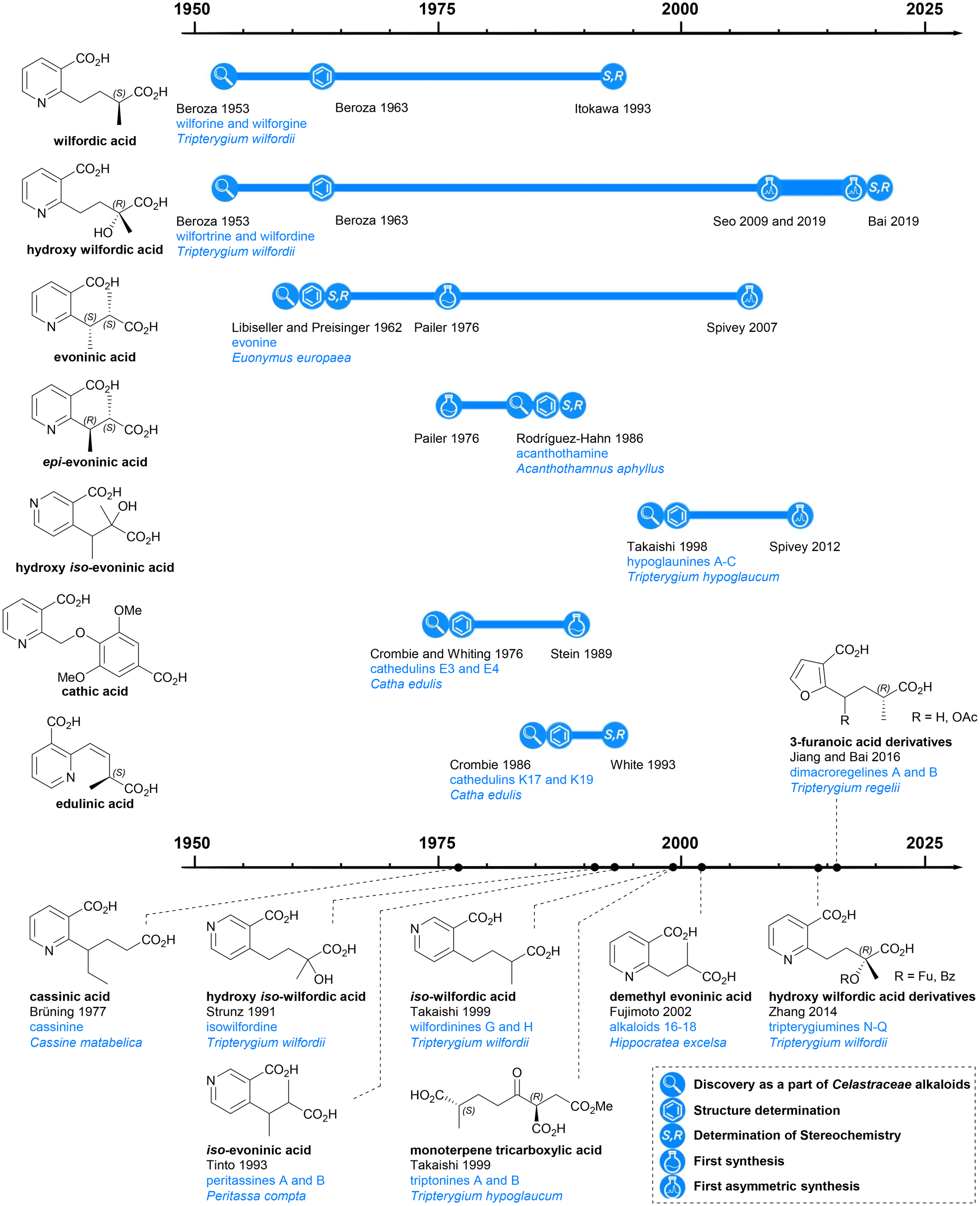 | ||
| Chart 2 Discovery and further studies related to the macrodilactone bridging ligands of Celastraceae sesquiterpene alkaloids. | ||
Discovery and classification of the macro-dilactone bridging ligands
The esterifying groups can be divided into two types in terms of their structures: nicotinic acid derivatives and 3-furoic acid derivatives. A monoterpene tricarboxylic acid has also been found with a bridging role. All the nicotinic acid derivatives have carboxylic acid-containing side chains at the 2- or 4-position of the pyridine ring: the wilfordates have butanoate side chains whereas evoninates have propanoate side chains. For the 3-furanoic acid derivatives, only the 2-position is substituted by a butanoate side chain. It is notable that each esterifying bridge only appears on either the upper or lower rim of the natural product cores, never both.Nicotinic acid derivatives
Among all macro-dilactone bridging ligands, the nicotinic acid derivatives are the most prevalent. This class includes wilfordates, evoninates and three other nicotinic acid derivatives. All these nicotinic acid derivatives with the exception of cathic acid, bridge between C-3 and C-13 across the lower rim of their dihydroagarofuran; cathic acid bridges on the upper rim between C-8 and C-14.Wilfordic acid derivatives
Wilfordic acid derivatives 1 to 4 (Chart 3a) were named after the species Tripterygium wilfordii, from which wilfordic acid (1) and hydroxy wilfordic acid (2a) were first isolated and characterized.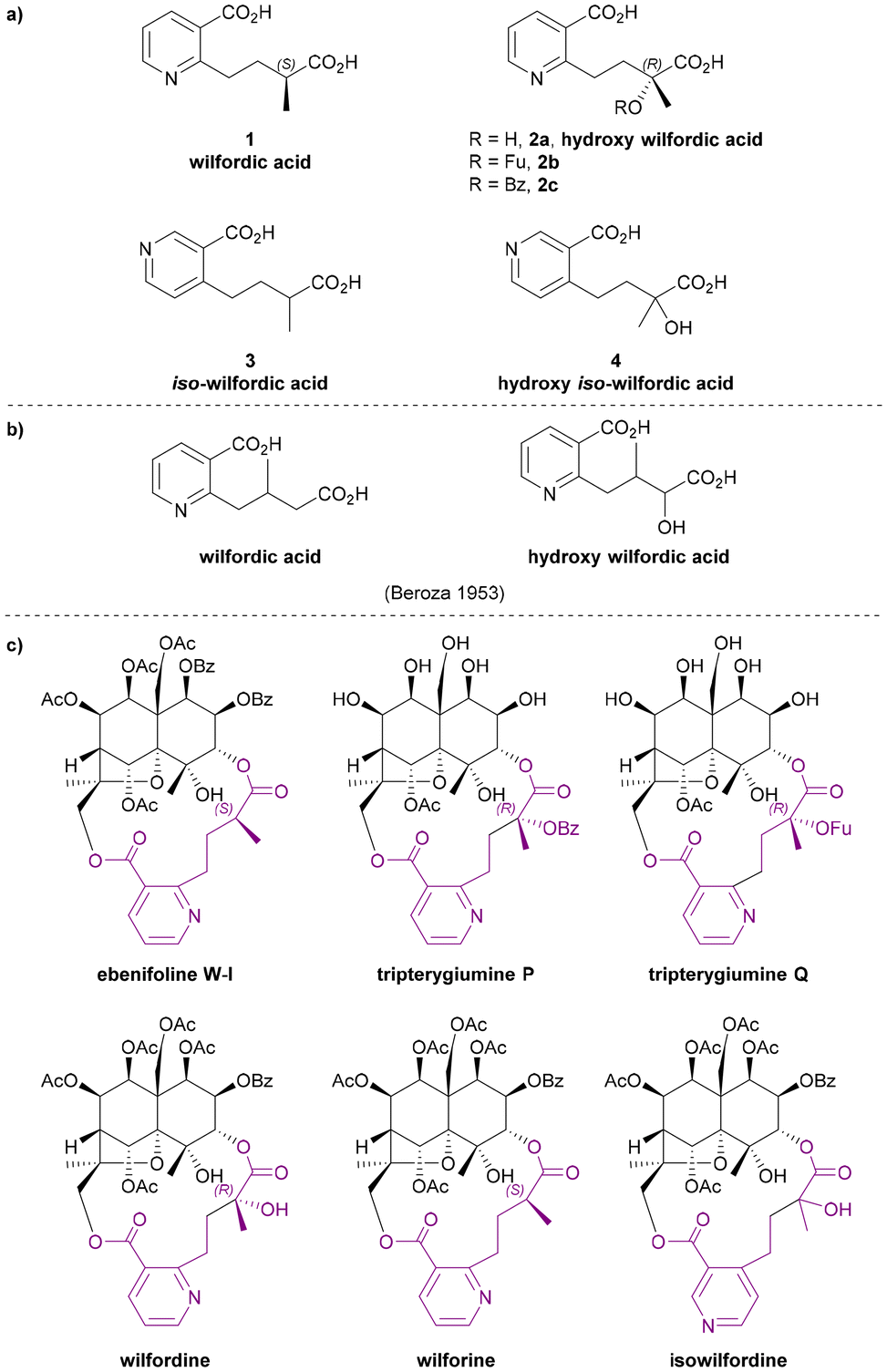 | ||
| Chart 3 (a) Wilfordic acid derivatives as the macrodilactone bridging ligands, (b) the structures for wilfordic acid and hydroxy wilfordic acid suggested by Beroza in 1953,11,12 (c) examples for Celastraceae sesquiterpene alkaloids that contributed to the studies of their bridging ligands. | ||
In 1950, Acree extracted an alkaloid wilfordine from Tripterygium wilfordii. The extract was believed to be a pure compound, as the melting point did not change after several recrystallizations. The formula was deduced, and wilfordine was identified as an ester alkaloid containing eight equivalents of acid upon saponification (including two equivalents of ‘steam non-volatile acid’).8 However shortly afterwards, Beroza followed up Acree's work, and proved that Acree's wilfordine was in fact a mixture of two structurally similar alkaloids differing by the presence/absence of a hydroxy group in the bridging ligand:9 the alcohol-containing congener was named wilforine, while the des-hydroxy one assigned the name wilfordine (Chart 3c).10 The structures were elucidated based on evidence from UV-vis absorption spectra, elemental analysis, X-ray diffraction (XRD) data and degradative reactions of these diacids. These structures as assigned in 1953 (Chart 3b), were subsequently revised in 1963 following the advent of NMR techniques (Chart 3a).11–14 Later, Beroza extracted three more alkaloids (wilforgine, wilfortrine, and wilforzine) from Tripterygium wilfordii, all of which had the same polyhydroxy core as wilforine and wilfordine. Several of these alkaloid were shown to possess significant insecticidal activity.15
Determination of the absolute configurations of these ligands was achieved only decades later. In the 1990s, Itokawa et al. studied some Amazonian medical plants belonging to the Maytenus genus, and isolated new sesquiterpenoids containing evoninate and wilfordate dilactone bridges.16–19 An XRD analysis on ebenifoline W-I (Chart 3c), a wilfordate type alkaloid from Maytenus ebenifolia, confirmed the structure of naturally occurring wilfordic acid 1 and revealed it to have an S-configuration.16 Hydroxy wilfordic acid (2a) as isolated from Tripterygium wilfordii Hook by Beroza in 1953, was determined to have an R-configuration in 2019 by both experimental and computational methods.20,21
Other wilfordic derivatives have been less intensively studied. The ester derivatives of hydroxy wilfordic acid 2b and 2c (Chart 3a) were only identified as constituents of alkaloids extracted from Tripterygium wilfordii in 2014; their stereochemistry was determined as being identical to hydroxy wilfordic acid (Chart 3c). The alkaloid containing 2b, named tripterygiumine Q, showed excellent immunosuppressive activity (IC50 8.67 μM against human peripheral mononuclear cells) but no cytotoxicity even at a high dose, making it a promising lead for drug development.22 iso-wilfordic acid (3), in which the aliphatic acid-containing side chain is situated at the 4- rather than 2-position of the nicotinic acid core, was first identified as a bridging ligand in three alkaloids (wilfordinines D-F) from the root xylem extract of Tripterygium wilfordii by Takaishi et al. in 1999.23
Hydroxy iso-wilfordic acid (4) was found in an isowilfordine (Chart 3c) – which was extracted by Li et al. in 1991 while re-examining alkaloids from this genera.24 The studies on both iso-wilfordic acid and hydroxy iso-wilfordic acid used NMR analysis for structure elucidation, and the absolute stereochemistry of the ligands were not determined.
Isotopic labelling studies relating to the biosynthesis of wilfordic and hydroxy wilfordic acids in Tripterygium wilfordii were reported in 1972![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 and in 2006, some structure activity relationships (SAR) studies established that the position of the carboxyalkyl chain in wilfordic vs. iso-wilfordic acids has negligible effect on their anti-HIV activity although the presence a hydroxy group in the carboxyalkyl side chain usually has a positive impact on activity.26
25 and in 2006, some structure activity relationships (SAR) studies established that the position of the carboxyalkyl chain in wilfordic vs. iso-wilfordic acids has negligible effect on their anti-HIV activity although the presence a hydroxy group in the carboxyalkyl side chain usually has a positive impact on activity.26
Evoninic acid derivatives
Evoninic acid derivatives 5 to 9 (Chart 4a) were named after the species Euonymus europaea. Probably the most well-known dilactone bridging ligand of the Celastraceae sesquiterpene alkaloids, evoninic acid (5) was discovered in 1962 by Libiseller, Preisinger, and Pailer.27–29 Studying the alkaloids from Euonymus europaea, ‘bases A, B, and C’ from Doebel's work in 194930 were identified as evorine, evozine, and evonine, respectively (Chart 4b). Upon the saponification of evonine, dimethyl evoninate was obtained. In 1976, Libiseller and Pailer suggested that evoninic acid (5) is a pyridine-containing dibasic acid with 2S,3S absolute configuration. They also determined that it is isomeric but not identical to wilfordic acid, as described by Beroza (Chart 6).28,29 Independently in 1971, Klásek et al. confirmed the structures of bases A (evorine), B (evozine) and C (evonine).31
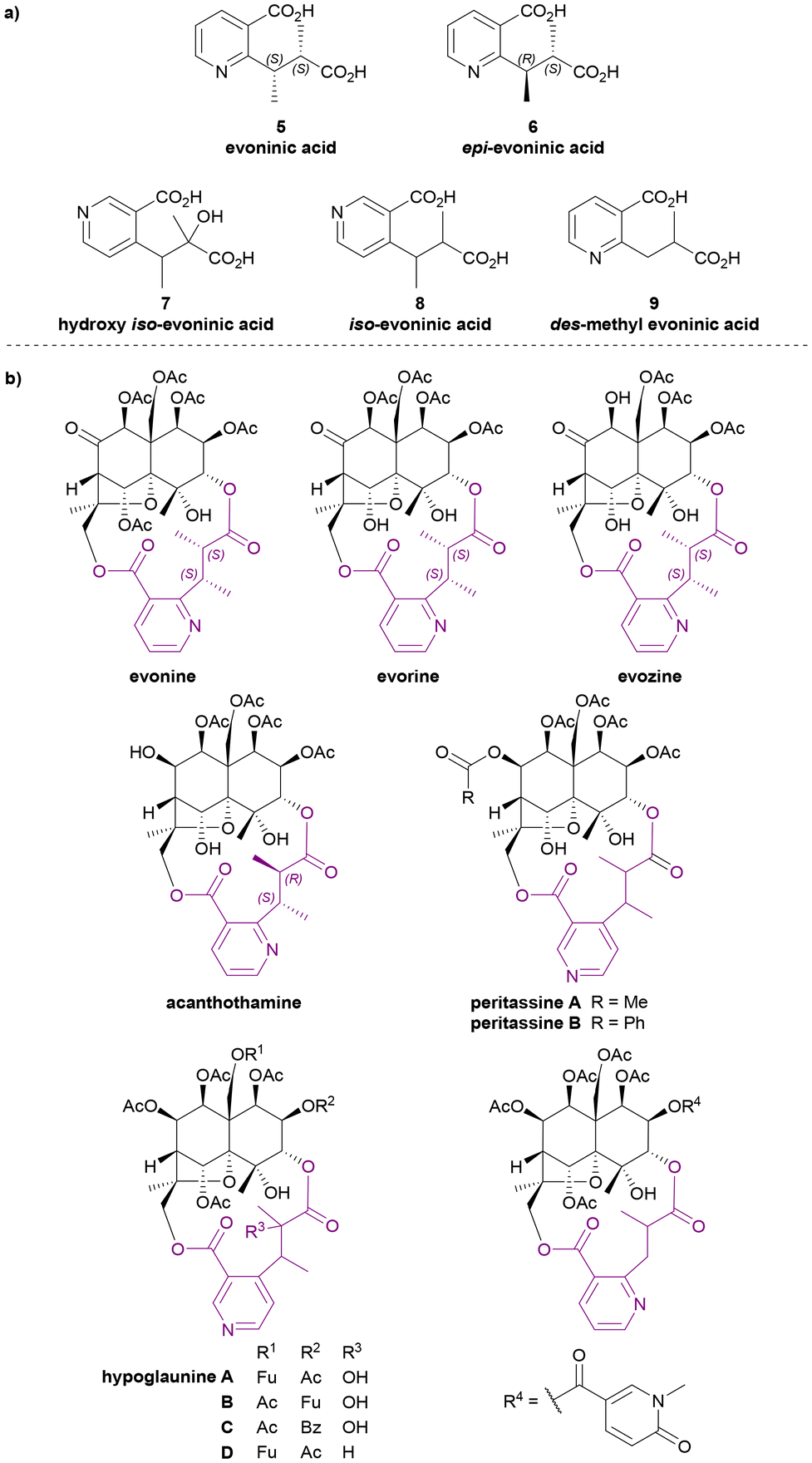 | ||
| Chart 4 (a) Evoninic acid derivatives as the macrodilactone bridging ligands, (b) examples for Celastraceae sesquiterpene alkaloids that contributed to the studies of their bridging ligands. | ||
In 1986, Rodríguez-Hahn et al. isolated a new alkaloid acanthothamine from Acanthothamnus aphyllus, which was identified as the first, and to date only, Celastraceae sesquiterpene alkaloid with an epi-evoninic acid (6) dilactone bridging ligand (Chart 4b). Though initially reported as ‘an iso-evoninic acid residue’, the authors noted that the 2R,3S configuration of epi-evoninic acid (6) is epimeric to evoninic acid (5), as shown by XRD analysis.32,33
In 1998, Takaishi et al. isolated four new sesquiterpene alkaloids hypoglaunines A, B, C and D (Chart 4b) from the root bark of Tripterygium hypoglaucum, and identified a novel bridging ligand, hydroxy iso-evoninic acid (7).34 In the same year, Li et al. independently isolated and characterised hypoglaunine A from the same plant, although the work was published a year later.35 More recently, a research group from Chongqing, China extracted hydroxy iso-evoninic acid (7) from the root bark of Tripterygium hypoglaucum. Their study found that this compound exhibits good immunosuppressive activity, anti-platelet aggregation activity and hypoglycaemic activity.36
In 1993 McLean et al. reported the presence of iso-evoninic acid (8) as a bridging ligand in peritassines A and B isolated from Peritassa compta (Chart 4b).37 In 2002, Fujimoto et al. reported des-methyl evoninic acid (9) (also named as nor-evoninic acid38) as a bridging ligand in three new sesquiterpene alkaloids isolated from Hippocratea excelsa.39 The absolute stereochemistries of neither 8 nor 9 were determined.
Other nicotinic acid derivatives
The three dilactone bridging ligands in this class (Chart 5a) are relatively rare compared to other Celastraceae sesquiterpene pyridine alkaloids: both cathic acid (10) and edulinic acid (11) were first found as bridging ligands in alkaloids from Catha edulis (khat), hence their names. Cassinic acid (12) has only been found as a bridging ligand in three Celastraceae plants, each belonging to different genera.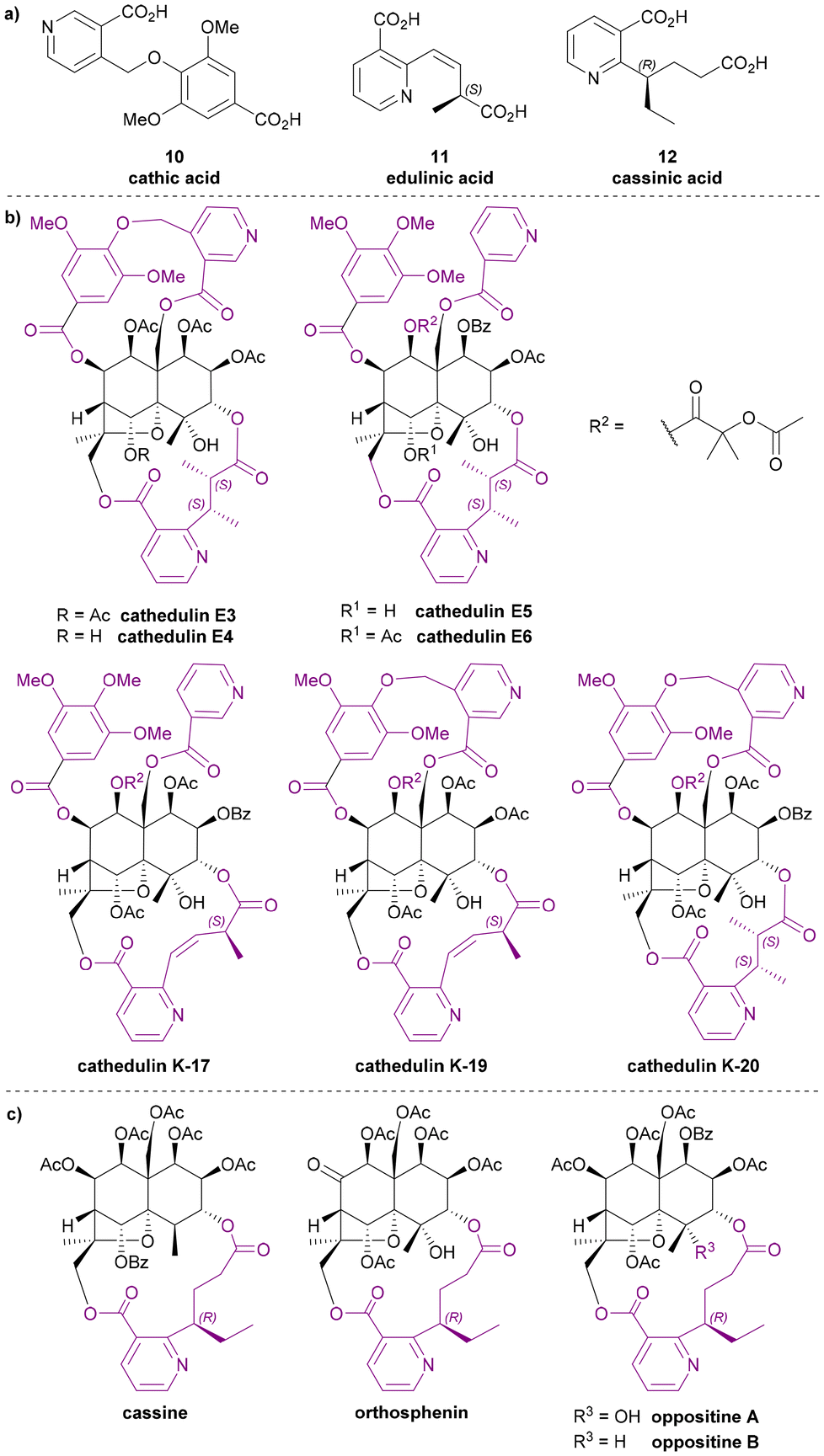 | ||
| Chart 5 (a) Other nicotinic acid derivatives as the macrodilactone bridging ligands for Celastraceae sesquiterpene alkaloids, (b) representative cathedulins, (c) all known Celastraceae sesquiterpene alkaloids with cassinic acid as the bridging ligands. | ||
From the late 1970s to late 1980s, a series of bridged sesquiterpenoids from khat sourced from Kenya, Ethiopia, and Yemen, received significant attention. These compounds were named cathedulins.40–45 Whiting and Crombie et al. isolated cathedulins 3 to 6 (later named cathedulins E3–E6 to designate their Ethiopian origin, Chart 5b) from khat leaves in 1976 and used NMR analysis and treatment with LiAlH4 to partially determine their structures. Cathedulins E3 and E4 were found to include a novel cathate ester unit, bridging between C-8 and C-14 of the core. Cathedulins E5 and E6 were shown to be congeners of cathedulins E3 and E4 but containing a gallate ester at C-8 and a nicotinate ester at C-14 – substituents that these authors speculated might undergo biosynthetic intramolecular radical macrocyclization to form the cathate dilactone bridge.41,46 In 1992, based on this hypothesis, Whiting et al. reported a radical process mimicking the proposed biosynthetic C–C bond formation.47 Among all dilactone bridging ligands of Celastraceae sesquiterpene alkaloids, cathic acid (10) is the only one without a stereogenic centre. In 1989, Stein and Nencini proposed a synthesis for dimethyl cathate (DMC) and performed the hydrolysis of DMC to obtain cathic acid (10). They also tested the bioactivity of DMC, showing that it appeared to protect mice from the convulsant effect of pentylenetetrazol.48
Whiting and Crombie et al. also identified edulinic acid (11) in cathedulins K17, K19 and K20 found in Kenyan khat leaves (Chart 5b). This diacid is the only macrodilactone bridging ligand with an unsaturated alkenyl side chain. Both cathedulins K17 and K19 were found to have an edulinic acid bridging ligands between C-3 and C-13 of their dihydroagarofuran cores. Interestingly, cathedulin K19 also has a cathate bridge between C-8 and C-14, making it highly topologically complex. The structure of edulinic acid (11) was elucidated using nOe experiments, showing that the alkene has a Z-configuration. The authors suggested edulinic acid (11) may have a similar biogenetic origin to evoninic acid (5), and therefore the stereogenic centre in edulinic acid (11) should be the same as found in evoninic acid (5).42 A synthesis (see later section) of (S)-edulinol was performed in 1993 by White et al.; this synthetic material was proven to be identical to the product obtained by reducing natural cathedulin K19. Hence, edulinic acid (11) was confirmed as having an S-configuration.49
Cassinic acid (12) was found in sesquiterpene alkaloids from three Celastraceae plants: cassinine from Cassine matabelica (1977),50,51 orthosphenin from Orthosphenia mexicana (1989),52 and oppositines A and B from Pleurostylia opposita (2006) (Chart 5c).53 In a 1977 study, Brüning et al. reported the XRD structure of cassinine, revealing its structural similarity to evonine and wilfordine.50
Monoterpene tricarboxylic acid
In 1999, Takaishi et al. extracted two novel sesquiterpene alkaloids, triptonines A and B (Chart 6b), from Tripterygium hypoglaucum.54 Later, the same group also found triptonine A in Tripterygium wilfordii.55 The structure elucidation of these two new alkaloids were completed using NMR and XRD analysis, which revealed a novel dilactone bridging ligand, the monoterpene tricarboxylic acid 13 (Chart 6a). The anti-HIV activity of triptonines A and B was tested: triptonine A exhibited potent anti-HIV activity (EC50 2.54 μg mL−1 for HIV replication in H9 lymphocytes) and relatively weak cell growth inhibition (IC50 >100 μg mL−1 against uninfected H9 cell).54,55 Later, in 2014, Zhang et al. isolated a third Celastraceae sesquiterpene alkaloid featuring this monoterpene tricarboxylic acid bridge, tripterygiumine A (Chart 6b).22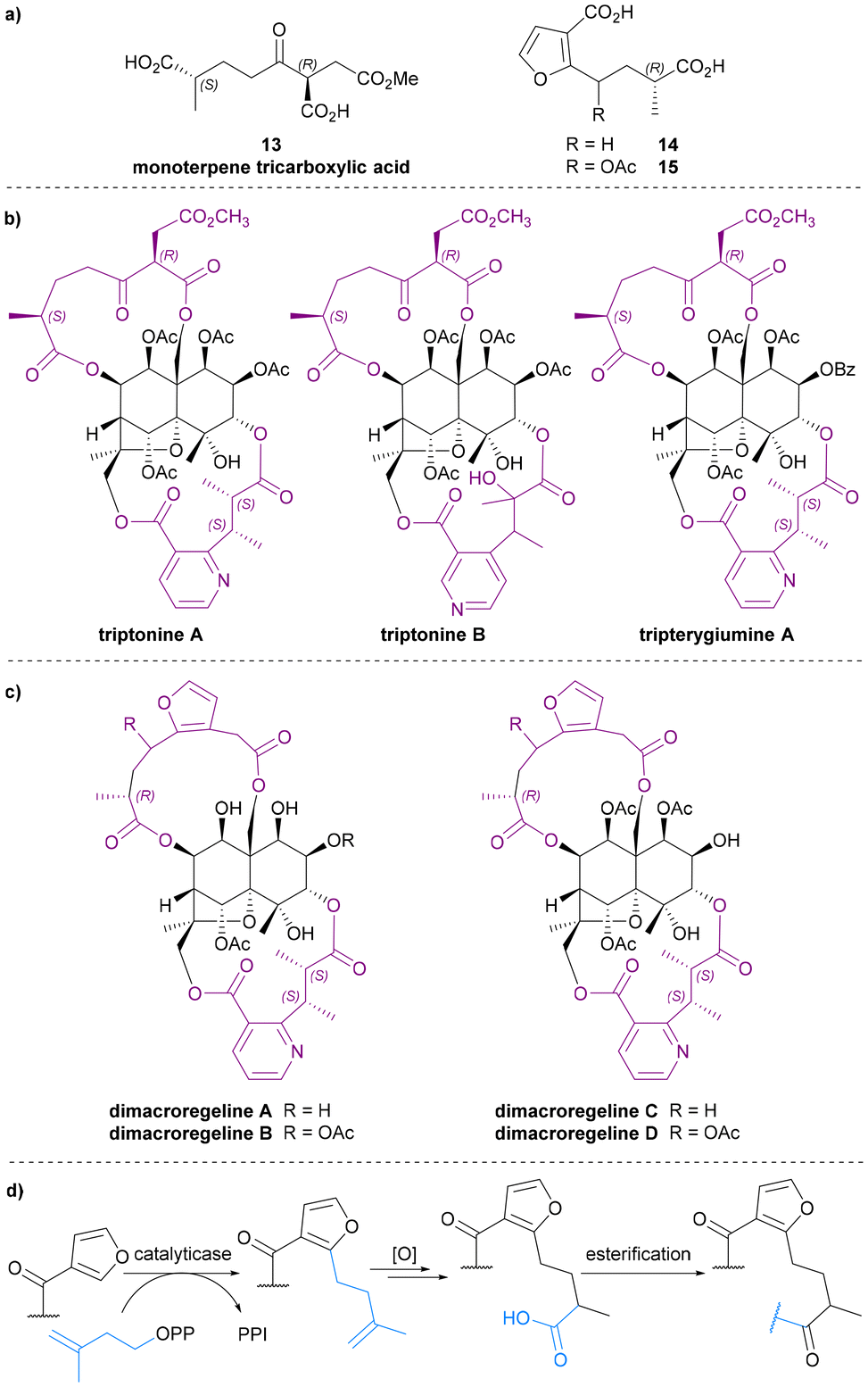 | ||
| Chart 6 (a) Monoterpene tricarboxylic acid and 3-furanoic acid derivatives, (b) all known Celastraceae sesquiterpene alkaloids with monoterpene tricarboxylic acid bridge, (c) all known Celastraceae sesquiterpene alkaloids with 3-furanoic acid derivatives as bridging ligands, (d) a plausible biosynthetic pathway for the biosynthesis of the 3-furanoic acid derivatives. | ||
Monoterpene tricarboxylic acid 13 is the only tricarboxylic acid dilactone bridging ligand to be discovered as a part of Celastraceae sesquiterpene alkaloids, and it only features in these di-macrocyclic derivatives on the bottom face of the dihydroagarofuran core, bridging between C-8 and C-14. Triacid 13 is the only known bridging ligand without an aromatic (i.e., pyridyl or furanyl) component.
3-Furanoic acid derivatives
3-Furanoic acid derivatives 14 and 15 (Chart 6a) are the most recently discovered dilactone bridging ligands. Like monoterpene tricarboxylic acid 13, they also bridge between C-8 and C-14 of the dihydroagarofuran core. Bai et al. extracted dimacroregelines A and B from Tripterygium regelii in 2016, and Kuang et al. extracted dimacroregelines C and D† from Tripterygium wilfordii in 2021 (Chart 6c).56,57By comparing NMR data, Bai et al. noticed the similarity in the structures of dimacroregelines A and B with triptonine A, which contains the monoterpene tricarboxylic acid 13. The difference was the appearance of a furan ring, and with further analysis using correlation methods, the structure of dilactone bridging ligands 14 and 15 were elucidated and reported for the first time. Kuang et al. confirmed the structures of ligands 14 and 15 based on the study of Bai et al., but the absolute configurations of these ligands were not established.56,57
Bai et al. also proposed a biosynthetic pathway by which these unprecedented 3-furanoic acid derivatives might plausibly be formed – involving regiospecific prenylation of a 3-furyl ester by a plant flavonoid prenyltransferase (Chart 6d).58 Due to their skeletal similarity, it is also possible that ligands 13 and 14 are biosynthetically related.
The synthesis of the macrodilactone bridging ligands and reactivities of Celastraceae sesquiterpene alkaloids
Compared to studies of dihydroagarofuran cores of Celastraceae sesquiterpene alkaloids and interest in their biological activity,59–62 studies directed at the structural elucidation and synthesis of their bridging ligands emerged later, reflecting advances in characterization techniques (e.g., NMR, XRD). The first synthesis of a bridging ligand was accomplished by Pailer in 1976,63 Just two further syntheses were reported in 1989 and 1993,48,49 before the 21st century saw the first stereoselective approaches reported.20,64–66The syntheses of evoninic acid (5) and epi-evoninic acid (6)
As is evident from the forgoing section, (S,S)-evoninic acid (5) is the bridging ligand of many bioactive Celastraceae sesquiterpenoids. The first synthesis of racemic evoninic acid (±-5), in 2% overall yield over 6 steps, was reported by Pailer and Pfleger in 1976 (Chart 7a).63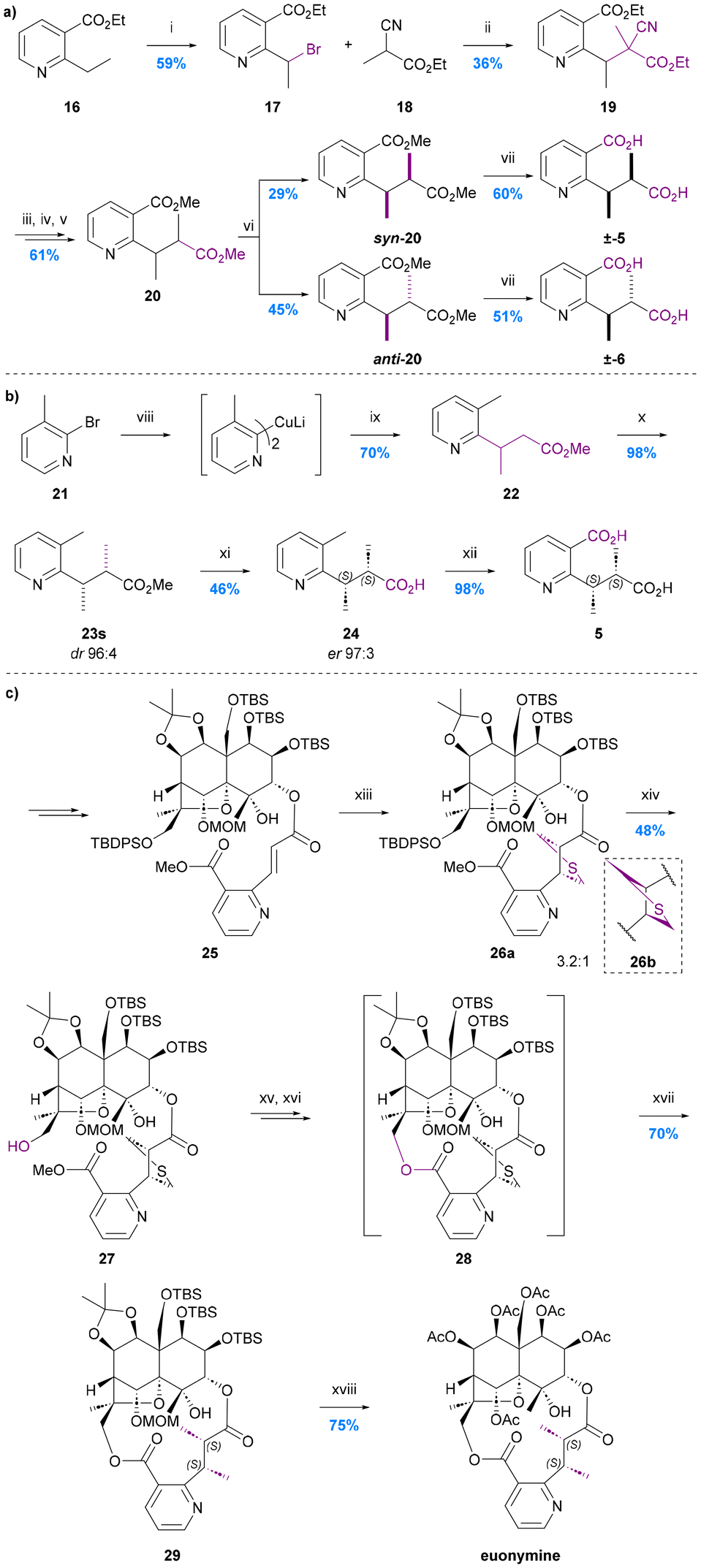 | ||
| Chart 7 The syntheses of evoninic acid 5. (a) Pailer and Pfleger 1976,63 i. N-Bromosuccinimide (1.9 eq.), benzoyl peroxide (0.04 eq.), CCl4, heating; ii. Na (1.0 eq.), 18 (1.0 eq.), EtOH; iii. conc. HCl, reflux; iv. NaOH, Et2O; v. CH2N2, MeOH/H2O 9:1; vi. Prep-TLC (iPr2O × 3); vii. 2 N KOH, reflux; (b) Spivey et al. 2007,65 viii. nBuLi, Et2O, −78 °C, then CuI, SBn2, Et2O, 0 °C; ix. methyl (E)-but-2-enoate, Et2O, rt; x. KHMDS, MeI, toluene, −78 °C; xi. lipase P. fluorescens, pH 7.0 buffer; xii. KMnO4, H2O, reflux; (c) Inoue et al. 2021,67 xiii. bis(trimethylsilylmethyl)sulfoxide (21 eq.), DMPU, 0 to 24 °C; xiv. nBu4NF, AcOH, DMF, 24 °C; xv. Me3SnOH, PhCl, 100 °C; xvi. PyBOP, DMAP, DMF, 80 °C; xvii. RANEY®-Ni, EtOH, 24 °C; xviii. AcOH, H2O, 100 °C, then Ac2O, Et3N, DMAP, CH2Cl2, 24 °C. | ||
Bromination of ethyl 2-nicotinate 16 using N-bromosuccinimide in the presence of sub-stoichiometric benzoyl peroxide, yielded bromide 17, to which was added the sodium salt of ethyl 2-cyanopropanoate prepared in situ. The resulting nitrile 19 was treated with conc. HCl under reflux to effect hydrolysis and decarboxylation, then neutralised and reacted with diazomethane in methanol/water to give a mix of stereoisomers of diester 20.
The syn and anti isomers of ester 20 were separated using preparative TLC and their relative stereochemistries assigned by reference to literature spectroscopic (IR, NMR, MS) and GC data for the dimethyl esters of naturally occurring evoninic acid. The corresponding racemic acids were obtained by basic hydrolysis. The overall yield of the syn- or (RS,RS)-isomer corresponding to evoninic acid (±-5) was just 2.3% from nicotinate 16 whereas the overall yield of the anti- or (RS,SR)-isomer corresponding to the (at that time, yet-to-be-isolated from a natural source) epi-evoninic acid (±-6) was 3.0%. The overall yield of this synthetic route was primarily compromised by the moderate yield of the key C–C bond forming enolate alkylation reaction (17 → 19, 36%) and the poor diastereoselectivity of the decarboxylation step (19 → 20, dr ∼29:45 syn:anti).
The first asymmetric total synthesis of evoninic acid (5), in 4 steps and 31% overall yield, was reported by Spivey et al. in 2007 (Chart 7b).65 Their synthesis started with the conjugate addition of a 2-pridinyl Gilman homocuprate 21 to (E)-methyl crotonate giving racemic ester 22. Subsequent diastereoselective methylation of the potassium enolate of this ester with MeI in toluene at low temperature gave the desired syn diastereoisomer 23s in 98% yield and 96:4 dr.
The purified syn-isomer was then subject to hydrolytic kinetic resolution using Pseudomonas fluorescens lipase in phosphate buffer (pH 7, 37 °C), which afforded the corresponding syn-acid 24 with an er of 97:3 in 46% yield. Finally, treatment with KMnO4 selectively oxidized the pyridine methyl group of the acid 24, to give (S,S)-evoninic acid 5 in almost quantitative yield. The absolute configuration was assigned by the conversion to the corresponding dimethyl ester and comparison with the literature data for the corresponding naturally derived material. Although both the diastereoselectivity and enantioselectivity obtained by this approach are good, the overall yield is compromised by the need for an enzymatic kinetic resolution step (23s → 24), which although efficient, necessarily means that more that 50% of the material is not progressed into the final product. The development of an asymmetric variant of the key cuprate conjugate addition step (21 → 22) would significantly improve the approach.
In 2021, Inoue et al. reported the first total synthesis of the evoninic acid bridged macrodilactone-containing sesquiterpene alkaloid euonymine; their route featured an unusual in situ construction of the evoninic acid substructure (Chart 7c).67 Thus, an aza-cinnamic acid precursor to the evoninate moiety was introduced at C-3 of an enantiopure advanced dihydroagarofuran intermediate via a standard esterification reaction giving ester 25 in 91% yield. The alkene therein was then heated with bis(trimethylsilylmethyl)sulfoxide (21 equiv.) and DBU to effect an intermolecular [3 + 2]-cycloaddition. This gave diastereomeric thiolanes 26a:26b in a 3.2:1 ratio, the major isomer of which was subsequently shown to lead to the required (S,S)-product after macrocyclization and hydrogenolysis. Interestingly, when macrocyclization was carried out prior to the [3 + 2]-cycloaddition, a thiolane corresponding to the undesired (R,R)-diastereoisomer was obtained exclusively. Although 26a and 26b were inseparable, separation could be achieved once the C13 alcohol had been deprotected using TBAF (i.e., compounds 27). Methyl ester hydrolysis, and then macrolactonization on the required diastereoisomer set the stage for the final thioether hydrogenolysis to reveal the required (S,S)-dimethyl motif of the evoninate bridge in protected euonymine 29.
Despite the large amount of bis(trimethylsilylmethyl)sulfoxide used and the high price of its commercially available precursor, the [3 + 2]-cycloaddition provided an elegant, albeit not very diastereoselective, way to introduce the two stereocentres of the evoninate bridge.
The synthesis of hydroxy iso-evoninic acid (8)
The first and only total synthesis of hydroxy iso-evoninic acid (8) was achieved via a benzylic ester rearrangement (BER) by Spivey and Warren in 2012 (Chart 8).66 They prepared a mixture of racemic syn and anti diastereoisomers and then used chiral stationary phase (CSP) HPLC to separate all four stereoisomers. Neither the relative nor absolute stereochemistry of naturally occurring iso-evoninic acid (8) is known.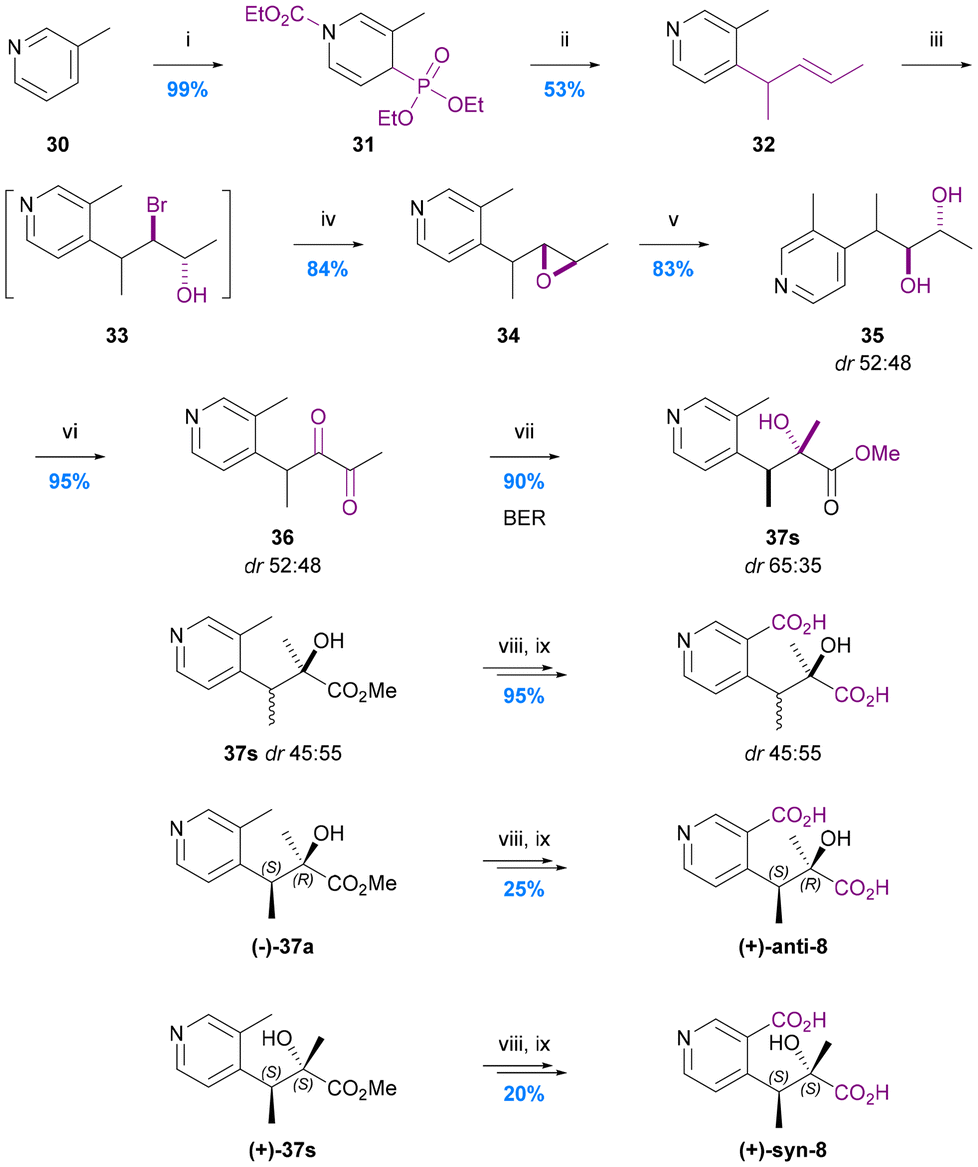 | ||
| Chart 8 The synthesis of hydroxy iso-evoninic acid 8. Spivey et al. 2012,66 (i) EtOCOCl, MeCN, then P(OEt)3, (ii) nBuLi (1 eq.), THF, then 2-bromopent-3-ene, then nBuLi (2 eq.), (iii) NBS, THF/H2O, (iv) 1 M NaOH, MeCN, (v) 6% HClO4 (aq.), MeCN, (vi) (COCl)2, DMSO, CH2Cl2, then Et3N, (vii) ZnCl2 (1 eq.), MeOH, 40 °C, (viii) 2 M NaOH, MeOH, (ix) 1 M NaOH, KMnO4. | ||
In the initial stage of the synthesis, N-carbamoylation of 3-picoline (30) and treatment with triethylphosphite gave the 1,4-dihydropyridine derivative 31 which was alkylated at the 4-position using n-BuLi (1 equiv.) and 2-bromopent-3-ene, and then re-aromatised to give 4-allyl-3-picoline 32 by treatment with n-BuLi (2 equiv.). This intermediate underwent epoxidation via a mixture of bromohydrins and subsequent hydrolytic ring-opening to give diol 35 (in an inconsequential dr of 52:48) which was converted to the 1,2-diketone 36via Swern oxidation. This labile 1,2-diketone was the substrate for the key BER which was promoted using zinc chloride in methanol at 40 °C. The BER afforded esters 37 in 90% yield with a dr of 35:65. A 76% yield of esters 37 with reverse diastereoselection (dr 66:34) could be obtained using 0.1 M NaOMe in methanol at 40 °C to promote the BER, but conditions that would deliver higher levels of diastereoselection favouring either diastereoisomer proved elusive. Consequently, all four stereoisomers were separated by CSP-HPLC and the dextro-rotatory syn and anti diastereoisomers progressed via saponification then benzylic oxidation using KMnO4 to give (2R,3S)-hydroxy-iso-evoninic acid [(+)-anti-8] and (2S,3S)-hydroxy-iso-evoninic acid [(+)-syn-8]. Although this 9-step synthesis gives access to a racemic mix of cis- and trans-hydroxy iso-evoninic acids 8 with ∼26% overall yield, the poor diastereoselectivity of the BER and need for CSP-HPLC separation of stereoisomers leaves much scope improvement. In particular, the development of an asymmetric BER concomitant with dynamic kinetic resolution (DKR)68 could constitute a very efficient ‘second generation’ approach.
The syntheses of (S)-hydroxy wilfordic acid ((S)-2a)
The total synthesis of hydroxy wilfordic acid has only been studied relatively recently. Seo et al. synthesized hydroxy wilfordic acid employing an asymmetric cyanosilylation as the key step in 2009, and the same research group reported an improved synthesis in 2019.20,64 The 2019 disclosure also demonstrated that naturally occurring hydroxy wilfordic acid 2a has the R-configuration, which is the antipode of their synthetic hydroxy wilfordic acid (S)-2a. The R-configuration of naturally occurring hydroxy wilfordic acid 2a had been proposed by Bai et al. earlier in 2019, based on the comparison between simulated and experimental ECD spectra.21In Seo's 2009 synthesis the methyl group in pyridine 21 was oxidised to aldehyde 38 and then a Horner–Wadsworth–Emmons (HWE) alkenylation was carried out to give enone 39 – the substrate for the key asymmetric cyanosilylation reaction (Chart 9a).64 Use of Jacobsen's commercially available amino thiourea catalyst69,70 led to the highest yield and enantioselectivity for this process giving silylated cyanohydrin 40 in 82% yield and 75:25 er. The mixture of enantiomeric nitriles was hydrolysed/esterified with 6 M HCl in MeOH, the alkene hydrogenated, and the benzylic methyl group oxidized to the corresponding benzylic acid with concomitant ester hydrolysis using aqueous KMnO4 to give hydroxy wilfordic acid (2a) as a crude mixture of enantiomers. This mixture was esterified and purified to give dimethyl hydroxy wilfordate 43 in 7 steps and 43% overall yield from bromopyridine 21. The major enantiomer of this 75:25 er product was shown to have an S-configuration via derivatisation of the acid corresponding to cyanohydrin 40 as an amide with (S)- and (R)-phenylglycine methyl esters (PGMEs) followed by hydrogenation of the alkene. These amides were then separated by HPLC to facilitate stereochemical assignment by NMR.
 | ||
| Chart 9 The synthesis of hydroxy wilfordic acid 2a. (a) Seo et al. 2009,64 (i) nBuLi, DMF, THF, (ii) MeCOCH2P(O)(OMe)2, NaH, THF, (iii) Jacobsen's amino-thiourea catalyst, TMSCN, CF3CH2OH, CH2Cl2, −78 °C, (iv) 6 M HCl, MeOH, reflux, (v) H2, Pd/C, MeOH, (vi) KMnO4, H2O, reflux, (vii) HCl, MeOH; (b) Seo et al. 2019,20 (viii) c. HCl, MeOH, 60 °C, (ix) TMSCHN2, MeOH, (x) H2, Pd/C, MeOH, (xi), LiOH·H2O, MeOH/H2O. | ||
Their second-generation 2019 route to hydroxy wilfordic acid, mirrored their initial route but started from methyl 2-chloronicotinate in order to avoid the late-stage benzylic oxidation (Chart 9b).20 This tactic fortuitously resulted in a much improved asymmetric cyanosilylation step to give silylated cyanohydrin 46s in 90% yield and 35:1 er using the same amino thiourea catalyst under unchanged conditions. Acid catalysed nitrile hydrolysis followed by hydrogenation gave the diester 47, which was saponified to give (S)-hydroxy wilfordic acid (S)-2a ([α]20 = +20.4; 93.3% ee) in 7 steps and 48% overall yield from methyl 2-chloronicotinate. A sample of the natural hydroxy wilfordic acid 2a ([α]24 = −24.1) was obtained by hydrolysing commercially available wilfortrine, thereby confirming natural hydroxy wilfordic acid as having an R-configuration.
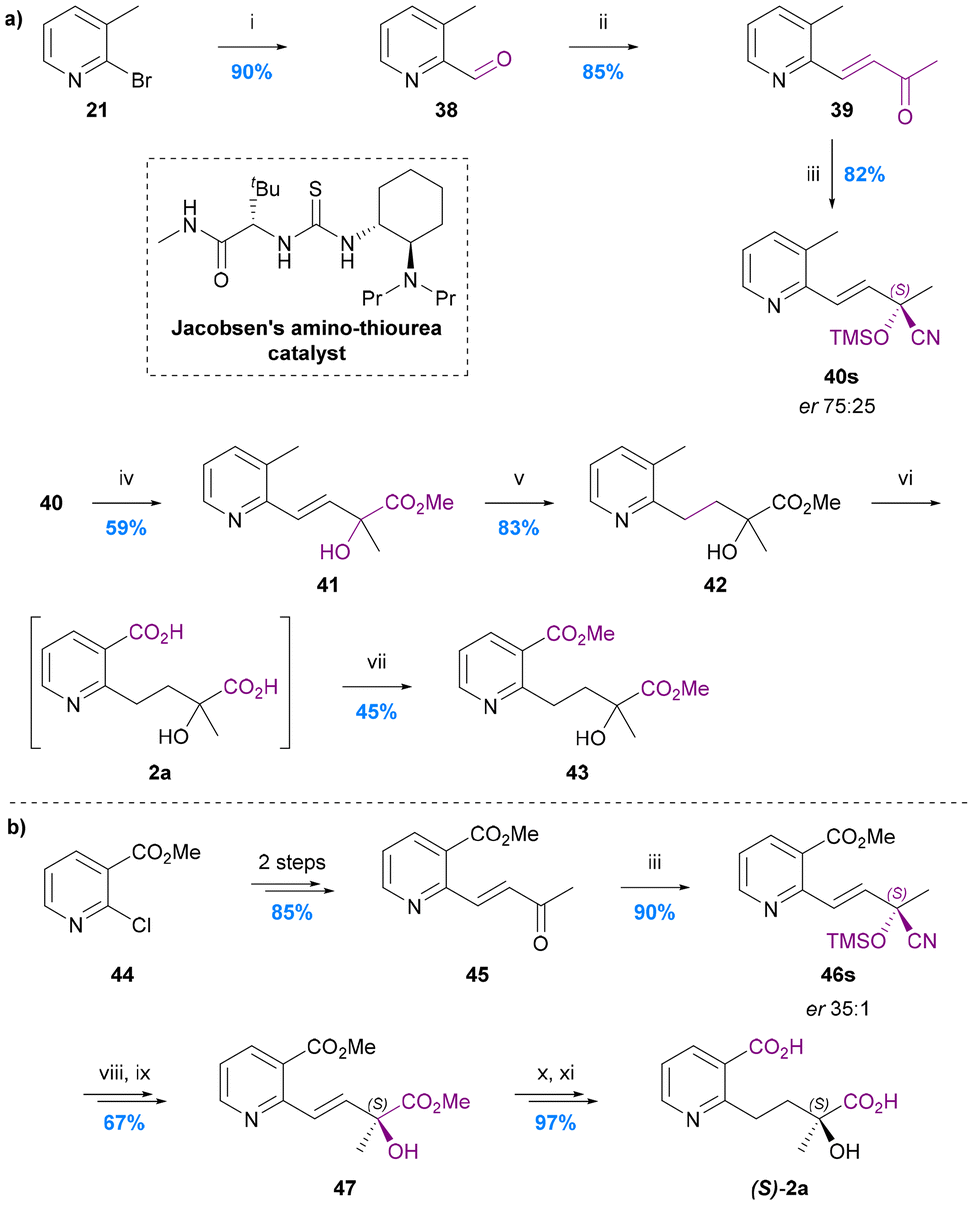
The synthesis of cathic acid (10)
The only total synthesis of cathic acid (10) was reported in 1989 by Stein and Nencini (Chart 10a).48 The synthesis commenced with regioselective methanolysis of 3,4-pyridinedicarboxylic acid anhydride 48, to give mono-methyl ester 49. LiAlH4 reduction of this ester to the benzylic alcohol 50 followed directly by treatment with POCl3/PCl5 and quenching with methanol gave 4-chloromethy methyl nicotinate (51). Alkylation of sodium methyl syringate (53) with this nicotinate derivative gave dimethyl cathate (DMC) 53. Saponification of the DMC gave cathic acid (10) in 75% yield, equating to an overall yield of ∼4% over 6 steps from ester 49. The yield and selectivity of the first step (48 to 49) was not reported, nor were any individual step yields up to DMC 53, which was the first purified product. The authors noted however that the syringate alkylation step was particularly inefficient, presumably because of the steric hindrance of the di-ortho-substituted nature of this phenol. An alternative, more efficient etherification protocol for syingate will likely need to be developed for any efficient next generation synthesis of cathic acid.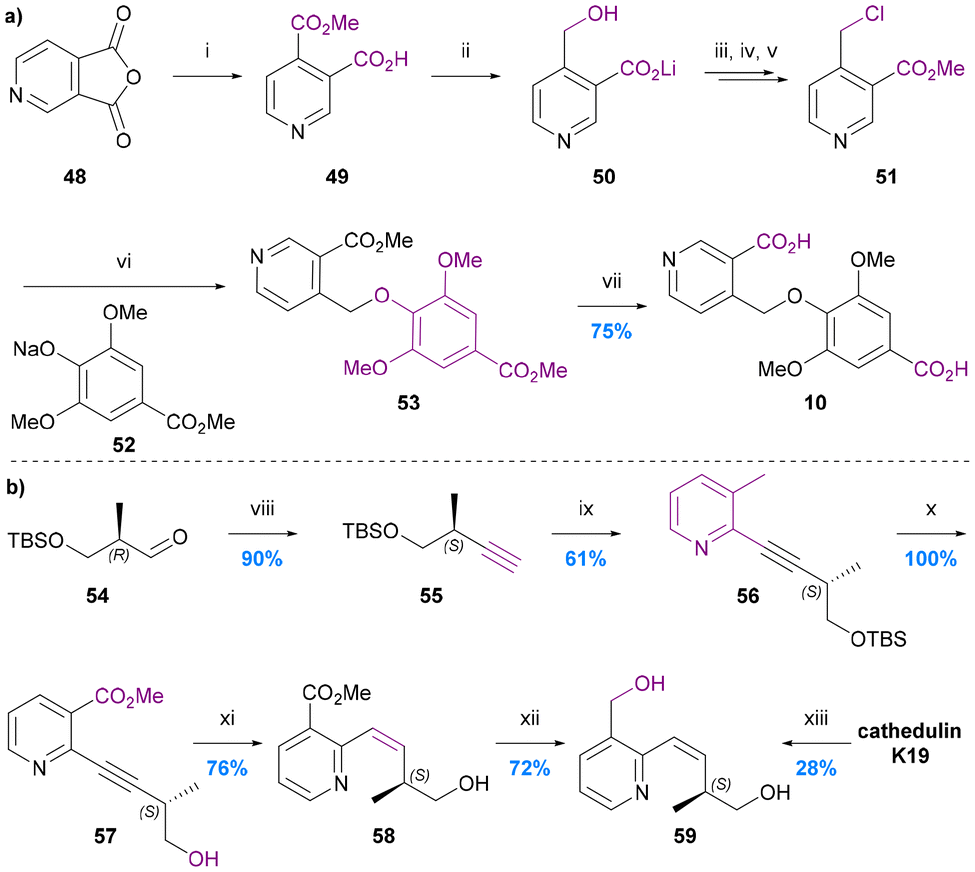 | ||
| Chart 10 The synthesis of cathic acid 10 and edulinol 59. (a) Stein and Nencini 1989,48 (i) MeOH, (ii) LiAlH4, THF, (iii) POCl3, PCl5, (iv) MeOH, (v) pH 7.4, (vi) MeOH, (vii) 4 N KOH, MeOH; (b) White et al. 1993,49 (viii) (MeO)2POCHN2, tBuOK, THF, (ix) methyl 2-chloronicotinate, (Ph3P)2PdCl2, CuI, Et2NH, (x) 5% HF/MeCN, (xi) H2, Lindlar cat., MeOG, (xii) LiAlH4, THF, 0 °C, (xiii) LiAlH4, THF/Et2O, 0 to 25 °C. | ||
The synthesis of edulinol (59)
No synthesis of edulinic acid 11 has been reported, but its reduced derivative edulinol 59 was synthesized by White in 1993 in order to establish the absolute configuration of edulinic acid from cathedulin K19 (Chart 10b).49The synthesis started with a Seyferth–Gilbert homologation of (2R)-3-siloxy-2-methylpropanalaldehyde 54 to furnish terminal alkyne 55. This alkyne then underwent Sonogashira coupling with methyl 2-chloronicotinate, deprotection and Lindlar's reduction to yield the cis-alkene 58, which was reduced to give edulinol 59 ([α]22 = +126) in 7 steps and 30% overall yield from aldehyde 54. Edulinol was also obtained upon reduction of cathedulin K19 ([α]22 = +210). The two edulinol samples had consistent IR, 1H and 13C NMR spectra and so the significant difference between the optical rotation values was attributed to the partial racemization of alkyne 57 during its syn-hydrogenation. 1H and 19F NMR spectroscopic analysis of its Mosher ester derivative indicated that alkene 58 had a 44% ee, suggesting that partial racemization also occurs during its isolation by reduction of cathedulin K19. There remains a need therefore for the future development of a synthesis of edulinic acid (11) that circumvents the racemisation-prone hydrogenation of an unsaturated C–C linkage between the stereogenic centre and the pyridine ring, either by introducing the stereogenic centre at a later stage of the synthesis or simply avoiding an unsaturated linkage.
Macrodilactone formation using these diacid ligands
To date there have been only two studies addressing the incorporation of diacid ligands into the macrodilactone structures of the Celastraceae alkaloids. In 1975 Yamada provided the first protocol for achieving this by the resynthesis of evonine from an evoninol core (obtained by degradation of natural evonine) and an evoninic acid derivative (Chart 11).62 Stepwise formation of the 14-membered dilactone ring with a modified core71 was accomplished by ester formation at the C-13 hydroxy group (62 to 63) followed by macrolactonization onto the C-3 hydroxy group (64 to 65). Remarkably, this remained the only report until the total syntheses of euonymine by Inoue et al. in 2021 (Chart 7c).67 In contrast to Yamada's resynthesis, Inoue first performed ester formation at the C-3 hydroxy group, and elegantly constructed the stereogenic centres of the diacid ligand before forging the 14-membered macro-dilactone ring with C-13 ester formation.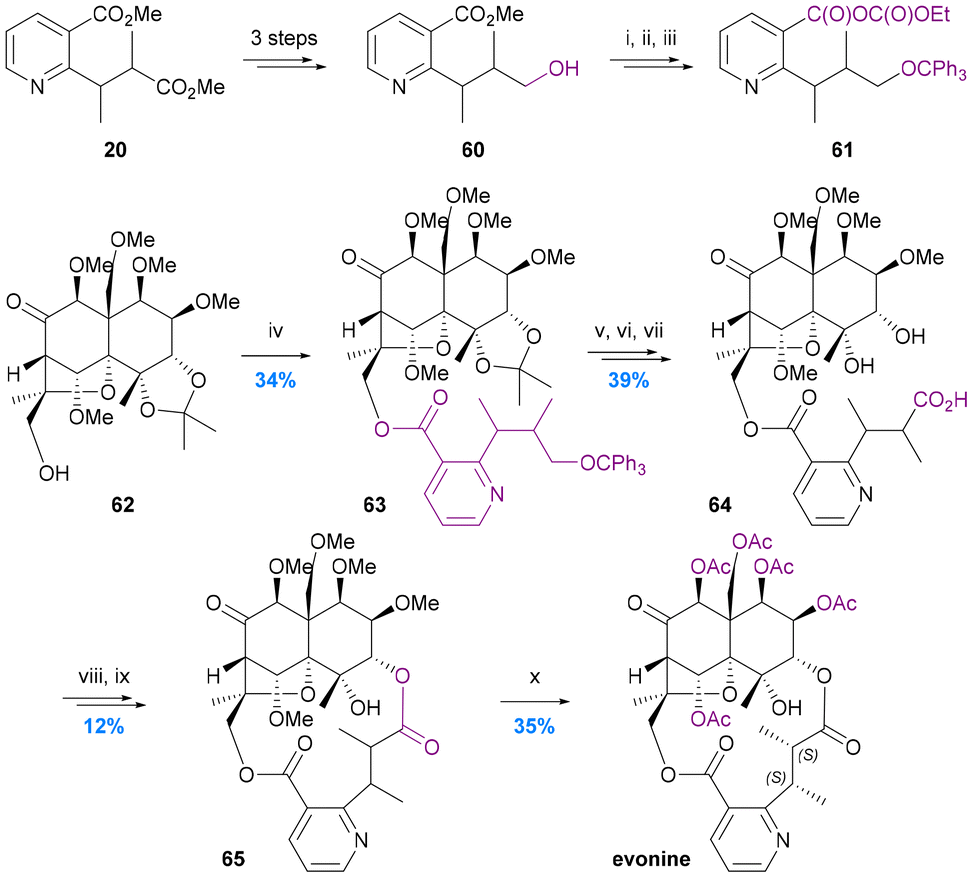 | ||
| Chart 11 Yamada's resynthesis of evonine. Yamada et al. 1975,62 (i) Ph3CCl-Py, 60 °C; (ii) 10% KOH–MeOH, 70 °C; (iii) ethyl chloroformate, Et3N, DME, RT; (iv) crude 61, DMAP, Et3N, DME, 90 °C; (v) 80% AcOH, 50 °C; (vi) CrO3-Py, 65 °C; (vii) 50% AcOH, 85 °C; (viii) CH2N2; (ix) NaH, DMF, RT; (x) BCl3–CH2Cl2, RT, and then AcO2-Py, 70 °C. | ||
Clearly, there is great scope for the development of creative new approaches to orchestrate the construction of the macrodilactone rings found in the natural product alkaloids. It is notable that there has been no work published to date for forging the C-8/C14 linked structures. Although the lactonisation reactions themselves are likely not inherently difficult, involving as they do primary (C13 and C14) and secondary (C3 and C8) alcohols, the main challenge will be constructing the dihydroagarofuran cores such that these specific positions can be reacted/activated selectively. This is a formidable endeavour given the complexity of the most prevalent core polyols, but the Herzon group has made important recent progress by preparing a selectively protected derivative of the nonahydroxylated dihydroagarofuran core (−)-euonyminol,72,73 so we can probably expect exploration of this chemistry in the near future.
Conclusions
More than 50 years have elapsed since the structure of the first macrodilactone containing sesquiterpene alkaloid was elucidated from a Celastraceae plant extract in 1971,74 and despite the fact that hundreds more have now been characterised and many display exciting biological activity, these structures remain formidable targets for synthesis. Indeed just one successful total synthesis has been described to date – that of euonymine by the Inoue group in 2021 (see Chart 7c).67 Whilst clearly the stereochemically rich polyhydroxylated dihydroagarofuran cores constitute the primary challenge for synthesis and have consequently attracted significant attention (and this work has been reviewed previously2–4), their elaboration to the bioactive natural products requires combination with the appropriate diacid bridging ligands that have been the focus of this review. Although progress has been made towards developing efficient methods to prepare these, there remain many ligands for which the relative and absolute stereochemistry remains unknown and for which no syntheses have been reported (Table 1). It is hoped that this review will spark interest in taking up the challenge of their synthesis, thereby paving the way for further total syntheses of the parent macrocyclic alkaloids and ultimately a better understanding of their biology and medicinal potential.| Bridging ligands | Stereochemistry | Synthesis |
|---|---|---|
| 1, wilfordic acid | ||
| 2a, hydroxy wilfordic acid | ✓ | ✓ |
| 2b, 2a derivative | ✓ | |
| 2c, 2a derivative | ✓ | |
| 3, iso-wilfordic acid | ||
| 4, hydroxy iso-wilfordic acid | ||
| 5, evoninic acid | ✓ | ✓ |
| 6, epi-evoninic acid | ✓ | ✓ |
| 7, hydroxy iso-evoninic acid | ||
| 8, iso-evoninic acid | ||
| 9, des-methyl evoninic acid | ✓ | |
| 10, cathic acid | N/A | ✓ |
| 11, edulinic acid | ✓ | |
| 12, cassinic acid | ||
| 13, monoterpene tricarboxylic acid | ✓ | |
| 14, furanoic acid derivative | ✓ | |
| 15, furanoic acid derivative |
Author contributions
CD and ACS wrote this manuscript jointly.Conflicts of interest
There are no conflicts to declare.References
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch, I. E. Orhan, M. Banach, J. M. Rollinger, D. Barreca, W. Weckwerth, R. Bauer, E. A. Bayer, M. Majeed, A. Bishayee, V. Bochkov, G. K. Bonn, N. Braidy, F. Bucar, A. Cifuentes, G. D’Onofrio, M. Bodkin, M. Diederich, A. T. Dinkova-Kostova, T. Efferth, K. El Bairi, N. Arkells, T.-P. Fan, B. L. Fiebich, M. Freissmuth, M. I. Georgiev, S. Gibbons, K. M. Godfrey, C. W. Gruber, J. Heer, L. A. Huber, E. Ibanez, A. Kijjoa, A. K. Kiss, A. Lu, F. A. Macias, M. J. S. Miller, A. Mocan, R. Müller, F. Nicoletti, G. Perry, V. Pittalà, L. Rastrelli, M. Ristow, G. L. Russo, A. S. Silva, D. Schuster, H. Sheridan, K. Skalicka-Woźniak, L. Skaltsounis, E. Sobarzo-Sánchez, D. S. Bredt, H. Stuppner, A. Sureda, N. T. Tzvetkov, R. A. Vacca, B. B. Aggarwal, M. Battino, F. Giampieri, M. Wink, J.-L. Wolfender, J. Xiao, A. W. K. Yeung, G. Lizard, M. A. Popp, M. Heinrich, I. Berindan-Neagoe, M. Stadler, M. Daglia, R. Verpoorte and C. T. Supuran, Natural products in drug discovery: advances and opportunities, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- A. C. Spivey, M. Weston and S. Woodhead, Celastraceae sesquiterpenoids: biological activity and synthesis, Chem. Soc. Rev., 2002, 31, 43–59 RSC.
- J.-M. Gao, W.-J. Wu, J.-W. Zhang and Y. Konishi, The dihydro-β-agarofuran sesquiterpenoids, Nat. Prod. Rep., 2007, 24, 1153–1189 RSC.
- O. Munoz, A. Penaloza, A. G. Gonzalez, A. G. Ravelo, I. L. Bazzocchi and N. L. Alvarenga, in Studies in Natural Products Chemistry, ed. R. Atta ur, Elsevier, 1996, vol. 18, pp. 739–783 Search PubMed.
- A. Beitter, Pharmakognostisch-chemische Untersuchung der Catha edulis, Arch. Pharm., 1901, 239, 17–33 CrossRef CAS.
- G. Trier, Die Alkaloide: Eine Monographie der natürlichen Basen, Gebrüder Borntraeger, 1931 Search PubMed.
- A. Orechoff, Untersuchung einiger Heil–und Giftpflanzen der UdSSR. auf deren Alkaloidgehalt, Arch. Pharm., 1934, 272, 673–691 CrossRef.
- F. Acree Jr. and H. Haller, Wilfordine, an insecticidal alkaloid from Tripterygium wilfordii Hook, J. Am. Chem. Soc., 1950, 72, 1608–1611 CrossRef.
- M. Beroza, Chromatographic separation employing ultraviolet absorbancy ratios, Anal. Chem., 1950, 22, 1507–1510 CrossRef.
- M. Beroza, Alkaloids from Tripterygium wilfordii Hook.—Wilforine and Wilfordine1, 2, J. Am. Chem. Soc., 1951, 73, 3656–3659 CrossRef CAS.
- M. Beroza, Alkaloids from Tripterygium wilfordii Hook. Isolation and structure of wilforzine, J. Am. Chem. Soc., 1953, 75, 2136–2138 CrossRef CAS.
- M. Beroza, Alkaloids from Tripterygium wilfordii Hook. The Structure of Wilforine, Wilfordine, Wilforgine and Wilfortrine, J. Am. Chem. Soc., 1953, 75, 44–49 CrossRef CAS.
- M. Beroza, Alkaloids from Tripterygium wilfordii Hook. The Chemical Structure of Wilfordic and Hydroxywilfordic Acids, J. Org. Chem., 1963, 28, 3562–3564 CrossRef CAS.
- M. Beroza, Alkaloids from Tripterygium wilfordii Hook: Wilforgine and Wilfortrine1, J. Am. Chem. Soc., 1952, 74, 1585–1588 CrossRef CAS.
- M. Beroza and G. Bottger, The insecticidal Value of Tripterygium wilfordii, J. Econ. Entomol., 1954, 47, 188–189 CrossRef.
- H. Itokawa, O. Shirota, H. Morita, K. Takeyaa and Y. Iitakab, Isolation, structural elucidation and conformational analysis of sesquiterpene pyridine alkaloids from Maytenus ebenifolia Reiss. X-Ray molecular structure of ebenifoline W-I, J. Chem. Soc., Perkin Trans. 1, 1993, 1247–1254 RSC.
- H. Itokawa, O. Shirota, H. Morita and K. Takeya, Sesquiterpene Pyridine Alkaloids from Maytenus ebenifolia, Heterocycles, 1992, 34, 885–889 CrossRef CAS.
- O. Shirota, H. Morita, K. Takeya and H. Itokawa, Sesquiterpene pyridine alkaloids from Maytenus ilicifolia, Heterocycles, 1994, 38, 383–389 CrossRef CAS.
- O. Shirota, A. Otsuka, H. Morita and K. Takeya, Sesquiterpene pyridine alkaloids from Maytenus chuchuhuasca, Heterocycles, 1994, 38, 2219–2219 CrossRef CAS.
- Y. Yuan, J.-W. Jung and S.-Y. Seo, Enantioselective synthesis and absolute configuration determination of hydroxywilfordic acid in sesquiterpene pyridine alkaloids, Org. Biomol. Chem., 2019, 17, 44–48 RSC.
- D. Fan, T. Li, Z. Zheng, G.-Y. Zhu, X. Yao, Z.-H. Jiang and L.-P. Bai, Macrolide sesquiterpene pyridine alkaloids from the stems of Tripterygium regelii, J. Nat. Med., 2019, 73, 23–33 CrossRef CAS PubMed.
- Y. Luo, X. Pu, G. Luo, M. Zhou, Q. Ye, Y. Liu, J. Gu, H. Qi, G. Li and G. Zhang, Nitrogen-Containing Dihydro-β-agarofuran Derivatives from Tripterygium wilfordii, J. Nat. Prod., 2014, 77, 1650–1657 CrossRef CAS PubMed.
- H. Duan, Y. Takaishi, Y. Jia and D. Li, Sesquiterpene alkaloids from extracts of Tripterygium wilfordii, Chem. Pharm. Bull., 1999, 47, 1664–1667 CrossRef CAS.
- Y. Li, G. M. Strunz and L. A. Calhoun, Isowilfordine: An alkaloid from Tripterygium wilfordii, Phytochemistry, 1991, 30, 719–721 CrossRef.
- H. J. Lee and G. R. Waller, Biosynthesis of pyridine alkaloids from Tripterygium wilfordii, Phytochemistry, 1972, 11, 2233–2240 CrossRef CAS.
- M. Horiuch, C. Murakami, N. Fukamiya, D. Yu, T.-H. Chen, K. F. Bastow, D.-C. Zhang, Y. Takaishi, Y. Imakura and K.-H. Lee, Tripfordines A–C, Sesquiterpene Pyridine Alkaloids from Tripterygium wilfordii, and Structure Anti-HIV Activity Relationships of Tripterygium Alkaloids, J. Nat. Prod., 2006, 69, 1271–1274 CrossRef CAS PubMed.
- R. Libiseller and A. Preisinger, Röntgenographische Untersuchung und Molekulargewichtsbestimmung von Evonin, einem Alkaloid aus Evonymus europaca L, Monatsh. Chem. Verw. Teile Anderer Wiss., 1962, 93, 417–419 CrossRef CAS.
- M. Pailer and R. Libiseller, Über Evonymus-Alkaloide, 1. Mitt.: Zur Reindarstellung und Konstitution des Evonins,(aus Evonymus europaea L.), Monatsh. Chem. Verw. Teile Anderer Wiss., 1962, 93, 403–416 CrossRef CAS.
- M. Pailer and R. Libiseller, Über Evonymus-Alkaloide, 2. Mitt.: Konstitution und absolute Konfiguration der Evoninsäure, einer Abbauverbindung des Evonins, Monatsh. Chem. Verw. Teile Anderer Wiss., 1962, 93, 511–516 CrossRef CAS.
- K. Doebel and T. Reichstein, Isolierung von drei krystallisierten Alkaloiden aus den Samen des Pfaffenhütchens, Evonymus europaea L, Helv. Chim. Acta, 1949, 32, 592–597 CrossRef CAS PubMed.
- A. Klásek, F. Šantavý, M. A. Duffield and T. Reichstein, Alkaloide aus Evonymus europaea L, Helv. Chim. Acta, 1971, 54, 2144–2158 CrossRef.
- A. A. Sánchez, J. Cárdenas, M. Soriano-García, R. Toscano and L. Rodríguez-Hahn, Acanthothamine, a sesquiterpenoid alkaloid from Acanthothamnus aphyllus, Phytochemistry, 1986, 25, 2647–2650 CrossRef.
- M. Soriano-García, R. A. Toscano, A. Sánchez and L. Rodríguez-Hahn, Crystal and molecular structure of acanthothamine monohydrate-methanol: A new alkaloid from Acanthothamnus aphyllus, J. Crystallogr. Spectrosc. Res., 1986, 16, 507–517 CrossRef.
- H. Duan and Y. Takaishi, Structures of sesquiterpene polyol alkaloids from Tripterygium hypoglaucum, Phytochemistry, 1998, 49, 2185–2189 CrossRef CAS.
- W. Li, B. Li and Y. Chen, Sesquiterpene alkaloids from Tripterygium hypoglaucum, Phytochemistry, 1999, 50, 1091–1093 CrossRef CAS.
- Y. Yang, Y. Guo, D. Yang, X. Qiao and H. Liu, CN112209874(A), 2021.
- J. Klass, W. F. Tinto, W. F. Reynolds and S. McLean, Peritassines A and B, new sesquiterpene alkaloids from Peritassa compta, J. Nat. Prod., 1993, 56, 946–948 CrossRef CAS.
- L. M. Liao, Sesquiterpene pyridine alkaloids, Alkaloids, 2003, 60, 287–344 CAS.
- M. Furukawa, M. Makino, T. Uchiyama, K. Ishimi, Y. Ichinohe and Y. Fujimoto, Sesquiterpene pyridine alkaloids from Hippocratea excelsa, Phytochemistry, 2002, 59, 767–777 CrossRef CAS PubMed.
- L. Crombie, W. M. L. Crombie, D. A. Whiting and K. Szendrei, Alkaloids of Catha edulis. Part 3. Structures of cathedulins K1, K2, K6, and K15; new Macrolide-bridged polyesters of euonyminol, J. Chem. Soc., Perkin Trans. 1, 1979, 2976–2981 RSC.
- R. L. Baxter, W. M. L. Crombie, L. Crombie, D. J. Simmonds, D. A. Whiting and K. Szendrei, Alkaloids of Catha edulis. Part 4. Structures of cathedulins E3, E4, E5, E6, and K12. Novel sesquiterpene alkaloids with mono-and bismacrolide bridges, J. Chem. Soc., Perkin Trans. 1, 1979, 2982–2989 RSC.
- L. Crombie, D. Toplis, D. A. Whiting, Z. Rozsá, J. Hohmann and K. Szendrei, New macrolide sesquiterpene alkaloids of Catha edulis: examples containing a novel dilactone bridge, J. Chem. Soc., Perkin Trans. 1, 1986, 531–534 RSC.
- L. Crombie, W. M. L. Crombie and D. A. Whiting, in The Alkaloids: Chemistry and Pharmacology, ed. A. Brossi, Academic Press, San Diego, 1990, vol. 39, ch. 4, pp. 139–164 Search PubMed.
- L. Crombie, The cathedulin alkaloids, Bull. Narc., 1980, 32, 37–50 CAS.
- L. Crombie, Natural products of cannabis and khat, Pure Appl. Chem., 1986, 58, 693–700 CrossRef CAS.
- R. L. Baxter, L. Crombie, D. J. Simmonds and D. A. Whiting, Extractives of Catha edulis(Khat): occurrence of celastraceaeous alkaloids having mono- and bis-macrolide bridges, J. Chem. Soc., Chem. Commun., 1976, 463–465 RSC.
- S. A. Ahmad-Junan, A. J. Walkington and D. A. Whiting, Aryloxymethyl radical cyclizations mimicking biological C–C bond formation to methoxy groups, J. Chem. Soc., Perkin Trans. 1, 1992, 2313–2320 RSC.
- M. Stein and P. Nencini, Dimethyl cathate: synthesis and pharmacological investigation, Farmaco, 1989, 44, 959–965 CAS.
- T.-S. Kim and J. D. White, The absolute configuration of edulinic acid, a constituent of the “Khat” alkaloid cathedulin K-19, Tetrahedron Lett., 1993, 34, 5535–5536 CrossRef CAS.
- H. Wagner, R. Brüning, H. Lotter and A. Jones, Die struktur von cassinin, einem neuen sesquiterpenalkaloid aus cassine matabelica, loes, Tetrahedron Lett., 1977, 18, 125–128 CrossRef.
- R. Brüning and H. Wagner, Übersicht über die celastraceen-inhaltsstoffe: Chemie, chemotaxonomie, biosynthese, pharmakologie, Phytochemistry, 1978, 17, 1821–1858 CrossRef.
- A. G. González, L. San Andrés, A. G. Ravelo, J. G. Luis, I. A. Jiménez and X. A. Domínguez, Minor Constituents from Orthosphenia mexicana, J. Nat. Prod., 1989, 52, 1338–1341 CrossRef.
- E. L. Whitson, S. M. V. D. Mala, C. A. Veltri, T. S. Bugni, E. D. de Silva and C. M. Ireland, Oppositines A and B, Sesquiterpene Pyridine Alkaloids from a Sri Lankan Pleurostylia opposita, J. Nat. Prod., 2006, 69, 1833–1835 CrossRef CAS PubMed.
- H. Duan, Y. Takaishi, M. Bando, M. Kido, Y. Imakura and K. Lee, Novel sesquiterpene esters with alkaloid and monoterpene and related compounds from Tripterygium hypoglaucum: A new class of potent anti-HIV agents, Tetrahedron Lett., 1999, 40, 2969–2972 CrossRef CAS.
- H. Duan, Y. Takaishi, H. Momota, Y. Ohmoto, T. Taki, Y. Jia and D. Li, Immunosuppressive sesquiterpene alkaloids from Tripterygium wilfordii, J. Nat. Prod., 2001, 64, 582–587 CrossRef CAS PubMed.
- D. Fan, G.-Y. Zhu, T. Li, Z.-H. Jiang and L.-P. Bai, Dimacrolide Sesquiterpene Pyridine Alkaloids from the Stems of Tripterygium regelii, Molecules, 2016, 21, 1146 CrossRef PubMed.
- H. L. Ye, Y. Liu, J. Pan, W. Guan, Y. Liu, X. M. Li, S. Y. Wang, A. M. Algradi, B. Y. Yang and H. X. Kuang, Three new sesquiterpenoid alkaloids from the roots of Tripterygium wilfordii and its cytotoxicity, Nat. Prod. Res., 2021, 36, 1–9 Search PubMed.
- R. Wang, R. Chen, J. Li, X. Liu, K. Xie, D. Chen, Y. Peng and J. Dai, Regiospecific Prenylation of Hydroxyxanthones by a Plant Flavonoid Prenyltransferase, J. Nat. Prod., 2016, 79, 2143–2147 CrossRef CAS PubMed.
- A. Klasek, Z. Samek and F. Ŝantavý, The structure of evozine, an alkaloid isolated from evonymus europaea L, Tetrahedron Lett., 1972, 13, 941–944 CrossRef.
- L. Crombie, P. Ham and D. Whiting, New sesquiterpenoid alkaloids from Euonymus europea, Phytochemistry, 1973, 12, 703–705 CrossRef CAS.
- Y. Shizuri, H. Wada, K. Yamada and Y. Hirata, The reactions of evonine and neoevonine alkaloids obtained from Euonymus Sieboldiana blume, Tetrahedron, 1973, 29, 1795–1800 CrossRef CAS.
- K. Sugiura, K. Yamada and Y. Hirata, Synthesis of Evonine from Evoninic Acid and an Evoninol Derivative, Chem. Lett., 1975, 4, 579–580 CrossRef.
- M. Pailer and K. Pfleger, Synthese der Racemate der beiden stereoisomeren Evoninsäuren, Monatsh. Chem., 1976, 107, 965–971 CrossRef CAS.
- J. S. Eun and S.-Y. Seo, Facile synthesis of hydroxy wilfordic acid, a esterifying unit of anti-HIV sesquiterpene pyridine alkaloids, Arch. Pharmacal Res., 2009, 32, 1673–1679 CrossRef CAS PubMed.
- A. C. Spivey, L. Shukla and J. F. Hayler, Conjugate Addition of 2- and 4-Pyridylcuprates: An Expeditious Asymmetric Synthesis of Natural (−)-Evoninic Acid, Org. Lett., 2007, 9, 891–894 CrossRef CAS PubMed.
- S. A. Warren, S. Stokes, C. S. Frampton, A. J. P. White and A. C. Spivey, Synthesis of anti and syn hydroxy-iso-evoninic acids, Org. Biomol. Chem., 2012, 10, 4685–4688 RSC.
- Y. Wang, T. Nagai, I. Watanabe, K. Hagiwara and M. Inoue, Total Synthesis of Euonymine and Euonyminol Octaacetate, J. Am. Chem. Soc., 2021, 143, 21037–21047 CrossRef CAS PubMed.
- H. Wu, Q. Wang and J. Zhu, Catalytic Enantioselective Benzilic Ester Rearrangement, Angew. Chem., Int. Ed., 2020, 59, 7261–7265 CrossRef CAS PubMed.
- S. J. Zuend and E. N. Jacobsen, Cooperative Catalysis by Tertiary Amino-Thioureas: Mechanism and Basis for Enantioselectivity of Ketone Cyanosilylation, J. Am. Chem. Soc., 2007, 129, 15872–15883 CrossRef CAS PubMed.
- D. E. Fuerst and E. N. Jacobsen, Thiourea-Catalyzed Enantioselective Cyanosilylation of Ketones, J. Am. Chem. Soc., 2005, 127, 8964–8965 CrossRef CAS PubMed.
- Y. Shizuri, H. Wada, K. Sugiura, K. Yamada and Y. Hirata, The structures of evonine and neoevonine alkaloids obtained from Euonymus Sieboldiana blume, Tetrahedron, 1973, 29, 1773–1794 CrossRef CAS.
- M. Tomanik, Z. Xu, F. Guo, Z. Wang, K. R. Yang, V. S. Batista and S. B. Herzon, Development of an Enantioselective Synthesis of (−)-Euonyminol, J. Org. Chem., 2021, 86, 17011–17035 CrossRef CAS PubMed.
- M. Tomanik, Z. Xu and S. B. Herzon, Enantioselective Synthesis of Euonyminol, J. Am. Chem. Soc., 2021, 143, 699–704 CrossRef CAS PubMed.
- Y. Shizuri, H. Wada, K. Sugiura, K. Yamada and Y. Hirata, Evonine, an alkaloid obtained from euonymus sieboldiana blume. II. The structure of evonine, Tetrahedron Lett., 1971, 12, 2659–2662 CrossRef.
Footnote |
| † NB. the stereochemistry of these alkaloids have been corrected – see: link: https://www.tandfonline.com/doi/epdf/10.1080/14786419.2021.1903460?needAccess=true&role=button. |
| This journal is © the Partner Organisations 2023 |