DOI:
10.1039/D2QO01534J
(Review Article)
Org. Chem. Front., 2023,
10, 237-266
Eosin Y mediated photo-catalytic C–H functionalization: C–C and C–S bond formation
Received
28th September 2022
, Accepted 22nd November 2022
First published on 23rd November 2022
Abstract
C–C and C–S bonds are valuable in organic transformation for synthesizing various bioactive heterocycles. Owing to the increasing awareness of the step and atom economy of a reaction, organic chemists have promulgated the idea of sustainable and green chemistry for organic synthesis. Hence, photoredox catalysis using organic dyes as photocatalysts has attracted constant attention over the last decade. Herein, recent advances and developments (over the previous ten years, 2012–2022) in eosin Y mediated photoredox catalyzed synthetic organic transformations for C–C and C–S bond formation along with their different mechanistic pathways (such as SET, HAT, and energy transfer) are discussed. This review is classified into two major sections, (i) C–C and (ii) C–S bond formations, which are further sub-categorized into various types of functionalizations including arylation, alkenylation, annulation, thionation, and sulfonylation.
 Dinesh Singla | Dinesh Singla was born in Punjab, India and graduated with B.Sc. from Punjabi University, Patiala, India. He obtained his M.Sc. (2016) in Pharmaceutical Chemistry from Punjabi University, Patiala, where he focused on organic synthesis and medicinal chemistry. Currently he is pursuing his PhD under the supervision of Dr Kamaldeep Paul at Thapar Institute of Engineering and Technology, Patiala, India, where he is focusing on C–H functionalization of biologically active heterocycles using organic dyes and transition metal catalysts. |
 Vijay Luxami | Dr Vijay Luxami received her PhD from the Department of Chemistry, Guru Nanak Dev University, Amritsar. She joined the School of Chemistry and Biochemistry, Thapar Institute of Engineering and Technology, Patiala as an Assistant Professor in 2010. She has received various awards including the DST Young Scientist, DST INSPIRE Faculty, and DST-DFG awards. She worked as a Visiting Scientist at the University of Bath, UK. Currently, she is working as a Professor at TIET, Patiala. Her area of research is organic supramolecular materials and their applications in molecular electronics. |
 Kamaldeep Paul | Dr Kamaldeep Paul received his Master's Degree from the Department of Pharmaceutical Sciences and PhD in Synthetic Organic and Medicinal Chemistry from the Department of Chemistry, Guru Nanak Dev University, Amritsar in 2006. Presently, he is working as a Professor in the School of Chemistry and Biochemistry, Thapar Institute of Engineering and Technology, Patiala, India. He has 100 research publications in peer-reviewed journals. His area of research is synthetic organic and medicinal chemistry, and his research is broadly focused on C–H functionalization and multistep synthesis of heterocyclic molecules and their in vitro evaluation for anticancer and antimicrobial activities. |
1. Introduction
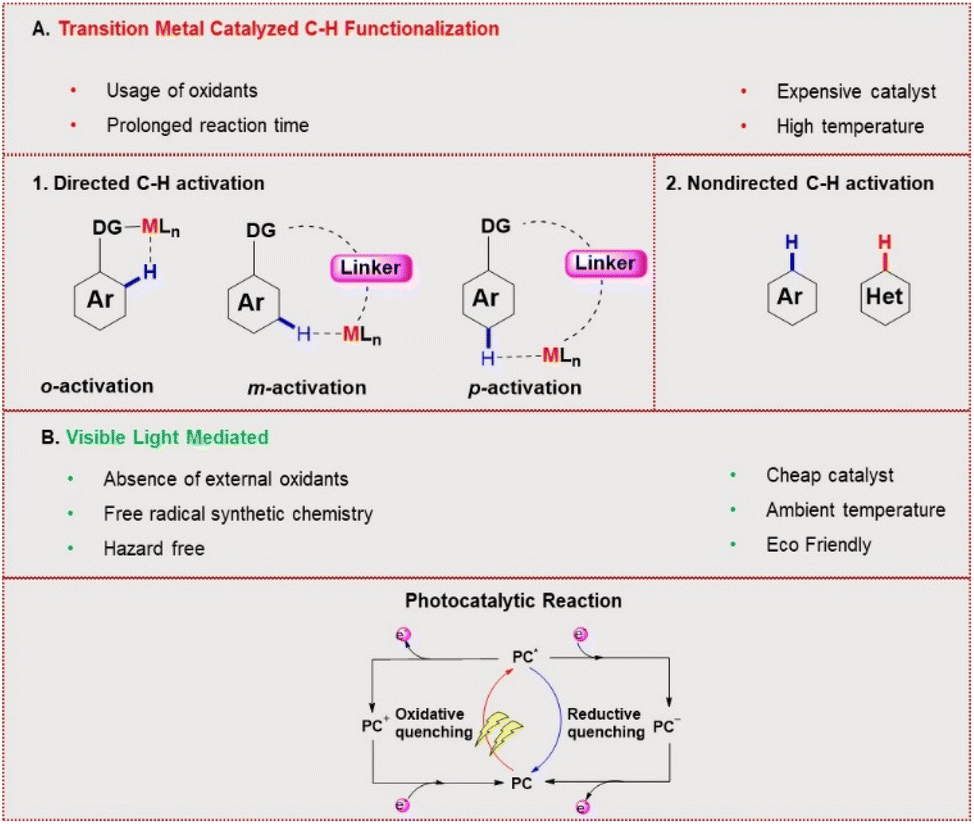
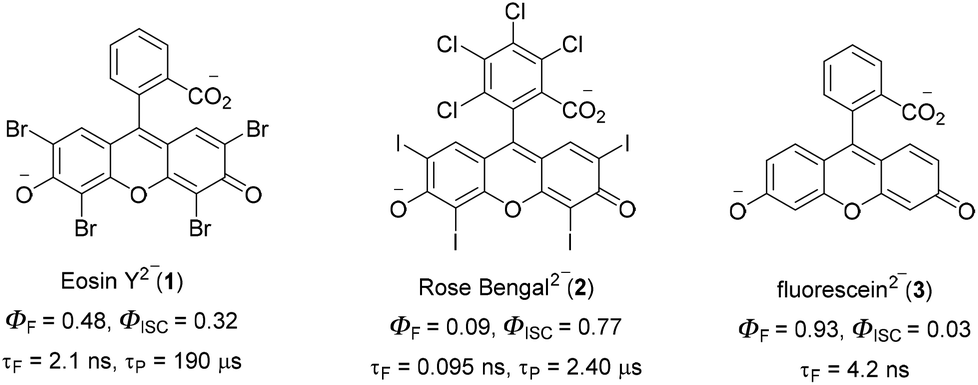
C–H functionalization for constructing valuable C–C and C–S bonds via sustainable methods has attracted incessant attention from organic chemists.1 In this context, visible light-mediated organophotoredox catalysis is an effective and novel synthetic route due to its sustainable properties.2 By introducing a number of organic transformations under mild reaction conditions, this approach has become a powerful tool and efficient alternative to transition metal-catalysed C–H functionalization (Fig. 1). The majority of photochemical reactions proceed through photoinduced single electron transfer (SET), energy transfer (ET), and hydrogen atom transfer (HAT) processes.3 The SET process is generally accomplished through two different cycles as oxidative and reductive quenching. These two cycles are commonly involved in the mechanism of photoredox catalysis. In this process, a photocatalyst (PC) initially undergoes excitation under visible light irradiation and leads to the generation of highly reactive radical ions from the substrate via SET oxidation or reduction. These unstable, reactive species readily combine in a controllable manner to give valuable compounds. In the last decade, various photocatalysts have been developed with these reaction methodologies. Among these, xanthene-related dyes such as eosin Y 1, rose bengal 2, and fluorescein 3 (Fig. 2) have been recognized as excellent organic photocatalysts.4
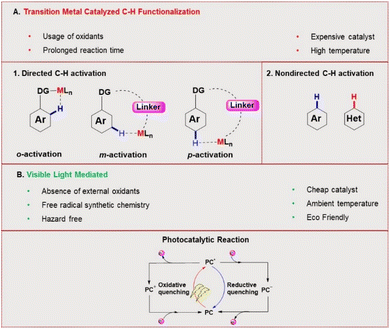 |
| | Fig. 1 Photoinduced SET pathway. | |
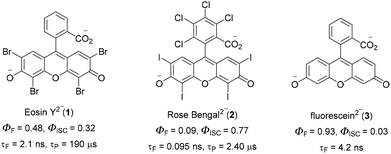 |
| | Fig. 2 Different organo-photocatalysts. | |
Eosin Y, a cheap and widely-used organic dye, has been employed as an economically and environmentally friendly substitute for many transition-metal-based photocatalysts. Depending on the pH of the solution, eosin Y has four different forms: spirocyclic form, monoanionic form, neutral form and di-anionic form.5 As eosin Y has heavy atoms such as Br, it has efficient intersystem crossing (φISC = 0.33) and strong oxidizing and reducing abilities.6 Eosin Y catalyzed reactions through the SET mechanism have some electrochemical requirements for the successful oxidation and reduction of a substrate, as the redox potential of the reacting substrate is comparable to that of eosin Y.7 In contrast, substrates having no comparability of redox potential could be functionalized through an energy transfer pathway or a hydrogen atom transfer mechanism.8 Photoinduced eosin Y catalyzed reactions that proceed through energy transfer pathways are not controlled by the electrochemical or redox potential of the substrate and mainly depend on three factors: (i) the triplet energy of the organic substrate (energy acceptor, EA) must be less than the energy of the photocatalyst (PC) energy donor (ED), (ii) the φISC of the ED must be greater than that of the EA, and (iii) energy must be released from the donor to the acceptor.9
Here the PC gets excited by absorption of visible light to the lowest singlet state S1 from its ground state S0. Next, the PC moves to its triplet state (T1) by efficient intersystem crossing (ISC), where it transfers its energy to the EA (substrate) and excites the substrate to its triplet state. After significant endeavours by several groups, it has been observed that eosin Y has the potential to carry out a broad range of organic reactions such as arylation, alkenylation, and sulphonation for the synthesis of important compounds by constructing C–C and C–X (where X = S, N, O, P) bonds.6
In this review, we mainly emphasize on eosin Y mediated photoinduced C–H functionalization for forming C–C and C–S bonds with mechanistic pathways of some selected reactions. We categorize the review into sections according to synthetic transformations and the goals achieved. Classification is mainly based on two sections: (i) C–C bond formation and (ii) C–S bond formation; these sections are further categorized into subsections based on organic transformations.
2. C–C bond formation
C–C bond formation is the fundamental prerequisite for the synthesis of organic compounds. C–H functionalization has revolutionized the synthesis of this valuable bond by various methods. Photoinduced C–C bond formation is a step economical and environment friendly process.
2.1. C–H arylation via aryl diazonium salts
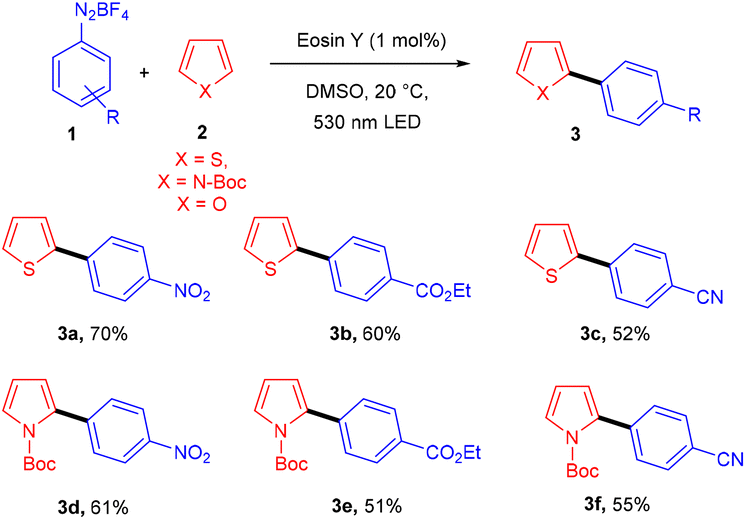
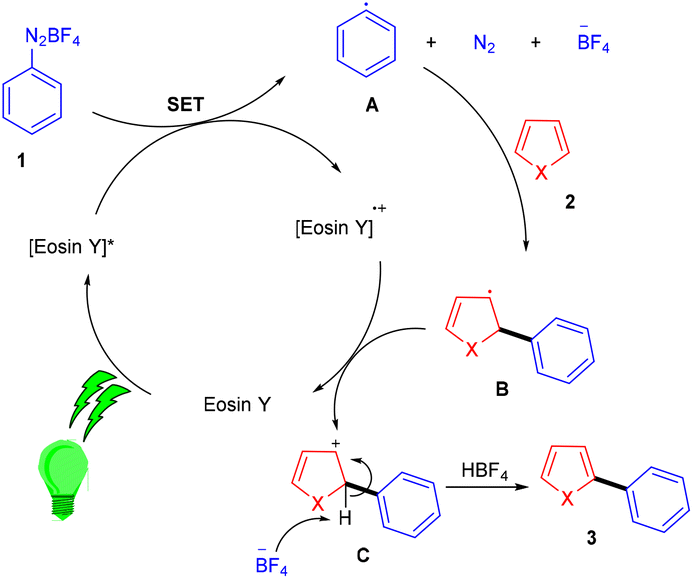
Over the last decade, aryl diazonium salts have attracted great attention as these have enormous potential for use as reagents or precursors of aryl radicals for several aromatic substitution reactions.10 König and coworkers in 2012 demonstrated the competency of aryldiazonium salts (1) as aryl radicals and functionalized heteroarenes (2) by direct C–H arylation under green light photoredox catalysis to overcome the shortcomings of the Meerwein arylation. During optimization they faced the problem of low product yield in the presence of a base, due to the direct reaction of the starting material with the base. Here, the reaction displayed complete regioselectivity with a broad substrate scope. Moreover, the methodology also showed broad functional group tolerance to nitro, ester, halogens, hydroxyl, and cyano groups (Scheme 1).11 Notably, electron-acceptor and neutral-diazonium salts have more compatibility than electron-donor-substituted salts. The reaction proceeded through the SET mechanism where an aryl diazonium salt was converted into an aryl radical (A) from photoexcited eosin Y followed by a reaction with heteroarene 2, where a radical intermediate B was obtained. Oxidation of intermediate Bvia an eosin Y radical cation gave carbocation C, and in the final stage, deprotonation of carbocation C afforded the final product 3 (Scheme 2).
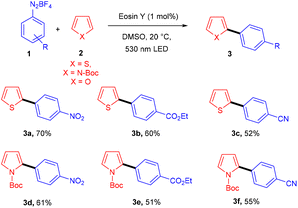 |
| | Scheme 1 Direct C–H arylation of heteroarenes. | |
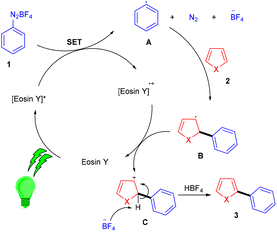 |
| | Scheme 2 Plausible mechanism for C–H arylation of heteroarenes. | |
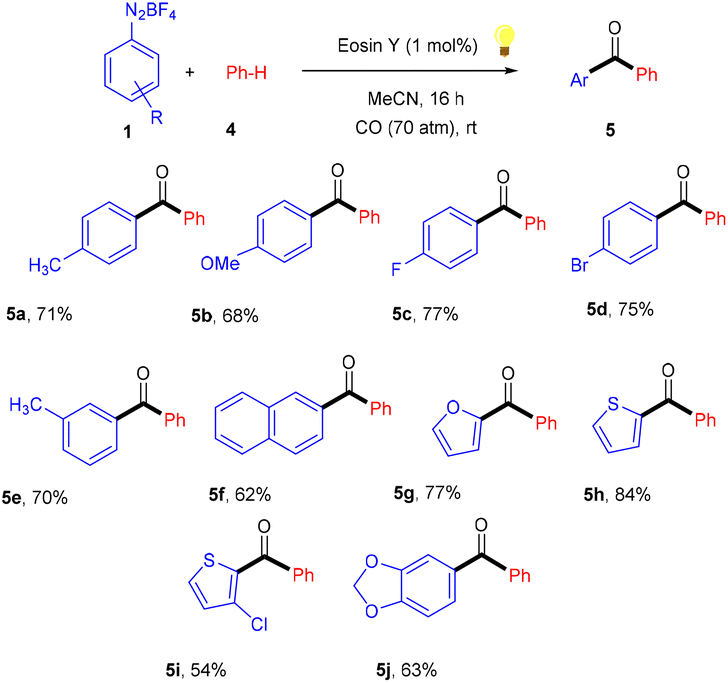
Similarly, aryl ketones are privileged motifs in natural products and are widely used as building blocks in synthesizing various pharmaceutical agents. In this context, aryl diazonium salts (1) have given access to aryl ketones (5) by photoredox carbonylation of arenes. Gu and his coworkers in 2015 used photoredox catalysis in the presence of eosin Y to establish a metal-free carbonylation process. The sequential functioning of SET reduction, carbonylation, and reverse electron transfer to produce an acylium cation is supported by mechanistic studies. At room temperature, aryl ketones (5) were synthesized from aryl diazonium salts (1), CO, and other heterocyclic arenes under mild reaction conditions. Several additional (hetero)arenes were likewise subjected to visible-light-induced acylation and gave the corresponding aryl ketones. Moreover, the authors also showed that the efficiency of the reaction was affected by steric effects. Notably, electron-rich aromatic rings have more compatibility and give the desired products in 63%–84% yields (Scheme 3).12 Like aryl ketones, benzoates are also privileged motifs in agrochemicals, materials, and various pharmaceuticals.
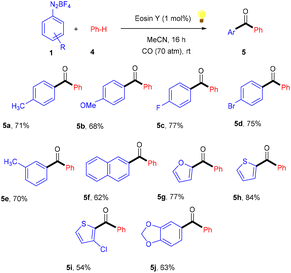 |
| | Scheme 3 Photoinduced carbonylation of arenes. | |
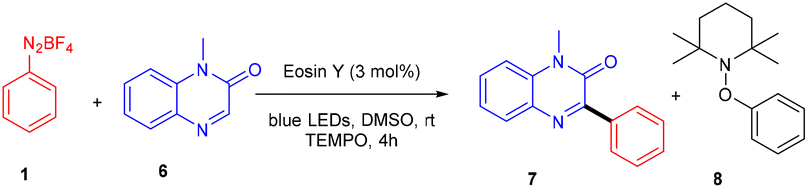
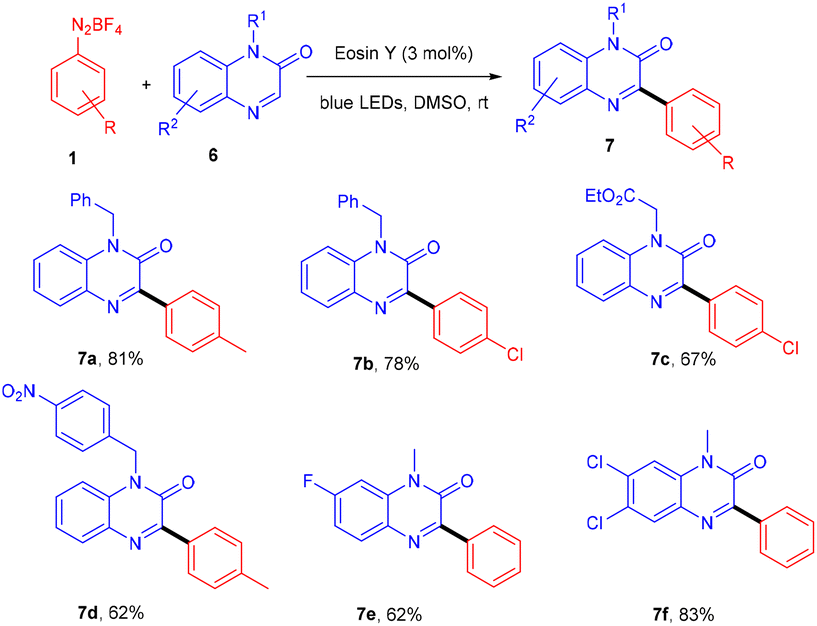
Kim et al. in 2018 demonstrated a direct C–C bond formation using a photoredox reaction where various 3-aryl quinoxaline-2(1H)-ones (7) were designed via the direct C–H arylation of quinoxaline-2(1H)-ones (6) with diazonium salts (1). A free radical trapping experiment (Scheme 4) was conducted using TEMPO as a radical scavenger, which indicated the SET mechanism. Due to steric hindrance, Kim's group reported that ortho-substituted diazonium salts were less compatible. Moreover, the electronic effects of quinoxaline-2(1H)-ones (6) were also investigated, where both electron-donating and withdrawing groups were consistent and gave good yields. On the other hand, various N-protecting groups were examined, and no significant effect on the reaction was observed (Scheme 5).13
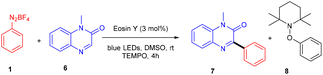 |
| | Scheme 4 Free radical trapping experiment. | |
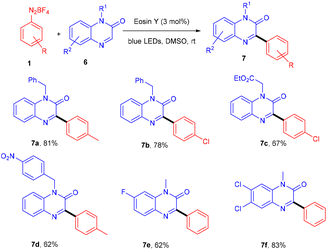 |
| | Scheme 5 Direct arylation of quinoxaline-2(1H)-ones. | |
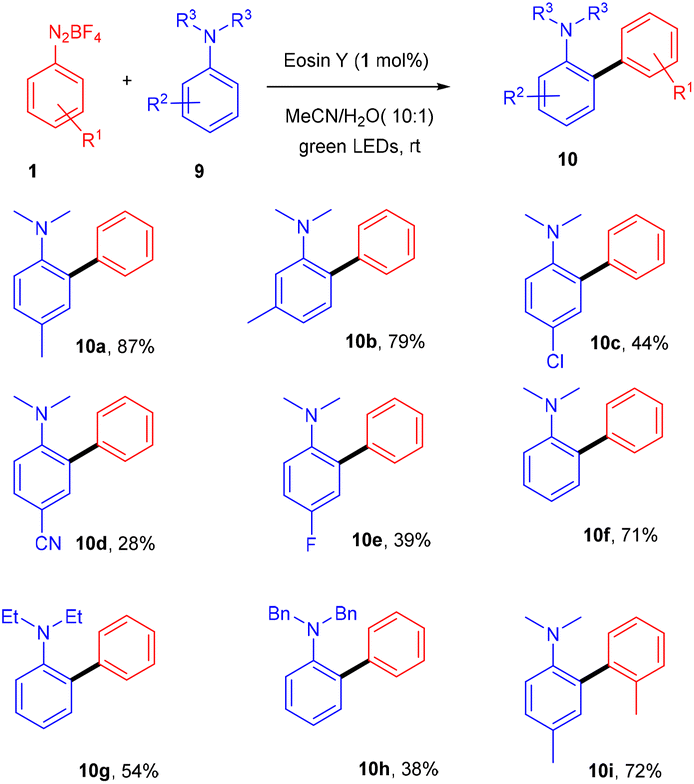
In the same year, the Yadav group extended Kim's method of direct arylation for the construction of the C–C bond to develop an efficient strategy for synthesizing amino-substituted biphenyl compounds by the straightforward installation of a phenyl ring on anilines; herein, they performed the reaction between anilines (9) and diazonium salts (1) in acetonitrile under green light photoredox catalysis. Substitution on aryldiazonium salts with alkyl, methoxy, fluoro, and cyano groups gave the corresponding diaryl anilines (10) in moderate to good yields. However, when cyano and fluoro groups were installed on N,N-dimethylaniline at the para-position, the corresponding compounds were obtained in low yields. Similarly, when N,N-diphenylaniline was used, again, a low yield of the final compound was obtained (Scheme 6).14 During investigation studies using TEMPO, they obtained a TEMPO-adduct which was further confirmed by HRMS, and indicated a radical pathway for the reaction.
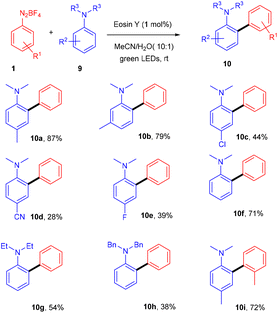 |
| | Scheme 6 Direct arylation of amines. | |
It is noteworthy that Na2–eosin Y (Na2EY) upon irradiation with visible light can get excited to its higher singlet state with t1/2 = 1.9 ns, whereas neutral eosin Y has t1/2 = 2.1 ns due to which Na2EY has more potential to undergo intersystem crossing (ISC) for the generation of its long-lived triplet state (lowest) compared to neutral eosin Y. Several studies have been reported using Na2EY as the photocatalyst having a high efficiency to improve the yield of the final product.15
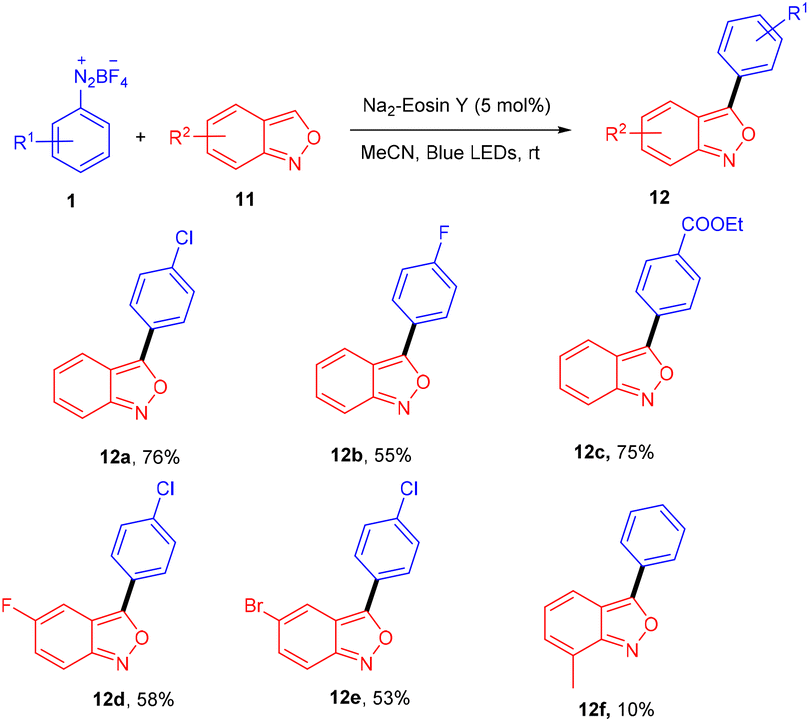
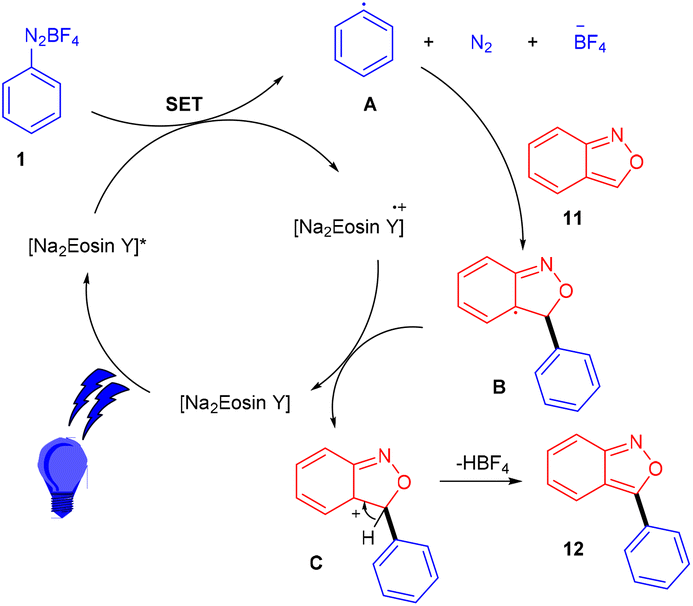
Hashmi and coworkers in 2020 revealed the C-3 direct arylation of valuable precursor anthranils (11) using various aryldiazonium salts (1) in the presence of the Na2EY photocatalyst at room temperature under the irradiation of blue light. Halogen (F, Br) substituted aryldiazonium salts reacted smoothly and afforded moderate yields, while substituents such as ester, acyl, and electron-withdrawing groups displayed better compatibility to afford good yields (Scheme 7). In addition, they showed the synthetic utility by synthesizing indole and quinoline scaffolds via the formation of α-imino gold carbenes, followed by their addition to alkynes.15 The authors performed radical trapping experiments and found an arene-TEMPO adduct that underwent a stress-free radical reaction pathway by a SET mechanism. The photoinduced triplet state (3[Na2EY]*) having a reduction potential (E1/2 ([Na2EY]/3[Na2EY]*) = −1.11 V vs. SCE) reduced the diazonium salt 1 to give aryl radical A. The attack of the aryl radical on anthranil gave radical intermediate B stabilized by benzisoxazole. SET by Na2EY˙− having an oxidation potential (E1/2 [Na2EY]˙−/[Na2EY] = 0.78 V) regenerated Na2EY along with the formation of a carbocation which gave the final product after deprotonation (Scheme 8).
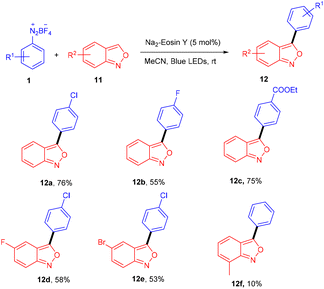 |
| | Scheme 7 Direct C3–H arylation of anthranils. | |
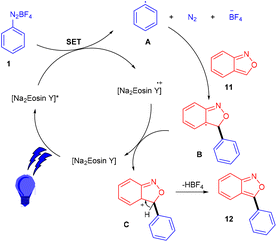 |
| | Scheme 8 Plausible mechanism for arylation. | |
2.2. C–H arylation using heteroarenes
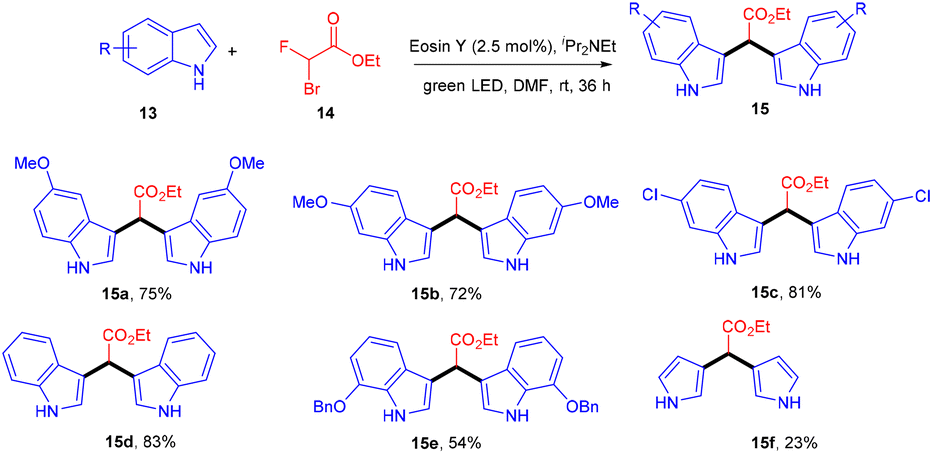
In 2015 Singh's group synthesized bisindolyl acetate scaffolds (15) by developing photoredox transformation. Ethyl bromofluoroacetate (14) was used as a radical precursor that reacted with nucleophilic arenes to afford valuable bisindole (15). Here, α,α-diarylated acetate compounds were obtained, following the construction of two C–C bonds. During the mechanistic investigation, the reaction did not proceed in the presence of TEMPO, which caused the trapping of the radical. This methodology can fill the gap in the respective area by offering an atom-economical route to access these molecules; using this approach, many substituted compounds were obtained in yields up to 79% (Scheme 9). However, free anilines and 3-substituted indoles did not provide the desired results.16
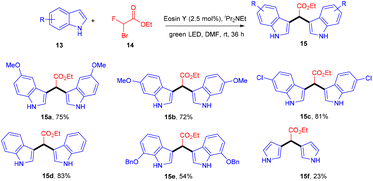 |
| | Scheme 9 Photoinduced activation of ethyl bromofluoroacetate. | |
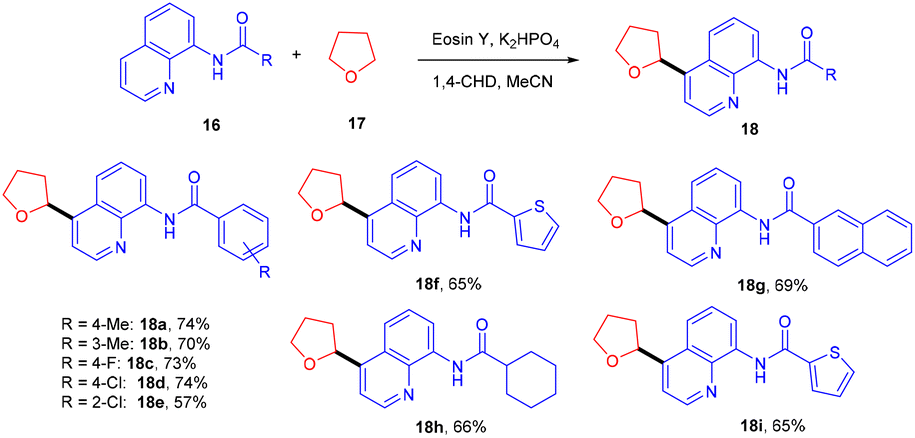
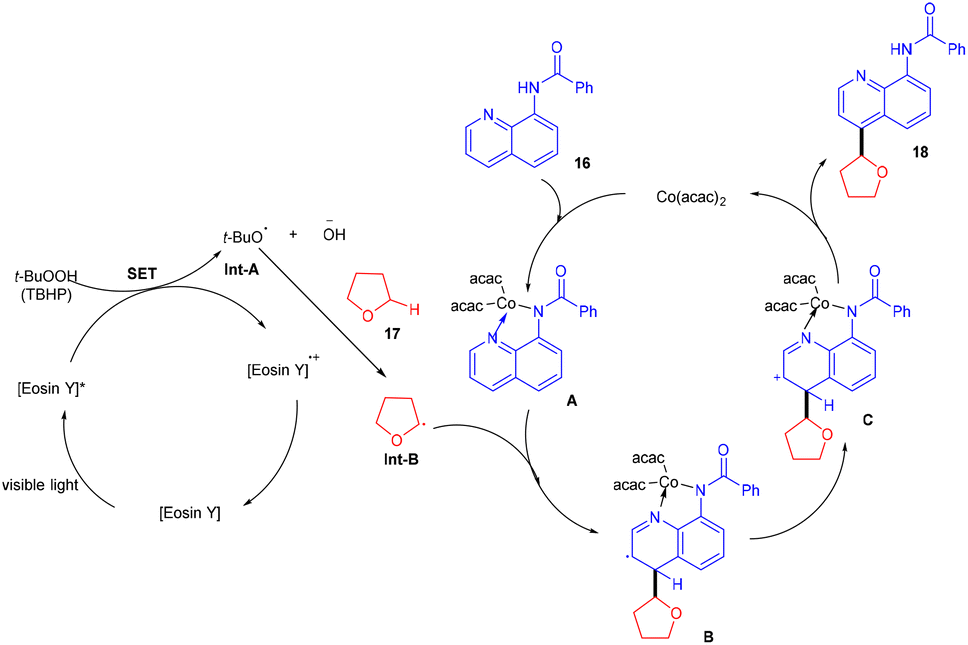
In 2018, Ranu and co-workers carried out C–H functionalization of cyclic ether for the formation of the C–C bond. They demonstrated remote C–H functionalization of 8-aminoquinoline amide by cyclic ethers under dual catalysis. They catalyzed the reaction under visible light using eosin Y as the main cocatalyst for the generation of free radicals of cyclic ether (17). It formed a cobalt chelate complex followed by interaction with the excited radical eosin Y and abstraction of H+ to give the desired product. They also demonstrated the hybridization of ether with the quinoline amide unit. This approach has synthesized several functionalized ethers (18) in good yields (Scheme 10).17 The reaction proceeded with the generation of a tert-butyl hydrogen radical (Int-A), generated by the dissociation of tert-butyl hydrogen peroxide using photoexcited eosin Y. The free radical abstracted the α-hydrogen of THF to give another radical (Int-B), which interacted with the cobalt–quinoline complex A and gave complex B. Cationic intermediate C was obtained from complex B followed by deprotonation of complex C and the final product was formed (Scheme 11).
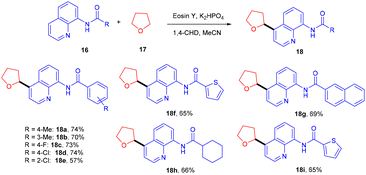 |
| | Scheme 10 Arylation of cyclic ether. | |
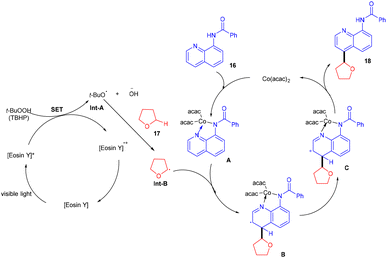 |
| | Scheme 11 Mechanism for remote C–H functionalization. | |
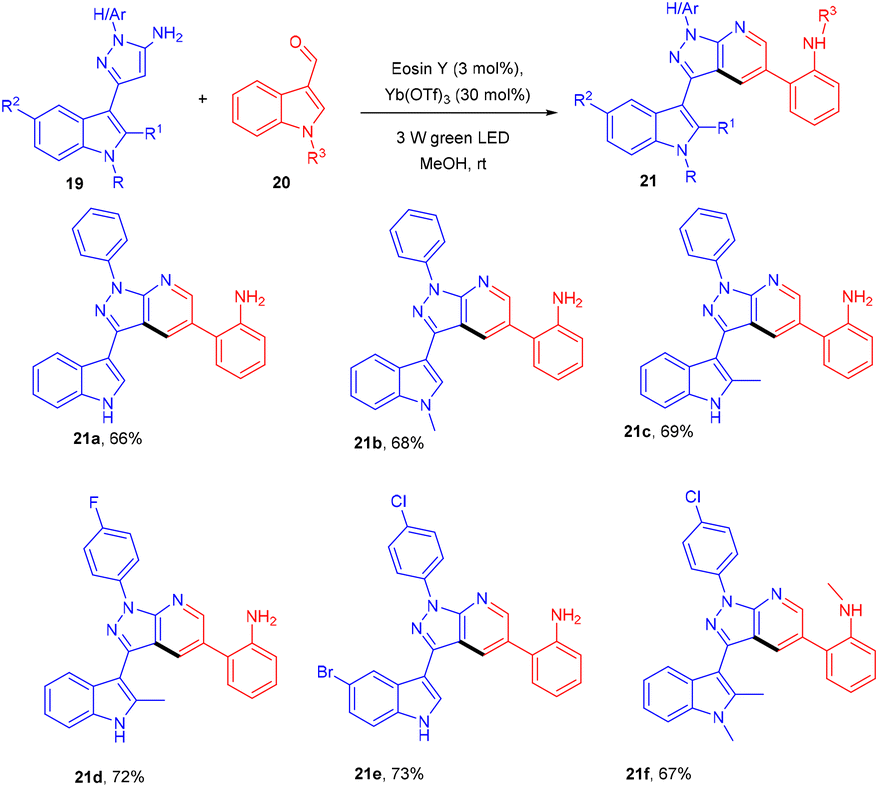
In the same year, the Yadav group developed an eosin Y-based dual catalytic system for synthesizing heterobiaryl-pyrazolo[3,4-b] pyridines (21). The authors demonstrated the efficiency of a catalytic system of eosin Y and Yb(OTf)3 (dual catalytic system) by carrying out the reaction of 3-(1H-indol-3-yl)-1-phenyl-1H-pyrazole-5-amine (19) with 1H-indole-3-carbaldehyde (20) for the synthesis of nitrogen-containing heterocyclic compounds. The reaction involved electrocyclization and opening of the indole ring under photoredox catalytic conditions (Scheme 12).18 The authors also demonstrated the TEMPO study and found complete reaction quenching, which means that the reaction involved a radical mechanism.
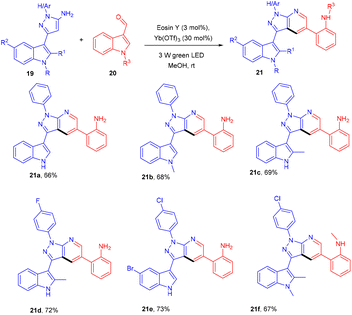 |
| | Scheme 12 Direct arylation of heteroarenes. | |
2.3. C–H arylation using arylhalides and arylamines
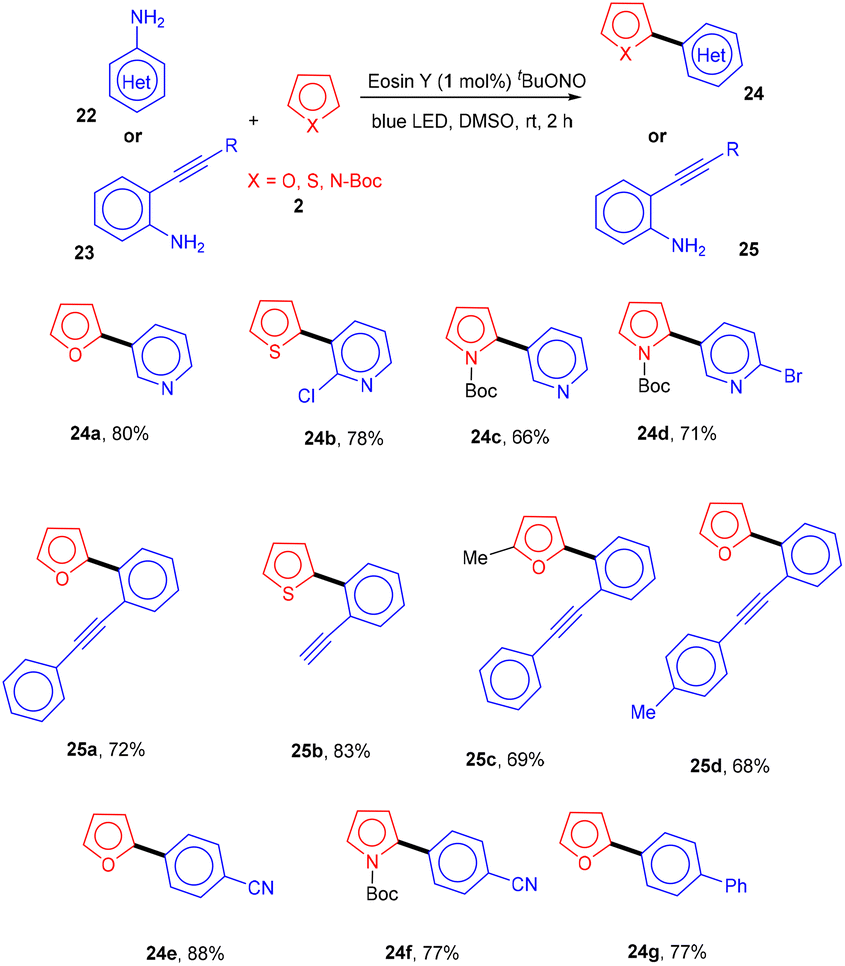
Inspired by König's work, the Ranu group in 2015 also carried out an arylation of heteroarenes (2) using substituted (hetero)anilines (22, 23) under photo-catalysis. Significantly, the heteroarylation by 2-ethynylanilines (23) progresses smoothly, which is not possible with other standard procedures. As a result, this procedure solves the substantial shortcomings of previous techniques for heteroarylation using heteroaryl amines that were not acceptable. Various heterocyclic arenes such as furan, –thiophene, and 2-methyl furan reacted efficiently with thiazolylamine, pyridinyl, and quinolinyl to afford the corresponding compounds (24, 25) in good yields. In addition, functional groups such as keto–carbonyl, methoxy, cyano, and nitro all showed compatibility for the reaction (Scheme 13).19 Here, tBuONO was used for diazonium salt formation from anilines, which underwent the SET mechanism and gave free radicals.
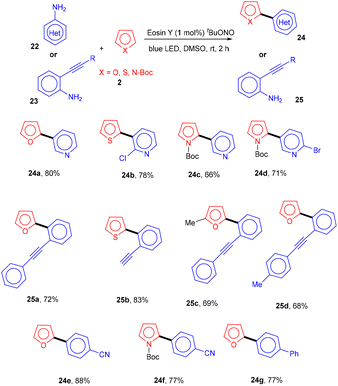 |
| | Scheme 13 Visible light-mediated synthesis of bi or bis-heteroaryls. | |
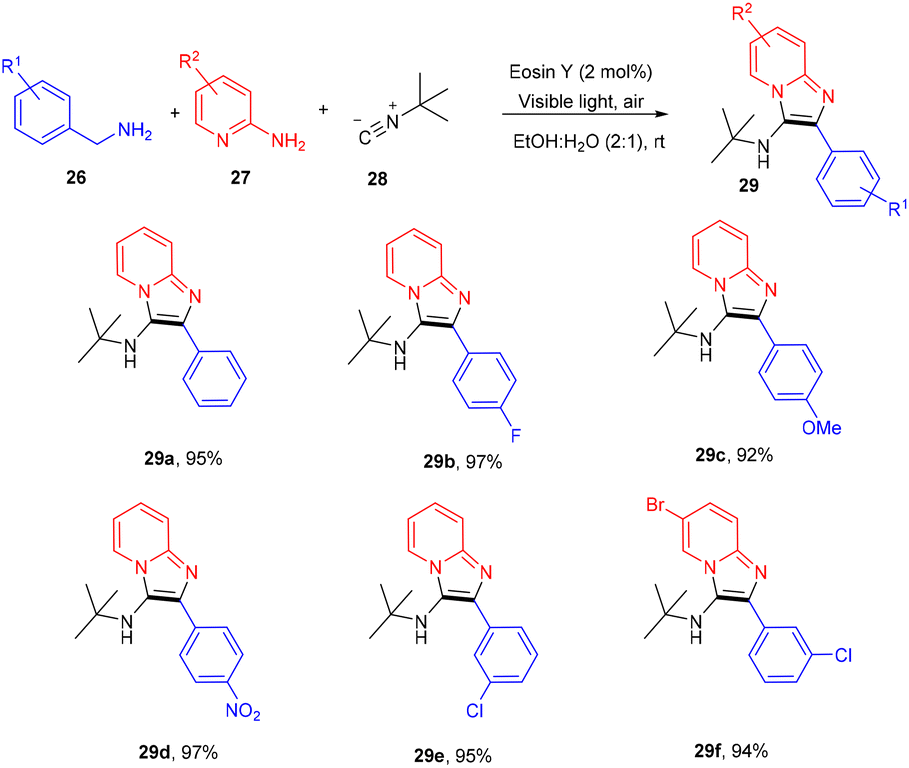
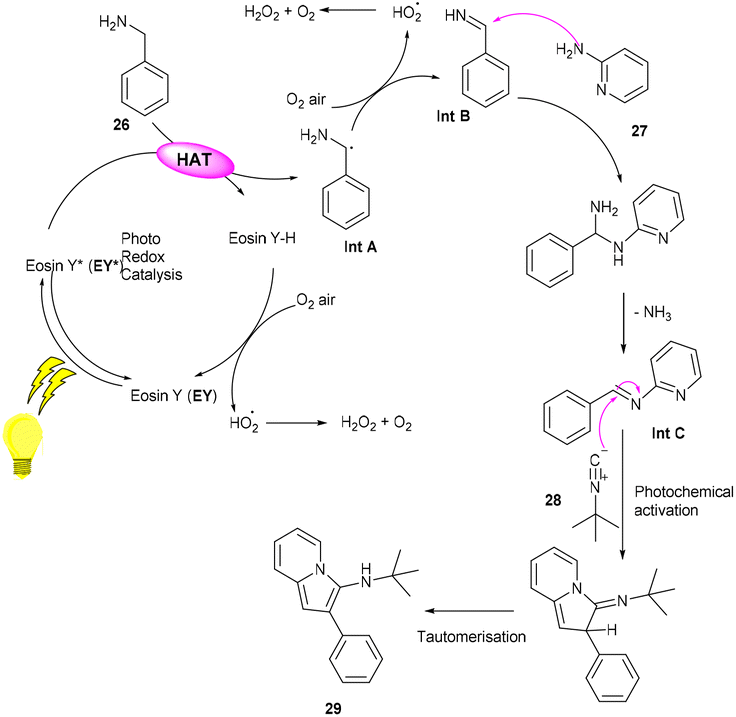
However, to access various 3-aminoimidazo[1,2-a]pyridines (29), a multicomponent photoinduced cascade transformation was developed in 2020 by Singh et al. They successfully performed the reaction with benzylamine (26), 2-aminopyridine (27), and t-butyl isocyanide (28) using eosin Y as the photocatalyst at room temperature. They demonstrated the formation of a C–C bond. This protocol has outstanding functional group tolerance to benzylamine and gave the desired products with notable yields. Electron-withdrawing groups such as nitro and fluoro gave higher yields than electron-donating groups (methyl and methoxy) (Scheme 14).20 Benzylamine is unable to undergo oxidation, due to the absence of a valid SET mode for this reaction. Here, eosin Y (EY) gets excited upon irradiation of visible light and gives excited eosin Y (EY*), responsible for hydrogen extraction from benzylamine to give an intermediate (Int A). Subsequently, intermediate Int B is formed by the oxidation of Int A. Reaction of 2-aminopyridine with Int B affords an imine intermediate (Int C) followed by the nucleophilic addition of isocyanide and rearrangement, where Int D is formed, and after tautomerism, the desired compound (29) is obtained (Scheme 15).
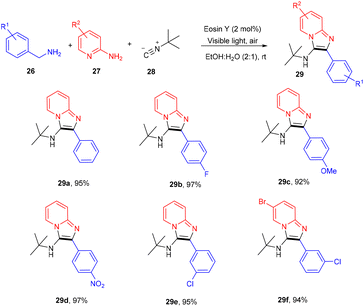 |
| | Scheme 14 Synthesis of 3-amino imidazopyridines. | |
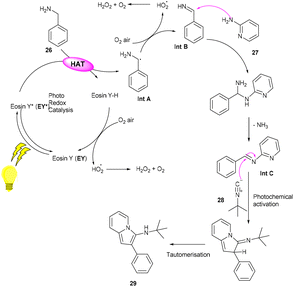 |
| | Scheme 15 Mechanism for the synthesis of 3-amino imidazopyridines. | |
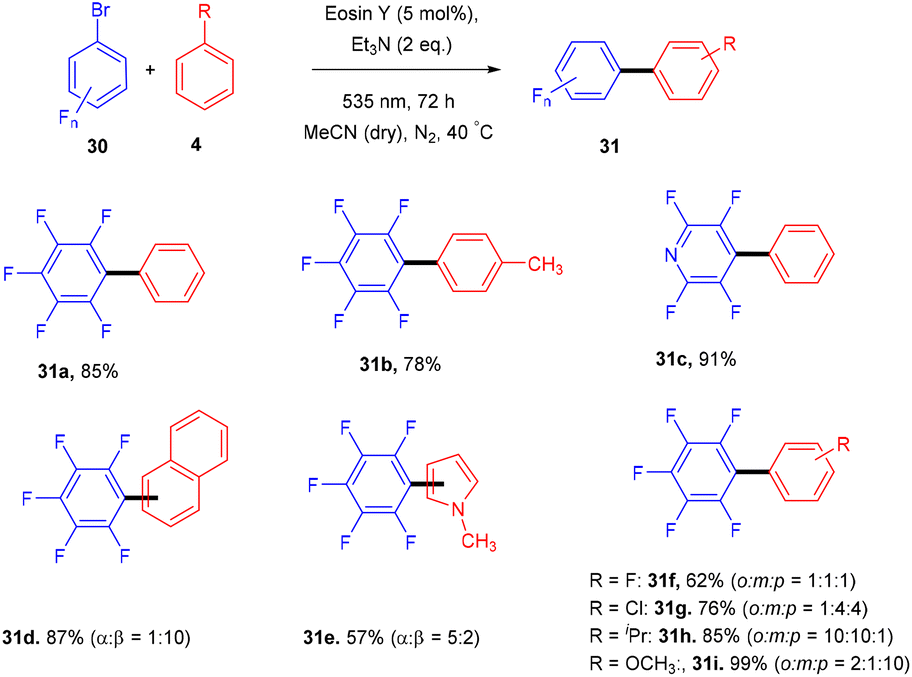
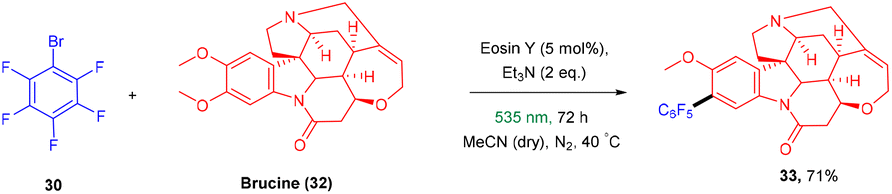
König and colleagues in 2016 reported the direct arylation of arenes for the synthesis of polyfluorinated biaryl compounds (31). Various arenes, having mono substitution, naphthalene and N-methyl-pyrrole gave the corresponding compounds in yields ranging from 57% to 91%. Moreover, electron-donating and withdrawing substituents were well tolerated to synthesize a number of halogenated biaryl compounds in the presence of eosin Y and showed tremendous significance over transition metal catalysts (Scheme 16).21 This approach has also been used for the late-stage functionalization of the natural product brucine (toxic alkaloid) (32) and demonstrated the functional group compatibility of this methodology (Scheme 17).21
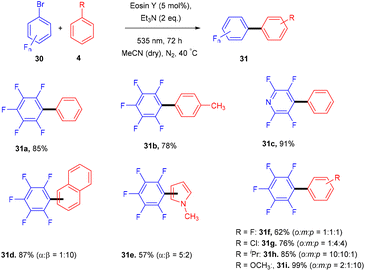 |
| | Scheme 16 Synthesis of polyfluorinated biaryls. | |
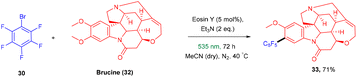 |
| | Scheme 17 Arylation of brucine. | |
It is noteworthy that the reaction proceeded through the reductive quenching cycle, where triethylamine was used as a sacrificial reductant which transferred electrons to the excited photocatalyst.
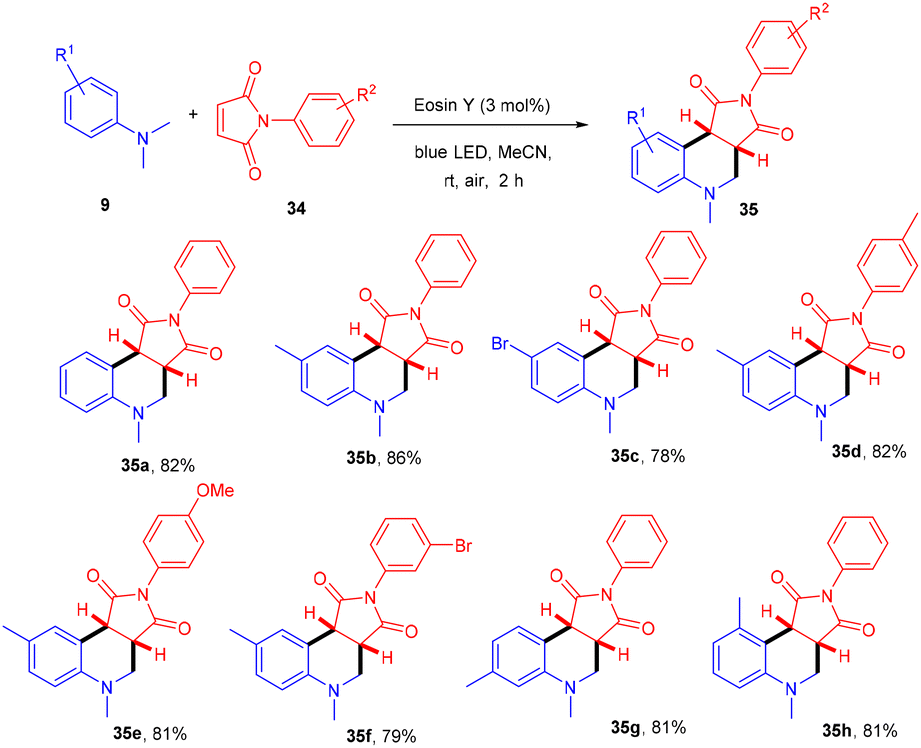
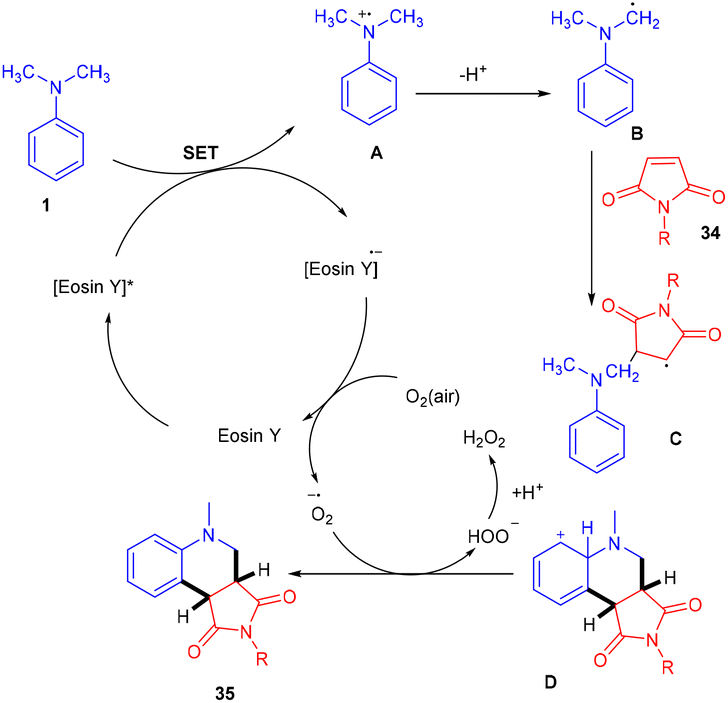
Zhan's group reported aerobic oxidative cyclization for synthesizing tetrahydroquinolines (35) using atmospheric oxygen and a metal-free photocatalyst. They carried out a one-pot reaction between N,N-dimethylanilines (9) and maleimides (34) under visible light as an energy source with 2 mol% loading of eosin Y (relatively low loading) at room temperature. Substituted N,N-dimethylanilines (9) with –Br and –CH3 groups reacted smoothly to afford the corresponding products in good yields. Furthermore, N-arylmaleimides (34) with –CH3, –OCH3, –Cl, and –Br gave related compounds with good yields (Scheme 18).22
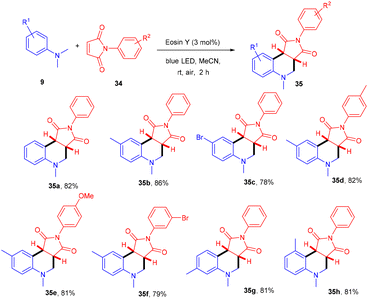 |
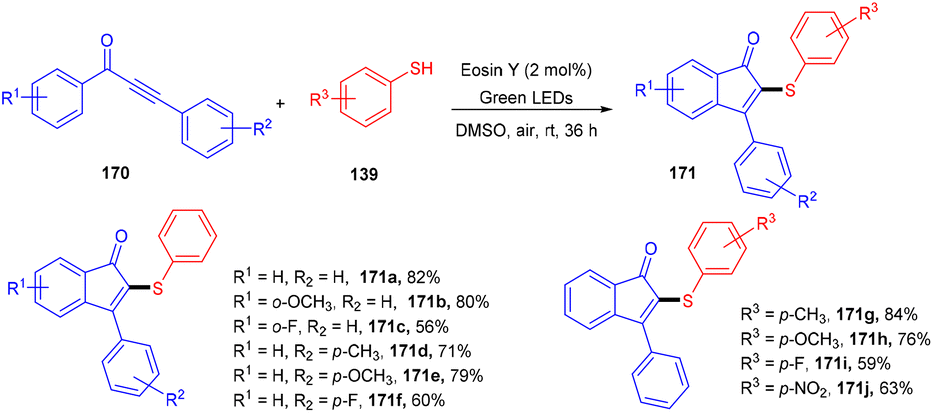
| | Scheme 18 Aerobic oxidative cyclization of N,N-dimethylanilines with maleimides. | |
Here the reaction started with electron transfer from amine to photoexcited eosin Y, which gave an amine radical cation along with [eosin Y]˙− which underwent SET in the presence of oxygen, was converted into neutral eosin Y and then formed a superoxide radical anion. Amine radical cation A underwent deprotonation to give α-aminoalkyl radical B, which further interacted with compound 34 and generated the corresponding radical C which underwent cyclization to produce cation intermediate D. The final product was obtained by proton and electron transfer during interaction between the superoxide radical anion and F along with HOO− which was further protonated to give H2O2 as the by-product (Scheme 19). H2O2 was confirmed using a KI/starch indicator.
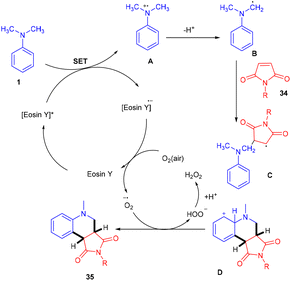 |
| | Scheme 19 Mechanism of aerobic oxidative cyclization. | |
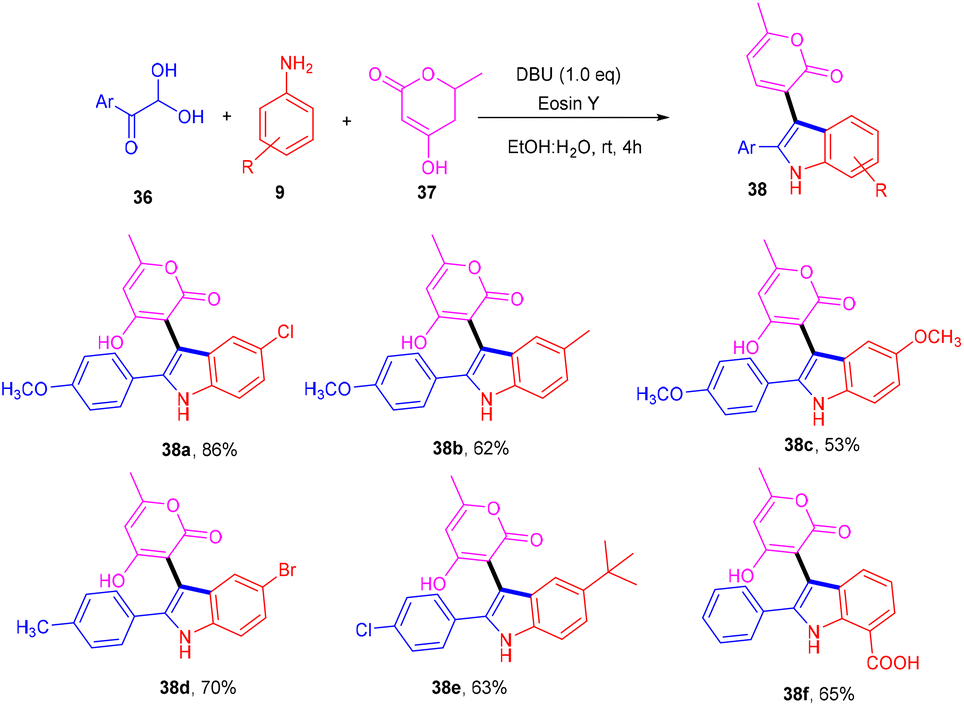
Siddiqui and his coworkers have recently reported a multicomponent one-pot synthesis of functionalized indole (38) using eosin Y as an organophotocatalyst. The reaction was performed using substituted anilines (9), 4-hydroxy-6(2H)-pyran-2-one (37) and 2,2-dihydroxy-1-(4-methoxyphenyl) ethan-1-one (36) and gave the corresponding compounds in moderate to good yields via the SET mechanism (Scheme 20).23 This methodology also reveals that eosin Y-mediated photocatalysis has the potential for the multicomponent reaction for synthesizing highly modified compounds.
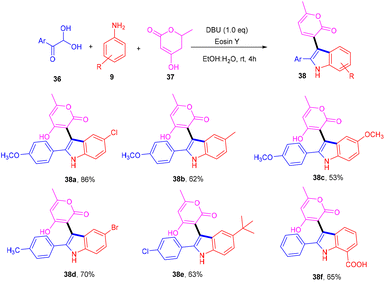 |
| | Scheme 20 Eosin Y mediated multicomponent reaction. | |
2.4. C–H arylation using sulfonyl chloride
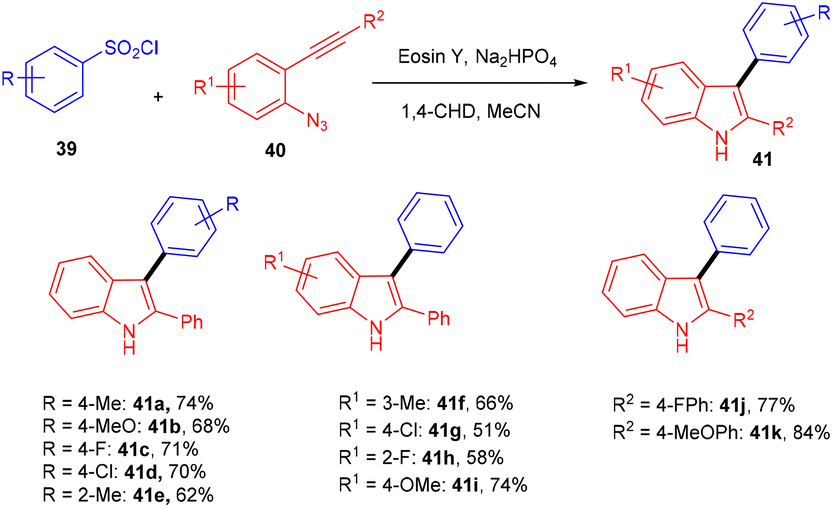
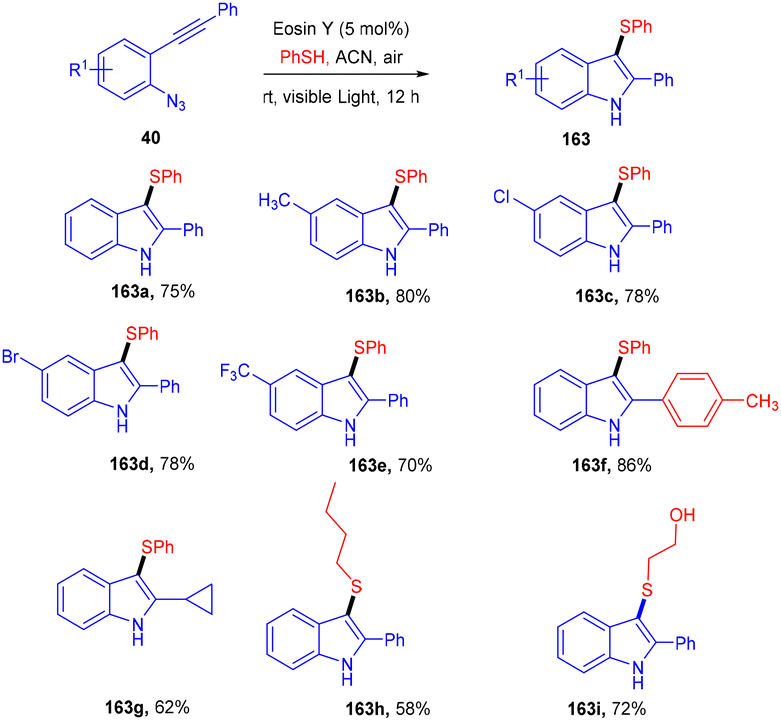
Inspired by Kim's work, Gu and his coworkers have developed an efficient photoredox eosin Y mediated methodology with aryl sulfonyl chloride (39). They performed direct annulation between o-azido aryl alkynes (40) and aryl sulfonyl chloride (39) to access various substituted unsymmetrical indoles (41) in good yields through cascade cyclization with high regioselectivity. During the screening of solvents, they found that the MeCN solvent gave a higher yield. The reaction was highly compatible with various sulfonyl chlorides (39) having different substituents at various positions. In addition, heteroaryl sulfonyl chlorides were well tolerated and gave good yields. The authors also performed radical trapping control experiments with TEMPO to depict the mechanism pathway and found that the reaction was completely quenched indicating a free radical pathway (Scheme 21).24
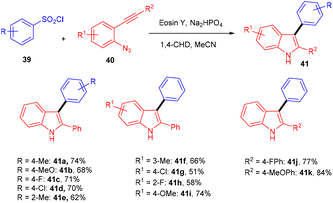 |
| | Scheme 21 Indole synthesis using aryl sulfonyl chloride. | |
2.5. C–C bond formation using carbonyl compounds as radical precursors
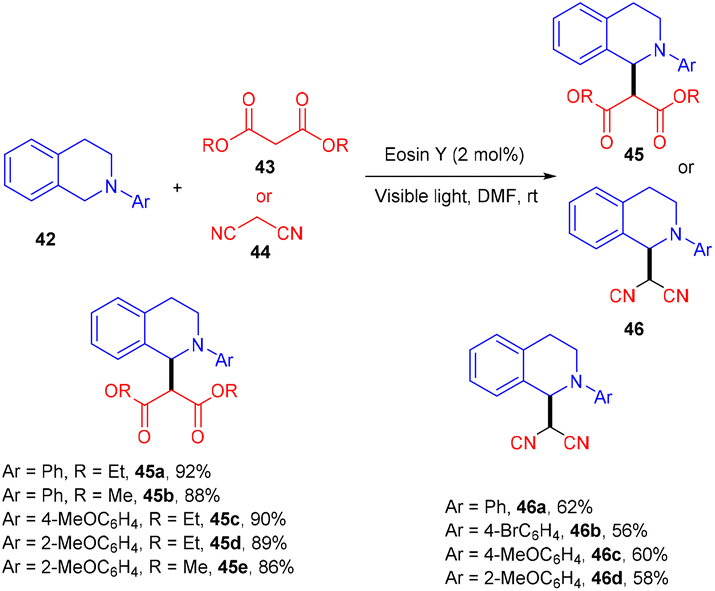
König's group developed various methodologies based on a visible light-mediated photoredox process catalyzed by the eosin Y photocatalyst. In 2011, they established photoinduced C–C bond formation to perform the C–H functionalization of tetrahydroisoquinoline without using any external oxidant. They used tetrahydroisoquinoline (42), dialkyl malonates (43), and malononitrile (44) for the envisioned hypothesis.25 During substrate scope screening, the authors found that malonates with methyl and ethyl groups were well reacted. In addition, substitutions such as –Br, –CH3, –OCH3 on the aromatic ring at different positions gave the corresponding scaffolds (45, 46) in good yields (Scheme 22). Notably, the reaction followed the SET mechanism, but photoexcited eosin Y was reduced by tetrahydroisoquinoline.
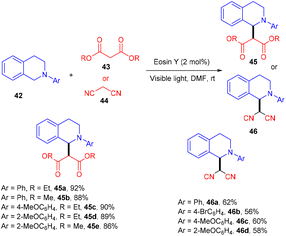 |
| | Scheme 22 Oxidative C–C bond formation. | |
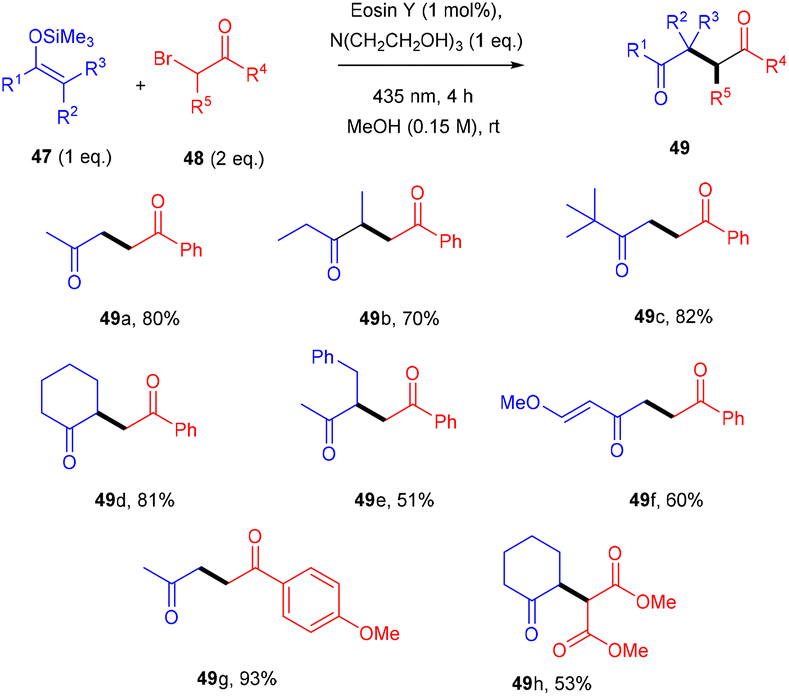
Yasuda and coworkers devised a feasible method for synthesizing 1,4-dicarbonyl compounds (49) by accelerating a reaction between α-halocarbonyls (48) and silyl enol ethers (47) with the inexpensive photocatalyst eosin Y under visible light irradiation. The chemoselectivity was optimal for the halo-substitution reaction (Scheme 23). Triethanolamine was discovered to be an effective reductant for the regeneration of eosin Y. This methodology was carried out with various silyl enol ethers and α-bromocarbonyl compounds. Furthermore, they envisioned secondary bromoketone, bromoamide, and bromoester, all giving the respective compounds (49) in moderate to good yields.26
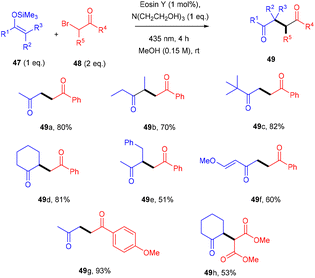 |
| | Scheme 23 Synthesis of 1,4-dicarbonyl compounds. | |
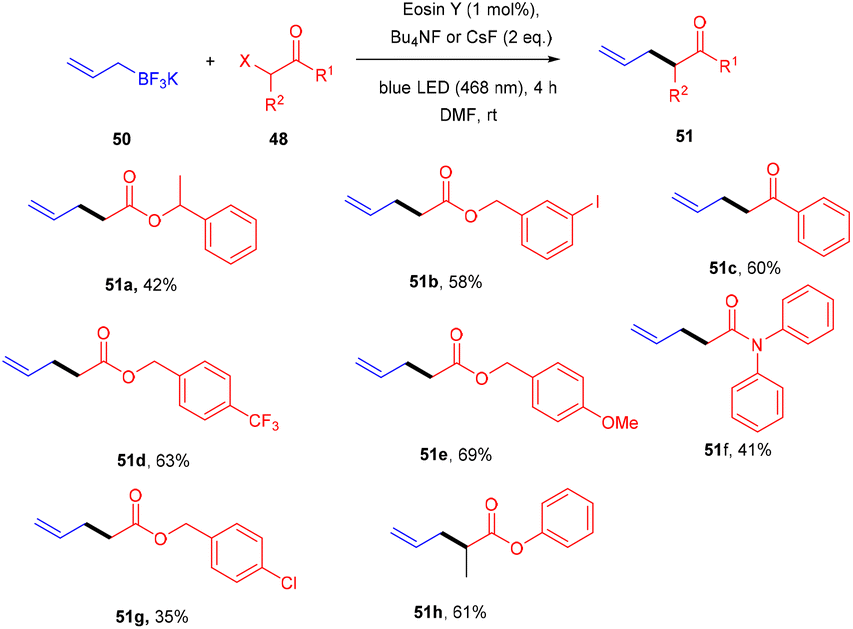
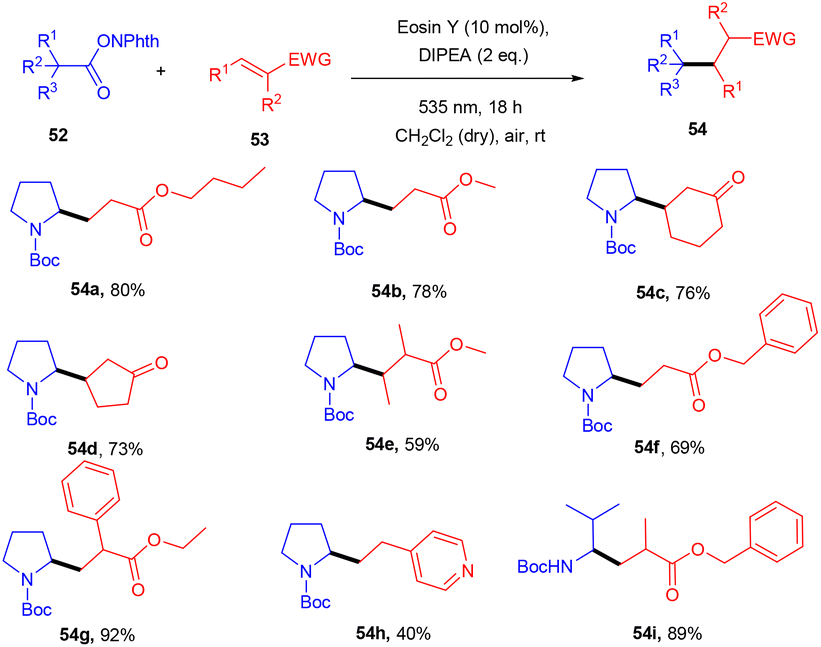
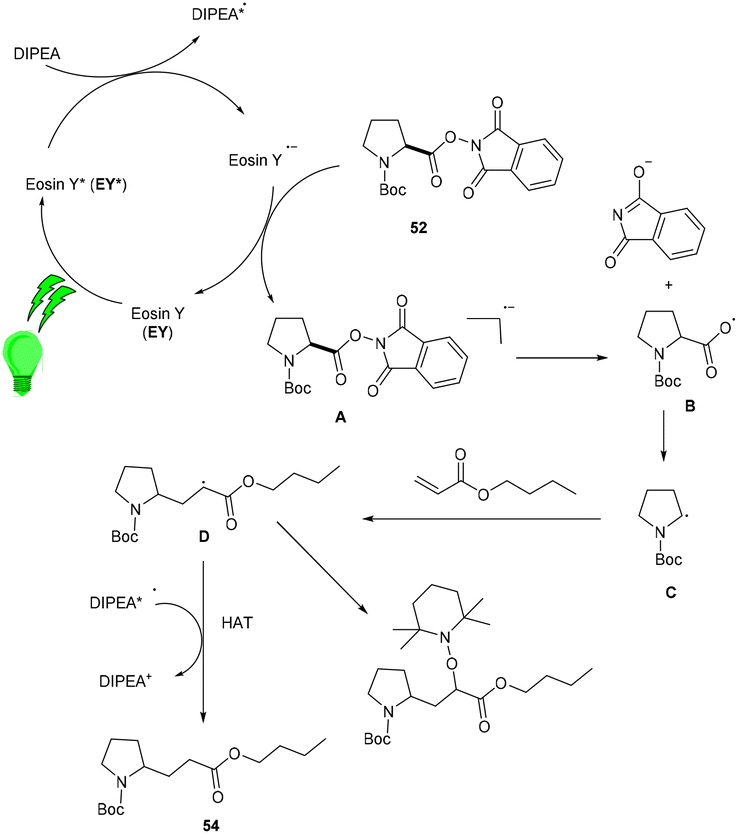
Yasuda's group also established exclusive selectivity for C–C bond formation in the reaction between α-bromocarbonyl (48) and allyl trifluoroborate (50) under photoredox catalysis (Scheme 24). In the presence of either Bu4NF or CsF, eosin Y catalyzed the reaction to form the desired product (51). Single-electron transfer (SET) from photoexcited eosin Y to halocarbonyls is aided by metal fluorides which behave as27 Lewis acids. The methodology was likewise successful when using allyl pinacol boronates as allylating reagents. For the allylation of α-halocarbonyls, this process is believed to be both economical and safe. In the same year, König's group also developed a unique approach in which metal-free eosin Y catalyzed decarboxylative alkylation for C–C bond formation. This approach was applied to α-oxy acids, amino acids, and various fatty acids and thus has a vast substrate scope (Scheme 25). Benzylic and non-benzylic α,β-unsaturated esters, and ketones (53) gave the corresponding compounds (54) in moderate to good yields. The reaction worked well for various carboxylic acids (such as amino acids, fatty acids, primary and tertiary acids). It has been demonstrated that several compounds could be synthesized from renewable biomass with the present methodology. Irradiation of visible light on eosin Y (EY) generated the excited photocatalyst (EY*), which underwent reductive quenching in the presence of the sacrificial reductant DIPEA to generate DIPEA˙+. Eosin Y (EY) regenerated by the reduction of N-(acyloxy)phthalimide gave the corresponding radical (B). In addition, decarboxylation gave an alkyl radical (C), which reacted with a Michael acceptor to form the respective intermediate (D) followed by hydrogen atom abstraction from DIPEA˙+, enabling the formation of the desired compound (54) (Scheme 26).28
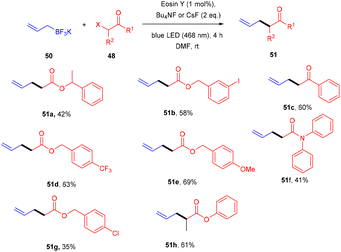 |
| | Scheme 24 Photoinduced allylation of α-halocarbonyls. | |
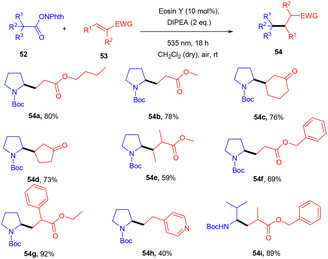 |
| | Scheme 25 Visible-light mediated decarboxylative alkylation. | |
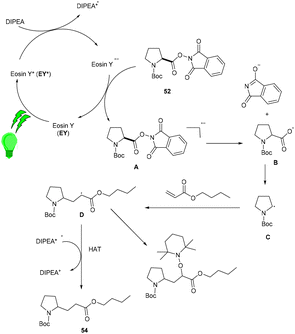 |
| | Scheme 26 Plausible mechanism of decarboxylative alkylation. | |
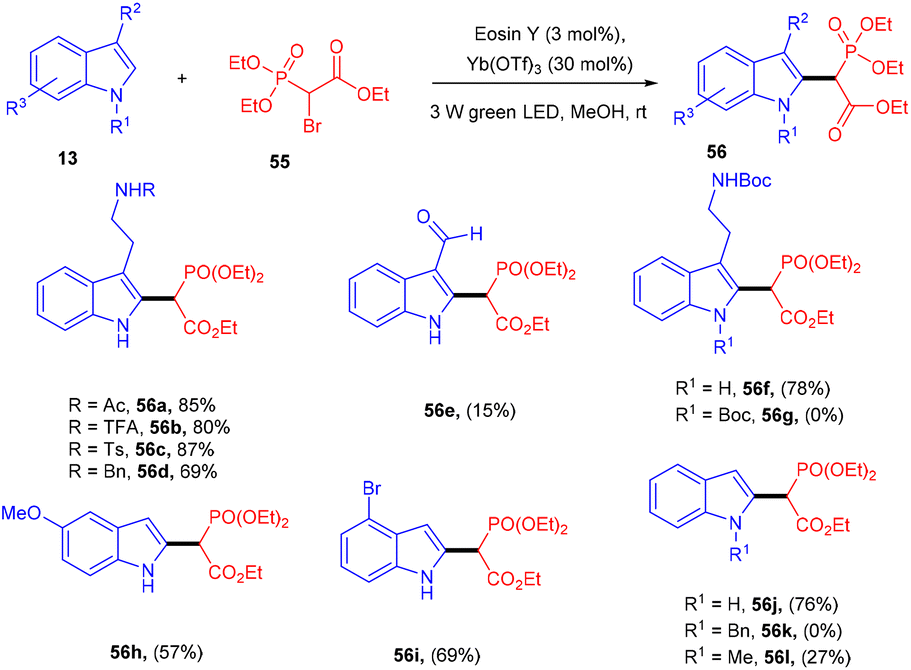
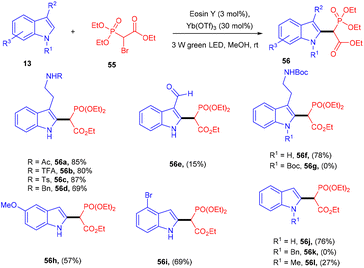
In one instance, Horner precursors were synthesized via C–C bond formation using triethyl 2-bromophosphonoacetate (56) and indoles (13) under photo-redox catalysis in 2018 by Opatz and co-workers. The authors used triethylphosphonoacetate as a radical precursor. N-Acylated or N-sulfonylated tryptamines reacted efficiently and gave a high yield of the corresponding compounds (56). However, 3-unsubstituted indoles afforded 2-substituted products with high regioselectivity. Due to steric effects, N-functionalized indoles were not converted into the corresponding products. This developed methodology has the potential for the total synthesis of various natural products and provides access to the late-stage modification of peptides (Scheme 27).29
 |
| | Scheme 27 Synthesis of Horner precursors. | |
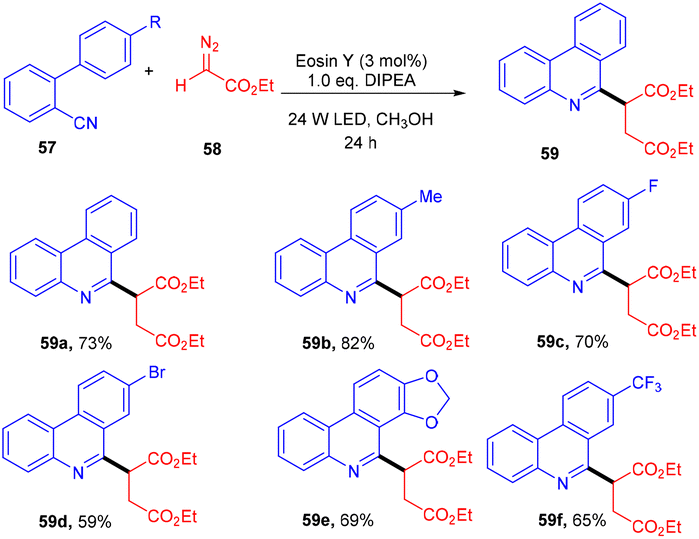
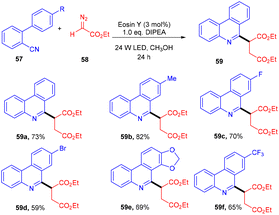
In 2018, Xuan and co-workers disclosed the synthesis of succinic ester (59) from diazo compounds (58) as 1,4-dicarbonyl precursors. During optimization studies, various solvents and bases were screened. Furthermore, control experiments also shed some light on the significant role of the photocatalyst and light source. Radical trapping experiments using TEMPO indicated that the reaction proceeded through a free radical pathway. In the presence of a deuterium source, a deuterated product was obtained which revealed proton-coupled electron transfer for the generation of an α-carbonyl methylene radical from diazoacetate. The reaction showed great compatibility with electron withdrawing and donating groups to afford the desired compounds in up to 82% yield (Scheme 28).30
 |
| | Scheme 28 Synthesis of succinic esters. | |
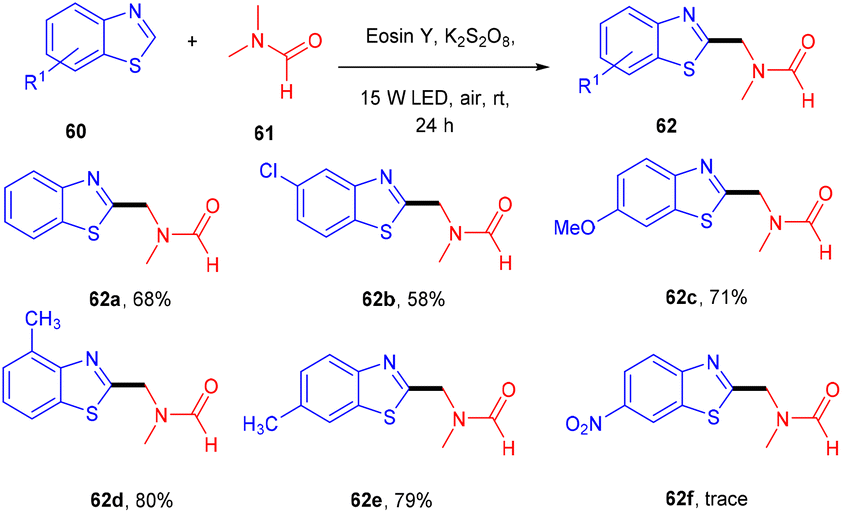
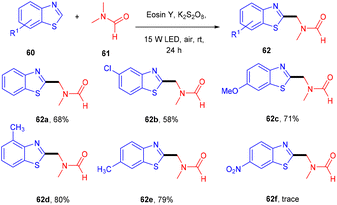
In 2019, Xu et al. performed the alkylation of benzothiazole (60) using N,N-dimethyl amides (61) via a xanthene-based dye-mediated photoredox system. The reaction was performed under solvent-free conditions using K2S2O8 as an oxidant. It was found that the substitution of benzothiazole at the 4 and 7 positions showed steric hindrance that could limit the potential of the reaction. In addition, they also found that electron-donating groups have more compatibility and give higher yields compared to electron-withdrawing groups (Scheme 29).31 During the introduction of TEMPO in the reaction, only a DMF-TEMPO adduct was obtained, revealing that the reaction proceeded through the free radical mechanism.
 |
| | Scheme 29 Eosin Y mediated alkylation of benzothiazole. | |
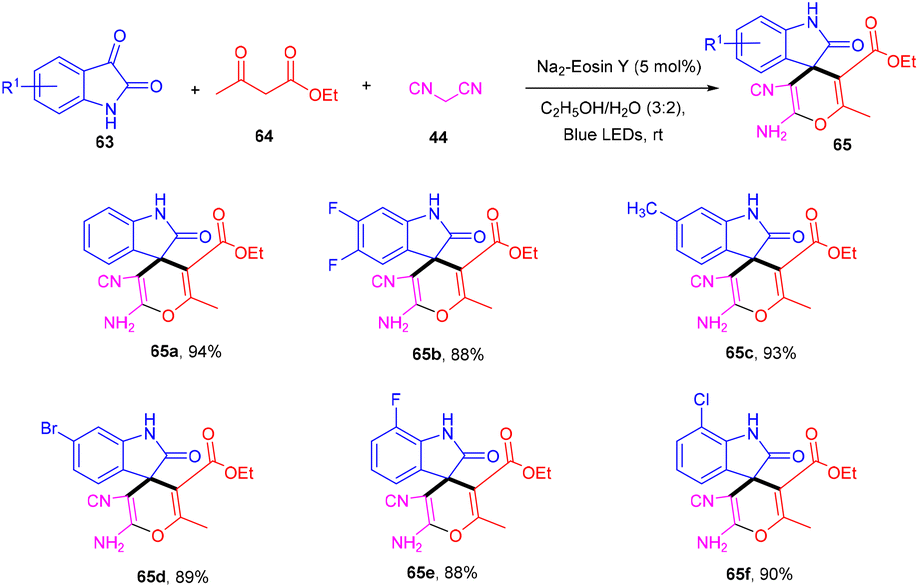
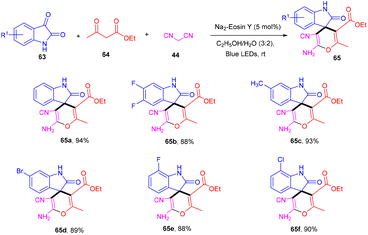
Zhang's group in 2020 developed an efficient synthesis of spiro[oxindole-3,4-(4H-pyran)] (65) scaffolds through the visible light-mediated one-pot multicomponent reaction and the formation of multiple C–C bonds. They performed a reaction between isatins (63), 1,3-dicarbonyl (64), and malononitrile (47) in the presence of eosin Y as an organo-photoredox catalyst to afford the final compounds (65) in excellent yields. They also explored substitution on the phenyl ring of isatin and revealed that both electron-donating and withdrawing groups have compatibility with the designed methodology (Scheme 30).32
 |
| | Scheme 30 Synthesis of spiro[oxindole-3,4-(4H-pyran)] under visible light. | |
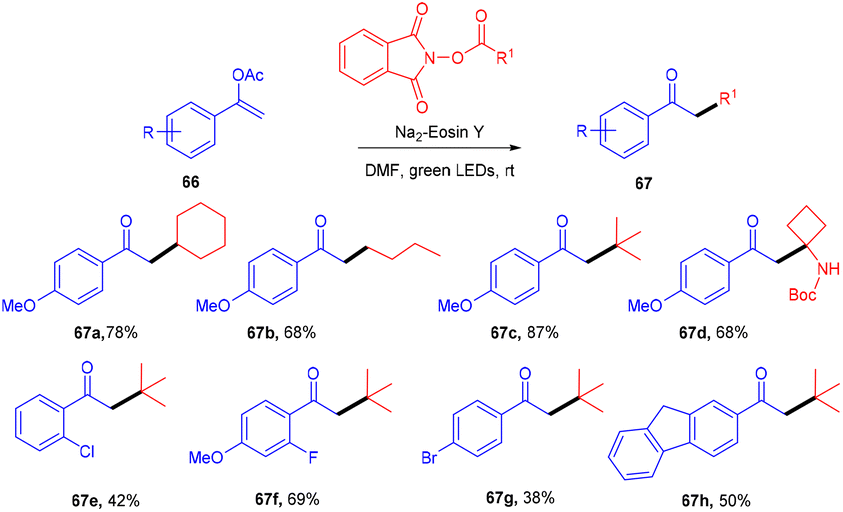
Leng's group in 2021 disclosed the practical synthesis of α-alkylated ketones (67) by eosin Y mediated alkylation of enol acetates through decarboxylation of N-hydroxyphthalimide acetate. A broad substrate scope and high site selectivity were the prominent features of this strategy. Enol acetate having nBuO, OBn, and OPh as functional groups were also well tolerated. In addition, they demonstrated the synthetic utility of this method by synthesizing dyclonine hydrochloride (Scheme 31).33 The reaction involving the radical decarboxylation addition of the alkyl radical to enol acetates followed by oxidation gave the desired products.
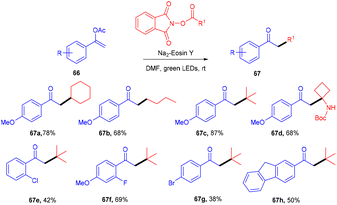 |
| | Scheme 31 Alkylation of enol acetates via radical decarboxylation. | |
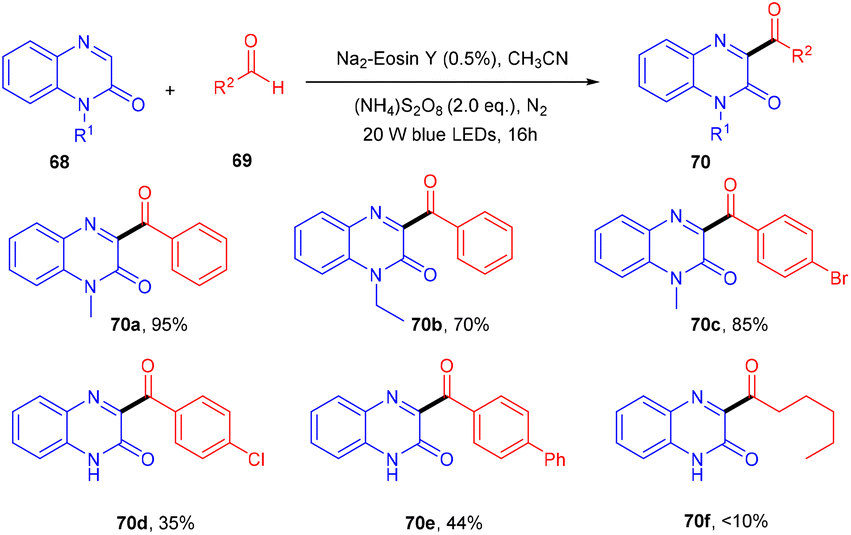
In the same year, direct C3-acylation of quinoxaline-2(1H)-one (68) under visible light using eosin Y as the catalyst was disclosed in 2021 by the Zhao group. Here, they started with readily available aldehydes (69), acting as radical precursors, generated by excited eosin Y, followed by the addition of the radical to the heterocyclic compound quinoxaline-2(1H)-one which underwent oxidation to afford the desired compounds (70) in excellent yields. This approach of acylation has a broad substrate scope. However, electron-donating and -withdrawing substitutions on aldehyde gave the corresponding compounds in lower yields. In addition, protected and unprotected quinoxaline-2(1H)-ones showed compatibility with the reaction (Scheme 32).34
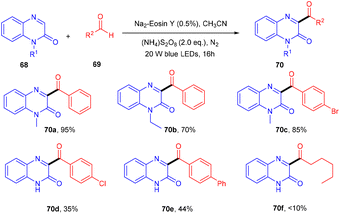 |
| | Scheme 32 Photoinduced C3-acylation of quinoxaline-2(1H)-ones. | |
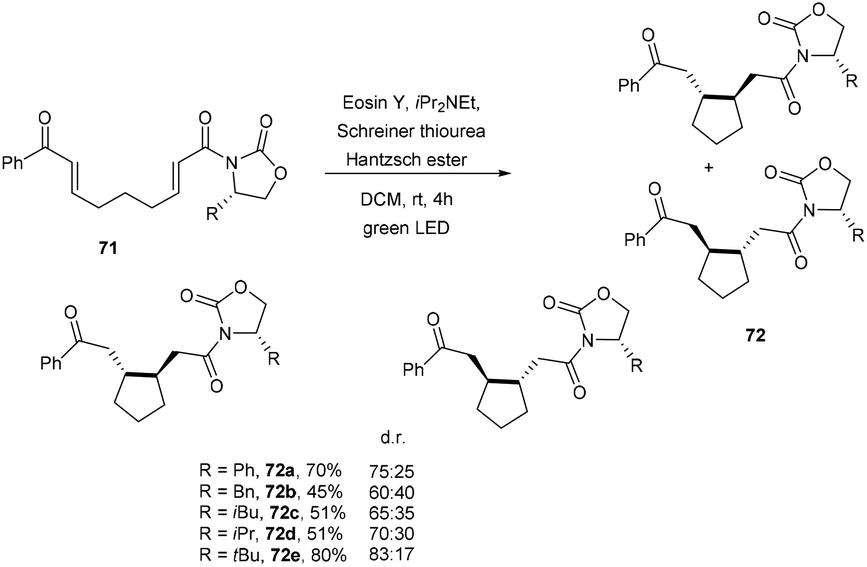
A promising technique to access cycloalkanes via the photoinduced cyclization of enones under the irradiation of visible light using an organic dye as a photocatalyst was developed by Benaglia and coworkers. They synthesized enantiomerically functionalized cyclopentane rings (72) using eosin Y. The reaction was performed in a homemade photoredox coil reactor and a 3D-printed meso reactor and obtained excellent yields of various scaffolds (Scheme 33).35
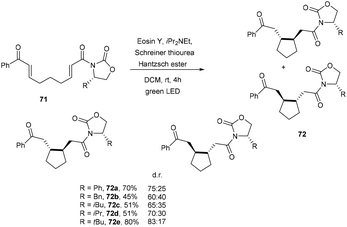 |
| | Scheme 33 Synthesis of the cyclopentane ring. | |
2.6. C–H alkylation using haloalkanes
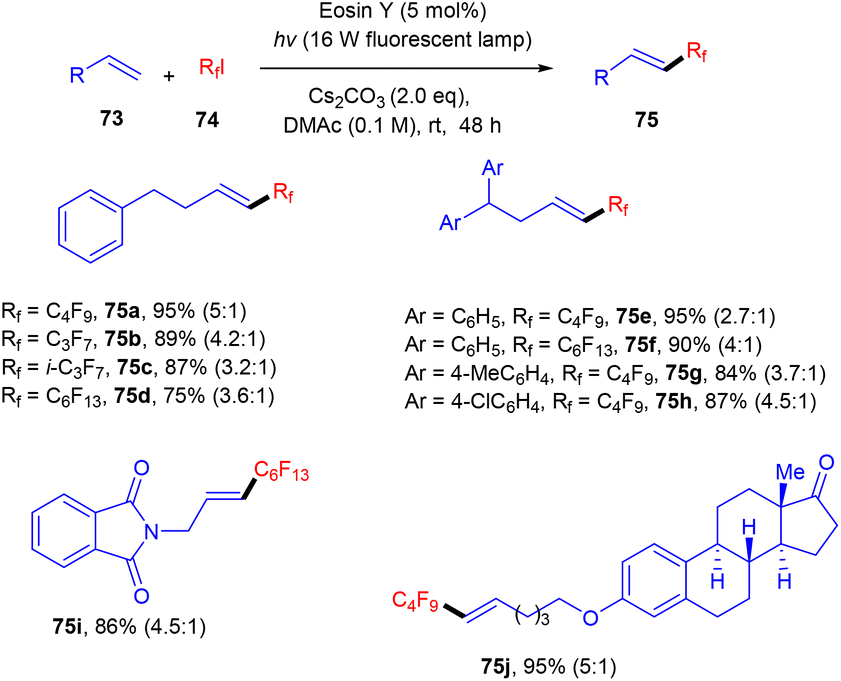
Bolm's group in 2017 synthesized perfluoroalkenes (75) directly from alkenes (73) in one step by C–C bond formation through an atom transfer followed by radical addition and elimination processes using photo-catalysis. They used eosin Y as the photocatalyst in the presence of a strong base Cs2CO3. The developed method was compatible with various substituted alkenes; electron-donating alkenes reacted faster than the electron-withdrawing ones to give high yields up to 92% (Scheme 34). Hence, this protocol provides access to the late-stage modification of highly decorated molecules.36
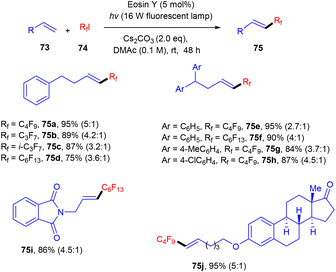 |
| | Scheme 34 Synthesis of perfluoroalkenes. | |
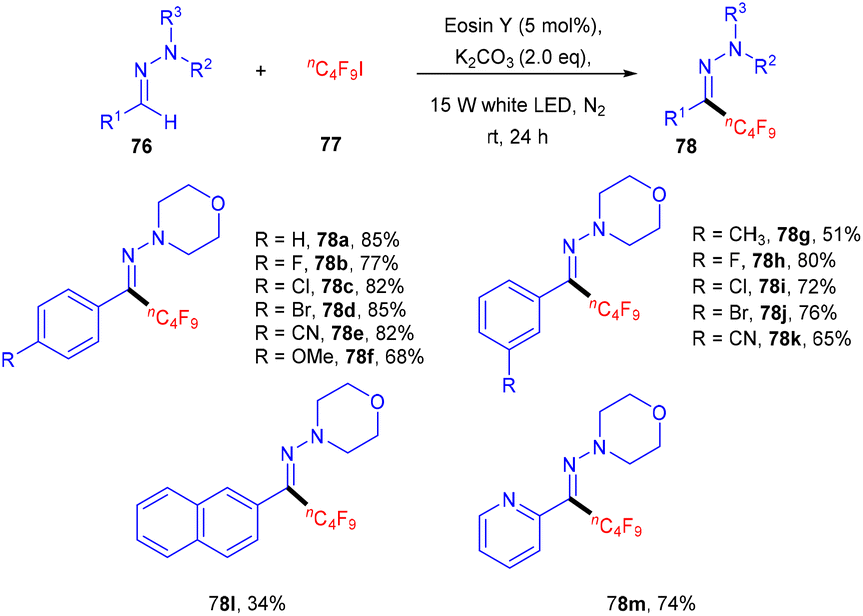
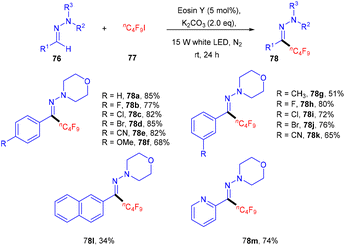
Hydrazones (76) can also be functionalized using photo-redox catalysis. Wang and his coworkers displayed one such report in 2019.37 They developed a photo-redox methodology for the perfluoroalkylation of hydrazones having wide functional group tolerance and a broad substrate scope (Scheme 35). During optimization, it has been observed that the base was an essential component of the reaction. In addition, the reaction was performed without a light source and organic dye, but no reaction efficacy was accomplished. Notably, hydrazones (76) having para-substituted phenyl rings with different halogens (F, Cl, Br) showed good reactivity. Hydrazones having an electron-withdrawing group (cyano, trifluoromethyl) gave a higher yield than the electron-donating group (methyl, methoxy). In addition, sterically hindered at the ortho-position gave the corresponding compounds (78) in good yields. Moreover, the author showed the methodology's utility at an industrial scale and the discovery of valuable scaffolds via a gram-scale reaction. The reaction proceeded through a single electron transfer mechanism.
 |
| | Scheme 35 Photoinduced perfluoroalkylation of hydrazones. | |
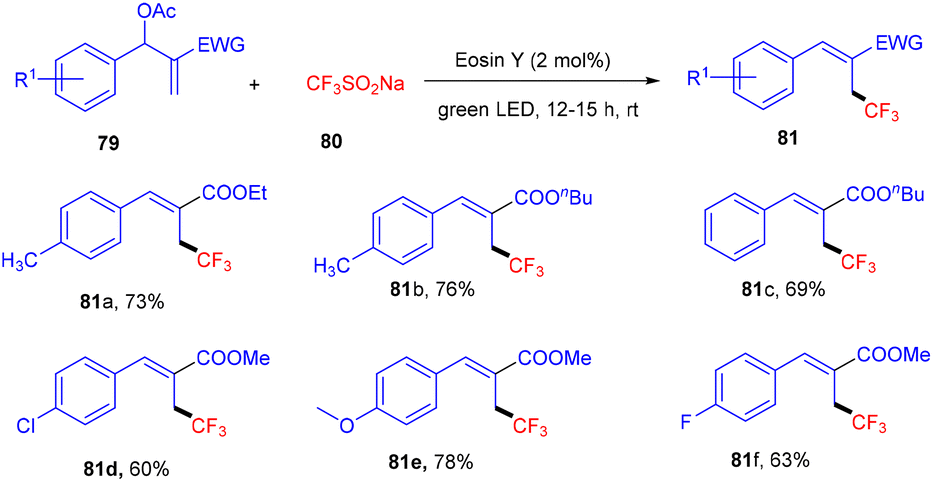
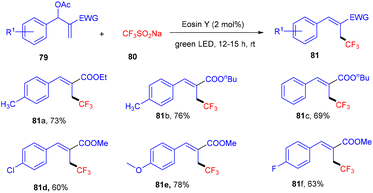
Allylic trifluoromethylation using CF3SO2Na (80) (bench stable Langlois reagent) of Baylis–Hillman acetates (79) under photoredox catalysis was developed by Yadav et al. This strategy was accomplished using eosin Y as the organophotocatalyst under the irradiation of green light at room temperature to give trisubstituted alkenes. During preliminary investigation, the authors found no significant effect of the atmosphere on the reaction, hence, reactions were performed in a sealed tube. In addition, in the presence of TEMPO, complete quenching was observed, which confirmed the radical pathway of the reaction. Among electron-withdrawing groups, the NO2 group has no compatibility for this reaction, while others and electron-donating groups have shown remarkable tunning with the developed protocol (Scheme 36).38
 |
| | Scheme 36 Trifluoromethylation of Baylis–Hillman acetates. | |
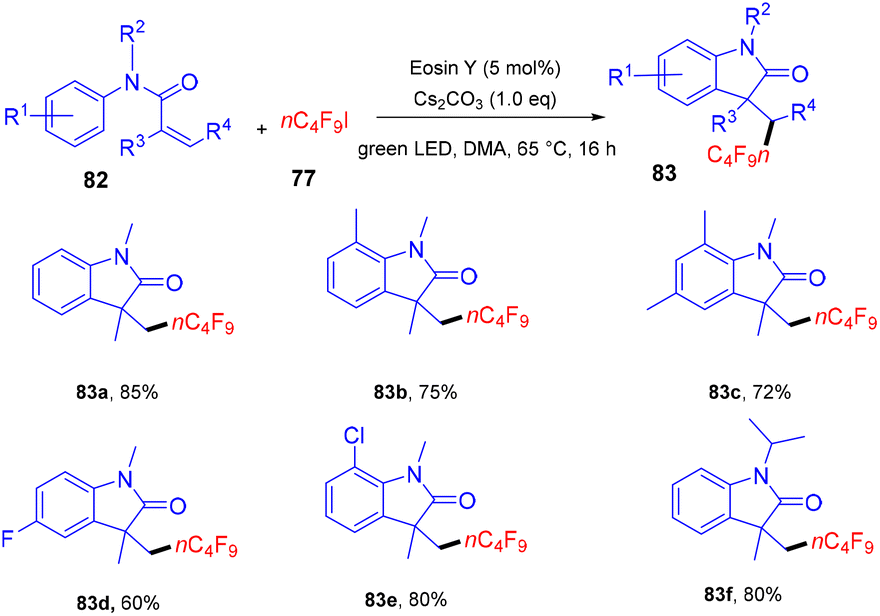
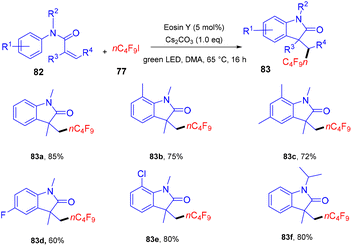
Yang and coworkers in 2019 developed a facile synthesis of perfluoroalkylated oxindoles (83) via a photoredox process. They have performed a reaction between N-methyl-N-arylmethacrylamide (82) with n-C4F9I (77). It was observed that both electron-withdrawing and -donating groups reacted smoothly (Scheme 37), and N-arylacrylamides having two methyl groups on a double bond also underwent the reaction smoothly. Still, this time, an anti-isomer of the product was observed. Substrates with N-protecting groups showed excellent compatibility and afforded the corresponding products (83) in good yields, while no efficacy was maintained with unprotected substrates. Based on control experiments and literature precedents, they proposed that the reaction proceeded through the SET mechanism.39
 |
| | Scheme 37 Synthesis of perfluoroalkylated oxindoles. | |
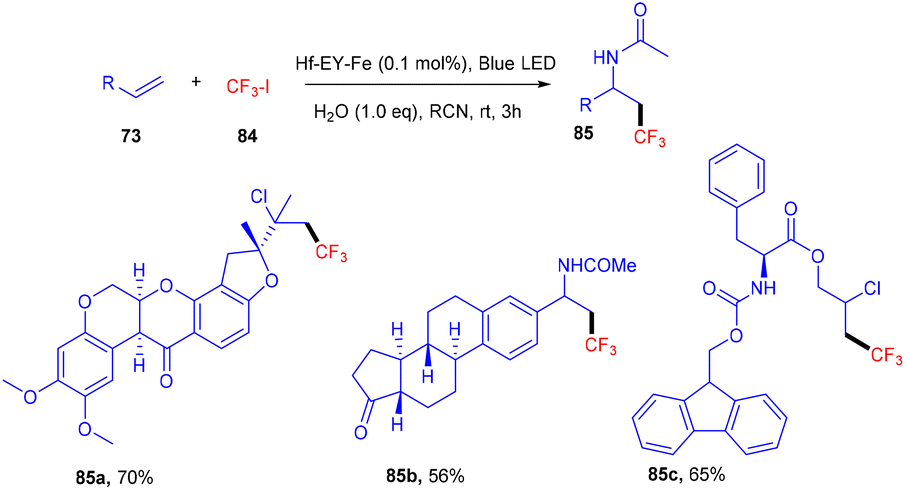
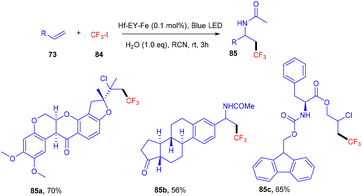
Recently eosin Y-based photoredox catalysis has brought a revolution in organic synthesis when the first MOL (metallic–organic layer) Hf–EY–Fe, containing eosin Y, was designed in 2020 by Lin and his coworkers. This MOL has provided an efficient 2D material for incorporating organic dyes for effective photoredox transformation. Hf–EY–Fe is compatible with large biomolecules and catalyzes various reactions effectively such as aminotrifluoromethylation, hydroxytrifluoromethylation, and chlorotrifluoromethylation. Furthermore, the designed methodology has allowed functionalization on bioactive molecules such as nootkatone, adapalene, estrone, etc. (Scheme 38).40
 |
| | Scheme 38 Synergistic photoredox catalysis. | |
2.7. C–H alkylarylation using alkenes
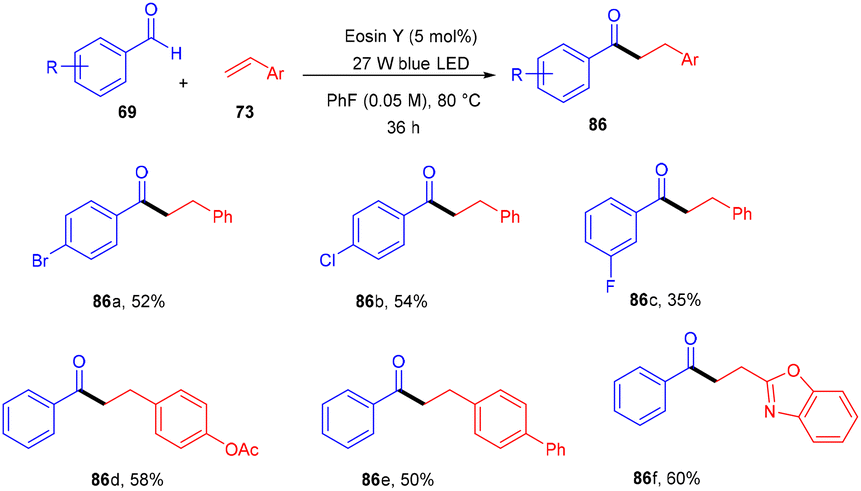
Wu and his coworkers in 2020 performed hydroacylation of styrene with anti-Markovnikov selectivity. The C–C bond has been constructed in a reaction between an aldehyde (69) and substituted styrenes (73) via a direct hydrogen atom transfer mechanism through eosin Y. Benzaldehyde having different functional groups such as halogens, ethers, or sulfides showed excellent tunning with the reaction. In contrast, ortho-substituted benzaldehyde gave lower yields of the corresponding compounds which might be due to steric hindrance. However, several styrenes (73) were also tested, having different groups at the different positions of the phenyl ring, giving products (86) in moderate to good yields (Scheme 39).41
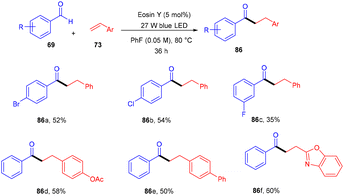 |
| | Scheme 39 Hydroacylation of styrene. | |
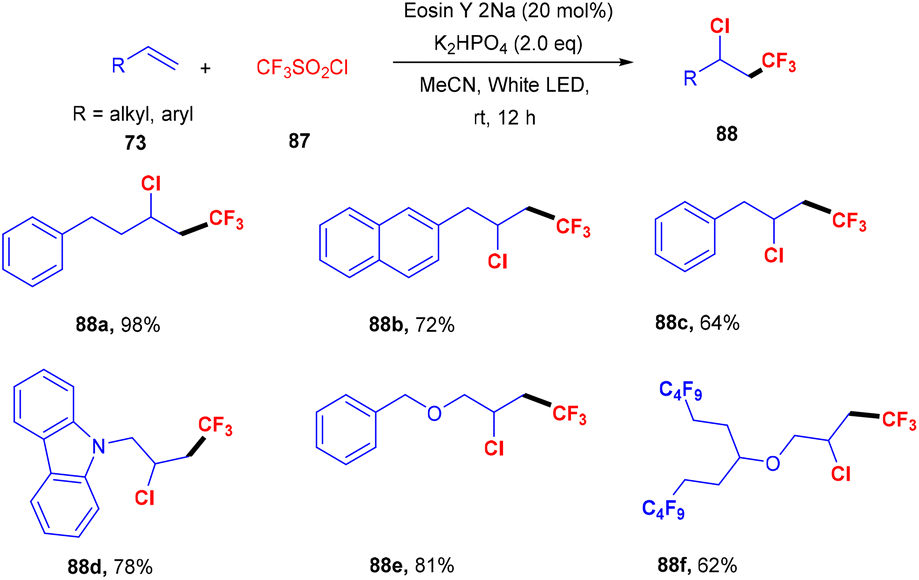
Similarly, Shin and coworkers developed a facile approach for the chlorotrifluoromethylation of terminal alkenes (73). They performed the reaction between various terminal alkenes (including aromatic heterocycles) and CF3SO2Cl (87), which was based on the SET mechanism; here free radicals formed from CF3SO2Cl by the interaction with photoexcited eosin Y. Several chlorotrifluoromethylated derivatives (88) were obtained in good yields (Scheme 40).42
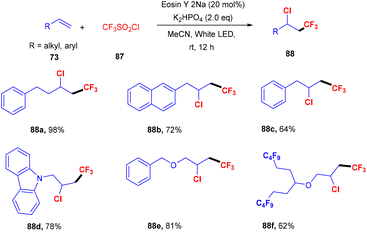 |
| | Scheme 40 Chlorotrifluoromethylation of terminal alkenes. | |
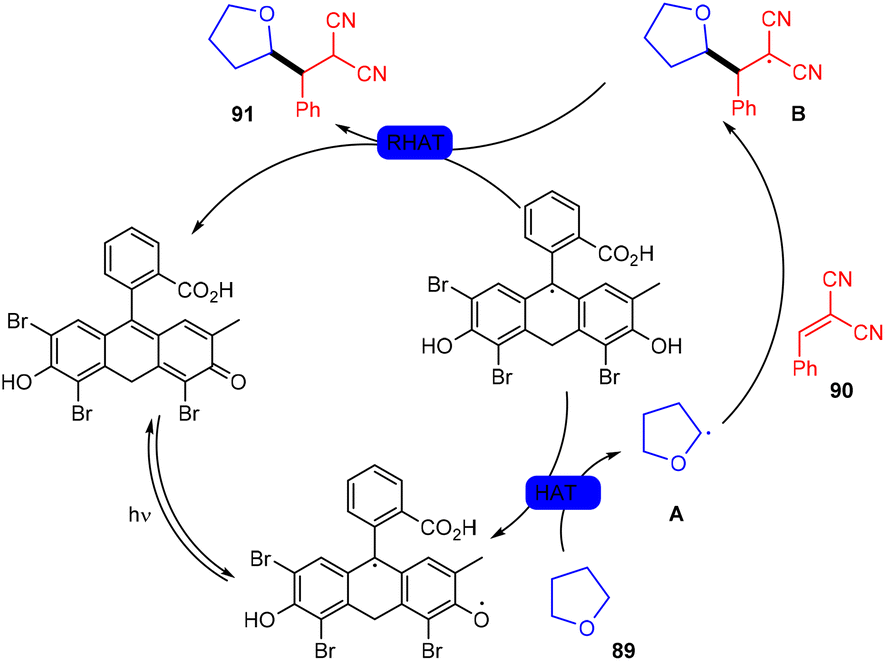
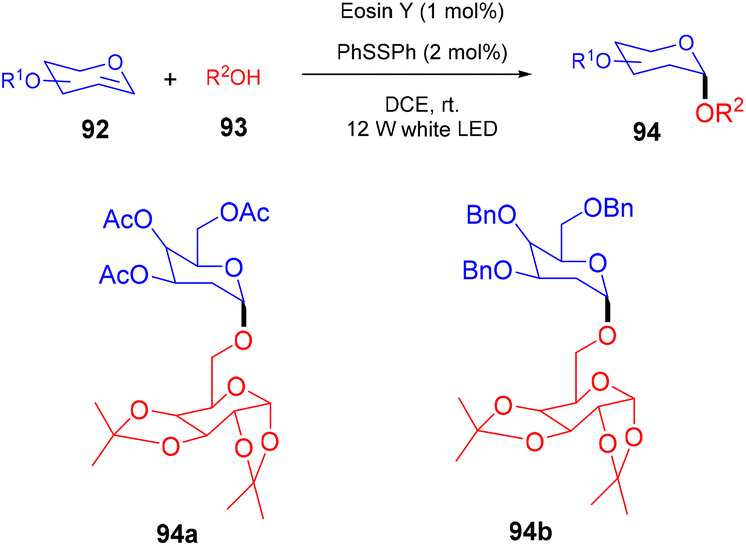
In 2018, the Wu group demonstrated for the first time C–H functionalization using a direct hydrogen-atom transfer photocatalyst and successfully formed a C–C bond. The reaction was performed using a xanthene-based dye (eosin Y) under mild reaction conditions and this methodology provided high selectivity by forming relatively stable intermediates. After the intensive screening of solvents, and catalyst loading, various functional groups (such as ethers, thioethers, alcohols, and aldehydes) were screened which showed great tunning with the reaction conditions (Scheme 41).43 Control experiments such as potential analysis and quenching studies suggested that C–H activation did not initiate the SET mechanism. Furthermore, a decrease in life time after adding alkene also supported the nonchain pathway. Here, the reaction proceeded through photoinduced eosin Y which helped in the formation of a carbon-centred radical (A) via the HAT process. This radical was trapped by olefin to give radical adduct B. Subsequent reactions between THF and radical B led to the reversible HAT process to give the desired alkylation product (91) (Scheme 42). In the same year, the Xiao group inspired by the initial work of the Wu group, also demonstrated C–H activation of glycals (92) via the hydrogen atom transfer (HAT) mechanism. They have synthesized several 2-deoxyglycosides (94) from glycals having broad functional group tolerance (Scheme 43).44
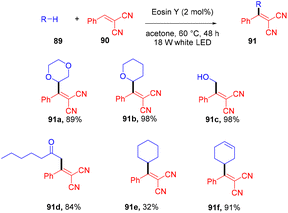 |
| | Scheme 41 Hydroacylation of styrene. | |
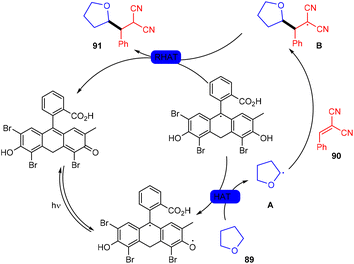 |
| | Scheme 42 Hydroacylation of styrene. | |
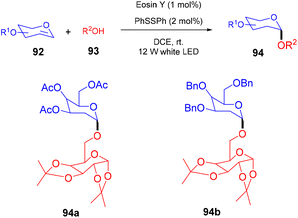 |
| | Scheme 43 Synthesis of 2-deoxygyclosides from glycals. | |
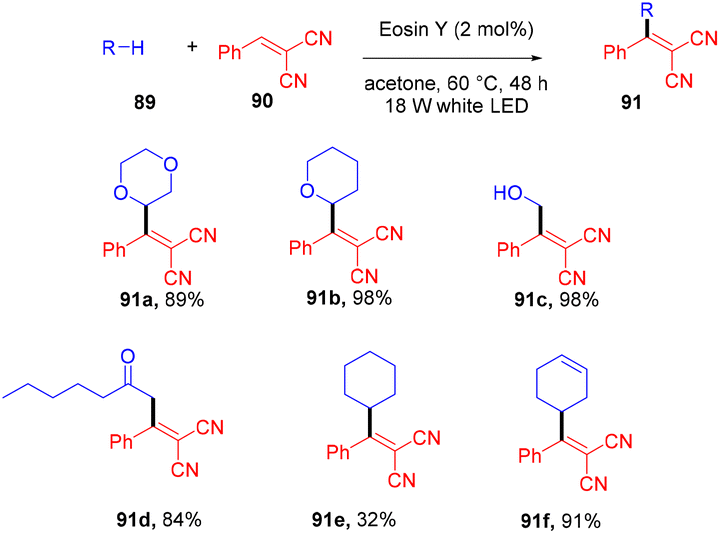
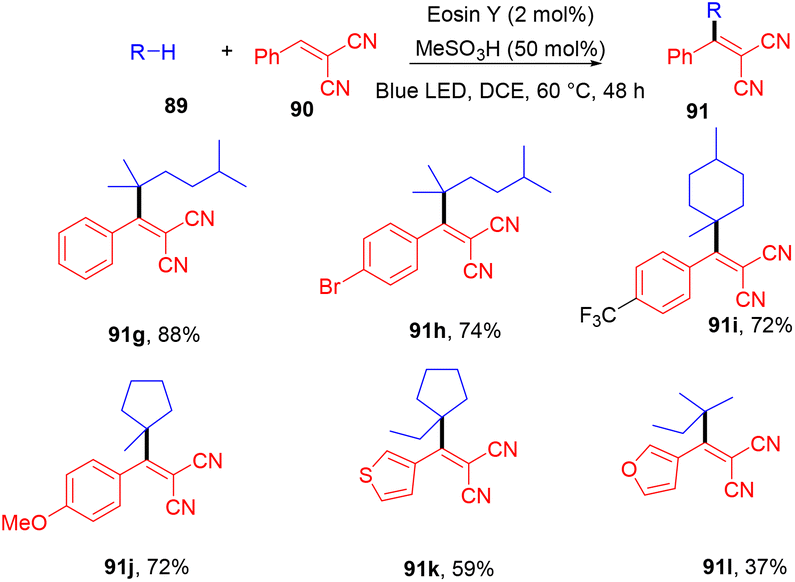
Very recently, Kong and coworkers demonstrated selective functionalization of unactivated C(sp3)–H bonds. As the transformation of unactivated C(sp3)–H bonds faces hurdles in synthetic organic chemistry, an effective methodology needs to be developed. In this context, the authors developed HAT photocatalysis. The reaction was performed using eosin Y as the photocatalyst in the presence of a Brønsted acid to promote the catalytic efficiency under an argon atmosphere at 60 °C for 48 h. The reaction showed wide substrate tolerance and functional group compatibility and gave the corresponding compounds (91) in moderate to good yields (Scheme 44). To find mechanistic insight they performed several control experiments that suggested the hydrogen atom transfer mechanism.45
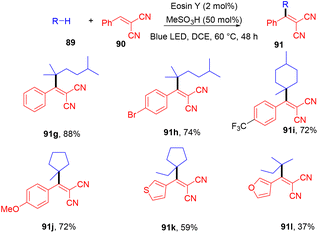 |
| | Scheme 44 Alkylation of unactivated C(sp3)–H bonds. | |
2.8. C–H alkylarylation using Katrizky salts
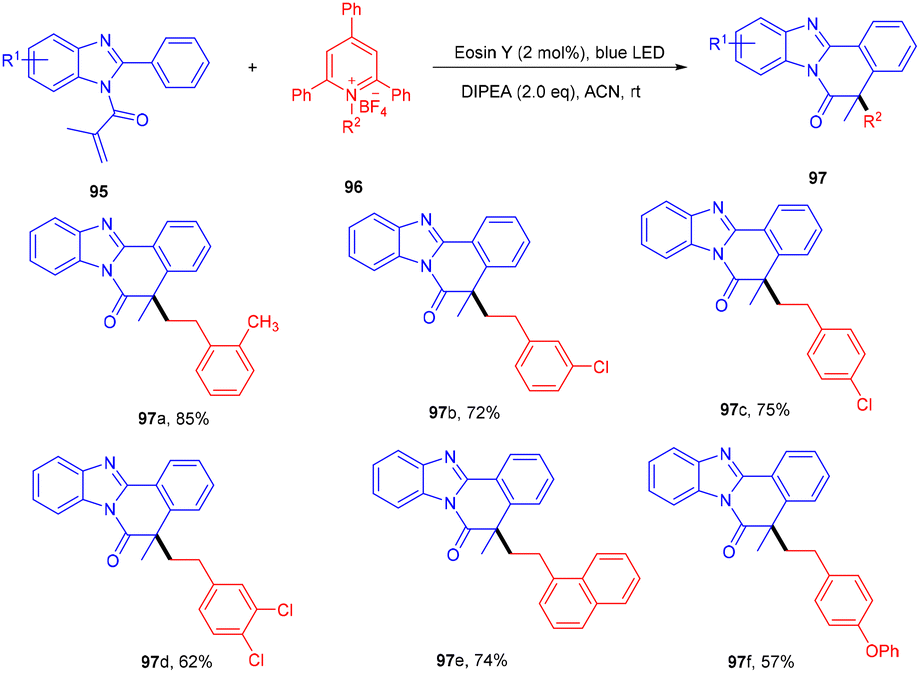
To access the benzo[4,5]imidazo[2,1-a]isoquinolin-6(5H)-one scaffold (95), the Adiyala group established a deaminative approach for the sequential operation of alkylation and cyclization between N-methacryloyl-2-phenyl benzimidazoles (95) and alkyl amine-based Katrizky salts (96). During optimization, the authors investigated various bases such as DABCO, DBU, Na2CO3, triethylamine, and DIPEA; only DIPEA showed desirable results. Notably, the reaction was performed using a continuous-flow microreactor, resulting in a shorter reaction time, good mixing, and higher yields over batch reactions. Control experiments using TEMPO and BHT (radical trapper) indicated a radical free pathway as traces of the final product were observed. An alkyl free radical was formed from Katrizky salts by interaction with photoinduced eosin Y followed by defragmentation along with an aromatic pyridine ring. The developed methodology was also applied to different amino acids that gave the final product (97) in good yields. Electron-donating (–tBu, –OMe) and -withdrawing groups (–CN and –NO2) on benzimidazole reacted smoothly and gave the respective compounds in good yields (Scheme 45). However, heteroarenes on benzimidazole were unable to react and thus did not give any desired product.46
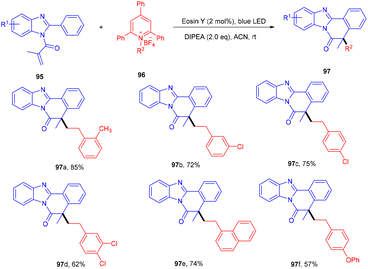 |
| | Scheme 45 Synthesis of benzo[4,5]imidazo[2,1-a]isoquinolin-6(5H)-one. | |
2.9. C–H alkylation using boronic acids
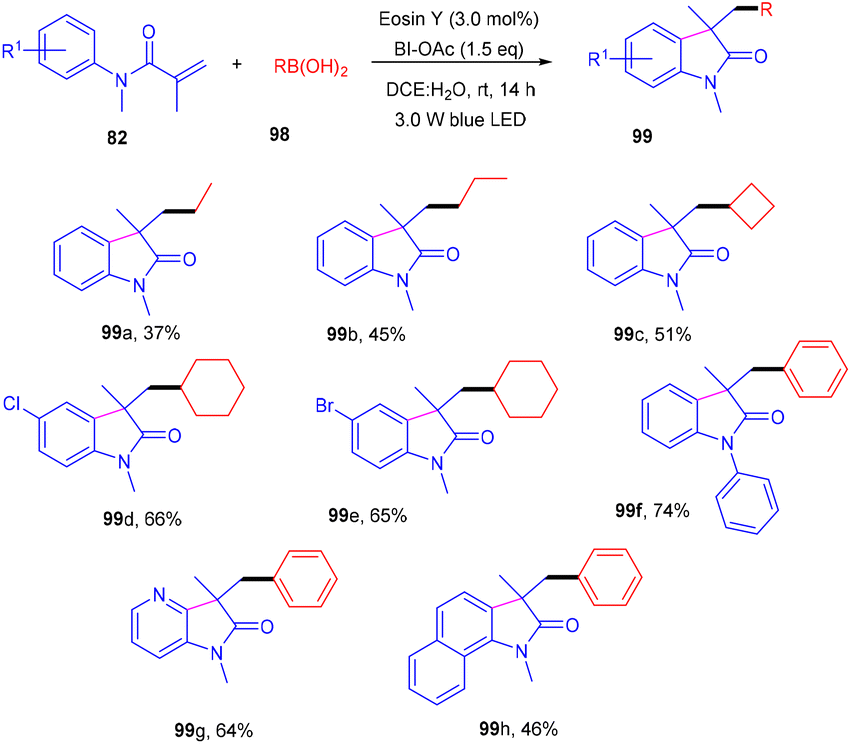
Photoinduced 3,3-disubstituted oxindoles (99) have been synthesized by Wang et al. Here, a photoinduced tandem approach was developed to carry out alkyl arylation of acrylamides (82) using a wide range of boronic acids (98) to access the corresponding derivatives in high yields (Scheme 46). During optimization, it was found that no product was formed without eosin Y, and the reaction efficiency was improved in a mixture of DCE and H2O. After solvent screening, several oxidants were employed, and only BI-OAc gave the desired results, as BI-OAc helped in the generation of free radicals from boronic acid. Substitution with alkyl substituents at the different positions of the benzene ring of N-acrylamides showed compatibility and gave the respective products in moderate yields; however, regioisomers were obtained in the case of meta substitution. The reaction was quenched in the presence of a TEMPO free radical scavenger, indicating a free radical mechanism for the developed strategy.47
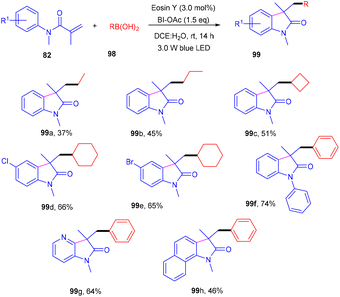 |
| | Scheme 46 Photoinduced deboronative alkyl arylation of acrylamides. | |
2.10. C–H alkylation via a dual catalytic system
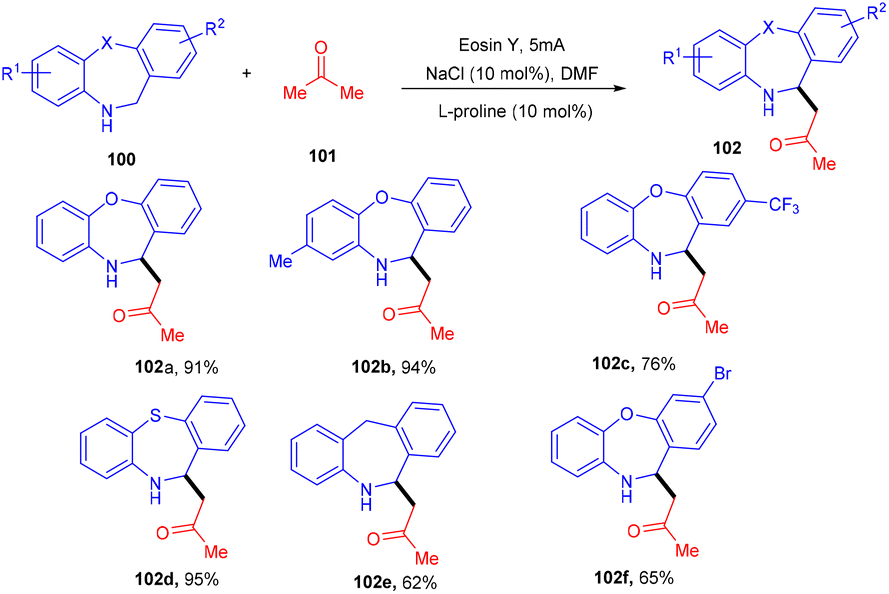
Recently, the Chauhan group developed a unique approach by merging an electrocatalyst with a photoredox catalyst for the stereoselective oxidative Mannich reaction. They demonstrated sp3–sp3 coupling between dihydro dibenzoxazepines (100) and cyclic/acyclic ketones (101). Oxazepines (102) were obtained by oxidative conversion of dihydro dibenzoxazepines by parallel utilization of eosin Y and graphite electrodes. The Mannich reaction catalyzed by L-proline with ketones gave the desired products in good yields (Scheme 47).48
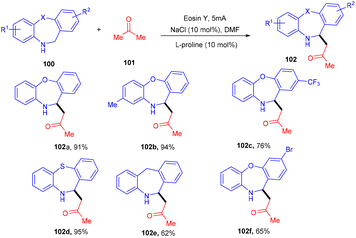 |
| | Scheme 47 Dual catalysis for the stereoselective oxidative Mannich reaction. | |
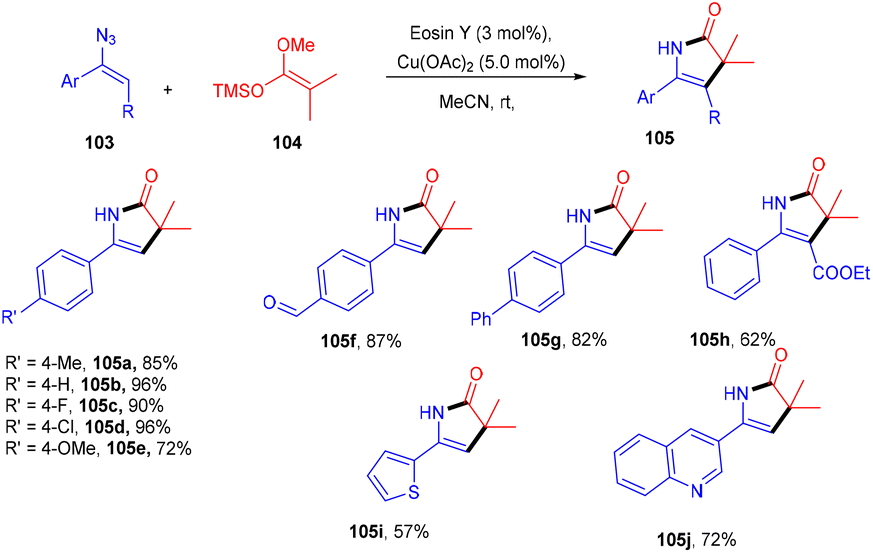
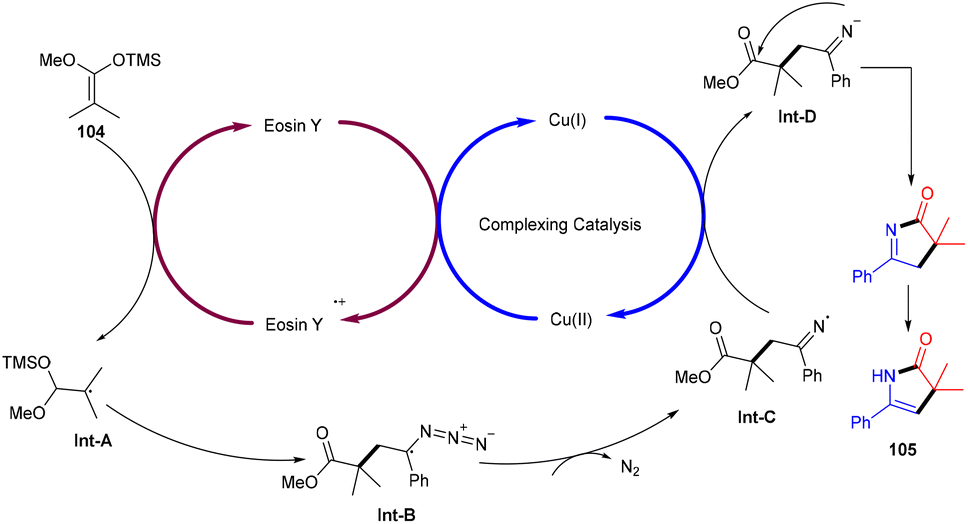
Ene-γ-lactam moieties are important synthons as they are an integral structural unit in important medicinal compounds. Liu's group reported that eosin Y, a standard photo-redox catalyst, has shown excellent properties to catalyze thermal redox reactions in the presence of a small amount of Cu(OAc)2. Vinyl azides (103) reacted with ketene silyl acetals (104) in the developed system via [3 + 2] cycloaddition in the dark to give the desired cycloadduct product (105). Electron-withdrawing groups (acetyl, fluoro) on the phenyl ring were more compatible and gave the desired product up to 94% yield (Scheme 48).49 Liu's group also proposed a mechanism for the developed method, as shown in Scheme 49. Initially, the generation of an eosin Y radical took place by the reaction of eosin Y and Cu(OAc)2, which oxidized ketene silyl acetals (KSA) (104) under SET to afford α-ester radicals (Int-A). Furthermore, an iminyl radical (Int-B) was formed via an attack of α-ester radicals on unsaturated vinyl azides. The iminyl radical underwent reduction to give the corresponding anion (Int-D), and then intramolecular rearrangements and isomerization delivered the desired product (105).
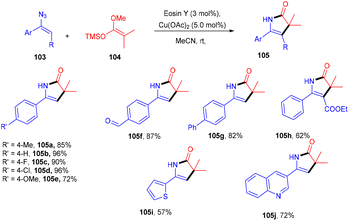 |
| | Scheme 48 Synthesis of ene-γ-lactams. | |
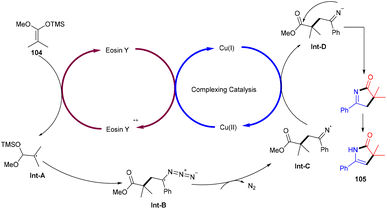 |
| | Scheme 49 Plausible mechanism for the synthesis of ene-γ-lactams. | |
2.11. C–H alkenylation/alkynylation
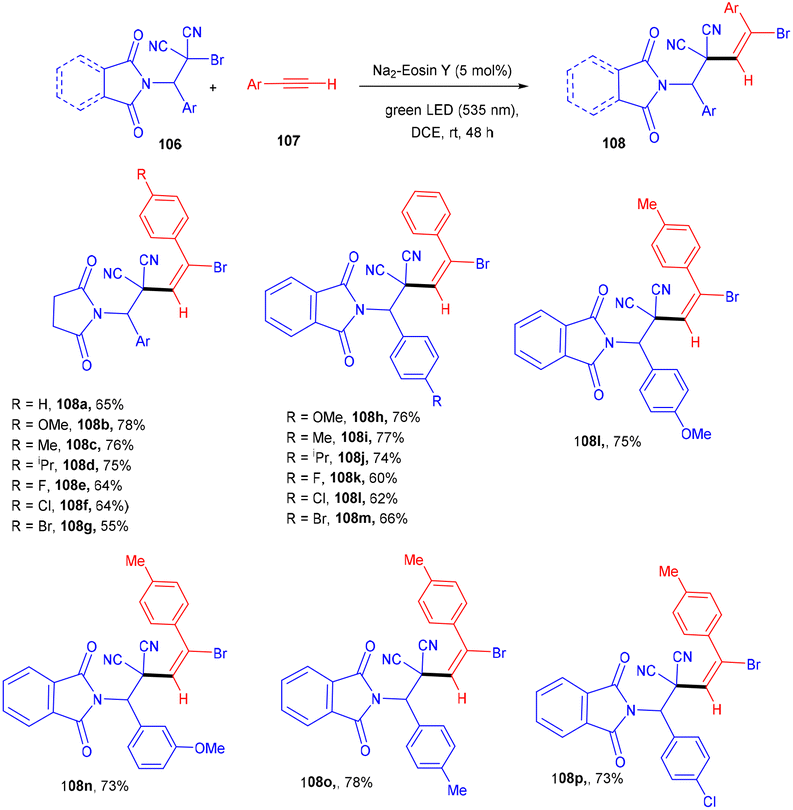
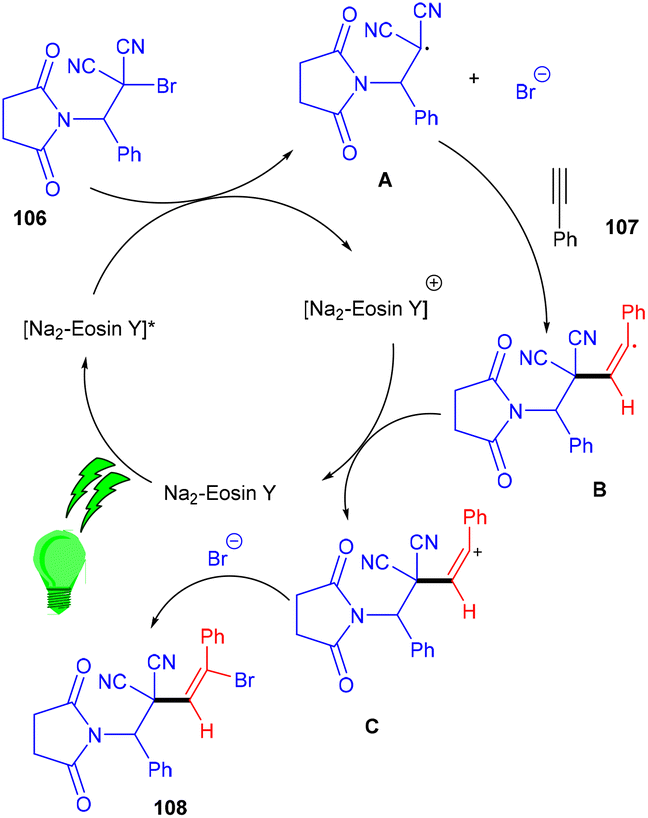
The Wang group in 2016 developed a stereoselective photoredox catalytic addition of highly decorated functionalized amino brominated alkyl bromides (106) to alkynes (107) for the synthesis of the corresponding E-isomers of vinyl bromides (108) via the formation of a C–C bond. The synthesis of vinyl bromides was done by functionalizing alkynes without a metal catalyst (Scheme 50). Substitution using –Me, –OMe, F, Cl, and Br at the different positions of the phenyl rings gave the desired products in good yields.
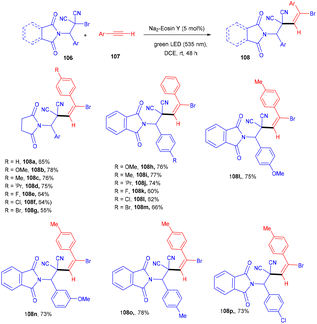 |
| | Scheme 50 Synthesis of vinyl bromides. | |
During control experiments, complete quenching of the reaction has been observed in the presence of TEMPO, indicating the free radical mechanism of the reaction. This reaction gives an atom-economical and efficient approach towards a bifunctional motif with a broad substrate scope.50 In this approach, the photocatalyst was excited under visible light and oxidatively quenched with 106 to give [Na2–eosin Y]+ and radical intermediate A, which underwent regioselective addition to alkynes and the formation of intermediate B. Vinyl carbocation C was obtained by oxidation of intermediate B which reacted with bromide anions and gave the final product vinyl bromides (108) (Scheme 51).
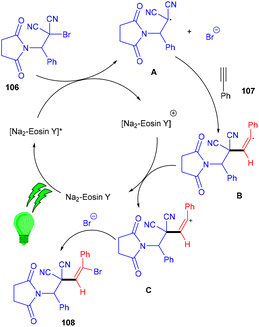 |
| | Scheme 51 Mechanism for the synthesis of vinyl bromides. | |
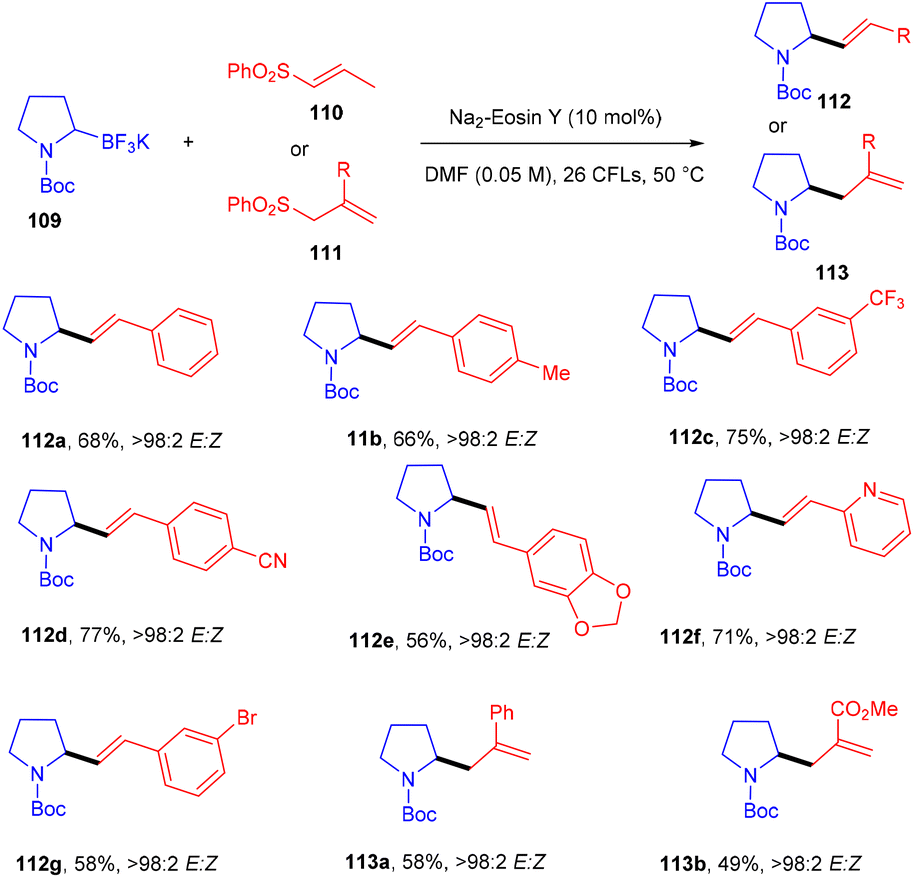
Molander and his coworkers used eosin Y as a photo-redox catalyst to protect allylic amines and alkyl nitriles by C–C bond formation. They carried out reactions between Boc-protected potassium α-aminomethyltrifluoroborates (109) and several alkenyl sulfones (110, 111). During optimization, a number of photocatalysts and light sources were examined, Na2–eosin Y and green LEDs showed the desired results. Addition of 2,6-lutidine as an additive to increase the yield was unsuccessful. In this reaction, several allylic amines (112, 113) were synthesized via the addition and elimination of an aminoethyl radical in moderate to good yields with high stereoselectivity (Scheme 52).51 But the reaction did not proceed in the case of α-aminomethyltrifluoroborates.
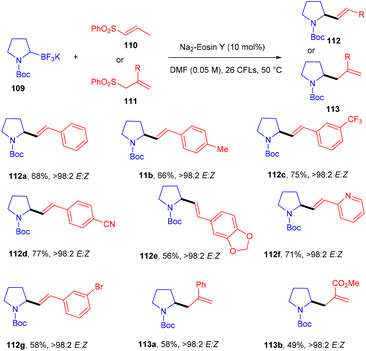 |
| | Scheme 52 Alkenylation and allylation of potassium alkyltrifluoroborates. | |
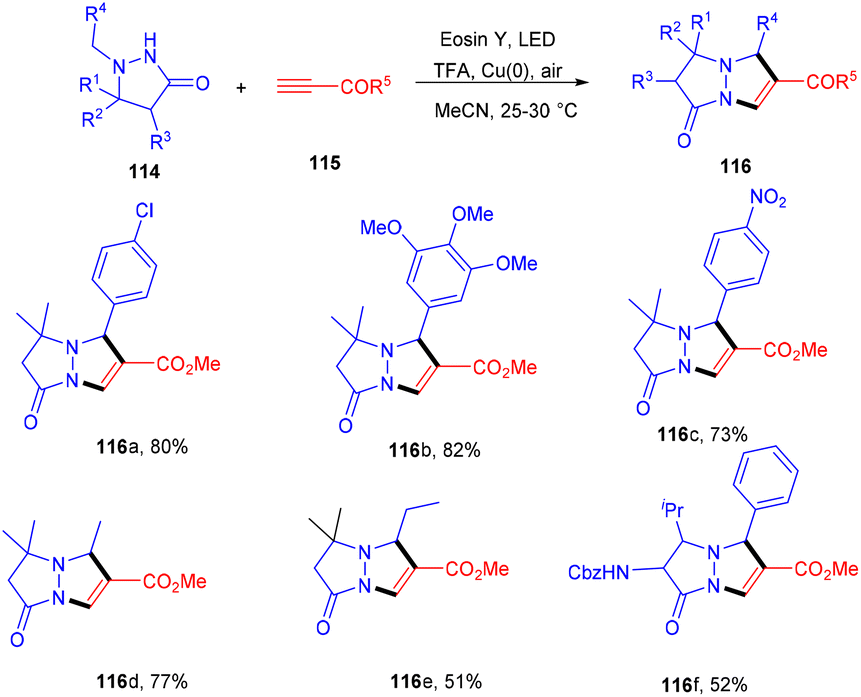
Multiple C–C bonds were formed in a one-pot reaction to synthesize pyrazolo[1,2-a] pyrazoles (116). The dual catalytic system comprising eosin Y and Cu(0) was developed in 2020 by Petek et al. They demonstrated the cycloaddition reaction between azomethine imines (114) and alkynes (115) in a dual catalytic system. The use of TFA under optimized reaction conditions provided acidic medium to eosin Y and the absorption maximum under these conditions was examined, which was at 470 nm. In addition, cyclic voltammetry of azomethine showed that E1/2(111˙+/111) = 0.73 V vs. SCE, hence, it could be oxidized by excited eosin Y having E1/2(3EY*/EY˙−) = 0.83 V vs. SCE. Furthermore, the reaction with TEMPO gave a TEMPO–pyrazolone adduct which was confirmed by HRMS analysis [(m/z); 349.2247 corresponding to C21H32ClN3O2], also suggesting the free radical pathway. This strategy also worked smoothly for both electron-donating and -withdrawing groups and gave the final products in excellent yields (Scheme 53).52
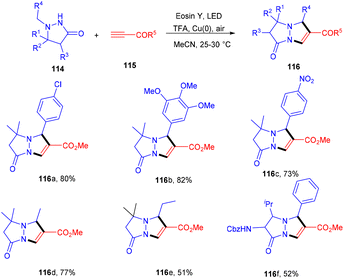 |
| | Scheme 53 Photoinduced aerobic transformation of pyrazolidine-3-one. | |
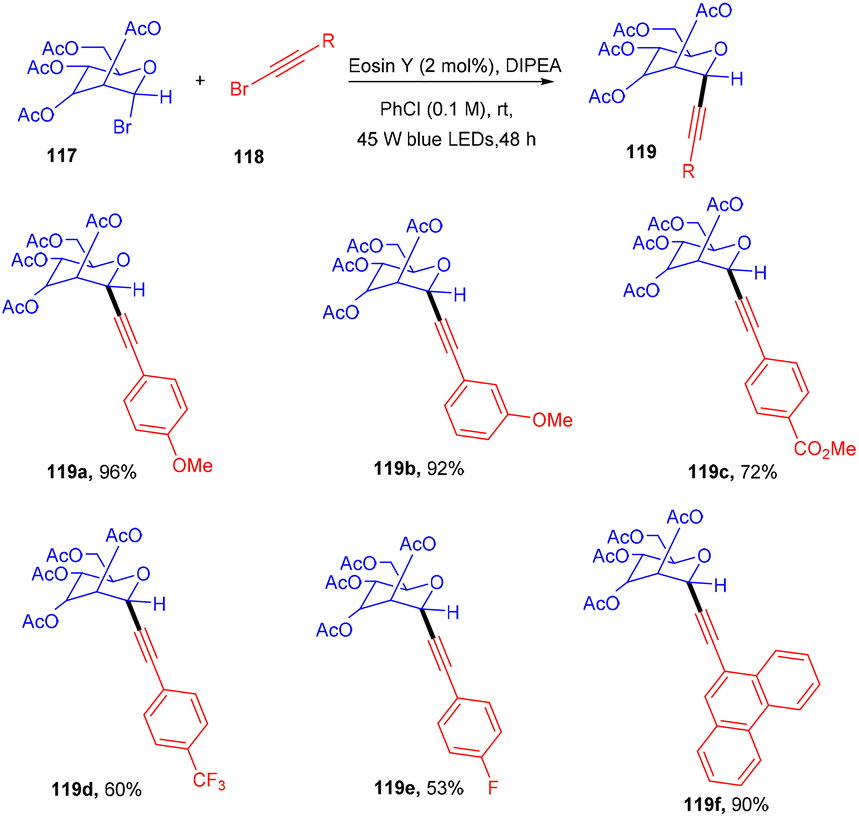
Recently the Yu group synthesized C-alkynyl glycosides via eosin-mediated photoinduced catalyzed reductive coupling between glycosyl bromides (117) and alkynyl bromides (118).
They performed the reaction in the presence of the organic base DIPEA, acting as a reducing agent at ambient temperature under blue light irradiation. Stern–Volmer fluorescence quenching data and radical trapping experiments supported the free radical pathway of the reaction. This methodology has a broad substrate scope and functional group tolerance (Scheme 54).53 Electron-donating groups (–Me, –OMe) and electron-withdrawing groups (CF3, NO2) at the different positions of the phenyl ring showed compatibility and gave products (119) in moderate to good yields. In addition, they demonstrated the synthetic versatility of the final compounds.
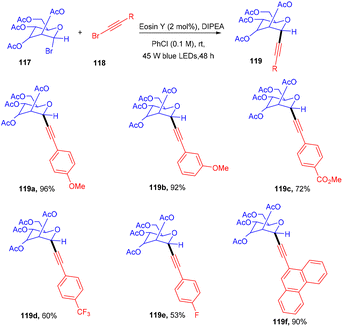 |
| | Scheme 54 Coupling between glycosyl bromide and alkynyl bromides. | |
2.12. C–H annulation reaction
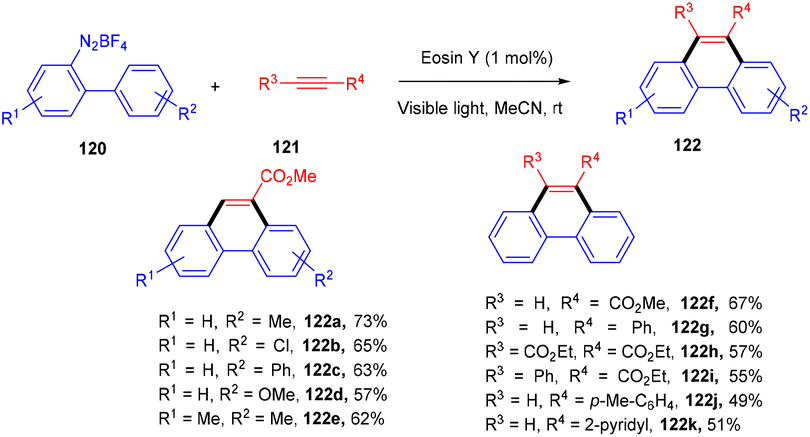
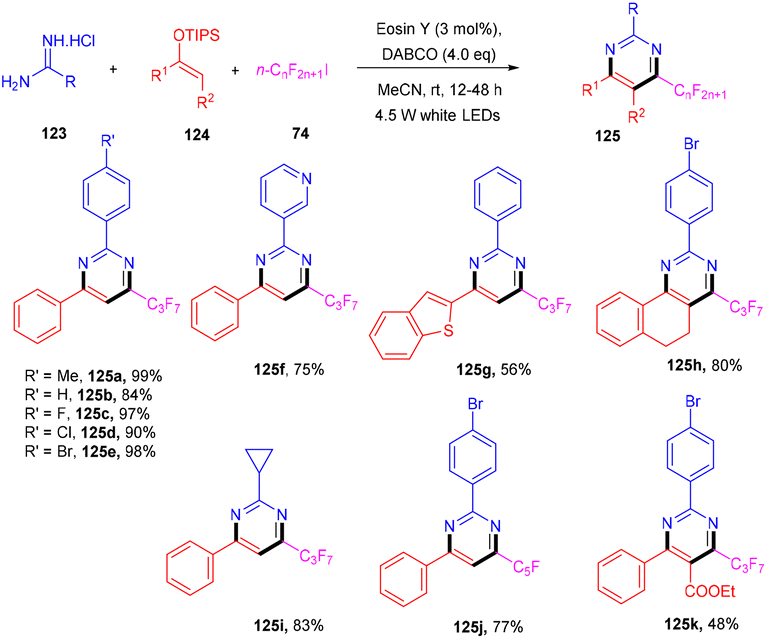
In 2012 Zhou and coworkers revealed the eosin Y mediated synthesis of substituted phenanthrenes. They showed photoinduced [4 + 2] benzannulation of substituted biaryldiazonium salts (120) using alkynes (121) via tandem radical addition and cyclization. They also tried dialkynes such as 1,4-diphenylbutadiyne and found that only one alkyne group reacted and gave the corresponding product (122) in a moderate yield. Furthermore, adding radicals to these alkynes with electron deficiency was more favorable (Scheme 55).54 However, Loh et al. have reported the facile synthesis of fluoroalkylated pyrimidine scaffolds (125) in a regio-defined pattern [3 + 2 + 1]. The multicomponent reaction started from readily available silyl enol ether (124), amidine (123), and alkyl halide (74) (fluoro substituted). The reaction proceeded through a single electron transfer mechanism and involved the coupling of radical fluoroalkylation, defluorination, and annulation, and the formation of new C–C bonds. The electronic effect of substituents also gave satisfactory results with this protocol (Scheme 56).55
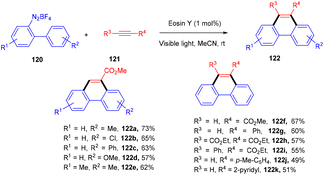 |
| | Scheme 55 Photoinduced [4 + 2] benzannulation. | |
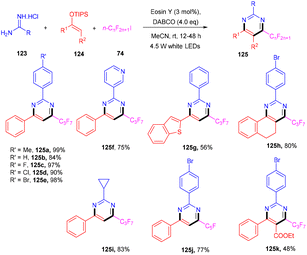 |
| | Scheme 56 Synthesis of fluoroalkylated pyrimidine scaffolds through the [3 + 2 + 1] pattern. | |
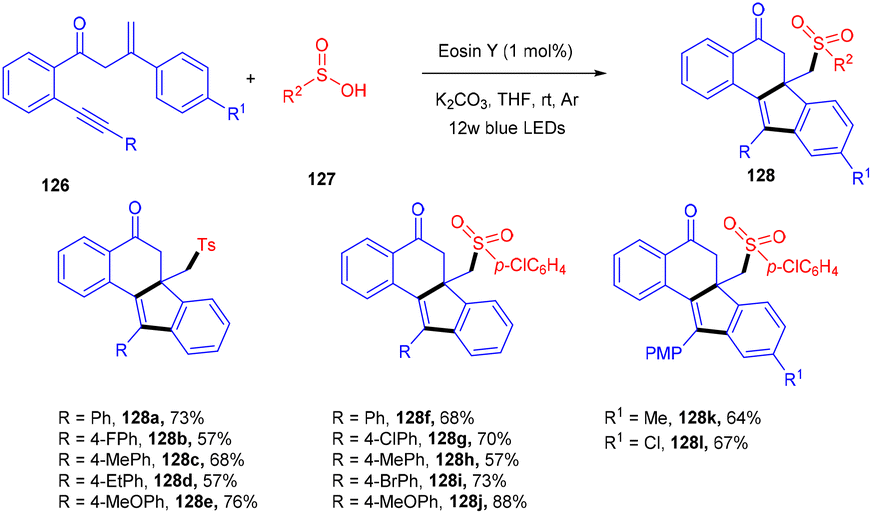
Jiang's group has demonstrated a unique approach for simultaneous formation of two C–C bonds along with the construction of a C–S bond. The developed method synthesized sulfone-containing benzo[a]fluoren-5-ones (128) by eosin Y mediated photocatalysis via cyclizing C(sp3)-tethered 1,7-enynes (126) with sulfinic acids (127). This approach has advantages of the potential formation of many functionalized compounds in good yields and high tolerance to a broad range of functional groups (Scheme 57).56 By constructing multiple bonds (C–C and C–S), this approach has triggered the efficient synthesis of polycyclic aryl sulfones.
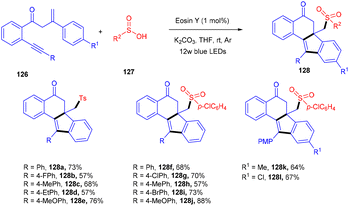 |
| | Scheme 57 Synthesis of sulfone-containing benzo[a]fluoren-5-ones. | |
3. C–S bond formation
Organosulfur compounds have gained significant interest from organic chemists due to their wide applications in medicinal chemistry, the pharmaceutical industries, materials science, etc. Subsequently, C–S bond formation is an essential prerequisite for synthesizing organosulfur compounds. C–H functionalization has revolutionized the synthesis of this valuable bond by adopting various methodologies. Photoinduced C–S bond formation is step-economical and environment friendly.
3.1. Sulphonylation
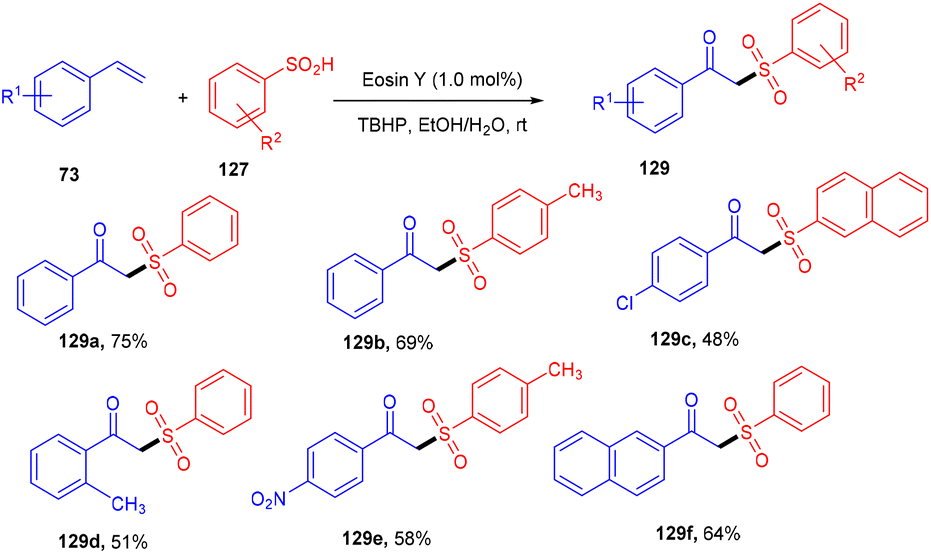
β-Ketosulfones are valuable compounds with sulfur atoms and are widely used as core structures for synthesizing natural products, polyfunctionalized compounds, and vinyl sulfones. These have comprehensive biological and medicinal activities. Owing to their properties, organic chemists have been engrossed in their synthesis; in 2016, the Wang group developed a metal-free and green approach to construct the C–S bond and provide access to β-ketosulfones. They carried out eosin Y catalyzed oxysulfonylation of alkenes (73) using sulfinic acids (127). During substrate scope investigation, the aryl ring of alkenes having both electron donating (–OMe, –Me) and withdrawing groups (–Cl, –Br, –NO2, –CN) showed compatibility and afforded the corresponding compounds (129) in good yields (Scheme 58).57 Furthermore, alkenes’ steric hindrance did not significantly affect the reaction. During the mechanistic study, complete quenching of the reaction in the presence of TEMPO (radical scavenger) was observed, suggesting that the reaction proceeded through a radical mechanism.
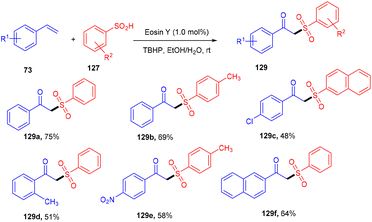 |
| | Scheme 58 Synthesis of β-ketosulfone. | |
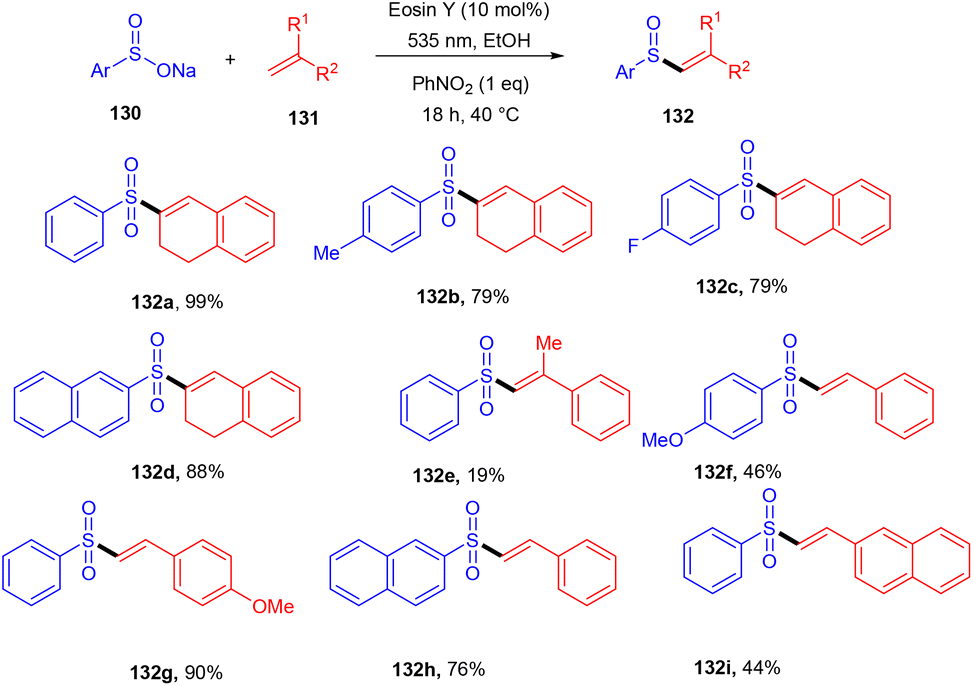
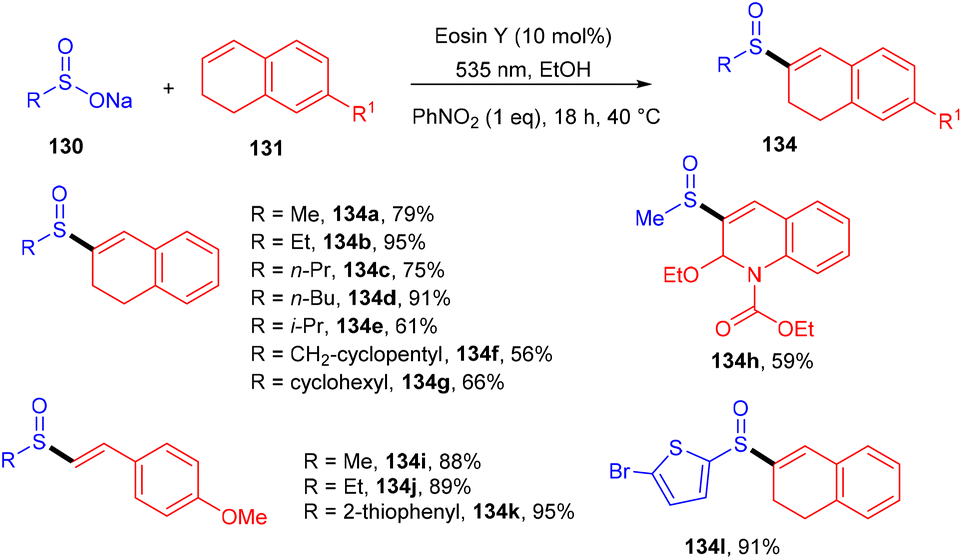
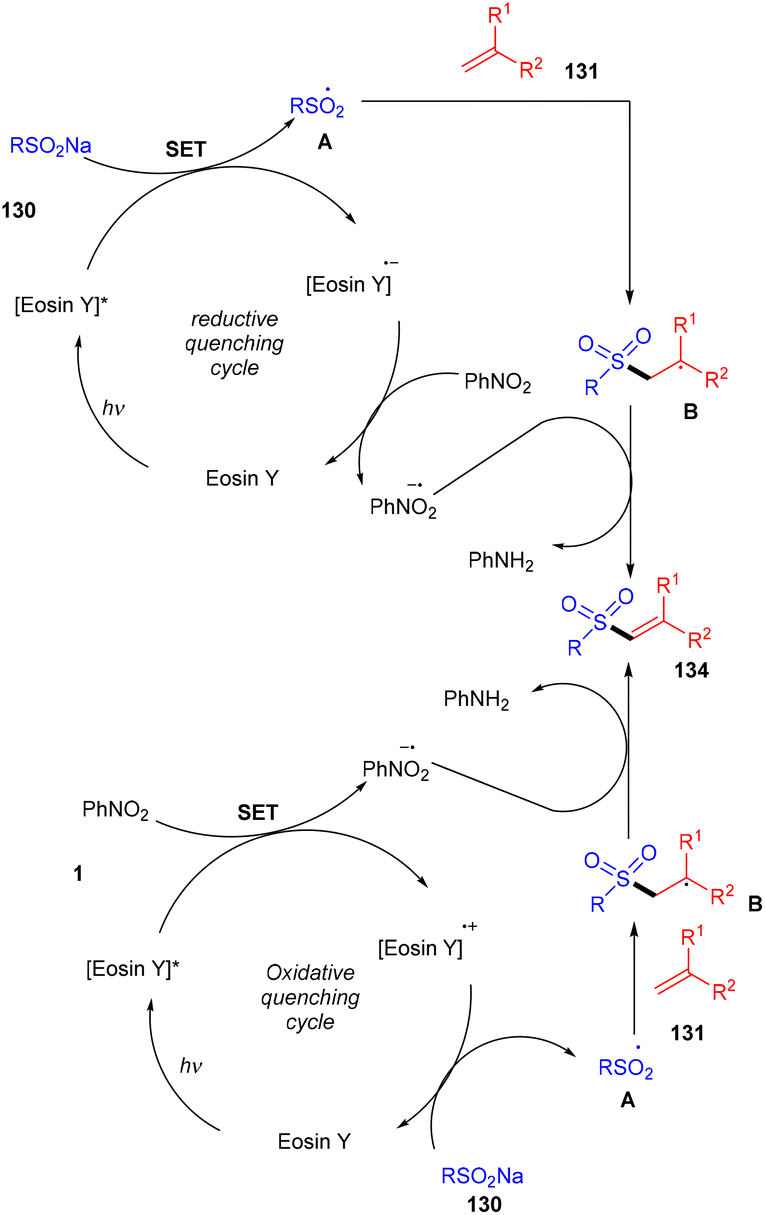
König's group synthesized vinyl sulfones (132) by photoredox catalysis of aryl sulfinates (130) in 2015. Notably, they synthesized only aryl vinyl sulfones in moderate to excellent yields (Scheme 59).58 However, after one year in 2016, they could achieve synthetic diversity and synthesized many alkyls and heteroaryl vinyl sulfones with high yields (Scheme 60). Various complex cyclic sulfones (134) were synthesized via two photocatalytic sequential steps to generate vinyl sulfones, followed by intramolecular cyclization.59 A broad substrate scope was explored and the corresponding compounds were obtained in good yields. In addition, complete regioselectivity was observed with mechanistic studies and gave two possible mechanisms: (i) reductive quenching cycle and (ii) oxidative quenching cycle as shown in Scheme 61. Here, EY could enter any of the two quenching cycles based on the electronic environment. This was confirmed by calculating the thermodynamics (Gibbs free energy) of both the mechanisms, indicating that both reductive and oxidative quenching cycles (photoinduced electron transfer) were equally exothermic (ΔGPeT = −60 kJ mol−1).
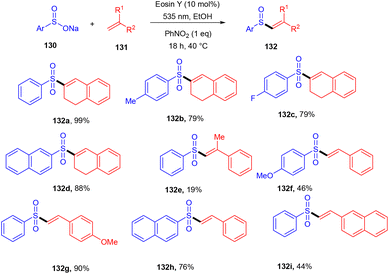 |
| | Scheme 59 Synthesis of aryl vinyl sulfones. | |
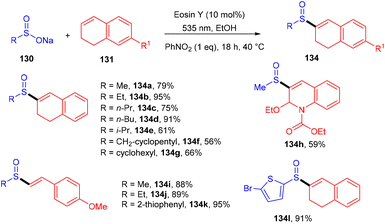 |
| | Scheme 60 Synthesis of heteroaryl sulfones. | |
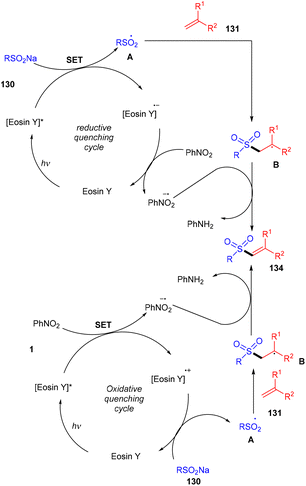 |
| | Scheme 61 Two different mechanism pathways. | |
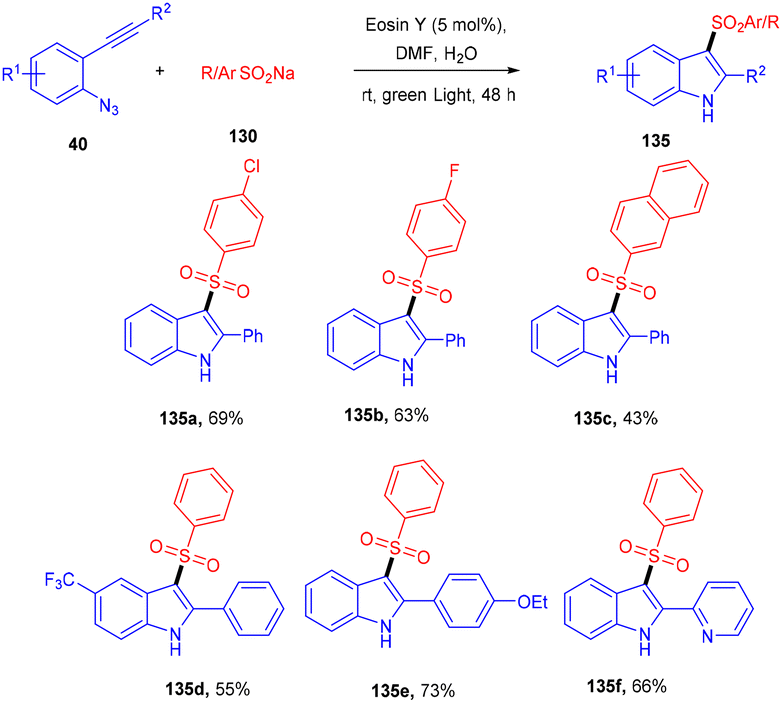
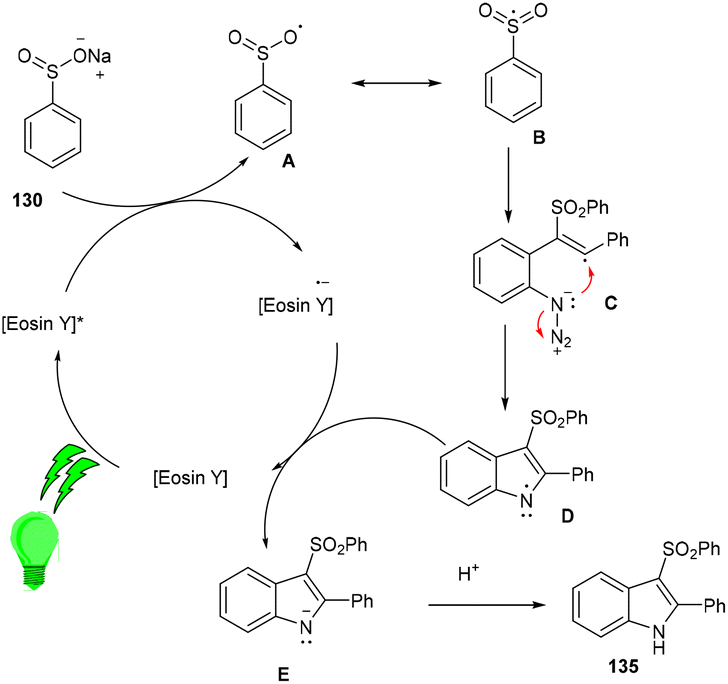
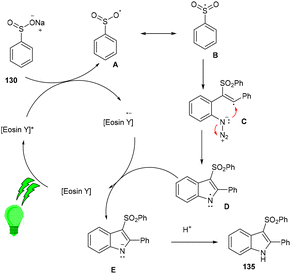
Inspired by Gu et al., Kshirsagar and co-workers performed regiospecific annulation of 2-alkynyl-azidoarenes (40) with sodium sulfinate salts (130) to synthesize 3-sulphonylindoles (135) via cascade cyclization with eosin Y as the photocatalyst. During optimization, combinations of solvents were used, and DMF/H2O gave the best results. Afterward, various azido benzenes having alkyl alkynyl or aryl alkynyl substituents were tested—substituted azidobenzenes with p-Me, p-Et, p-Bu, and p-Br afforded the respective products in good yields. A number of compounds were also synthesized with a variety of sodium sulfinate salts in moderate to good yields (Scheme 62). This reaction proceeded by the reductive quenching cycle, where oxidation of the sulfinate anion by excited eosin Y (EY*) gave an S-centred radical (B). After the regioselective attack of the sulfonyl radical on the alkyne, an alkene radical intermediate (C) was obtained, followed by intramolecular cyclization; an N-centred intermediate (E) was also obtained, which underwent proton abstraction to give the desired compound (135) (Scheme 63).60
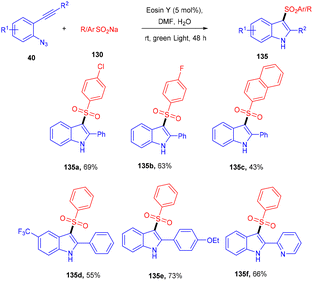 |
| | Scheme 62 Synthesis of 3-sulphonylindoles. | |
 |
| | Scheme 63 Plausible mechanism for the synthesis of 3-sulphonylindoles. | |
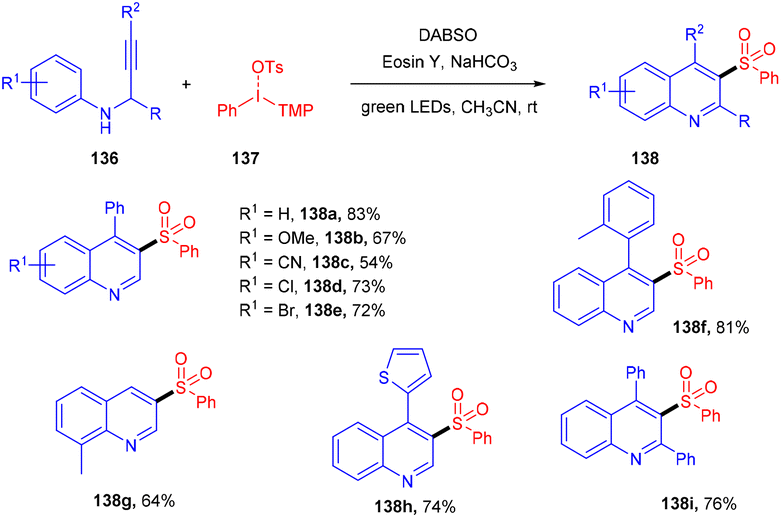
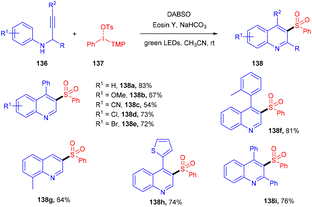
The direct synthesis of aryl sulfonyl quinolines (138) via multicomponent one-pot cycloaddition of N-propargyl arylamines (136), SO2, and diaryliodonium salts (137) was achieved by Zhang's group in 2018.61 Based on control experiments of radical trapping it was concluded that the reaction proceeded through the free radical mechanism. The role of DABSO was crucial for SO2 incorporation. Here, the direct formation of the C–S bond afforded the decorated quinoline in one step. Moreover, this approach also exhibited broad functional group tolerance and a broad substrate scope, giving the corresponding sulfur-containing quinolines in moderate to good yields (Scheme 64).
 |
| | Scheme 64 Synthesis of aryl sulfonyl quinolines. | |
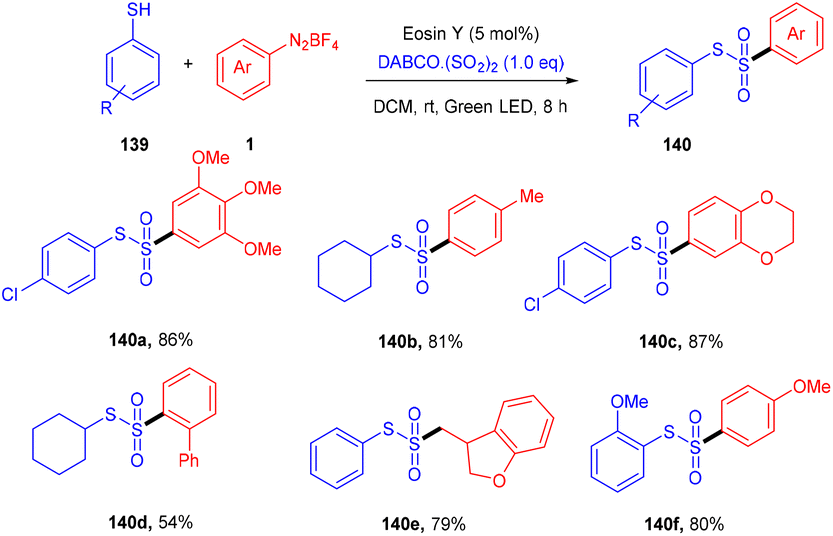
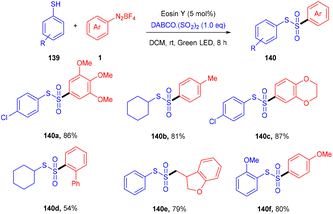
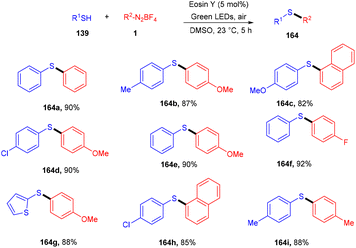
Volla's group in the same year reported the synthesis of thiosulfonates (140) by introducing the SO2 group. The reaction occurred through cross-coupling photoinduced sulfenyl radicals from thiols (139), aryl sulfonyl radicals from aryl diazonium salts (1), and DABCO·(SO2)2. This coupling was accomplished through complete regioselectivity. Furthermore, this protocol also demonstrated that allyl tethered aryldiazonium salts reacted smoothly with thiols to form dihydro benzofuran thiosulfonates (Scheme 65).62 It was also observed that aliphatic thiols have significant compatibility compared to aromatic thiols and gave excellent yields. In addition, the substitution of diazonium salts with electron-donating and -withdrawing groups revealed that all reacted smoothly, but electron-donating groups have a better reaction profile.
 |
| | Scheme 65 Synthesis of thiosulfonates. | |
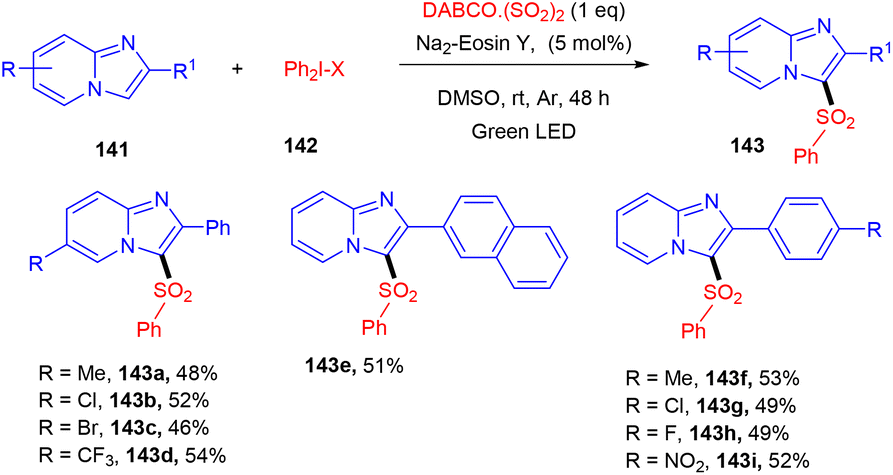
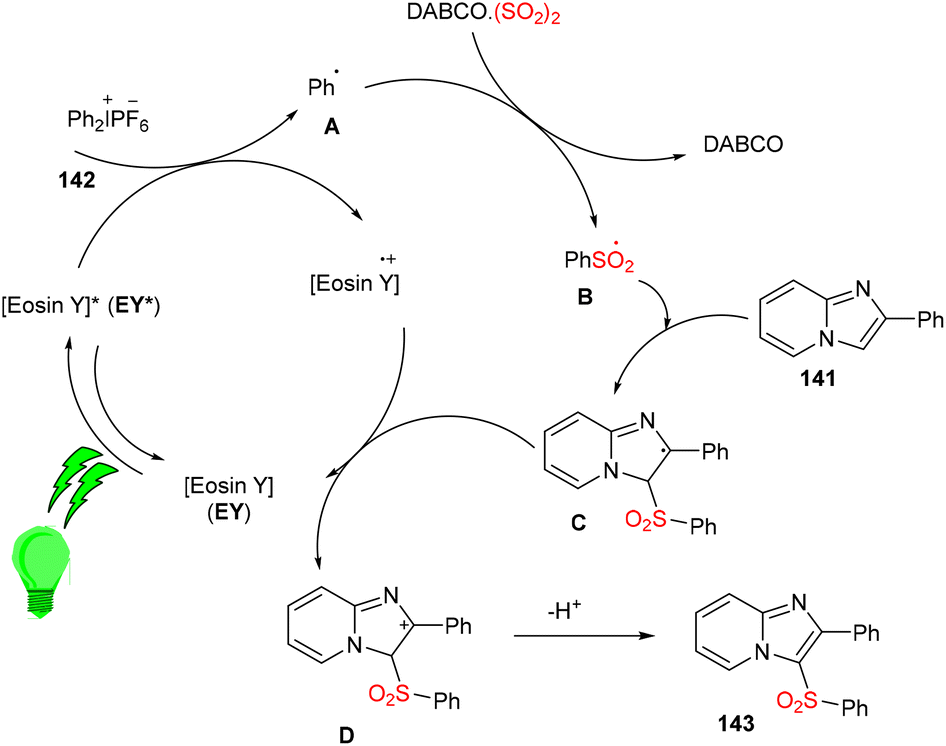
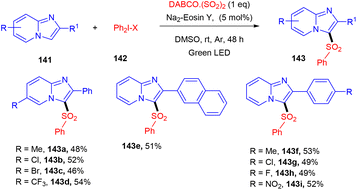
Multicomponent sulfonylation of imidazoheterocycles via the formation of two C–S bonds has been developed by the Piguel group. C-3 sulfonylated imidazoheterocycles (143) were synthesized from different imidazopyridines (141) and diaryliodonium salts (142) through a straightforward synthesis, demonstrating an efficient C–H functionalization using eosin Y as the photo-redox catalyst and DABCO·SO2 as the SO2 surrogate. This strategy gave the desired compounds in moderate to good yields with a broad substrate scope. Various electron-donating and -withdrawing groups on the aryl ring of imidazopyridines and aryl diazonium salts were well tolerated to afford the respective products in notable yields (Scheme 66).63
 |
| | Scheme 66 Synthesis of C-3 sulfonylated imidazopyridines. | |
Under the irradiation of green light, photoexcited eosin Y (EY*) was formed and transferred one electron by a SET mechanism to a diphenyl iodonium salt to afford a phenyl radical (A). Then, SO2 trapped the phenyl radical and gave a sulfonyl radical (B), which reacted regioselectively with 2-phenylimidazo[1,2-a]pyridine (141) to give a radical intermediate (C), which transferred the electron to the eosin Y cation and was converted into a carbocation (D), followed by re-aromatization via the loss of a proton and gave the desired sulfone 143 (Scheme 67).
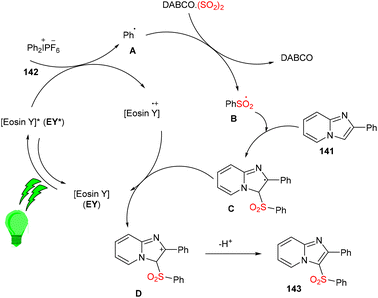 |
| | Scheme 67 Mechanism for the synthesis of C-3 sulfonylated imidazopyridines. | |
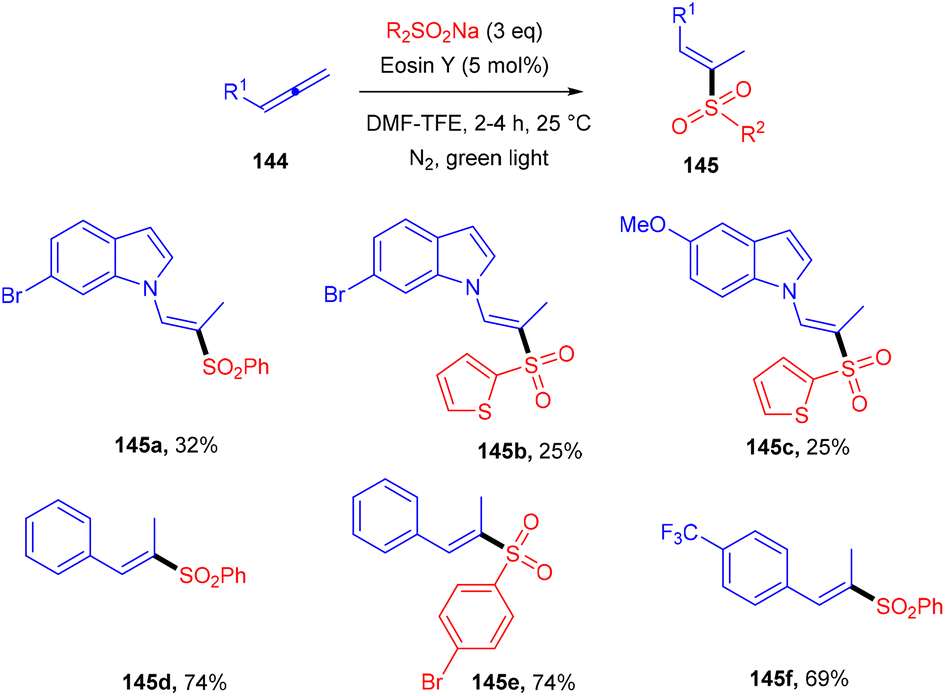
Hydrosulfonylation of allenes was elaborated using organophotoredox catalysis by Voskressensky's group. They performed the reaction using allenes (144) with sodium p-tolylsulfinate in the presence of eosin Y as a photoinduced catalyst. Control experiments suggested the free radical pathway. The authors also demonstrated a deuterium labelled experiment and observed deuterium exchange at the α-position. Furthermore, addition of acetic acid could fasten the reaction and a mixture of vinyl sulfones (regio-isomers) was obtained. Substituted indoles gave a lower yield of the final product compared to substituted sulfinates. The authors also revealed that aliphatic allenes have very low compatibility for the developed method due to the instability of free radicals (Scheme 68).64
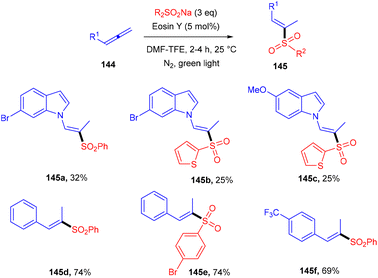 |
| | Scheme 68 Photoinduced hydrosulfonylation of allenes. | |
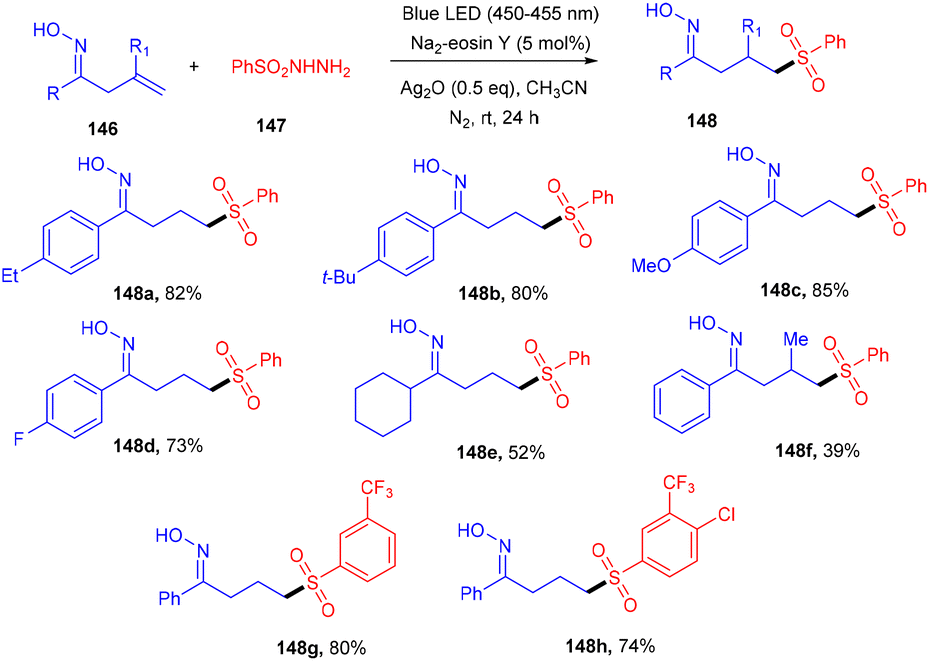
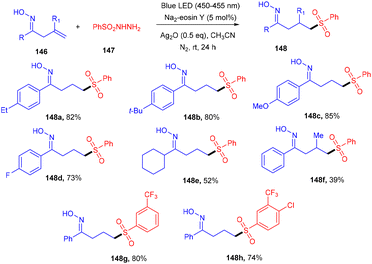
Oxime having an active OH unit has attracted significant attention from organic chemists for its number of applications in synthesizing organic compounds. Inspired by the work of Loh's group,55 Li's group explored a 1,5-hydrogen radical shift after adding a sulfur-centered radical to the double bond of the oxime. The sulfonylation of alkenes (146) was developed, assisted by oxime with sulfonyl hydrazides (147) using eosin Y as the photocatalyst at room temperature. Notably, the addition of Ag2O under the reaction conditions was important for the generation of a free radical from sulfonyl hydrazide which was captured by TEMPO and further confirmed by HRMS analysis. This strategy established excellent regioselectivity with wide functional group tolerance (Scheme 69).65
 |
| | Scheme 69 Regioselective sulfonylation of alkenes assisted by oxime. | |
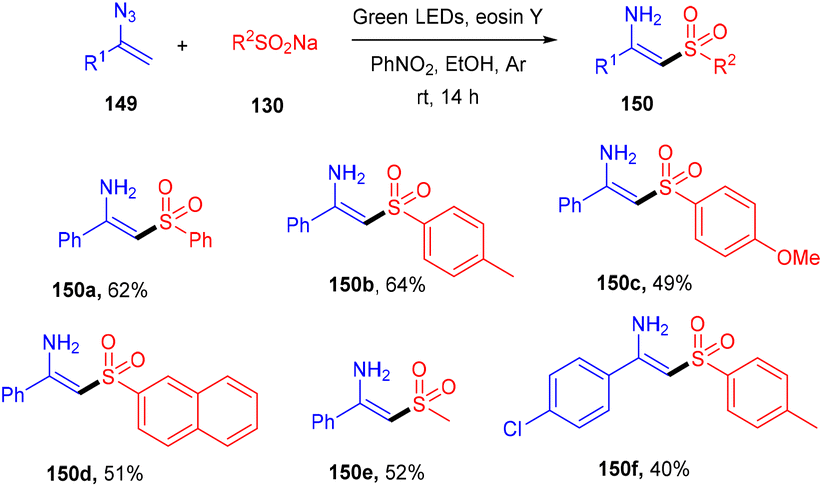
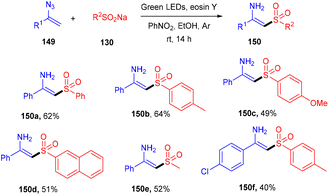
N-Substituted enamisulfones (150) were accessed by reacting vinyl azides (149) with sodium sulfinates (130) under photoredox catalysis. This strategy was developed by Mulina et al. in 2021. The reaction proceeded with a number of steps viz., radical sulfonyl formation, combination with the double bond followed by the generation of an iminyl radical and then reduction, which led to the final product. Electron-donating groups on the sulfinate phenyl ring showed better results than electron-withdrawing groups. In addition, various alkyl sulfinates have also been investigated, and the products were found in good yields (Scheme 70).66
 |
| | Scheme 70 Synthesis of N-substituted enamisulfones. | |
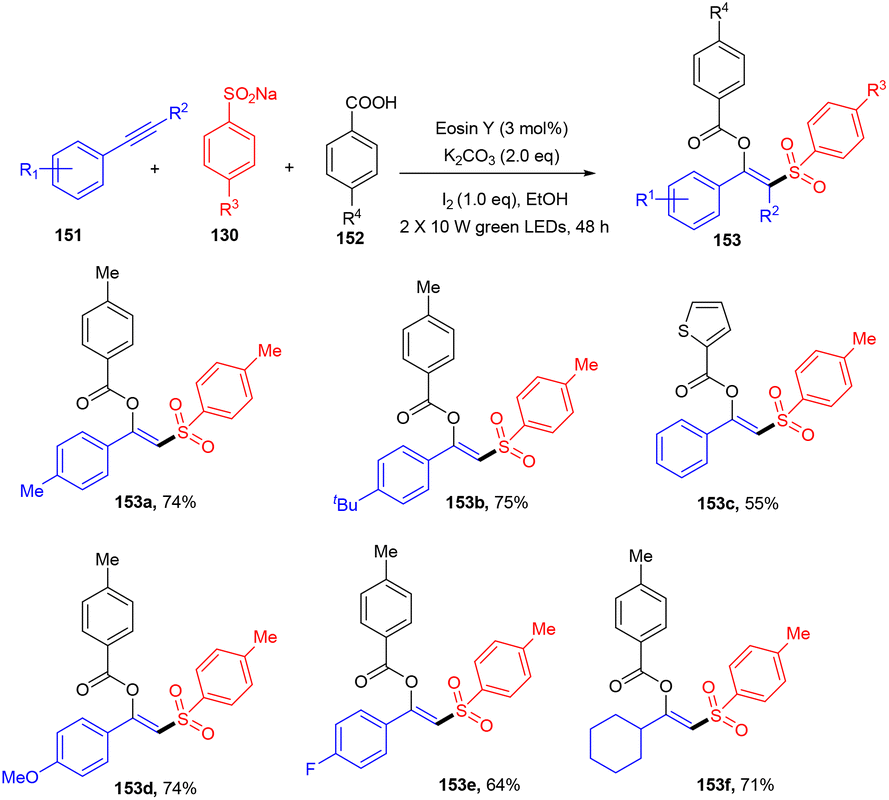
Inspired by the work of Nevado et al., a new strategy that involved an organophotoredox catalyst was developed by Patel and his co-workers in 2021, which resulted in an efficient synthesis of β-substituted vinyl sulfones (153). Regioselective difunctionalization of aryl alkynes (151) was carried out in the presence of sodium aryl sulfonates (130) and carboxylic acids (152). During optimization, it was found that the use of KI in the reaction was crucial as an iodinating agent. In addition, initial investigation of eosin Y in ethanolic solution also suggested the use of green light. To confirm the radical nature of the developed protocol, various radical trapping and Stern–Volmer fluorescence quenching experiments were performed which confirmed the radical pathway. It has been revealed that this strategy worked smoothly with a steric bulk substrate (Scheme 71).67 Formation of the C–S bond and simultaneous introduction of different functional groups such as ester and sulfone made this strategy superior to other previously reported approaches.
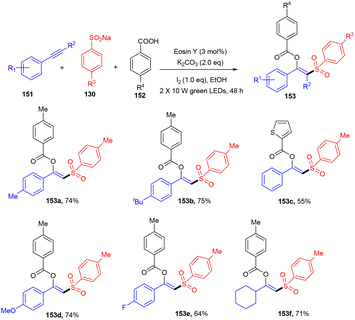 |
| | Scheme 71 Synthesis of β-substituted vinyl sulfones. | |
3.2. Thionation
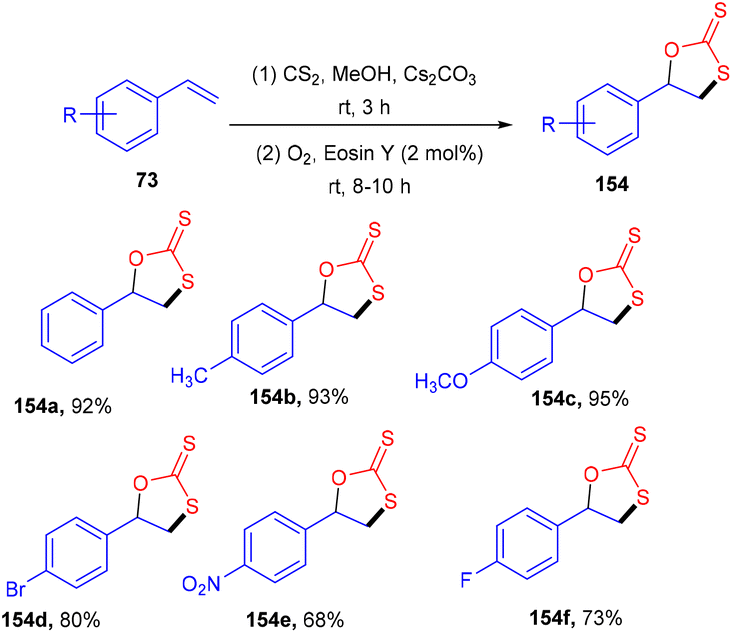
Yadav's group revealed a one-pot multicomponent synthesis of 1,3-oxathiolane-2-thiones (154). Low-cost and readily available styrene (73) was used as the starting material in the presence of CS2, and Cs2CO3 and eosin Y were used as photoredox catalysts at ambient temperature. Electron donating substituted styrenes with –Me, –OMe groups showed more tolerance and reacted faster under this protocol (Scheme 72). However, aliphatic alkenes did not work well due to the low stability of the free radical intermediate.68 Moreover, the reaction in the presence of TEMPO was completely quenched, which suggested that the reaction proceeded via a radical pathway.
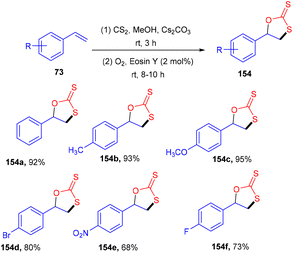 |
| | Scheme 72 Synthesis of 1,3-oxathiolane-2-thiones. | |
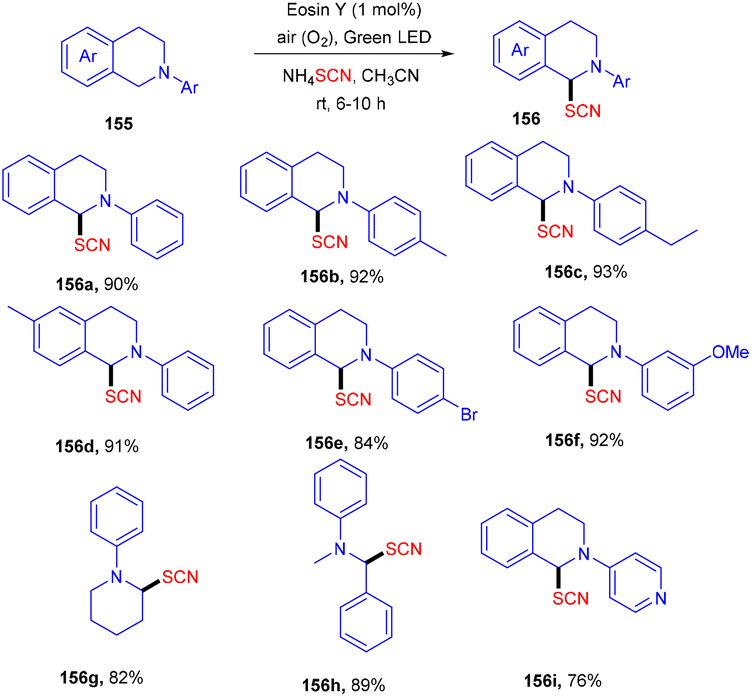
Using easily accessible and affordable NH4SCN as the source of SCN, Yadav and co-workers developed an efficient process for the synthesis of α-C(sp3)–H thiocyanate of N-disubstituted amines at ambient temperature. Tertiary amines also showed significant compatibility for this protocol. This approach was also used to produce a C-thiocyanate product from the aza-Baylis–Hillman adduct; the scope of nucleophiles amenable for iminium ion chemistry is expanded. The reaction required 1 mol% of eosin Y, while the greenest and most sustainable reagents are ambient oxygen and visible light. The current technique provides a new methodology for constructing C–S bonds (Scheme 73).69
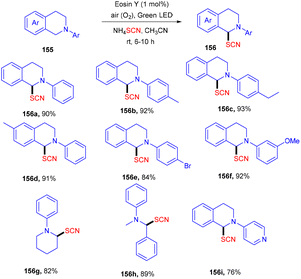 |
| | Scheme 73 Eosin Y mediated thiocyanation of tertiary amines. | |
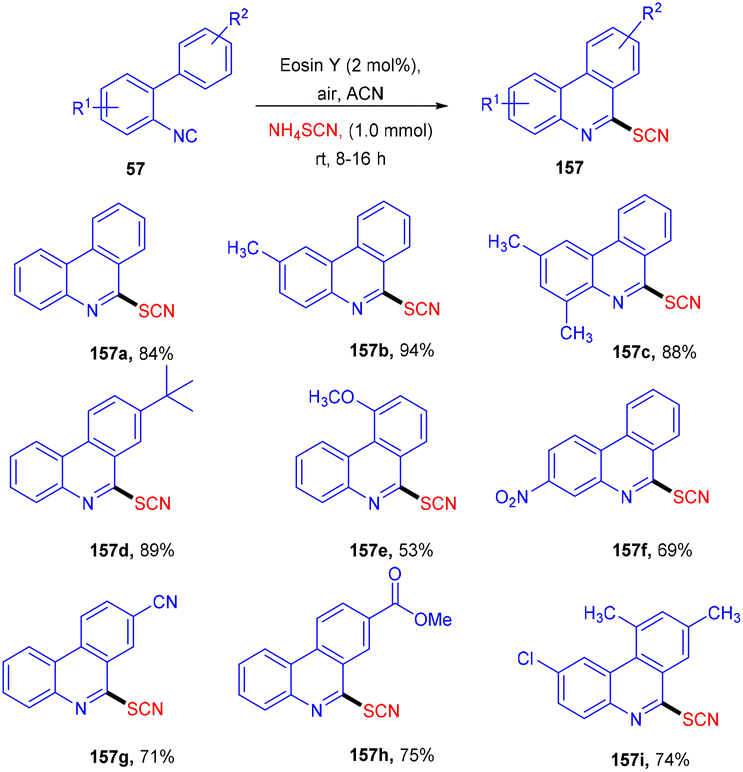
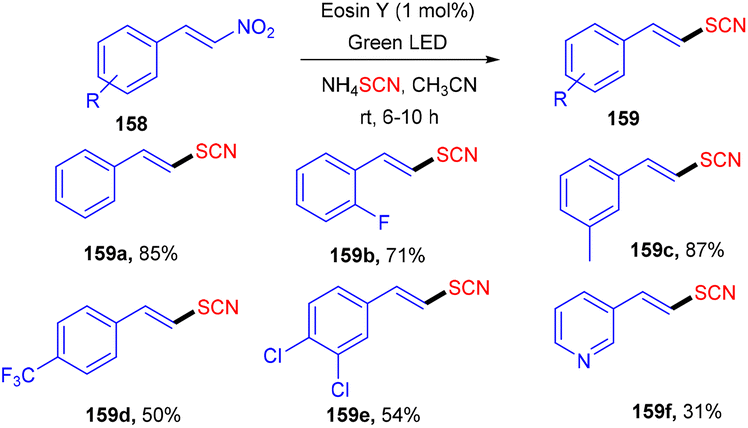
Later on, the authors also developed a photoredox methodology for synthesizing 6-thiocyanatophenanthridines (157) through a sequential reaction of radical cyclization followed by aromatization of isocyanobiphenyls (57) with ammonium thiocyanate (Scheme 74).70 It is noteworthy that in all developed, convenient and efficient strategies with photoredox catalysts, Yadav's group has used eosin Y as an inexpensive photoredox catalyst, which indicated its potential for thiolation reaction. Recently, they developed a methodology to catalyze the aerobic photoinduced formation of the C–S bond via denitration of β-nitrostyrene (158) using ammonium thiocyanate to give (E)-vinylthiocyanates (159) in up to 87% yield (Scheme 75).71 Substitution at the different positions of the aryl ring with electron-donating groups showed tunning with the reaction and gave the corresponding compounds in good yields. However, substitution with electron-withdrawing groups only at the ortho-position gave compounds in moderate to good yields, while substitution at the meta- and -para positions with these groups gave lower yields of the final compounds.
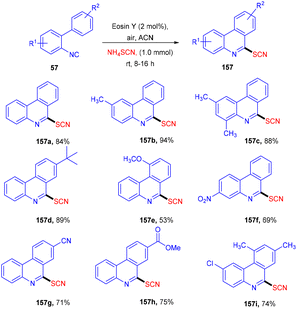 |
| | Scheme 74 Synthesis of 6-thiocyanatophenanthridines. | |
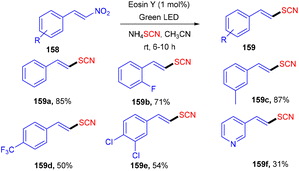 |
| | Scheme 75 Denitration of β-nitrostyrene. | |
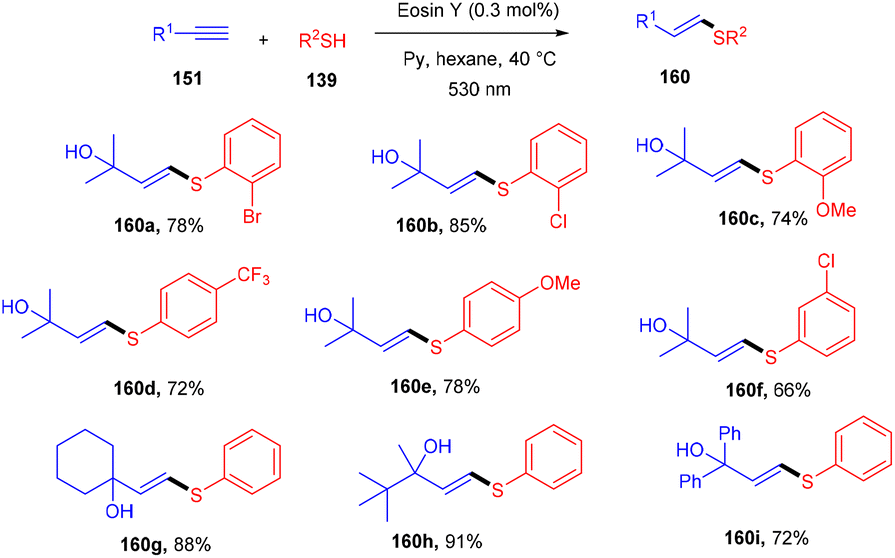
A click reaction between thiol (139) and alkyne (151) for C–S bond formation via a photo-redox reaction using eosin Y as a catalyst was reported by the Ananikov group in 2016. They demonstrated regioselectivity and broad tolerance of aryl thiols, giving valuable sulfur-containing scaffolds in moderate to good yields. Electron-donating groups (–Me, –OMe) on thiols gave higher yields of the final products (160). However, ortho-substituted thiols with halogens (–Cl, –Br) showed more compatibility and gave better yields. Notably, hexane has been used as a solvent to overcome the degradation of eosin Y. They also designed a photoreactor that could potentially improve the reactivity of the catalytic system (Scheme 76).72
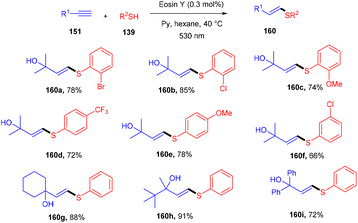 |
| | Scheme 76 Photoinduced thiol–alkyne click reaction. | |
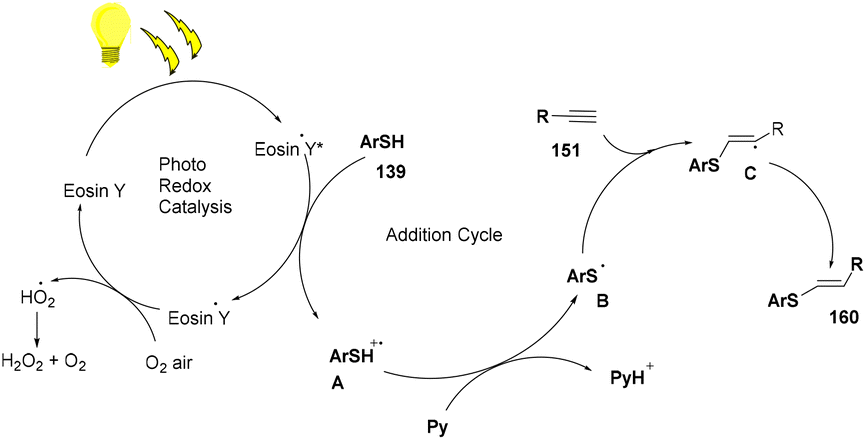
The reaction proceeded through the excitation of eosin Y generating excited eosin Y (EY*), which underwent oxidation and helped in the generation of a radical cation (A) of thiophenol. Subsequently, abstraction of a proton took place from this species by pyridine to give a thiyl radical (B). This radical is reacted with alkyne (151) to provide the desired product (160) (Scheme 77).
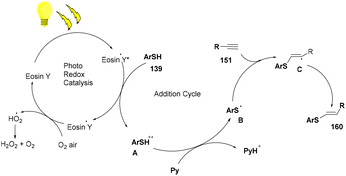 |
| | Scheme 77 Mechanism of the photoinduced thiol–alkyne click reaction. | |
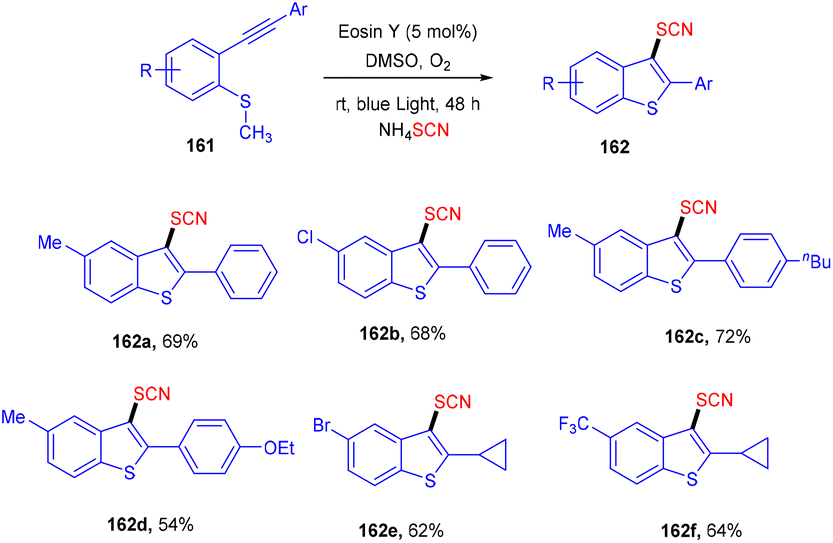
However, the Krishnagar group in 2018 gave access to 3-thiocyanatobenzothiophenes (162) by forming multiple C–S bonds. An efficient cascade methodology has been designed, starting with methyl(2-(phenylethynyl)phenyl) sulfane (161) which underwent cascade annulation on reaction with ammonium thiocyanate under visible light-mediated photoredox catalysis of eosin Y. They screened other thiocyanates as well, but NH4SCN showed superior results. Control experiments with TEMPO indicated a radical free mechanism for this reaction. This strategy was favourable for a number of alkynylarylsulfanes and gave good yields of the products. It was also found that the yields dropped significantly under a nitrogen atmosphere (Scheme 78).73
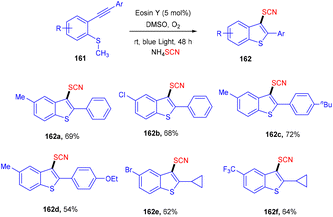 |
| | Scheme 78 Synthesis of 3-thiocyanobenzothiophenes. | |
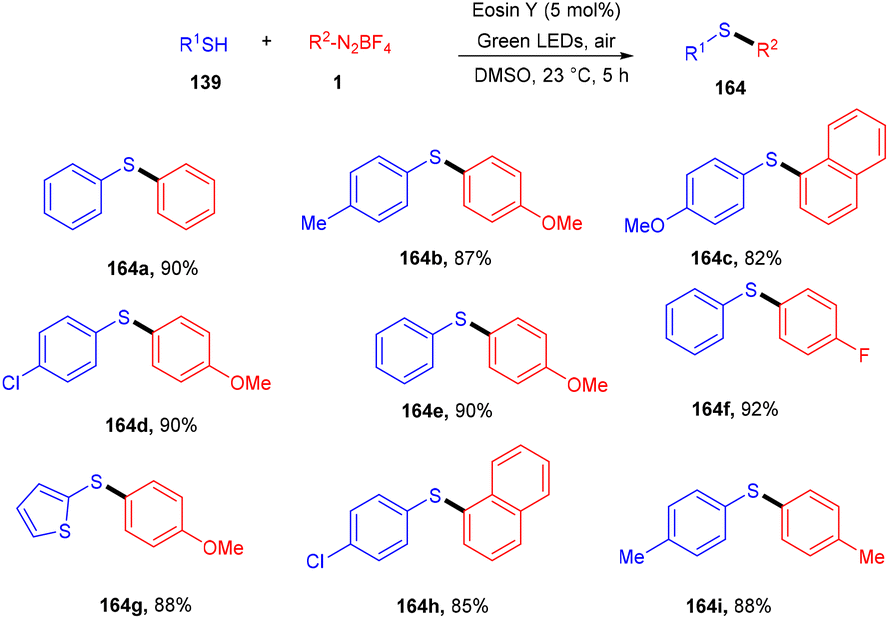
In the same year, 3-sulfenylindolines (163) were also synthesized by thioamination of alkynes. Here, they performed photoinduced radical cascade annulation by reacting 2-alkynyl-azidoarenes (40) with aryl thiols (Scheme 79).74 This reaction also proceeded through the free radical mechanism. However, diaryl sulfides (162) were synthesized in 2017 by the Lee group, in which the reaction was carried out between thiol (139) and aryl diazonium salts (1) using eosin Y as a photocatalyst. A C–S bond formation took place which gave access to the desired sulfides; electron-donating and electron-withdrawing substituted thiols were equally compatible with this approach and had excellent yields up to 92%. They further transformed diaryl sulfide to sulfone, which might be used as an amyloid aggregate inhibitor (Scheme 80).75 A control experiment has also been performed for the mechanistic studies and it was observed that the reaction proceeds through a reductive quenching cycle.
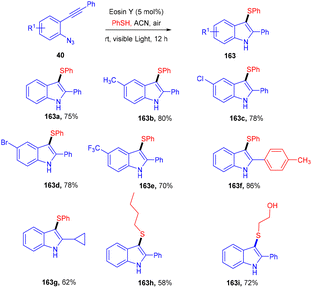 |
| | Scheme 79 Eosin Y catalyzed vicinal thioamination of alkynes. | |
 |
| | Scheme 80 Synthesis of diaryl sulfides. | |
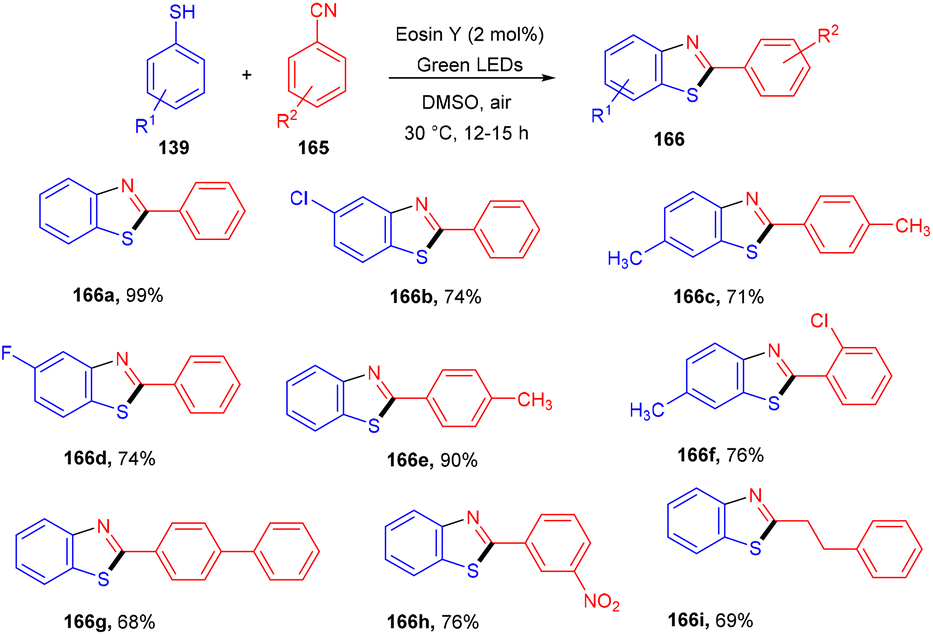
Natarajan and co-workers reported a simple and efficient synthesis strategy of benzothiazole by constructing a C–S bond using eosin Y as a photocatalyst. In this strategy, thiophenol (139) reacted with nitriles (165) to afford the corresponding benzothiazole (166). A number of derivatives were synthesized in good yields. The reaction proceeded through a combination of a radical of thiophenol and nitriles to give an iminyl radical, which underwent intramolecular cyclization and loss of a hydrogen radical to give the final compounds (Scheme 81).76
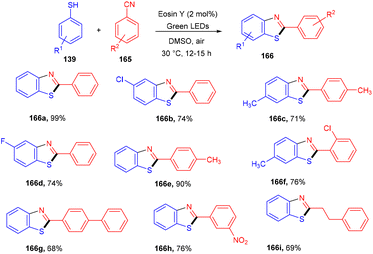 |
| | Scheme 81 Synthesis of 2-phenylbenzothiazoles. | |
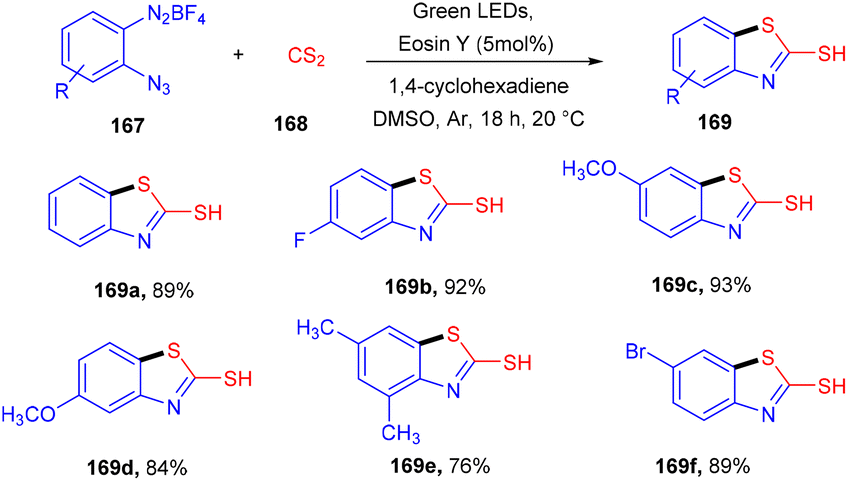
However, in 2020, they developed the synthesis of 2-mercaptobenzothiazoles (169) by reacting 2-azidoarenediazonium salts (167) and CS2 (168). Generation of an azido arene radical via the SET mechanism and reduction of the azido arene diazonium ion followed by CS2 insertion and intramolecular cyclization gave the desired products in high yields (Scheme 82).77
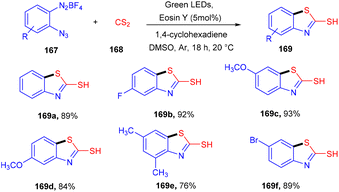 |
| | Scheme 82 Synthesis of 2-mercapto benzothiazole. | |
In another work they demonstrated that the reaction proceeded through the addition of a self-centred radical, generated from thiophenol by the SET mechanism followed by the attack of 1,3-diarylpropynones (170); the reaction was compatible with several thiophenols (139) having p-CH3, p-OCH3, p-NO2, and p-F groups as well as 1,3-diarylpropynones (Scheme 83).78 Notably, the reaction was carried out in open air without using any base and external oxidant.
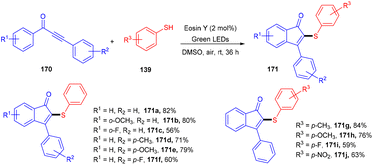 |
| | Scheme 83 Synthesis of 2-sulfenylindenones. | |
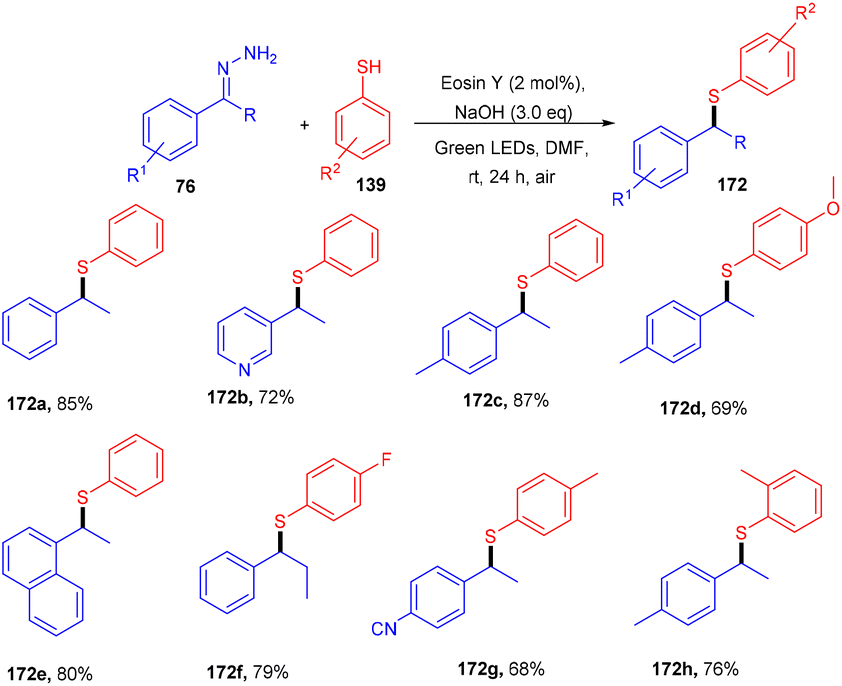
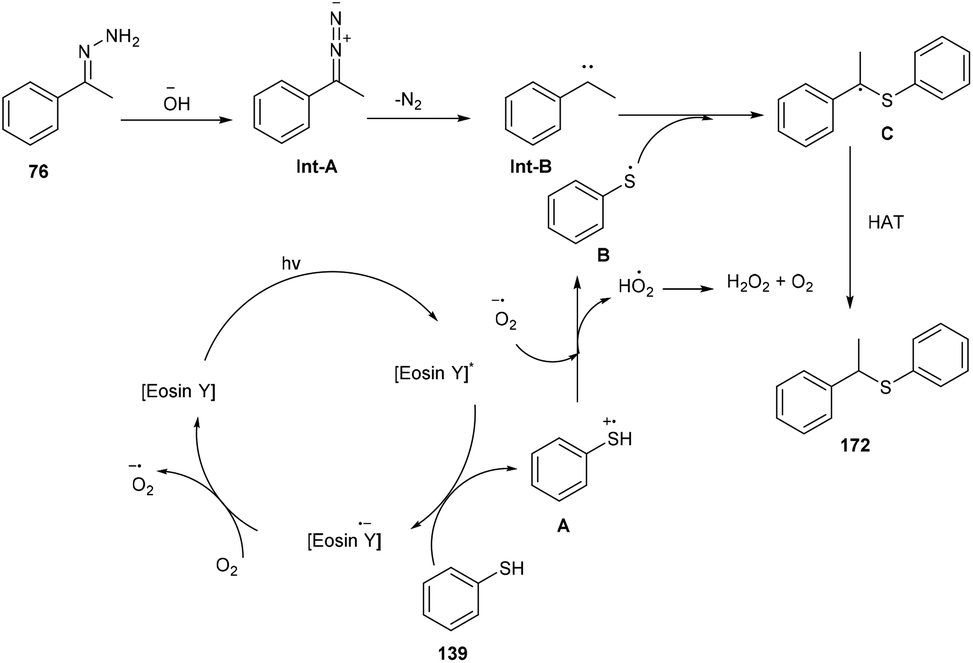
In the same year, the Singh group reported the sustainable and efficient synthesis of thioethers (172) using photoredox catalysis. The authors proposed that the reaction proceeded through a single electron transfer mechanism. They constructed a direct C–S bond by the reaction of substituted thiols (139) and a number of functionalized hydrazones (76) to access various thioethers (172) in good yields. In addition, a low amount of base and thiol might decrease the yield of the final compound (Scheme 84).79 The cyclic voltammogram of thiophenol showed an oxidation potential of −0.37 V and a reduction potential of −1.3 V, clearly indicating that eosin Y could allow the oxidation of thiophenol via the SET mechanism. Based on control experiments, it was proposed that photoinduced eosin Y oxidized thiol and gave the corresponding thiyl radical cation A and underwent arial oxidation to give neutral eosin Y along with a superoxide radical. Proton abstraction from the thiyl radical cation using the superoxide radical gave thiyl radical B along with the generation of a hydroperoxyl radical (H2O˙). Reaction of hydrazone with a hydroxide ion afforded diazo compound Int-A, which further gave carbene Int-B by the removal of N2; subsequent coupling between carbene Int-B and thiyl radical B gave intermediate C. The final product was obtained by the hydrogen abstraction from C (Scheme 85).
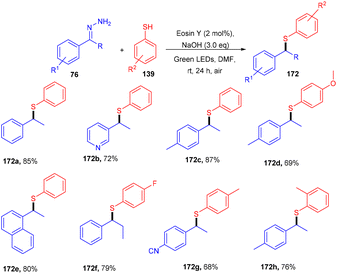 |
| | Scheme 84 Synthesis of thioethers. | |
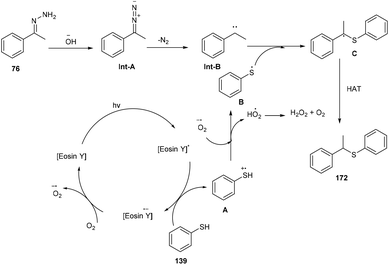 |
| | Scheme 85 Plausible mechanism to access thioethers. | |
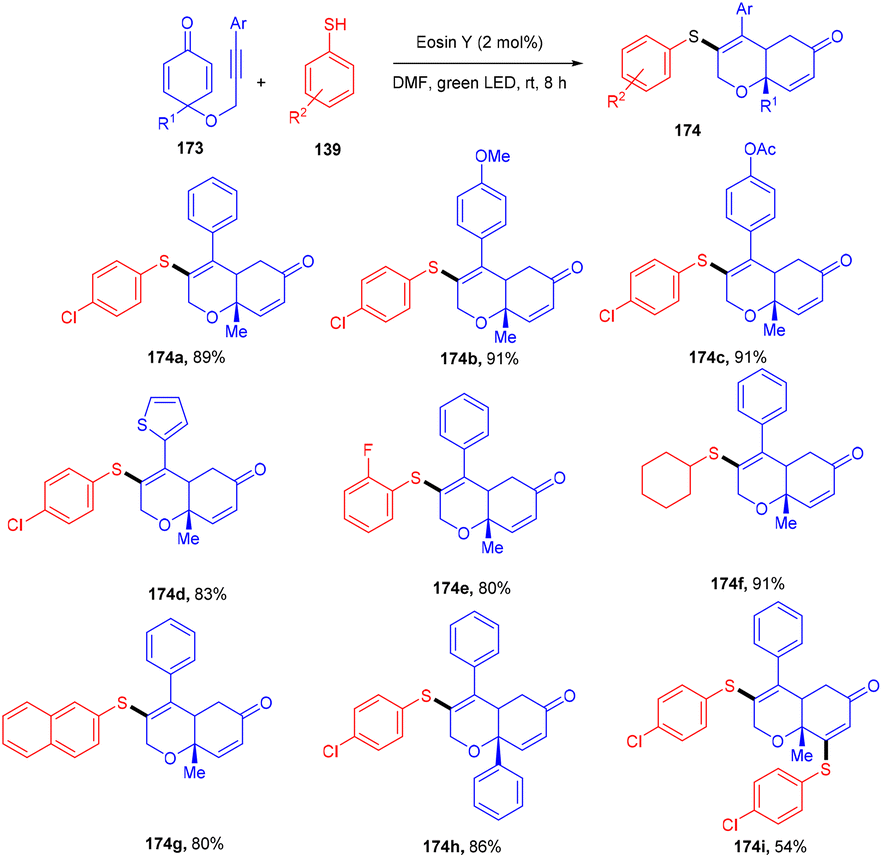
To access highly decorated cis-dihydrochromenone compounds (174), the Volla group has developed an efficient protocol for tandem sulfenylation annulation of the alkyne chain containing cyclohexadienones (173). This protocol demonstrated high diastereoselectivities, a broad substrate scope, and good yields (Scheme 86).80 In addition, in the presence of excess thiophenol and a prolonged time up to 16 h, bisulfenylated scaffolds were synthesized as a single diastereomer. Moreover, various tricyclic derivatives were obtained through self-centred radical addition and the intramolecular Michael addition of amines.
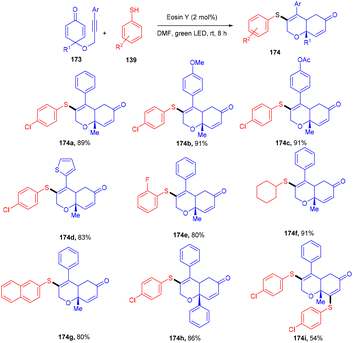 |
| | Scheme 86 Tandem sulfenylation annulation of alkynes. | |
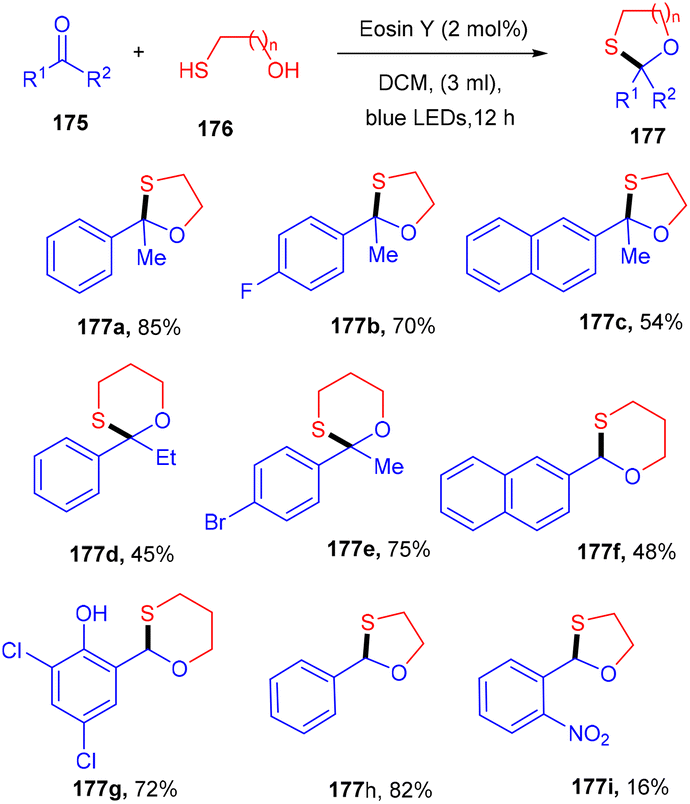
In 2020, the protection of ketones with mercapto alcohols under organophotoredox catalysis was demonstrated by Lee and co-workers. An effective strategy has been developed for the oxathiacetalization of aldehydes and ketones (175) and demonstrated the formation of the C–S bond. The employed conditions were also compatible with various aldehydes such as aromatic, aliphatic, and α,β-unsaturated aldehydes. Based on control experiments it was proposed that the reaction followed a free radical pathway. The eminent feature of the developed strategy was high functional group compatibility (Scheme 87).81
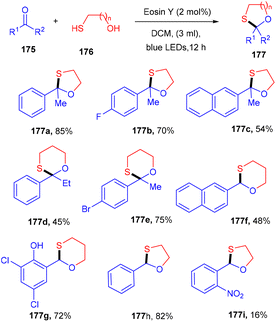 |
| | Scheme 87 Photoinduced oxathiacetalization of aldehydes and ketones. | |
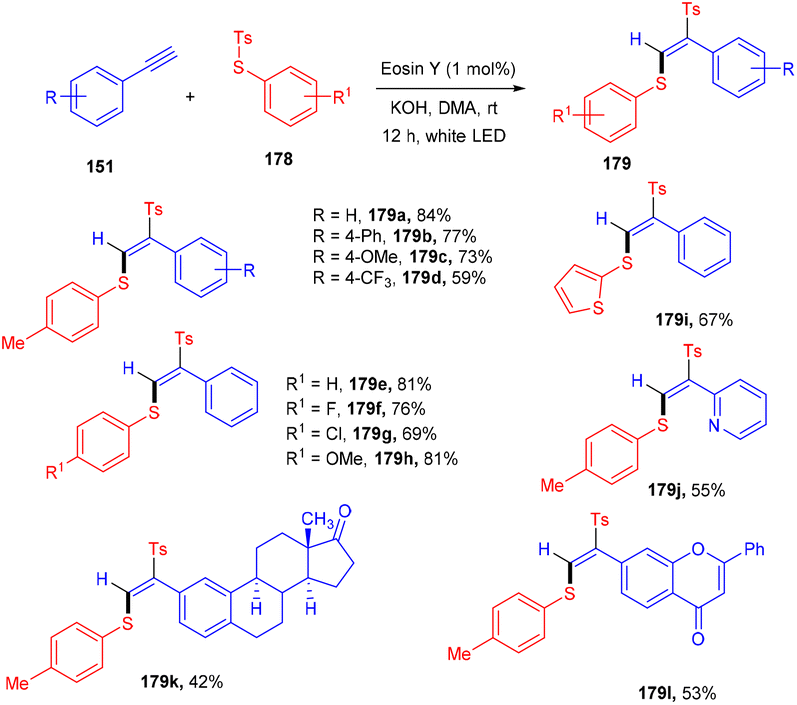
Jia's group has recently developed a photoinduced Atom Transfer Radical Addition (ATRA) reaction for accessing (E)-β-aryl sulfonyl vinyl sulfides (179) with high regio- and stereo-selectivities. They started with aryl alkynes (148) and thiosulfonates (178) under eosin Y catalysis at room temperature. The authors demonstrated the application of the developed strategy by late-stage modification of naturally occurring bioactive motifs (Scheme 88). Based on investigation studies it was revealed that thiyl radicals formed SET and followed the free radical pathway.82
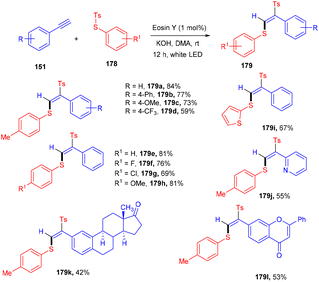 |
| | Scheme 88 Radical addition of thiosulfonates and aryl alkynes. | |
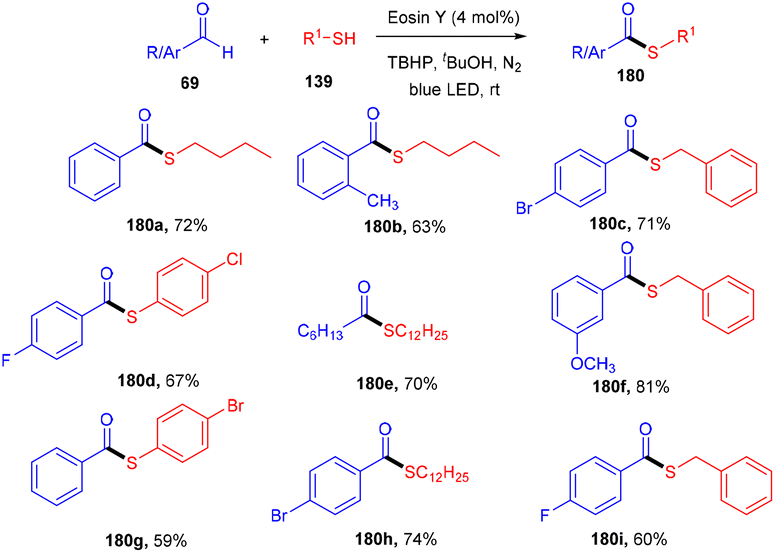
Roy et al. in 2021 developed a prominent synthesis of thioester (180) by forming a C–S bond through photoinduced cross dehydrogenative coupling (PCDC) between aldehydes and thiols at ambient temperature. This strategy proceeded smoothly with both aryl/alkyl aldehydes (69) and alkyl/aryl-thiols (139) to give the final motifs in good to high yields (Scheme 89). Furthermore, during mechanistic studies, it has been revealed that the generation of an acyl radical from an aldehyde was intercepted by a disulfide intermediate to afford the desired thioester.83
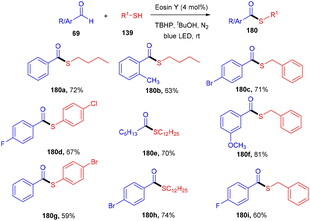 |
| | Scheme 89 Metal-free synthesis of thioesters. | |
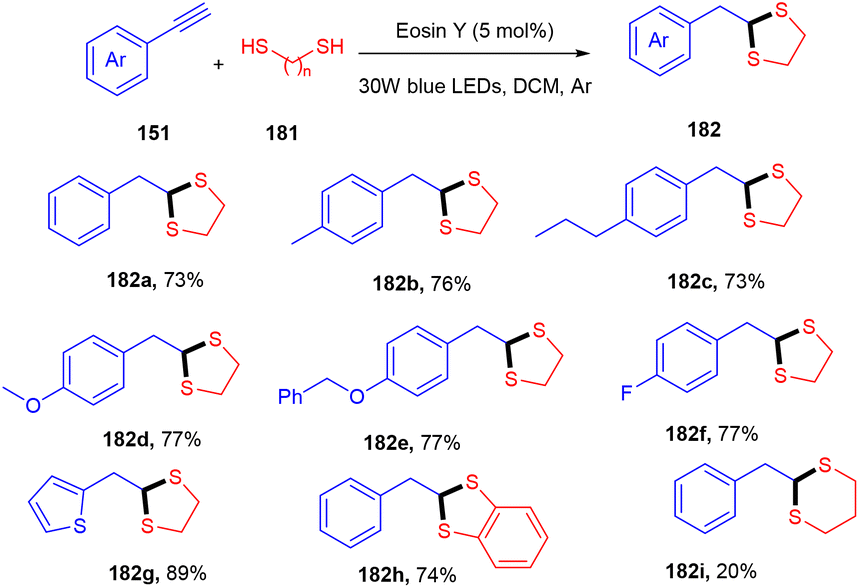
Recently, Khade et al. have reported the practical synthesis of 1,3-dithiolanes (182). These scaffolds were obtained without using any base and external additives from terminal aromatic and heteroaromatic alkynes (151) via the photoinduced catalyst eosin Y. Various electron-donating, withdrawing, and deactivating groups showed compatibility with the employed protocol and gave the final products in good yields (Scheme 90).84 The authors also performed various control experiments such as cyclic voltammetry and Stern–Volmer constant calculation to gain mechanistic insight and concluded the free radical pathway via SET oxidation.
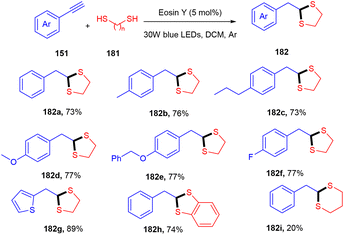 |
| | Scheme 90 Direct synthesis of 1,3-dithiolanes. | |
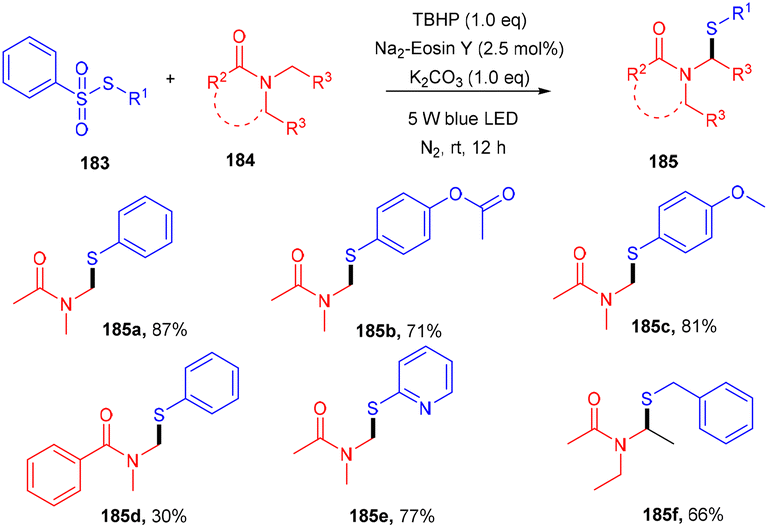
Zhang et al. performed direct sulfenylation to construct highly sulfenylated amides (185) via a reaction between thiosulfonates (183) and alkyl/aryl/heteroaryl amides (184) in the presence of TBHP and potassium carbonate, with eosin Y being used as the photocatalyst under mild reaction conditions. The reaction in the presence of TEMPO (radical trapping experiment) confirmed the radical free pathway of the reaction. Different sulfenyl amides were obtained from various amides and sulfones via direct construction of the C–S bond in moderate to good yields (Scheme 91).85
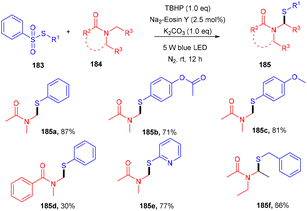 |
| | Scheme 91 Photoinduced sulfenylation of the C–H bond in amide. | |
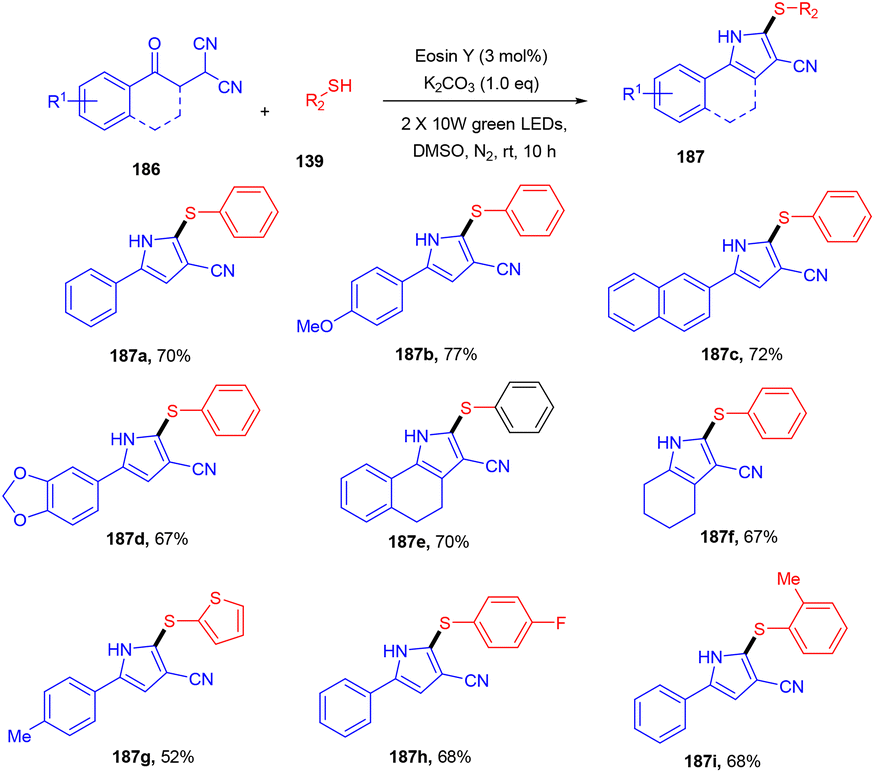
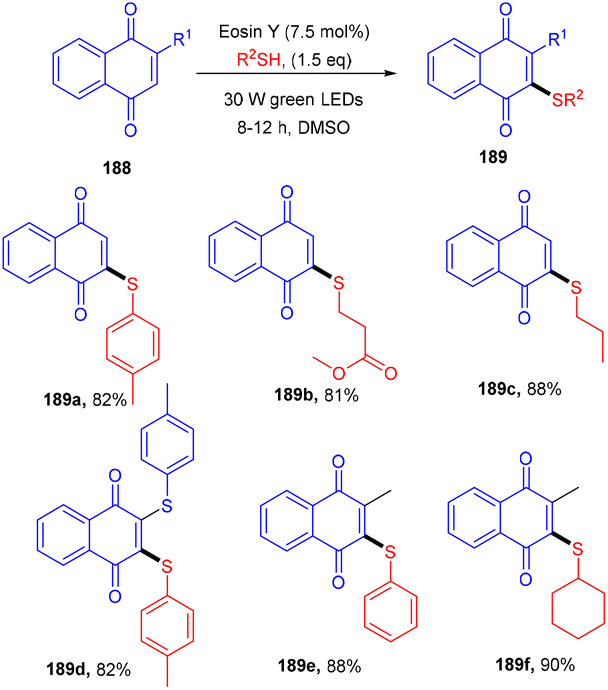
Recently, the Patel group synthesized thio-functionalized pyrroles (187) using visible light and an organophotocatalyst. They gave an inimitable example of visible-light-induced access to thio-functionalized pyrroles. They established the methodology by reacting β-ketodinitriles (186) with thiophenols (139) using a xanthene-based dye (eosin Y) as the photocatalyst. Here the authors also revealed the industrial utility of this large-scale reaction and its synthetic utility by further modification of the final products (Scheme 92).86 Dhar and coworkers demonstrated the thiolation of 1,4-naphthoquinones (188) under photoredox catalysis. They showed the direct formation of the C–S bond and synthesized several thiol substituted naphthoquinones (189) in excellent yields. Various aromatic and aliphatic thiols have been investigated and the radical generation pathway has also been demonstrated by HRMS analysis of the TEMPO-adduct (Scheme 93).87
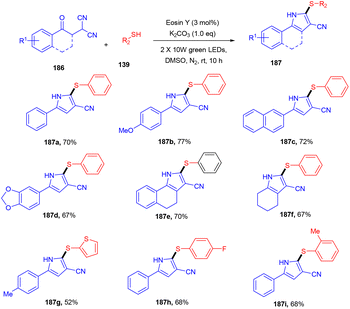 |
| | Scheme 92 Synthesis of thiol functionalized pyrroles. | |
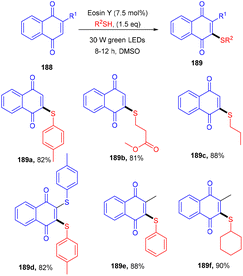 |
| | Scheme 93 Photoinduced thiolation of 1,4-naphthoquinones. | |
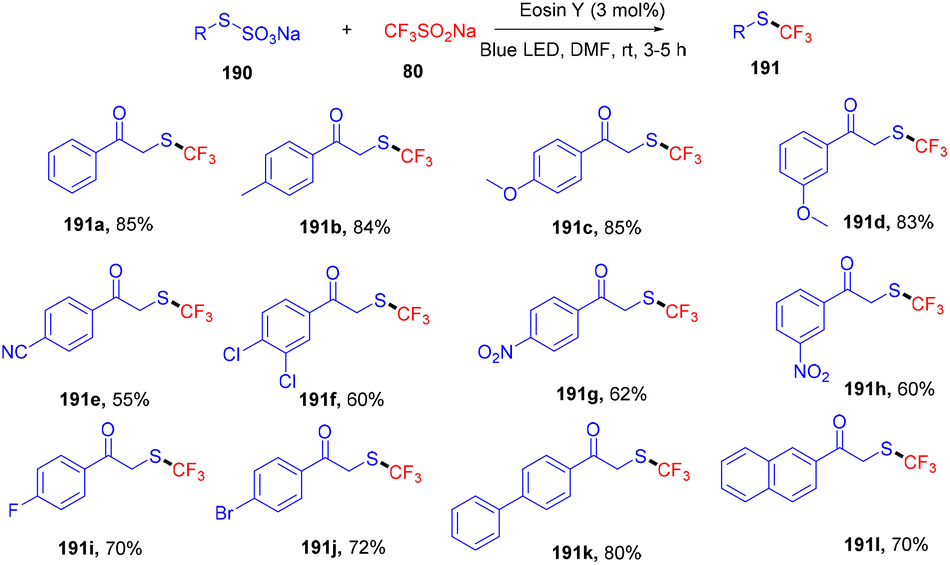
Recently, Singh and coworkers demonstrated the trifluoromethylation of Bunte salts through the direct formation of the C–S bond under visible light irradiation using eosin Y as the photocatalyst. During control experiments it was observed that only green LEDs gave the desired product in a good yield. Various other dyes were also employed but no efficacy was maintained. In addition, several electron-donating and -withdrawing groups were investigated and it was found that electron-rich (–CH3, –OCH3) groups on the aryl ring afforded high yields, while electron-deficient groups (p-CN, p-NO2) showed less compatibility (Scheme 94).88 Furthermore, the reaction was performed in the presence of a radical trapping scavenger (TEMPO) and complete quenching of the reaction was observed, suggesting the free radical pathway of the reaction.
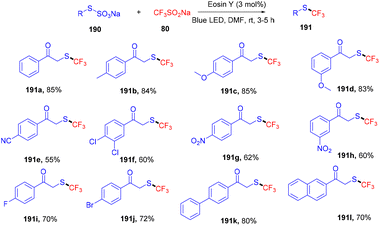 |
| | Scheme 94 Photoinduced trifluoromethylation of Bunte salts. | |
4. Conclusions and future perspectives
Eosin Y mediated photoredox C–H functionalization has been developed as a powerful tool for synthesizing organic molecules containing C–C and C–S bonds. The main advantages of eosin Y catalyzed reactions involved transition metal-free process, mild reaction conditions, sustainable chemistry, step and atom economy, good functional group tolerance, and high industrial applicability. Here, we described the formation of C–C and C–S bonds using a transition metal-free organophotocatalyst under three different mechanistic pathways. Eosin Y-based transformations have the potential to provide access to diversely functionalized organic compounds. Several valuable synthons and bioactive compounds (such as Bunte salts, β-lactams, diazonium salts, indoles, quinones, Baylis–Hillman acetates, etc.) can be accessed through eosin Y as the photocatalyst.
Despite unprecedented developments, many challenges still exist in this field. Firstly, in some reports, the authors claimed sustainability of the reaction, while the use of solvents such as DMSO, DCE, and DMF limited their sustainability. Secondly, the use of a base in some reactions completely quenches the reaction; as a consequence, the yield of the final product is lowered. Notably, in the present era, regioselective C–H functionalization has attracted incessant attention from the organic community, hence the developed methodologies must have fulfilled the condition of regioselectivity. In addition, it was found that ortho-substituted aryl diazonium salts were less active for the free radical mechanism, which could limit the substrate scope. Various methodologies to accomplish remote C–H functionalization have merged metal catalysis with photocatalysis and hence limited the concept of green chemistry.
Furthermore, reactions of various functional groups such as alcohols, urea, etc. are rare and not much explored for novel coupling partners or biologically active heterocyclic scaffolds. Single electron transfer and hydrogen atom transfer mechanisms are the basic mechanisms followed by most reactions catalyzed by eosin Y. However, very rare organic transformations using eosin Y as a photocatalyst follow the energy transfer mechanism. Hence, there is a significant scope to form C–C and C–S bonds using eosin Y via an energy transfer mechanism. Secondly, more effective methodologies need to be developed to avoid the degradation of eosin Y, as photodegradation is a common process. In this context, efficient immobilization with a covalent organic framework may solve this issue. Thirdly, there is a need for specific light for single electron reduction; in this vein, visible light-mediated catalysis is more favourable. Last but foremost, more profound knowledge of photoinduced organic transformation is demanded. New theories, methodologies, and mechanisms should be developed to deal with the current issues. There is also a need to develop reactions which can make use of water as the solvent for the concept of green chemistry and atom economy. Undoubtedly, a promising development of photoinduced organic transformation for forming C–C and C–S bonds will take place in the future to advance sustainable and green chemistry.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge SERB (CRG/2018/002159) and CSIR (02(0310)/17/EMR-II) for financial support.
References
-
(a) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Visible light mediated photoredox catalytic arylation reactions, Acc. Chem. Res., 2016, 49, 1566 CrossRef CAS;
(b) C. Shen, P. Zhang, Q. Sun, S. Bai, T. A. Hor and X. Liu, Recent advances in C–S bond formation via C–H bond functionalization and decarboxylation, Chem. Soc. Rev., 2015, 44, 291 RSC.
- L. Zheng, K. Tao and W. Guo, Recent Developments in Photo–Catalyzed/Promoted Synthesis of Indoles and Their Functionalization: Reactions and Mechanisms, Adv. Synth. Catal., 2021, 363, 62 CrossRef CAS.
- L. Capaldo, L. L. Quadri and D. Ravelli, Photocatalytic hydrogen atom transfer: the philosopher's stone for late-stage functionalization?, Green Chem., 2020, 22, 3376 RSC.
-
(a) A. Singh, K. Teegardin, M. Kelly, K. S. Prasad, S. Krishnan and J. D. Weaver, Facile synthesis and complete characterization of homoleptic and heteroleptic cyclometalated Iridium(III) complexes for photocatalysis, J. Organomet. Chem., 2015, 776, 51 CrossRef CAS;
(b) D. Ravelli, M. Fagnoni and A. Albini, Photoorganocatalysis. What for?, Chem. Soc. Rev., 2013, 42, 97 RSC;
(c) M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Organic photocatalysts for the oxidation of pollutants and model compounds, Chem. Rev., 2012, 112, 1710 CrossRef CAS;
(d) M. Reckenthaeler and A. G. Griesbeck, Photoredox catalysis for organic syntheses, Adv. Synth. Catal., 2013, 355, 2727 CrossRef CAS.
-
(a) D. P. Hari and B. König, Synthetic applications of eosin Y in photoredox catalysis, Chem. Commun., 2014, 50, 6688 RSC;
(b) V. Srivastava and P. P. Singh, Eosin Y catalysed photoredox synthesis: a review, RSC Adv., 2017, 7, 31377 RSC;
(c) A. Bosveli, T. Montagnon, D. Kalaitzakis and G. Vassilikogiannakis, Eosin: a versatile organic dye whose synthetic uses keep expanding, Org. Biomol. Chem., 2021, 19, 3303 RSC.
- J. D. Bell and J. A. Murphy, Recent advances in visible light-activated radical coupling reactions triggered by (i) ruthenium, (ii) iridium and (iii) organic photoredox agents, Chem. Soc. Rev., 2021, 50, 9540 RSC.
- L. Marzo, S. K. Pagire, O. V. Reiser and B. König, Visible–light photocatalysis: does it make a difference in organic synthesis?, Angew. Chem., Int. Ed., 2018, 57, 10034 CrossRef CAS PubMed.
- L. Capaldo and D. Ravelli, Hydrogen atom transfer (HAT): a versatile strategy for substrate activation in photocatalyzed organic synthesis, Eur. J. Org. Chem., 2017, 2056 CrossRef CAS PubMed.
- Q. Q. Zhou, Y. Q. Zou, L. Q. Lu and W. J. Xiao, Visible–light–induced organic photochemical reactions through energy–transfer pathways, Angew. Chem., Int. Ed., 2019, 58, 1586 CrossRef CAS PubMed.
- D. P. Hari and B. Konig, The Photocatalyzed Meerwein Arylation: Classic Reaction of Aryl Diazonium Salts in a New Light, Angew. Chem., Int. Ed., 2013, 52, 4734 CrossRef CAS PubMed.
- D. P. Hari, P. Schroll and B. König, Metal-free, visible-light-mediated direct C–H arylation of heteroarenes with aryl diazonium salts, J. Am. Chem. Soc., 2012, 134, 2958 CrossRef CAS PubMed.
- L. Gu, C. Jin and J. Liu, Metal-free, visible-light-mediated transformation of aryl diazonium salts and (hetero) arenes: an efficient route to aryl ketones, Green Chem., 2015, 17, 3733 RSC.
- S. J. Kwon, H. I. Jung and D. Y. Kim, Visible light photoredox–catalyzed arylation of quinoxalin–2(1H)–ones with aryldiazonium salts, ChemistrySelect, 2018, 3, 5824 CrossRef CAS.
- R. Kapoor, R. Chawla and L. D. S. Yadav, Visible-light-mediated Gomberg-Bachmann reaction: An efficient photocatalytic approach to 2-aminobiphenyls, Tetrahedron Lett., 2019, 60, 805 CrossRef CAS.
- T. Adak, C. Hu, M. Rudolph, J. Li and A. S. K. Hashmi, Metal-Free, Visible-Light-Enabled Direct C3–H Arylation of Anthranils, Org. Lett., 2020, 22, 5640 CrossRef CAS.
- S. D. Jadhav, D. Bakshi and A. Singh, Visible light mediated organocatalytic activation of ethyl bromofluoroacetate: Coupling with indoles and anilines, J. Org. Chem., 2015, 80, 10187 CrossRef CAS.
- T. Ghosh, P. Maity and B. C. Ranu, Cobalt-Catalyzed Remote C-4 Functionalization of 8-Aminoquinoline Amides with Ethers via C–H Activation under Visible-Light Irradiation. Access to α-Heteroarylated Ether Derivatives, Org. Lett., 2018, 20, 1011 CrossRef CAS PubMed.
- S. Singh, P. Chauhan, M. Ravi and P. P. Yadav, Eosin Y–Yb (OTf) 3 catalyzed visible light mediated electrocyclization/indole ring opening towards the synthesis of Heterobiaryl-Pyrazolo [3, 4-B] pyridines, New J. Chem., 2018, 42, 6617 RSC.
- P. Maity, D. Kundu and B. C. Ranu, Visible–Light–Photocatalyzed Metal–Free C–H Heteroarylation of Heteroarenes at Room Temperature: A Sustainable Synthesis of Biheteroaryls, Eur. J. Org. Chem., 2015, 1727 CrossRef CAS.
- H. K. Singh, A. Kamal, S. Kumari, D. Kumar, S. K. Maury, V. Srivastava and S. Singh, Eosin Y-catalyzed synthesis of 3-aminoimidazo [1, 2-a] pyridines via the HAT process under visible light through formation of the C–N bond, ACS Omega, 2020, 5, 29854 CrossRef CAS.
- A. U. Meyer, T. Slanina, C. J. Yao and B. König, Metal-free perfluoroarylation by visible light photoredox catalysis, ACS Catal., 2016, 6, 369 CrossRef CAS.
- Z. Liang, S. Xu, W. Tian and R. Zhang, Eosin Y-catalyzed visible-light-mediated aerobic oxidative cyclization of N, N-dimethylanilines with maleimides, Beilstein J. Org. Chem., 2015, 11, 425 CrossRef CAS.
- N. Yadav, M. D. Ansari, V. B. Yadav, A. Verma, S. K. Tiwari, S. Ansari and I. R. Siddiqui, Metal-free visible-light-mediated organophotoredox catalysis: synthesis of 3-functionalized indole via C–C, C–N bond formation, Mol. Divers., 2021, 25, 1103 CrossRef CAS PubMed.
- L. Gu, C. Jin, J. Liu, H. Ding and B. Fan, Transition-metal-free, visible-light induced cyclization of arylsulfonyl chlorides with 2-isocyanobiphenyls to produce phenanthridines, Chem. Commun., 2014, 50, 4643 RSC.
- D. P. Hari and B. König, Eosin Y catalyzed visible light oxidative C–C and C–P bond formation, Org. Lett., 2011, 13, 3852 CrossRef CAS PubMed.
- N. Esumi, K. Suzuki, Y. Nishimoto and M. Yasuda, Synthesis of 1, 4-dicarbonyl compounds from silyl enol ethers and bromocarbonyls, catalyzed by an organic dye under visible-light irradiation with perfect selectivity for the halide moiety over the carbonyl group, Org. Lett., 2016, 18, 5704 CrossRef CAS PubMed.
- I. Suzuki, N. Esumi and M. Yasuda, Photoredox α–Allylation of α–Halocarbonyls with Allylboron Compounds Accelerated by Fluoride Salts under Visible Light Irradiation, Asian J. Org. Chem., 2016, 5, 179 CrossRef CAS.
- J. Schwarz and B. König, Metal-free, visible-light-mediated, decarboxylative alkylation of biomass-derived compounds, Green Chem., 2016, 18, 4743 RSC.
- M. M. Nebe, D. Loeper, F. Fürmeyer and T. Opatz, Visible–Light Organophotoredox–Catalyzed Synthesis of Precursors for Horner–Type Olefinations, Eur. J. Org. Chem., 2018, 2471 CrossRef CAS.
- H. B. Ye, X. Y. Zhou, L. Li, X. K. He and J. Xuan, Photochemical synthesis of succinic ester-containing phenanthridines from diazo compounds as 1,4-dicarbonyl precursors, Org. Lett., 2022, 24, 6018 CrossRef CAS PubMed.
- J. Q. Weng, W. X. Xu, X. Q. Dai, J. H. Zhang and X. H. Liu, Alkylation reactions of benzothiazoles with N, N-dimethylamides catalyzed by the two-component system under visible light, Tetrahedron Lett., 2019, 60, 390 CrossRef CAS.
- M. N. Chen, J. Q. Di, J. M. Li, L. P. Mo and Z. H. Zhang, Eosin Y-catalyzed one-pot synthesis of spiro [4H-pyran-oxindole] under visible light irradiation, Tetrahedron, 2020, 76, 131059 CrossRef CAS.
- Y. Nie, Z. Wang, Z. Feng, B. Dong, Y. Bai, Y. Leng and J. Wu, Na2 Eosin Y Catalyzed Alkylation of Enol Acetates by Radical Decarboxylation of N–Hydroxyphthalimide Esters, Asian J. Org. Chem., 2021, 10, 1675 CrossRef CAS.
- H. Ni, Y. Li, X. Shi, Y. Pang, C. Jin and F. Zhao, Eosin Y as a direct hydrogen-atom transfer photocatalyst for the C3-H acylation of quinoxalin-2 (1H)-ones, Tetrahedron Lett., 2021, 68, 152915 CrossRef CAS.
- F. Medici, S. Resta, P. Presenti, L. Caruso, A. Puglisi, L. Raimondi and M. Benaglia, Stereoselective Visible–Light Catalyzed Cyclization of Bis (enones): A Viable Approach to the Synthesis of Enantiomerically Enriched Cyclopentane Rings, Eur. J. Org. Chem., 2021, 4521 CrossRef CAS.
- D. P. Tiwari, S. Dabral, J. Wen, J. Wiesenthal, S. Terhorst and C. Bolm, Organic Dye-Catalyzed Atom Transfer Radical Addition–Elimination (ATRE) Reaction for the Synthesis of Perfluoroalkylated Alkenes, Org. Lett., 2017, 19, 4295 CrossRef CAS.
- M. D. Zhou, Z. Peng, L. Li and H. Wang, Visible-light-promoted organic dye catalyzed perfluoroalkylation of hydrazones under mild conditions, Tetrahedron Lett., 2019, 60, 151124 CrossRef CAS.
- A. K. Yadav, A. K. Sharma and K. N. Singh, Visible light enabled γ-trifluoromethylation of Baylis–Hillman acetates: stereoselective synthesis of trisubstituted alkenes, Org. Chem. Front., 2019, 6, 989 RSC.
- Z. Yang and A. Tang, Synthesis of perfluoroalkyl-substituted oxindoles through organophotoredox-catalyzed perfluoroalkylation of N-arylacrylamides with perfluoroalkyl iodides, Synlett, 2019, 1061 CrossRef CAS.
- Y. Quan, W. Shi, Y. Song, X. Jiang, C. Wang and W. Lin, Bifunctional metal–organic layer with organic dyes and iron centers for synergistic photoredox catalysis, J. Am. Chem. Soc., 2021, 143, 3075 CrossRef CAS.
- H. Liu, F. Xue, M. Wang, X. Tang and J. Wu, Neutral-Eosin Y-Catalyzed Regioselective Hydroacylation of Aryl Alkenes under Visible-Light Irradiation, Synlett, 2021, 406 CAS.
- V. R. Rao, K. T. Patil, D. Kumar, S. Sebastian, M. K. Gupta and D. S. Shin, Facile metal-free visible-light-mediated chlorotrifluoromethylation of terminal alkenes, Monatsh. Chem., 2022, 153, 495 CrossRef CAS.
- X. Z. Fan, J. W. Rong, H. L. Wu, Q. Zhou, H. P. Deng, J. D. Tan and J. Wu, Eosin Y as a direct hydrogen–atom transfer photocatalyst for the functionalization of C–H bonds, Angew. Chem., Int. Ed., 2018, 57, 8514 CrossRef CAS PubMed.
- D. M. Yan, J. R. Chen and W. J. Xiao, New roles for photoexcited eosin Y in photochemical reactions, Angew. Chem., Int. Ed., 2019, 58, 378 CrossRef CAS PubMed.
- H. Cao, D. Kong, L. C. Yang, S. Chanmungkalakul, T. Liu, J. L. Piper and J. Wu, Brønsted acid-enhanced direct hydrogen atom transfer photocatalysis for selective functionalization of unactivated C (sp3)–H bonds, Nat. Synth., 2022, 1, 794 CrossRef.
- V. Ramesh, M. Gangadhar, J. B. Nanubolu and P. R. Adiyala, Visible-light-induced deaminative alkylation/cyclization of alkyl amines with N-methacryloyl-2-phenylbenzoimidazoles in continuous-flow organo-photocatalysis, J. Org. Chem., 2021, 86, 12908 CrossRef CAS PubMed.
- X. Li, M. Y. Han, B. Wang, L. Wang and M. Wang, Visible-light-induced deboronative alkylarylation of acrylamides with organoboronic acids, Org. Biomol. Chem., 2019, 17, 6612 RSC.
- Y. Hussain, D. Sharma, N. Kotwal, I. Kumar and P. Chauhan, Stereoselective Oxidative Mannich Reaction of Ketones with Dihydrodibenzo–Oxazepines via a Merger of Photoredox–/Electro–Catalysis with Organocatalysis, ChemSusChem, 2022, e202200415 CAS.
- W. L. Lei, K. W. Feng, T. Wang, L. Z. Wu and Q. Liu, Eosin Y-and copper-catalyzed dark reaction to construct
ene-γ-lactams, Org. Lett., 2018, 20, 7220 CrossRef CAS.
- K. Wang, L. G. Meng and L. Wang, Visible-light-initiated Na2-Eosin Y catalyzed highly regio-and stereoselective difunctionalization of alkynes with alkyl bromides, J. Org. Chem., 2016, 81, 7080 CrossRef CAS PubMed.
- D. R. Heitz, K. Rizwan and G. A. Molander, Visible-light-mediated alkenylation, allylation, and cyanation of potassium alkyltrifluoroborates with organic photoredox catalysts, J. Org. Chem., 2016, 81, 7308 CrossRef CAS PubMed.
- N. Petek, U. Grošelj, J. Svete, F. Požgan, D. Kočar and B. Štefane, Eosin Y-catalyzed visible-light-mediated aerobic transformation of pyrazolidine-3-one derivatives, Catalysts, 2020, 10, 981 CrossRef CAS.
- L. Xia, M. Jin, Y. Jiao and S. Yu, Synthesis of C-Alkynyl Glycosides by Photoredox-Catalyzed Reductive Coupling of Alkynyl Bromides with Glycosyl Bromides, Org. Lett., 2021, 24, 364 CrossRef PubMed.
- T. Xiao, X. Dong, Y. Tang and L. Zhou, Phenanthrene synthesis by eosin Y–catalyzed, visible light–induced [4 + 2] benzannulation of biaryldiazonium salts with alkynes, Adv. Synth. Catal., 2012, 354, 3195 CrossRef CAS.
- X. Q. Chu, T. Xie, L. Li, D. Ge, Z. L. Shen and T. P. Loh, Combining Fluoroalkylation and Defluorination to Enable Formal [3 + 2 + 1] Heteroannulation by Using Visible-Light Photoredox Organocatalysis, Org. Lett., 2018, 20, 2749 CrossRef CAS PubMed.
- M. H. Huang, Y. L. Zhu, W. J. Hao, A. F. Wang, D. C. Wang, F. Liu and B. Jiang, Visible–light photocatalytic bicyclization of 1, 7–enynes toward functionalized sulfone–containing benzo [a] fluoren–5–ones, Adv. Synth. Catal., 2017, 359, 2229 CrossRef CAS.
- D. Yang, B. Huang, W. Wei, J. Li, G. Lin, Y. Liu and H. Wang, Visible-light initiated direct oxysulfonylation of alkenes with sulfinic acids leading to β-ketosulfones, Green Chem., 2016, 18, 5630 RSC.
- A. U. Meyer, S. Jaeger, D. Prasad Hari and B. Koenig, Visible Light–Mediated Metal–Free Synthesis of Vinyl Sulfones from Aryl Sulfinates, Adv. Synth. Catal., 2015, 357, 2050 CrossRef CAS.
- A. U. Meyer, K. Straková, T. Slanina and B. König, Eosin Y (EY) photoredox–catalyzed sulfonylation of alkenes: scope and mechanism, Chem. – Eur. J., 2016, 22, 8694 CrossRef CAS PubMed.
- R. S. Rohokale, S. D. Tambe and U. A. Kshirsagar, Eosin Y photoredox catalyzed net redox neutral reaction for regiospecific annulation to 3-sulfonylindoles via anion oxidation of sodium sulfinate salts, Org. Biomol. Chem., 2018, 16, 536 RSC.
- D. Sun, K. Yin and R. Zhang, Visible-light-induced multicomponent cascade cycloaddition involving N-propargyl aromatic amines, diaryliodonium salts and sulfur dioxide: rapid access to 3-arylsulfonylquinolines, Chem. Commun., 2018, 54, 1335 RSC.
- A. M. Nair, S. Kumar, I. Halder and C. M. Volla, Visible-light mediated sulfonylation of thiols via insertion of sulfur dioxide, Org. Biomol. Chem., 2019, 17, 5897 RSC.
- C. Breton-Patient, D. Naud-Martin, F. Mahuteau-Betzer and S. Piguel, Three–Component C–H Bond Sulfonylation of Imidazoheterocycles by Visible–Light Organophotoredox Catalysis, Eur. J. Org. Chem., 2020, 6653 CrossRef CAS.
- O. A. Storozhenko, A. A. Festa, G. I. Detistova, V. B. Rybakov, A. V. Varlamov, E. V. Van der Eycken and L. G. Voskressensky, Photoredox-Catalyzed Hydrosulfonylation of Arylallenes, J. Org. Chem., 2019, 85, 2250 CrossRef PubMed.
- S. Yang, L. Wang, L. Wang and H. Li, Visible-Light Photoredox-Catalyzed Regioselective Sulfonylation of Alkenes Assisted by Oximes via [1, 5]–H Migration, J. Org. Chem., 2019, 85, 564 CrossRef.
- O. M. Mulina, A. I. Ilovaisky, T. Opatz and A. O. Terent'ev, Photoredox-catalyzed synthesis of N-unsubstituted enaminosulfones from vinyl azides and sulfinates, Tetrahedron Lett., 2021, 64, 152737 CrossRef CAS.
- A. K. Sahoo, A. Dahiya, B. Das, A. Behera and B. K. Patel, Visible-Light-Mediated Difunctionalization of Alkynes: Synthesis of β-Substituted Vinylsulfones Using O-and S-Centered Nucleophiles, J. Org. Chem., 2021, 86, 11968 CrossRef CAS PubMed.
- A. K. Yadav and L. D. S. Yadav, Eosin Y catalyzed difunctionalization of styrenes using O 2 and CS 2: a direct access to 1, 3-oxathiolane-2-thiones, Green Chem., 2016, 18, 4240 RSC.
- A. K. Yadav and L. D. S. Yadav, Visible-light-induced direct α-C (sp3)–H thiocyanation of tertiary amines, Tetrahedron Lett., 2015, 56, 6696 CrossRef CAS.
- M. Singh, A. K. Yadav, L. D. S. Yadav and R. K. P. Singh, Synthesis of 6-thiocyanatophenanthridines by visible-light-and air-promoted radical thiocyanation of 2-isocyanobiphenyls, Synlett, 2018, 176 CAS.
- R. Kapoor, R. Chawla and L. D. S. Yadav, Denitrative thiocyanation of β-nitrostyrenes through visible light photoredox catalysis: An easy access to (E)-vinyl thiocyanates, Tetrahedron Lett., 2020, 61, 152505 CrossRef CAS.
- S. S. Zalesskiy, N. S. Shlapakov and V. P. Ananikov, Visible light mediated metal-free thiol–yne click reaction, Chem. Sci., 2016, 7, 6740 RSC.
- S. D. Tambe, M. S. Jadhav, R. S. Rohokale and U. A. Kshirsagar, Metal–Free Synthesis of 3–Thiocyanatobenzothiophenes by Eosin Y Photoredox–Catalyzed Cascade Radical Annulation of 2–Alkynylthioanisoles, Eur. J. Org. Chem., 2018, 4867 CrossRef CAS.
- S. D. Tambe, R. S. Rohokale and U. A. Kshirsagar, Visible–Light–Mediated Eosin Y Photoredox–Catalyzed Vicinal Thioamination of Alkynes: Radical Cascade Annulation Strategy for 2–Substituted–3–sulfenylindoles, Eur. J. Org. Chem., 2018, 2117 CrossRef CAS.
- B. Hong, J. Lee and A. Lee, Visible-light-promoted synthesis of diaryl sulfides under air, Tetrahedron Lett., 2017, 58, 2809 CrossRef CAS.
- P. Natarajan, Manjeet, Muskan, N. K. Brar and J. J. Kaur, Visible light photoredox catalysis: conversion of a mixture of thiophenols and nitriles into 2-substituted benzothiazoles via consecutive C–S and C–N bond formation reactions, Org. Chem. Front., 2018, 5, 1527 RSC.
- P. Natarajan and N. Kumar, Eosin Y–Catalyzed Visible–Light–Mediated Synthesis of 2–Mercaptobenzothiazoles from 2–Azidoarenediazonium Tetrafluoroborates and Carbon Disulfide, ChemistrySelect, 2020, 5, 4862 CrossRef CAS.
- C. Chen, Q. Xiong and J. Wei, Synthesis of 2-sulfenylindenones by visible-light-mediated addition of sulfur-centered radicals to 1, 3-diarylpropynones, Synth. Commun., 2019, 49, 869 CrossRef CAS.
- S. Chand, A. K. Pandey, R. Singh, S. Kumar and K. N. Singh, Eosin–Y–Catalyzed Photoredox C–S Bond Formation: Easy Access to Thioethers, Chem. – Asian J., 2019, 14, 4712 CrossRef CAS PubMed.
- A. M. Nair, S. Kumar and C. M. Volla, Visible Light Mediated Sulfenylation–Annulation Cascade of Alkyne Tethered Cyclohexadienones, Adv. Synth. Catal., 2019, 361, 4983 CrossRef CAS.
- Y. C. Liu, D. M. Reddy, X. A. Chen, Y. C. Shieh and C. F. Lee, Blue LED–Promoted Oxathiacetalization of Aldehydes and Ketones, Eur. J. Org. Chem., 2020, 2542 CrossRef CAS.
- Z. Peng, H. Yin, H. Zhang and T. Jia, Regio-and Stereoselective Photoredox-Catalyzed Atom Transfer Radical Addition of Thiosulfonates to Aryl Alkynes, Org. Lett., 2020, 22, 5885 CrossRef CAS PubMed.
- V. J. Roy, P. P. Sen and S. Raha Roy, Visible-Light-Mediated Cross Dehydrogenative Coupling of Thiols with Aldehydes: Metal-Free Synthesis of Thioesters at Room Temperature, J. Org. Chem., 2021, 86, 16965 CrossRef CAS PubMed.
- V. V. Khade, A. S. Thube, P. D. Dharpure and R. G. Bhat, Direct synthesis of 1, 3-dithiolanes from terminal alkynes via visible light photoredox catalysis, Org. Biomol. Chem., 2022, 20, 1315 RSC.
- W. Z. Bi, W. J. Zhang, C. Y. Li, L. H. Shao, Q. P. Liu, S. X. Feng and L. B. Qu, Photoexcited sulfenylation of C (sp3)–H bonds in amides using thiosulfonates, Org. Biomol. Chem., 2022, 20, 3902 RSC.
- A. K. Sahoo, A. Rakshit, A. Dahiya, A. Pan and B. K. Patel, Visible-Light-Mediated Synthesis of Thio-Functionalized Pyrroles, Org. Lett., 2022, 24, 1918 CrossRef CAS PubMed.
- B. Nagar and B. B. Dhar, Visible Light-Mediated Thiolation of Substituted 1, 4-Naphthoquinones Using Eosin Y as a Photoredox Catalyst, J. Org. Chem., 2022, 87, 3195 CrossRef PubMed.
- N. S. Prabhakar, S. Kumar, P. K. Gupta and K. N. Singh, Visible-Light-Induced Photocatalytic Trifluoromethylation of Bunte Salts: Easy Access to Trifluoromethylthiolated Synthons, J. Org. Chem., 2022, 87, 11112 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Kamaldeep
Paul
and
Kamaldeep
Paul