DOI:
10.1039/D2QO01670B
(Research Article)
Org. Chem. Front., 2023,
10, 150-156
Rational design and organocatalytic enantioselective [1 + 4]-annulations of MBH carbonates with modified enones†
Received
21st October 2022
, Accepted 14th November 2022
First published on 16th November 2022
Abstract
According to the frontier molecular orbital theory, two types of modified enones have been designed and successfully applied in the chiral phosphine-catalyzed stereoselective [1 + 4]-annulation of Morita–Baylis–Hillman (MBH) carbonates for the first time. The reaction proceeds smoothly under mild conditions and exhibits excellent functional group tolerance, furnishing a broad scope of enantioenriched 2,3-dihydrofurans with high efficiency. DFT calculations have been applied to provide guidance for the design of additional enones and understand the origin of stereoselectivity. Importantly, this protocol further explores the scope of enones and enriches the chemistry of [1 + 4]-annulations of MBH carbonates for preparation of optically active multifunctional 2,3-dihydrofurans.
Introduction
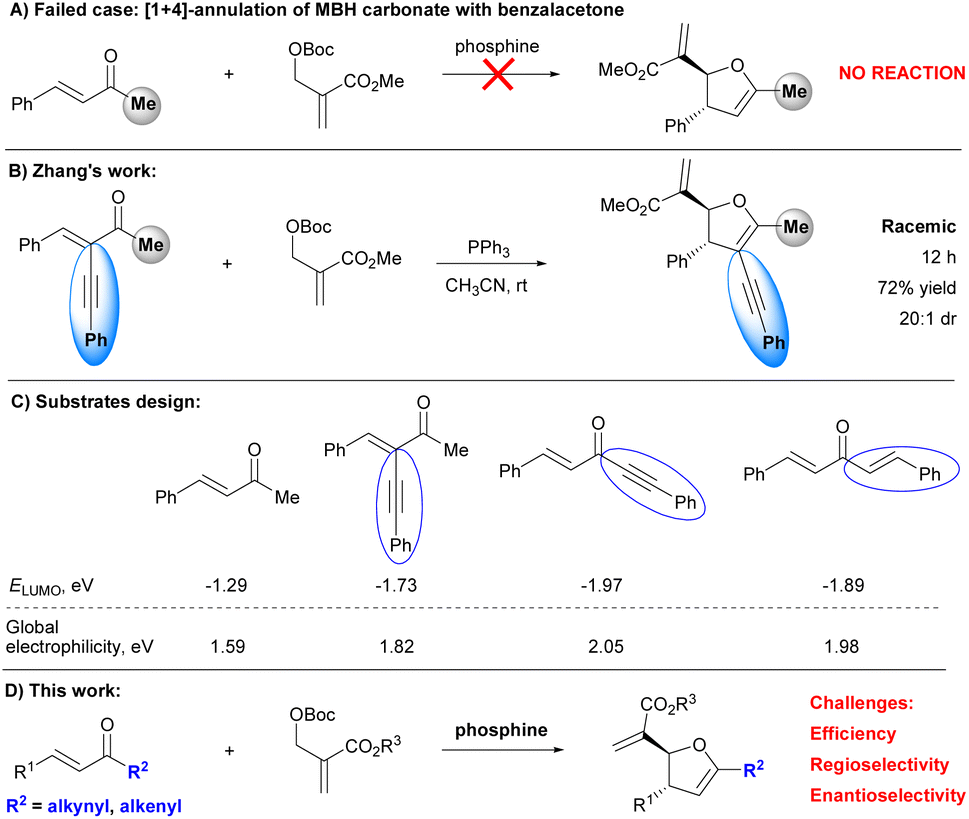
Since the seminal work from Lu's group,1 Morita–Baylis–Hillman (MBH) carbonates have emerged as a class of versatile building blocks in organocatalytic stereoselective annulations, providing a robust method for the asymmetric construction of functionalized heterocycles or carbocycles.2 However, in sharp contrast with well-established [3 + 2]-annulations, the organocatalytic enantioselective [1 + 4]-annulation of MBH carbonates remains underdeveloped.3–7 To enrich the chemistry of MBH carbonates as C1-synthons, we have successfully established organocatalytic asymmetric [1 + 4]-annulations of MBH carbonates with a series of electron-deficient olefins such as β,γ-unsaturated α-keto esters,8a chalcones,8ao-quinone methides,8b 2-enoylpyridines,8c α,β-unsaturated imines,8d 2-enoylpyridine N-oxides,8e and thiazolyl enones.8f However, we failed to achieve the organocatalytic enantioselective [1 + 4]-annulation of MBH carbonate with benzalacetone (Scheme 1A). Notably, Zhang et al., introduced an alkynyl group to the α-position of benzalacetone to improve the reactivity of the enone by lowering the energy of the lowest unoccupied molecular orbital (LUMO), successfully realizing a PPh3-mediated [1 + 4]-annulations of MBH carbonate with active enone (Scheme 1B).9 Although racemic 2,3-dihydrofurans were obtained, Zhang's work indicated that proper modification of enone enabled the occurrence of [1 + 4]-annulations with MBH carbonates. Accordingly, we hypothesized that lowering the LUMO energy and increasing the global electrophilicity of enones could lead to high reactivities. To this end, we modified the structure of benzalacetones, by respectively replacing methyl with alkynyl and alkenyl to furnish 1,5-diphenylpent-1-en-4-yn-3-one and 1,5-diphenylpenta-1,4-dien-3-one (Scheme 1C) and aimed to apply these enones in the organocatalytic stereoselective [1 + 4]-annulation of MBH carbonates (Scheme 1D).
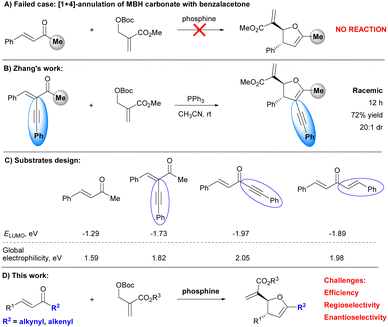 |
| | Scheme 1 Related [1 + 4]-annulations and substrates design. | |
Results and discussion
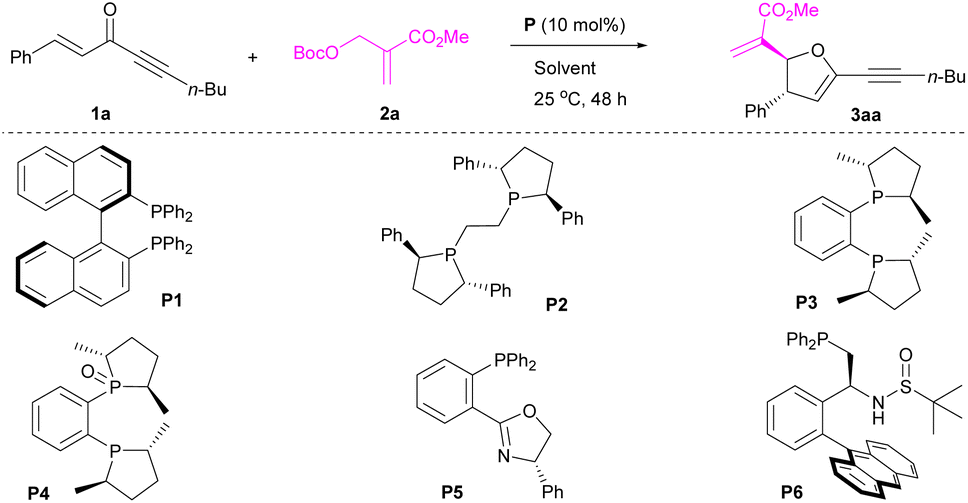
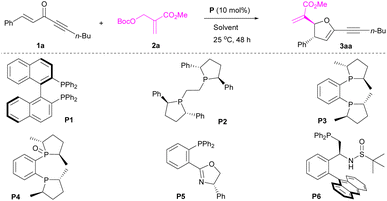
We started our investigation with the model reaction between 1,5-diphenylpent-1-en-4-yn-3-one 1a and MBH carbonate 2a (Table 1). It was confirmed that enone 1a could act as a four-atom synthon in [1 + 4]-annulation of MBH carbonate 2a, although initial survey resulted in poor results (Table 1, entry 1). Pleasingly, after further screening of chiral phosphines (Table 1, entries 2–6), both yield and enantioselectivity were essentially improved. Particularly, catalyst P4 containing a hemilabile ligand was identified as a suitable catalyst to afford the desired product 3aa in 72% yield with 88% ee and >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr (Table 1, entry 4).10 Compared with P3, P4, The examination of reaction media (Table 1, entries 7–10) disclosed that MeCN enabled the formation of product 3aa in 65% yield with 92% ee and >20:1 dr (Table 1, entry 10). Further adjusting reactant ratio (Table 1, entries 11–15) could improve the yield of product 3aa to 85% without comprising the stereoselectivity (Table 1, entry 14). Optimization of reaction time indicated that product 3aa could be obtained in 89% yield with 93% ee and >20:1 dr in 36 h (Table 1, entry 17).
:1 dr (Table 1, entry 4).10 Compared with P3, P4, The examination of reaction media (Table 1, entries 7–10) disclosed that MeCN enabled the formation of product 3aa in 65% yield with 92% ee and >20:1 dr (Table 1, entry 10). Further adjusting reactant ratio (Table 1, entries 11–15) could improve the yield of product 3aa to 85% without comprising the stereoselectivity (Table 1, entry 14). Optimization of reaction time indicated that product 3aa could be obtained in 89% yield with 93% ee and >20:1 dr in 36 h (Table 1, entry 17).
Table 1 Optimization for the reaction of enone 1a with MBH carbonate 2a
|
|
| Entrya |
P
|
Solvent |
Yieldb [%] |
eec [%] |
drd |
|
Unless noted, a mixture of 1a (0.10 mmol), 2a (0.10 mmol) and P (10.0 mol%) in the solvent (1.0 mL) was stirred at 25 °C for 48 h.
Isolated yield.
Determined by chiral-phase HPLC analysis.
Determined by 1H NMR analysis.
2a (0.15 mmol).
1a (0.15 mmol).
1a (0.20 mmol).
1a (0.30 mmol).
1a (0.40 mmol).
|
| 1 |
P1
|
CH2Cl2 |
3aa, 16 |
10 |
>20:1 |
| 2 |
P2
|
CH2Cl2 |
3aa, 13 |
12 |
>20:1 |
| 3 |
P3
|
CH2Cl2 |
3aa, 25 |
87 |
>20:1 |
| 4 |
P4
|
CH2Cl2 |
3aa, 72 |
88 |
>20:1 |
| 5 |
P5
|
CH2Cl2 |
3aa, 58 |
13 |
>20:1 |
| 6 |
P6
|
CH2Cl2 |
3aa, 13 |
44 |
>20:1 |
| 7 |
P4
|
EtOAc |
3aa, 42 |
80 |
>20:1 |
| 8 |
P4
|
Toluene |
3aa, 31 |
73 |
>20:1 |
| 9 |
P4
|
THF |
3aa, 34 |
77 |
>20:1 |
| 10 |
P4
|
MeCN |
3aa, 65 |
92 |
>20:1 |
| 11e |
P4
|
MeCN |
3aa, 54 |
94 |
>20:1 |
| 12f |
P4
|
MeCN |
3aa, 55 |
94 |
>20:1 |
| 13g |
P4
|
MeCN |
3aa, 75 |
94 |
>20:1 |
| 14h |
P4
|
MeCN |
3aa, 85 |
94 |
>20:1 |
| 15i |
P4
|
MeCN |
3aa, 85 |
94 |
>20:1 |
| 16h |
P4
|
MeCN (24 h) |
3aa, 72 |
93 |
>20:1 |
| 17h |
P4
|
MeCN (36 h) |
3aa, 89 |
93 |
>20:1 |
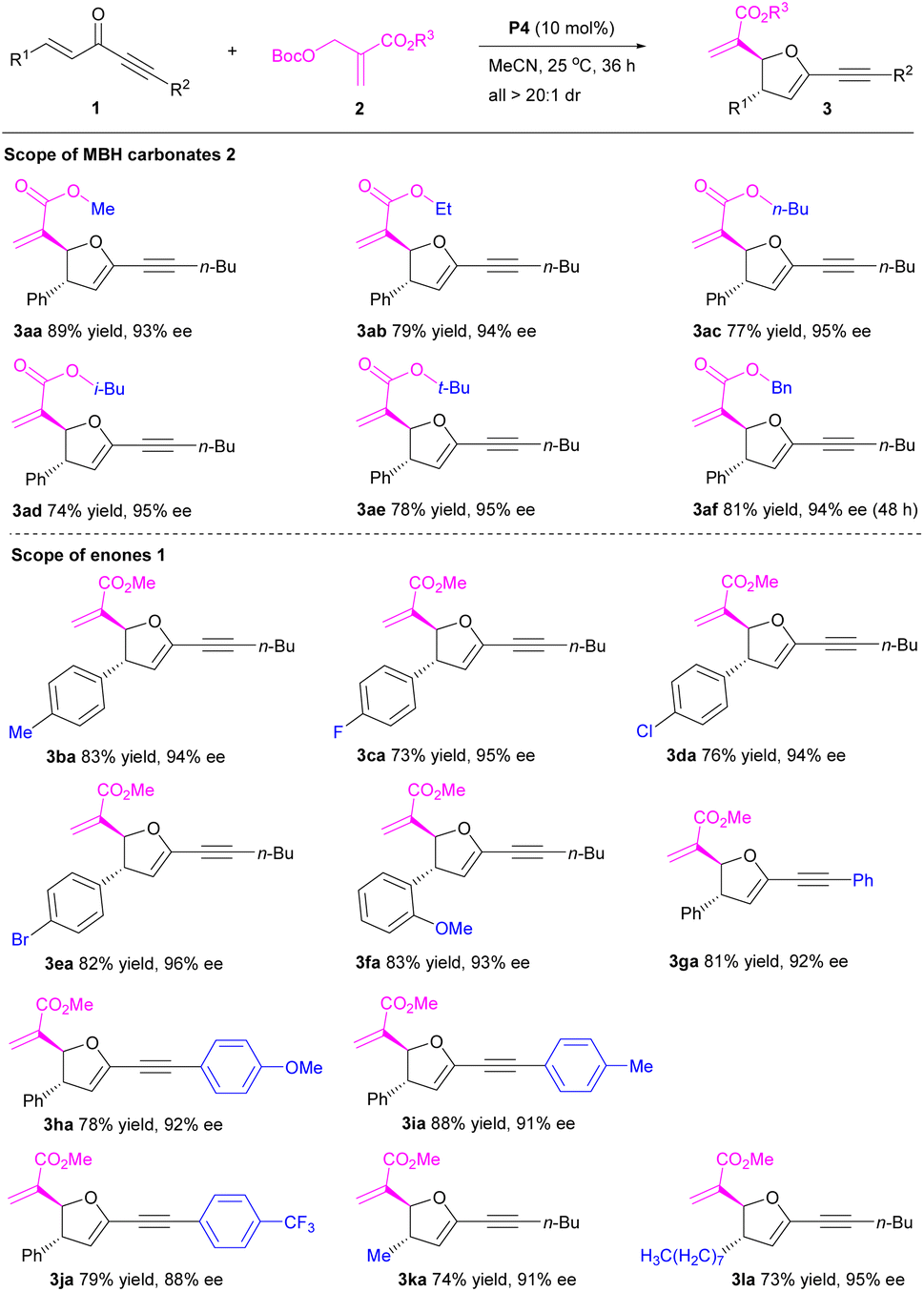
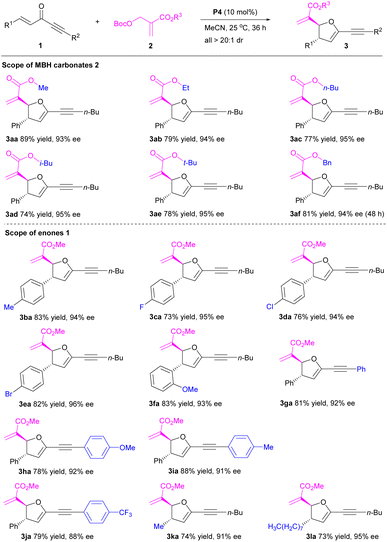
With the established optimal conditions in hand, we examined the scope of the P4-catalyzed [1 + 4]-annulation of MBH carbonates 2 with enones 1 (Table 2). Importantly, a series of MBH carbonates with different ester groups (R3) 2a–f reacted smoothly with enone 1a to afford the corresponding 2,3-dihydrofurans 3aa–af in generally high yields with excellent stereoselectivities (Table 2, entries 1–6). Obviously, these ester groups had a slight effect on the reaction. On the other hand, a range of enones 1 with different aromatic rings (R1/R2) were compatible, furnishing 2,3-dihydrofurans 3ba–ja in 73–88% yields with 88–96% ee and >20:1 dr (Table 2, entries 7–15). Furthermore, the reactions of enones with aliphatic groups (R1/R2) 1k–l also generated the desired 2,3-dihydrofurans 3ka–la in 73–74% yields with 91–95% ee and >20:1 dr (Table 2, entries 16–17). These encouraging data indicated that neither electronic effect nor steric hindrance had a significant influence on the reaction. Notably, the substrate scope of organocatalytic enantioselective [1 + 4]-annulation of MBH carbonates has been successfully extended to pent-1-en-4-yn-3-ones.
Table 2 Scope of reaction between enones 1 and MBH carbonates 2a
|
Reaction condition: a mixture of 1 (0.30 mmol), 2 (0.10 mmol) and P4 (10.0 mol%) in MeCN (1.0 mL) was stirred at 25 °C for 36 h. All dr > 20:1, determined by 1H NMR analysis. Products were obtained in isolated yields. The ee were determined by chiral-phase HPLC analysis.
|
|
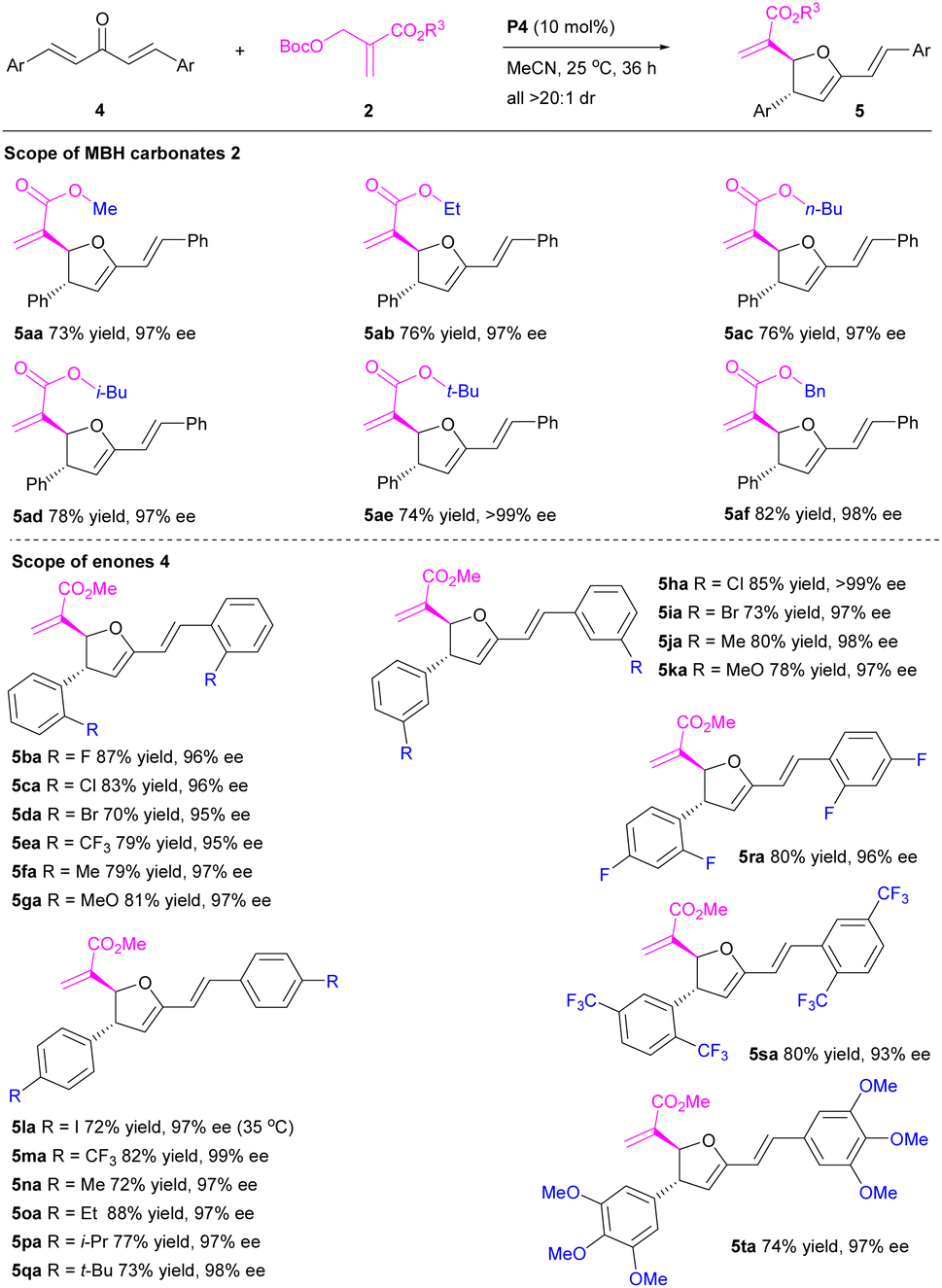
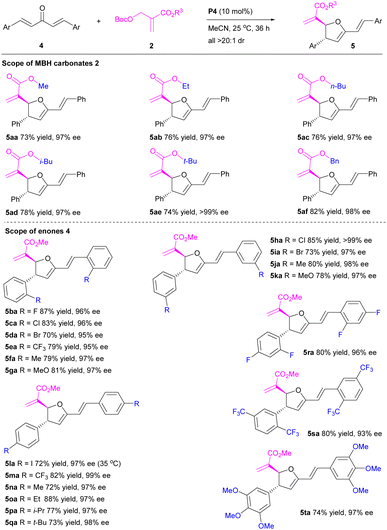
Subsequently, we turned our attention to the organocatalytic enantioselective [1 + 4]-annulation of MBH carbonates 2 with penta-1,4-dien-3-ones 4 under the established optimal conditions (Table 3). Gracefully, all the probed [1 + 4]-annulations proceeded smoothly to generate the desired 2,3-dihydrofurans in good to high yields with excellent diastereo- and enantioselectivities. In detail, the reactions of MBH carbonates with different ester groups (R3) 2a–f furnished the desired 2,3-dihydrofurans 5aa–af in 73–82% yields with 97–>99% ee and >20:1 dr. Notably, the effect of ester groups was negligible. Varied aromatic substrates (Ar) with either electron withdrawing groups or electron-donating groups 4b–q were all tolerated to afford the corresponding product 5ba–qa in 70–88% yields with 95–>99% ee and >20:1 dr. Particularly, [1 + 4]-annulations of the bulked enones 4r–t also proceeded very well, affording the desired products 5ra–ta in 74–80% yields with 93–97% ee and >20:1 dr. Neither essential steric-hindrance effect nor significant electronic effect was observed. Obviously, penta-1,4-dien-3-ones have been successfully applied in the organocatalytic enantioselective [1 + 4]-annulation of MBH carbonates.
Table 3 Scope of reaction between enones 4 and MBH carbonates 2a
|
Reaction condition: a mixture of 4 (0.30 mmol), 2 (0.10 mmol), P4 (10.0 mol%) in the MeCN (1.0 mL) was stirred at 25 °C for 36 h. All dr > 20:1, determined by 1H NMR analysis. Products were obtained in isolated yields. The ee were determined by chiral-phase HPLC analysis.
|
|
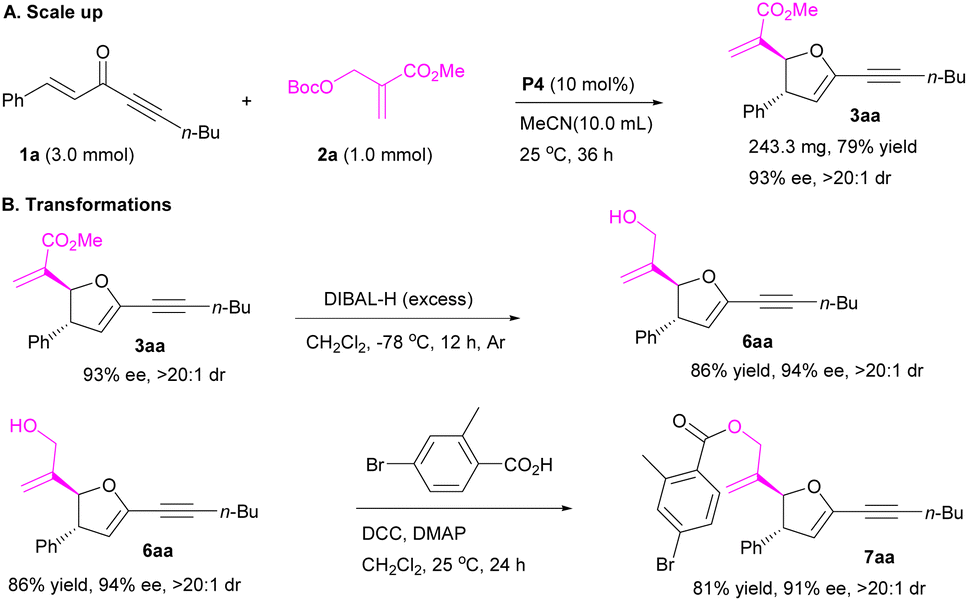
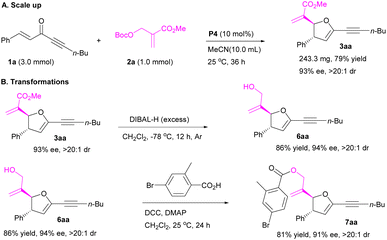
To further demonstrate the utility of the protocol, the [1 + 4]-annulation of MBH carbonate 2a with enone 1a was scaled up to 1.0 mmol, affording product 3aa in 79% yield with 93% ee and >20:1 dr (Scheme 2A). Selective reduction of ester group of 2,3-dihydrofuran 3aa afforded product 6aa bearing a hydroxyl group in 86% yield without losing stereoselectivity (Scheme 2B). Esterification of 6aa gave product 7aa in 81% yield (Scheme 2B).
 |
| | Scheme 2 Further investigations. | |
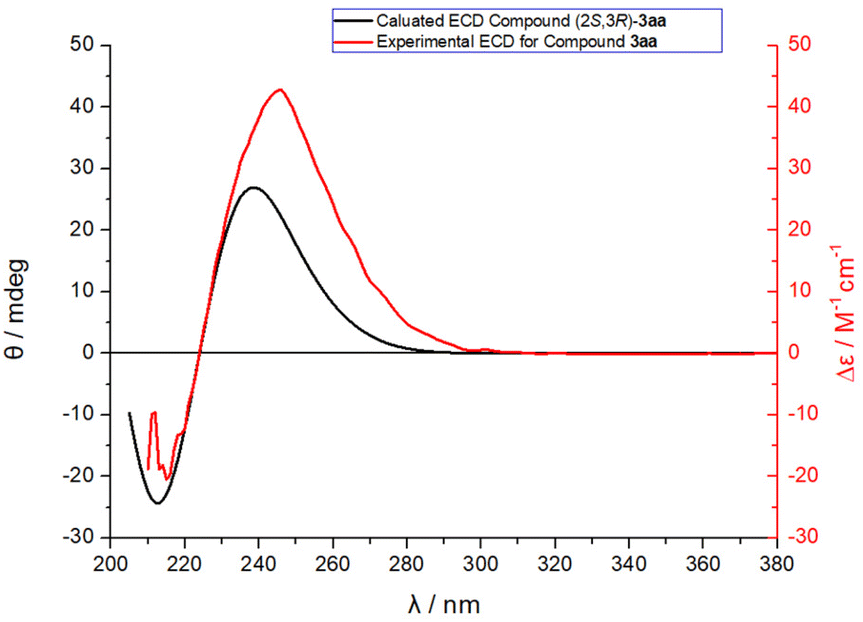
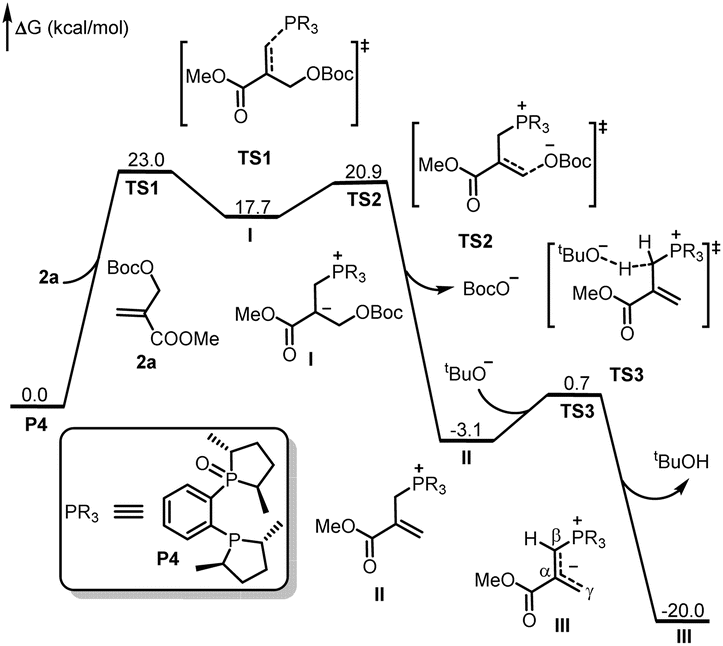
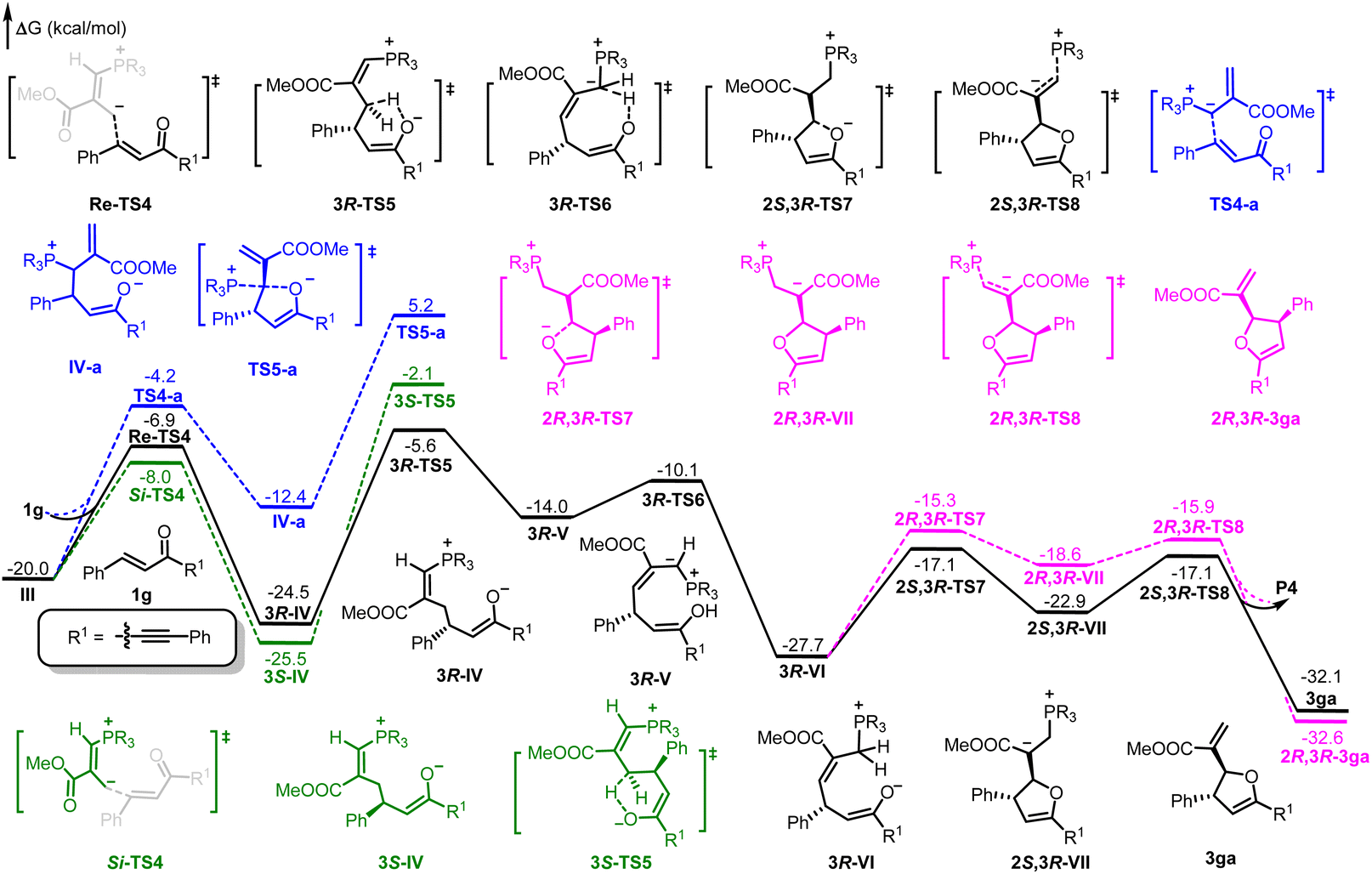
As shown in Fig. 1, the absolute configuration of chiral 2,3-dihydrofuran 3aa was determined by ECD (see ESI for details†), which was in accordance with our previous work.8a To further elucidate the mechanism and origin of stereoselectivity, we performed density functional theory (DFT) calculations based on the P4-mediated reaction of enone 1g with MBH carbonate 2a affording the major enantiomer 2,3-dihydrofuran (2S,3R)-3ga. As shown in Fig. 2, the reaction was initiated by nucleophilic addition of P4 to 2avia transition state TS1 with an overall energy barrier of 23.0 kcal mol−1 to generate zwitterionic intermediate I. The subsequent removing the BocO− anion through C–O bond cleavage delivered the phosphonium intermediate II associated with an energy barrier of 3.2 kcal mol−1, followed by deprotonation by in situ generated strong base t-BuO− anion via transition state TS3 with a very low energy barrier of 3.8 kcal mol−1 to give the key allylic phosphorus dipole intermediate III. Subsequently, the γ-selective Michael addition of intermediate III to enone 1g formed intermediate IV, which underwent an intramolecular 1,5-proton transfer to afford intermediate V (Fig. 3). After a systematic conformational search, we identified the most stable Michael addition transition states Re-TS4 and Si-TS4 and 1,5-proton transfer transition states 3R-TS5 and 3S-TS5, respectively (see SI for details). The calculated results indicated that the energy barrier of Michael addition via transition state Re-TS4 (ΔG‡ = 13.1 kcal mol−1) is slightly higher than that of transition state Si-TS4 (ΔG‡ = 12.0 kcal mol−1), but the energy barrier of 1,5-proton transfer via transition state 3R-TS5 (ΔG‡ = 18.9 kcal mol−1) is lower than that of 3S-TS5 (ΔG‡ = 23.4 kcal mol−1). Accordingly, the pathway associated with Re-face attack is more energetically favorable. Moreover, the free energy barrier of intramolecular 1,5-proton transfer (18.9 kcal mol−1) is lower than that assisted by water (28.5 kcal mol−1), which ruled out the water-assisted proton transfer process (see ESI for details†).11 Alternatively, the β-selective addition of intermediate III to enone 1g generated intermediate IV-avia transition state TS4-a with a relatively high free energy barrier. Moreover, the subsequent SN2 attack triggered ring-closing reaction furnished the desired product 3gavia transition state TS5-a with a high free energy barrier of 25.2 kcal mol−1 relative to intermediate III, which indicated the process was unfavorable. The intramolecular 1,7-proton transfer enabled the formation of intermediate 3R-VI from intermediate 3R-Vvia transition state 3R-TS6 with a barrier of 3.9 kcal mol−1. The preferred intramolecular nucleophilic addition proceeded via transition state 2S,3R-TS7 with a free energy barrier of 10.6 kcal mol−1 to afford five-member ring intermediate 2S,3R-VII, followed by the dissociation of catalyst P4 to give the final product 3gavia transition state 2S,3R-TS8 (ΔG‡ = 5.8 kcal mol−1).
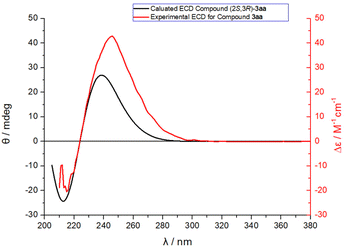 |
| | Fig. 1 Comparison of the calculated ECD of compound (2S,3R)-3aa with the experimental one of compound 3aa. | |
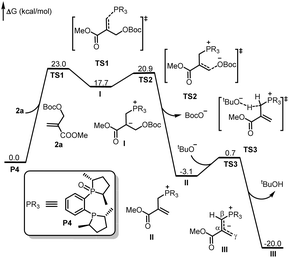 |
| | Fig. 2 DFT-computed free energy profile for P4 catalyzed the reaction of 2a. The calculations were performed via DFT/M06-2X method in acetonitrile, and the relative energies are given in kcal mol−1. | |
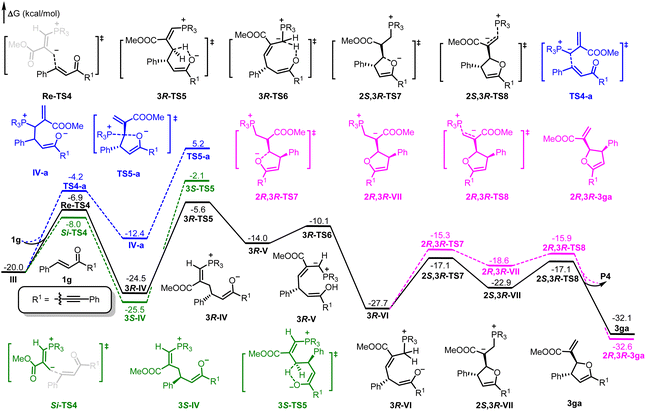 |
| | Fig. 3 DFT-computed free energy profile for P4 catalyzed the reaction of 2a with 1g. The calculations were performed via DFT/M06-2X method in acetonitrile, and the relative energies are given in kcal mol−1. | |
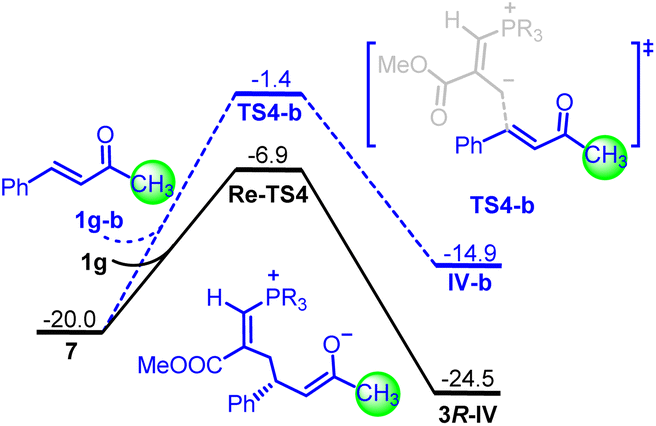
To corroborate the substituent effects from computed LUMO energies and nucleophilicity index (Scheme 1C), the barriers for two different enones were also compared (Fig. 4). In striking contrast, the energy barrier of transition state TS4-b for the reaction of intermediate III with benzalacetone 1g–b was 5.5 kcal mol−1 higher than that of transition state Re-TS4 for the reaction between intermediate III and enone 1g. Furthermore, the Gibbs free energy of the enolate intermediate IV-b was 9.6 kcal mol−1 higher than that of the intermediate 3R-IV. These results indicated that the P4-catalyzed [1 + 4] annulation reaction of benzalacetone was energetically unfavorable in terms of both kinetics and thermodynamics. Accordingly, the introduction of either alkynyl or alkenyl group into enones not only improved the reactivity but also stabilized the corresponding enolate anion intermediate.
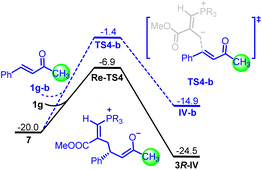 |
| | Fig. 4 DFT-computed free energy profile for P4 catalyzed the reaction of intermediate III with 1g and 1g–b. The calculations were performed via DFT/M06-2X method in acetonitrile, and the relative energies are given in kcal mol−1. | |
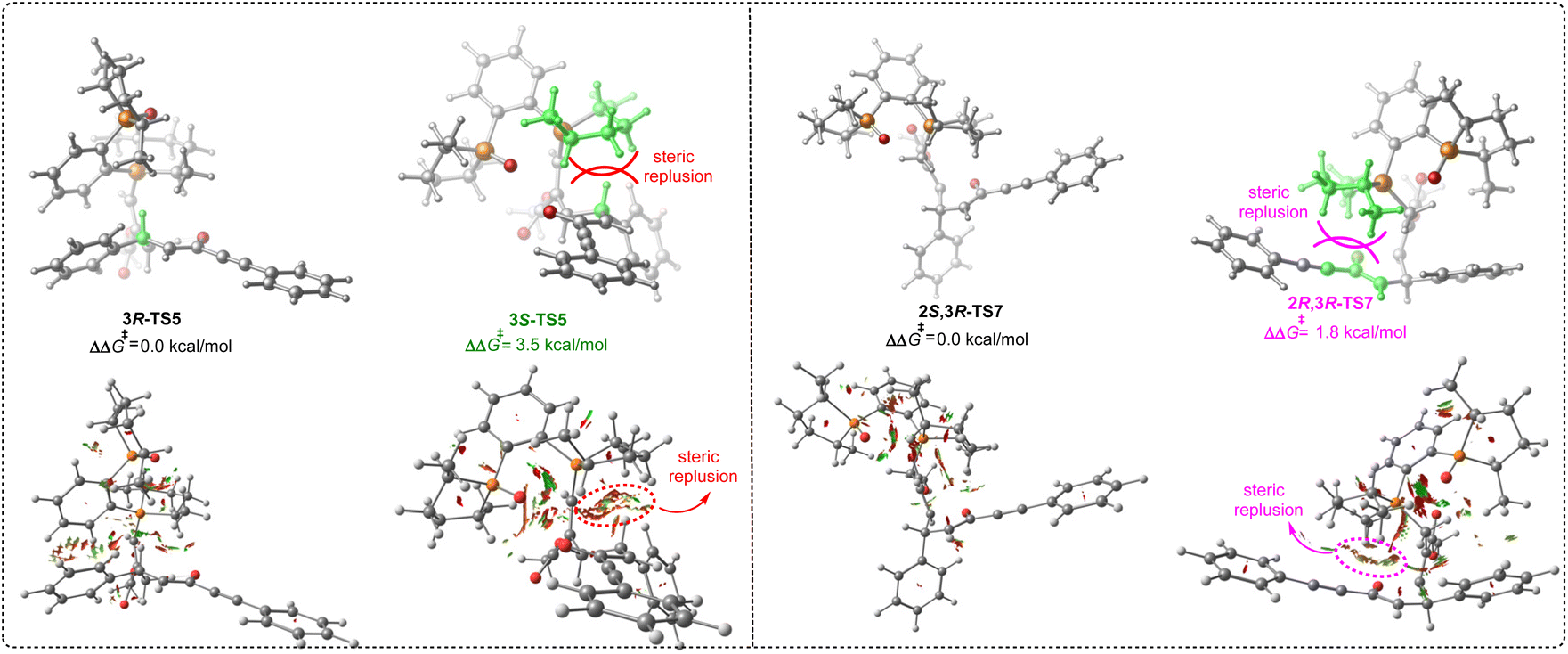
We then turned attention to disclosing the origin of stereoselectivity. As discussed above, the intramolecular 1,5-proton transfer controlled the stereoselectivity of intermediate IV. Accordingly, the noncovalent interaction (NCI) analysis for the 1,5-proton transfer transition states 3R-TS5 and 3S-TS5 was carried out (Fig. 5). Notably, there is a significant steric hindrance between hydrogen atom of the reactant and the 2,5-dimethylphospholane group of the phosphine catalyst P4 in transition state 3S-TS5, while there was a less steric repulsion in transition state 3R-TS5. On the other hand, the intramolecular nucleophilic addition process dominated the stereoselectivity of intermediate VII. Similarly, there was a significant steric hindrance between the enolate group of the reactant and the 2,5-dimethylphospholane group of the phosphine catalyst in transition state 2R,3R-TS7 and a less steric repulsion in transition state 2S,3R-TS7. The NCI analysis revealed that the stereoselectivity was mainly controlled by the steric effect between reactant and 2,5-dimethylphospholane group of the phosphine catalyst, suggesting that chiral phosphine catalyst with bulky substituent is important for controlling stereoselectivity.
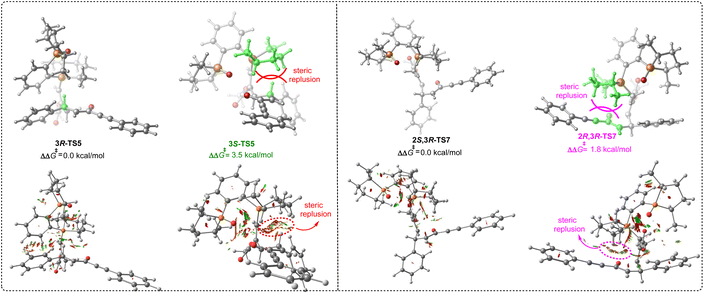 |
| | Fig. 5 The noncovalent interaction (NCI) analysis of 3R-TS5, 3S-TS5, 2S,3R-TS7 and 2S,3R-TS7. | |
Conclusions
In conclusion, we have successfully developed chiral phosphine-catalyzed regio- and enantioselective [1 + 4]-annulations of MBH carbonates with pent-1-en-4-yn-3-ones and penta-1,4-dien-3-ones based on the frontier molecular orbital theory. Compared with Zhang's work, two types of modified enones were independently developed, and applied in the stereoselective [1 + 4]-annulations. This work further explored the substrate scope of organocatalytic asymmetric [1 + 4]-annulations of MBH carbonates and enriched the chemistry of enantioenriched multifunctional 2,3-dihydrofurans.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the financial support from National Natural Science Foundation of China (21871128), Guangdong Innovative Program (2019BT02Y335), and the Guangdong Provincial Key Laboratory of Catalysis (2020B121201002). The authors acknowledge the assistance of SUSTech Core Research Facilities, Xiaoyong Chang (X-ray), Yang Yu (HRMS). Computational work was supported by Center for Computational Science and Engineering at SUSTech, and the CHEM high-performance supercomputer cluster (CHEM-HPC) located at the Department of Chemistry, SUSTech.
References
- Y. Du, X. Lu and C. Zhang, A Catalytic Carbon–Phosphorus Ylide Reaction: Phosphane-Catalyzed Annulation of Allylic Compounds with Electron-Deficient Alkenes, Angew. Chem., Int. Ed., 2003, 42, 1035–1037 CrossRef CAS.
- For selected reviews, see:
(a) L.-W. Ye, J. Zhou and Y. Tang, Phosphine-triggered Synthesis of Functionalized Cyclic Compounds, Chem. Soc. Rev., 2008, 37, 1140–1152 RSC;
(b) T.-Y. Liu, M. Xie and Y.-C. Chen, Organocatalytic Asymmetric Transformations of Modified Morita–Baylis–Hillman Adducts, Chem. Soc. Rev., 2012, 41, 4101–4112 RSC;
(c) Y. C. Fan and O. Kwon, Advances in Nucleophilic Phosphine Catalysis of Alkenes, Allenes, Alkynes, and MBHADs, Chem. Commun., 2013, 49, 11588–11619 RSC;
(d) Y. Wei and M. Shi, Recent Advances in Organocatalytic Asymmetric Morita-Baylis-Hillman/aza-Morita-Baylis-Hillman Reactions, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS;
(e) P. Xie and Y. Huang, Morita-Baylis-Hillman Adduct Derivatives (MBHADs): Versatile Reactivity in Lewis Base-promoted Annulation, Org. Biomol. Chem., 2015, 13, 8578–8595 RSC;
(f) T. Wang, X. Han, F. Zhong, W. Yao and Y. Lu, Amino Acid-Derived Bifunctional Phosphines for Enantioselec-tive Transformations, Acc. Chem. Res., 2016, 49, 1369–1378 CrossRef CAS;
(g) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Phosphine Organocatalysis, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed;
(h) N.-J. Zhong, Y.-Z. Wang, L. Cheng, D. Wang and L. Liu, Recent Advances in the Annulation of Morita-Baylis-Hillman Adducts, Org. Biomol. Chem., 2018, 16, 5214–5227 RSC;
(i) Z.-C. Chen, Z. Chen, W. Du and Y.-C. Chen, Transformations of Modified Morita-Baylis-Hillman Adducts from Isatins Catalyzed by Lewis Bases, Chem. Rec., 2020, 20, 541–555 CrossRef CAS PubMed.
-
(a) X.-N. Zhang, H.-P. Deng, L. Huang, Y. Wei and M. Shi, Phosphine-catalyzed Asymmetric [4 + 1] Annulation of Morita-Baylis-Hillman Carbonates with Dicyano-2-methylenebut-3-enoates, Chem. Commun., 2012, 48, 8664–8666 RSC;
(b) F.-L. Hu, Y. Wei and M. Shi, Phosphine-catalyzed Asymmetric [4 + 1] Annulation of Activated α,β-Unsaturated Ketones with Morita-Baylis-Hillman Carbonates: Enantioselective Synthesis of Spirooxindoles Containing Two Adjacent Quaternary Stereocenters, Chem. Commun., 2014, 50, 8912–8914 RSC.
-
(a) H. Li, J. Luo, B. Li, X. Yi and Z. He, Enantioselective [4 + 1]-Annulation of α,β-Unsaturated Imines with Allylic Carbonates Catalyzed by a Hybrid P-Chiral Phos-phine Oxide-Phosphine, Org. Lett., 2017, 19, 5637–5640 CrossRef CAS PubMed;
(b) J. Luo, R. Chen, X. Fan, J. Gong, J. Han and Z. He, Catalytic Asymmetric (4 + 1) Annulation of Nitroalkenes with Allylic Acetates: Stereoselective Synthesis of Isoxazoline N-Oxides, Org. Biomol. Chem., 2019, 17, 6989–6993 RSC;
(c) H. Li, B. Li, J. Zeng, C. Jiang and Z. He, Enantiopure Chiral Phosphines Bearing a Sulfinyl Group and their Application in Catalytic Enantiodivergent Synthesis of Pol-ysubstituted Pyrrolines, Adv. Synth. Catal., 2020, 362, 2760–2766 CrossRef CAS;
(d) H. Li and Z. He, Chiral Phosphine-catalyzed Asymmetric [4 + 1] Annulation of Polar Dienes with Allylic Derivatives: Enantioselective Synthesis of Substituted Cyclopentenes, Tetrahedron Lett., 2021, 67, 152863 CrossRef CAS.
- G. Zhan, M.-L. Shi, Q. He, W.-J. Lin, Q. Ouyang, W. Du and Y.-C. Chen, Catalyst-Controlled Switch in Chemo- and Diastereoselectivities: Annulations of Morita–Baylis–Hillman Carbonates from Isatins, Angew. Chem., Int. Ed., 2016, 55, 2147–2151 CrossRef CAS.
- F. Jiang, G.-Z. Luo, Z.-Q. Zhu, C.-S. Wang, G.-J. Mei and F. Shi, Application of Naphthylindole-Derived Phosphines as Organo-catalysts in [4 + 1] Cyclizations of o-Quinone Methides with Morita-Baylis-Hillman Carbonates, J. Org. Chem., 2018, 83, 10060–10069 CrossRef CAS PubMed.
- T. Zhou, T. Xia, Z. Liu, L. Liu and J. Zhang, Asymmetric Phosphine-Catalyzed [4 + 1] Annulations of o-Quinone Methides with MBH Carbonates, Adv. Synth. Catal., 2018, 360, 4475–4479 CrossRef CAS.
-
(a) Y. Chen, Y. Han and P. Li, Organocatalytic Enantioselective [1 + 4] Annulation of Morita-Baylis-Hillman Carbonates with Electron-Deficient Olefins: Ac-cess to Chiral 2,3-Dihydrofuran Derivatives, Org. Lett., 2017, 19, 4774–4777 CrossRef;
(b) Y. Cheng, Z. Fang, W. Li and P. Li, Phosphine-mediated Enantioselective [4 + 1] Annulations Between ortho-Quinone Methides and Morita-Baylis-Hillman Car-bonates, Org. Chem. Front., 2018, 5, 2728–2733 RSC;
(c) T. Wang, P. Zhang, W. Li and P. Li, Phosphine-mediated Enantioselective [1 + 4]-Annulation of Morita-Baylis-Hillman Carbonates With 2-Enoylpyridines, RSC Adv., 2018, 8, 41620–41623 RSC;
(d) C. Qian, P. Zhang, W. Li and P. Li, Phosphine-Catalyzed Enantioselective [1 + 4] Annulation of Mori-ta-Baylis-Hillman Carbonates with α,β-Unsaturated Imines, Asian J. Org. Chem., 2019, 8, 242–245 CAS;
(e) P. Zhang, X. Guo, C. Liu, W. Li and P. Li, Enantioselective Construction of Pyridine N-Oxides Featuring 2,3-Dihydrofuran Motifs via Phosphine-Catalyzed [4 + 1]-Annulation of 2-Enoylpyridine N-Oxides with Morita-Baylis-Hillman Carbonates, Org. Lett., 2019, 21, 152–155 CrossRef CAS;
(f) C. Liu, P. Zhang and P. Li, Asymmetric Organocatalytic Enantioselective [1 + 4]-Annulation of Morita-Baylis-Hillman Carbonates with Thiazolyl Enones for Assembling of Dihydrofurans Featuring Thiazole Skeleton, Chem. J. Chin. Univ., 2020, 41, 2272–2278 CAS.
- Z. Chen and J. Zhang, An Unexpected Phosphine-Catalyzed Regio- and Diastereoselec-tive [4 + 1] Annulation Reaction of Modified Allylic Compounds with Activated Enones, Chem. – Asian J., 2010, 5, 1542–1545 CrossRef CAS PubMed.
- A. A. Boezio, J. Pytkowicz, A. Côté and A. B. Charette, Asymmetric, Catalytic Synthesis of α-Chiral Amines Using a Novel Bis(phosphine) Monoxide Chiral Ligand, J. Am. Chem. Soc., 2003, 125, 14260–14261 CrossRef CAS PubMed.
- Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z.-X. Yu, An Unexpected Role of a Trace Amount of Water in Catalyzing Proton Transfer in Phosphine-Catalyzed (3 + 2) Cycloaddition of Allenoates and Alkenes, J. Am. Chem. Soc., 2007, 129, 3470–3471 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, spectroscopic data. See DOI: https://doi.org/10.1039/d2qo01670b |
| ‡ These authors contributed equally. |
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b,
Peiyuan
Yu
*b,
Peiyuan
Yu