
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA π-extended phenanthrene-fused aza[7]helicenium as a novel chiroptically-active architecture in organic and aqueous media†
Céline
Olivier
 *a,
Nao
Nagatomo
bc,
Tadashi
Mori
d,
Nathan
McClenaghan
a,
Gediminas
Jonusauskas
e,
Brice
Kauffmann
f,
Yutaka
Kuwahara
bc,
Makoto
Takafuji
bc,
Hirotaka
Ihara
bg and
Yann
Ferrand
*h
*a,
Nao
Nagatomo
bc,
Tadashi
Mori
d,
Nathan
McClenaghan
a,
Gediminas
Jonusauskas
e,
Brice
Kauffmann
f,
Yutaka
Kuwahara
bc,
Makoto
Takafuji
bc,
Hirotaka
Ihara
bg and
Yann
Ferrand
*h
aInstitut des Sciences Moléculaires UMR 5255, CNRS Université de Bordeaux, 351 cours de la Libération, 33405 Talence, France. E-mail: celine.olivier@u-bordeaux.fr
bFaculty of Advanced Science and Technology, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto 860-8555, Japan
cIROAST, Kumamoto University, Japan
dDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan
eLaboratoire Ondes et Matière d'Aquitaine CNRS UMR 5798, Université de Bordeaux, 351 cours de la Libération, 33405 Talence, France
fUniversité de Bordeaux, CNRS, INSERM, UMS3033, IECB, 2 rue Robert Escarpit, 33600 Pessac, France
gNational Institute of Technology, Okinawa College, 905 Henoko, Nago, Okinawa 905-2192, Japan
hInstitut CBMN (UMR5248), Université de Bordeaux, CNRS, IPB, 2 rue Escarpit, 33600 Pessac, France. E-mail: yann.ferrand@u-bordeaux.fr
First published on 13th December 2022
Abstract
The synthesis and characterization of an original π-extended cationic azahelicene is reported. The phenanthrene-fused aza[7]helicene derivative encompasses a total of ten aromatic fused rings leading to a dissymmetric yet helically folded structure, as revealed by NMR and X-ray diffraction analyses. The polyaromatic and cationic nature of the new azahelicenium makes it soluble in both organic and aqueous media, which allowed photophysical studies in solvents of different polarities. The extended chromophoric species demonstrates a broad absorption over the whole visible range and orange-red fluorescence emission. Chiral resolution of the racemate was performed subsequently, affording two optically pure and configurationally stable azahelicenium enantiomers. Multi-band circular dichroism and long-wavelength circularly polarized emission were observed, associated with remarkable absorption and luminescence dissymmetry factors, both in organic and aqueous media.
Introduction
Among chiral organic small molecules, helicenes are undoubtedly the most extensively harnessed mainly because of their appealing helical shape combined with inherent electronic delocalization.1–4 In recent years, the use of chiral luminophores, and among them helicenes,5 has been widely acknowledged as an ideal approach to directly generate circularly polarized light (CPL).6 The development of CPL-active architectures is growing continuously as various applications emerge such as circularly-polarized electroluminescent devices,7 optical information encoding or processing,8 and chemical and biological chirality sensing.9 The latter being still rather unexplored due to the poor number of bio-compatible CPL-active systems.The major asset of small organic systems towards practical applications is the possibility of fine-tuning their photophysical properties through molecular engineering. The modulation of helicene opto-electronic properties can be envisaged by two means: i. via the introduction of one or more heteroatoms in the π-conjugated structure, resulting in heterohelicenes such as phosphahelicenes, azahelicenes, borahelicenes and silicahelicenes with strongly modified chiroptical response compared to carbohelicenes;10 ii. by enhancing their specific chiroptical properties through the design of multiple helicenes11 or π-extended systems,12 thereby promoting large π-electron delocalization and affording unique optoelectronic properties. However, the increase in the number of fused aromatic rings may give rise to solubility issues and limit their study to organic media exclusively. The introduction of a cationic chain or motif enhances the solubilization of polyaromatic systems in aqueous media. The cationic analogues of helicenes, i.e. helicenium derivatives, may be anticipated to present good solubility in an aqueous phase thus making helicenes applicable in water,13 notably towards bio-related applications as in sensors or bio-imaging. However, despite some examples of azoniahelicenes,14 azahelicenoids15 and helquats,16 the field of cationic azahelicenes remains rather overlooked, and even more so with regard to their chiroptical properties.
Herein we report a simple synthesis route towards a new type of cationic azahelicenia, from triarylamine derivatives, using Vilsmeier–Haack conditions. This route is an alternative to more traditional methods such as photocyclodehydrogenation and [2 + 2 + 2] cycloisomerization of alkynes.17 The first example of an extended azahelicenium described in the present study encompasses ten aromatic fused rings and an endocyclic quaternary nitrogen atom. The optical and chiroptical properties of 1 were assessed in both organic and aqueous media and further rationalized through quantum chemical calculations.
Results and discussion
Design and synthesis
The strategy to prepare 1 is directly inspired by a straightforward two-step synthesis of non-helical dibenzoacridinium salts from triarylamine precursors that we recently developed (Scheme 1a).18 For example, treating triarylamine 2 under Vilsmeier–Haack conditions led to intramolecular cycloaromatization and simultaneous quaternarization of the nitrogen atom to form the dibenzoacridinium derivative 3. We envisaged that using similar conditions for 5, bearing two 2-benzo[c]phenanthrene moieties, would afford an expanded aza[9]helicenium with an ortho–ortho–meta–ortho–ortho fused backbone (Scheme 1, Fig. S1†). Therefore, 5 was readily prepared from bromobenzo[c]phenanthrene 4.19 Surprisingly, when using the Vilsmeier–Haack conditions, the expected diphenanthro-acridinium could not be isolated but instead one could observe a subsequent intramolecular cyclisation occurring via the cyclodehydrogenation of one of the phenanthrene arms and the neighbouring pyridinium ring to form a benzo[ghi]perylene motif (Scheme 1b). Overall, the double cyclization process yielded the dissymmetrical π-extended azahelicenium 6 comprising ten aromatic fused-rings. Finally, anion metathesis with an excess of KPF6 afforded racemic helicenium 1. The synthesis of 1 is detailed in the Experimental section. | ||
| Scheme 1 (a) Synthesis of an N-phenyl-dibenzoacridinium salt from a triarylamine precursor. The heteroaromatic ring in purple is formed during the reaction.16 (b) Synthesis of rac-1 (only the P-enantiomer is shown). The purple rings are formed successively during the reaction. (i) POCl3, DMF, 90 °C; (ii) Pd(OAc)2, P(tBu)3, Cs2CO3, PhMe, 90 °C; and (iii) KPF6, MeOH. | ||
Crystallography
Single crystals were grown by slow liquid–liquid diffusion of hexane into a dichloromethane solution of rac-1. The structure of the polyaromatic salt was solved in the orthorhombic and centrosymmetric Pna2(1) space group with P and M enantiomers coexisting in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in the unit cell (see the ESI†). The crystalline structure revealed a helical conformation with perfect overlapping of the terminal benzene rings (Fig. 1a). The polycondensed aromatic backbone displays a total of ten 6-membered aromatic fused rings, including one aza-aromatic ring. The longest ortho-fused sequence is composed of seven aromatic rings (Fig. 1c, A–G) merged with three additional benzene rings (H–J), forming a π-extended phenanthrene-fused aza[7]helicenium moiety. The polyaromatic core revealed a high degree of distortion with a dihedral angle (i.e., the angle between the two mean planes defined by the edge rings A and G) of 27.65° and a helical pitch of 3.48 Å (Fig. 1b) indicating strong intramolecular π–π interactions. The sum of the five torsion angles derived from the seven C–C bonds that form the inner rim is equal to 108.27°, which is significantly higher than those of other hetero[7]helicenes.20 This and the perfect overlapping of the terminal benzene rings are consistent with a high stability against racemization.
:1 ratio in the unit cell (see the ESI†). The crystalline structure revealed a helical conformation with perfect overlapping of the terminal benzene rings (Fig. 1a). The polycondensed aromatic backbone displays a total of ten 6-membered aromatic fused rings, including one aza-aromatic ring. The longest ortho-fused sequence is composed of seven aromatic rings (Fig. 1c, A–G) merged with three additional benzene rings (H–J), forming a π-extended phenanthrene-fused aza[7]helicenium moiety. The polyaromatic core revealed a high degree of distortion with a dihedral angle (i.e., the angle between the two mean planes defined by the edge rings A and G) of 27.65° and a helical pitch of 3.48 Å (Fig. 1b) indicating strong intramolecular π–π interactions. The sum of the five torsion angles derived from the seven C–C bonds that form the inner rim is equal to 108.27°, which is significantly higher than those of other hetero[7]helicenes.20 This and the perfect overlapping of the terminal benzene rings are consistent with a high stability against racemization.
 | ||
| Fig. 1 X-ray structure of 1 (only the P enantiomer is shown for clarity): (a–c) different views in stick representation. In (c), the ortho-aza[7]helicene motif is colored in red, whereas the fused phenanthrene unit is shown in light orange; (d) front view in CPK representation. The counter anion is omitted for clarity. (e) 1H NMR spectrum of 1 in CD2Cl2 (700 MHz, 298 K, 1 mM). | ||
NMR studies
Further characterization of rac-1 was performed using NMR spectroscopy to observe its behaviour in solution. Unequivocal chemical shift assignment of 1 was obtained using a combination of 1D and 2D NMR techniques (Fig. S2–S7†). The 1H NMR spectrum of 1 showed sharp signals in CD2Cl2 (Fig. 1e) and increasing the concentration did not lead to any sort of signal broadening which is indicative of the absence of non-specific aggregation. The lack of symmetry within the helix was confirmed as each of the resonances integrates for a single proton. Unlike 3, which exhibits a low field resonance (δ = 11.5 ppm in DMSO-d6) for the proton in the para position to the nitrogen of the pyridinium ring, the 1H NMR spectrum of 1 is devoid of any signal. Finally, 1H–1H NOESY experiments allowed the identification of strong NOE contacts between the two ends of the helix (i.e., rings A and G).Chiral resolution
The helical nature of 1 prompted us to resolve its enantiomers. We anticipated that 1, due to its aza[7]helicene structure, should present a high tolerance towards racemization as reported by several studies.10,21 Chiral resolution of rac-1 was achieved by chiral HPLC (details and chromatograms are given in the ESI, Fig. S8 and S9†). It is noteworthy that despite the notorious difficulty in resolving the chiral ionic species, the separation was performed without prior reduction or any other chemical modification. The first eluted fraction showed a negative CD signal at 300 nm, whereas the second eluted peak yielded a mirror-image positive signal.Absorption and emission properties
The absorption and emission properties of rac-1 in various solvents were studied in order to investigate the effect of the cationic nature of the helical fused aromatic core on electronic transitions. In solution, 1 revealed a red-wine colour with panchromatic absorption properties starting in the ultraviolet region and covering the whole visible range. Its electronic absorption spectrum in chloroform (Fig. 2b and S10†) displayed multiple transitions from 220 nm to 650 nm that can be partitioned into three domains. An intense absorption is observed in the 220–320 nm region (ε = 40000–60000 M−1 cm−1) associated with π–π* and n–π* electronic transitions. A set of well-structured bands of moderate intensity (ε = 20000 to 10000 M−1 cm−1) is observed in the 320–420 nm region, with maxima at 356, 380 and 404 nm. This vibrational hyper-fine structure is characteristic of rigid polycyclic aromatic hydrocarbons (PAHs).22 Finally, a prominent feature of 1 compared to neutral aza-helicenes is the presence of moderately strong (ca. 10000 M−1 cm−1), yet very broad absorption bands covering the whole visible range from 420 nm to 685 nm, with main transitions at 454, 487, 560 and 610 nm, which exhibit intramolecular charge transfer (ICT) character. Such an unusually broad absorption spectrum could therefore be ascribed to the polyaromatic and cationic nature of 1. The electronic absorption spectrum in an aqueous medium shows similar features (Fig. S11†) although with a slight diffusion effect presumably due to the aggregation phenomena occurring in water at the concentration required for absorption spectroscopy.
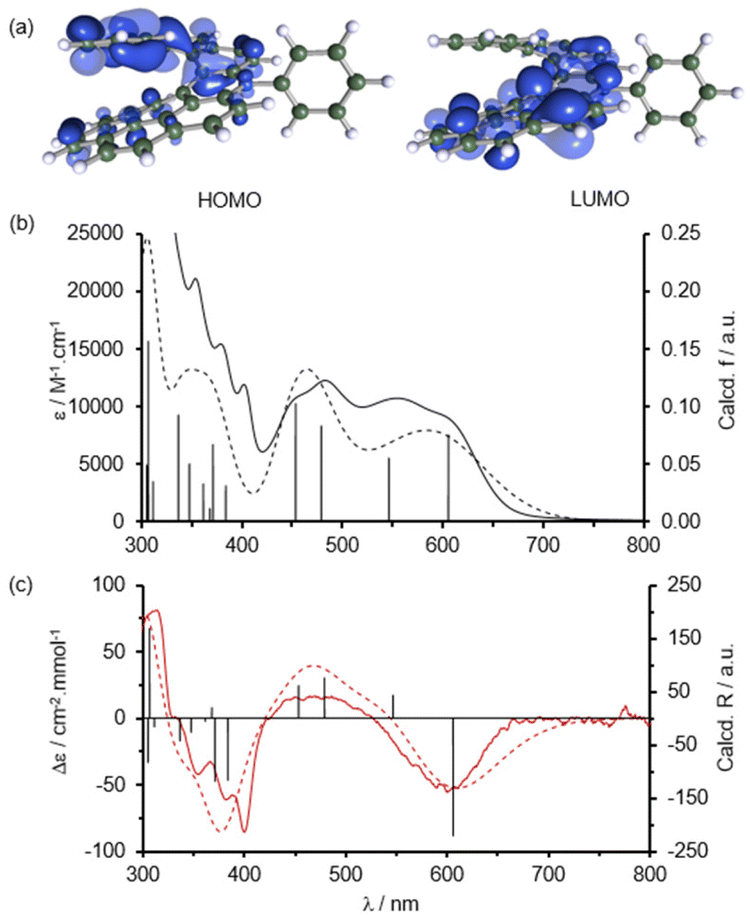 | ||
| Fig. 2 Quantum chemical calculations. (a) Frontier molecular orbitals of 1. HOMO: highest-occupied molecular orbital; LUMO: lowest-unoccupied molecular orbital. Comparison of the computed (dotted) and experimental (plain) absorption (b) and ECD (c) spectra. Black bars show the contribution of different electronic transitions. Simulated spectra were obtained by empirically applying 0.2 eV half-width at half-maximum (HWHM) for Gaussian expansion. | ||
The emission properties of 1 were evaluated in dilute solutions (∼10−6 M) at room temperature (see the ESI†). The excitation-independent emission and the similitude between the absorption and excitation spectra in dilute solution indicate the purity of the sample and the absence of aggregates. In chloroform, orange-red fluorescence was observed, characterized by a broad emission band with the maximum at λem = 692 nm (Fig. S10†). A fluorescence quantum yield of Φlum = 0.08 was calculated, in line with the values reported for other π-extended aza[7]helicenes.23 The steady-state emission studies were then carried out in different solvents: chloroform, acetonitrile, methanol and a water/methanol mixture (9:1, v/v). No significant solvent dependence of the main emission was observed (Fig. S12†). In aqueous solution, the emission band maximum was also located at λem = 695 nm, however, with a lower fluorescence quantum yield of Φlum = 0.01 attributed to enhanced non-radiative deactivation processes in polar protic solvents.
Computational studies
Quantum chemical calculations were performed to get deeper insights into the optical and chiroptical properties of 1. The details of theoretical calculations are given in the ESI.† The frontier molecular orbitals (MOs) of 1 are presented in Fig. 2a. The calculations show major localization of the LUMO on the aza-benzo[ghi]perylene core indicating that the positive charge is preferentially delocalized over the benzo[ghi]perylene part, while the HOMO is localized on the opposite benzo[c]phenanthrene part. This distinct localization of the frontier MOs strongly favors low-energy electronic transitions. The calculated UV-vis absorption spectrum perfectly matches the experimental spectrum as shown in Fig. 2b. The lowest energy transitions located at 605 nm and 546 nm were attributed to the HOMO → LUMO and HOMO−1 → LUMO transitions, respectively. These transitions give rise to the large absorption bands observed in the 500–700 nm range. The bands observed at 400–500 nm were attributed to higher energy transitions, i.e. HOMO−2 → LUMO and HOMO−3 → LUMO (see the ESI†). Moreover, the calculations related to the M-enantiomer of 1 correctly predicted almost all the important features of the experimental ECD of the second eluted peak (Fig. 2c) allowing us to unambiguously determine the absolute configuration of the two enantiomers.Thus, according to the simulated ECD spectrum, the first and second HPLC fractions correspond to the P and M enantiomers, respectively. The sign of the ECD signal at the lowest energy is therefore consistent with that commonly reported for hetero[n]helicenes, i.e. a positive CD signal for the P-enantiomer and a negative CD signal for the M-enantiomer.9,10,12d
Chiroptical properties
The experimental ECD spectra of the two HPLC fractions recorded in chloroform solution (Fig. 3a) were perfect mirror images corresponding to the P- and M-enantiomers of 1. The ECD spectra displayed multiple Cotton effects at high energies as well as in the long wavelength range, with three distinct sign inversions at λ = 521, 425 and 332 nm. From low to high energies, the first eluted HPLC peak demonstrated a positive CD signal at 606 nm (Δε = 67 mmol−1 cm−2 and gabs = 6.4 × 10−3) followed by a negative CD signal at 475 nm (Δε = −21 mmol−1 cm−2 and gabs = −1.7 × 10−3). Well-structured positive Cotton bands were then observed with maxima at 400, 382 and 355 nm (Δε = 84, 65, and 40 mmol−1 cm−2; gabs = 7.1 × 10−3, 4.1 × 10−3, and 1.9 × 10−3), followed by an intense negative CD signal at 312 nm (Δε = −90 mmol−1 cm−2 and gabs = −2.3 × 10−3) and a less intense band at 255 nm (Δε = −45 mmol−1 cm−2). The second eluted HPLC peak exhibited an exact opposite ECD profile (Fig. 3a). Interestingly, the ECD spectra recorded in aqueous solution (H2O/MeOH, 9:1, v/v) were perfectly superimposable with those recorded in organic solution (Fig. S16†).
 | ||
| Fig. 3 ECD (a) and CPL (b) spectra of the separated enantiomers of 1 recorded in CHCl3. Blue: first eluted peak (P-1); red: second eluted peak (M-1). Inset: calculated glum factor. | ||
The chirality of the excited state was investigated by means of circularly polarized luminescence (CPL) spectroscopy (Fig. 3b). The two enantiomers of 1 showed CPL signals in chloroform solution at wavelengths corresponding to the fluorescence emission. The sign of the CPL signals corresponded to that of the ECD signals at low energies. Hence, the first eluted HPLC peak (and conversely for the other enantiomer) showed a broad and rather intense positive CPL signal in the long-wavelength range, with the maximum intensity at 685 nm and corresponding glum = 6.0 × 10−3. It should be noted that this value is equal to the absorption dissymmetry factor of the lowest energy transition determined by ECD (gabs = 6.4 × 10−3). This tends to indicate that there is no significant geometry change between the ground and excited states. It is also noteworthy that the CPL response of 1 extends over a broad long-wavelength range, showing a tail up to 780 nm. The CPL brightness, BCPL, calculated at the maximum CPL intensity was 2.8, in line with those of some cationic helicenoid structures,24 yet the current study uniquely reports the BCPL value for azahelicenium. Remarkably, the CPL spectra of the two enantiomers of 1 also displayed broad signals in aqueous solution (Fig. S14†), comparable to those recorded in an organic medium, extending from 630 nm to the NIR region (780 nm), with the maximum signal intensity at 690 nm and the corresponding |glum| = 6.0 × 10−3. Thus it appears here that unlike total photoluminescence, i.e. fluorescence, the relative intensity difference between the left and right circularly polarized emission, i.e. CPL, is independent of the solvent nature and polarity. This property has been rarely reported, except in the case of the water-soluble cationic helicenoids reported by Lacour et al., which exhibit the same glum factor in water and acetonitrile.15 This property, together with long-wavelength emission, i.e. in the red domain, at the threshold of the biological window, could make cationic aza-helicenes interesting candidates for bio-imaging applications. In particular CPL microscopy, although still in its infancy, represents an advanced technology with high application potential in the field of chiral interaction studies on a sub-cellular level.25
Conclusions
In summary, a convenient and modular synthesis route is reported for a new type of cationic polyaromatic nitrogen-based structure, starting from simple triarylamine derivatives. The π-extended aza-helicenium 1 encompasses a total of ten aromatic fused rings. Resolution of the enantiomers was performed and their photophysical properties were studied both in organic and aqueous media. Structural analyses revealed a stable helical backbone and no racemization could be observed even after several days in solution. The extended azahelicenium showed multi-band electronic circular dichroism and remarkable long-wavelength CPL emission, with high absorption and luminescence dissymmetry factors both in organic and aqueous media. We believe that the new type of synthetically-accessible π-extended cationic aza-helicene reported herein represents a versatile platform to design future chiral molecular systems with specific physical and chiroptical properties, in particular, for bio-related applications. Further studies on other azahelicenia and their applications are currently in progress in our laboratories.Experimental section
Methods for chemical synthesis
All reactions were carried out under a dry nitrogen atmosphere. Commercial reagents were purchased from Sigma-Aldrich, Alfa-Aesar and TCI and used without further purification unless otherwise specified. Chloroform (CHCl3) and diisopropylethylamine (DIPEA) were dried over calcium hydride (CaH2) and distilled prior to use. Toluene was dried over sodium and distilled prior to use. Bromobenzo[c]phenanthrene 4 was obtained as described in the literature.19:2, v/v) to afford 2 as a pale yellow powder (3 g, 8.7 mmol, 87% yield). 1H NMR (300 MHz, CDCl3): δ = 7.80 (d, 2H, 3JH–H = 7.1 Hz), 7.77 (d, 2H, 3JH–H = 9.1 Hz), 7.61 (d, 2H, 3JH–H = 7.1 Hz), 7.50 (d, 2H, 4JH–H = 2.2 Hz), 7.46–7.29 (m, 8H), 7.21 (d, 2H, 3JH–H = 8.7 Hz), 7.11 (t, 1H, 3JH–H = 7.2 Hz). 13C NMR (75 MHz, CDCl3): δ = 147.8, 145.5, 134.5, 130.3, 129.4, 129.0, 127.7, 127.1, 126.4, 124.8, 124.7, 124.6, 123.2, 120.6. HR-MS ESI+ (m/z): 345.1514 [M+]+ (calcd 345.1517 for [C26H19N]+).
:1, v:v). The target compound precipitated as a highly coloured solid by the addition of ethyl acetate. The crude product was purified on a silica gel column (CH2Cl2/MeOH (9:1, v/v)) to afford 3 as a bright yellow powder (1.4 g, 3.6 mmol, 92% yield). 1H NMR (300 MHz, dmso-d6): δ = 11.50 (s, 1H), 9.85 (d, 2H, 3JH–H = 8.3 Hz), 8.64 (d, 2H, 3JH–H = 9.6 Hz), 8.34 (d, 2H, 3JH–H = 7.3 Hz), 8.19 (t, 2H, 3JH–H = 7.2 Hz), 8.06–7.97 (m, 5H), 7.89 (m, 2H), 7.37 (d, 2H, 3JH–H = 9.6 Hz). 13C NMR (75 MHz, dmso-d6): δ = 141.6, 140.2, 137.8, 137.3, 131.7, 131.3, 130.5, 130.1, 129.9, 129.8, 128.3, 127.8, 125.1, 124.8, 117.11. HR-MS ESI+ (m/z): 356.1451 [M]+ (calcd 356.1439 for [C27H18N]+).
:2, v/v) to afford 5 as a pale yellow powder (205 mg, 0.37 mmol, 70% yield). 1H NMR (300 MHz, CDCl3): δ = 8.66 (s, 2H), 8.50 (d, 2H, 3JH–H = 8.6 Hz), 7.99 (d, 2H, 3JH–H = 8.7 Hz), 7.90–7.86 (m, 5H), 7.84–7.74 (m, 6H), 7.64 (dd, 2H, 3JH–H = 8.7 Hz), 7.46–7.39 (m, 6H), 7.13 (t, 2H, 3JH–H = 7.1 Hz). 13C NMR (75 MHz, CDCl3): δ = 147.8, 145.5, 134.5, 130.3, 129.4, 129.0, 127.7, 127.1, 126.4, 124.8, 124.7, 124.6, 123.2, 120.6. HR-MS ESI+ (m/z): 546.2217 [M + H]+ (calcd 546.2222 for [C42H28N]+).
:1, v/v)) to afford 6 as a dark purple powder (52 mg, 0.09 mmol, 32% yield). 1H NMR (300 MHz, CD2Cl2): δ = 8.95 (d, 1H, 3JH–H = 9.3 Hz), 8.82 (d, 1H, 3JH–H = 8.3 Hz), 8.70 (d, 1H, 3JH–H = 8.3 Hz), 8.53 (d, 1H, 3JH–H = 9.1 Hz), 8.34 (d, 1H, 3JH–H = 7.4 Hz), 8.26 (d, 1H, 3JH–H = 8.8 Hz), 8.21–7.95 (m, 8H), 7.84 (d, 1H, 3JH–H = 8.6 Hz), 7.83 (d, 1H, 3JH–H = 9.2 Hz), 7.68 (d, 1H, 3JH–H = 9.1 Hz), 7.62 (d, 2H, 3JH–H = 8.5 Hz), 7.55 (d, 1H, 3JH–H = 6.6 Hz), 7.22 (d, 1H, 3JH–H = 8.1 Hz), 7.13 (t, 1H, 3JH–H = 7.9 Hz), 6.50 (t, 1H, 3JH–H = 7.1 Hz), 5.69 (t, 1H, 3JH–H = 7.3 Hz).
:v) at RT. After solvent removal, the crude was dissolved back in pure CH2Cl2, filtered and evaporated to dryness to obtain 1.
1H NMR (300 MHz, CD2Cl2): δ = 8.95 (d, 1H, 3JH–H = 9.3 Hz), 8.82 (d, 1H, 3JH–H = 8.3 Hz), 8.70 (d, 1H, 3JH–H = 8.3 Hz), 8.53 (d, 1H, 3JH–H = 9.1 Hz), 8.34 (d, 1H, 3JH–H = 7.4 Hz), 8.26 (d, 1H, 3JH–H = 8.8 Hz), 8.21–7.95 (m, 8H), 7.84 (d, 1H, 3JH–H = 8.6 Hz), 7.83 (d, 1H, 3JH–H = 9.2 Hz), 7.68 (d, 1H, 3JH–H = 9.1 Hz), 7.62 (d, 2H, 3JH–H = 8.5 Hz), 7.55 (d, 1H, 3JH–H = 6.6 Hz), 7.22 (d, 1H, 3JH–H = 8.1 Hz), 7.13 (t, 1H, 3JH–H = 7.9 Hz), 6.50 (t, 1H, 3JH–H = 7.1 Hz), 5.69 (t, 1H, 3JH–H = 7.3 Hz). 13C NMR (75 MHz, CDCl3): δ = 152.8, 141.6, 139.3, 137.4, 133.8, 133.5, 133.4, 132.9, 132.1, 131.9, 131.4, 131.3, 130.7, 129.9, 129.5, 129.3, 129.1, 129.0, 128.5, 128.4, 127.4, 127.2, 126.0, 125.3, 125.1, 124.8, 124.2, 124.0, 121.8, 119.4, 117.8, 1117.0, 110.9. HR-MS ESI+ (m/z): 554.1897 [M]+ (calcd 554.1903 for [C43H24N]+).
Author contributions
C. O. conducted the experiments, analysed the data and wrote the manuscript. G. J. and N. McC conducted photophysical studies and assisted with manuscript preparation. T. M. performed the computational studies and assisted with manuscript preparation. B. K. determined the crystal structure. N. N., Y. K., M. T. and H. I. performed the chiral resolution of 1. Y. F. designed the work, conducted the NMR studies and finalised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
YF and CO thank the IdEx University of Bordeaux, Grand Research Program “GPR LIGHT” for financial support. YK, MT and HI thank Grants-in-Aid for Scientific Research, KAKENHI (JSPS), for financial support. TM thanks the CREST (JST, Grant number JPMJCR2001) and KAKENHI (JSPS) for financial support.References
- C.-F. Chen and Y. Shen, in Helicene Chemistry: From Synthesis to Applications, Springer-Verlag GmbH, Berlin Heidelberg, 2017 Search PubMed.
- J. Crassous, Circularly polarized luminescence in helicene and helicenoid derivatives, in Circularly Polarized Luminescence of Isolated Small Organic Molecules, ed. T. Mori, Springer, Singapore, 2020, pp. 53–97 Search PubMed.
- E. M. Sanchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Circularly Polarized Luminescence from Simple Organic Molecules, Chem. – Eur. J., 2015, 21, 13488 CrossRef CAS.
- H. Tanaka, Y. Inoue and T. Mori, Circularly Polarized Luminescence and Circular Dichroisms in Small Organic Molecules: Correlation between Excitation and Emission Dissymmetry Factors, ChemPhotoChem, 2018, 2, 386 CrossRef CAS.
- W.-L. Zhao, M. Li, H.-Y. Lu and C.-F. Chen, Advances in Helicene Derivatives with Circularly Polarized Luminescence, Chem. Commun., 2019, 55, 13793 RSC.
- Y. Deng, M. Wang, Y. Zhuang, S. Liu, W. Huang and Q. Zhao, Circularly Polarized Luminescence from Organic Micro-/Nano-Structures, Light: Sci. Appl., 2021, 10, 76 CrossRef CAS PubMed.
- D.-W. Zhang, M. Li and C.-F. Chen, Recent Advances in Circularly Polarized Electroluminescence Based on Organic Light-Emitting Diodes, Chem. Soc. Rev., 2020, 49, 1331 RSC.
- H. Li, H. Li, W. Wang, Y. Tao, S. Wang, Q. Yang, Y. Jiang, C. Zheng, W. Huang and R. Chen, Stimuli-Responsive Circularly Polarized Organic Ultralong Room Temperature Phosphorescence, Angew. Chem., Int. Ed., 2020, 59, 4756 CrossRef CAS PubMed.
- (a) Y. Imai, Y. Nakano, T. Kawai and J. Yuasa, A Smart Sensing Method for Object Identification Using Circularly Polarized Luminescence from Coordination-Driven Self-Assembly, Angew. Chem., Int. Ed., 2018, 57, 8973 CrossRef CAS PubMed; (b) K. Staszak, K. Wieszczycka, V. Marturano and B. Tylkowski, Lanthanides Complexes – Chiral Sensing of Biomolecules, Coord. Chem. Rev., 2019, 397, 76 CrossRef CAS; Y. Zhao, D. Niu, J. Tan, Y. Jiang, H. Zhu, G. Ouyang and M. Liu, Small Methods, 2020, 4, 2000493 Search PubMed.
- K. Dhbaibi, L. Favereau and J. Crassous, Enantioenriched Helicenes and Helicenoids Containing Main-Group Elements (B, Si, N, P), Chem. Rev., 2019, 119, 8846 CrossRef CAS PubMed.
- (a) K. Nakamura, S. Furumi, M. Takeuchi, T. Shibuya and K. Tanaka, Enantioselective Synthesis and Enhanced Circularly Polarized Luminescence of S-Shaped Double Azahelicenes, J. Am. Chem. Soc., 2014, 136, 5555 CrossRef CAS PubMed; (b) V. Berezhnaia, M. Roy, N. Vanthuyne, M. Villa, J.-V. Naubron, J. Rodriguez, Y. Coquerel and M. Gingras, Chiral Nanographene Propeller Embedding Six Enantiomerically Stable [5]Helicene Units, J. Am. Chem. Soc., 2017, 139(51), 18508–18511 CrossRef CAS PubMed; (c) T. Mori, Chiroptical Properties of Symmetric Double, Triple, and Multiple Helicenes, Chem. Rev., 2021, 121, 2373 CrossRef CAS PubMed.
- (a) C. M. Cruz, S. Castro-Fernandez, E. Maçoas, J. M. Cuerva and A. G. Campana, Undecabenzo[7]superhelicene: A Helical Nanographene Ribbon as a Circularly Polarized Luminescence Emitter, Angew. Chem., Int. Ed., 2018, 57, 14782 CrossRef CAS PubMed; (b) Y. Hu, G. M. Paternò, X.-Y. Wang, X.-C. Wang, M. Guizzardi, Q. Chen, D. Schollmeyer, X.-Y. Cao, G. Cerullo, F. Scotognella, K. Müllen and A. Narita, π-Extended Pyrene-Fused Double [7]Carbohelicene as a Chiral Polycyclic Aromatic Hydrocarbon, J. Am. Chem. Soc., 2019, 141, 12797 CrossRef CAS PubMed; (c) Y. Chen, C. Lin, Z. Luo, Z. Yin, H. Shi, Y. Zhu and J. Wang, Double p-Extended Undecabenzo[7]helicene, Angew. Chem., Int. Ed., 2021, 60, 7796 CrossRef CAS PubMed; (d) C. Shen, G. Zhang, Y. Ding, N. Yang, F. Gan, J. Crassous and H. Qiu, Oxidative Cyclo-Rearrangement of Helicenes into Chiral Nanographenes, Nat. Commun., 2021, 12, 2786 CrossRef CAS PubMed; (e) W. Zheng, T. Ikai, K. Oki and E. Yashima, Consecutively Fused Single-, Double-, and Triple-Expanded Helicenes, Nat. Sci., 2022, e20210047 Search PubMed.
- L. Latterini, E. Galletti, R. Passeri, A. Barbafina, L. Urbanelli, C. Emiliani, F. Elisei, F. Fontana, A. Mele and T. Caronna, Fluorescence Properties of Aza-Helicenium Derivatives for Cell Imaging, J. Photochem. Photobiol., A, 2011, 222, 307 CrossRef CAS.
- Z. Wang, L. Jiang, J. Ji, F. Zhou, J. Lan and J. You, Construction of Cationic Azahelicenes: Regioselective Three-Component Annulation Using In Situ Activation Strategy, Angew. Chem., Int. Ed., 2020, 59, 23532 CrossRef CAS PubMed.
- S. Pascal, C. Besnard, F. Zinna, L. Di Bari, B. Le Guennic, D. Jacquemin and J. Lacour, Zwitterionic [4]Helicene: a Water-Soluble and Reversible pH-Triggered ECD/CPL Chiroptical Switch in the UV and Red Spectral Regions, Org. Biomol. Chem., 2016, 14, 4590 RSC.
- P. E. Reyes-Gutiérrez, M. Jirásek, L. Severa, P. Novotná, D. Koval, P. Sázelová, J. Vávra, A. Meyer, I. Císařová, D. Šaman, R. Pohl, P. Štěpánek, P. Slavíček, B. J. Coe, M. Hájek, V. Kašička, M. Urbanová and F. Teplý, Functional Helquats: Helical Cationic Dyes with Marked, Switchable Chiroptical Properties in the Visible Region, Chem. Commun., 2015, 51, 1583 RSC.
- I. G. Stará and I. Starý, Helically Chiral Aromatics: The Synthesis of Helicenes by [2 + 2 + 2] Cycloisomerization of π-Electron Systems, Acc. Chem. Res., 2020, 53, 144 CrossRef PubMed.
- S. Amrane, M. L. Andreola, C. Olivier and Y. Ferrand, Novel dibenzoacridinium derivatives, their process of preparation and their use for treating viral infections, WO2022053704A1, 2022.
- H. R. Talele, M. J. Gohil and A. V. Bedekar, Synthesis of Derivatives of Phenanthrene and Helicene by Improved Procedures of Photocyclization of Stilbenes, Bull. Chem. Soc. Jpn., 2009, 82, 1182 CrossRef CAS.
- T. Otani, T. Sasayama, C. Iwashimizu, K. S. Kanyiva, H. Kawai and T. Shibata, Short-step Synthesis and Chiroptical Properties of polyaza[5]–[9]helicenes with Blue to Green-Colour Emission, Chem. Commun., 2020, 56, 4484 RSC.
- K. Nakano, Y. Hidehira, K. Takahashi, T. Hiyama and K. Nozaki, Stereospecific Synthesis of Hetero[7]helicenes by Pd-Catalyzed Double N-Arylation and Intramolecular O-Arylation, Angew. Chem., Int. Ed., 2005, 44, 7136 CrossRef CAS PubMed.
- (a) R. Rieger and K. Mullen, Forever Young: Polycyclic Aromatic Hydrocarbons as Model Cases for Structural and Optical Studies, J. Phys. Org. Chem., 2010, 23, 315 CAS; (b) D. Reger, P. Haines, K. Y. Amsharov, J. A. Schmidt, T. Ullrich, S. Bçnisch, F. Hampel, A. Görling, J. Nelson, K. E. Jelfs, D. M. Guldi and N. Jux, A Family of Superhelicenes: Easily Tunable, Chiral Nanographenes by Merging Helicity with Planar π Systems, Angew. Chem., Int. Ed., 2021, 60, 18073 CrossRef CAS PubMed.
- (a) G. M. Upadhyay, H. R. Talele and A. V. Bedekar, Synthesis and Photophysical Properties of Aza[n]helicenes, J. Org. Chem., 2016, 81, 7751 CrossRef CAS PubMed; (b) K. Uematsu, C. Hayasaka, K. Takase, K. Noguchi and K. Nakano, Transformation of Thia[7]helicene to Aza[7]helicenes and [7]Helicene-like Compounds via Aromatic Metamorphosis, Molecules, 2022, 27, 606 CrossRef CAS PubMed.
- L. Arrico, L. Di Bari and F. Zinna, Quantifying the Overall Efficiency of Circularly Polarized Emitters, Chem. – Eur. J., 2021, 27, 2920 CrossRef CAS PubMed.
- P. Stachelek, L. MacKenzie, D. Parker and R. Pal, Circularly Polarized Luminescence Laser Scanning Confocal Microscopy to Study Live Cell Chiral Molecular Interactions, Nat. Commun., 2022, 13, 553 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic and calculation details. CCDC 2183137. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qo01942f |
| This journal is © the Partner Organisations 2023 |