
Asymmetric total synthesis of (+)-tubingensin A†
Hongbin
Zhai
 *abcd,
Dong
Liu
a and
Taimin
Wang
*c
*abcd,
Dong
Liu
a and
Taimin
Wang
*c
aThe State Key Laboratory of Chemical Oncogenomics, Guangdong Provincial Key Laboratory of Nano-Micro Materials Research, School of Chemical Biology and Biotechnology, Shenzhen Graduate School of Peking University, Shenzhen 518055, China
bShenzhen Bay Laboratory, Shenzhen 518055, China
cInstitute of Marine Biomedicine, Shenzhen Polytechnic, Shenzhen 518055, China
dCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, China
First published on 18th November 2022
Abstract
A nine-step asymmetric total synthesis of (+)-tubingensin A has been realized. The synthesis features (a) an intramolecular Heck reaction to install the C20 quaternary carbon center thus establishing the key vicinal quaternary stereocenter motif present in the natural product and (b) a late-stage Zweifel olefination applied to side chain elongation.
Tubingensins A (1, Fig. 1) and B (2) were isolated from the fungus Aspergillus tubingensis by the Gloer group over 30 years ago.1 Both of the structural isomers belong to the carbazole-containing natural products of great singnificance.2 They have been demonstrated to possess antiviral, anticancer, and insecticidal activities.1 Structurally, tubingensin A (1) contains a disubstituted carbazole moiety fused to a densely functionalized cis-decalin backbone that is imbedded with four continuous stereocenters, two of which are vicinal quaternary stereocenters. Based upon the biosynthetic proposals reported, both 1 and 2 might be derived from anominine (3), an indole diterpenoid.3
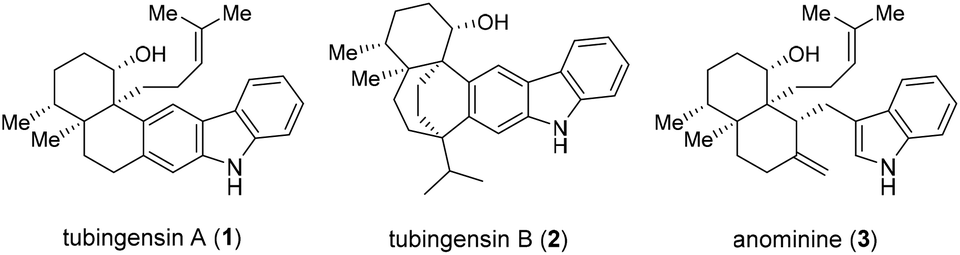 | ||
| Fig. 1 Tubingensin A (1) and related family members 2 and 3. | ||
Owing to their intriguing architecture and excellent biological activities, this family of natural products such as tubingensin A (1) and anominine (3) have attracted considerable attention from the synthetic community. In 2010, Bonjoch and co-workers reported the first total synthesis of anominine featuring a series of chemoselective transformations controlled by the bicyclic core and the development of a new, efficacious synthesis of Wieland–Miescher ketone-type compounds with N-Ts-(Sa)-binam-L-Pro as the organocatalyst under solvent-free conditions.4 Li and Nicolaou accomplished an elegant bio-inspired total synthesis of 1 employing an Ueno–Stork radical cyclization and a Sc(OTf)3-mediated Mukaiyama aldol reaction for the assembly of the stereogenic centers and a CuOTf-promoted 6π-electrocyclization/aromatization sequence for the construction of the pentacyclic skeleton in 2012.3a Garg and co-workers achieved a concise nine-step enantiospecific total synthesis of 1via an aryne cyclization to introduce the vicinal quaternary stereocenters in 2014;5a however, the final reduction of the C19 carbonyl group delivered a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of tubingensin A (minor) and its C19 epimer (major) in their work. Thus, introduction of the correct stereochemistry at C19 at an earlier stage would represent one of the efficient solutions to circumventing this problem. In addition, aryne generation and the subsequent cyclization involved the use of a base such as sodium amide, which would pose potential functional group incompatibility in some cases. In 2017, the total synthesis of tubingensin B (2) was completed essentially via the same strategy by the same group.5b
:2 ratio of tubingensin A (minor) and its C19 epimer (major) in their work. Thus, introduction of the correct stereochemistry at C19 at an earlier stage would represent one of the efficient solutions to circumventing this problem. In addition, aryne generation and the subsequent cyclization involved the use of a base such as sodium amide, which would pose potential functional group incompatibility in some cases. In 2017, the total synthesis of tubingensin B (2) was completed essentially via the same strategy by the same group.5b
The intriguing chemical structure and promising biological properties of tubingensin A (1) coupled with its scarcity in Nature prompted us to embark on a novel, efficacious chemical synthesis of this alkaloid.
The retrosynthetic analysis for tubingensin A is outlined in Scheme 1. We envisioned that the side chain at C20 of 1 might be installed from pentacycle 4via late-stage functionalization. The quaternary stereocenter at C20 and the cis-decalin framework in 4 would be accessible through an intramolecular Heck reaction of bromide 5, which could be obtained from cyclohexane derivative 7 and carbazole 6 through a Suzuki–Miyaura coupling reaction. Fragment 7 could be prepared from enone (+)-86via a Michael addition/aldol reaction/Luche reduction sequence.
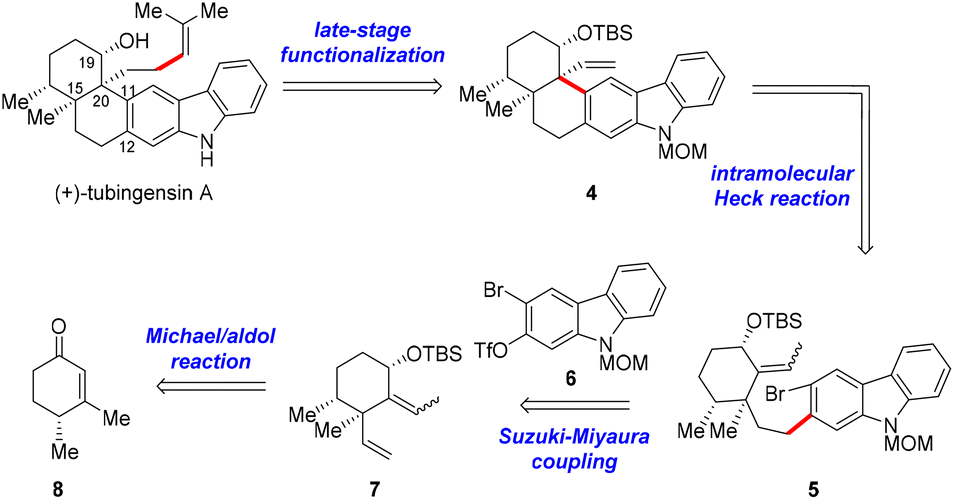 | ||
| Scheme 1 Retrosynthetic analysis of tubingensin A (1). | ||
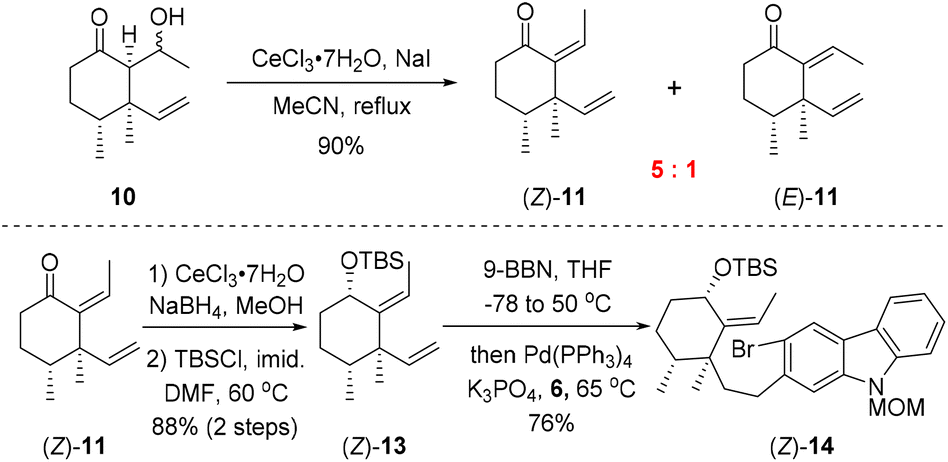
As shown in Scheme 2, our synthesis commenced with preparation of carbazole triflate 6 and cyclohexyl derivative (E)-13. Commercially available 2-hydroxycarbazole was first brominated to deliver compound 9 by adopting a known procedure.7 Sequential O-triflation and N-protection of 9 afforded triflate 6 in 50% yield over three steps. Cyclohexenone was elaborated to form known enone 86 through a five-step reaction sequence.8 Vinyl cuprate addition to 8 followed by an aldol reaction9 furnished β-hydroxyl ketone 10. Dehydration of 10 with PTSA in refluxing benzene generated (E)-11 and (Z)-11 in 80% and 5% yields, respectively. The latter ((Z)-11) could be converted into (E)-11 upon heating. Luche reduction of ketone (E)-11 followed by hydroxyl protection gave compound (E)-13.
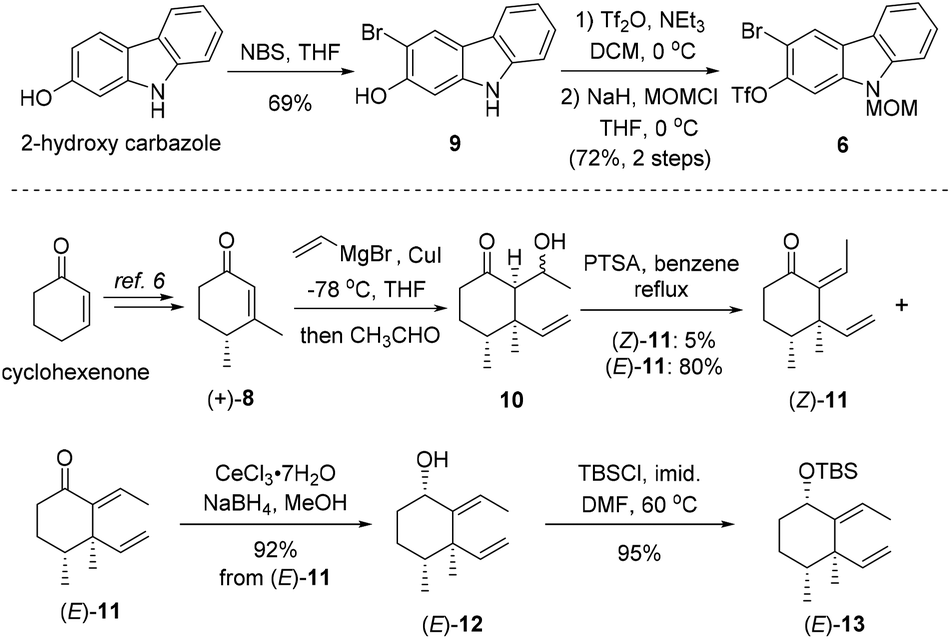 | ||
| Scheme 2 Synthesis of compounds 6 and (E)-13. | ||
With carbozole triflate 6 and diene (E)-13 in hand, the Suzuki–Miyaura coupling of the two fragments and the ensuing intramolecular Heck reaction were pursued (Scheme 3). Treatment of (E)-13 with 9-BBN10 led to chemo- and regioselective hydroboration of the terminal olefin moiety. Palladium-catalyzed cross-coupling11 of the resultant organoboron intermediate with triflate 6 proceeded smoothly to lead to aryl bromide (E)-14. Unfortunately, attempts to effect the intramolecular Heck reaction12 of (E)-14 were unsuccessful. Although various Pd catalysts in combination with different ligands and bases were tested (see the ESI† for details), the major product of the reaction was determined to be the dehalogenation product 16 rather than pentacycle 15.
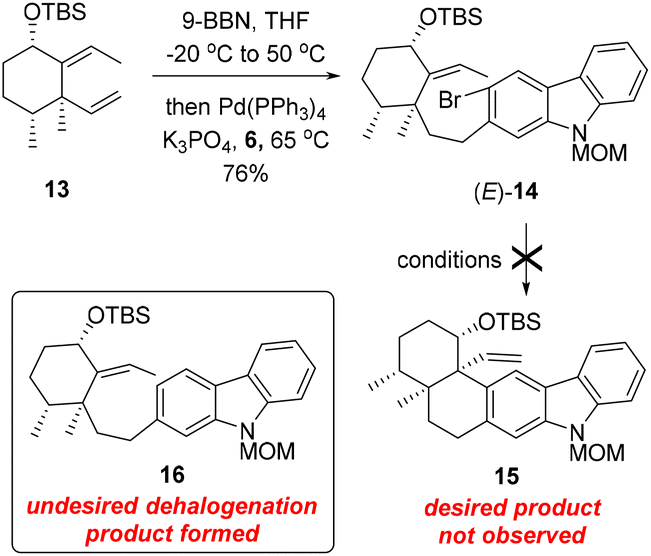 | ||
| Scheme 3 Preparation and attempted Heck reaction of (E)-14. | ||
The failure of the cyclization reaction might be attributed to the steric hindrance exerted by the (E)-alkene moiety in (E)-14. As mentioned above, dehydration of 10 with PTSA furnished (E)-11 as the major product and (Z)-11 was formed in only 5% yield (Scheme 2). In order to gain (Z)-11 more efficiently, different dehydration conditions were explored on alcohol 10, although (E)-11 was generated under most of the common reaction conditions as it was the thermodynamically favored product. Fortunately, exposure of 10 to CeCl3·7H2O/NaI13 in refluxing acetonitrile for one hour provided alkenes (Z)-11 and (E)-11 with a ratio up to 5:1 (Scheme 4). Thus, bromoarene (Z)-14 containing an (Z)-alkene moiety was synthesized from (Z)-11 by an analogous reaction sequence consisting of Luche reduction, hydroxyl protection, and sequential hydroboration/Suzuki–Miyaura coupling with carbozole triflate 6.
 | ||
| Scheme 4 Preparation of compound (Z)-14. | ||
With a sufficient quantity of (Z)-14 in hand, the intramolecular Heck reaction was examined with different Pd catalysts (10 mol%), bases (5 equiv), and ligands (20 mol%) (Table 1). Exposure of (Z)-14 to Pd(OAc)2, PPh3, and Et3N in DMF at 100 °C for 12 h effected the desired transformation and produced pentacycle 15 in 15% yield (entry 1). The maximum yield of 15 did not exceed 36% when the reaction was performed at 100 °C in the precence of a non-Pd(PPh3)4 palladium catalyst (Pd(OAc)2, Pd(PPh3)2Cl2, Pd(TFA)2, Pd(dba)2, or Pd2(dba)3) with Et3N as a base and PPh3 as a ligand (entries 1–5). However, treatment of (Z)-14 with Pd(PPh3)4 and Et3N in DMF at 100 °C resulted in the formation of 15 in 60% yield (entry 6). Subsequently, several other bases such as PMP, DIPEA, Ag3PO4, and Cs2CO3 were screened (entries 7–10), and triethylamine was found to perform better than all other bases tested for the reaction. In addition, higher loading (20 mol%) of Pd(PPh3)4 further enhanced the reaction efficiency (entry 11). With prolonged reaction time (24 h), the cyclization product 15 was afforded in 75% yield (entry 12). By contrast, conducting the intramolecular Heck reaction at 90 and 120 °C led to pentacycle 15 in 10% and 70%, respectively (entries 13 and 14). The structure of 15 was confirmed via single-crystal X-ray diffraction analysis after derivatization (see the ESI† for details).
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | Ligand | Base | T (°C) | Yieldb (%) |
| a Reaction conditions: (Z)-14 (0.01 mmol), [Pd] catalyst (10 mol%), ligand (20 mol%), base (5 equiv), DMF (1 mL), Ar, T, 12 h. b Isolated yields. c TBAI was added. d 20 mol% of Pd(PPh3)4 was used. e Reaction time was extended to 24 h. PMP = 1,2,2,6,6-pentamethylenepiperidine, DIPEA = N,N-diisopropylethylamine. | |||||
| 1 | Pd(OAc)2 | PPh3 | Et3N | 100 | 15 |
| 2 | Pd(PPh3)2Cl2 | PPh3 | Et3N | 100 | 36 |
| 3 | Pd(TFA)2 | PPh3 | Et3N | 100 | 19 |
| 4 | Pd(dba)2 | PPh3 | Et3N | 100 | 16 |
| 5 | Pd2(dba)3 | PPh3 | Et3N | 100 | 23 |
| 6 | Pd(PPh3)4 | — | Et3N | 100 | 60 |
| 7 | Pd(PPh3)4 | — | PMP | 100 | 54 |
| 8 | Pd(PPh3)4 | — | DIPEA | 100 | 41 |
| 9c | Pd(PPh3)4 | — | Ag3PO4 | 100 | 37 |
| 10c | Pd(PPh3)4 | — | Cs2CO3 | 100 | 35 |
| 11d | Pd(PPh3)4 | — | Et3N | 100 | 66 |
| 12 , | Pd(PPh 3 ) 4 | — | Et 3 N | 100 | 75 |
| 13d,e | Pd(PPh3)4 | — | Et3N | 90 | 10 |
| 14d,e | Pd(PPh3)4 | — | Et3N | 120 | 70 |
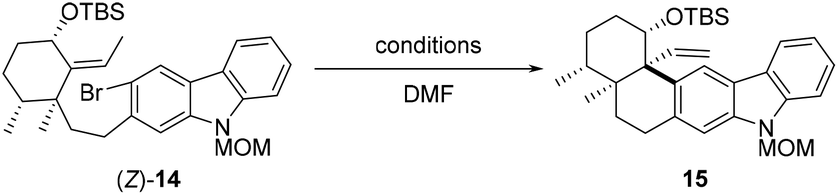
As demonstrated by our experiment results, alkene (E)-14 failed to undergo the intramolecular Heck reaction to deliver pentacycle 15, while (Z)-14 was a suitable substrate for the cyclization. According to the literature,14 the configuration of the double bond involved in Heck reaction affects both the yield and stereoselectivity of the product. Within the catalytic cycle, the migratory insertion step might not be favored due to the presence of 1,3-allylic strain for (E)-14, while it was not the case for (Z)-14.
After pentacycle 15 was constructed, the remaining tasks of the total synthesis of tubingensin A (1) would be installment of the complete side chain at C20 and global deprotection (Scheme 5). Desilylation of 15 with TBAF led to alcohol 17, homologization of which on the side chain was realized via a two-step Zweifel olefination protocol,15 affording trisubstituted alkene 18 in 61% yield. Finally, removal of the MOM group of 18 in acidic media furnished tubingensin A (1) in 78% yield.
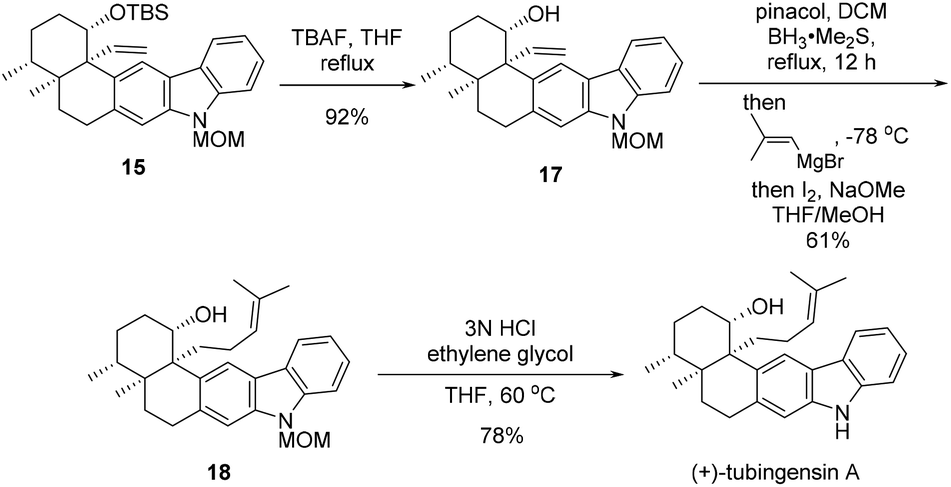 | ||
| Scheme 5 Completion of the total synthesis of tubingensin A (1). | ||
Conclusions
We have realized an efficeint total synthesis of (+)-tubingensin A from known enone (+)-8, proceeding in nine steps for the longest linear sequence and with 11% overall yield. The present synthesis features (a) an intramolecular Heck reaction with high functional group compatibility under mild reaction conditions to install the C20 quaternary carbon center thus establishing the key vicinal quaternary stereocenter motif present in the natural product and (b) a late-stage Zweifel olefination applied to side chain elongation. Introduction of the correct stereochemistry at C19 at an earlier stage ensured the total synthesis with better stereoselectivity and higher overall yield. Our current endeavors may facilitate future biological studies of (+)-tubingensin A and analogues thereof.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Shenzhen Science and Technology Innovation Committee (GXWD20201231165807007-20200812100115001, JCYJ20180504165454447), Shenzhen Science and Technology Program (Grant No. KQTD20190929174023858), The Shenzhen Bay Laboratory Open Research Program (SZBL2019062801010), National Natural Science Foundation of China (21871018, 21732001, 21901014, 22231003, 22271008), Guangdong Science and Technology Program (2017B030314002), Industry and Information Technology Bureau of Shenzhen Municipality (201806151622209330), Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (2019SHIBS0004), the Third Foster Plan in 2019 “Molecular Imaging Probe Preparation and Characterization of Key Technologies and Equipment” (ZDKJ20190305003) for the Development of Key Technologies and Equipment in Major Science and Technology Infrastructure in Shenzhen, and the National Ten Thousand Talent Program (the Leading Talent Tier) for the financial support. We are grateful to Prof. Huifei Wang of Ningbo University and Prof. Zhiqiang Ma of South China University of Technology for enlightening discussion.References
- (a) J. B. Gloer, B. L. Rinderknecht, D. T. Wicklow and P. F. Dowd, Nominine: a new insecticidal indole diterpene from the sclerotia of Aspergillus nomius, J. Org. Chem., 1989, 54, 2530 CrossRef CAS; (b) M. R. TePaske, J. B. Gloer, D. T. Wicklow and P. F. Dowd, Tubingensin A: an antiviral carbazole alkaloid from the sclerotia of Aspergillus tubingensis, J. Org. Chem., 1989, 54, 4743 CrossRef CAS; (c) J. B. Gloer, Antiinsectan natural products from fungal sclerotia, Acc. Chem. Res., 1995, 28, 343 CrossRef CAS.
- (a) A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Occurrence, biogenesis, and synthesis of biologically active carbazole alkaloids, Chem. Rev., 2012, 112, 3193 CrossRef CAS; (b) H.-J. Knölker and K. R. Reddy, Isolation and synthesis of biologically active carbazole alkaloids, Chem. Rev., 2002, 102, 4303 CrossRef; (c) J. Bergman and B. Pelcman, Synthesis of carbazole alkaloids, Pure Appl. Chem., 1990, 62, 1967 CrossRef CAS.
- For biosynthetic proposals, see: (a) M. Bian, Z. Wang, X. Xiong, Y. Sun, C. Matera, K. C. Nicolaou and A. Li, Total syntheses of anominine and tubingensin A, J. Am. Chem. Soc., 2012, 134, 8078 CrossRef CAS; (b) I. S. Marcos, R. F. Moro, I. Costales, M. A. Escola, P. Basabe, D. Díez and J. G. Urones, Synthesis of hexahydrocarbazoles by cyclisation of 3-(but-3-enyl) indole derivatives, Tetrahedron, 2009, 65, 10235 CrossRef CAS.
- B. Bradshaw, G. Etxebarria-Jardí and J. Bonjoch, Total synthesis of (−)-anominine, J. Am. Chem. Soc., 2010, 132, 5966 CrossRef CAS.
- (a) A. E. Goetz, A. L. Silberstein, M. A. Corsello and N. K. Garg, Concise enantiospecific total synthesis of tubingensin A, J. Am. Chem. Soc., 2014, 136, 3036 CrossRef CAS; (b) M. A. Corsello, J.-J. Kim and N. K. Garg, Total synthesis of (–)-tubingensin B enabled by the strategic use of an aryne cyclization, Nat. Chem., 2017, 9, 944 CrossRef CAS.
- J. D. White, U. M. Grether and C.-S. Lee, (R)-(+)-3,4-Dimethyl-2-cyclohexen-1-one, Org. Synth., 2005, 82, 108 CrossRef CAS.
- S. M. Bonesi, M. A. Ponce and R. J. Erra-Balsells, A study of substituent effect on 1H and 13C nmr spectra of N- and C- substituted carbazoles, Heterocycl. Chem., 2004, 41, 161 CrossRef CAS.
- (a) D. Enders and H. Eichenauer, Asymmetric synthesis of α-substituted ketones by metalation and alkylation of chiral hydrazones, Angew. Chem., Int. Ed. Engl., 1976, 15, 549 CrossRef; (b) M. Schwaebe and R. D. Little, Asymmetric reductive cyclization. Total synthesis of (−)-C10-desmethy-arteannuin B, J. Org. Chem., 1996, 61, 3240 CrossRef CAS.
- (a) A. B. Smith III, E. G. Nolen, R. Shirai, F. R. Blasé, M. Ohta, N. Chida, R. A. Hartz, D. M. Fitch, W. M. Clark and P. A. Sprengeler, Tremorgenic Indole Alkaloids. 9. Asymmetric construction of an advanced F-G-H-ring lactone precursor for the synthesis of penitrem D, J. Org. Chem., 1995, 60, 7837 CrossRef; (b) X. Lei, C. Li, X. Li and X. Wang, Diversity-oriented synthesis of bicyclic ring systems via a conjugate addition/aldol/RCM process, Sci. China: Chem., 2013, 56, 337 CrossRef; (c) M.-J. Dai, Y. Yang, C. W. Haskins, W. Zhang and P. Low, Divergent total syntheses of lyconadins A and C, Angew. Chem., Int. Ed., 2014, 53, 3922 CrossRef PubMed.
- N. Miyaura, T. Ishiyama, H. Sasaki, M. Ishikawa, M. Satoh and A. Suzuki, Palladium-catalyzed inter- and intramolecular cross-coupling reactions of B-alkyl-9-borabicyclo[3.3.1]nonane derivatives with 1-halo-1-alkenes or haloarenes. Syntheses of functionalized alkenes, arenes, and cycloalkenes via a hydroboration-coupling sequence, J. Am. Chem. Soc., 1989, 111, 314 CrossRef CAS.
- S. R. Chemler, D. Trauner and S. J. Danishefsky, The B-alkyl Suzuki-Miyaura cross-coupling reaction: development, mechanistic study, and applications in natural product synthesis, Angew. Chem., Int. Ed., 2001, 40, 4544 CrossRef CAS.
- For selected reviews on Heck reaction, see: (a) A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Pd metal catalysts for cross-couplings and related reactions in the 21st century: A critical review, Chem. Rev., 2018, 118, 2249 CrossRef CAS PubMed; (b) I. P. Beletskaya and A. V. Cheprakov, The Heck reaction as a sharpening stone of palladium catalysis, Chem. Rev., 2000, 100, 3009 CrossRef CAS; (c) A. B. Dounay and L. E. Overman, The asymmetric intramolecular Heck reaction in natural product total synthesis, Chem. Rev., 2003, 103, 2945 CrossRef CAS.
- G. Bartoli, M. Bosco, R. Dalpozzo, A. Giuliani, E. Marcantoni, T. Mecozzi, L. Sambri and E. Torregiani, An efficient procedure for the preparation of (E)-α-alkylidenecycloalkanones mediated by a CeCl3·7H2O-NaI system. Novel methodology for the synthesis of (S)-(−)-pulegone, J. Org. Chem., 2002, 67, 9111 CrossRef CAS.
- A. Ashimori, T. Matsuura, D. J. Poon and L. E. Overman, Catalytic asymmetric synthesis of either enantiomer of physostigmine. Formation of quaternary carbon centers with high enantioselection by intramolecular Heck reactions of (Z)-2-butenanilides, J. Org. Chem., 1993, 58, 6949 CrossRef CAS.
- (a) G. Zweifel, H. Arzoumanian and C. C. Whitney, A convenient stereoselective synthesis of substituted alkenes via hydroboration-iodination of alkynes, J. Am. Chem. Soc., 1967, 89, 3652 CrossRef CAS; (b) R. J. Armstrong, W. Niwetmarin and V. K. Aggarwal, Synthesis of functionalized alkenes by a transition-metal-free Zweifel coupling, Org. Lett., 2017, 19, 2762 CrossRef CAS; (c) R. J. Armstrong and V. K. Aggarwal, 50 years of Zweifel olefination: a transition-metal-free coupling, Synthesis, 2017, 49, 3323 CrossRef CAS; (d) X. Yu, L. Xiao, Z. Wang and T. Luo, Scalable total synthesis of (−)-vinigrol, J. Am. Chem. Soc., 2019, 141, 3440 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2215805. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qo01540d |
| This journal is © the Partner Organisations 2023 |