
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShape-shifting p-cyclophanes as portals to switching, sensing, delivery and logic operations in water†
Chao-Yi
Yao
a,
Hong-Yu
Lin
 a,
Brian
Daly
a,
Ze-Qing
Chen
a,
Hannah S. N.
Crory
a,
H. Q. Nimal
Gunaratne
a,
Eric V.
Anslyn
ab and
A. Prasanna
de Silva
a
a,
Brian
Daly
a,
Ze-Qing
Chen
a,
Hannah S. N.
Crory
a,
H. Q. Nimal
Gunaratne
a,
Eric V.
Anslyn
ab and
A. Prasanna
de Silva
a
aSchool of Chemistry and Chemical Engineering, Queen's University, Belfast BT9 5AG, Northern Ireland, UK. E-mail: a.desilva@qub.ac.uk
bDepartment of Chemistry, University of Texas at Austin, 100 E 24th Street, Norman Hackerman Building (Room 114A), Austin, TX 78712, USA
First published on 26th January 2023
Abstract
The hybridization state of carbon atoms between phenylene units in p-cyclophanes is shown to control the guest binding properties of these hosts. Dimeric cyclophanes can be shape-switched by redox so that small aromatics can be captured or released. Trimeric cyclophanes perform similar functions on polypyridineRu(II) complexes. Controlled capture and release are key to delivery of such cargos. The luminescence and electrochemical signals which are modulated during these processes serve as sensing functions. Some of these modulations are so strong that Boolean logic schemes can be applied. Small-scale serial integration of sequential and combinational logic is seen.
Introduction
p-Cyclophanes have a 40-year history of binding molecular guests in water1–6 but there have only been a few instances of them being endowed with switchable properties.7–9 Some of these hosts have a pair of diphenylmethane ‘corner sections’ with a sp3-hybridized carbon ‘corner’ each which orient their phenylene units perpendicular to the mean macrocycle plane so that these ‘wall’ sections create a hydrophobic cavity.1–6 These diphenylmethane ‘corner sections’ are most commonly connected by two oligomethylene linkers to create the macrocycle. In other words, such hosts are alternating cyclodimers of a linker and a diphenylmethane ‘corner section’ which serve as co-monomers.What if these corners were changed to sp2-hybridized carbon atoms? The phenylene units should then change their orientation so that their planes are in the mean macrocycle plane, as far as cross-conjugation allows. This should make the macrocycle cavity smaller, shallower and with less aromatic π-cloud lining, which should be less inviting for guests suited to the original cyclophane host (Fig. 1A). Guest binding should then be switched from ‘on’ to ‘off’. Such guest capture and release should enable delivery of guest from one place to another. Switching operations10 lie within the field of molecular logic, especially when several switching actions are articulated.11–26
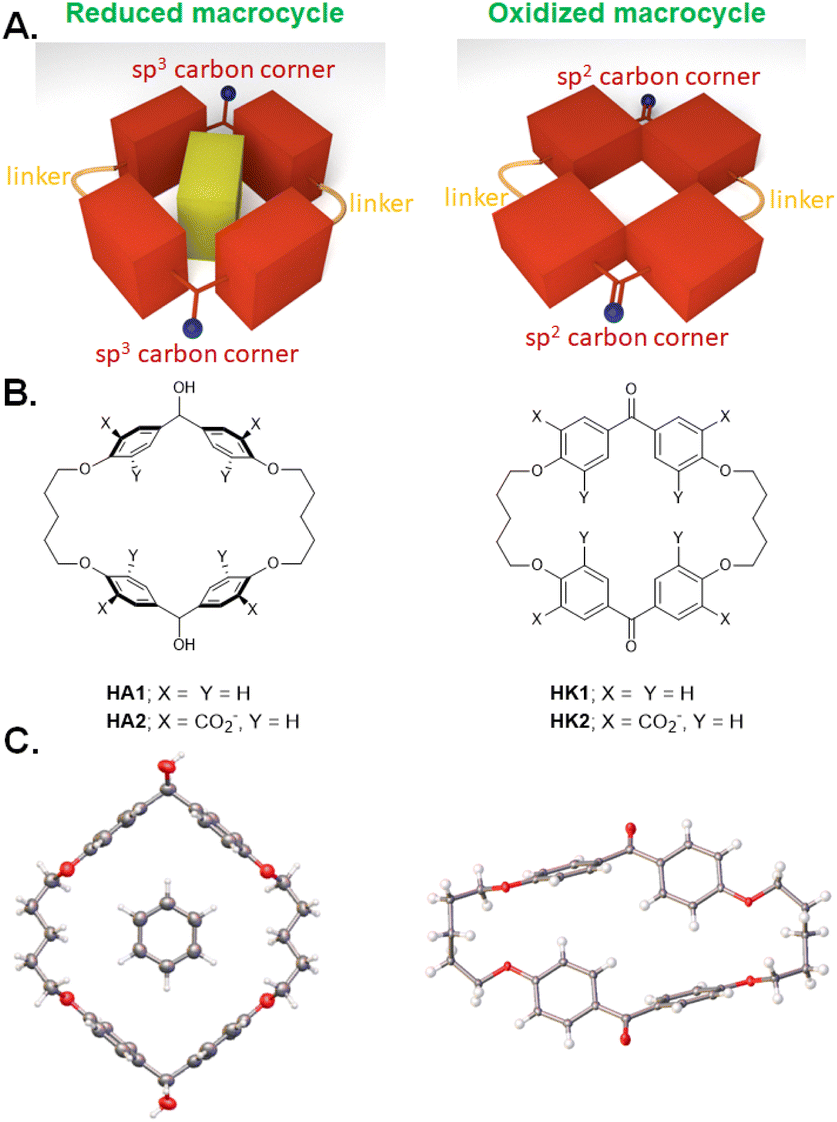 | ||
| Fig. 1 A. Model of redox-switchable shape-shifting dimeric cyclophanes for the capture/release of aromatic guests. B. Molecular structures of such dimeric cyclophanes in their reduced (HA1 & HA2) and oxidized (HK1 & HK2) forms. C. X-ray crystallographic structures of HA1 (left) and HK1 (right). The structure of HA1 also includes a molecule of G1 as a guest. Adapted from ref. 27. | ||
So, the diphenylmethane ‘corner section’ can be altered to a benzophenone to produce the sp2-hybridized carbon ‘corner’. A classical ketone reduction, e.g. with aqueous NaBH4, would give a benzhydrol, a sec-alcohol with its sp3-hybridized carbon ‘corner’. This has the advantage of permitting an equally classical alcohol oxidation back to the ketone, e.g. with aqueous KMnO4. Redox interconversion of sec-alcohols and ketones becomes a practical implementation of this approach. When available, the diphenylmethane version serves as a reference for the benzhydrol carrying sp3-hybridized carbon ‘corners’.
Dimeric cyclophanes
Synthesis and X-ray crystallography
We began with analogues of Koga's original structures,4 but found it convenient to start with a macrocyclization of 4,4′-dihydroxybenzophenone and 1,5-diiodopentane via ether formation under basic conditions.27 Although insoluble in water, diketone HK1 (Fig. 1B) and its reduction product dialcohol HA1 provided an early test of parts of the hypothesis given above. X-ray crystallography showed the macrocycle cavity of diketone HK1 had only two phenylenes presenting their π-faces towards the cavity interior (Fig. 1C). The other two phenylenes thrust their C–H units into the cavity so that free space is constricted. In contrast, dialcohol HA1 had the planes of its four phenylene rings orthogonal to the mean macrocycle plane so that a cavity of considerable depth was created, cf. that seen in diketone HK1. As a bonus, the X-ray structure of dialcohol HA1 contained a molecule of the crystallizing solvent G1 (Fig. 2A) nestled in the cavity by means of a set of C–H–π interactions28 where the π functionalities originate in the macrocycle and the C–H units belong to the guest.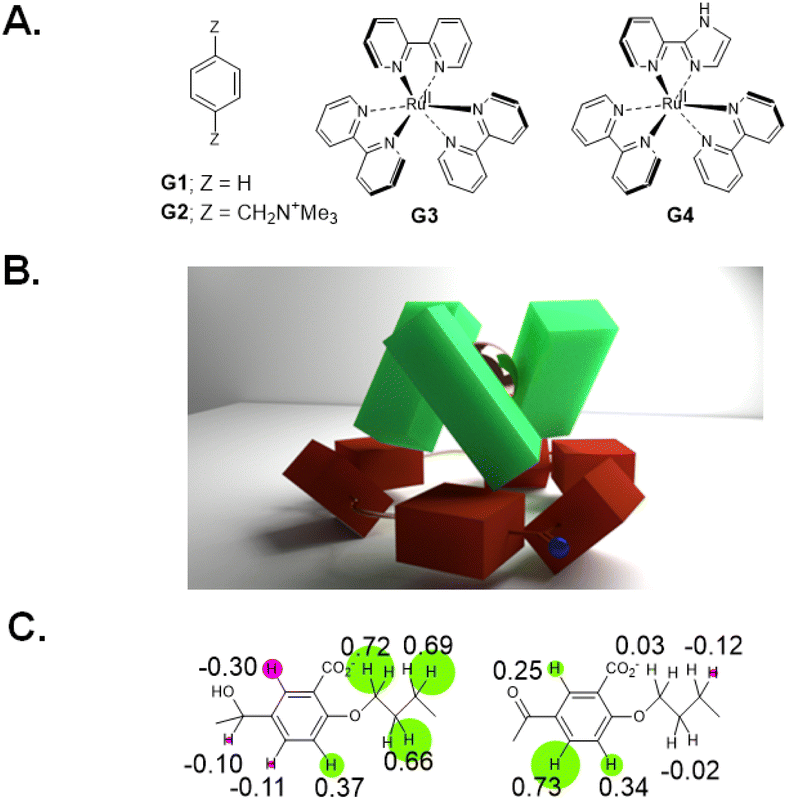 | ||
| Fig. 2 A. Molecular structures of prospective guests G1, G2, G3 & G4. B. Model of guest G3 perching on oxidized form of trimeric cyclophane HK4. C. Δδ maps of hosts HA4 (left) and HK4 (right) when bound to guest G3. Δδ values are given on partial molecular structures and their relative magnitudes are illustrated by the radii of circles centred on each proton. Negative or positive Δδ values are shown by green or red circles respectively. | ||
Water-solubility of the diketone macrocycle was arranged by outfitting it with a carboxylate group at each 2-position of the phenylene units. This relied on a remarkable Ag(I)-promoted iodination at these 2-positions,29 followed by a Pd(0)-catalyzed carbonylation.30 This gave diketonetetracarboxylate HK2 (Fig. 1B). Aqueous NaBH4 reduced HK2 into dialcoholtetracarboxylate HA2. Aqueous KMnO4 smoothly oxidized dialcoholtetracarboxylate HA2 back to diketonetetracarboxylate HK2. Thus, redox interconversion of the two macrocycles HA2 and HK2 was secured.
1H NMR and fluorescence spectroscopy
The stage was set for testing the capture/release of prospective guest G2 (Fig. 2A) in water. 1H NMR spectroscopy showed significant complexation-induced chemical shift changes (Δδ values) for most protons of host and guest. The relative magnitudes of these Δδ values allowed the conclusion that G2 orients axially within the cavity of dialcohol HA2. Concentration-dependence of Δδ values fitted a binding constant (β) of around 104 M−1. In contrast, near-zero Δδ values were found for all protons in the potential diketone host (HK2)-guest (G2) system showing no evidence of binding, in keeping with our hypothesis. The idea of a wall collapse or erection causing mobility or restriction of individuals seen in molecules e.g.HA2/HK2 and G2,27 can also be found in human behaviour in personal and social situations.Guest binding/unbinding behaviour when the macrocycle is reduced (HA1 & HA2) or oxidized (HK1 & HK2) corresponds to a computer memory at the molecular scale.15,27,31 A computer memory holds its state for an indefinite period until a specific command is given to change to the other state. Alcohol/ketone systems, e.g.HA1/HK1 or HA2/HK2, maintain the alcohol state when a reducing agent is introduced. The alcohol state is preserved even if more reductant is added or if the reductant is later removed. This alcohol state shifts to the ketone state only when an oxidizing agent is introduced. The ketone state is preserved even if more oxidant is added or if the oxidant is removed subsequently. A simple computer memory of this type, known as a RS flip-flop,15,27,31 is symbolically pictured in Fig. 3A in terms of an array of OR and NOT gates.
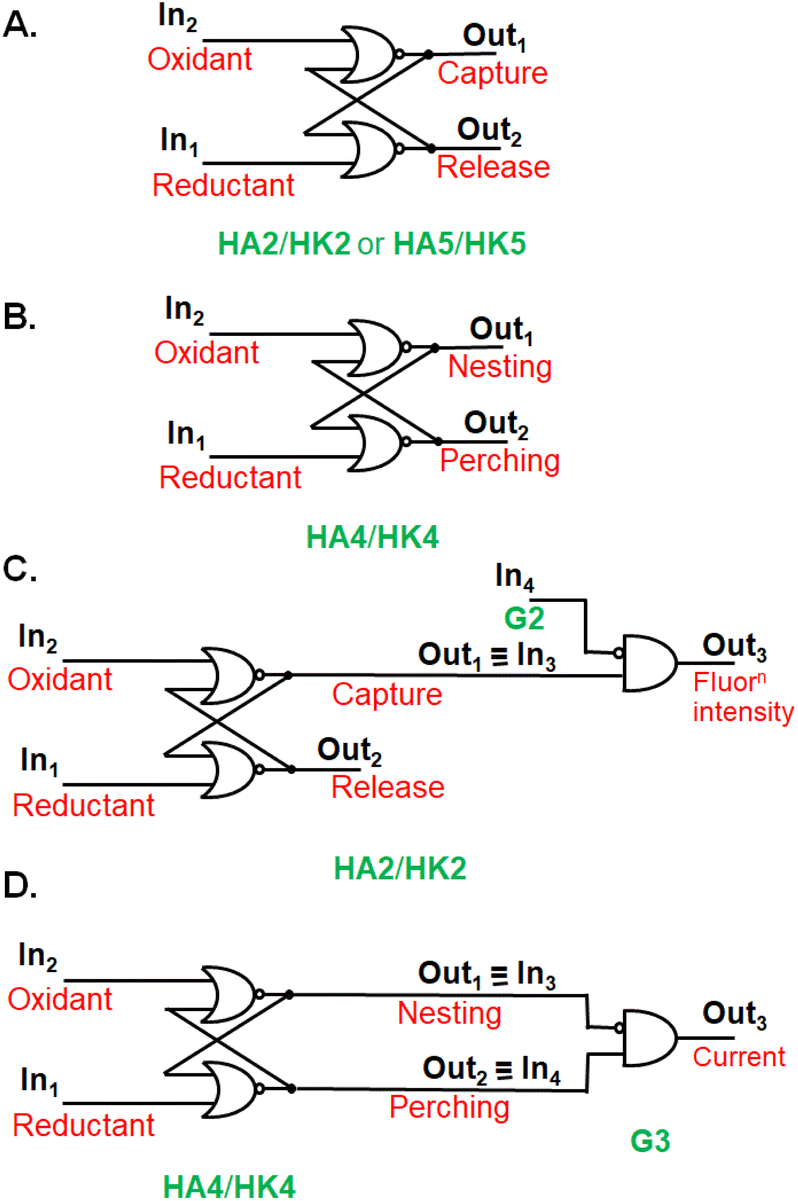 | ||
| Fig. 3 A. Electronic representation of a RS flip-flop (HA2/HK2 or HA5/HK5) where capture or release of a guest occurs. B. A RS flip-flop (HA4/HK4) where the guest binding mode switches between perching and nesting. C. The fluorescence output3 of HA2/HK2 host system behaving as a RS flip-flop integrated with an INHIBIT logic gate, where the disabling input4 is the guest G2. D. The current output3 of guest G3 corresponding to a RS flip-flop (HA4/HK4) physically integrated with an INHIBIT logic gate (G3). The role of diversity in logic inputs and outputs15 is on view in these cases. | ||
Such identification of computing phenomena within (bio)chemical reactions and interactions is one of the goals of molecular logic-based computation.11–26 This is the Boolean approach to molecular information processing where supramolecular chemistry has much to offer. The title of a review written by Lehn soon after his Nobel prize, ‘Perspectives in supramolecular chemistry – from molecular recognition towards molecular information-processing and self-organization’,32 recognized this general path.
Coupling guest binding/unbinding behaviour with other follow-up actions can offer more complex logic arrays, beside creating opportunities for sensing. For instance, complexation of guest G2 by cyclophane dialcoholtetracarboxylate HA2 is signalled by quenching of the fluorescence of HA2. Photoinduced electron transfer (PET)33,34 is thermodynamically feasible here. This means that output1 of the RS flip-flop serves as input3 of an INHIBIT gate receiving G2 as the disabling input4. When input3 = 1 and input4 = 0, the fluorescence output3 is 1. When input3 = 1 and input4 = 1, the fluorescence output3 becomes 0 (Fig. 3C).
It is interesting that our neurons release stored neurotransmitters so that downstream neural processing events can occur.35 Fluorescence ‘on–off’ signalling of the occupation of a receptor, as discussed above, shows this concatenation of information storage and information processing (Fig. 3C). In digital electronics, information storage concerns sequential logic whereas the history-independent information processing represents combinational logic.
Trimeric cyclophanes
Synthesis and X-ray crystallography
During the synthesis of diketone HK1, traces of triketone HK3 (Fig. 4B) were identified in the reaction mixture. Since linear co-polymers are commonly found during macrocyclizations under practical high-dilution conditions, the formation of HK3 can be understood as step-growth of a linear alternating co-trimer, followed by cyclization at that stage.36HK1 would have been formed by a similar cyclization of the corresponding co-dimer. By careful adjustment of the conditions of dropping the reactants into the solvent and base, the triketone HK3 became the major non-polymeric product.37 X-ray crystallography of triketone HK3 showed all the phenylene units aligned in the mean macrocycle plane. None of the phenylenes presented their π-faces towards the cavity interior (Fig. 4C). The X-ray crystallographic packing diagram showed the macrocycles were stacked to create channels. Such packing could have influenced the high-degree of π-conjugation between the carbonyl groups and their flanking phenylenes which was observed.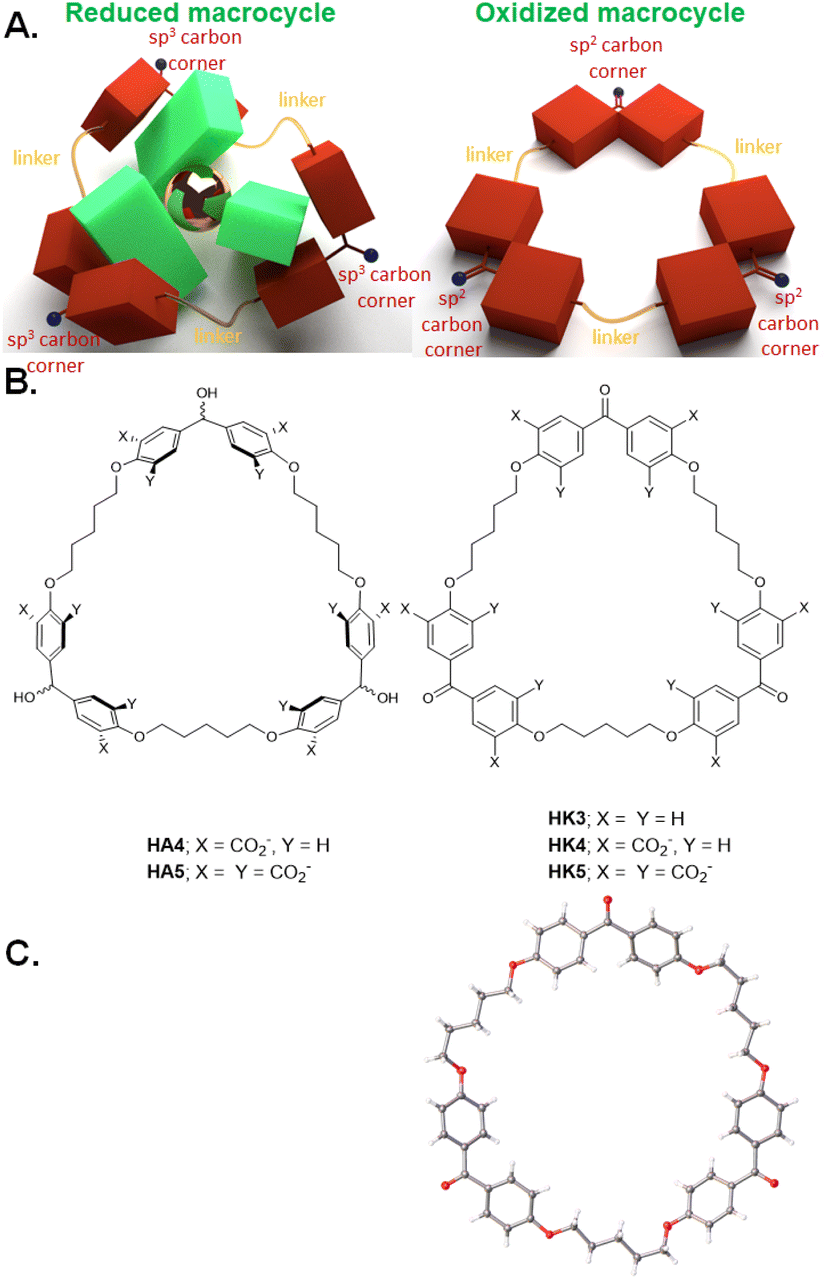 | ||
| Fig. 4 A. Model of redox-switchable shape-shifting trimeric cyclophanes for the capture/release of polypyridine metal complexes. B. Molecular structures of such trimeric cyclophanes in their reduced (HA4 & HA5) and oxidized (HK3, HK4 & HK5) forms. C. X-ray crystallographic structure of HK3. Adapted from ref. 37. | ||
Functionalization of triketone HK3, as done for the dimeric cyclophanes, proceeded smoothly to give triketonehexacarboxylate HK4 (Fig. 4B). Aqueous NaBH4 reduced HK4 into trialcoholhexacarboxylate HA4. Aqueous KMnO4 oxidized trialcoholhexacarboxylate HA4 back to triketonehexacarboxylate HK4.
1H NMR and luminescence spectroscopy
Since these trimeric cyclophanes were unprecedented, we needed to explore potential guests. Fortunately, tris(bipyridine)Ru(II) (G3) proved to be a suitable guest according to 1H NMR Δδ values. We could conclude from the relative magnitudes of these Δδ values that each bipyridine blade of G3 was cradled within the wedge of each benzhydrol unit of HA4. A binding constant (β) of over 106 M−1 was estimated from concentration-dependent studies. An accurate assessment (107.3 M−1) was done by analyzing the enhancement of luminescence intensity of G3 as a function of increasing host HA4 concentration. By forming a nesting complex of guest G3 inside host HA4, the hydration-induced deexcitation of the metal-to-ligand charge transfer (MLCT) triplet excited state of G3 was supressed.Would switching the host trialcoholhexacarboxylate HA4 to triketonehexacarboxylate HK4 result in the binding of G3 being switched from ‘on’ to ‘off’? This dream schematized in Fig. 4A was not observed, but a remarkable switching of the binding mode was found. 1H NMR Δδ values showed that guest G3 perched on the host HK4. The ends of each bipyridyl blade of G3 engaged in C–H–π interactions with a phenylene of each benzophenone corner of host HK4 (Fig. 2B). This was supplemented by the presence of a hydrophobic patch in each phenylene to overlap with a hydrophobic region on the outer edge of each bipyridine ligand of G3. Indeed, the Δδ map across all protons of the triketonehexacarboxylate host HK4 showed the opposite pattern to that seen with trialcoholhexacarboxylate host HA4 (Fig. 2C). Large Δδ values were noticed for the aliphatic protons in the linkers of host HA4, along with tiny Δδ values found for the host aromatic protons. The reverse situation showed up in host HK4. The binding constant (β) of the perching complex was measured from concentration-dependent 1H NMR spectroscopy to be 105 M−1. As might be anticipated, perching binding is of a lower affinity than nesting binding between related host–guest pairs. Fig. 3B is the overall logic representation.
Switching the binding of G3 from ‘on’ to ‘off’ should become possible if perching binding to the host could be prevented, e.g. in a host like triketonedodecacarboxylate HK5 (Fig. 4B) which has minimized the hydrophobic regions for C–H–π interactions. HK5's redox partner, trialcoholdodecacarboxylate HA5, has an equatorial belt of hydrophobicity to allow the nesting binding of guest G3 with significant affinity.
Trialcoholdodecacarboxylate HA5 was prepared by starting with analogues of Koga's original materials,4 and by macrocyclizing a linear alternating co-trimer via ether formation under basic conditions.37 Triketonedodecacarboxylate HK5 was the synthetic precursor of HA5. As seen before with analogues, these two compounds could be interconverted with classical redox reagents like KMnO4 and NaBH4.
1H NMR spectroscopy showed nesting binding of guest G3 by trialcoholdodecacarboxylate host HA5, with an association constant of 104.4 M−1. In contrast, no complexation-induced chemical shift differences were found for a mixture of triketonedodecacarboxylate HK5 and G3. The switching of binding of G3 from ‘on’ to ‘off’ was therefore demonstrated and the dream of Fig. 4A was realized. Luminescence spectroscopy also confirmed this remarkable result by showing a significant enhancement of intensity of G3 only in the presence of host HA5.
Cyclic voltammetry of G3
Electrochemical measurements of guest G3 also produced evidence of complexation by the cyclophane hosts. Cyclic voltammetry of G3 gave a 20 mV cathodic shift of the Ru(II)/Ru(III) couple upon perching binding by triketonehexacarboxylate host HK4 without a significant decrease in current. On the other hand, the nesting binding of G3 by trialcoholhexacarboxylate HA4 suppressed the current by a factor of ca. 5, because of the relative inaccessibility of G3 to the electrode. The 20 mV cathodic shift was still seen, because of the rather hydrophobic environment of G3 upon complexation. Such large host-induced effects are very suitable for Boolean representation, as shown in Fig. 3D.In situ redox switching
Experiments on switchable binding of guests with hosts are best conducted in situ, i.e. when both components are present in the same solution. This was especially important because guest G3 was known to be more redox active than hosts HA4 and HK4. Indeed, many applications of G3 depended on its redox activity. Therefore, we had to choose redox switching protocols with some care. In practice, the oxidation of trialcohol HA4 with KMnO4 gave triketone HK4 while converting G3 to its Ru(III) form. However, treatment of the mixture with the mild reductant methanol recovered G3 in its original Ru(II) form, while leaving triketone HK4 untouched. Also, reduction of triketone HK4 to trialcohol HA4 with NaBH4 did not disturb G3. Once these conditions were established, smooth cycling between the two redox forms of the hosts HA4/HK4, and also HA5/HK5, could be demonstrated in the presence of guest G3.Phenolate quenching of luminescence of G3
Although the ‘on’/‘off’ binding of guest G3 with switchable hosts influenced its luminescence and electrochemical properties, influencing of photochemical reactions was yet to be demonstrated. G3 was known to be a photosensitizer for phenolate oxidation.38 The shielding of G3 by hosts should therefore hinder such reactions. The quenching of luminescence of G3 by phenolates was found to be suppressed by nearly two orders of magnitude upon nesting binding by trialcoholhexacarboxylate HA4 or by trialcoholdodecacarboxylate HA5. As might be imagined, perching binding by triketonehexacarboxylate HK4 had a smaller, but still significant, suppression of reactivity, whereas triketonedodecacarboxylate HK5 had almost no effect at all.Steady-state luminescence spectroscopy of G4
Following the successful binding and unbinding of guest G3 with hosts HA4/HK4 and HA5/HK5, we turned to analogues of G3 with extra functionality. Ru(II) complex G4 was interesting in this regard because of its ionizable proton.39–42 In order to proceed at significant rates, such ionizations require sufficient hydration, which might not be available in rather hydrophobic cyclophane cavities and environments. In particular, proton ionization in the excited state would have to compete with luminescence. The effect of hosts on luminescence intensity-pH profiles proved to be dramatic,37,43 in that the luminescence of G4 was switched ‘on’ at pH 7 due to nesting binding by trialcoholdodecacarboxylate host HA5, and to a slightly lesser extent by nesting host trialcoholhexacarboxylate HA4. Naturally, perching binding due to host HK4 produced a smaller effect. The non-binding cyclophane HK5 caused no effect at all. Binding of DNA to certain Ru(II) complexess is known to produce similar switching ‘on’ of luminescence.44Time-resolved luminescence spectroscopy of G4
Luminescence lifetimes of guest G4 with cyclophane hosts also showed significant lengthening.43 The combination of such lifetime data with quantum yield results obtained from steady state luminescence measurements allowed the extraction of radiative and nonradiative rate constants. The latter confirmed the conclusions of the previous paragraph, i.e. that the paucity of water in the cyclophane environment retarded proton ionization. The radiative rate constant was also significantly increased because the π-electron rich cyclophane environment was more polarizable than water. Corresponding experiments on guest G3 showed a similar host effect on the radiative rate constants.Conclusions and outlook
Outfitting the classical Koga cyclophanes with alcohol moieties did not alter their host properties towards small aromatics in water. However, facile oxidation of these alcohol units into ketones suppressed the host properties essentially completely. These host properties were recovered upon reduction of the ketones into alcohol moieties. The redox-induced alteration of the macrocycle cavity in terms of size, shape and nature was held responsible for the switching of host behaviour. Sensing applications were indicated by the fluorescence of the reduced form of the host being switched ‘off’ upon guest binding.The serendipitous discovery of trimeric relatives of the classical Koga cyclophanes was developed into a useful synthesis so that their redox-switched host properties could be investigated. Serendipity intervened again when polypyridineRu(II) species were found to be suitable guests, opening up the control of many celebrated properties of these iconic metal complexes. Three states of nesting binding, perching binding and release were elucidated with two sets of trimeric cyclophanes carrying differing hydrophobic regions. Varying degrees of exposure of the polypyridineRu(II) complexes to water were achieved in this way so that their electrochemical and photochemical properties were modulated accordingly.
Such control of properties is an aspect of input-output systems which can receive Boolean descriptions.11–26 In the present instances, molecular memories, with or without downstream processors are found. Molecular logic-based computation15 is rarely seen in macrocyclic capture/release systems, although important examples of polymeric material versions do exist.45,46 So, the current line of research should develop into a broad avenue within molecular logic besides the more common avenue of fluorescent/luminescent sensing.
We value this opportunity to offer a general observation on molecular logic. Molecular logic-based computation originated from fluorescence sensing.11,15 Since it is an extremely wide field, its most vibrant growth is occurring in two populations essentially insulated from each other – supramolecular/analytical scientists12,15,22 and molecular/synthetic biologists.15,22,47 The challenge will be for these two communities which speak different languages and have different philosophical positions to find ways to appreciate their common origin and to share best practice for progressing towards common goals. One of the latter will be to recognize molecular logic within living systems and another will be to apply molecular logic devices to probe/perturb those systems.
This article has summarized the first steps in the command and control of elementary properties of some organic and coordination compounds by exposing or hiding them in shape-switchable cyclophanes.27,37,43 Since electron/proton-transfer in ground/excited states of iconic molecules like G3 and derivatives like G4 have already been controlled in this way for the first time, following studies should allow the control of applications of these compounds and relatives in photocatalysis, electrocatalysis and luminescence sensing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Meilan Huang, Paul Kavanagh, Steven Bell, Peter Nockemann, Andre Alves-Areias, John Malone and Yikai Xu (Queen's University Belfast), Warispreet Singh (Northumbria University), Wesley Browne (University of Groningen), Philip Morgenfurt and Tia Keyes (Dublin City University) for their collaboration. We also thank Queen's University Belfast, Department of Employment and Learning of Northern Ireland, Engineering and Physical Sciences Research Council UK, China Scholarship Council and the Leverhulme Trust (RPG-2019-314), Gaoming Yao, Fafeng Huang, Yixiao Lin, Qin Zhang, Ming Chen, Mei Chen and Amila Norbert for their support. EVA thanks the Welch Regents Chair for support (F-0046).References
- K. Odashima, A. Itai, Y. Iitaka and K. Koga, Host–guest complex-formation between a water-soluble polypara-cyclophane and a hydrophobic guest molecule, J. Am. Chem. Soc., 1980, 102, 2504 CrossRef CAS
.
-
F. Diederich, Cyclophanes, Royal Society of Chemistry, Cambridge, 1991 Search PubMed
- A. R. Bernardo, J. F. Stoddart and A. E. Kaifer, Cyclobis(paraquat-p-phenylene) as a synthetic receptor for electron-rich aromatic-compounds - electrochemical and spectroscopic studies of neurotransmitter binding, J. Am. Chem. Soc., 1992, 114, 10624 CrossRef CAS
- M. Miyake, M. Kirisawa and K. Koga, Biomimetic studies using artificial systems .6. design and synthesis of novel cyclophanes having 8 carboxyl groups on the aromatic rings, Chem. Pharm. Bull., 1993, 41, 1211 CrossRef CAS
- R. Meric, J.-P. Vigneron and J.-M. Lehn, Efficient complexation of quaternary ammonium-compounds by a new water-soluble macrobicyclic receptor molecule, J. Chem. Soc., Chem. Commun., 1993, 129 RSC
- K. S. Cameron, L. Fielding, R. Mason, L. W. Muir, D. C. Rees, S. Thorn and M. Q. Zhang, Anionic cyclophanes as potential reversal agents of muscle relaxants by chemical chelation, Bioorg. Med. Chem. Lett., 2002, 12, 753 CrossRef CAS PubMed
- E. M. Seward, R. B. Hopkins, W. Sauerer, S. W. Tam and F. Diederich, Redox-dependent binding ability of a flavin cyclophane in aqueous-solution - hydrophobic stacking versus cavity-inclusion complexation, J. Am. Chem. Soc., 1990, 112, 1783 CrossRef CAS
- H. Kanazawa, M. Higuchi and K. Yamamoto, An electric cyclophane: cavity control based on the rotation of a paraphenylene by redox switching, J. Am. Chem. Soc., 2005, 127, 16404 CrossRef CAS PubMed
- H. Wu, Y. Chen, L. Zhang, O. Anamimoghadam, D. K. Shen, Z. C. Liu, K. Cai, C. Pezzato, C. L. Stern, Y. Liu and J. F. Stoddart, A dynamic tetracationic macrocycle exhibiting photoswitchable molecular encapsulation, J. Am. Chem. Soc., 2019, 141, 1280 CrossRef CAS PubMed
-
Molecular Switches, ed. B. L. Feringa and W. R. Browne, Wiley-VCH, Weinheim, 2nd edn, 2011 Search PubMed
- A. P. de Silva, H. Q. N. Gunaratne and C. P. McCoy, A molecular photoionic and gate based on fluorescent signaling, Nature, 1993, 364, 42 CrossRef
-
Molecular and Supramolecular Information Processing, ed. E. Katz, Wiley-VCH, Weinheim, 2012 Search PubMed
-
Biomolecular Information Processing, ed. E. Katz, Wiley-VCH, Weinheim, 2012 Search PubMed
-
K. Szacilowski, Infochemistry, Wiley, Chichester, 2012 Search PubMed
-
A. P. de Silva, Molecular Logic-based Computation, Royal Society of Chemistry, Cambridge, 2013 Search PubMed
-
E. Katz, Enzyme-based Computing Systems, Wiley-VCH, Weinheim, 2019 Search PubMed
-
V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines, VCH, Weinheim, 2nd edn, 2008 Search PubMed
- A. P. de Silva, Y. Leydet, C. Lincheneau and N. D. McClenaghan, Chemical approaches to nanometre-scale logic gates, J. Phys.: Condens. Matter, 2006, 18, S1847 CrossRef
- J. Andreasson and U. Pischel, Molecules with a sense of logic: a progress report, Chem. Soc. Rev., 2015, 44, 1053 RSC
- B. Daly, J. Ling, V. A. D. Silverson and A. P. de Silva, Taking baby steps in molecular logic-based computation, Chem. Commun., 2015, 51, 8403 RSC
- S. Erbas-Cakmak, S. Kolemen, A. C. Sedgwick, T. Gunnlaugsson, T. D. James, J. Y. Yoon and E. U. Akkaya, Molecular logic gates: the past, present and future, Chem. Soc. Rev., 2018, 47, 2228 RSC
- C. Y. Yao, H. Y. Lin, H. S. N. Crory and A. P. de Silva, Supra-molecular agents running tasks intelligently (SMARTI): recent developments in molecular logic-based computation, Mol. Syst. Des. Eng., 2020, 5, 1325 RSC
- J. Andreasson and U. Pischel, Light-stimulated molecular and supramolecular systems for information processing and beyond, Coord. Chem. Rev., 2021, 429, 213695 CrossRef CAS
- F. Nicoli, E. Paltrinieri, M. T. Bakic, M. Baroncini, S. Silvi and A. Credi, Binary logic operations with artificial molecular machines, Coord. Chem. Rev., 2021, 428, 213589 CrossRef CAS
- D. C. Magri, Logical sensing with fluorescent molecular logic gates based on photoinduced electron transfer, Coord. Chem. Rev., 2021, 426, 213598 CrossRef CAS
- L. Liu, P. Liu, L. Ga and J. Ai, Advances in applications of molecular logic gates, ACS Omega, 2021, 6, 30189 CrossRef CAS PubMed
- B. Daly, T. S. Moody, A. J. M. Huxley, C. Y. Yao, B. Schazmann, A. Alves-Areias, J. F. Malone, H. Q. N. Gunaratne, P. Nockemann and A. P. de Silva, Molecular memory with downstream logic processing exemplified by switchable and self-indicating guest capture and release, Nat. Commun., 2019, 10, 49 CrossRef PubMed
- C. A. Hunter and J. K. M. Sanders, The nature of pi-pi interactions, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef CAS
- D. T. Racys, S. A. I. Sharif, S. L. Pimlott and A. Sutherland, Silver(I)-catalyzed iodination of arenes: tuning the lewis acidity of n-iodosuccinimide activation, J. Org. Chem., 2016, 81, 772 CrossRef CAS PubMed
- N. A. Bumagin, K. V. Nikitin and I. P. Beletskaya, Palladium-catalyzed carbonylation of aryl iodides in aqueous-media, J. Organomet. Chem., 1988, 358, 563 CrossRef CAS
- J. Andreasson and U. Pischel, A simplicity-guided approach toward molecular set-reset memories, New J. Chem., 2010, 34, 2701 RSC
- J.-M. Lehn, Perspectives in supramolecular chemistry - from molecular recognition towards molecular information-processing and self-organization, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304 CrossRef
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Signaling recognition events with fluorescent sensors and switches, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed
- B. Daly, J. Ling and A. P. de Silva, Current developments in fluorescent PET (photoinduced electron transfer) sensors and switches, Chem. Soc. Rev., 2015, 44, 4203 RSC
-
E. R. Kandel, J. D. Koester, S. H. Mack and S. A. Siegelbaum, Principles of Neural Science, McGraw-Hill, New York, 6th edn, 2021 Search PubMed
- M. A. Winnik, Cyclization and the conformation of hydrocarbon chains, Chem. Rev., 1981, 81, 491 CrossRef CAS
- C. Y. Yao, H. Y. Lin, B. Daly, Y. Xu, W. Singh, H. Q. N. Gunaratne, W. R. Browne, S. E. J. Bell, P. Nockemann, M. Huang, P. Kavanagh and A. P. de Silva, Taming tris(bipyridine)ruthenium(II) and its reactions in water by capture/release with shape-switchable symmetry-matched cyclophanes, J. Am. Chem. Soc., 2022, 144, 4977 CrossRef CAS PubMed
- T. X. Nguyen, S. Landgraf and G. Grampp, Kinetics of photoinduced electron transfer reactions of ruthenium(II) complexes and phenols, tyrosine, n-acetyl-tyrosine and tryptophan in aqueous solutions measured with modulated fluorescence spectroscopy, J. Photochem. Photobiol., B, 2017, 166, 28 CrossRef CAS PubMed
- M.-A. Haga, Synthesis and protonation-deprotonation reactions of ruthenium(II) complexes containing 2,2′-bibenzimidazole and related ligands, Inorg. Chim. Acta, 1983, 75, 29 CrossRef CAS
- K. M. Lancaster, J. B. Gerken, A. C. Durrell, J. H. Palmer and H. B. Gray, Electronic structures, photophysical properties, and electrochemistry of ruthenium(II)(bpy)(2) pyridylimidazole complexes, Coord. Chem. Rev., 2010, 254, 1803 CrossRef CAS
- J. G. Vos, Excited-state acid-base properties of inorganic-compounds, Polyhedron, 1992, 11, 2285 CrossRef CAS
- A. Pannwitz and O. S. Wenger, Proton coupled electron transfer from the excited state of
a ruthenium(II) pyridylimidazole complex, Phys. Chem. Chem. Phys., 2016, 18, 11374 RSC
- C. Y. Yao, H. Y. Lin, P. Morgenfurt, T. E. Keyes and A. P. de Silva, Multiple molecular logic gate arrays in one system of (2-(2 ‘-pyridyl)imidazole)Ru(II) complexes and trimeric cyclophanes in water, Chem. Sci., 2022, 13, 10856 RSC
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, Molecular light switch for DNA - Ru(bpy)2(dppz)2+, J. Am. Chem. Soc., 1990, 112, 4960 CrossRef CAS
- H. Komatsu, S. Matsumoto, S. Tamaru, K. Kaneko, M. Ikeda and I. Hamachi, Supramolecular hydrogel exhibiting four basic logic gate functions to fine-tune substance release, J. Am. Chem. Soc., 2009, 131, 5580 CrossRef CAS PubMed
- B. A. Badeau, M. P. Comerford, C. K. Arakawa, J. A. Shadish and C. A. DeForest, Engineered modular biomaterial logic gates for environmentally triggered therapeutic delivery, Nat. Chem., 2018, 10, 251 CrossRef CAS PubMed
-
DNA- and RNA-Based Computing Systems, ed. E. Katz, Wiley-VCH, Weinheim, 2021 Search PubMed
Footnote |
| † In memoriam John Fossey. |
| This journal is © the Partner Organisations 2023 |