DOI:
10.1039/D2QO01582J
(Review Article)
Org. Chem. Front., 2023,
10, 216-236
Visible light metallaphotoredox catalysis in the late-stage functionalization of pharmaceutically potent compounds
Received
7th October 2022
, Accepted 4th November 2022
First published on 8th November 2022
Abstract
The late stage functionalization (LSF) of complex biorelevant chemicals is a valuable approach for accelerating the discovery of structure–activity relationships (SARs) and optimising ADME (absorption, distribution, metabolism, and excretion) profiles. To this end, visible-light metallaphotocatalysis provides a unique opportunity to develop smooth and clean drug functionalization by realising site-specific reactivities under very mild reaction conditions. This study provides a comprehensive evaluation of the current research, highlighting recent advances in the discipline while emphasising predicted future advancements and prospective applications along with visible-light metallaphotocatalytic synthesis methods for the LSF of medicines and pharmaceutically potent compounds that are now accessible.
 Praveen P. Singh | Praveen P. Singh is working as Assistant Professor in the Department of Chemistry, United College of Engineering and Research, Prayagraj, India. He obtained his B.Sc. degree and M.Sc. degree in organic chemistry from T. D. P. G. College (V. B. S. Purvanchal University), Jaunpur and D.Phil. from the Department of Chemistry, University of Allahabad, India. His current research interests include the development of synthetic receptors for the recognition of biological target structures and the application of visible light chemical photocatalysis towards organic synthesis and nanophotocatalysis. |
 Pravin K. Singh | Pravin K. Singh is working as Assistant Professor in the Department of Chemistry, C.M.P. College (constituent P.G. college of Central University of Allahabad), Prayagraj, India. Dr Singh is actively engaged in advanced research work for the development of environmentally benign, new synthetic routes for various bioactive heterocyclic compounds. He obtained his B.Sc., M.Sc., doctorate (D.Phil.) and post-doctorate (D.Sc.) degrees from the University of Allahabad, India. |
 Vishal Srivastava | Vishal Srivastava is working as Assistant Professor in the Department of Chemistry, C.M.P. College (constituent P.G. college of Central University of Allahabad), Prayagraj, India. He obtained his B.Sc., M.Sc. in organic chemistry and doctoral degree (D.Phil.) from the Department of Chemistry, University of Allahabad, India. His current research work involves the designing of novel biologically active photoredox catalysed synthesis of organic compounds. |
1. Introduction
Visible-light-driven photochemistry is well adapted to explore biological environments as many common and photoredox catalysts absorb light at longer wavelengths than the biomolecules found within biological systems.1,2 The Nobel Prizes in 2001 (to Knowles, Noyori, and Sharpless for stereoselective catalysis),3 2005 (to Chauvin, Grubbs, and Schrock for olefin metathesis),4 and 2010 (to Heck, Negishi, and Suzuki for palladium-catalyzed cross-coupling)5 demonstrate the importance of transition metal catalysis in science and society. These techniques all have one thing in common that they potentially permit the rapid synthesis of important organic molecules from relatively simple ingredients, although their development required several years of research. More importantly, transition metal chemistry has consistently produced new forms of chemical activation that permit bond-forming or bond-breaking routes that are frequently unprecedented within organic chemistry. Transition metal catalysis has been shown to be a useful platform for organic synthesis and, more broadly, the synthesis of organic compounds for societal benefit due to its extraordinary capacity for ‘novel reactivity’.
Synthetic chemists have always been interested in selective functionalization of similar and chemically comparable chemical bonds.6 The addition of transition-metal catalysts in this case normalises the pre-existing barrier, allowing selective bond formation to occur. A properly designed directing group can coordinate with the transition metal to generate an intermediate, allowing for effective functionalization of the target chemical bond.7 Under mild conditions, metallaphotocatalysis enables selective carbon–heteroatom and carbon–carbon cross-coupling reactions.8 Modulation of the oxidation state of transition metal complexes by SET reactions, radical addition, or photosensitization is crucial to success. This strategy has been extensively investigated using nickel complexes9 and has since been extended to a variety of other transition metals10,11 including first row metals like cobalt,12 copper,13 and iron.14,15 Several 4d transition metals (Pd, Rh, Ru) as well as 3d transition metals (Cu, Co, Ni, Fe) have been applied for accomplishing ortho-C–H functionalization.16–18 Many studies on ortho-C–H arylation catalysed by transition metals have been published, and they have already been discussed in previous reviews.19,20a Transition metal-catalyzed processes in conjugation with photocatalysis are changing the way in which chemists approach the building of organic compounds.20b–d More recently, the synthetic community has focused on the application of photoredox catalysis in the field of transition metal catalysis, which has resulted in the invention of a number of valuable novel C–C and C–heteroatom bond-forming protocols.21–23
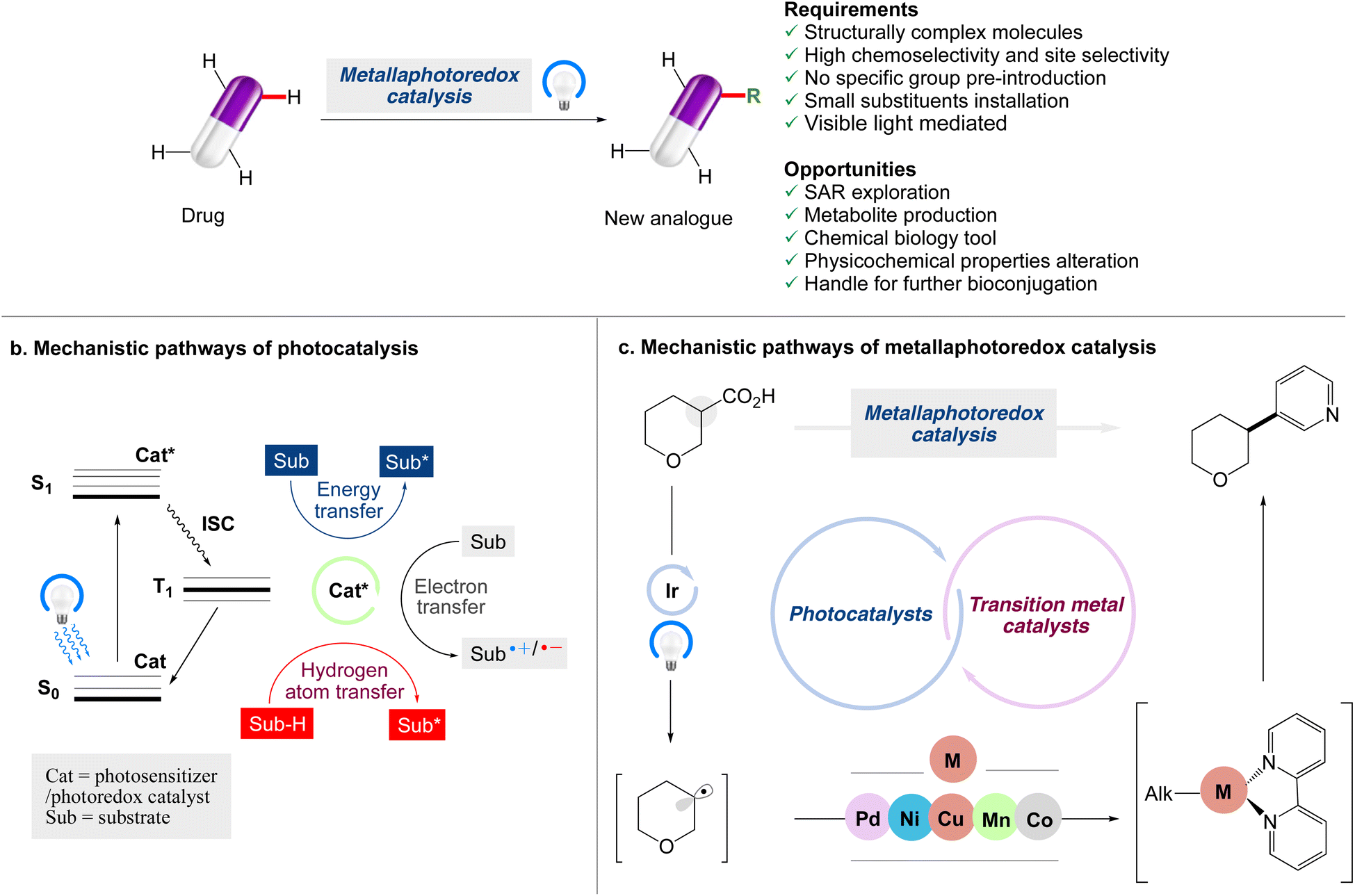
Although visible light photocatalytic synthesis is a relatively new discipline of organic chemistry, it has quickly become an important tool in the arsenal of synthetic chemists. The benefits of visible light photocatalysis have led to a variety of medicinal chemistry applications, such as drug discovery, bioconjugation, late-stage C–H functionalization, and isotopic labelling.24,25 The concept of employing C–H bond functionalization to make mimics of complex chemical structures has been identified as a game-changing platform.26–30 The emergence of late-stage functionalization (LSF) techniques has given the landscape of molecular synthesis a huge boost over the last decade. Medicinal chemists have been able to include LSF tactics into their drug discovery programmes by the implementation of a variety of new methodologies that have been effectively applied to the late stage functionalization of drug molecules (Scheme 1).
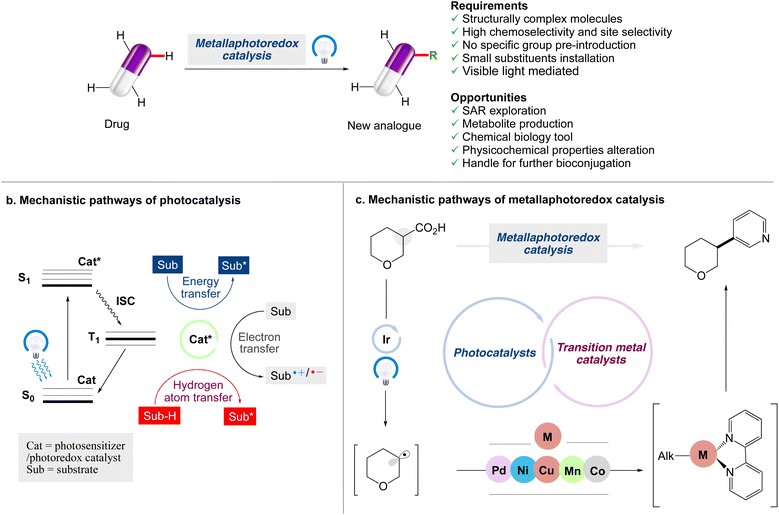 |
| | Scheme 1 Opportunities for modern chemistry to enable late-stage functionalization strategies in drug discovery. (a) Main reaction manifolds enabling LSF in drug discovery, along with the requirements and opportunities of such strategies. (b) Jablonski diagram highlighting mechanisms of photoinduced energy transfer, electron transfer and hydrogen atom transfer pathways. (c) Mechanistic pathways of metallaphotoredox catalysis. | |
However, important improvements in organic synthesis have only subsequently made the late-stage functionalization (LSF) of complex scaffolds possible, particularly as a result of the advent of chemoselective methods with extensive functional group tolerance.31–35 The term ‘LSF’ is now frequently applied in the field of molecular chemistry36 as a result of the advent of such techniques and other types of methodological approaches, such as metallaphotoredox-catalyzed cross-couplings37,38 that can be considered for LSF as well. As the small molecule synthesis relies heavily on metallaphotocatalysis, which is at the forefront of light-mediated processes, and in continuation of our work on photocatalysed organic synthesis,39,40 in this review article, the recent applications of visible light induced metallaphotoredox catalysed trends for the late stage functionalization of organic compounds via different modes of chemical bond formation are reviewed. This review might be particularly useful for those who are already involved in the area of organic synthesis to carry out visible light mediated photocatalysed reactions by the application of metallaphotocatalysis.
2. Nickel metallaphotocatalysis
In recent years, a variety of valuable transformations for gaining access to important organic compounds have been realised, with the help of photoredox and nickel dual catalysis. Due to the availability of nickel catalysts in a variety of its oxidation states, photoredox and nickel dual catalysis have been applied to the deoxygenative arylation of alcohols, synthesis of substituted purine nucleoside analogues, aminoalkylation of aryl halides, carboarylation of alkenes, cross electrophile coupling and cross coupling arylation reactions.
2.1. Metallaphotoredox-enabled deoxygenative arylation of alcohols
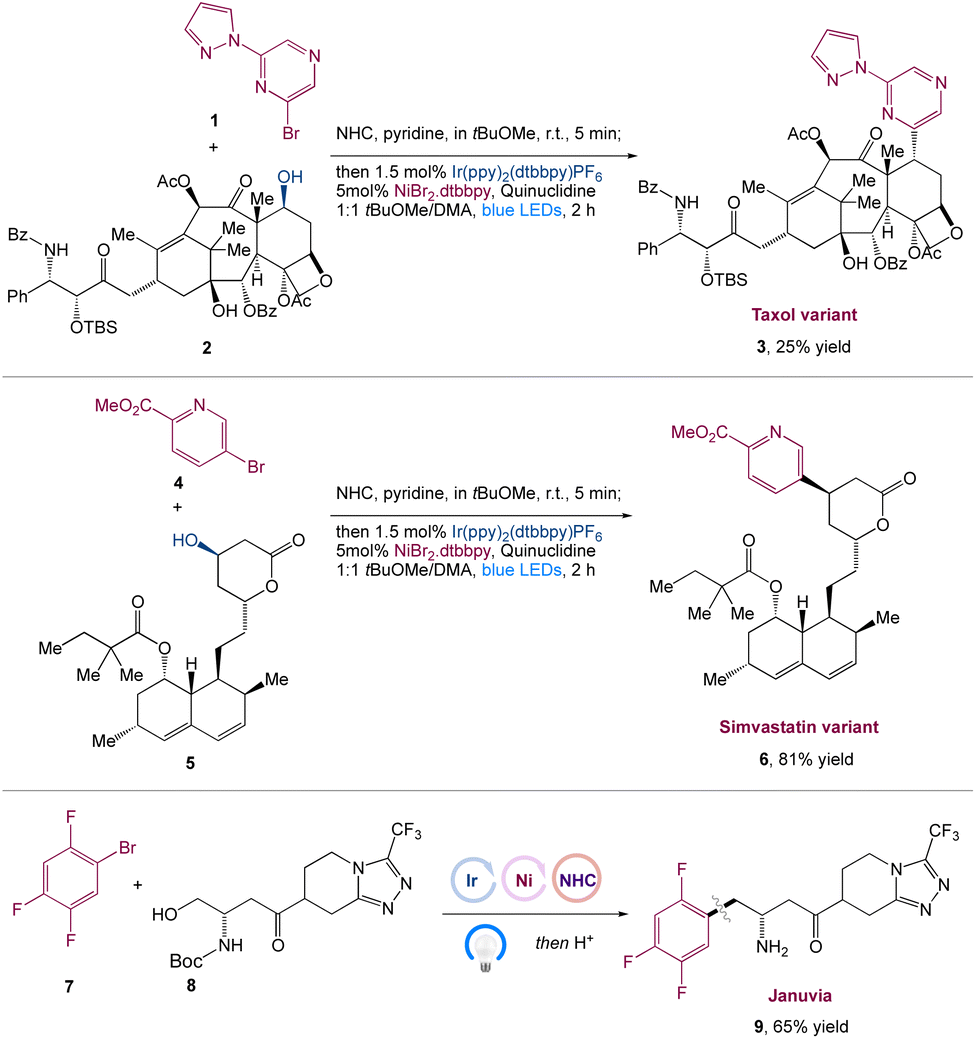
MacMillan et al.41 reported the in situ activation of free alcohols by N-heterocyclic carbene salts for carbon–carbon bond synthesis with aryl halide coupling partners in a metallaphotoredox-based cross-coupling platform. This approach is mild, robust, selective, and can accommodate a wide range of primary, secondary, and tertiary alcohols, as well as pharmaceutically relevant aryl and heteroaryl bromides and chlorides. This transformation's power had been proved in a number of complex scenarios, including the late-stage functionalization of Taxol 3 and a modular synthesis of the antidiabetic drug Januvia 9. This method is a generic approach for combining in situ alcohol activation with transition metal catalysis (Scheme 2).
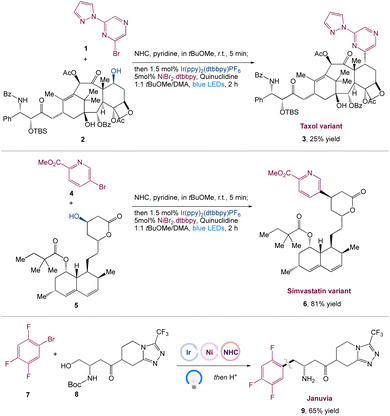 |
| | Scheme 2 Synthesis of Januvia and its variants, as well as to the late-stage functionalization of pharmaceutical variants, such as Taxol and Simvastatin.41 | |
2.2. Synthesis of C6-substituted purine nucleoside analogues
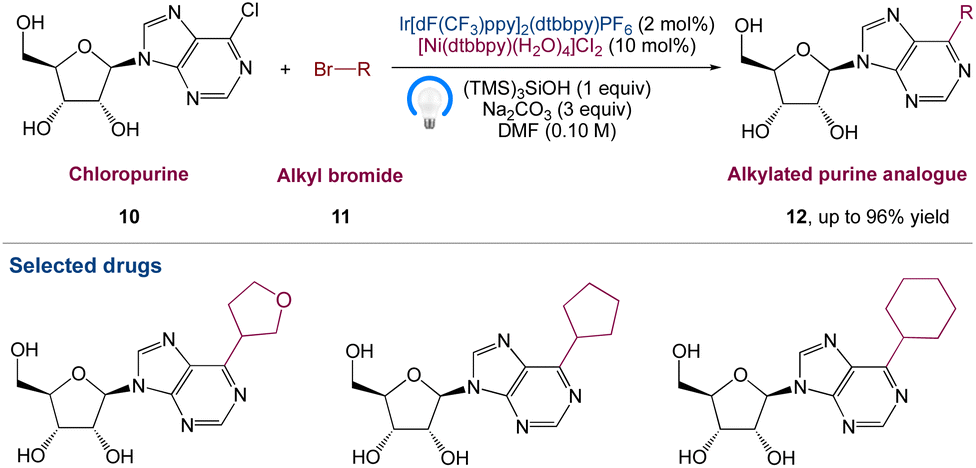
Nucleoside analogues have always been and will continue to be crucial chemicals in medicinal development. Under optimum reaction conditions, Marrouni et al.42 proposed a technique for the synthesis of C6-alkylated purine analogues by metallaphotoredox catalysed sp2–sp3 cross-electrophile coupling of chloropurine 10 with a wide range of readily available primary and secondary alkyl bromides 11. The unprotected nucleosides functionalized efficiently, yielding products 12, allowing for diversification at the end of the synthesis if needed. They anticipated that by using this method, they would be able to speed up SAR investigation in drug development and it would be particularly useful in concurrent medicinal chemistry endeavours (Scheme 3).
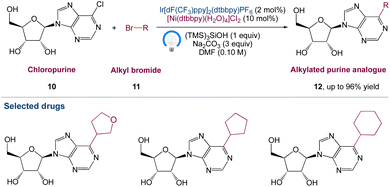 |
| | Scheme 3 Metallaphotoredox catalysed synthesis of C6-substituted purine nucleoside analogues.42 | |
2.3. Aminoalkylation of aryl halides
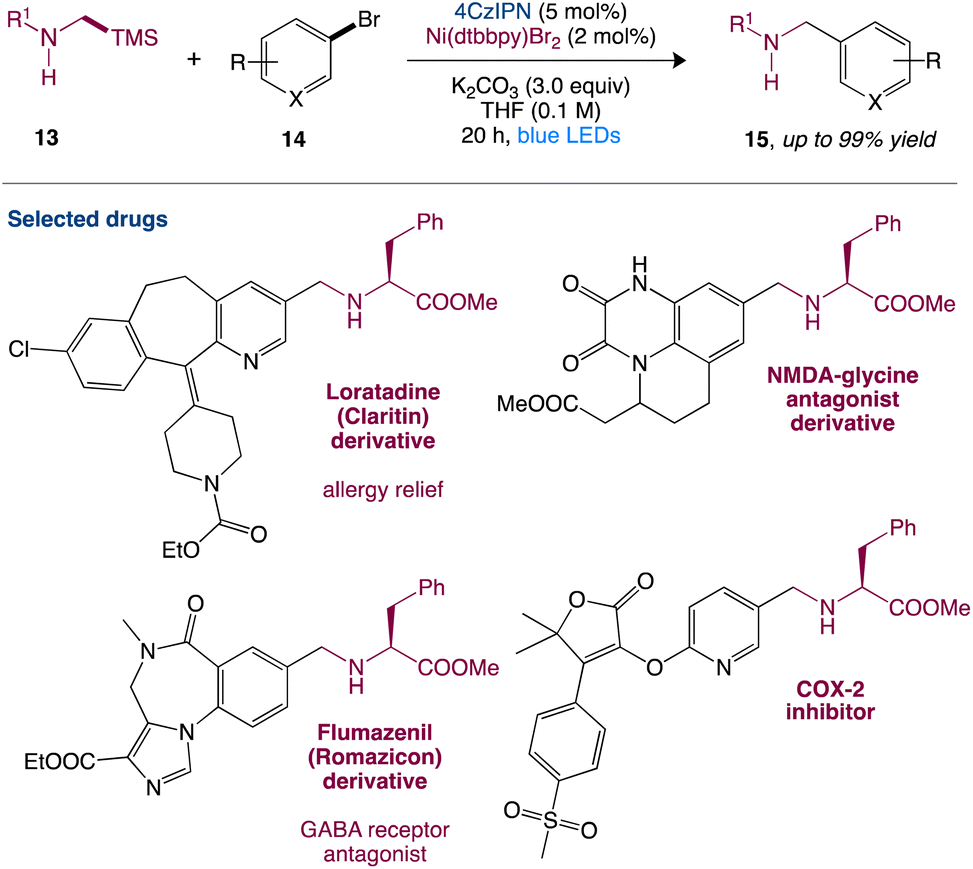
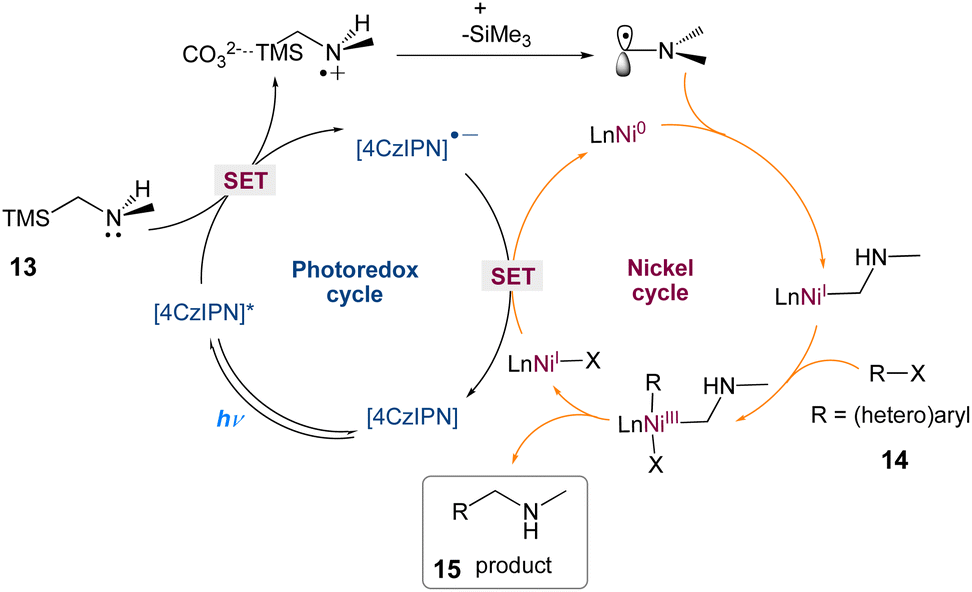
In nickel-mediated C–C coupling, Molander et al.43 reported a general aminoalkylation of aryl halides, overcoming the intolerance of free amines. This transformation has a high efficiency and a wide functional group tolerance. Amino radicals are created regioselectively using the quick desilylation of silylamines assisted by a carbonate during single-electron transfer (SET), which then participate in nickel-mediated C–C coupling (Scheme 4). This reaction has high chemoselectivity towards the production of C–C over C–N bond formation. Pharmacophores and peptides with high functionalization can also be used.
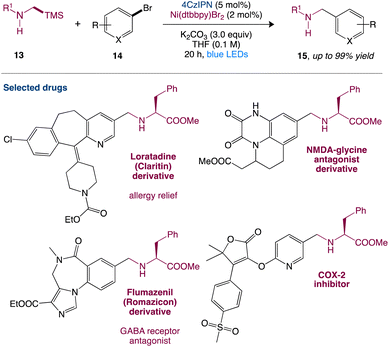 |
| | Scheme 4 Metallaphotoredox catalysed aminoalkylation of aryl halides.43 | |
In this mechanism postulated by the authors, a dual photoredox/nickel-catalyzed process is proposed (Scheme 5). The excitation of 4CzIPN is enabled by blue LED irradiation, which then oxidises α-silylamine via SET. The facile α-desilylation facilitated by a carbonate promotes the formation of an α-amino radical, which could then be intercepted by Ni(0), yielding an alkyl-Ni(I) species, with subsequent oxidative addition of an aryl halide. The Ni(III) complex that results is easily reductively eliminated, yielding the C–C coupled product and Ni(I). Finally, SET from 4CzIPN's reduced form to Ni(I) regenerates 4CzIPN's ground state and Ni(0) for the next catalytic cycle.
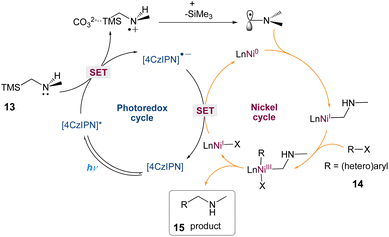 |
| | Scheme 5 Proposed mechanism for metallaphotoredox catalysed aminoalkylation of aryl halides.43 | |
2.4. General method for enantioselective three-component carboarylation of alkenes enabled by metallaphotoredox catalysis
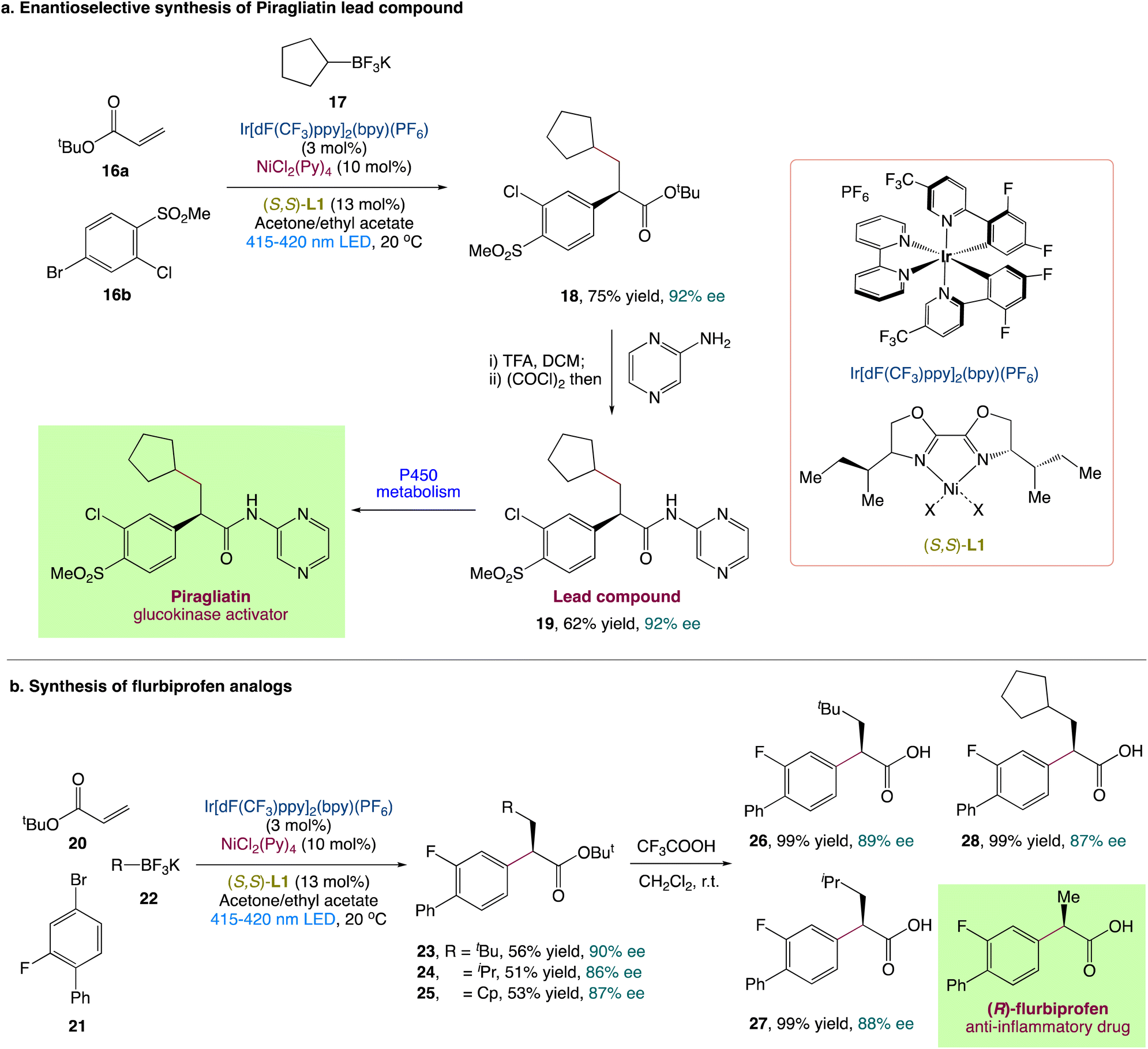
L. Chu et al.44 described an enantioselective three-component carboarylation of alkenes with tertiary and secondary alkyltrifluoroborates and aryl bromides using a visible-light-promoted photoredox/nickel protocol (Scheme 6). From easily available starting materials, this redox-neutral protocol allows for the easy and divergent access to a wide range of enantioenriched β-alkyl-α-arylated carbonyls, phosphonates, and sulfones in high yields and with excellent enantioselectivities. To illustrate synthetic utility, they also reported a modular and enantioselective synthesis of flurbiprofen analogues and piragliatin as the lead chemical compound. To explore more about the mechanism and origin of chemo and enantioselectivity, researchers conducted experimental and computational mechanistic studies.
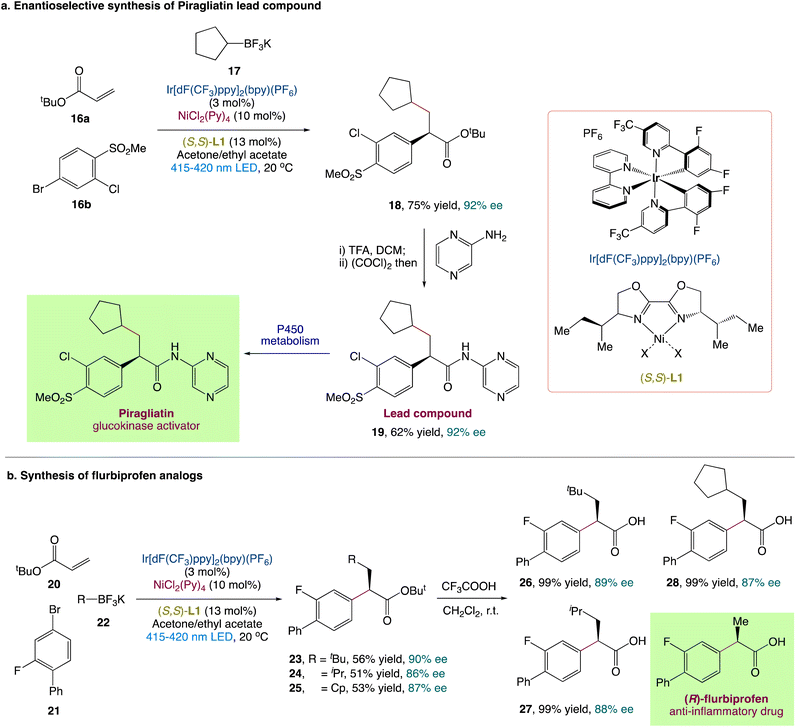 |
| | Scheme 6 (a) Synthesis of lead compound of Piragliatin and (b) synthesis of flurbiprofen analogues.44 | |
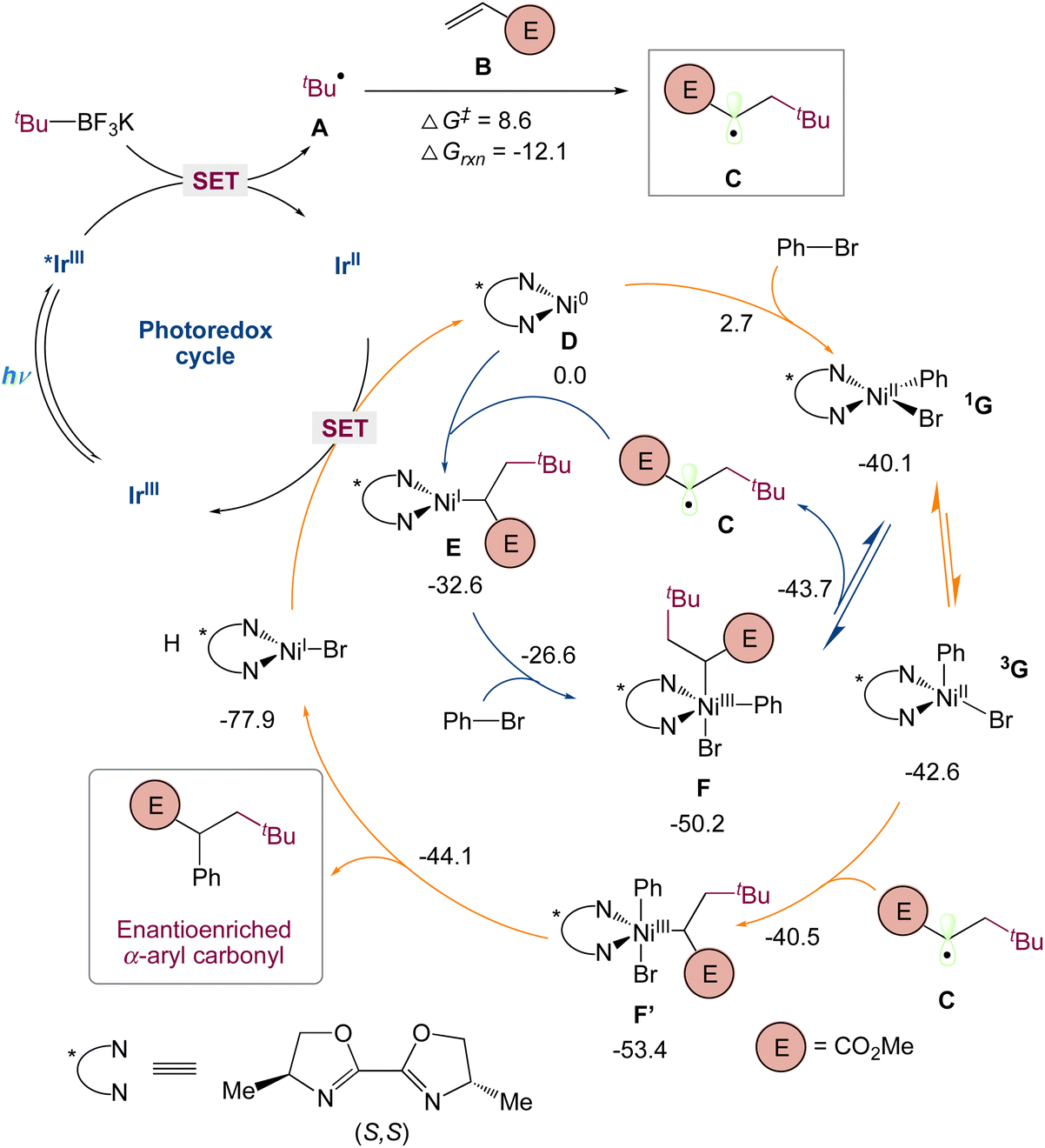
They used quantum mechanical computations in the mechanism proposed by the authors. Specifically, they used dispersion-corrected DFT to identify (1) the preference for the three-component carboarylation over direct arylation of the generated alkyl radical and (2) the factors controlling the enantioselectivity. Calculations show that alkyl radical A, probably resulted from the favorable single-electron oxidation of alkyl trifluoroborate reagent with Ir* I, that can undergo fast and irreversible radical addition to methyl acrylate B (via A-TS-C, barrier of 8.6 kcal mol−1) to form the α-ester radical C, which is 12.1 kcal mol−1 smaller in energy (Scheme 7). To understand the destiny of the alkyl radical C, they looked at Ni(0)/Ni(I)/Ni(III) and Ni(0)/Ni(II) as well as alternative processes. As found with the bipyridyl-Ni systems (blue), a chiral Ni(0) D species could undergo rapid oxidative addition (barrier of only 2.7 kcal mol−1) with phenyl bromide to give a square planar singlet Ni(II) species 1G. In turn, this species can then equilibrate with a triplet tetrahedral Ni(II) 3G before interacting with the alkyl radical C to undergo productive C–C bond formation (vide infra). Alternatively, as indicated in red, the alkyl radical C can be intercepted by Ni(0) D, resulting in a Ni(I)-alkyl species E with 32.6 kcal mol−1 lower energy. As observed with related transformations, Ni(I) E can then undergo oxidative addition with phenyl bromide to afford the Ni(III) species F, which, as previously found before for related transformations, undergoes reversible radical dissociation (barrier of 6.5 kcal mol−1) to form a square planar singlet Ni(II) species 1G. Thus, irrespective of the species that intercepts the alkyl radical C, these routes will converge at a singlet square planar Ni(II) before irreversibly forming C–C bonds (vide infra). Specifically, they found that the Ni(II) 1G can undergo isomerization and intersystem crossing to form the more stable tetrahedral triplet spin state Ni(II) 3G species. Many groups had previously reported on this singlet triplet conversion of NiII species in the photoredox/Ni system in related nickel transformations. In turn, 3G can then rapidly and irreversibly intercept the α-ester radical C to form the productive Ni(III) F′ species. Finally, subsequent reductive elimination will lead to the observed three-component alkylarylation product and Ni(I) H which after SET will restart the dual catalytic cycle.
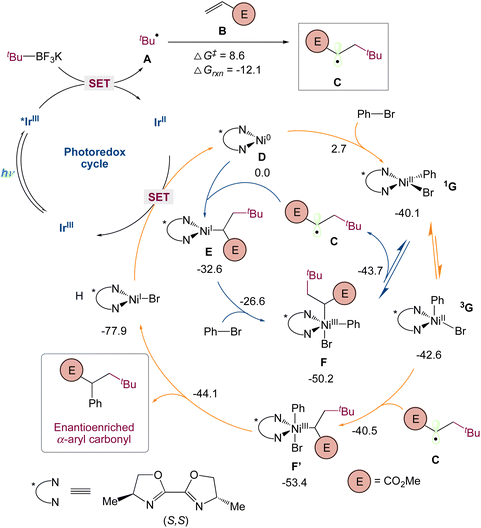 |
| | Scheme 7 Mechanistic proposal based on computations [UB3LYP-D3(BJ)/def2-SVP-CPCM(THF)] for enantioselective three-component carboarylation of alkenes enabled by metallaphotoredox catalysis. All free energies are in kcal mol−1 (298 K).44 | |
2.5. Metallaphotoredox catalysed cross-electrophile coupling of aziridines with pyridin-1-ium salts via dearomatization
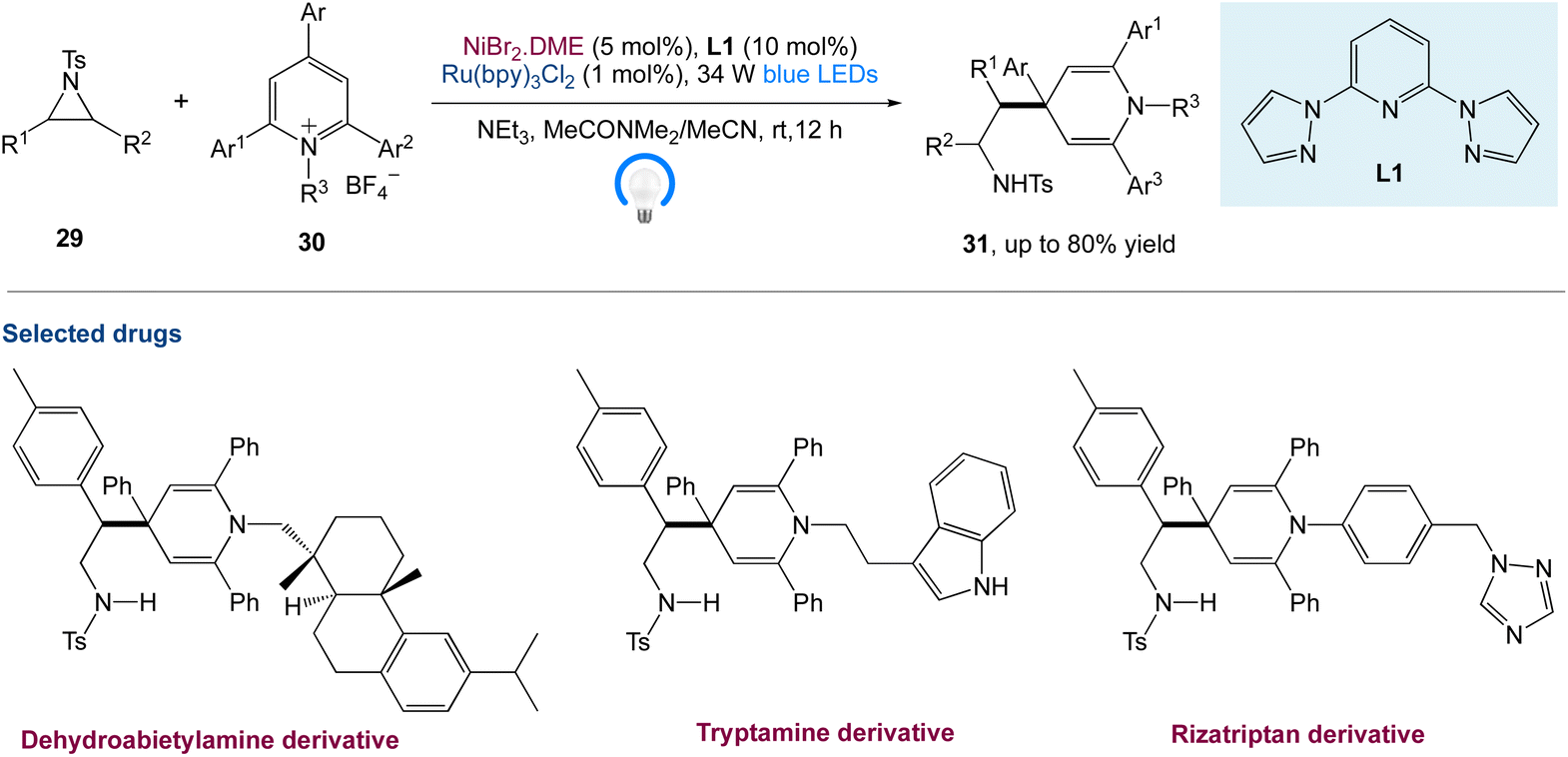
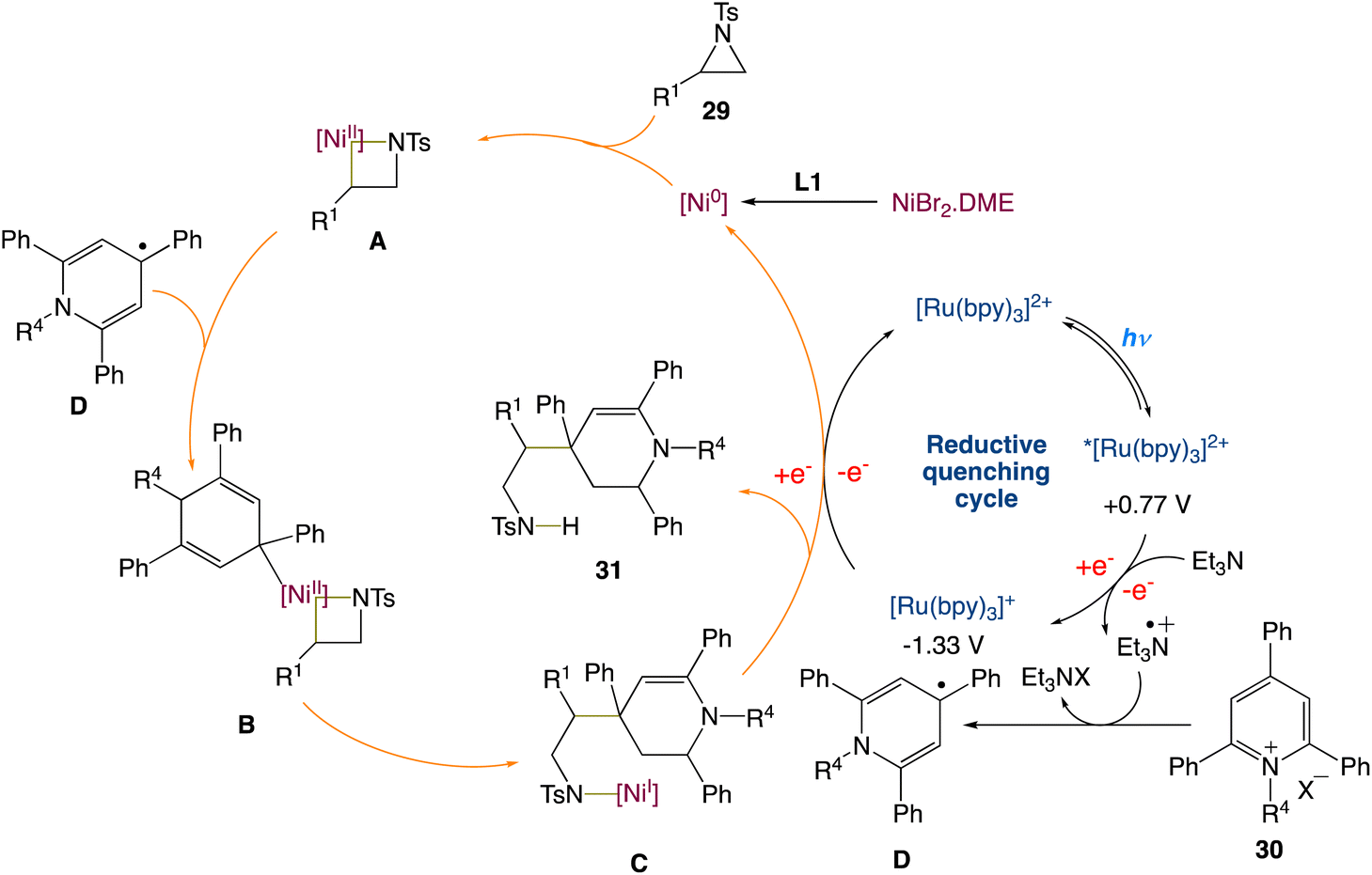
W. Deng et al.45 reported a new protocol to access β-(1,4-dihydropyridin-4-yl)-ethylamines via a metallaphotoredox catalysed cross-electrophile coupling of aziridines 29 with pyridin-1-ium salts 30via dearomatization. A wide range of aziridines (e.g., 2-aryl aziridines, 2-vinyl aziridines, 2,3-disubstituted aziridines) and pyridin-1-ium salts can be used in this approach. Moreover, this method enables the synthesis of highly valuable β-aryl-β-(1,4-dihydro-pyridin-4-yl)-ethyl-amine 31 (Scheme 8) building blocks and bioactive molecule-based analogues from readily available starting materials, which display encouraging potential application areas in natural product and drug synthesis.
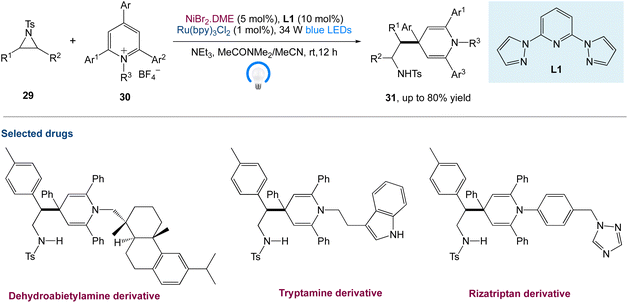 |
| | Scheme 8 Metallaphotoredox catalysed cross-electrophile coupling of aziridines with pyridin-1-ium salts via dearomatization.45 | |
On the basis of the current results and the precedent literature, the possible mechanism for this cross-electrophile coupling protocol was proposed. Initially, coordination of the NiIIBr2·DME species with a ligand L1 affords the reactive Ni0 species, which then undergoes the regioselective oxidative insertion of the C2–N bond of aziridine 29 to give the NiII intermediate A, due to the electronic and steric hindrance effect of the ligand and substrates. Reaction of the intermediate A with the 1,4-dihydropyridin-4-yl radical D, that is generated from the reaction between pyridin-1-ium salt 30 and the radical cation of NEt3via a single electron transfer (SET) process, delivers the NiIII intermediate B. Reductive elimination of the intermediate B results in the formation of the NiI intermediate C. Finally, SET reduction of the intermediate C by the reduced species Ru(I) affords product 31 and regenerates the active Ni(0) species/the Ru(II) photocatalyst (Scheme 9).
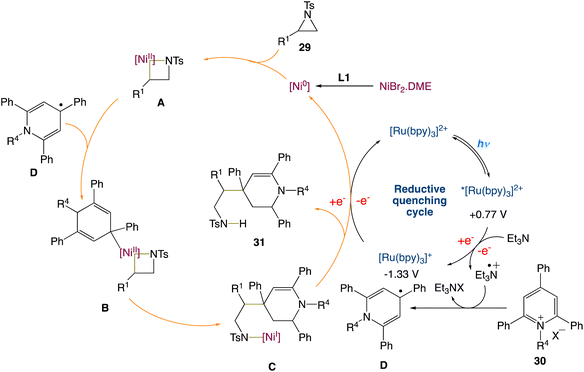 |
| | Scheme 9 Proposed mechanism for metallaphotoredox catalysed cross-electrophile coupling of aziridines with pyridin-1-ium salts via dearomatization.45 | |
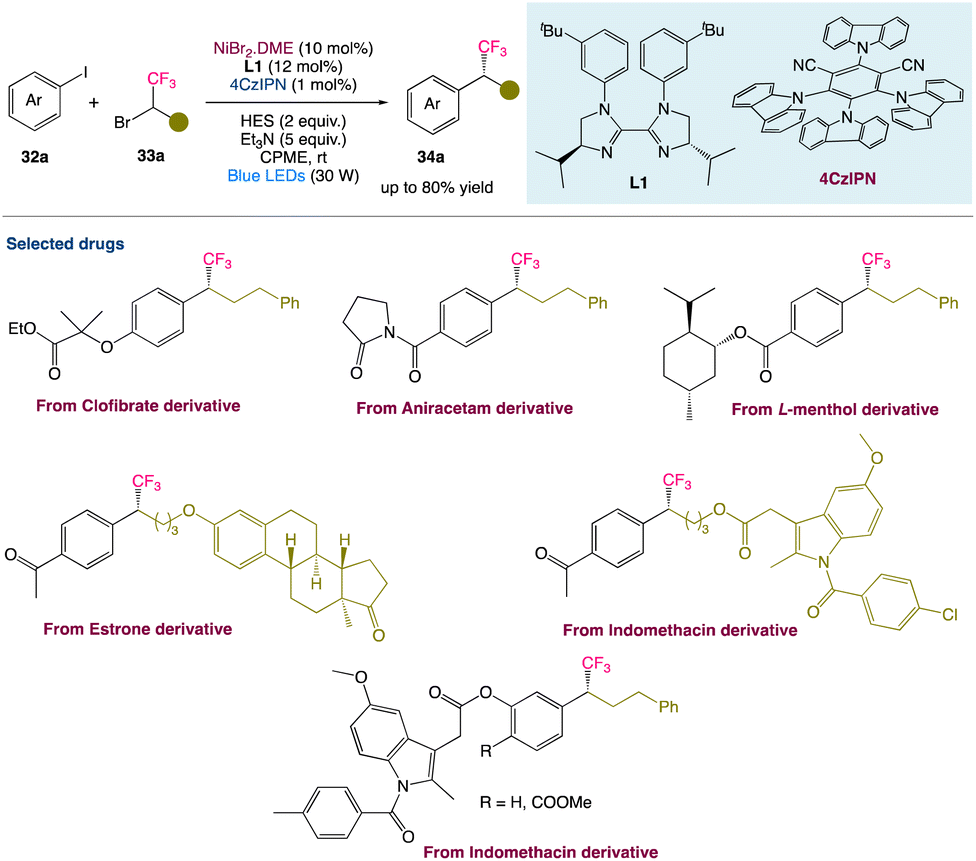
2.6. Metallaphotoredox catalysed reductive cross-coupling to access chiral trifluoromethylated alkanes
T. Xu et al.46 described a metallaphotoredox catalysed regime for enantioconvergent reductive cross-couplings of aryl halides 32a with CF3− substituted racemic alkyl electrophiles 33a. In comparison with the existing methods for making enantiomerically enriched trifluoromethylated molecule 34a, this method may be carried out under mild conditions (visible light, room temperature, no strong base) and has a high catalytic efficiency. It can accommodate a wide range of functional groups, emphasising the method's utility in the late-stage functionalization of complex compounds (Scheme 10).
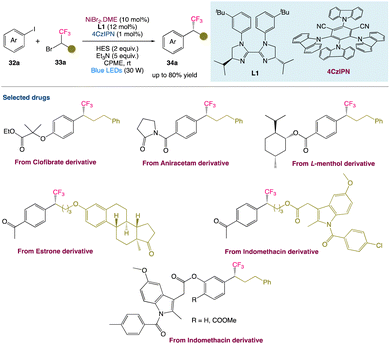 |
| | Scheme 10 Metallaphotoredox catalysed reductive cross-coupling to access chiral trifluoromethylated alkanes.46 | |
2.7. Cross-coupling arylation
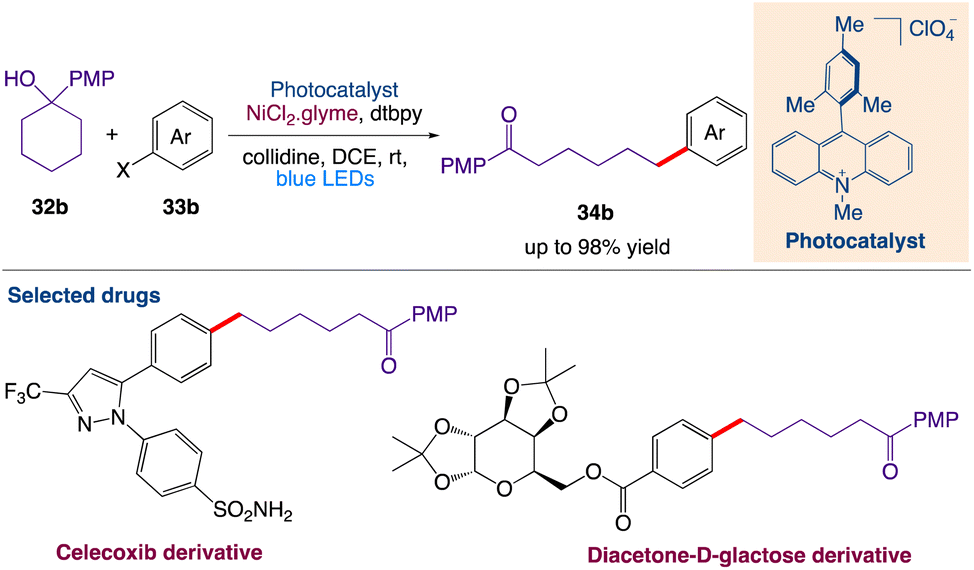
Huang et al.47 reported a mild and general strategy via the combination of photoredox-mediated multisite concerted proton–electron transfer (MS-PCET) and nickel catalysis for the activation of a diverse set of readily available cyclic alcohols for the remote and site-specific arylation of ketones. This protocol includes cross-coupling with the generation of an alkoxy radical utilizing bond-dissociation free energy (BDFE) as the thermodynamic driving force. Subsequently, the resulting remote carbon-centered radicals formed by C–C cleavage merge with the nickel catalytic cycle to create the challenging C(sp3)–C(sp2) bonds (Scheme 11).
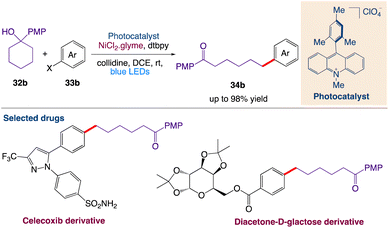 |
| | Scheme 11 Cross-coupling arylation via proton-coupled electron transfer-enabled C–C bond cleavage.47 | |
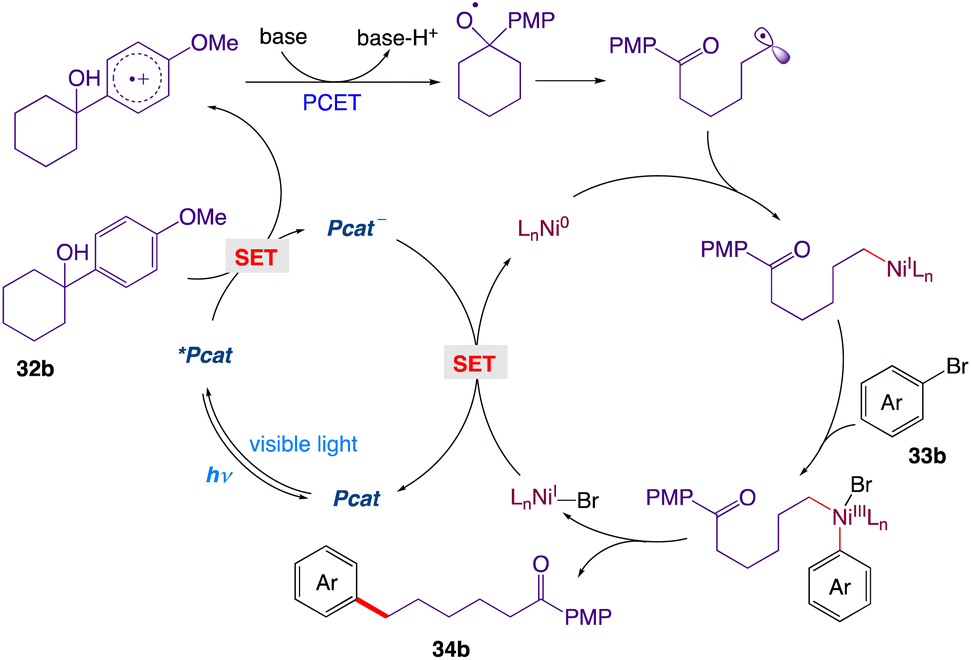
The proposed cross-coupling with a detailed mechanism is depicted in Scheme 12. Upon irradiation with visible light, the ground-state of Mes-Acr-Me+ is promoted to its highly oxidizing singlet excited state *Mes-Acr-Me+, which undergoes a single electron transfer with the tertiary alcohol 32b, furnishing the corresponding arene radical cation along with the reduced form of the photocatalyst Mes-Acr-Me˙. Subsequent deprotonation (PT) and intramolecular ET reaction between the alkoxide and the radical cation would give the key alkoxyl radical species, which readily breaks into a carbonyl moiety and a distal carbon-centered radical through scission of the neighboring C–C bond at the β position. The alkyl radical is then captured by the Ni(0) and gives an alkylnickel(I) intermediate, which is expected to rapidly undergo oxidative addition with an aryl halide 33b, generating a Ni(III) species. Reductive elimination at this stage would deliver the cross-coupled product 34b and the Ni(I) intermediate; the latter can be reduced by the reduced form of the photocatalyst and can regenerate both simultaneously, thus completing the catalytic cycle.
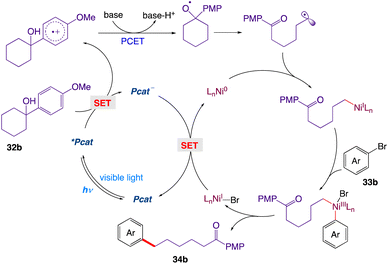 |
| | Scheme 12 Mechanistic proposal for the MS-PCET-enabled C–C cleavage/cross-coupling via dual photoredox nickel catalysis.47 | |
3. Copper metallaphotocatalysis
Copper photoredox catalysts are powerful photocatalysts used for organic transformation reactions. Copper salts have gained popularity as photoredox catalyst materials because of their availability, affordability, and capacity to produce potent photoexcited reducing power. Their effectiveness is dependent on the potent reducing power of copper complexes, their ability to coordinate substrates, or to bind reactive intermediates. The applications of photoinduced copper-catalyzed reactions include decarboxylative alkynylation, coupling reaction, trifluoromethylation, cyanation and decarboxylative radical sulfonylation.
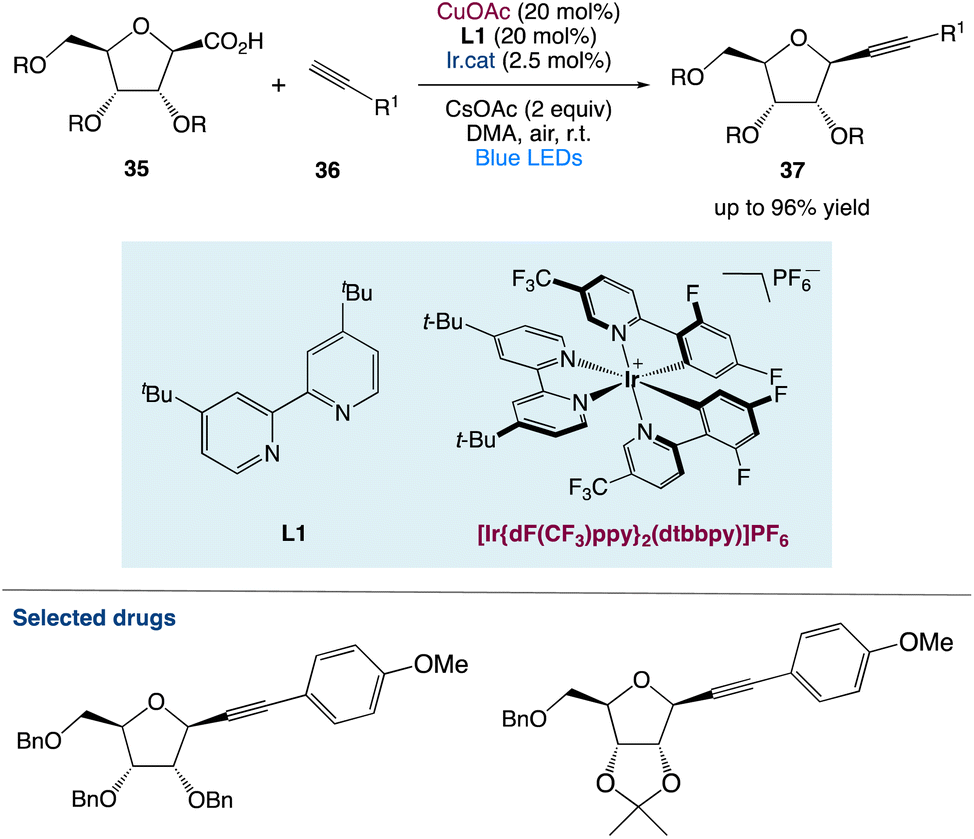
3.1. Metallaphotoredox catalysed diastereoselective decarboxylative alkynylation of anomeric carboxylic acids
S. Messaoudi et al.48 used a metallaphotoredox catalysed approach to alkynylate anomeric furanosyls 35 with different terminal alkynes 36. With great efficiency and diastereoselectivity, this approach gives unparalleled access to alkynyl C-nucleosides under moderate conditions. They considered that this procedure not only provides a diverse and convergent approach for the synthesis of complicated C-nucleosides through subsequent functionalizations of the triple bond but also opens a novel avenue for the catalytic alkynylation of furanosides (Scheme 13).
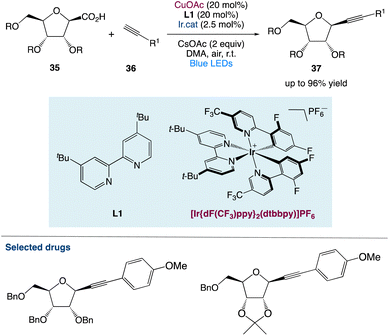 |
| | Scheme 13 Metallaphotoredox catalysed diastereoselective decarboxylative alkynylation of anomeric carboxylic acids.48 | |
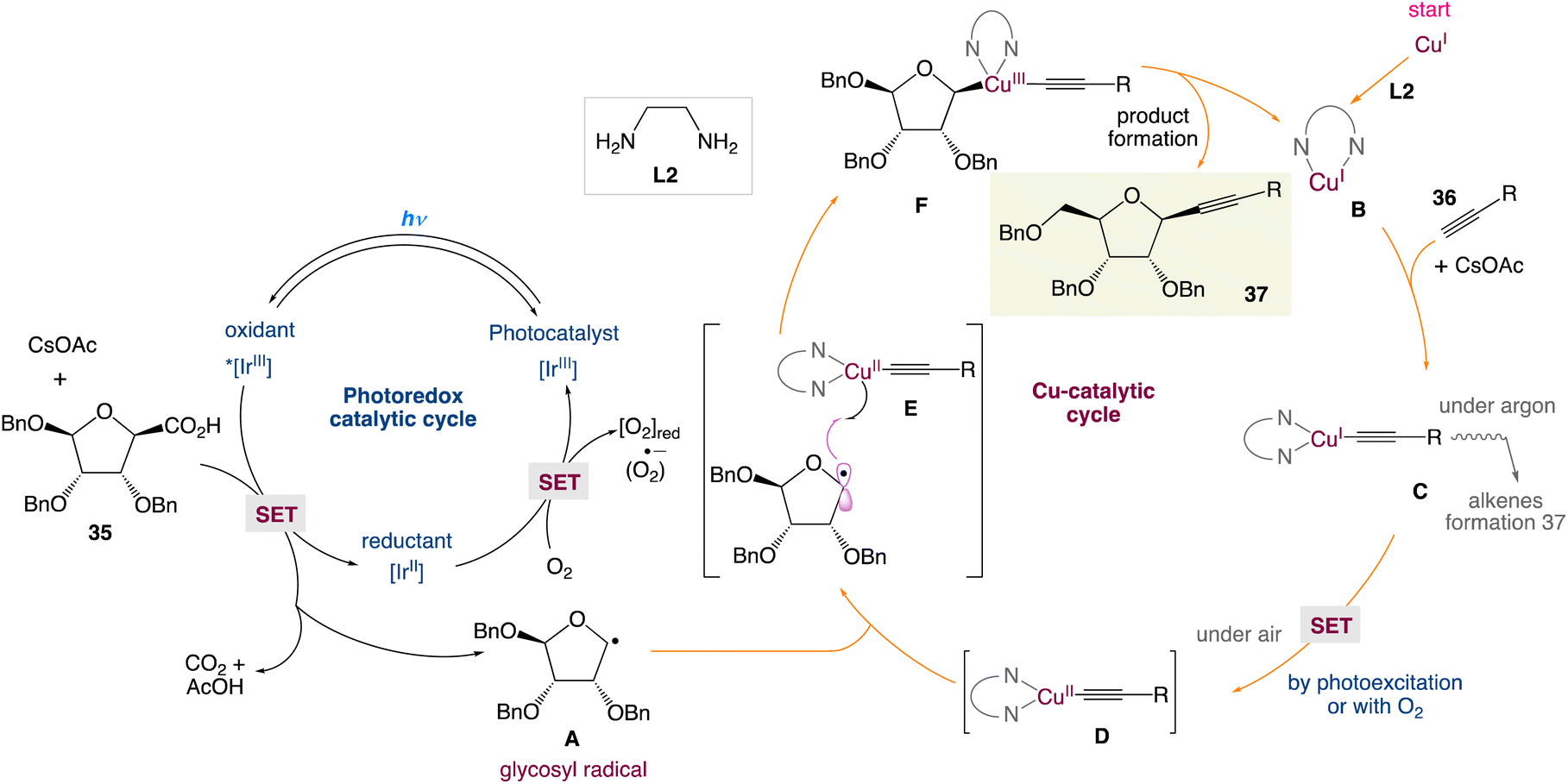
In the mechanism postulated by the authors, the photocatalytic cycle would start by the activation of the iridium catalyst to the excited state Ir* by means of visible light followed by the oxidation of carboxylic acid (or cesium carboxylate) leading to the formation of the anomeric radical A and reduced iridium(II) complex. Simultaneously, the reaction of the Cu(I) catalyst and alkyne 36 in the presence of the base gives the monomeric Cu(I)–acetylene complex C. Then, single-electron transfer (SET) of this later led to the Cu(II) complex D. At this stage, two pathways may be involved: (i) oxidation of the Cu(I) complex by oxygen as a terminal niddle or (ii) by the means of photoexcitation of [LCu(I)] to [LCu(I)*] followed by an electron-transfer mechanism. Later, the anomeric radical A can be captured by LCu(II)-acetylene D to form the high-valent transient Cu(III) species Evia the radical relay mechanism. Reductive elimination would deliver the final β-alkynyl nucleoside 37 and regenerate the Cu(I) catalyst for the next catalytic cycle. The reoxidation of the iridium(II) complex by a molecule of dioxygen to form the ground-state Ir-photocatalyst and conclude the catalytic cycle would be the final crucial step of the cycle (Scheme 14).
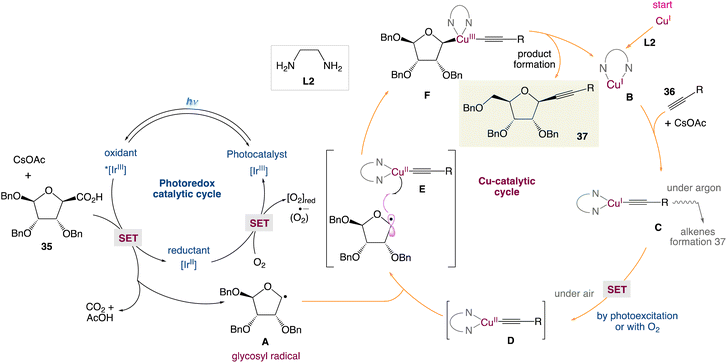 |
| | Scheme 14 Proposed mechanism for the metallaphotoredox catalysed alkynylation of anomeric radicals.48 | |
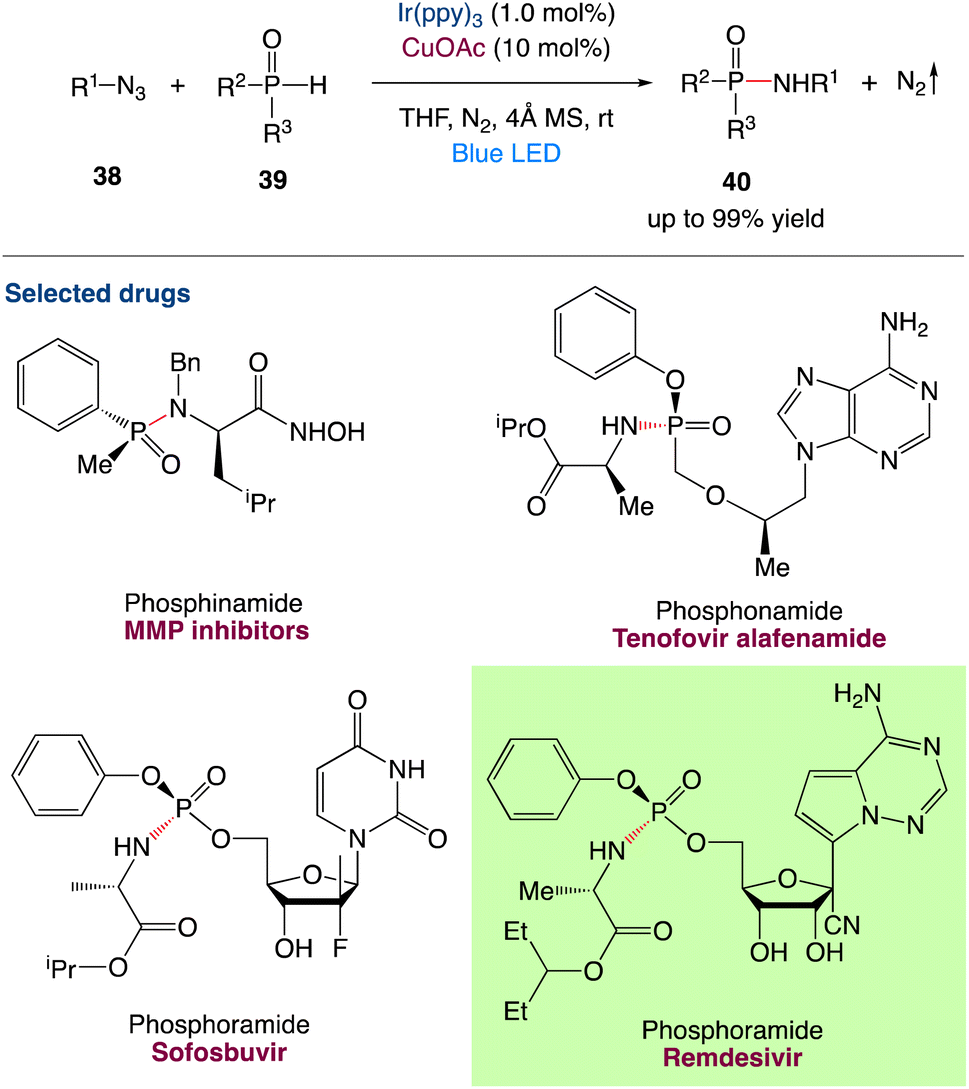
3.2 Redox neutral P(O)–N coupling between P(O)–H compounds and azides via metallaphotocatalysis
H. Lu et al.49 reported a practical synthetic strategy for the redox-neutral construction of P(O)–N bond (Scheme 15). The reaction combines photoredox and copper catalysis to allow for the coupling reactions of organic azides 38 and P(O)H compounds 39, resulting in versatile phosphinamides, phosphonamides, and phosphoramides with only N2 as a byproduct. The use of versatile P(O)–H compounds including diphenyl-, dialkyl-, alkylphenyl, alkoxylphenyl, dialkoxyl, diphenoxyl-phosphine oxides, and organic azides including aryl and alkyl azides, reveals the ubiquitous nature of this method. This reaction is chemoselective and tolerates a wide range of nucleophilic functional groups, including phenol, benzoic acid, alkyl carboxylic acid, amide, aniline, and alkylamines. The utilisation of a microchannel device to demonstrate late-stage functionalization of azido-bearing synthetic intermediates of bioactive substances, possible uses in asymmetric versions, and flow chemistry are also presented. The reaction may go through visible-light-induced EnT and SET processes, as well as a Cu(I)-catalytic cycle, according to mechanistic studies. It differs from existing approaches in terms of mechanism, and it has a lot of potential in the synthesis of pharmacological compounds and natural products.
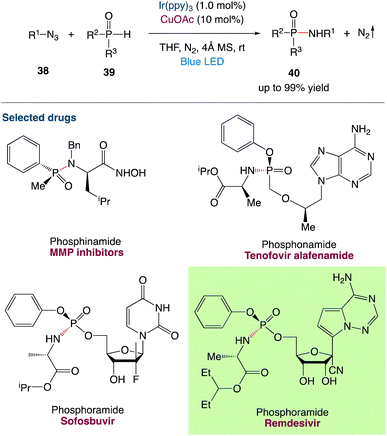 |
| | Scheme 15 Redox-neutral P(O)–N coupling via metallaphotocatalysis.49 | |
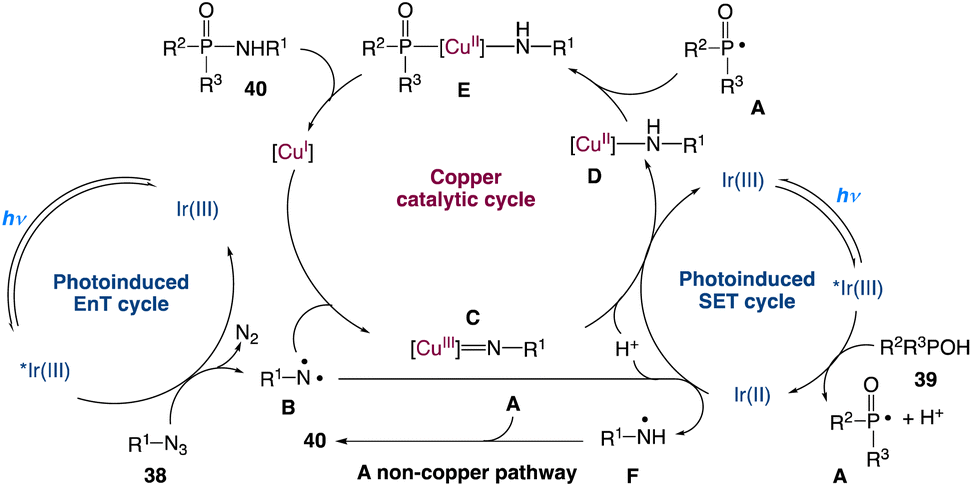
A plausible mechanism with three catalytic cycles is proposed (Scheme 16). Visible light induces [Ir(ppy)3]3+ to produce [Ir(ppy)3]3+* which participates in two catalytic cycles: a SET process with 39 to obtain [Ir(ppy)3]2+ and formation of a phosphoryl radical A accompanied by proton dissociation, and an EnT process involving 38, resulting in the loss of N2 and the generation of the triplet nitrene B. B is captured by Cu(I) to generate a Cu(III) nitrene intermediate C. After undergoing the SET process and protonation, C is converted to a Cu(II) complex D which is then coupled with A to give the Cu(III) complex E. Reductive elimination of E forms 40 and regenerates the Cu(I) catalyst. Cu(II) species might be reduced to Cu(I) species by an excess amount of P(O)–H compound 39 under the reaction conditions, which is the reason why Cu(OAc)2 could also promote the reaction. As a minor reaction pathway, B can convert to the nitrogen radical F directly via the SET process and protonation. In the absence of CuOAc, the radical coupling of A and F leads to the formation of product 40.
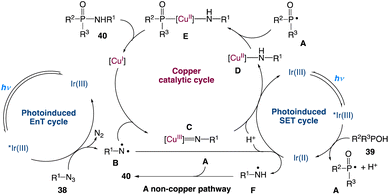 |
| | Scheme 16 Proposed mechanism for the redox-neutral P(O)–N coupling via metallaphotocatalysis.49 | |
3.3. Metallaphotoredox catalysed direct C(sp3)–H trifluoromethylation
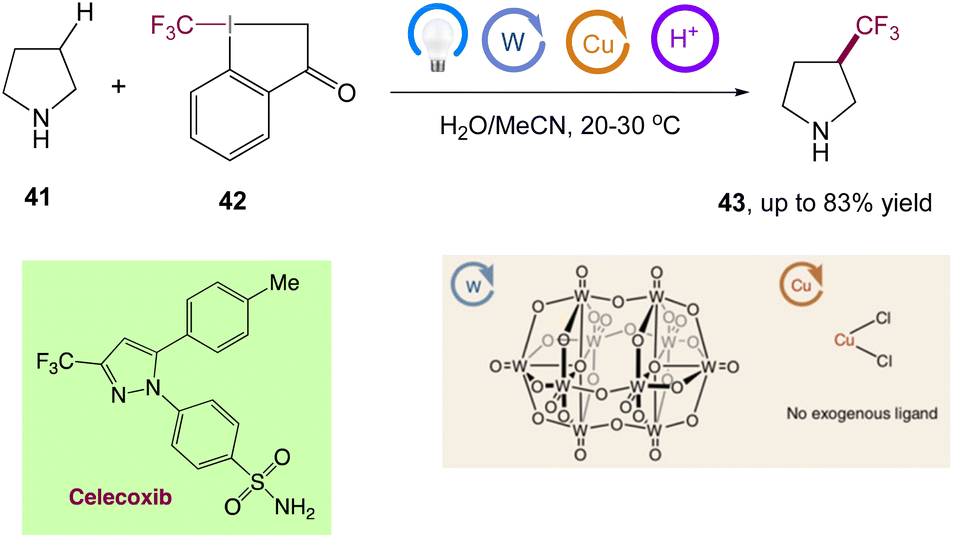
Through the combination of light-driven, decatungstate-catalyzed hydrogen atom transfer and copper catalysis, MacMillan et al.50 reported a dual-catalytic C(sp3)–H trifluoromethylation. Using a bench-stable, commercially available trifluoromethylation reagent, this metallaphotoredox approach allows for the direct conversion of both strong aliphatic and benzylic C–H bonds into the corresponding C(sp3)–CF3 products in a single step. In this reaction only one equivalent of substrate is required, and the reaction proceeds with high selectivity for positions distal to unprotected amines. They have directly derivatized a wide range of licenced pharmaceuticals and natural items to develop useful trifluoromethylated analogues to highlight the value of this innovative technology for late-stage functionalization (Scheme 17).
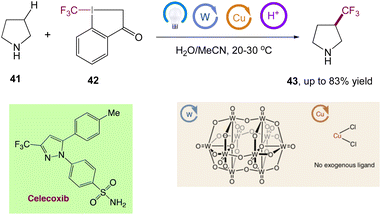 |
| | Scheme 17 Metallaphotoredox catalysed direct C(sp3)–H trifluoromethylation and application to pharmaceuticals. Reproduced from ref. 50 with permission from [Springer Nature], copyright [2020]. | |
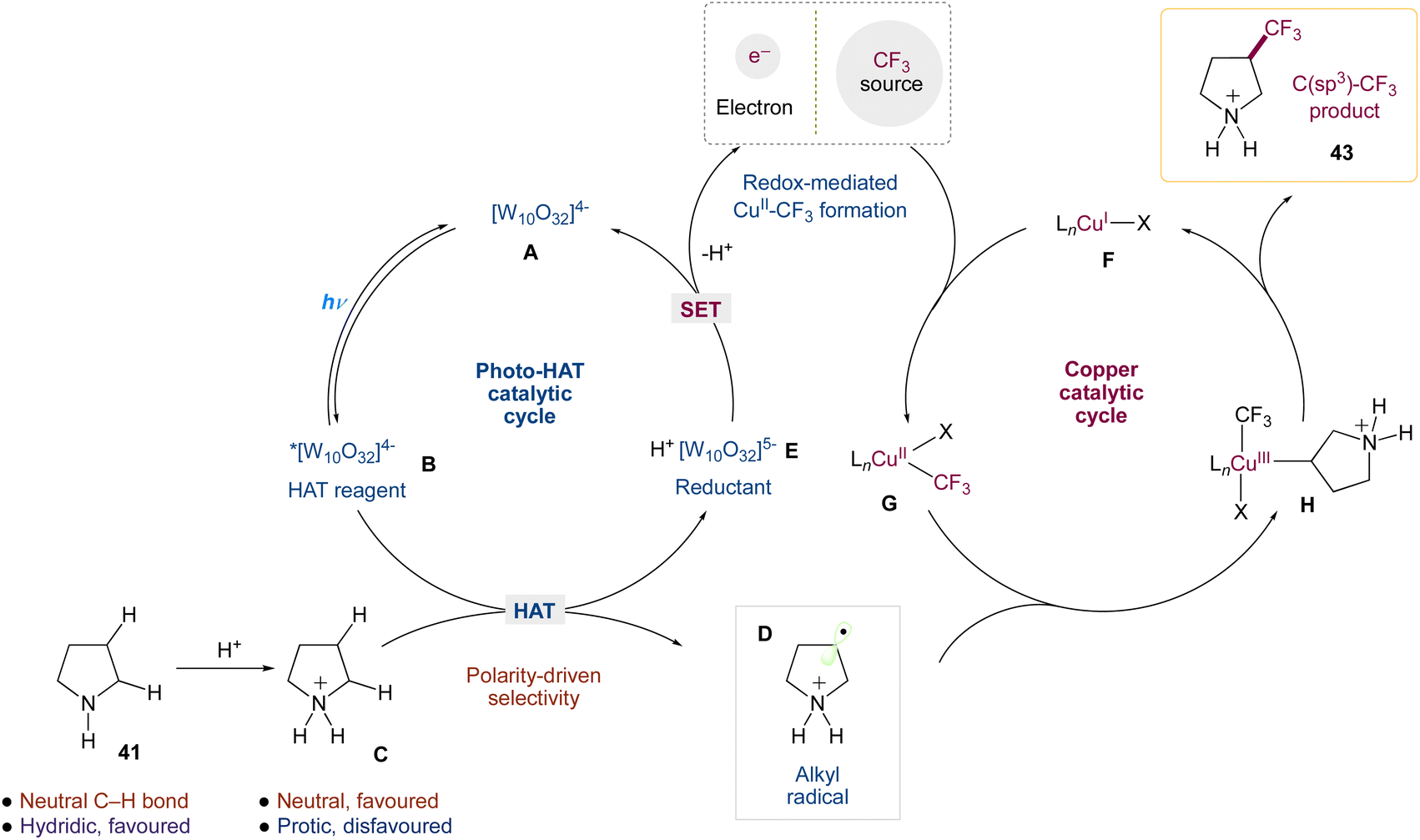
Authors wrapped up their research by analysing the mechanism of this dual-catalytic system. These studies provided substantial evidence for a mechanism in which ‘Cu–CF3 complexes’ are generated via the interaction of copper(I) with Togni reagent II, and that the copper catalyst is involved in the critical C(sp3)–CF3 bond-forming step, which is consistent with this mechanistic design (Scheme 18). Key components of this evidence include (1) the privileged nature of copper as a catalyst for this transformation; (2) the generation of Cu–CF3 species via the reaction of copper(I) with Togni reagent II, the competency of these species in our transformation, and the observation of such species under reaction conditions; (3) the observation of enantioinduction and improvements to diastereoselectivity in the presence of a chiral ligand.
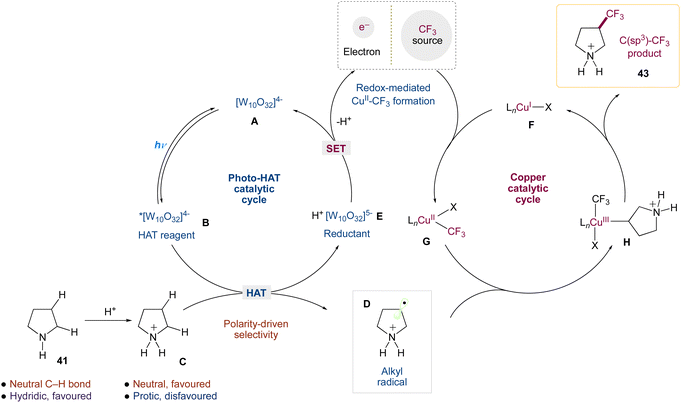 |
| | Scheme 18 Proposed mechanism for direct C(sp3)–H trifluoromethylation via decatungstate and copper catalysis.50 | |
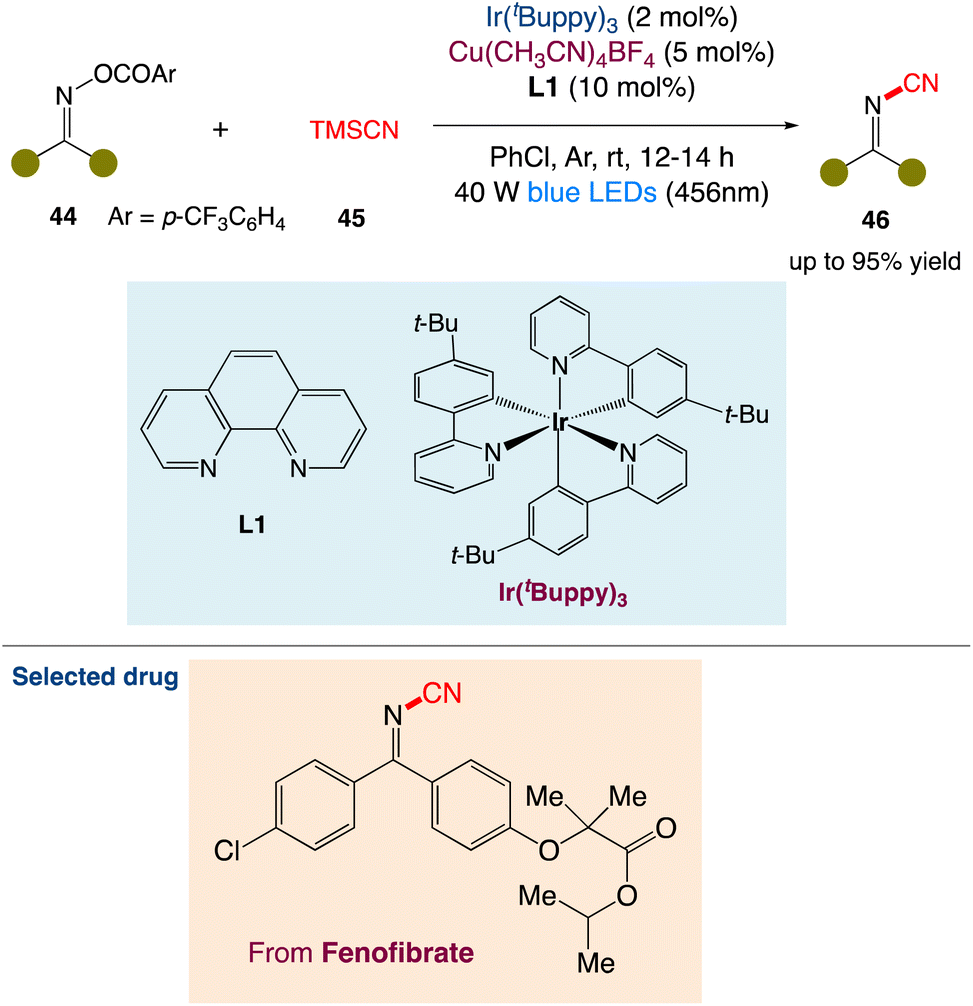
3.4. Metallaphotoredox catalysed cyanation of O-acyl oximes
S. Yu et al.51 described a simple method for synthesising pharmaceutically relevant and synthetically useful cyanoimines and cyanamides. Dual photoredox/copper-catalyzed cyanation of O-acyl oximes or O-acyl hydroxamides enables this method. The present protocol for cyanoimines and cyanamides includes features like readily available starting materials, mild reaction conditions, good functional group tolerance, and operational simplicity. The resultant cyanoimines are synthetically valuable, which is revealed by several transformations of the cyanoimines to structurally diverse and functionally important N-containing heterocycles (Scheme 19).
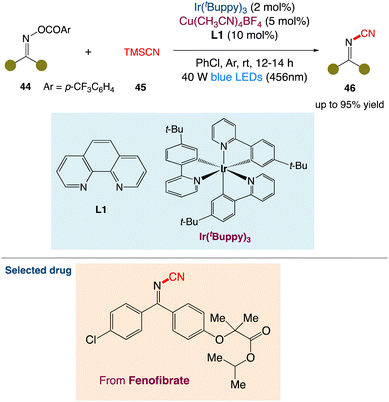 |
| | Scheme 19 Metallaphotoredox catalysed cyanation of O-acyl oximes.51 | |
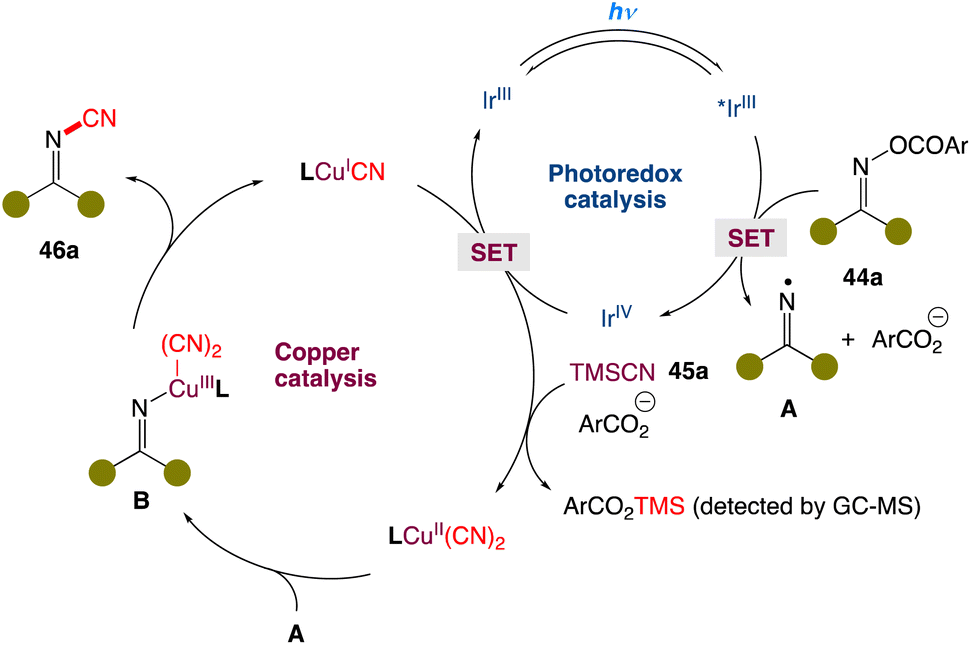
In the mechanism postulated by the authors, the reaction is initiated by the reduction of O-acyl oxime 44a by the excited IrIII* species via single-electron transfer. Fragmentation of reduced 44a generates the iminyl radical A, along with carboxylic anion (ArCO2−) and IrIV. The resultant IrIV species oxidizes the LCuICN complex to afford the oxidized LCuII(CN)2 complex in the presence of TMSCN and the carboxylic anion with concurrent regeneration of the IrIII species. The CuII complex could capture iminyl radical A to furnish the high-valent CuIII complex B. Finally, reductive elimination affords the cyanoimine 46a with concurrent regeneration of the catalytic CuI species to close the copper catalytic cycle (Scheme 20).
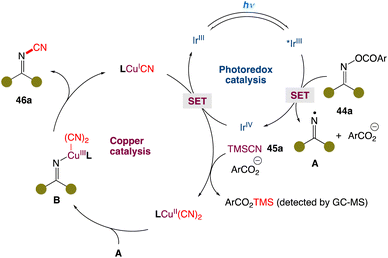 |
| | Scheme 20 Proposed mechanism for metallaphotoredox catalysed cyanation of O-acyl oximes.51 | |
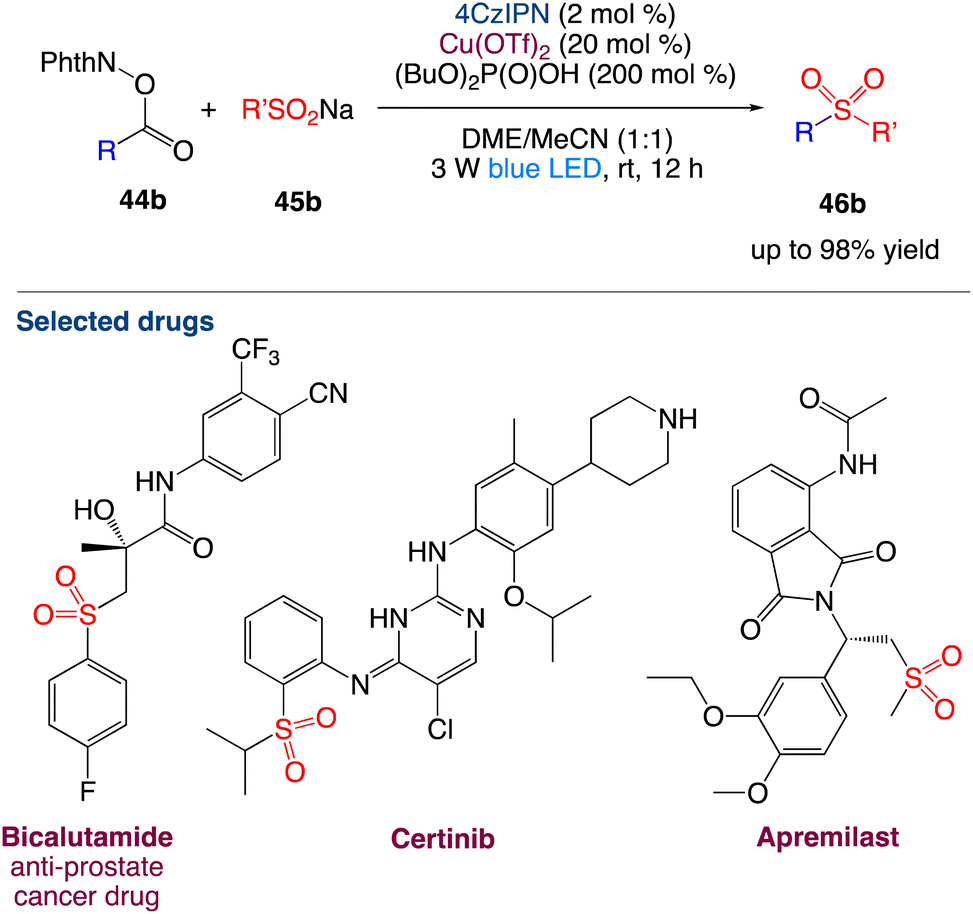
3.5. Decarboxylative radical sulfonylation
Li et al.52 reported reported the decarboxylative radical sulfonylation with sulfinates. With the merger of 4CzIPN (1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene) and Cu(OTf)2 as catalysts, the visible-light-induced reaction of redox-active esters of aliphatic carboxylic acids 44b with organosulfinates 45b at room temperature provides the corresponding decarboxylative sulfonylation products 46b in satisfactory yields (Scheme 21). This redox-neutral protocol exhibits broad substrate scope and wide functional group compatibility, enabling the late-stage modification of complex natural products and bioactive pharmaceuticals. The synthetic utility of this protocol has been further demonstrated by the improved synthesis of the anti-prostate cancer drug bicalutamide.
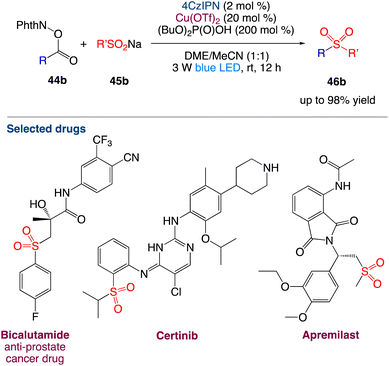 |
| | Scheme 21 Decarboxylative radical sulfonylation.52 | |
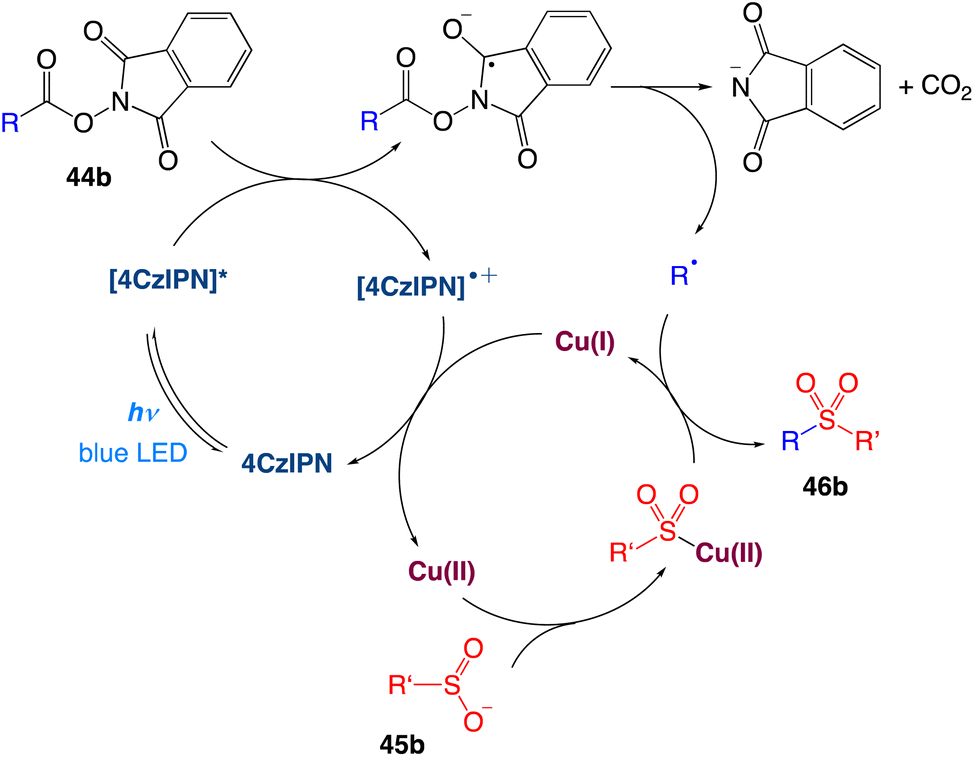
The authors proposed a plausible mechanism as depicted in Scheme 22. Excitation of photocatalyst 4CzIPN generates the long-lived triplet-excited-state [4CzIPN]* that undergoes single electron transfer with a NHPI (N-hydroxyphthalimide) ester 44b to give the 4CzIPN radical cation and the NHPI ester radical anion. The N–O bond cleavage takes place for the NHPI ester radical anion to produce a carboxyl radical, which upon extrusion of CO2 generates the corresponding alkyl radical. In the meantime, transmetalation of sulfinate anion 45b forms the Cu(II)–SO2R intermediate. The alkyl radical is then intercepted by Cu(II)–SO2R to provide the sulfone product 46b and Cu(I). Finally, Cu(I) is oxidized by 4CzIPN radical cation, leading to the regeneration of both Cu(II) and 4CzIPN catalysts.
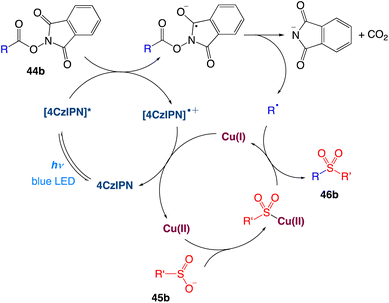 |
| | Scheme 22 Proposed mechanism of decarboxylative sulfonylation.52 | |
4. Palladium metallaphotocatalysis
By combining photoredox and palladium catalysis, several organic reactions could be conducted at ambient temperature and in the absence of strong external oxidants, resulting in higher functional group tolerance than would be exhibited using traditional reaction conditions. The applications of photoinduced palladium-catalyzed reactions include olefination of arenes, decarboxylative hydroaminoalkylation of allenes, carboxylation reaction, dehydrogenative acylation and arylation reaction.
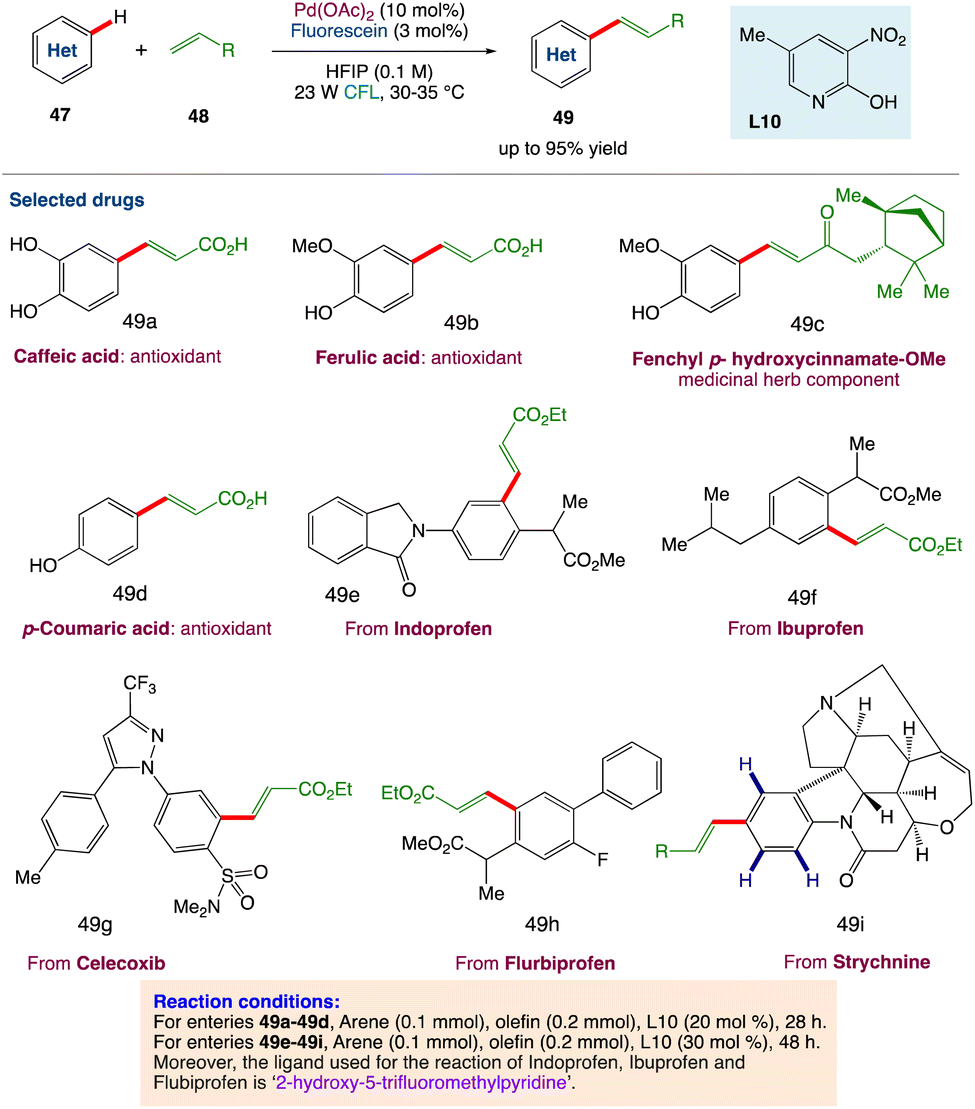
4.1. Metallaphotoredox catalysed olefination of arenes
D. Maiti et al.53 described a photoredox catalytic system based on a palladium/organo-photocatalyst (PC) merger that enables explicit regioselective oxidative olefination with a variety of arenes and heteroarenes. Without the use of silver salts or thermal energy, visible light plays an important role in executing the “regioresolved” Fujiwara–Moritani reactions. The catalytic system can also be used for proximal and distal olefination with the help of the corresponding directing groups (DGs), emphasizing the protocol's adaptability in addressing the complete spectrum of C(sp2)H olefination. Streamlining the synthesis of natural products, chiral compounds, and medicines, as well as diversification through late-stage functionalizations, all add to the value of this long-term protocol. Through control reactions and kinetic studies, the photoinduced accomplishment of this regioselective transformation has been mechanistically proved (Scheme 23).
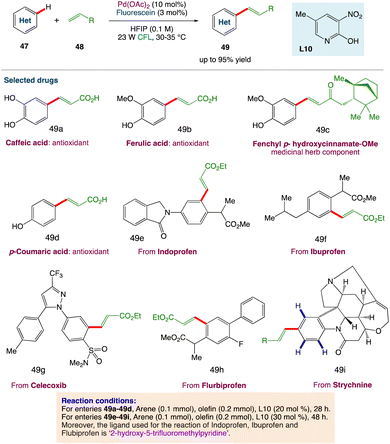 |
| | Scheme 23 Metallaphotoredox catalysed olefination of arenes.53 | |
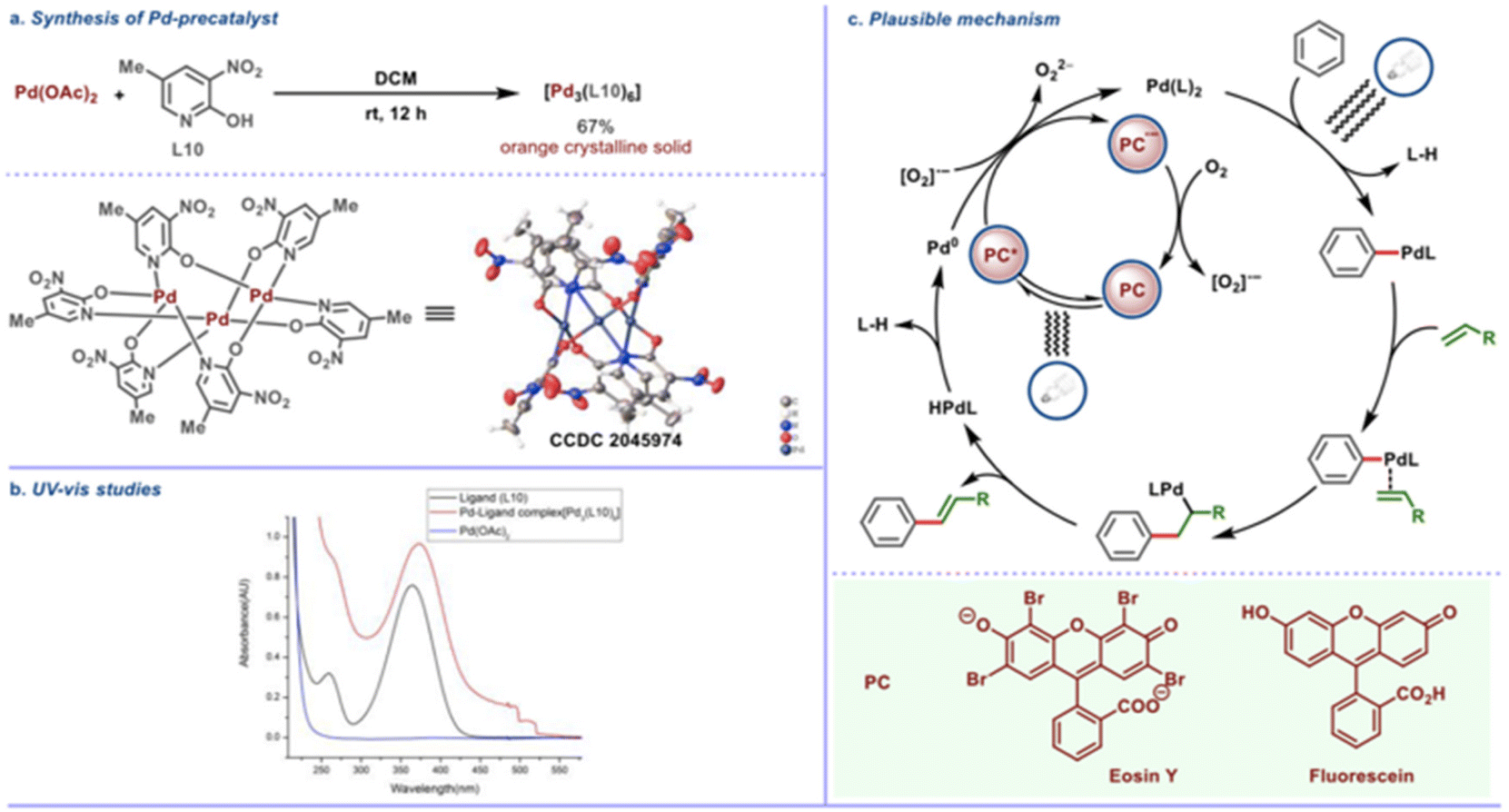
The precatalyst trimeric palladium complex [Pd3(L10)6] with the pyridone ligand (L10) was synthesized and structurally characterized by X-ray crystallography (Scheme 24a). This complex was found to have absorption maxima in the visible light region (Scheme 24b) and to be catalytically competent with this transformation, implying that the trimeric complex uses photochemical energy to activate the desired C–H bond. Aside from that, an on/off experiment was used to track the reaction's conversion profile. When CFL was turned off, the product formation stopped, showing that an effective transformation requires ongoing photo-irradiation. Based on the findings of the mechanistic studies, an intuitive mechanistic postulate is proposed, according to which direct excitation of the in situ generated Pd complex is expected to favour the CH activation step, whereas precedents suggest that photocatalyst excitation would allow for mild reoxidation of Pd(0) to Pd(II) in the presence of oxygen (Scheme 24c). Alternatively to the showcased oxidation step (Scheme 24c), it is also possible that upon regeneration of the photocatalyst from its reduced state, molecular oxygen (O2) is reduced to form [O2]˙−, while in the other oxidation step [from Pd(0) to Pd(II)], O2 is involved in a similar single electron reduction process, to generate [O2]˙−. This radical anion [O2]˙− species, being exceedingly short-lived, would undergo rapid protonation.
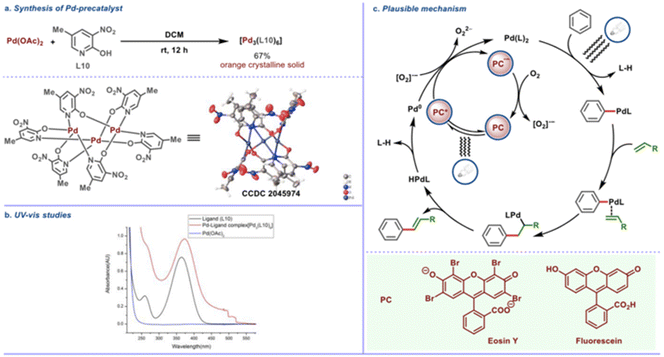 |
| | Scheme 24 Plausible mechanism of photoinduced regioselective olefination. (a) Crystal structure of Pd-precatalyst. (b) UV absorption studies of reaction components. (c) Mechanistic hypothesis for the photoinduced Fujiwara–Moritani reaction. Reproduced from ref. 53 with permission from [American Chemical Society], copyright [2022]. | |
4.2. Metallaphotoredox catalysed decarboxylative hydroaminoalkylation of allenes
Breit et al.54a described an asymmetric dual photoredox/palladium-catalyzed decarboxylation of alkoxylallenes to produce synthetically relevant protected vinyl 1,2-amino alcohol compounds with good regio- and enantioselectivities. A wide range of common protective groups, as well as a diverse range of protected amino acids with various functional groups, were used (Scheme 25).
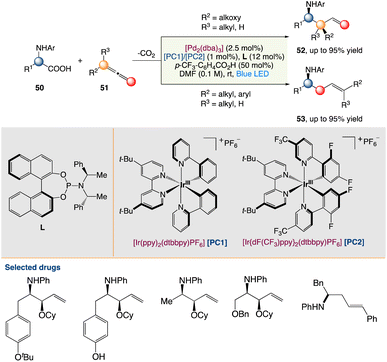 |
| | Scheme 25 Metallaphotoredox catalysed decarboxylative hydroaminoalkylation of allenes.54a | |
In the mechanism proposed by the authors, this transformation begins from the oxidative addition of palladium catalyst with amino acid furnishing the palladium hydride species A. Then, migratory insertion of the intermediate A to the allene gives the π-allyl palladium species B, which then undergoes competing pathways between a fast reductive elimination leading to an ester intermediate E and a relatively slow decarboxylation via a reductive quenching process of the blue-light-excited Ir(III)* complex. According to recent research from Tunge's group,54b a single electron transfer from the carboxylate to Pd in intermediate C can speed up the decarboxylation process, and the newly produced carbon radical then rebounds with the π-allyl-Pd species, forming intermediate D. The π-allyl palladium(III) species D undergoes reductive elimination forging the formation of a new C–C bond and a Pd(I) species. In this catalytic cycle, Pd(0) is regenerated at the end by reducing Pd(I) with the Ir(II) complex (Scheme 26).
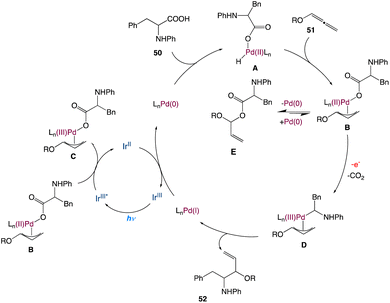 |
| | Scheme 26 Proposed mechanism for metallaphotoredox catalysed decarboxylative hydroaminoalkylation of allenes.54a | |
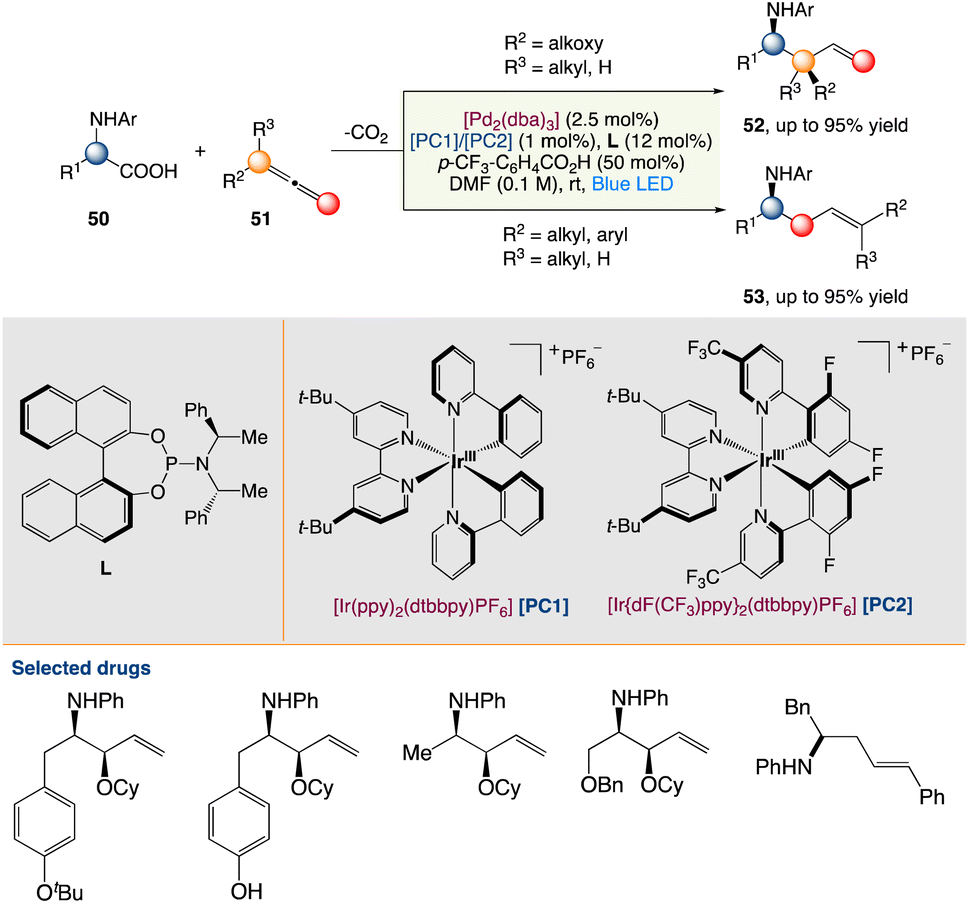
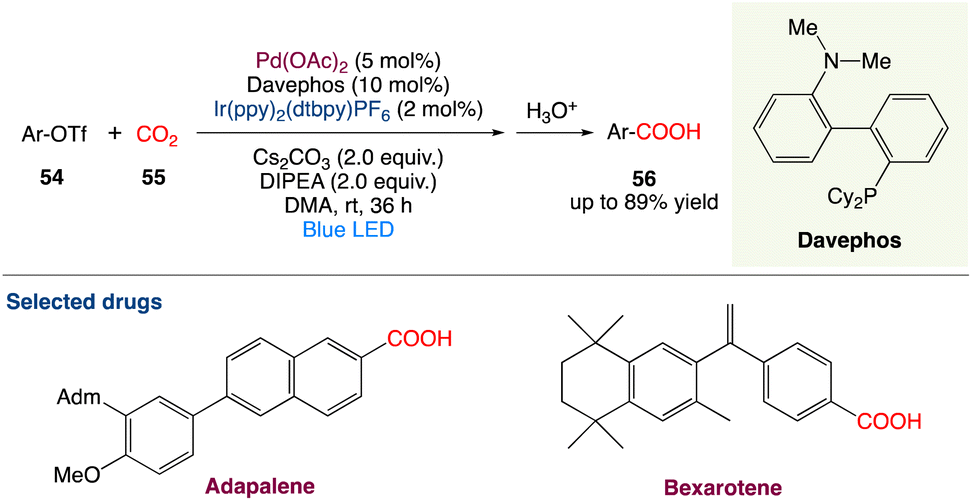
4.3. Metallaphotoredox catalysed carboxylation of aryl triflates with CO2
R. Jana et al.55 described a feasible carboxylation of readily accessible aryl triflates with CO2 at ambient temperature and pressure using palladium and visible-light-iridium(III) dual catalysis. This moderate and chemoselective approach is excellent for late-stage carboxylation of estrone and the production of adapalene and bexarotene medicines. Furthermore, in an H-type closed vessel, an interesting decarboxylation–carboxylation reaction has been demonstrated, which is a novel concept for the strategic sectors in chemical industries for sustainable development (Scheme 27).
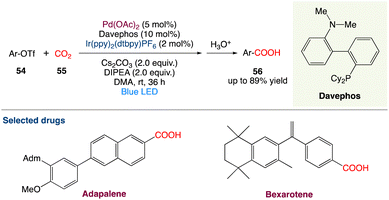 |
| | Scheme 27 Metallaphotoredox catalysed carboxylation of aryl triflates with CO2.55 | |
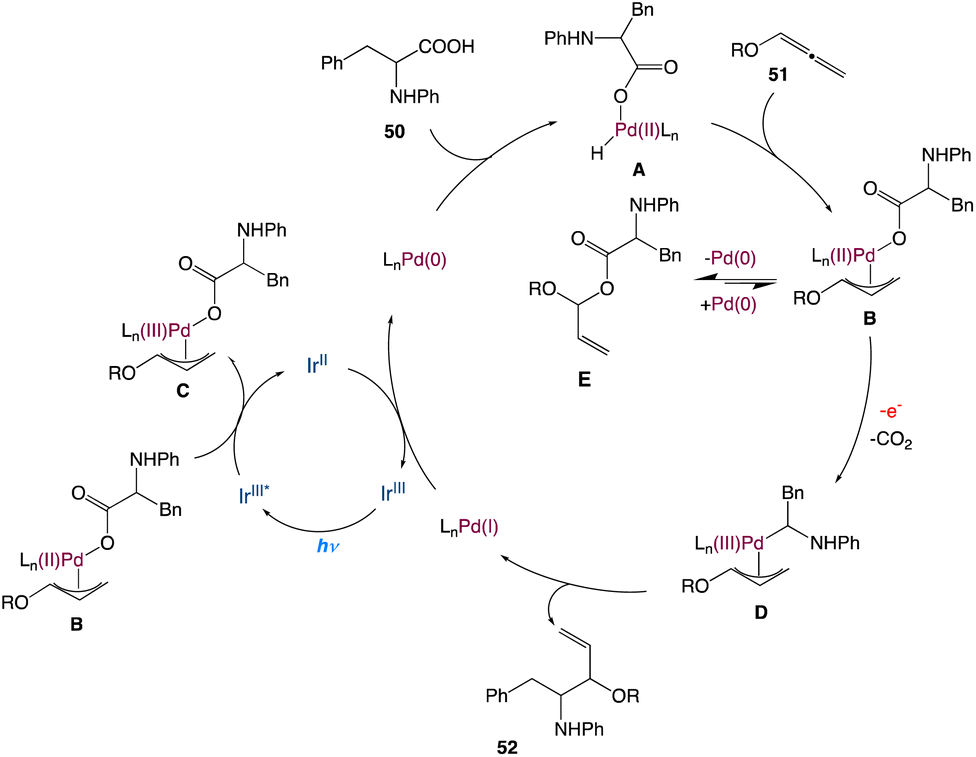
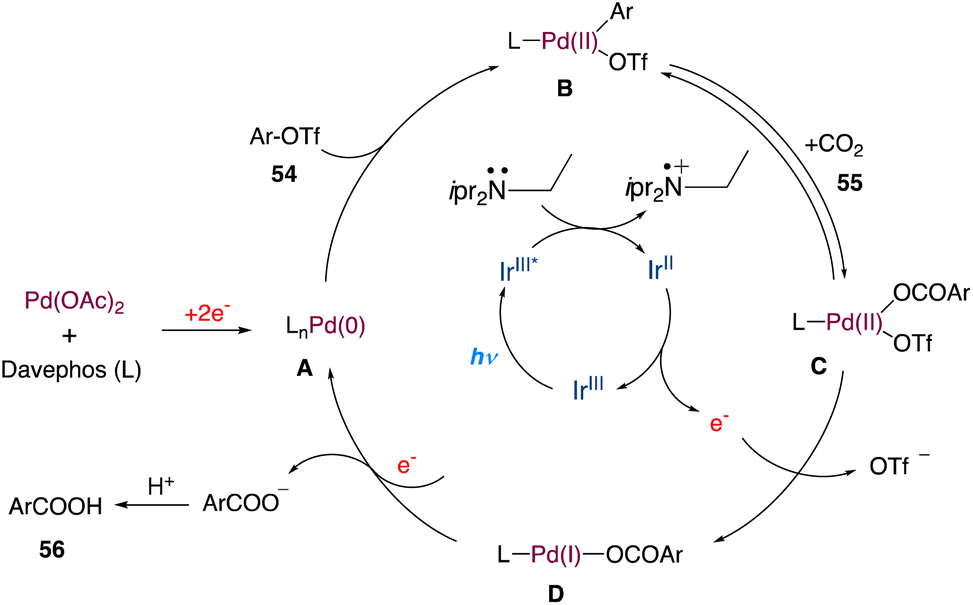
In the mechanism as proposed by the authors, this mechanism is closely related to that proposed by Iwasawa and Martin; initially, a Pd(0) species is formed, which undergoes oxidative addition to aryl triflates providing intermediate B (Scheme 28). In a reversible manner, it may undergo carboxylation with CO2 to form the intermediate C. Following a single-electron reduction by Ir(II), intermediate D was produced, which was then reduced by one more electron to produce aryl carboxylate and Pd(0) for further runs.
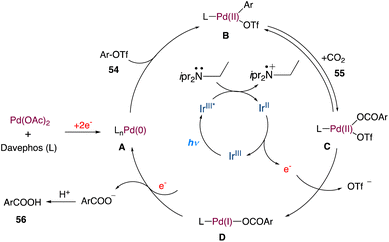 |
| | Scheme 28 Proposed mechanism for metallaphotoredox catalysed carboxylation of aryl triflates with CO2.55 | |
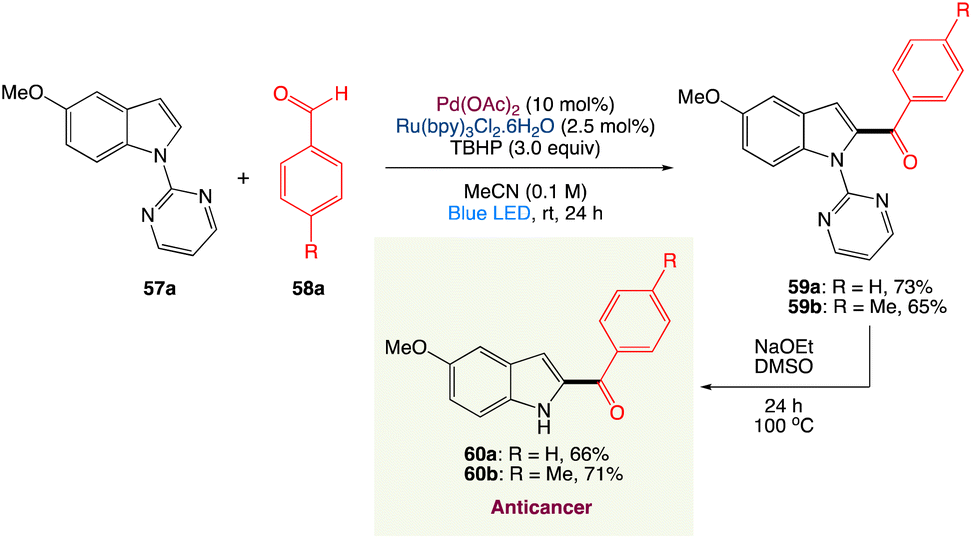
4.4. Metallaphotoredox catalysed dehydrogenative C2-acylation of indoles
R. Jana et al.56 have described a simple method for C2-acylation of N-pyrimidine-protected indoles that incorporated visible light photoredox and palladium(II) catalysts. The reaction proceeds at room temperature via the radical pathway, yielding a high-quality acylation product with good regioselectivity. The approach was used to synthesise tubulin inhibitors D-64131 and D-68144 as well as late-stage functionalization of tryptophan. It was also shown that carbazole can be monoacylated selectively (Scheme 29).
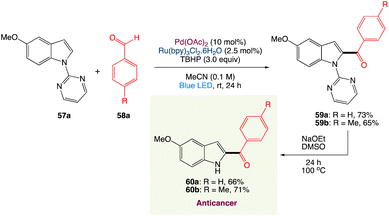 |
| | Scheme 29 Synthesis of anticancer D-64131 and D-68144.56 | |
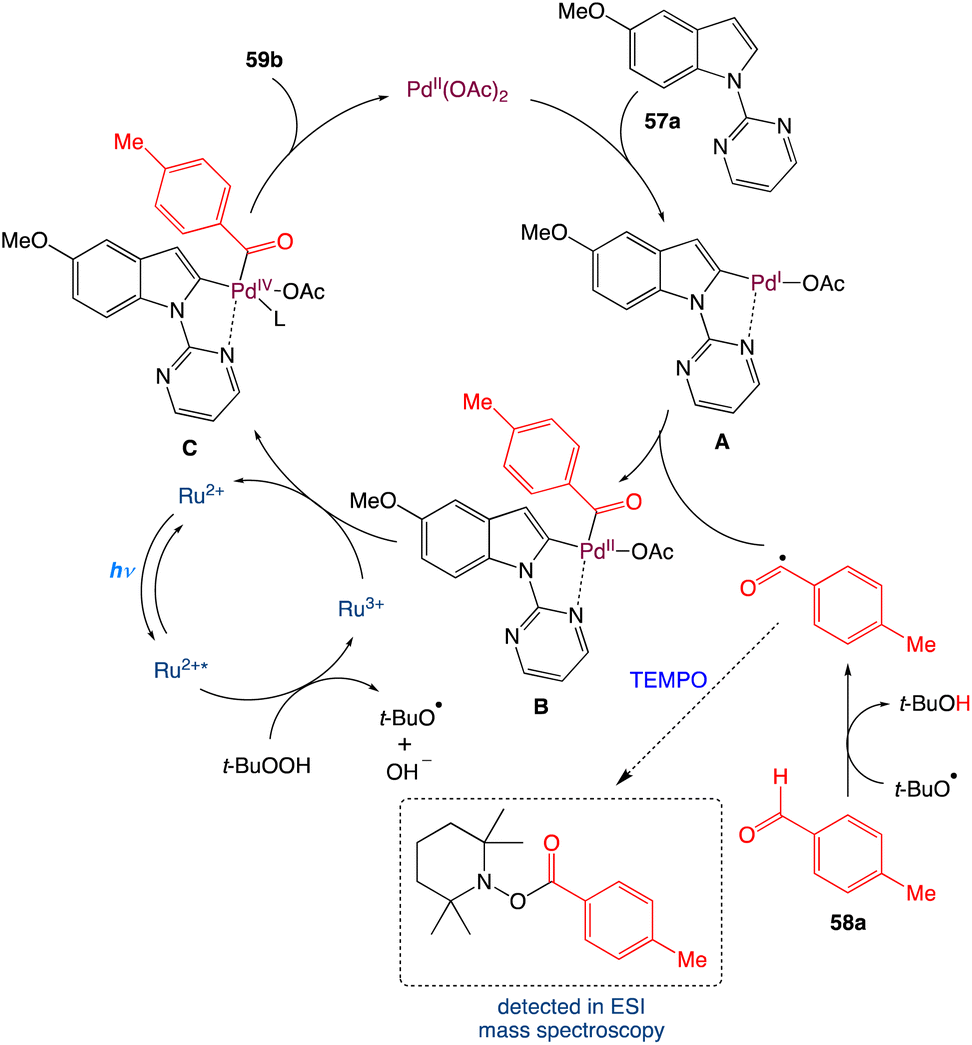
In the mechanism as proposed by the authors, first in presence of light the Ru2+ is excited to Ru2+* from its ground state. It transfers a single electron to t-BuOOH which cleaves the weak O–O bond to produce OH− and t-BuO˙ and itself oxidised to Ru3+. The generated t-BuO˙ abstracts a hydrogen atom from aldehyde to form t-BuOH and an acyl radical. On the other hand, N-pyrimidyl indole undergoes a directed palladation to the C2 position of indole and forms a five membered palladacycle cycle intermediate A. Furthermore, on oxidative addition with the generated acyl radical it furnishes a cyclopalladated Pd(III) species B which may be oxidised to a Pd(IV) species Cvia a single electron transfer to Ru3+. By single electron transfer Ru3+ reduced to Ru2+ and the catalytic cycle is completed for the photoredox catalyst. The Pd(IV) species C undergoes reductive elimination to generate the corresponding acylated product and Pd(II) species (Scheme 30).
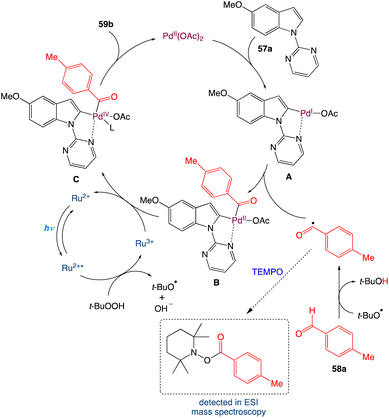 |
| | Scheme 30 Proposed mechanism for metallaphotoredox catalysed dehydrogenative C2-acylation of indoles.56 | |
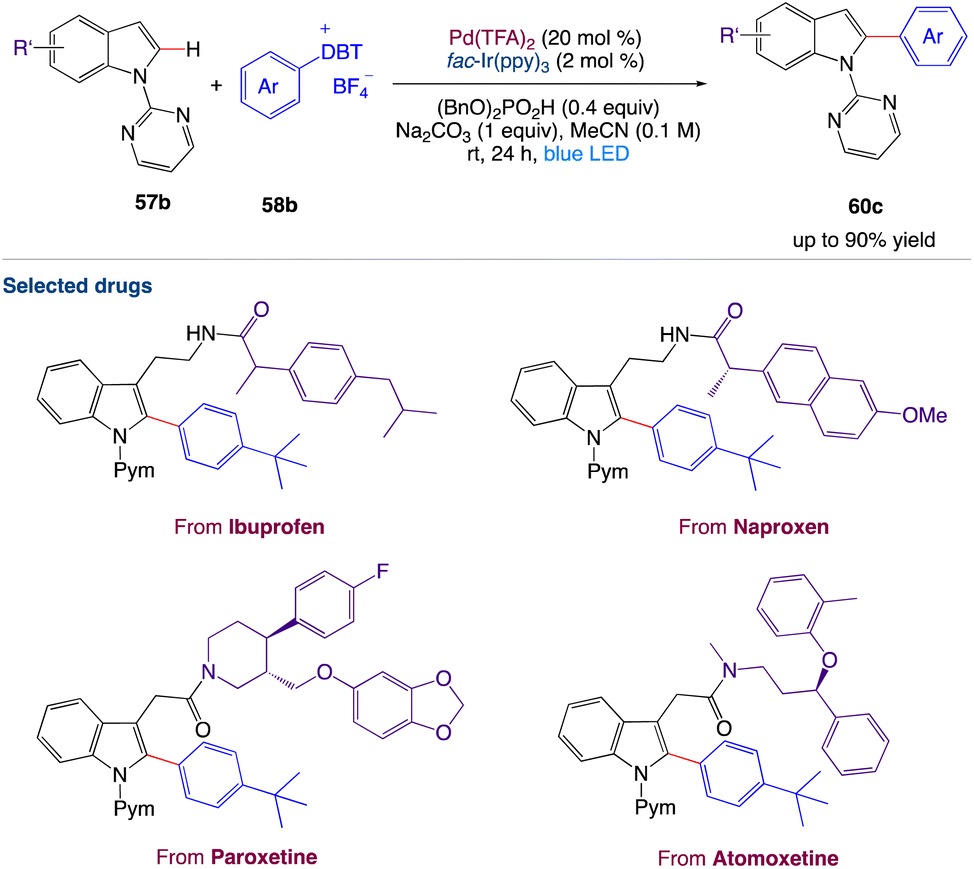
4.5. Metallaphotoredox catalyzed 2-arylation of indole derivatives
Wang et al.57 reported a two-step synthetic method for C(sp2)–H/C(sp2)–H cross-coupling reactions to synthesize 2-arylindole derivatives 60e by combining palladium catalysis and photocatalysis (Scheme 31). This mild, dual-catalysis method displayed good functional group tolerance, wide substrate scope and could be used for late-stage functionalization of oligopeptides, drugs, and natural products.
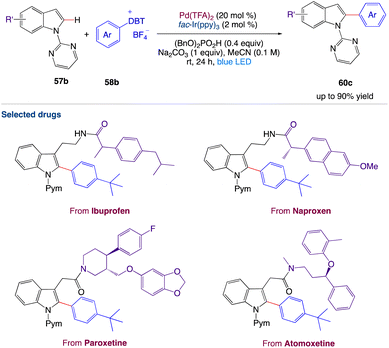 |
| | Scheme 31 Metallaphotoredox catalyzed 2-arylation of indole derivatives.57 | |
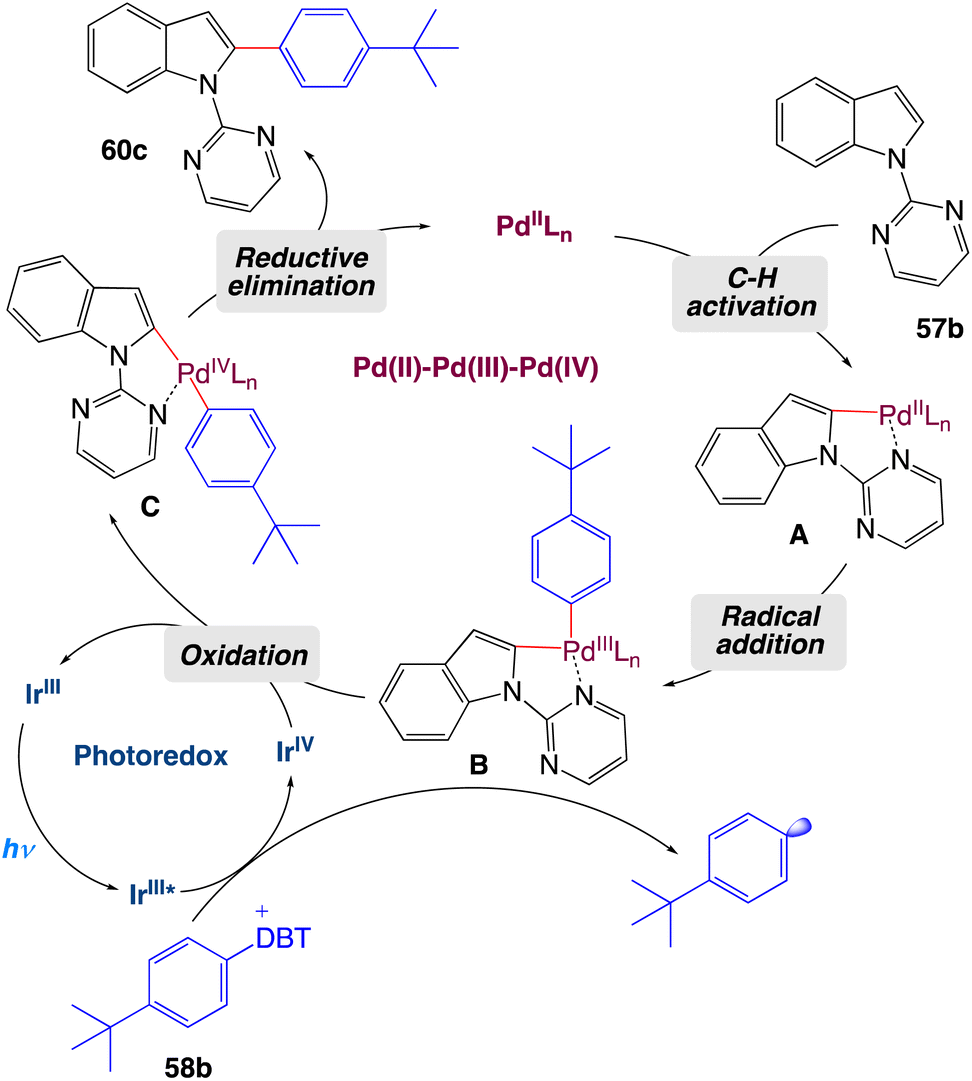
On the basis of mechanistic studies, the authors proposed a plausible catalytic cycle as shown in Scheme 32. Initially, palladacycle A forms from N-pyrimidylindole 57b and the palladium(II) catalyst, and an aryl radical is generated by a photocatalytic process in which the excited photosensitizer is oxidatively quenched by aryl-dibenzothiophenium salt 58b. Then the aryl radical is captured by A to form palladium complex B, which is oxidized by the high-valence photocatalyst to deliver intermediate C and complete the photoredox cycle. Finally, C undergoes a reductive elimination reaction to release arylated product 60c and regenerate the palladium(II) catalyst.
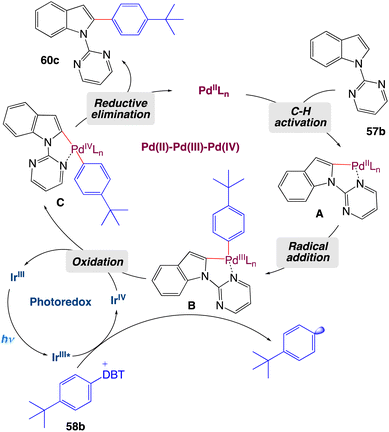 |
| | Scheme 32 Proposed mechanism of metallaphotoredox catalyzed 2-arylation of indole derivatives.57 | |
5. Titanocene metallaphotocatalysis
Titanocene/photoredox radical based dual catalysed transformations have been used more frequently in organic synthesis during the past few years as a result of their distinguishing qualities, including ease of generation, mild reaction conditions, and wide functional group compatibility. Titanocene, a moderate single-electron transfer (SET) reagent, is regarded as a powerful tool in modern radical chemistry because of its capacity to support a variety of fundamental radical-based transformations. Titanocene/photoredox dual catalysis has been utilized to carry out spirocyclization of epoxides and barbier allylation of aldehydes and ketones.
5.1. Metallaphotoredox catalysed spirocyclization of epoxides
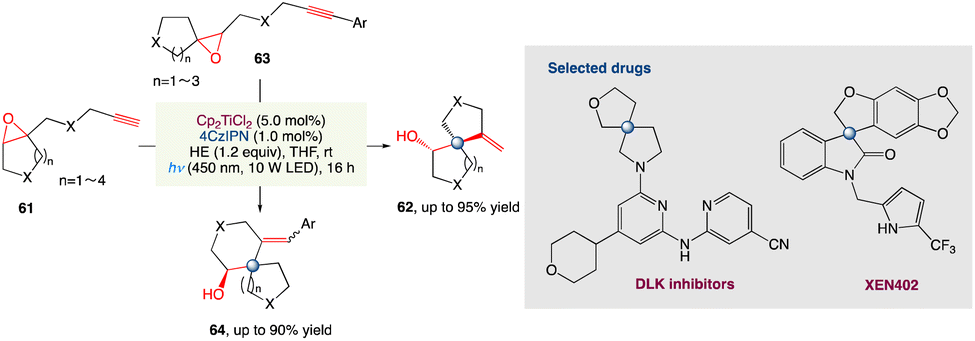
For the radical spirocyclization of epoxides under photocatalytic conditions, L. Shi et al.58 proposed a novel and greener dual catalyst system comprised of titanocene and the organic dye 4CzIPN. This method presents a strategically different way for the construction of heterospirocycles with a spiro all-carbon quaternary stereocenter that is operationally easy, scalable, and efficient. Unlike in metal reduction catalytic conditions, mechanistic studies reveal that Ti III is present in photo-catalytic conditions as Cp2TiCl which is readily accessible to epoxides. The photocatalytic conditions for the assembly of heterospirocycles showed exceptional high reactivity, as expected. The photocatalytic approach complements the metal reduction approach in this regard (Scheme 33).
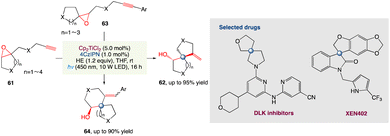 |
| | Scheme 33 Metallaphotoredox catalysed spirocyclization of epoxides.58 | |
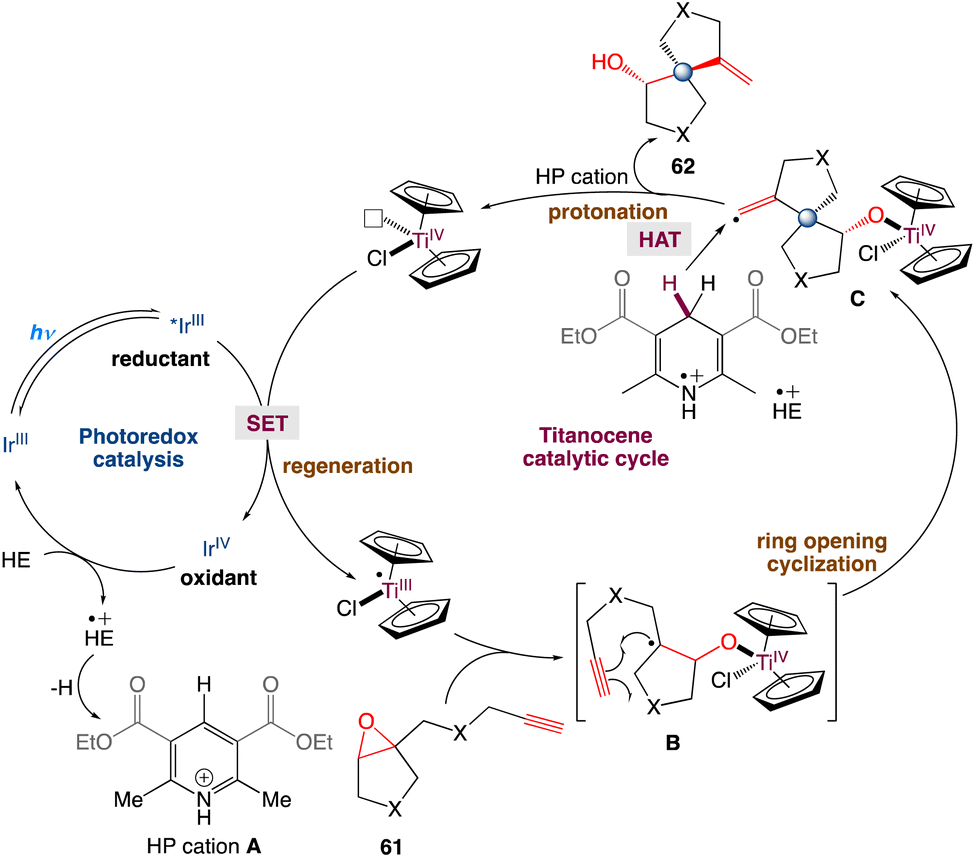
In the mechanism proposed by the authors, first, Cp2TiCl2 is reduced to Cp2TiIIICl by a photocatalyst (such as excited IrIII). The strong oxidant IrIV species is then reduced by HE to IrIII and produces HE+˙ concurrently. Cp2TiIIICl promoted the reductive opening of the epoxides. The resulting carbon radical B intramolecularly adds to the pendant alkyne to form the spirocyclic structure. The newly generated vinyl radical C can abstract a C4–H hydrogen atom of the HE radical cation (HE+˙). This is because HE+˙ has a low homolytic bond-dissociation free energy for the C4–H bond. Deuterium labeling experiments also exhibits that THF may offer a H source for hydrogen atom transfer (Scheme 34). The in situ produced Ti–oxygen bond is protonated by the HP cation and TiIV is released as well as the cyclization product. Finally, IrIII can regenerate Cp2TiIIICl for the next catalytic cycle.
 |
| | Scheme 34 Proposed mechanism for metallaphotoredox catalysed spirocyclization of epoxides.58 | |
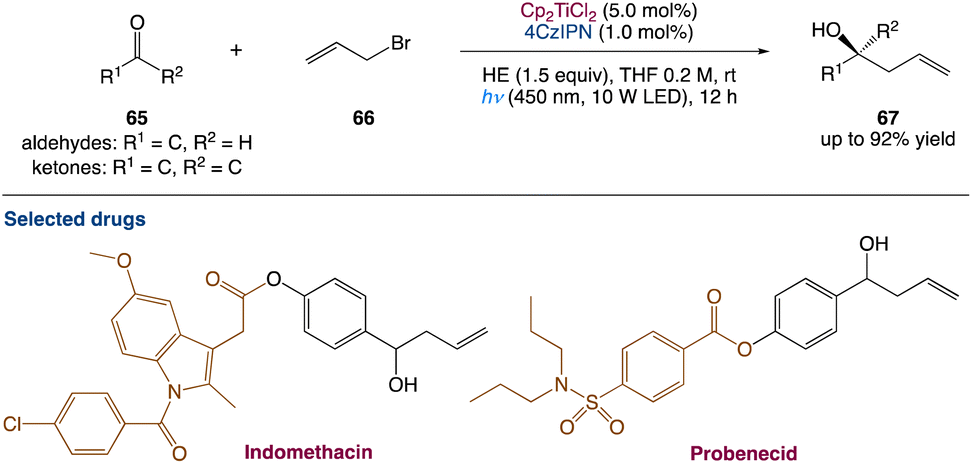
5.2. Metallaphotoredox catalysed barbier allylation of aldehydes and ketones
L. Shi et al.59 used dual titanium and photoredox catalysis to perform a photocatalytic Barbier-type allylation of different aldehydes and ketones with allyl halides for the synthesis of homoallylic alcohols. This environmentally benign process uses the organic dye 2,4,5,6-(9H-carbazol-9-yl)isophthalonitrile (4CzIPN) as a photocatalyst and Hantzsch ester (HE) as an electron donor instead of stoichiometric metallic reductants. Propargylation, benzylation, and prenylation reactions are used to demonstrate applicability of this method (Scheme 35).
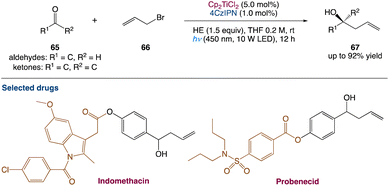 |
| | Scheme 35 Metallaphotoredox catalysed barbier allylation of aldehydes and ketones.59 | |
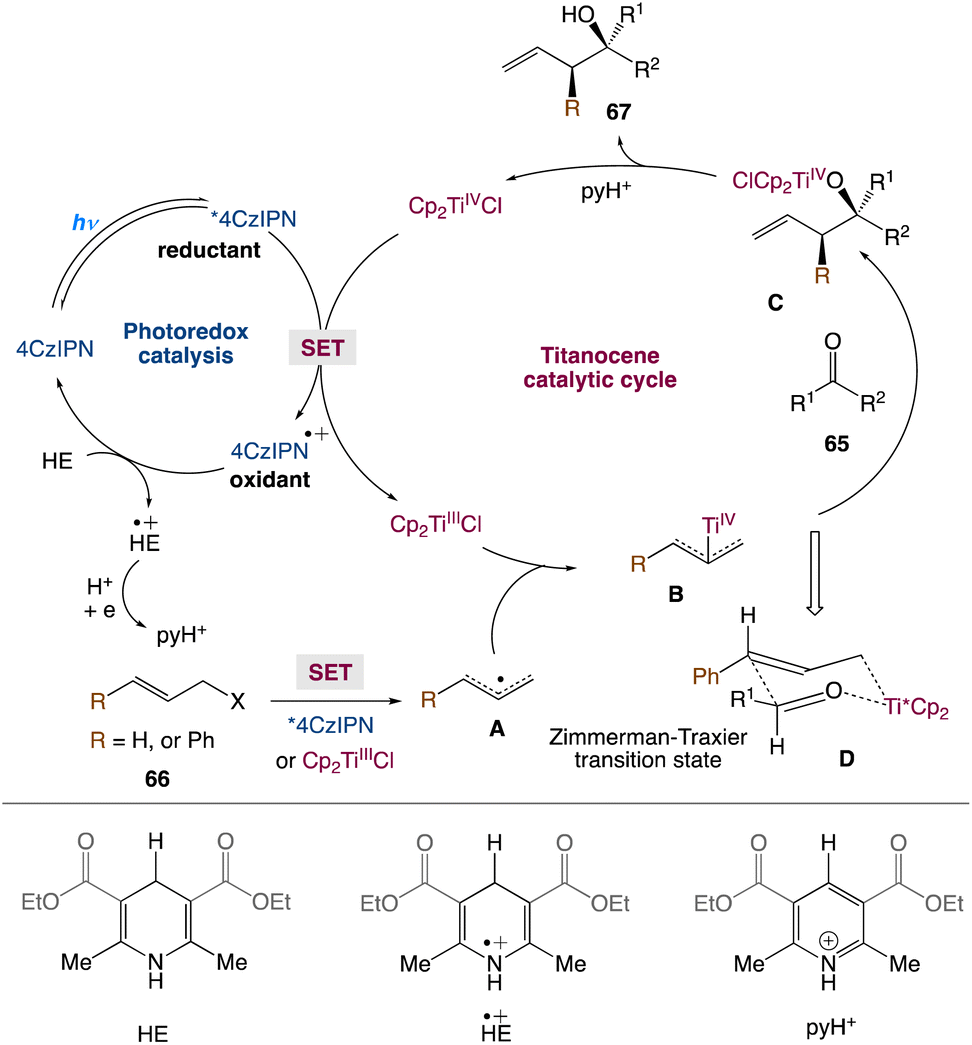
In the mechanism proposed by the authors, first, Cp2TiCl2 is reduced to Cp2TiIIICl via a photoexcited 4CzIPN (Scheme 36). The strong oxidant 4CzIPN+˙ species is then reduced by HE to produce HE+˙, which can further participate in electron transfer events and ultimately produce pyH+. The allyl bromide would be reduced by photoexcited 4CzIPN* to yield an allyl radical A. Alternatively, A can also be produced via the reaction of allyl bromide with Cp2TiIIICl. The resulting A can react with Cp2TiIIICl and yield nucleophilic allyltitanium species B. The coupling of the allyltitanium complex B with the carbonyl group would form a new C–C bond in the alkoxytitanium product C. Protonation of the Ti–oxygen bond by pyH+, which acts as a proton source, releases TiIV as well as the homoallylic alcohol product. Eventually, 4CzIPN can regenerate Cp2TiIIICl for the next catalytic cycle. The Zimmerman-Traxler transition state D, in which the oxygen of the aldehyde coordinates with the titanium, explains the anti-selectivity of homoallylic alcohols. The anti isomer is formed when the R group of an aldehyde and the phenyl group preferentially adopt an equatorial orientation.
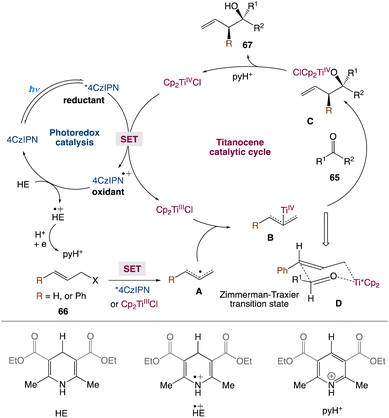 |
| | Scheme 36 Proposed mechanism for metallaphotoredox catalysed barbier allylation of aldehydes and ketones.59 | |
6. Chromium metallaphotocatalysis
Despite being far less studied in synthetic chemistry, the Cr/photoredox dual catalytic system offers a variety of opportunities to carry out challenging radical-to-polar crossover-based transformations. The applications of photoinduced chromium-catalyzed reactions included carbonyl propargylation, synthesis of amino alcohols and allylation of aldehydes.
6.1. Metallaphotoredox catalysed radical carbonyl propargylation
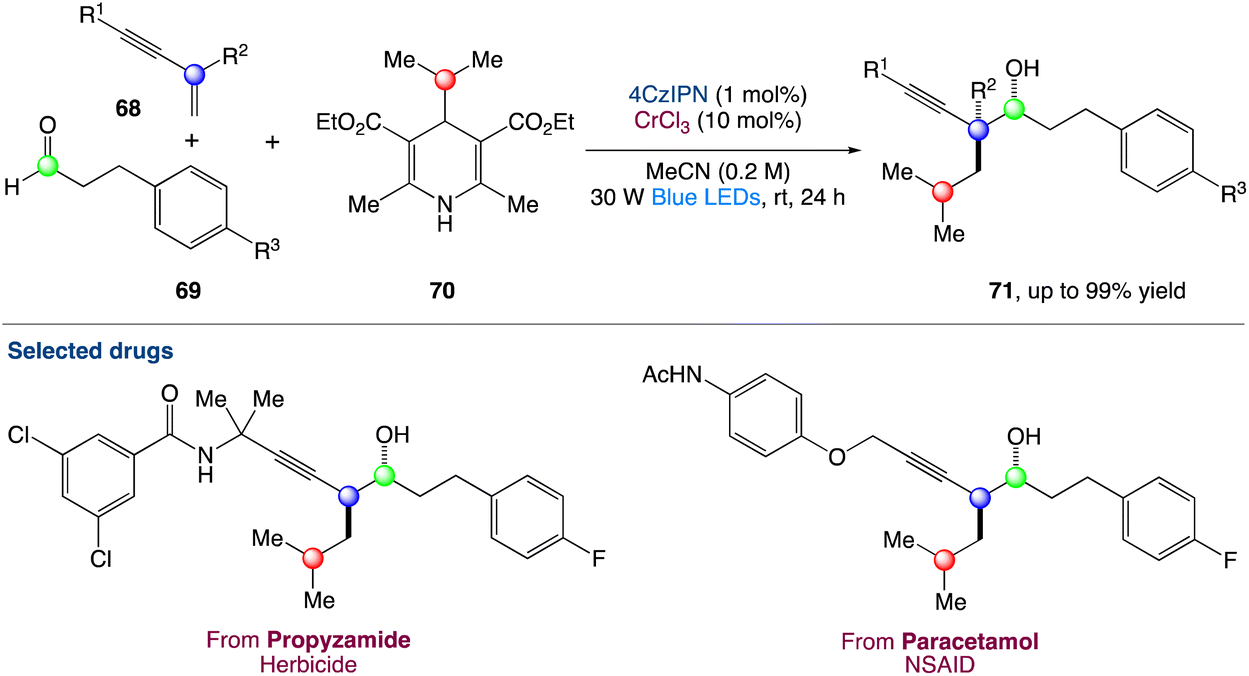
The radical carbonyl propargylation by dual chromium/photoredox catalysis was described by F. Glorius et al.60 A catalytic radical three-component coupling of 1,3-enynes, aldehydes, and appropriate radical precursors could yield a library of valuable homopropargylic alcohols with all-carbon quaternary centres. This multi-component redox-neutral reaction occurs under relatively mild conditions and has a high functional group tolerance. Interestingly, CrCl3 could be used as a chromium supply because it is bench-stable, non-toxic, and affordable (Scheme 37).
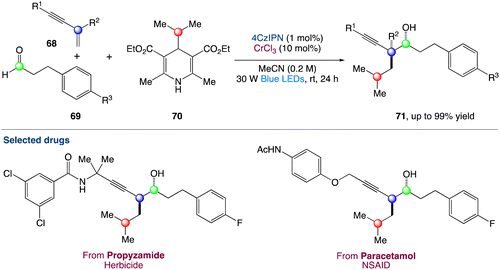 |
| | Scheme 37 Metallaphotoredox catalysed radical carbonyl propargylation.60 | |
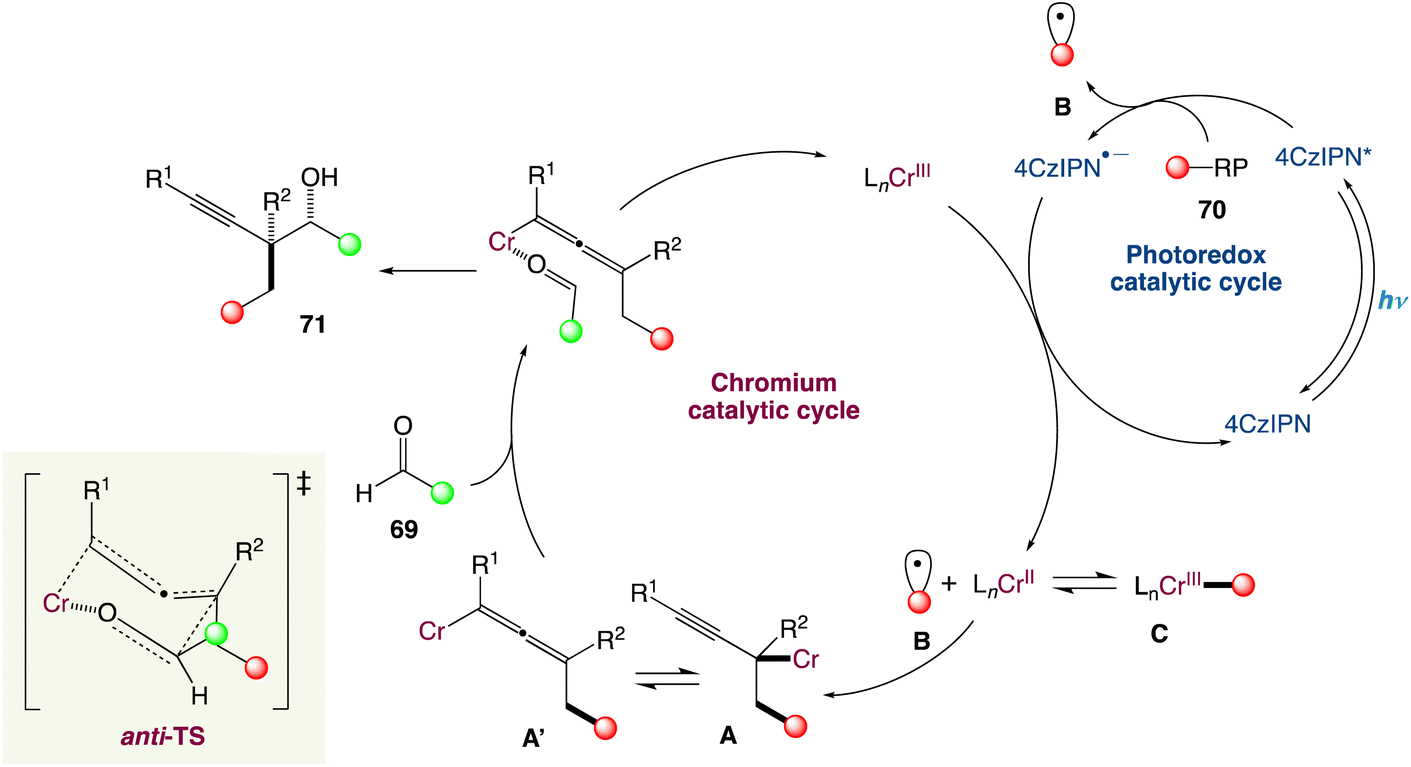
In the mechanism proposed by the authors, the alkyl radical B is generated from a suitable radical precursor following reductive quenching of the photoexcited 4-CzIPN (Scheme 38). Then the LnCr(III) species is reduced in situ to LnCr(II) (E1/2 = −0.65 V vs. SCE in H2O, E1/2 = −0.51 V vs. SCE in DMF) by the reduced photocatalyst PC˙− (for instance, organic dye 4CzIPN, E1/2 (PC/PC˙−) = −1.21 V vs. SCE in MeCN; PC-1, E1/2(IrII/IrIII) = −1.37 V vs. SCE in MeCN). The alkyl radical VII could either reversibly add to a low-valent LnCr(II) species to form the resting state Cr(III)-alkyl complex C or add to 1,3-enynes to generate a propargylic radical in equilibrium with its allenyl equivalent. After radical capture by the LnCr(II) species, propargylic Cr(III) intermediate IV and allenic species Cr(III) IV′ are generated. Interestingly, when attempting to obtain macrocyclic alcohols using a designed 1,3-enyne containing an aldehyde functional group in the end through cascade reaction by dual chromium/photoredox catalysis, no macrocyclization could be observed. Instead, they obtained a combination of acyclic alkyne and allene radical addition products, implying that intermediates A and A′. Finally, through an anti-transition state, either A or A′ can react with appropriate aldehydes 69 to give the final homopropargylic products 71, accounting for the observed diastereoselectivity. The decreased diastereoselectivity of the reaction when polysubstituted enynes are reacted can be attributed to a lower energy difference between the anti- and syn-transition states due to greater steric requirement of the substituents.
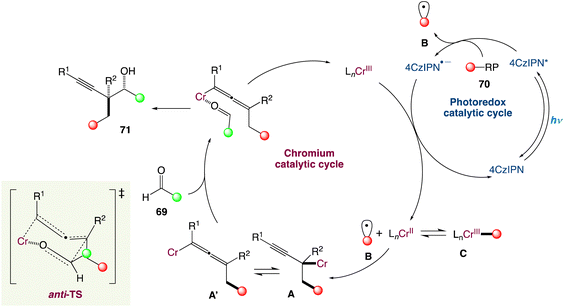 |
| | Scheme 38 Proposed mechanism for metallaphotoredox catalysed radical carbonyl propargylation.60 | |
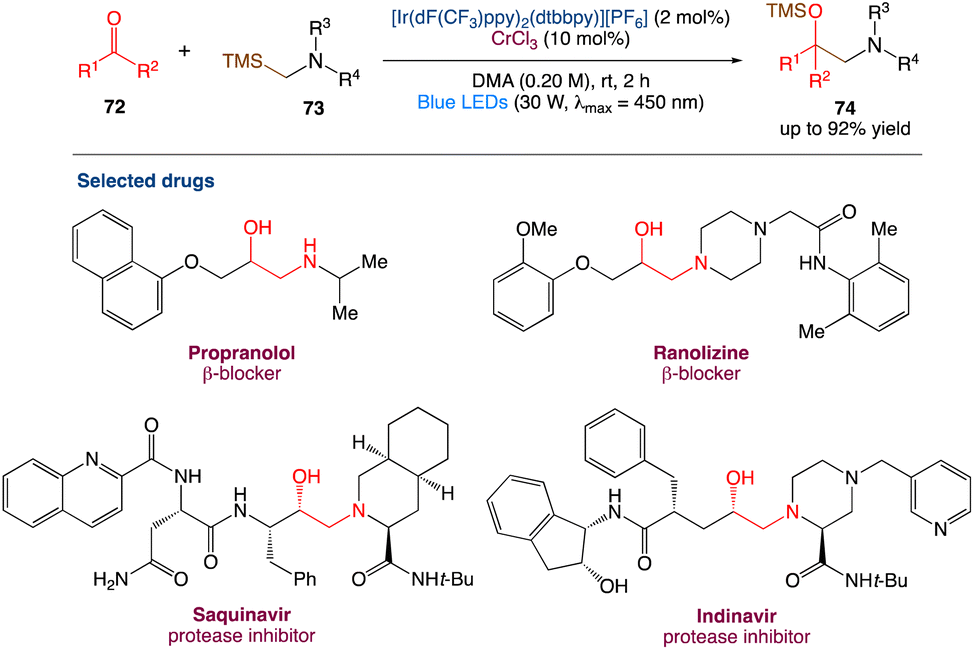
6.2. Metallaphotoredox catalysed synthesis of 1,2-amino alcohol
Protected 1,2-amino alcohols were also synthesised by F. Glorius et al.61 using carbonyl compounds (mostly aldehydes) and α-silyl amines. A Cr/photoredox dual catalytic system enables the in situ synthesis of α-amino carbanion equivalents, which act as nucleophiles, in this process. The aminoalkylation of ketones and an acyl silane, two classes of electrophiles that were previously unresponsive to the addition of alkyl–Cr reagents, was used to demonstrate this reaction. Overall, this reaction extends the range of Cr-mediated carbonyl alkylations and reveals a previously unknown retrosynthetic strategy for 1,2-amino alcohols (Scheme 39).
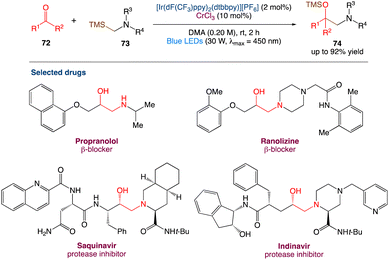 |
| | Scheme 39 Metallaphotoredox catalysed synthesis of 1,2-amino alcohol.61 | |
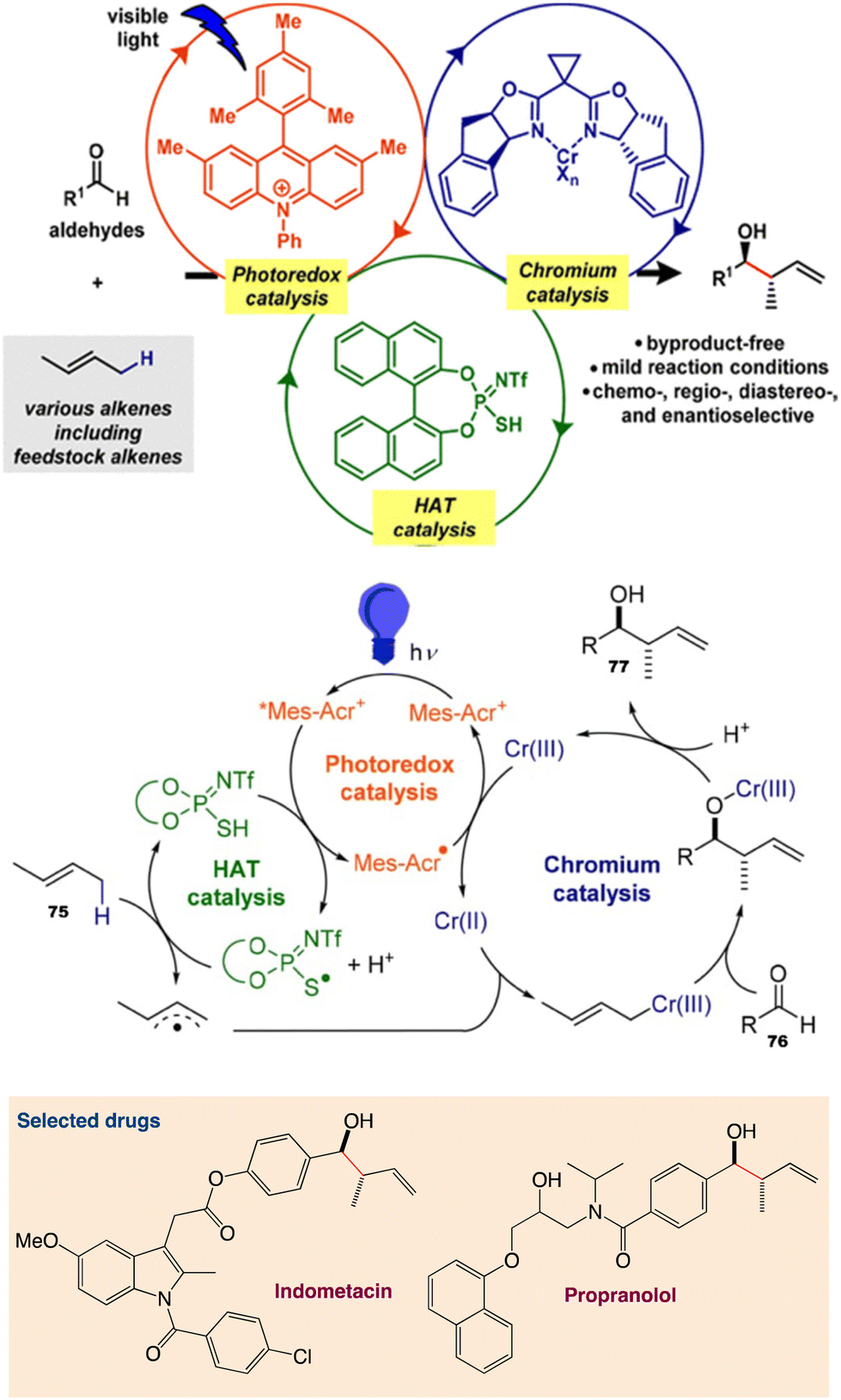
6.3. Allylation of aldehydes using unactivated alkenes
Kanai et al.62 reported a ternary hybrid catalyst system comprising a photoredox catalyst, a hydrogen-atom-transfer (HAT) catalyst, and a chromium complex catalyst, enabling catalytic allylation of aldehydes with simple alkenes, including feedstock lower alkenes. The reaction proceeded under visible-light irradiation at room temperature and with high functional group tolerance. The reaction was extended to an asymmetric variant by employing a chiral chromium complex catalyst (Scheme 40). The incorporation of a proper HAT catalytic cycle significantly extended the substrate scope of alkenes.
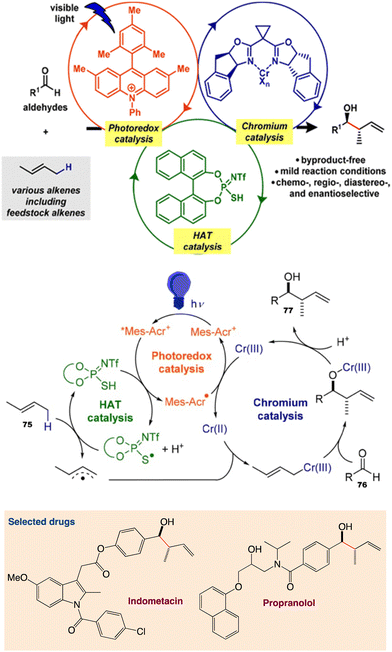 |
| | Scheme 40 Catalytic allylation of aldehydes using simple unactivated alkenes and proposed catalytic cycle. Reproduced from ref. 62 with permission from [American Chemical Society], copyright [2020]. | |
7. Conclusions
The synthetic community has recently been attracted towards a new dual catalytic platform generated via the fusion of photoredox and transition metal catalysis. Photoredox catalysis provides access to a wide variety of carbon and heteroatom-centered radicals, which have been used as nucleophilic coupling junctions in metallaphotocatalytic organic transformations. The merger of photoredox with transition metals provides an opportunity to produce transformations that were not conceivable earlier. The complementary nature of these two domains, combining them to create enhanced automated experimentation platforms, is quite promising in the pharmaceutical industry, and can be examined through developing LSF methodologies. Furthermore, LSF techniques have the potential to create a significant impact on sectors other than drug discovery programmes and can also be beneficial for the introduction of novel applications in agrochemistry, process chemistry, and chemical biology. As a result, we believe that the field of metallaphotocatalysis will grow and establish a new dimension in this area of research.
Conflicts of interest
There are no conflicts to declare.
References
-
A. Rodger, Encyclopedia of Biophysics, ed. G. C. K. Roberts, Springer, 2013, pp. 2714–2718 Search PubMed.
- S. Rovio, J. Yli-Kauhaluoma and H. Siren, Determination of neutral carbohydrates by CZE with direct UV detection, Electrophoresis, 2007, 28, 3129–3135 CrossRef CAS PubMed.
- A. Ault, The Nobel prize in chemistry for 2001, J. Chem. Educ., 2002, 79, 572–577 CrossRef CAS.
- C. P. Casey, 2005 Nobel prize in chemistry. Development of the E–olefin metathesis method in organic synthesis, J. Chem. Educ., 2006, 83, 192–195 CrossRef CAS.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Palladium–catalyzed cross– coupling: a historical contextual perspective to the 2010 Nobel prize, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- S. Reischauer and B. Pieber, Emerging concepts in photocatalytic organic synthesis, iScience, 2021, 24(3), 102209 CrossRef CAS.
- S. K. Sinha,
et al., Toolbox for distal C–H bond functionalizations in organic molecules, Chem. Rev., 2022, 122(6), 5682–5841 CrossRef CAS.
-
(a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, Photoredox catalysis in organic chemistry, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed;
(b) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS;
(c) N. W. Dow, A. Cabre and D. W. C. MacMillan, A general N-alkylation platform via copper metallaphotoredox and silyl radical activation of alkyl halides, Chem, 2021, 7, 1827–1842 CrossRef CAS.
- J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Alkyl carbon–carbon bond formation by nickel/photoredox cross-coupling, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed.
- M. N. Hopkinson, B. Sahoo, J. L. Li and F. Glorius, Dual catalysis sees the light: combining photoredox with organo-, acid, and transition- metal catalysis, Chem. – Eur. J., 2014, 20, 3874–3886 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Dual catalysis strategies in photochemical synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- M. Kojima and S. Matsunaga, The merger of photoredox and cobalt catalysis, Trends Chem., 2020, 2, 410–426 CrossRef CAS.
- A. Hossain, A. Bhattacharyya and O. Reiser, Copper's rapid ascent in visible-light photoredox catalysis, Science, 2019, 364, eaav9713 CrossRef.
- M. Neumeier, U. Chakraborty, D. Schaarschmidt, V. de la Pena O'Shea, R. Perez-Ruiz and A. Jacobi von Wangelin, Combined photoredox and iron catalysis for the cyclotrimerization of alkynes, Angew. Chem., Int. Ed., 2020, 59, 13473–13478 CrossRef CAS PubMed.
- X.-H. Ouyang, Y. Li, R.-J. Song, M. Hu, S. Luo and J. H. Li, Intermolecular dialkylation of alkenes with two distinct C(sp3)—H bonds enabled by synergistic photoredox catalysis and iron catalysis, Sci. Adv., 2019, 5, eaav9839 CrossRef CAS.
- D.-H. Wang, T.-S. Mei and J.-Q. Yu, Versatile Pd(II)-catalyzed C–H activation/aryl−aryl coupling of benzoic and phenyl acetic acids, J. Am. Chem. Soc., 2008, 130, 17676–17677 CrossRef CAS.
- O. Daugulis and V. G. Zaitsev, Anilide ortho-arylation by using C–H activation methodology, Angew. Chem., Int. Ed., 2005, 44, 4046–4048 CrossRef CAS.
- S. Yang, B. Li, X. Wan and Z. Shi, Ortho-arylation of acetanilides
via Pd(II)-catalyzed C–H functionalization, J. Am. Chem. Soc., 2007, 129, 6066–6067 CrossRef CAS.
- Y. Yun, J. Yang, Y. Miao, J. Sun and X. Wang, Recent advances in palladium(II)-catalyzed activation of aromatic ring C–H bonds, J. Saudi Chem. Soc., 2020, 24, 151–185 CrossRef CAS.
-
(a) J. Grover, G. Prakash, N. Goswami and D. Maiti, Traditional and, sustainable approaches for the construction of C-C bonds by harnessing C-H arylation, Nat. Commun., 2022, 13, 1085 CrossRef CAS PubMed;
(b) W. J. Zhou, Y. H. Zhang, Y. Y. Gui, L. Sun and D. G. Yu, Merging Transition-Metal Catalysis with Photoredox Catalysis: An Environmentally Friendly Strategy for C–H Functionalization, Synthesis, 2018,(17), 3359–3378 CrossRef CAS;
(c) L. Guillemard and J. W. Delord, When metal-catalyzed C–H functionalization meets visible-light photocatalysis, Beilstein J. Org. Chem., 2020, 16, 1754–1804 CrossRef CAS PubMed;
(d) A. Lipp, S. O. Badir and G. A. Molander, Stereoinduction in Metallaphotoredox Catalysis, Angew. Chem., Int. Ed., 2021, 60(4), 1714–1726 CrossRef CAS.
- J. Twilton, C. (Chip) Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The merger of transition metal and photocatalysis, Nat. Rev. Chem., 2017, 1(7), 0052, DOI:10.1038/s41570-017-0052.
- C. Vila, Merging visible–light–photoredox and nickel catalysis, ChemCatChem, 2015, 7, 1790–1793 CrossRef CAS.
- M. D. Levin, S. Kim and F. D. Toste, Photoredox catalysis unlocks single–electron elementary steps in transition metal catalyzed cross–coupling, ACS Cent. Sci., 2016, 2, 293–301 CrossRef CAS PubMed.
- J. J. Douglas, M. J. Sevrin and C. R. J. Stephenson, Visible light photocatalysis: applications and new disconnections in the synthesis of pharmaceutical agents, Org. Process Res. Dev., 2016, 20, 1134–1147 CrossRef CAS.
-
(a) P. Li, J. A. Terrett and J. R. Zbieg, Visible-light photocatalysis as an enabling technology for drug discovery: a paradigm shift for chemical reactivity, ACS Med. Chem. Lett., 2020, 11, 2120–2130 CrossRef CAS PubMed;
(b) R. Cannalire, S. Pelliccia, L. Sancineto, E. Novellino, G. C. Tron and M. Giustiniano, Visible light photocatalysis in the late-stage functionalization of pharmaceutically relevant compounds, Chem. Soc. Rev., 2021, 50, 766–897 RSC.
- R. G. Bergman, C–H activation, Nature, 2007, 446, 391–393 CrossRef CAS.
- L. Ackermann, R. Vicente and A. R. Kapdi, Transition- metal-catalyzed direct arylation of (hetero)arenes by C–H bond cleavage, Angew, Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS.
- L. McMurray, F. O'Hara and M. J. Gaunt, Recent developments in natural product synthesis using metal-catalysed C–H bond functionalization, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, C–H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS.
- L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Late-stage C–H functionalization offers new opportunities in drug discovery, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS.
- W. R. Gutekunst and P. S. Baran, C–H functionalization logic in total synthesis, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- J. Wencel-Delord and F. Glorius, C–H bond activation enables the rapid construction and late-stage diversification of functional molecules, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- E. J. E. Caro-Diaz, M. Urbano, D. J. Buzard and R. M. Jones, C–H activation reactions as useful tools for medicinal chemists, Bioorg. Med. Chem. Lett., 2016, 26, 5378–5383 CrossRef CAS PubMed.
- D. J. Abrams, P. A. Provencher and E. J. Sorensen, Recent applications of C–H functionalization in complex natural product synthesis, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC.
- B. Hong, T. Luo and X. Lei, Late-stage diversification of natural products, ACS Cent. Sci., 2020, 6, 622–635 CrossRef CAS.
- J. Börgel and T. Ritter, Late-stage functionalization, Chem, 2020, 6, 1877–1887 Search PubMed.
- J. C. Tellis,
et al., Single-electron transmetalation via photoredox/nickel dual catalysis: unlocking a new paradigm for sp3−sp2 cross-coupling, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS.
- J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Alkyl carbon–carbon bond formation by nickel/photoredox cross-coupling, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS.
-
(a) V. Srivastava, P. K. Singh and P. P. Singh, Novel one-pot Facile Synthesis of Thiopyranopyrazole Using [H mim]HSO4 Catalyst, Croat. Chem. Acta, 2014, 87(2), 91–95 CrossRef;
(b) V. Srivastava, P. K. Singh and P. P. Singh, Microwave-mediated InCl3catalysed three component efficient annelation of thiadiazinyl ring on 4-Amino-4H-[1,2,4]triazole-3-thiol, Chem. Heterocycl. Compd., 2014, 50(4), 573–578 CrossRef CAS;
(c) V. Srivastava, P. K. Singh and P. P. Singh, Eosin Y Catalyzed Visible-light-promoted One –Pot Facile Synthesis of 1,3,4- Thiadiazole, Croat. Chem. Acta, 2015, 88(1), 59–65 CrossRef CAS;
(d) V. Srivastava, P. K. Singh and P. P. Singh, Eosin Y Catalysed Visible light promoted aerobic oxidative cyclization of 2-Aminobenzothiazole, Croat. Chem. Acta, 2015, 88(3), 227–233 CrossRef CAS;
(e) V. Srivastava, P. K. Singh and P. P. Singh, Trifluoromethylation of Disubstituted Morpholines by Metal-Free Visible Light Photoredox Catalysis, Asian J. Chem., 2016, 28(10), 2159–2163 CrossRef CAS;
(f) V. Srivastava, P. K. Singh and P. P. Singh, [bmim]OH Catalyzed Four Component One-Pot Synthesis of Imidazo[4,5-C]Pyrazole-2-Thione-N-Nucleosides, Rev. Roum. Chim., 2016, 61(10), 755–761 Search PubMed;
(g) V. Srivastava, P. K. Singh and P. P. Singh, Visible Light Promoted Allylic C–H Oxidation, Croat. Chem. Acta, 2017, 90(3), 435–441 CrossRef CAS;
(h) V. Srivastava, P. K. Singh, S. Kanaujia and P. P. Singh, Photoredox catalysed synthesis of amino alcohol, New J. Chem., 2018, 42, 688 RSC;
(i) P. K. Singh, P. P. Singh and V. Srivastava, Facile Aerobic Photo-Oxidative Synthesis of Sulfinic Esters, Croat. Chem. Acta, 2018, 91(3), 383–387 CrossRef;
(j) V. Srivastava, P. K. Singh and P. P. Singh, Visible light photoredox catalysed amidation of carboxylic acids with amines, Tetrahedron Lett., 2019, 60, 40–43 CrossRef CAS;
(k) V. Srivastava, P. K. Singh and P. P. Singh, Photocatalysed eosin Y mediated C(sp3)-H alkylation of amine substrates via direct HAT, Tetrahedron Lett., 2019, 60, 1333–1336 CrossRef CAS;
(l) V. Srivastava, P. K. Singh and P. P. Singh, Eosin Y catalysed visible-light mediated aerobic oxidation of tertiary amines, Tetrahedron Lett., 2019, 60, 151041 CrossRef CAS;
(m) V. Srivastava, P. K. Singh and P. P. Singh, Visible Light Promoted Synthesis of Disubstituted 1,2,3-Thiadiazoles, Rev. Roum. Chim., 2020, 65(3), 221–226 CrossRef.
-
(a) V. Srivastava and P. P. Singh, Eosin Y catalyzed photoredox synthesis: a review, RSC Adv., 2017, 7, 31377–31392 RSC;
(b) V. Srivastava, P. K. Singh, A. Srivastava and P. P. Singh, Recent application of visible-light induced radicals in C–S bond formation, RSC Adv., 2020, 10, 20046 RSC;
(c) A. Srivastava, P. K. Singh, A. Ali, P. P. Singh and V. Srivastava, Recent Applications of Rose Bengal Catalysis in N-heterocycles: a short review, RSC Adv., 2020, 10, 39495 RSC;
(d) V. Srivastava, P. K. Singh, A. Srivastava and P. P. Singh, Synthetic applications of flavin photocatalysis: a review, RSC Adv., 2021, 11, 14251–14259 RSC;
(e) V. Srivastava and P. P. Singh, Recent advances of 4DPAIPN in photocatalytic transformations, Org. Biomol. Chem., 2021, 19, 313–321 RSC;
(f) P. P. Singh, P. K. Singh, M. Z. Beg, A. Kashyap and V. Srivastava, Recent applications of photoredox catalysis in O-heterocycles: A short review, Synth. Commun., 2021, 51(20), 3033–3058 CrossRef CAS;
(g) V. Srivastava, P. K. Singh, A. Srivastava, S. Sinha and P. P. Singh, Recent Advances of Dicyanopyrazine (DPZ) in Photoredox Catalysis, Photochemistry, 2021, 1, 237–246 Search PubMed;
(h) V. Srivastava, P. K. Singh, S. Tivari and P. P. Singh, Visible light photocatalysis in the synthesis of pharmaceutically relevant heterocyclic scaffolds, Org. Chem. Front., 2022, 9, 1485 RSC;
(i) V. Srivastava, P. K. Singh and P. P. Singh, Recent advances of visible-light photocatalysis in the functionalization of organic compounds, J. Photochem. Photobiol., C, 2022, 50, 100488 CrossRef CAS;
(j) V. Srivastava and P. P. Singh, Recent advances in visible-light graphitic carbon nitride (g-C3N4) photocatalysts for chemical transformations, RSC Adv., 2022, 12, 18245 RSC;
(k) P. P. Singh, S. Sinha, G. Pandey and V. Srivastava, Molybdenum disulfide (MoS2) based photoredox catalysis in chemical transformations, RSC Adv., 2022, 12, 29826 RSC.
- Z. Dong and D. W. C. MacMillan, Metallaphotoredox-enabled deoxygenative arylation of alcohols, Nature, 2021, 598, 451–456 CrossRef CAS.
- J. J. Perkins, V. W. Shurtleff, A. M. Johnson and A. E. Marrouni, Synthesis of C6-Substituted Purine Nucleoside Analogues via Late- Stage Photoredox/Nickel Dual Catalytic Cross-Coupling, ACS Med. Chem. Lett., 2021, 12, 662–666 CrossRef CAS PubMed.
- W. Dong, S. O. Badir, X. Zhang and G. A. Molander, Accessing Aliphatic Amines in C−C Cross-Couplings by Visible Light/Nickel Dual Catalysis, Org. Lett., 2021, 23, 4250–4255 CrossRef CAS PubMed.
- L. Guo, M. Yuan, Y. Zhang, F. Wang, S. Zhu, O. Gutierrez and L. Chu, General Method for Enantioselective Three-Component Carboarylation of Alkenes Enabled by Visible-Light Dual Photoredox/Nickel Catalysis, J. Am. Chem. Soc., 2020, 142(48), 20390–20399 CrossRef CAS PubMed.
- C. H. Xu, J. H. Li, J. N. Xiang and W. Deng, Merging Photoredox/Nickel Catalysis for Cross-Electrophile Coupling of Aziridines with Pyridin-1-ium Salts via Dearomatization, Org. Lett., 2021, 23, 3696–3700 CrossRef CAS.
- P. Zhou, X. Li, D. Wang and T. Xu, Dual Nickel- and Photoredox-Catalyzed Reductive Cross-Coupling to Access Chiral Trifluoromethylated Alkanes, Org. Lett., 2021, 23, 4683–4687 CrossRef CAS PubMed.
- L. Huang, T. Ji and M. Rueping, Remote Nickel-Catalyzed Cross-Coupling Arylation via Proton-Coupled Electron Transfer-Enabled C–C Bond Cleavage, J. Am. Chem. Soc., 2020, 142(7), 3532–3539 CrossRef CAS PubMed.
- M. Zhu and S. Messaoudi, Diastereoselective Decarboxylative Alkynylation of Anomeric Carboxylic Acids Using Cu/Photoredox Dual Catalysis, ACS Catal., 2011, 11, 6334–6342 CrossRef.
- Y. Wu, K. Chen, X. Ge, P. Ma, Z. Xu, H. Lu and G. Li, Redox-Neutral P(O)−N Coupling between P(O)−H Compounds and Azides via Dual Copper and Photoredox Catalysis, Org. Lett., 2020, 22(15), 6143–6149 CrossRef CAS PubMed.
- P. J. Sarver, V. Bacauanu, D. M. Schultz, D. A. DiRocco, Y. Lam, E. C. Sherer and D. W. C. MacMillan, The merger of decatungstate and copper catalysis to enable aliphatic C(sp3)–H trifluoromethylation, Nat. Chem., 2020, 12, 459–467 CrossRef CAS PubMed.
- H. Zhang, Z. Wei, A. H. Zhang and S. Yu, Access to Cyanoimines Enabled by Dual Photoredox/Copper- Catalyzed Cyanation of O–Acyl Oximes, Org. Lett., 2020, 22(18), 7315–7320 CrossRef CAS.
- J. He, G. Chen, B. Zhang, Y. Li, J. R. Chen, W. J. Xiao, F. Liu and C. Li, Catalytic Decarboxylative Radical Sulfonylation, Chem, 2020, 6, 1149–1159 CAS.
- A. Saha, S. Guin, W. Ali, T. Bhattacharya, S. Sasmal, N. Goswami, G. Prakash, S. K. Sinha, H. B. Chandrashekar, S. Panda, S. S. Anjana and D. Maiti, Photoinduced Regioselective Olefination of Arenes at Proximal and Distal Sites, J. Am. Chem. Soc., 2022, 144, 1929–1940 CrossRef CAS PubMed.
-
(a) J. Zheng, A. Nikbakht and B. Breit, Dual Palladium/Photoredox-Catalyzed Enantioselective and Regioselective Decarboxylative Hydroaminoalkylation of Allenes, ACS Catal., 2021, 11, 3343–3350 CrossRef CAS;
(b) K. C. Cartwright and J. A. Tunge, Organophotoredox/Palladium Dual Catalytic Decarboxylative Csp3-Csp3 Coupling of Carboxylic Acids and π-electrophiles, Chem. Sci., 2020, 11, 8167–8175 RSC.
- S. K. Bhunia, P. Das, S. Nandi and R. Jana, Carboxylation of Aryl Triflates with CO2 Merging Palladium and Visible-Light-Photoredox Catalysts, Org. Lett., 2019, 21(12), 4632–4637 CrossRef CAS PubMed.
- M. K. Manna, G. Bairy and R. Jana, Dual Visible-light Photoredox and Palladium(II) Catalysis for Dehydrogenative C2-Acylation of Indoles at Room Temperature, Org. Biomol. Chem., 2017, 15, 5899–5903 RSC.
- X. Wang, X. Xun, H. Song, Y. Liu and Q. Wang, Palladium Metallaphotoredox-Catalyzed 2−Arylation of Indole Derivatives, Org. Lett., 2022, 24, 4580–4585 CrossRef CAS PubMed.
- S. Lin, Y. Chen, F. Li, C. Shi and L. Shi, Visible-light-driven spirocyclization of epoxides via dual titanocene and photoredox catalysis, Chem. Sci., 2020, 11, 839–844 RSC.
- F. Li, Y. Chen, S. Lin, C. Shi, X. Li, Y. Sun, Z. Guo and L. Shi, Visible-light-mediated Barbier allylation of aldehydes and ketones via dual titanium and photoredox catalysis, Org. Chem. Front., 2020, 7, 3434–3438 RSC.
- H. M. Huang, P. Bellotti, C. Daniliuc and F. Glorius, Radical Carbonyl Propargylation by Dual Catalysis, Angew. Chem., Int. Ed., 2020, 60(5), 2464–2471 CrossRef PubMed.
- J. L. Schwarz, R. Kleinmans, T. O. Paulisch and F. Glorius, 1,2-Amino Alcohols via Cr/Photoredox Dual Catalyzed Addition of α- Amino Carbanion Equivalents to Carbonyls, J. Am. Chem. Soc., 2020, 142(5), 2168–2174 CrossRef CAS PubMed.
- S. Tanabe, H. Mitsunuma and M. Kanai, Catalytic Allylation of Aldehydes Using Unactivated Alkenes, J. Am. Chem. Soc., 2020, 142(28), 12374–12381 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Pravin K.
Singh
b and
Vishal
Srivastava
a,
Pravin K.
Singh
b and
Vishal
Srivastava