
Ruthenium-catalyzed divergent deaminative and denitrative C–N cleavages: facile access to quinoxalines†
Shanshan
Liu
 *a,
Jiahui
Liang
a,
Pingjun
Zhang
a,
Zhenzhen
Li
a,
Lin-Yu
Jiao
*b,
Wei
Jia
a,
Yangmin
Ma
a and
Michal
Szostak
*ac
*a,
Jiahui
Liang
a,
Pingjun
Zhang
a,
Zhenzhen
Li
a,
Lin-Yu
Jiao
*b,
Wei
Jia
a,
Yangmin
Ma
a and
Michal
Szostak
*ac
aShaanxi Key Laboratory of Chemical Additives for Industry, College of Chemistry and Chemical Engineering, Shaanxi University of Science and Technology, Xi'an 710021, P. R. China. E-mail: liushanshan@sust.edu.cn
bSchool of Chemical Engineering, Northwest University, Xi'an, Shaanxi 710069, P. R. China. E-mail: lyjiao@nwu.edu.cn
cDepartment of Chemistry, Rutgers University, 73 Warren Street, Newark, New Jersey 07102, USA. E-mail: michal.szostak@rutgers.edu
First published on 16th November 2022
Abstract
Divergent deaminative and denitrative C–N cleavages and cascade transformations using amines and nitroalkanes have been developed under a highly effective and environmentally-friendly catalytic system (RuCl3/TFE). This protocol provides a versatile toolbox for the construction of quinoxalines under redox-neutral and mild conditions. Specifically, Ru-catalyzed acceptorless dehydrogenation and hydrogen borrowing strategies have been successfully implemented in the activation of amines, which is distinct from the dehydrogenation of frequently used alcohol as a hydrogen donor, enabling the deaminative coupling of amines with 2-arylanilines and 2-nitroarylpyrroles in the absence of additives. Crucially, unprecedented Ru-catalyzed denitrative C–NO2 cleavages have been realized, wherein nitroalkanes were explored as unconventional electrophilic C synthons to accomplish [5 + 1] annulation. This identified process features exceedingly simple, environmentally-friendly conditions, ease of operation, a broad substrate scope, and the capacity to synthesise antileishmanial agents and bipolar host materials for OLEDs.
Nitrogen containing compounds are ubiquitous in many organic molecules and biomacromolecules.1 Therefore, catalytic cleavages of C–N bonds and ensuing the utilization of newly formed C-fragments open the door for the facile preparation of compounds with high structural diversity and complexity.2 In this respect, amides and amines have been extensively employed as carbon sources via C–N bond disconnection.3–6 Generally, the non-carbonyl C–N bond is considered to be less reactive than the amide C–N bond.7a In this context, tremendous efforts have been devoted to activating amines through transition-metal-catalyzed oxidative addition, imine or iminium species formation, and ammonium species formation.7 Among the established protocols, oxidative deamination via imine or iminium species is a common and effective strategy for C–N bond cleavage,2b where the oxidation of the C–H bond adjoining the nitrogen atom typically requires the presence of oxidants (Scheme 1a).2b,8 Conversely, oxidant-free catalytic dehydrogenations or borrowing hydrogen methodologies are significantly attractive from the viewpoint of green chemistry due to their excellent redox economy and high atom-efficiency.9 However, they are usually applied to alcohols as hydrogen donors,10 and are seldom explored in the activation of amines,11 especially toward the synthesis of heterocyclic frameworks (Scheme 1b). A few reports for transition-metal-catalyzed dehydrogenative and deaminative annulation of amines have been disclosed, which allowed for the construction of select architectures using amines as hydrogen donors and C synthons (Scheme 1b, left and bottom).12 In these transformations, the metal catalyst abstracts the hydrogen atom from the amine, leading in situ to a metal-hydride species and reactive imine intermediate without the aid of oxidants (Scheme 1b, top). In particular, hydrogen borrowing deaminative coupling of amines and nitroaromatics, which in some cases involves the utilization of hydrogen from the dehydrogenation process for the reduction of nitroaromatics, has shown high step-economy since anilines are usually obtained by reducing nitroaromatics. Benefitting from this strategy, [3 + 3], [4 + 1], and [5 + 1] annulations have been achieved for the assembly of N-heterocycles catalyzed by Ru, Pd, and Fe (Scheme 1b, left and bottom).13 The sole example of activated carbon-catalyzed [5 + 1] annulation, which only allowed benzylic primary deamination via the hydrogen transfer pathway, has been demonstrated (Scheme 1b, right).14 Despite these achievements, the aforementioned transformations suffer from a narrow substrate scope and the requirement of Lewis acid additives, external hydrogen acceptors, or extremely high temperatures, which limit their practical applications. Thus, it would be highly desirable to explore new and general methods for the activation and transformation of C–N bonds to accommodate a broad range of nitrogen containing substrates under redox-neutral and mild conditions. More significantly, compared to deaminative disconnection via the C–N bond amine cleavage, the denitrative cleavage of C–NO2 has been rarely accomplished.15 To date, Ru-catalyzed denitrative transformations have not been realized, and especially the catalytic denitrative annulation using nitroalkanes as an electrophilic C synthon has been entirely unprecedented.
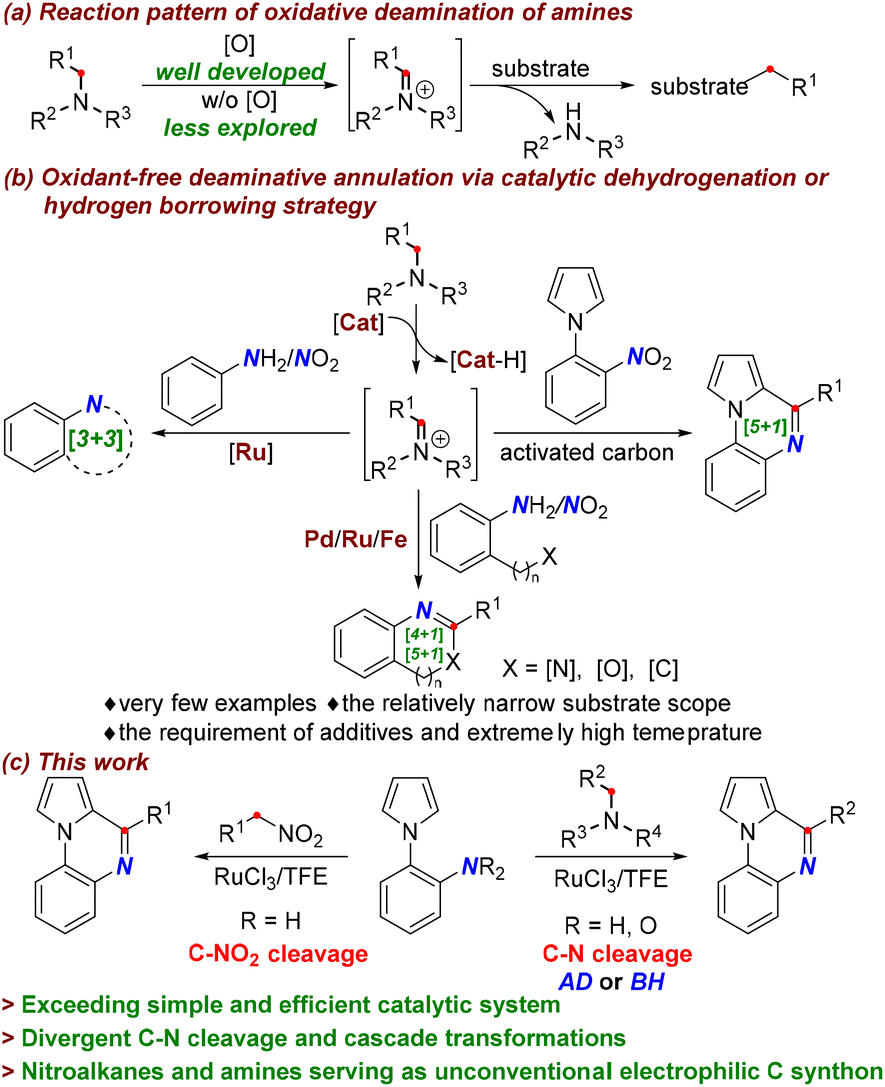 | ||
| Scheme 1 Activation and transformation of C–N bonds. | ||
Quinoxalines are privileged molecular scaffolds that exhibit various pharmacological and biological activities. Consequently, various methods for the synthesis of quinoxalines from 2-arylanilines have been established using carbon synthons such as aldehydes, alcohols, ethers, polyethylene glycol, and so on.16 Despite the great progress that has been made, most previous methods generally rely on the requirement of harsh acidic conditions or excess amounts of oxidants. With our continuing interest in developing green and practical synthetic methods for the construction of this scaffold,17 herein, we communicate the Ru-catalyzed divergent [5 + 1] annulation for the cascade synthesis of quinoxalines using amines and nitroalkanes as unconventional and inert electrophilic carbon sources by C–N activation (Scheme 1c). Notably, deaminative coupling of amines with 2-arylanilines and 2-nitroarylpyrroles has been achieved under redox-neutral conditions via acceptorless dehydrogenation (AD) and borrowing hydrogen (BH) strategies. The RuCl3 catalyst with the aid of TFE (TFE = 2,2,2-trifluoroethanol) establishes an effective catalytic system that can tolerate numerous readily available amine feedstocks without any additional additives, which is distinct from oxidative deaminations requiring oxidants or various additives. More strikingly, the unprecedented Ru-catalyzed denitrative C–NO2 cleavage has been realized, which provides a new and attractive avenue to access quinoxalines by employing nitroalkanes as electrophilic C1 synthons.
We commenced this study with a model reaction between 2-pyrrolylaniline (1a) and triethylamine (2a) using a ruthenium catalyst with the assistance of AgTFA. Fortunately, the desired product was obtained in 41% yield in the absence of a sacrificial hydrogen acceptor (Table 1, entry 1). Subsequently, a range of solvents were investigated to further improve the yield. An obvious enhancement in the yield was observed with TFE or HFIP (entries 5 and 6), while all other solvents led to a decreased yield of varying degrees (entries 1–4). Interestingly, the product with 10% yield was observed only in the presence of TFE (entry 8). These findings implied that the acidic alcohol solvent might not only serve as a ligand, but its acidity was also beneficial for dehydrogenation. With these results in hand, the reaction was conducted in TFE without additives; however, the yield dropped significantly (entry 7). Hence, further effort was made on catalyst screening to circumvent the need for additives. It was found that RuCl3 could restore the high yield (entry 13), which was attributed to the increase in the nucleophilicity of Ru(III) to facilitate the dehydrogenation process. Furthermore, the reaction temperature and atmosphere were examined; the results showed that the transformation could proceed well at 120 °C in air without the requirement of much higher temperature and inert conditions (entries 14–16). Importantly, the control reactions showed that the yield did not drop greatly with the substoichiometric amount of amine, which revealed that triethylamine could be reused through multiple C–N cleavages to release ammonia in this process. Gratifyingly, the combination of RuCl3 and TFE established a highly efficient system to promote the reaction in excellent yield with straightforward, environmentally-friendly conditions and operational simplicity. The optimal result obtained by making variations in the reaction conditions is outlined in entry 16 of Table 1.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | Yield (%) |
| Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), catalyst (5 mol%), additive (20 mol%), solvent (0.5 mL), 100 °C, Ar, 24 h.a Isolated yield.b 120 °C.c 140 °C.d Air instead of Ar, 120 °C.e 0.3 equivalents of triethylamine were used. | ||||
| 1 | [Ru(p-cymene)Cl2]2 | AgTFA | 1,4-Dioxane | 41 |
| 2 | [Ru(p-cymene)Cl2]2 | AgTFA | i-PrOH | 22 |
| 3 | [Ru(p-cymene)Cl2]2 | AgTFA | DMSO | Trace |
| 4 | [Ru(p-cymene)Cl2]2 | AgTFA | THF | 40 |
| 5 | [Ru(p-cymene)Cl2]2 | AgTFA | HFIP | 82 |
| 6 | [Ru(p-cymene)Cl2]2 | AgTFA | TFE | 84 |
| 7 | [Ru(p-cymene)Cl2]2 | — | TFE | 45 |
| 8 | — | — | TFE | 10 |
| 9 | Ru(PPh3)3Cl2 | — | TFE | 30 |
| 10 | [Cp*IrCl2]2 | — | TFE | 54 |
| 11 | Ru3(CO)12 | — | TFE | 63 |
| 12 | [Cp*RhCl2]2 | — | TFE | 68 |
| 13 | RuCl3 | — | TFE | 80 |
| 14b | RuCl3 | — | TFE | 89 |
| 15c | RuCl3 | — | TFE | 84 |
| 16d | RuCl3 | — | TFE | 87 (72e) |

Under the optimal conditions, the generality of the deaminative coupling was investigated with diverse amines (Scheme 2). In general, a wide range of amines including primary, secondary, and tertiary amines 2a–2m could be smoothly converted into the corresponding products in moderate to excellent yields. Intriguingly, the activations of rigid amines such as 1,3,5-trimethyl-hexahydro-s-triazine 2c and hexamethylenetetramine 2d were realized, where both acted as methane sources to provide the same quinoxaline scaffold 3ac. However, a lower yield was obtained with 2d since it underwent two C–N cleavages. Importantly, the unsymmetrical amine 2g consisting of a differently substituted moiety resulted in a mixture of products predominantly through benzylic C–N cleavage mode. Remarkably, triallylamine served as an internal hydrogen acceptor to deliver the reduction product 3ab through the hydrogen transfer pathway. Curiously, diisopropylamine 2j was well compatible to afford the dihydroquinoxaline product. Indeed, the steric effect was evident as it gave an inferior result to that obtained by the less sterically-hindered diethylamine 2i. Moreover, primary amines are more challenging substrates due to the poor leaving ability of the free NH2 group, which led to a slightly lower yield compared to secondary and tertiary amines. Satisfyingly, the dehydrogenative coupling method allowed for benzylic α-primary and tertiary deamination, furnishing the products with the incorporation of aryl and thienyl functionalities (3al and 3am).
 | ||
| Scheme 2 Substrate scope of amines. | ||
The scope was further extended to ortho-indolyl/pyrrolyl anilines 1b–1x by fine-tuning the electronic properties and the position of the substituents (Scheme 3). A similar electronic effect was observed in both cases with the preference for electron-rich substrates, although lower yields were generally obtained with indole substrates. In addition, the detrimental effect of steric hindrance was observed in 1f and 1r. Notably, the hydride-iodination product was obtained using the iodo-pyrrolylaniline substrate in this catalytic system, while other halogen substituents remained intact 1kvs.1l–1n. Considering that TFE could function as a hydrogen donor to promote the Ru-catalyzed hydrogenation of nitroaromatics, the deaminative coupling was devised using 2-nitroaryl pyrroles 4a–4f directly. Gratifyingly, a variety of pyrrolylquinoxalines were obtained in good yields via hydrogen transfer annulation under the same Ru catalytic system. The substrates bearing electron donating groups were superior to the electron withdrawing substituents on the phenyl ring.
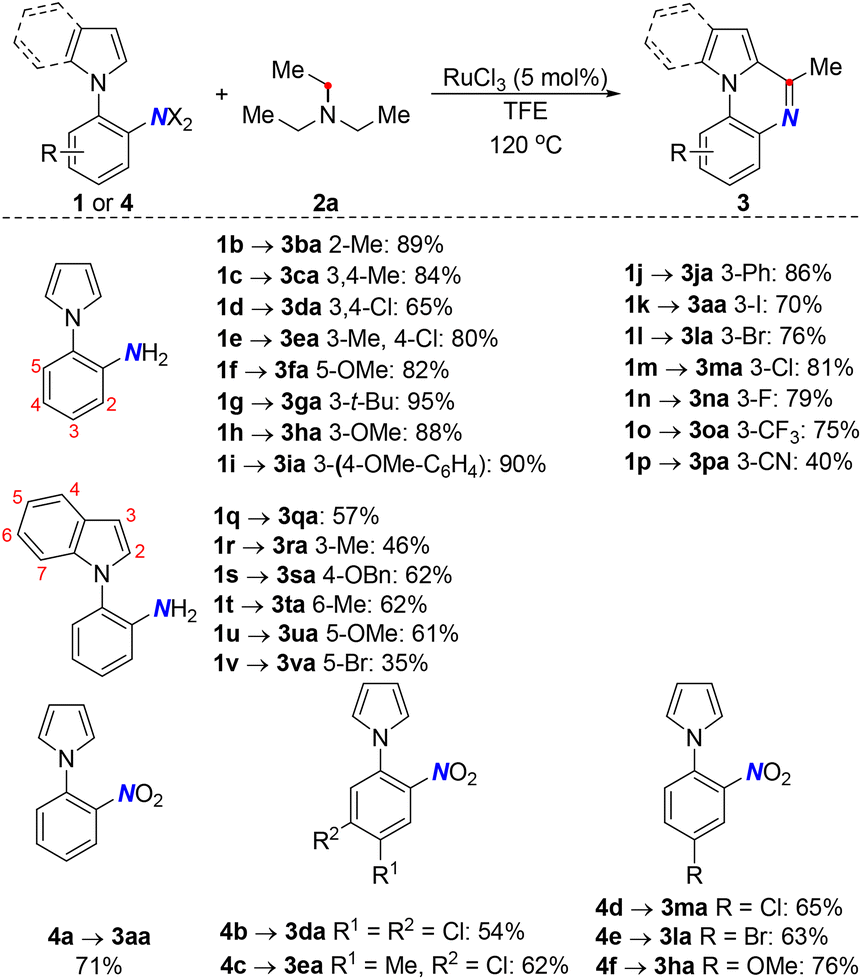 | ||
| Scheme 3 Substrate scope of 2-arylanilines and 2-nitroaryl pyrroles. | ||
Unlike the deaminative C–N activation of amines, denitrative C–N activation of nitroalkanes as an electrophilic carbon source remains difficult due to the inherent α-C-nucleophilicity. Nitromethane can be converted to iminium cations in the presence of zinc triflate, which serves as an electrophilic nitrile source.18 We envisioned that nitroalkanes could be activated by RuCl3 as inverted electrophiles to participate in the denitrative C–N transformation. To our delight, the coupling of pyrrolylaniline with various nitroalkanes was unprecedently realized, leading to the denitrative formation of quinoxalines in modest to good yields under Ru catalysis (Scheme 4). In this transformation, nitromethyl arenes 5c–5h afforded higher yields than other aliphatic nitro compounds 5a–5b due to the formation of a stable benzylic imine ion intermediate.
 | ||
| Scheme 4 Substrate scope of nitroalkanes. | ||
Next, we embarked on assessing the practicality and synthetic utility of this novel method. To our delight, this methodology was applicable to enable facile access to an antileishmanial agent 3hn from tripentylamine (Scheme 5a). Moreover, further derivatizations of several products were performed. Upon treatment with benzaldehyde, the methyl group of 3aa could be easily transformed into styryl functional units, leading to the formation of an antileishmanial agent 7 (Scheme 5b). Furthermore, given the unique photophysical, thermal, and electrochemical properties of quinoxaline-based bipolar host materials, we established a synthetic method to introduce carbazole and triphenylamine as electron donors on the quinoxaline skeleton through the Ullmann reaction and Suzuki cross-coupling (Scheme 5c and d). Moreover, UV-vis absorption, photoluminescence (PL), and cyclic voltammetry (CV) measurements were performed for both compounds (see the ESI†). Interestingly, the new compound 10 showed enhancement of the fluorescence intensity compared to an organic light-emitting diode (OLED) material 8, clearly indicating its potential use as a host material for highly efficient PhOLEDs.
 | ||
| Scheme 5 The synthetic utility of this protocol. | ||
From the viewpoint of cost efficiency and sustainability, our effort was devoted to recycle the catalyst in a 2 mmol scale reaction. After completion of the reaction, the catalyst was filtered and reused four times with 80%, 70%, 62%, and 55% yields, respectively. The quantitative green metrics of 3aa in this work and representative references,16a,d,e,18 including atom economy (AE), E-factor, carbon efficiency (CE), reaction mass efficiency (RME), mass intensity (MI), solvent intensity (SI), and mass productivity (MP), were next evaluated (Scheme 6). All values were calculated from the same starting material 2-nitrobenzyl pyrrole 4a. The CE and RME values were higher in this protocol calculated by a separate step (this work 1) compared to other methods calculated in the same way, while the AE value was lower compared to other methods. To our delight, better AE and EI values were observed for the borrowing hydrogen strategy applied in this work, indicating that this protocol is more efficient and more beneficial for the environment.
 | ||
| Scheme 6 Comparison of the green metrics of 3aa between this work and representative references. This work 1: the values were calculated from 2-nitrobenzyl pyrrole by a separate step. This work 2: the values were calculated from 2-nitrobenzyl pyrrole in one step through the hydrogen borrowing strategy. | ||
A series of control experiments were performed to shed light on the mechanism of this divergent deaminative/denitrative C–N functionalization (Scheme 7). Firstly, hydrogen gas rather than ethyl aldehyde was detected by gas chromatography (GC) when triethylamine was subjected to the standard reaction conditions (eqn (1)). To further verify if aldehyde was involved in the deaminative process, 20 equivalents of water were added to facilitate hydrolysis of the iminium ion, which led to a reduced yield (eqn (2)). These findings imply that the imine ion was initially formed via dehydrogenation, while it has not transformed into aldehyde through deamination. In addition, 11 gave rise to the desired product in 95% yield, which showed that it was also the possible intermediate in the deaminative coupling (eqn (3)). Subsequently, the hydrogen source for the deaminative coupling of nitroaromatics was investigated. It was found that 4a showed almost no conversion when TFE was employed in the reaction. In comparison, the desired product was obtained only in 16% yield in the absence of TFE accompanied by 75% of recovered nitroaromatics (eqn (4)).
 | ||
| Scheme 7 Control experiments. | ||
These results indicated that triethylamine was the preferable hydrogen source, where TFE significantly promoted the borrowing hydrogen process. Finally, the study on the denitrative coupling was carried out, and benzyl aniline was not observed (<5% conversion) when 5c was employed in the reaction system, which excludes the involvement of the hydrogen transfer pathway in this reaction (eqn (5)). Likewise, a comparable yield in the competition experiments revealed that C–NO2 cleavage is not sensitive to the electronic effects of the substituents (eqn (6)). Furthermore, the yield dropped dramatically only in the presence of an acidic solvent (eqn (7)). Thus, the inverted electrophilic characteristic of nitroalkanes presumably resulted from the formation of electrophilic imine ions activated by the Ru catalyst.
Based on the experimental observations and previous reports, a plausible mechanism is depicted for the deaminative coupling of amines (Scheme 8). Initially, the combination of RuCl3 and TFE promotes the dehydrogenation of triethylamine to generate the imine ion species A. Then, nucleophilic attack on the imine ion by pyrrolylaniline gave intermediate B. Subsequently, C–N cleavage occurred to provide the annulated compound by the elimination of amine. Importantly, the amine can be reused as a C synthon through multiple dehydrogenations and C–N cleavages to release ammonia at the end. Ultimately, the desired product was formed by the release of hydrogen gas (Scheme 8, middle). In the case of deaminative coupling of nitroaromatics with amines, the reaction involves the dehydrogenation of triethylamine (2a), providing imine ions and ruthenium-hydride species, which reduced 2-nitroarylpyrrole into 2-pyrrolylaniline in situ (Scheme 8, left). Afterward, pyrrolylaniline and imine ions were involved in the same process (Scheme 8, B → C → 3aa).
 | ||
| Scheme 8 Proposed reaction mechanism. | ||
On the other hand, the denitration pathway starts with the activation of nitroalkanes by RuCl3 and tautomerization to the imine cation D, which acted as a C-electrophile.19,20 Accordingly, nucleophilic attack by pyrrolylaniline affords intermediate E, followed by elimination of water to give oxime F, and finally the sequential intramolecular cyclization and C–N cleavage. The final product was formed with the concomitant loss of NH2OH (Scheme 8, right).
In conclusion, we provided a divergent platform for modular access to structurally diverse quinoxalines through Ru-catalyzed [5 + 1] annulation using amines and nitroalkanes as unconventional and inert electrophilic carbon sources through deaminative and denitrative C–N cleavages. The combination of RuCl3 and TFE established a highly efficient catalytic system to promote divergent C–N cleavages and cascade transformations under redox-neutral conditions. Specifically, benefitting from Ru-catalyzed acceptorless dehydrogenation and hydrogen borrowing methods, deaminative coupling of amines with both nitroarenes and amines has been achieved using amines as hydrogen donors and carbon synthons, which is distinct from the dehydrogenation of frequently used alcohols. This established method tolerates numerous readily available amine feedstocks without any additives, in contrast to the oxidative aminations requiring oxidants or additives. In addition, the reduction of alkenes and deiodination was also realized in this catalytic system via the borrowing hydrogen strategy. Remarkably, nitroalkanes were explored as C-synthons to furnish quinoxaline scaffolds in a straightforward manner, wherein Ru-catalyzed denitrative C–NO2 cleavages are involved. This developed protocol features an environmentally-friendly, exceedingly simple system, operational-simplicity, and a broad substrate scope. The synthetic utility of this new deaminative transformation has been exemplified by the succinct syntheses of antileishmanial agents and bipolar host materials for OLEDs.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (NSFC: 21901150) for their financial support. As a Tang Scholar, L.-Y. Jiao is indebted to the Cyrus Tang Foundation for support.Notes and references
- (a) D. K. Markus, Electrochemical strategies for C–H functionalization and C–N bond formation, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (b) R. Li, S.-X. Jiang, H.-L. Zhneg and G.-J. Deng, Iron-catalyzed indolo[2,3-c]quinoline synthesis from nitroarenes and benzylic alcohols/aldehydes promoted by elemental sulfur, Green Synth. Catal., 2022, 3, 95–101 CrossRef; (c) X. Tian, X.-F. Xu, Z.-H. Kang and W.-H. Hu, Enantioselective formal carbene insertion into C–N bond of aminal as a concise track to chiral α-amino-β2,2-amino acids and synthetic applications, Green Synth. Catal., 2021, 2, 337–344 CrossRef; (d) Y.-J. Wu, G. Liao and B.-F. Shi, Stereoselective construction of atropisomers featuring a C–N chiral axis, Green Synth. Catal., 2022, 3, 117–136 CrossRef; (e) B.-K. Yu, B. Gao, X.-X. Zhang and H.-M. Huang, Palladium-Catalyzed Aminomethylation of Nitrodienes and Dienones via Double C–N Bond Activation, Chin. J. Chem., 2021, 39, 566–570 CrossRef CAS.
- (a) J. Liu, Y. Yang, K. Ouyang and W.-X. Zhang, Transition-metal-catalyzed transformations of C–N single bonds: Advances in the last five years, challenges and prospects, Green Synth. Catal., 2021, 2, 87–122 CrossRef; (b) K. J. Berger and M. D. Levin, Reframing primary alkyl amines as aliphatic building blocks, Org. Biomol. Chem., 2021, 19, 11–36 RSC.
- G. Meng, J. Zhang and M. Szostak, Acyclic Twisted Amides, Chem. Rev., 2021, 121, 12746–12783 CrossRef CAS.
- G. G. Meng and M. Szostak, General Olefin Synthesis by the Palladium-Catalyzed Heck Reaction of Amides: Sterically Controlled Chemoselective N–C Activation, Angew. Chem., Int. Ed., 2015, 54, 14518–14522 CrossRef CAS.
- G. Li, S. Ma and M. Szostak, Amide Bond Activation: The Power of Resonance, Trends Chem., 2020, 2, 914–928 CrossRef CAS.
- (a) P. T. K. Arachchige, S. Handunneththige, M. R. Talipov, N. Kalutharage and C. S. Yi, Scope and Mechanism of the Redox-Active 1,2-Benzoquinone Enabled Ruthenium-Catalyzed Deaminative α-Alkylation of Ketones with Amines, ACS Catal., 2021, 11, 13962–13972 CrossRef; (b) K. J. Berger, J. L. Driscoll, M.-B. Yuan, B. D. Dherange, O. Gutierrez and M. D. Levin, Direct Deamination of Primary Amines via Isodiazene Intermediates, J. Am. Chem. Soc., 2021, 143, 17366–17373 CrossRef CAS; (c) H.-T. Qin, W.-S. Cai, S. Wang, T. Guo, G.-G. Li and H.-J. Lu, N-Atom Deletion in Nitrogen Heterocycles, Angew. Chem., Int. Ed., 2021, 60, 20678–20683 CrossRef CAS.
- (a) K. Ouyang, W. Hao, W.-X. Zhang and Z. F. Xi, Transition-Metal-Catalyzed Cleavage of C–N Single Bonds, Chem. Rev., 2015, 115, 12045–12090 CrossRef CAS PubMed; (b) H. M. Huang, Q. J. Wang, Y. J. Su and L. X. Li, Transition-metal catalysed C–N bond activation, Chem. Soc. Rev., 2016, 45, 1257–1272 RSC.
- (a) N. J. Turner, Enantioselective Oxidation of C–O and C–N Bonds Using Oxidases, Chem. Rev., 2011, 111, 4073–4087 CrossRef CAS PubMed; (b) C. Wang, Y. Li, R. Guo, J. J. Tian, C. Tao, B. Cheng, H. Y. Wang, J. Zhang and H. B. Zhai, Iodine-Catalyzed Facile Synthesis of Pyrrolo- and Indolo[1,2-a]quinoxalines, Asian J. Org. Chem., 2015, 4, 866–869 CrossRef CAS; (c) X. F. Wang, H. H. Liu, C. X. Xie, F. Y. Zhou and C. Ma, Terminal Methyl as a One-Carbon Synthon: Synthesis of Quinoxaline Derivatives via Radical-type Transformation, New J. Chem., 2020, 44, 2465–2470 RSC; (d) S. Tang, M. Rauch, M. Montag, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Catalytic Oxidative Deamination by Water with H2 Liberation, J. Am. Chem. Soc., 2020, 142, 20875–20882 CrossRef CAS; (e) C. Q. Rao, S. Y. Mai and Q. L. Song, Cu-Catalyzed Synthesis of 3-Formyl Imidazo[1,2-a]pyridines and Imidazo[1,2-a]pyrimidines by Employing Ethyl Tertiary Amines as Carbon Sources, Org. Lett., 2017, 19, 4726–4729 CrossRef CAS; (f) Q. H. Gao, H. J. Yan, M.-M. Wu, J.-J. Sun and A. X. Wu, Direct synthesis of 2-methylpyridines via I2-triggered [3+2+1] annulation of aryl methyl ketoxime acetates with triethylamine as the carbon source, Org. Biomol. Chem., 2018, 16, 2342–2348 RSC.
- (a) Y.-L. Zheng, Y. Long, H.-H. Gong, J.-X. Xu, C.-C. Zhang, H.-Y. Hu, H. Chen and R.-X. Li, Ruthenium-Catalyzed Divergent Acceptorless Dehydrogenative Coupling of 1,3-Diols with Aryl-hydrazines: Synthesis of Pyrazoles and 2-Pyrazolines, Org. Lett., 2022, 24, 3878–3883 CrossRef CAS PubMed; (b) X. Q. Hu, Y.-X. Hou, Z.-K. Liu and Y. Gao, Ruthenium-catalysed C–H/C–N bond activation: facile access to isoindolinones, Org. Chem. Front., 2021, 8, 915–921 RSC; (c) M. A. Stevens and A. L. Colebatch, Cooperative approaches in catalytic hydrogenation and dehydrogenation, Chem. Soc. Rev., 2022, 51, 1881–1898 RSC; (d) A. Corma, J. Navas and M. J. Sabater, Advances in One-Pot Synthesis through Borrowing Hydrogen Catalysis, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS; (e) K.-M. Li, Q. Zhang, Z.-M. Xu, R. Chen, T.-T. Liu, J.-Y. Luo, Y.-W. Wu, Y.-B. Huang and Q. Lu, Tunable mono- and di-methylation of amines with methanol over bimetallic CuCo nanoparticle catalysts, Green Chem., 2022, 24, 5965–5977 RSC; (f) H. Li, H. Guo, Z. Fang, T. M. Aida and R. L. Smith, Jr., Cycloamination strategies for renewable N-heterocycles, Green Chem., 2020, 22, 582–611 RSC.
- (a) K.-K. Sun, H.-B. Shan, G.-P. Lu, C. Cai and M. Beller, Synthesis of N-Heterocycles via Oxidant-Free Dehydrocyclization of Alcohols Using Heterogeneous Catalysts, Angew. Chem., Int. Ed., 2021, 60, 25188–25202 CrossRef CAS; (b) N. Hofmann and K. C. Hultzsch, Borrowing Hydrogen and Acceptorless Dehydrogenative Coupling in the Multicomponent Synthesis of N-Heterocycles: A Comparison between Base and Noble Metal Catalysis, Eur. J. Org. Chem., 2021, 6206–6223 CrossRef CAS; (c) W.-K. Hu, Y.-L. Zhang, H.-Y. Zhu, D.-D. Ye and D.-W. Wang, Unsymmetrical triazolyl-naphthyridinyl-pyridine bridged highly active copper complexes supported on reduced graphene oxide and their application in water, Green Chem., 2019, 21, 5345–5351 RSC; (d) S. Bera, L. M. Kabadwai and D. Banerjee, Recent advances in transition metal-catalyzed (1,n) annulation using (de)-hydrogenative coupling with alcohols, Chem. Commun., 2021, 57, 9807–9819 RSC.
- (a) Z. W. Yin, H. S. Zeng, J. Wu, S. P. Zheng and G.-Q. Zhang, Cobalt-Catalyzed Synthesis of Aromatic, Aliphatic, and Cyclic Secondary Amines via a “Hydrogen-Borrowing” Strategy, ACS Catal., 2016, 6, 6546–6550 CrossRef CAS; (b) D. Hollmann, S. Bähn, A. Tillack and M. A. Beller, General Ruthenium-Catalyzed Synthesis of Aromatic Amines, Angew. Chem., Int. Ed., 2007, 46, 8291–8294 CrossRef CAS; (c) O. Saidi, A. J. Blacker, M. M. Farah, S. P. Marsden and J. M. J. Williams, Selective Amine Cross-coupling Using Iridium-catalysed Borrowing Hydrogen Methodology, Angew. Chem., Int. Ed., 2009, 48, 7375–7378 CrossRef CAS; (d) D. W. Wang, K. Y. Zhao, C. Y. Xu, H. Y. Miao and Y. Q. Ding, Synthesis, Structures of Benzoxazolyl Iridium(III) Complexes, and Applications on C–C and C–N Bond Formation Reactions under Solvent-Free Conditions: Catalytic Activity Enhanced by Noncoordinating Anion without Silver Effect, ACS Catal., 2014, 4, 3910–3918 CrossRef CAS; (e) L. Neubert, D. Michalik, W. Holla and M. Beller, Ruthenium-Catalyzed Selective α,β-Deuteration of Bioactive Amines, J. Am. Chem. Soc., 2012, 134, 12239–12244 CrossRef CAS.
- (a) C. S. Cho, B. H. Oh and S. C. Shim, Synthesis of quinolines by ruthenium-catalyzed heteroannulation of anilines with 3-amino-1-propanol, J. Heterocycl. Chem., 1999, 36, 1175 CrossRef CAS; (b) C. S. Cho, B. H. Oh, J. S. Kim, T.-J. Kim and S. C. Shim, Synthesis of quinolines via ruthenium-catalysed amine exchange reaction between anilines and trialkylamines, Chem. Commun., 2000, 1885–1886 RSC; (c) M. Pizzetti, E. D. Luca, E. Petricci, A. Porcheddu and M. A. Taddei, General Approach to Substituted Benzimidazoles and Benzoxazoles via Heterogeneous Palladium-Catalyzed Hydrogen-Transfer with Primary Amines, Adv. Synth. Catal., 2012, 13, 2353–2464 Search PubMed; (d) T. Pandula, K. Arachchige and C. S. Yi, Synthesis of Quinazoline and Quinazolinone Derivatives via Ligand-Promoted Ruthenium-Catalyzed Dehydrogenative and Deaminative Coupling Reaction of 2-Aminophenyl Ketones and 2-Aminobenzamides with Amines, Org. Lett., 2019, 21, 3337–3341 CrossRef.
- (a) C. S. Cho, T. K. Kim, T.-J. Kim and S. C. Shim, Ruthenium-Catalyzed Synthesis of Quinolines via Reductive Heteroannulation of Nitroarenes with 3-Amino-1-propanols, J. Heterocycl. Chem., 2002, 39, 291 CrossRef CAS; (b) C. S. Cho, N. Y. Lee, H. J. Choi, T. G. Kim and S. C. Shim, Ruthenium-catalyzed formation of quinolines via reductive cyclization of nitroarenes with tris(3-hydroxypropyl)amine in an aqueous medium, J. Heterocycl. Chem., 2003, 40, 929–932 CrossRef CAS; (c) C. S. Cho, T. K. Kim, B. T. Kim, T. G. Kim and S. C. Shim, Ruthenium-catalyzed reductive cyclization of nitroarenes with trialkylamines leading to quinolones, J. Organomet. Chem., 2002, 650, 65–68 CrossRef CAS; (d) T. B. Nguyen, J. L. Bescont, L. Ermolenko and A.-M. Ali, Cobalt- and Iron-Catalyzed Redox Condensation of o-Substituted Nitrobenzenes with Alkylamines: A Step- and Redox-Economical Synthesis of Diazaheterocycles, Org. Lett., 2013, 15, 6218–6221 CrossRef CAS PubMed; (e) L. Tang, P. F. Wang, Y. Fan, X. K. Yang, C. F. Wan and Z. G. Zha, Heterogeneous Palladium-Catalyzed Hydrogen-Transfer Cyclization of Nitroacetophenones with Benzylamines: Access to C–N Bonds, ChemCatChem, 2016, 8, 3565–3569 CrossRef CAS.
- Q. Sun, L. Y. Liu, Y. Yang, Z. G. Zha and Z. Y. Wang, Unexpected activated carbon-catalyzed pyrrolo[1,2-a]quinoxalines synthesis in water, Chin. Chem. Lett., 2019, 30, 1379–1382 CrossRef CAS.
- (a) M. Kashihara and Y. Nakao, Cross-Coupling Reactions of Nitroarenes, Acc. Chem. Res., 2021, 54, 2928–2935 CrossRef CAS PubMed; (b) J. Feng, J. X. Yao, L. Yu and W. G. Duan, Palladium-catalyzed denitrative N-arylation of nitroarenes with pyrroles, indoles, and carbazoles, Org. Chem. Front., 2022, 9, 2351–2356 RSC; (c) Z. R. Li, Y.-G. Peng and T. Wu, Palladium-Catalyzed Denitrative α-Arylation of Ketones with Nitroarenes, Org. Lett., 2021, 23, 881–885 CrossRef CAS; (d) M. K. Reddy, S. Mallik, I. Ramakrishna and M. Baidya, Nitrosocarbonyl–Henry and Denitration Cascade: Synthesis of α-Ketoamides and α-Keto Oximes, Org. Lett., 2017, 19, 1694–1697 CrossRef CAS PubMed; (e) B. A. Vara and J. N. Johnston, Enantioselective Synthesis of β-Fluoro Amines via β-Amino α-Fluoro Nitroalkanes and a Traceless Activating Group Strategy, J. Am. Chem. Soc., 2016, 138, 13794–13797 CrossRef CAS PubMed; (f) T. Nakano, K. Miyazaki and A. Kamimura, A. Preparation of 2,3-Dihydrofurans via a Double Allylic Substitution Reaction of Allylic Nitro Compounds, J. Org. Chem., 2014, 79, 8103–8109 CrossRef CAS PubMed.
- (a) Z.-Y. An, L.-B. Zhao, M.-Z. Wu, J.-X. Ni, G.-Q. Yu and R.-L. Yan, FeCl3-Catalyzed synthesis of pyrrolo[1,2-a]quinoxaline derivatives from 1-(2-aminophenyl)pyrroles through annulation and cleavage of cyclic ethers, Chem. Commun., 2017, 53, 11572–11575 RSC; (b) H.-R. Huo, X.-Y. Tang and Y.-F. Gong, Metal-Free Synthesis of Pyrrolo[1,2-a]quinoxalines Mediated by TEMPO Oxoammonium Salts, Synthesis, 2018, 2727–2740 CAS; (c) C.-C. Ding, S.-C. Li, K.-L. Feng and C. Ma, PEG-400 as a carbon synthon: highly selective synthesis of quinolines and methyl-quinolines under metal-free conditions, Green Chem., 2021, 23, 5542–5548 RSC; (d) C.-X. Xie, L. Feng, W.-L. Li, X.-J. Ma, X.-K. Ma, Y.-H. Liu and C. Ma, Efficient synthesis of pyrrolo[1,2-a]quinoxalines catalyzed by a Brønsted acid through cleavage of C–C bonds, Org. Biomol. Chem., 2016, 14, 8529–8535 RSC; (e) M. Viji, M. Vishwantha, J. Sim, Y. J. Park, C. Jung, H. Lee, K. H. Lee and J.-K. Jung, α-Hydroxy acid as an aldehyde surrogate: metal-free synthesis of pyrrolo[1,2-a]quinoxalines, quinazolinones, and other N-heterocycles via decarboxylative oxidative annulation reaction, RSC Adv., 2020, 10, 37202–37208 RSC.
- S.-S. Liu, P.-J. Zhang, Y.-Y. Zhang, J. Nan and Y.-M. Ma, Bifunctional acidic ionic liquid-catalyzed decarboxylative cascade synthesis of quinoxalines in water under ambient conditions, Org. Chem. Front., 2021, 8, 5858–5865 RSC.
- J.-X. Li, J.-L. Zhang, H.-M. Yang and G.-X. Jiang, A Green Aerobic Oxidative Synthesis of Pyrrolo[1,2-a]quinoxalines from Simple Alcohols without Metals and Additives, J. Org. Chem., 2017, 82, 765–769 CrossRef CAS PubMed.
- Y. Nagase, T. Sugiyama, S. Nomiyama, K. Yonekura and T. Tsuchimoto, Zinc-Catalyzed Direct Cyanation of Indoles and Pyrroles: Nitromethane as a Source of a Cyano Group, Adv. Synth. Catal., 2014, 356, 347–352 CrossRef CAS.
- S. G. Zlotin, I. L. Dalinger, N. N. Makhova and V. A. Tartakovsky, Nitro compounds as the core structures of promising energetic materials and versatile reagents for organic synthesis, Russ. Chem. Rev., 2020, 89, 1–54 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra for corresponding products. See DOI: https://doi.org/10.1039/d2qo01572b |
| This journal is © the Partner Organisations 2023 |