
Recent advances in the electrochemical generation of 1,3-dicarbonyl radicals from C–H bonds
 *abc
*abc
Abstract
Electrochemistry, which employs electrons as reagents and avoids the use of traditional redox reagents, becomes a powerful tool in oxidative dehydrogenation cross-coupling reactions. 1,3-Dicarbonyl derivatives are common and easily available synthetic blocks and are widely used for the construction of active molecules in organic synthesis. 1,3-Dicarbonyl radicals can be generated by direct anodic oxidation or indirect anodic oxidation with the assistance of a redox reagent and can undergo addition to arenes, double bonds or triple bonds to build multifarious organic molecules. In this review, we succinctly summarize the recent advances in the electrochemical generation of 1,3-dicarbonyl radicals from C–H-based 1,3-dicarbonyl derivatives and focus on the mechanistic insights and synthetic applications of these methods and transformations.
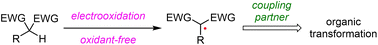
- This article is part of the themed collections: 2023 Organic Chemistry Frontiers HOT articles and 2023 Organic Chemistry Frontiers Review-type Articles
 Please wait while we load your content...
Please wait while we load your content...

