
A modular and divergent approach for the total synthesis of Elaeocarpus alkaloids†
Guang
Tian
 ab,
Yi-Chi
Zhang
b,
Chuanguang
Qin
*a and
Jie
Wang
*bc
ab,
Yi-Chi
Zhang
b,
Chuanguang
Qin
*a and
Jie
Wang
*bc
aDepartment of Chemistry, Shanxi Key Laboratory of Polymer Science & Technology, MOE Key Laboratory of Supernomal Material Physics & Chemistry, School of Chemical & Chemical Engineering, Northwestern Polytechnical University, Xi'an 710129, China. E-mail: qinchg@nwpu.edu.cn
bDepartment of Medicinal Chemistry, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: jiewang@simm.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 8th November 2022
Abstract
Herein we report a unified strategy for the divergent total synthesis of six Elaeocarpus alkaloids in 6–10 steps from commercially available materials. This modular approach relied on the rapid construction of the tetrahydrobenzopyran-4-one framework through an aldol/dehydration/oxa-Michael process to set all carbons and functional groups ready for the core construction. A key NbCl5-mediated intramolecular Mannich reaction was used to build the C ring and secure both stereoisomers existing in these natural products. Finally, a diversification was achieved through the intermediacy of thioamide, with the side chain of elaeocarfoline A/B installed by an Eschenmoser sulfide contraction/reduction sequence.
Introduction
Plants of the Elaeocarpus genus produce a rich array of novel indolizidine alkaloids along with some pyrrolidine and phenethylamine-containing natural products.1 To date, more than 40 Elaeocarpus alkaloids have been isolated,2 amongst which the most representative structure is characterized to be a 6/6/6/5 tetracyclic ring system with a phenyl (A ring, dearomatized in some cases) and an indolizidine (C/D ring) fused by a tetrahydropyran-4-one (B ring) framework, as found in elaeocarpine (1), isoelaeocarpine (2),3 oxoelaeocarpine (3),2 oxoisoelaeocarpine (4),4 and many others (Fig. 1).5Elaeocarpus ganitrus Roxb., the stony endocarp of which is known as Rudraksha, is traditionally used to treat cough, bronchitis, migraine, mental disorders, and many other diseases in Hindu countries.1a Pharmacological activities were widely studied on Elaeocarpus herbal extracts but few were directly on isolated chemical ingredients.6 In a high-throughput screening drug discovery program led by Carroll et al., Elaeocarpus alkaloids including elaeocarpine (1), isoelaeocarpine (2), grandisine A (5) and several others were found to bind to the human δ-opioid receptor (DOR) with certain selectivity, which is of great interest in analgesic drug discovery with minimized side effects.7 Recently, two new alkaloids elaeocarfoline A (7) and B (8) were isolated by Wang and co-workers from Elaeocarpus angustifolius, which represented the hitherto heaviest members of this class.2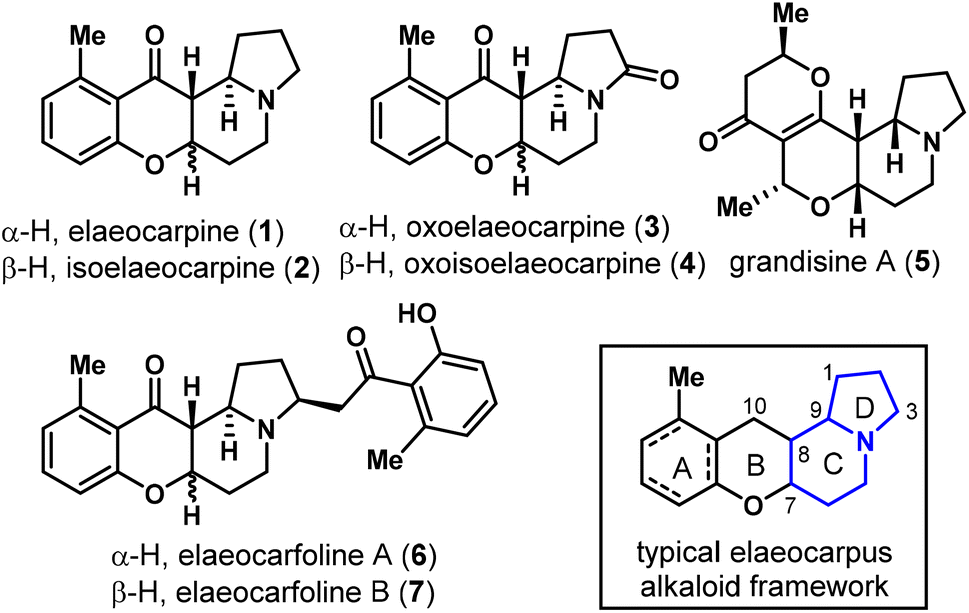 | ||
| Fig. 1 Representative structures of the Elaeocarpus alkaloids. | ||
Due to their structural diversity and unique biological activities, Elaeocarpus alkaloids have attracted considerable attention from the synthetic community. In 1970, Tanaka and co-workers accomplished the total synthesis of elaeocarpine (1) and isoelaeocarpine (2) for the first time utilizing copper catalyzed diazoketone-pyrrol insertion and the Dieckmann condensation as key steps.8 Thereafter, Onaka reported a very short synthesis of 1/2 through reductive condensation of dehydroindolizidine and salicylaldehyde, albeit with low yields.9 Tufariello and co-workers disclosed an elegant synthesis of 1/2 in 1979 employing a nitrone-olefin cycloaddition reaction as the key strategy.10 From 2007, the synthetic interest was revived due to Carroll's discovery of the unique biological activities, leading to the successful total synthesis of grandisines A (5), B, D and F by Danishefsky,11 Tamura,12 Taylor13 and their co-workers. The Tamura group also prepared elaeocarpenine and its analogs to evaluate the opioid receptor binding affinity.14
We became interested in Elaeocarpus alkaloids particularly due to the apparent correlation between the DOR binding potency and the dihedral orientation of B/D rings of these natural products, reflected by their relative stereochemistry in C7/8/9 conjunction (Scheme 1a). With the B ring being directed in an orthogonal orientation to the D ring, compounds 5 (cis–cis in C7/8/9) and 2 (cis–trans) exhibit a higher binding affinity than 1 (2.7 and 13.6 μM vs. 86.4 μM), whose B ring is located almost in the same plane as the D ring (trans–trans). Recognizing that basic nitrogen and extended aromatic substituents are important features for selective DOR binders,15 the ideal scaffolds from this class would be molecules possessing both cis–cis configuration and an aromatic A ring, e.g., 8-epi-1, which is unnatural and not easy to construct according to Danishefsky's studies.11,16 In addition, some newly isolated natural products, such as 3, 4, 6 and 7, have not been synthesized yet. Many of the Elaeocarpus alkaloids are also extremely scarcely available from their natural sources, with 3, 6 and 7 all being isolated in less than 1 ppm from the corresponding dried plants, which hampers the detailed biological evaluations of these natural products. In this regard, we intended to devise a stereodivergent approach to synthesize natural Elaeocarpus alkaloids and create novel analogs in a modular fashion17 and explore their potential as analgesic agents.
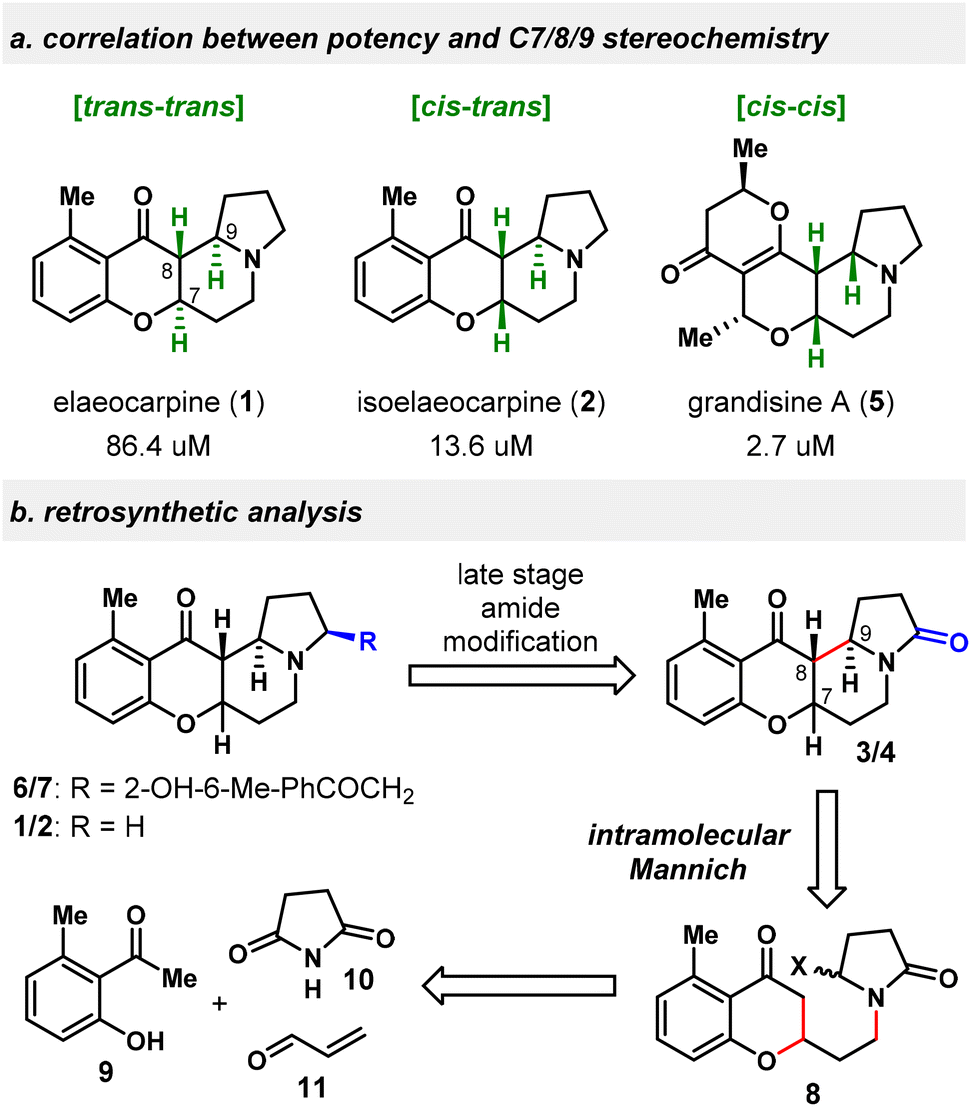 | ||
| Scheme 1 (a) Correlation of the DOR binding affinity with the stereochemistry and (b) retrosynthetic analysis. | ||
Our retrosynthetic analysis is shown in Scheme 1b. The C7/8/9 relative stereochemistry is the same for 1, 3 and 6 as trans/trans, while in 2, 4 and 7 it is cis–trans. Therefore, 3/4 were designated as the gateway compounds in our synthetic plan, which would connect to 1/6 and 2/7 by late-stage selective modification of the amide carbonyl group. Considering that both C7/8/9 trans/trans and cis–trans stereochemistry are desired for the target natural products and that the cis–cis isomer is also of interest for biological evaluations, the C ring was strategically disconnected at C8–C9 by an intramolecular Mannich reaction18 through the intermediacy of an N-acyliminium ion. The precursor 8 could be in turn broken into three commercially available fragments 9, 10, and 11. Although not necessarily representing the biosynthetic pathway,5c,7a,9 the current retrosynthesis was inspired by the hypothesized biosynthetic intermediates proposed by Onaka.9
Results and discussion
Our synthesis commenced with the connection between acetophenone 9 and aldehyde 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 which was readily prepared in one step from succinimide 10 and acrolein 11 (Scheme 2). Initial attempts employing the Wittig or HWE type reactions, either with the free phenol form or O-methyl ether protected 9, all proved to be fruitless due to the instability of 12. Thus, the direct aldol reaction using 2.0 equivalents of LDA at −78 °C provided the desired product smoothly, which was involved in a dehydration/oxa-Michael process20 (see the ESI† for details) catalysed by p-TsOH at a high temperature and furnished chromanone 13 in 75% yield. Next, the imide group needed to be reduced selectively with the ketone group remaining unaltered. Under conventional conditions (NaBH4, LiBH4, hydrosilanes,21 and LiEt3BH22), only a global reduction or selective ketone reduction was observed. We then explored the Stoltz's three-step sequence for the intramolecular Mannich cyclization through an N-acyliminium ion.23 However, no cyclized product was observed, with the only product isolated being the hemiaminal 14a. Normally, a simple unactivated ketone is a poor nucleophile in the N-acyliminium ion chemistry and needs to be pre-activated to the corresponding enol ether or 1,3-dicarbonyl to achieve high efficiency.24 In 2010, Duñach and co-workers discovered an extremely active Lewis acid, namely Sn(NTf2)4, to efficiently catalyse the substitution of unprotected hemiaminals with various nucleophiles25 and the system could be easily applied to simple unactivated ketone nucleophiles.26 Encouraged by these results, we set out to try the cyclization reaction directly on 14a.
19 which was readily prepared in one step from succinimide 10 and acrolein 11 (Scheme 2). Initial attempts employing the Wittig or HWE type reactions, either with the free phenol form or O-methyl ether protected 9, all proved to be fruitless due to the instability of 12. Thus, the direct aldol reaction using 2.0 equivalents of LDA at −78 °C provided the desired product smoothly, which was involved in a dehydration/oxa-Michael process20 (see the ESI† for details) catalysed by p-TsOH at a high temperature and furnished chromanone 13 in 75% yield. Next, the imide group needed to be reduced selectively with the ketone group remaining unaltered. Under conventional conditions (NaBH4, LiBH4, hydrosilanes,21 and LiEt3BH22), only a global reduction or selective ketone reduction was observed. We then explored the Stoltz's three-step sequence for the intramolecular Mannich cyclization through an N-acyliminium ion.23 However, no cyclized product was observed, with the only product isolated being the hemiaminal 14a. Normally, a simple unactivated ketone is a poor nucleophile in the N-acyliminium ion chemistry and needs to be pre-activated to the corresponding enol ether or 1,3-dicarbonyl to achieve high efficiency.24 In 2010, Duñach and co-workers discovered an extremely active Lewis acid, namely Sn(NTf2)4, to efficiently catalyse the substitution of unprotected hemiaminals with various nucleophiles25 and the system could be easily applied to simple unactivated ketone nucleophiles.26 Encouraged by these results, we set out to try the cyclization reaction directly on 14a.
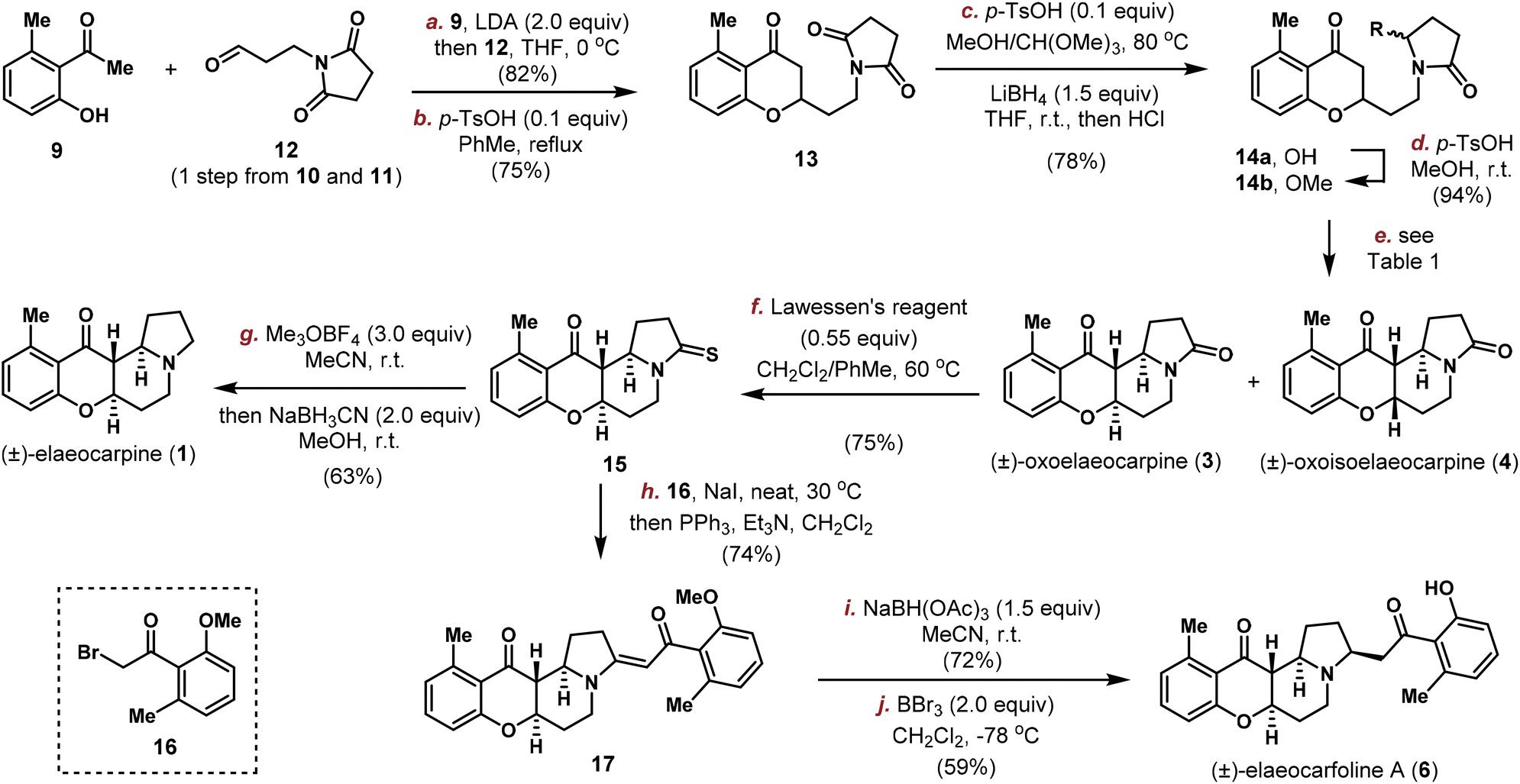 | ||
| Scheme 2 Total synthesis of elaeocarpine (1), oxoelaeocarpine (3), oxoisoelaeocarpine (4) and elaeocarfoline A (6). | ||
First, a modified procedure (methyl enol ether was obtained with catalytic p-TsOH in MeOH/CH(OMe)3, the imide group was reduced with LiBH4 in THF and then worked up with HCl) was carried out in one-pot to produce 14a in 78% overall yield on gram-scale. Unfortunately, with 1 mol% of Sn(NTf2)4 as the catalyst, no cyclization product was observed on our substrate 14a (entry 1, Table 1). We then tested several other Lewis and Brønsted acids in different solvents. While Sc(OTf)3, NbCl5 and HNTf2 in CH3CN proved to be unsuccessful (entries 2–4), catalytic p-TsOH in MeOH under refluxing conditions furnished the cyclized product in 55% yield (entry 5). After careful characterization, the product was determined to be a 1:1.2 mixture of the natural product (±)-oxoisoelaeocarpine (4) and its cis–cis isomer 18 whose structure was confirmed unambiguously by X-ray crystallographic analysis. The observed solvent preference inspired us to assume that the hemiaminal O-methyl ether might be formed in situ and it is important for the cyclization reaction to take place, with the diastereoselectivity possibly being tuned by the solvent and catalyst to obtain the trans/trans isomer required in other natural products. To this end, hemiaminal 14a was converted into its corresponding O-methyl ether 14b in 94% yield with p-TsOH in In(OTf)3 and no or a trace amount of product was obtained (entries 6–10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Lewis or Brønsted acid | Solvent | Temperature | Yield (3/4/18, %)b |
| a Reaction conditions: a solution of 14a or 14b (0.1 mmol) and catalyst in the indicated solvent (1.0 mL) was stirred for 24 hours. b Yield determined by crude 1H NMR using 1,1,2,2-tetrachloroethane as the internal standard. c Isolated yield. ND = not detected. | |||||
| 1 | 14a | Sn(NTf2)4 (1 mol%) | CH3CN | 60 °C | ND |
| 2 | 14a | Sc(OTf)3 (1 mol%) | CH3CN | 60 °C | ND |
| 3 | 14a | NbCl5 (60 mol%) | CH2Cl2 | r.t. | ND |
| 4 | 14a | HNTf2 (5 mol%) | CH3CN | 60 °C | ND |
| 5 | 14a | TsOH (10 mol%) | MeOH | Reflux | 0/25(23c)/30(30c) |
| 6 | 14b | I2 (10 mol%) | CCl4, or MeOH | r.t. | ND |
| 7 | 14b | Cu(OTf)2 (10 or 60 mol%) | CH2Cl2 | r.t. | <5% |
| 8 | 14b | In(OTf)3 (10 or 60 mol%) | CH2Cl2 | r.t. | <5% |
| 9 | 14b | FeCl3 (10 or 60 mol%) | CH2Cl2 | r.t. | ND |
| 10 | 14b | Sc(OTf)3 (10 or 60 mol%) | CH2Cl2 | r.t. | <5% |
| 11 | 14b | NbCl5 (60 mol%) | MeOH | 60 °C | 0/65/26 |
| 12 | 14b | NbCl5 (60 mol%) | DMF or DMSO or CH3CN | 60 °C | ND |
| 13 | 14b | NbCl5 (60 mol%) | Acetone | 60 °C | 29/28/27 |
| 14 | 14b | NbCl5 (60 mol%) | CH2Cl2 | r.t. | 41(39c)/36(36c)/0 |

|
|||||
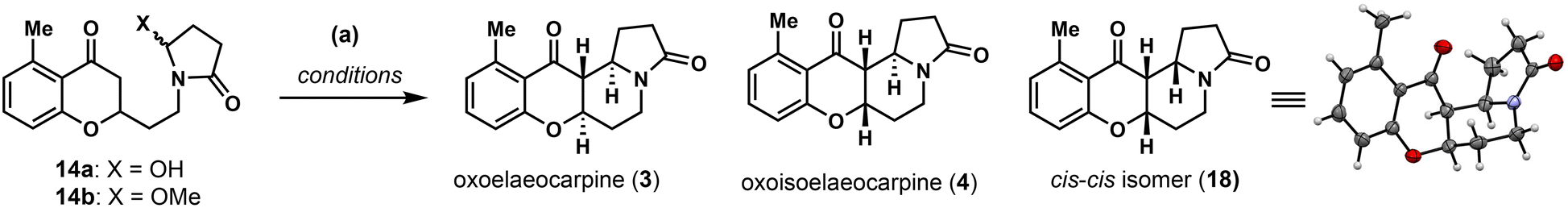
NbCl5 turned out to be an efficient mediator, a substoichiometric amount (0.6 equivalent) of which in MeOH produced 4 and 18 in a 2.5:1 ratio and 91% yield (entry 11).27 The diastereoselectivity was found to alter in different solvents. In acetone, the natural product (±)-oxoelaeocarpine (3) could be produced along with 4 and 18 in a 1:1:1 ratio (entry 13). Finally, the cis–cis isomer could be completely avoided by switching the solvent to CH2Cl2 (entry 14) and the desired natural products 3 and 4 were obtained in 39% and 36% isolated yield respectively (separable by flash column chromatography).
Throughout the conditions of screening, three stereoisomers out of four were observed including the cis–cis isomer 18 with a rather congested conformation, but not the trans–cis isomer 19. This prompted our interest to perform a conformational analysis of the reaction transition states (TS) (Table 1b). In both trans–trans (TS1) and cis–trans (TS2) configuration that exist in natural sources, the side chain on the pyran ring occupies an equatorial position and subsequently undergoes a chair TS to form cyclized products. In the cis–cis (TS3) scenario, with the side chain switching from the equatorial to the axial position, a chair TS can also be obtained. However, in order to form the trans–cis isomer, the side chain has to occupy an equatorial position and form the subsequent boat TS (TS4), which is high in energy and kinetically unfavored. The remarkable solvent effects for the observed 3/4/18 selectivity could also be explained with these TSs by dipole interaction. Since the axial conformation might exhibit a higher dipole moment than the equatorial,28 the lowest dipole moment was generated in TS1 (starting from the e form and all three substituents being in the e,e,e form in the newly formed C ring), whereas the highest of which was generated in TS3 (starting from the a form and all the substituents being in the e,a,e form) and TS2 behaved somewhat in between (starting from the e form and all the substituents being in the a,e,e form). As the transition state with a higher dipole is expected to be more favoured in the solvent with a larger dielectric constant,28TS1 was the least favourable in MeOH and TS3 the least favourable in CH2Cl2, which agreed well with our results that 3 was not observed in MeOH, 18 was not observed in CH2Cl2, and that all the three isomers (3, 4, and 18) were obtained in the acetone solvent with a medium dielectric constant.
The Nb-mediated intramolecular Mannich reaction could be implemented on a gram-scale to produce 3 and 4 in quantities with unaltered efficiency, and the stage is now setting for the divergent synthesis of other Elaeocarpus alkaloids. To achieve this goal, oxoelaeocarpine (3) was first converted into the corresponding thioamide 15 in 75% yield using Lawessen's reagent (Scheme 2). After activation with Meerwein's salt and reduction using NaBH3CN, (±)-elaeocarpine (1) was synthesized successfully in 63% yield. To attach the side chain to C3 as required in elaeocarfoline A (6), metal-catalyzed diazocarbonyl-thioamide coupling was initially attempted. While copper29 did not provide any useful product, ruthenium catalysis30 produced the desired enaminone 17 successfully. However, the yield was very low (<20%) and could not be improved after an extensive optimization, suffering from a low conversion rate due to steric hindrance from both coupling partners. This problem was solved employing the canonical Eschenmoser sulfide contraction reaction by modifying Michael31 and Russowsky's32 protocols. By mixing 15, bromide 16 and NaI in a minimum amount of CH2Cl2 and then evaporating off CH2Cl2, the mixture was rendered in a neat form and stirred for two days to achieve a high conversion. Followed by treatment with PPh3 and Et3N in CH2Cl2, enaminone 17 was obtained as a single isomer in 74% yield. The double bond geometry was tentatively assigned to be E, based on Russowsky's studies.32 With 17 in hand, reduction of the enaminone was achieved diastereoselectively from the convex face using NaBH(OAc)3 in CH3CN providing a single isomer in 72% yield, the stereochemical outcome of which was confirmed by demethylation with BBr3 to furnish (±)-elaeocarfoline A (6) in 59% yield.
Similarly, as shown in Scheme 3, oxoisoelaeocarpine (4) was converted into the corresponding thioamide 20 and then activated and reduced under Me3OBF4/NaBH3CN conditions to furnish the natural product (±)-isoelaeocarpine (2). A three-step sequence (sulfide contraction, reduction and demethylation) was also applied on 20 to produce (±)-elaeocarfoline B (7) uneventfully in 29% overall yield. The spectroscopic data for synthetic 1, 2, 3, 4, 6 and 7 are all in good agreement with those reported from the isolation literature.
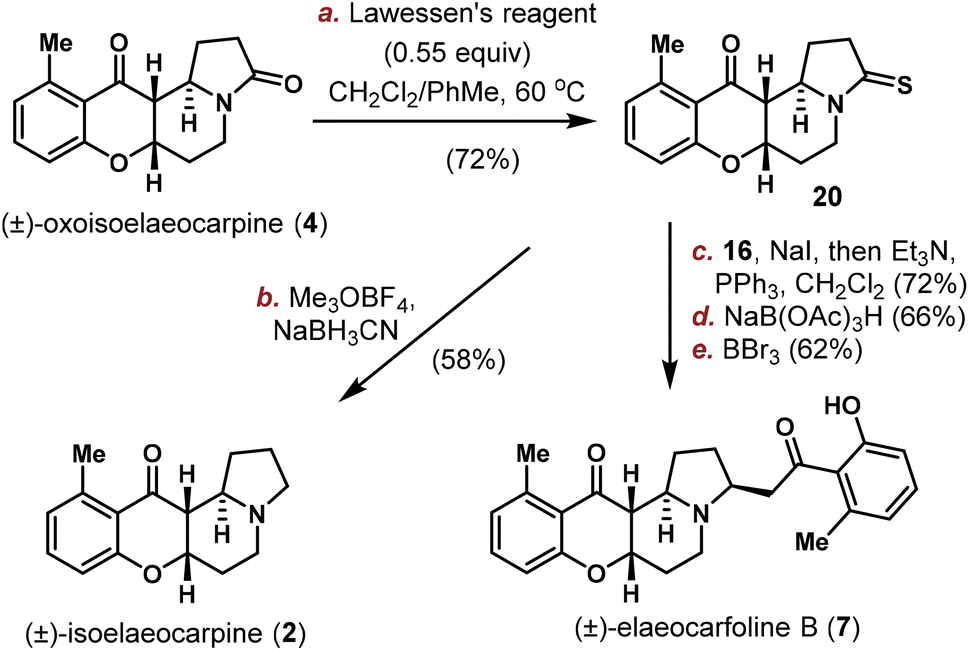 | ||
| Scheme 3 Total synthesis of isoelaeocarpine (2) and elaeocarfoline B (7). | ||
Conclusion
In summary, a unified strategy was developed for the divergent total synthesis of six Elaeocarpus alkaloids in 6–10 steps from commercially available materials 9, 10 and 11 in a modular manner, amongst which 3, 4, 6 and 7 have been synthesized for the first time. This route features (i) the rapid construction of the tetrahydrobenzopyran-4-one framework through an aldol/dehydration/oxa-Michael sequence, (ii) a NbCl5-mediated intramolecular Mannich reaction with a simple ketone and an N-acyliminium ion to form the C ring and secure all the stereoisomers for the synthesis of both natural products and unnatural analogs, and (iii) diversification through the intermediacy of thioamide, enabling connection of the six natural products with different oxidation states and side chains. Efforts to use this approach for analog synthesis and DOR binding activity studies is ongoing and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (Grant No. 22101290 and 21572180), the Intergovernmental Science and Technology Cooperation and Exchange Program between P. R. China and Romania (Grant No. 43-24-20180510), the Chinese Academy of Sciences and Shanghai Institute of Materia Medica.References
- (a) B. Singh, M. P. S. Ishar, A. Sharma, R. Arora and S. Arora, Phytochemical and biological aspects of Rudraksha, the stony endocarp of Elaeocarpus ganitrus (Roxb.): a review, Isr. J. Plant Sci., 2015, 62, 265–276 Search PubMed; (b) M. C. Ezeoke, P. Krishnan, D. S. Sim, S. H. Lim, Y. Y. Low, K. W. Chong and K. H. Lim, Unusual phenethylamine-containing alkaloids from Elaeocarpus tectorius, Phytochemistry, 2018, 146, 75–81 CrossRef CAS PubMed.
- W. Hong, Y. Zhang, J. Yang, M. Y. Xia, J. F. Luo, X. N. Li, Y. H. Wang and J. S. Wang, Alkaloids from the Branches and Leaves of Elaeocarpus angustifolius, J. Nat. Prod., 2019, 82, 3221–3226 CrossRef CAS.
- S. R. Johns, J. A. Lamberton, A. A. Sioumis and J. A. Wunderlich, Alkaloids of a new type from Elaeocarpus polydactylus Schl. (family elaeocarpaceae), Chem. Commun., 1968, 290–291 RSC.
- C.-X. Zhou, X.-Y. Wang, J.-X. Mo, J. Zhang and L.-S. Gan, Optical Resolution and Structure Determination of New Indolizidine Alkaloids from Elaeocarpus sphaericus, Helv. Chim. Acta, 2011, 94, 347–354 CrossRef CAS.
- (a) S. R. Johns, J. A. Lamberton and A. A. Sioumis, Three new indolizidine alkaloids related to elaeocarpine and isoelaeocarpine, Chem. Commun., 1968, 1324–1325 RSC; (b) S. R. Johns, J. A. Lamberton, A. A. Sioumis and R. I. Willing, Elaeocarpus alkaloids. I. The structures of (±)-elaeocarpine, (±)-isoelaeocarpine, and (±)-isoelaeocarpicine, three new indolizidine alkaloids from Elaeocarpus polydactylus, Aust. J. Chem., 1969, 22, 775–792 CrossRef CAS; (c) S. R. Johns, J. A. Lamberton and A. A. Sioumis, Elaeocarpus alkaloids. II. (+)-Elaeocarpiline and (-)-isoelaeocarpiline, new indolizidine alkaloids from Elaeocarpus dolichostylis, Aust. J. Chem., 1969, 22, 793–800 CrossRef CAS; (d) S. R. Johns, J. A. Lamberton, A. A. Sioumis, H. Suares and R. I. Willing, The structures and absolute configurations of seven alkaloids from Elaeocarpus sphaericus, J. Chem. Soc. D, 1970, 804–805 RSC; (e) S. R. Johns, J. A. Lamberton, A. A. Sioumis, H. Suares and R. I. Willing, Elaeocarpus alkaloids. IV. The alkaloids of Elaeocarpus sphaericus, Aust. J. Chem., 1971, 24, 1679–1694 CrossRef CAS; (f) A. B. Ray, L. Chand and V. B. Pandey, Rudrakine, a new alkaloid from Elaeocarpus ganitrus, Phytochemistry, 1979, 18, 700–701 CrossRef CAS.
- P. Prasannan, Y. Jeyaram, A. Pandian, R. Raju and S. Sekar, A Review on Taxonomy, Phytochemistry, Pharmacology, Threats and Conservation of Elaeocarpus L. (Elaeocarpaceae), Bot. Rev., 2020, 86, 298–328 CrossRef.
- (a) A. R. Carroll, G. Arumugan, R. J. Quinn, J. Redburn, G. Guymer and P. Grimshaw, Grandisine A and B, novel indolizidine alkaloids with human delta-opioid receptor binding affinity from the leaves of the Australian rainforest tree Elaeocarpus grandis, J. Org. Chem., 2005, 70, 1889–1892 CrossRef CAS PubMed; (b) P. L. Katavic, D. A. Venables, P. I. Forster, G. Guymer and A. R. Carroll, Grandisines C-G, indolizidine alkaloids from the Australian rainforest tree Elaeocarpus grandis, J. Nat. Prod., 2006, 69, 1295–1299 CrossRef CAS PubMed; (c) P. L. Katavic, D. A. Venables, T. Rali and A. R. Carroll, Indolizidine alkaloids with delta-opioid receptor binding affinity from the leaves of Elaeocarpus fuscoides, J. Nat. Prod., 2007, 70, 872–875 CrossRef CAS PubMed; (d) P. L. Katavic, D. A. Venables, T. Rali and A. R. Carroll, Habbemines A and B, pyrrolidine alkaloids with human delta-opioid receptor binding affinity from the leaves of Elaeocarpus habbemensis, J. Nat. Prod., 2007, 70, 866–868 CrossRef CAS; (e) P. L. Katavic, D. A. Venables, G. P. Guymer, P. I. Forster and A. R. Carroll, Alkaloids with human delta-opioid receptor binding affinity from the Australian rainforest tree Peripentadenia mearsii, J. Nat. Prod., 2007, 70, 1946–1950 CrossRef CAS.
- T. Tanaka and I. Iijima, Total synthesis of dl-elaeocarpine and dl-isoelaeocarpine, Tetrahedron Lett., 1970, 11, 3963–3966 CrossRef.
- T. Onaka, Two-step synthesis of -elaeocarpine, utility of dihydropyridines as a versatile synthetic intermediate, Tetrahedron Lett., 1971, 12, 4395–4398 CrossRef.
- J. J. Tufariello and S. A. Ali, Elaeocarpus Alkaloids - Synthesis Using Nitrones, J. Am. Chem. Soc., 1979, 101, 7114–7116 CrossRef CAS.
- D. J. Maloney and S. J. Danishefsky, Conformational locking through allylic strain as a device for stereocontrol–total synthesis of grandisine A, Angew. Chem., Int. Ed., 2007, 46, 7789–7792 CrossRef CAS.
- (a) H. Kurasaki, I. Okamoto, N. Morita and O. Tamura, A flexible approach to grandisine alkaloids: total synthesis of grandisines B, D, and F, Chem. – Eur. J., 2009, 15, 12754–12763 CrossRef CAS; (b) H. Kurasaki, I. Okamoto, N. Morita and O. Tamura, Total synthesis of grandisine D, Org. Lett., 2009, 11, 1179–1181 CrossRef CAS PubMed.
- J. D. Cuthbertson, A. A. Godfrey and R. J. Taylor, The preparation of (-)-grandisine B from (+)-grandisine D; a biomimetic total synthesis or formation of an isolation artefact?, Org. Lett., 2011, 13, 3976–3979 CrossRef CAS.
- H. Kurasaki, I. Okamoto, N. Morita and O. Tamura, Synthesis and evaluation of opioid receptor-binding affinity of elaeocarpenine and its analogs, Bioorg. Med. Chem. Lett., 2010, 20, 1601–1603 CrossRef CAS PubMed.
- F. Li, C. Yin, J. Chen, J. Liu, X. Xie and A. Zhang, Synthesis and SAR study of opioid receptor ligands: mono- and bis-indolomorphinans, Chem. Biol. Drug Des., 2009, 74, 335–342 CrossRef CAS.
- S. J. Danishefsky and D. J. Maloney, Studies toward the Total Synthesis of Grandisine A: Synthesis of 9-epi-Grandisine A, Heterocycles, 2007, 72, 167–174 CrossRef PubMed.
- A. W. Sun, S. Lackner and B. M. Stoltz, Modularity: Adding New Dimensions to Total Synthesis, Trends Chem., 2019, 1, 630–643 CrossRef CAS.
- Y. Shi, Q. Wang and S. Gao, Recent advances in the intramolecular Mannich reaction in natural products total synthesis, Org. Chem. Front., 2018, 5, 1049–1066 RSC.
- D. F. Taber, R. S. Hoerrner and M. D. Hagen, A practical preparation of the indolizidine nucleus: synthesis of (.+-.)-elaeokanine A, J. Org. Chem., 1991, 56, 1287–1289 CrossRef CAS.
- D. Iguchi, R. Erra-Balsells and S. M. Bonesi, Formation of 2,2-dimethylchroman-4-ones during the photoinduced rearrangement of some aryl 3-methyl-2-butenoate esters. A mechanistic insight, Tetrahedron, 2016, 72, 1903–1910 CrossRef CAS.
- G. Ding, C. Li, Y. Shen, B. Lu, Z. Zhang and X. Xie, Potassium Hydroxide-Catalyzed Chemoselective Reduction of Cyclic Imides with Hydrosilanes: Synthesis of ω-Hydroxylactams and Lactams, Adv. Synth. Catal., 2016, 358, 1241–1250 CrossRef CAS.
- G. Lemiere, S. Sedehizadeh, J. Toueg, N. Fleary-Roberts and J. Clayden, A general synthetic approach to the amnesic shellfish toxins: total synthesis of (-)-isodomoic acid B, (-)-isodomoic acid E and (-)-isodomoic acid F, Chem. Commun., 2011, 47, 3745–3747 RSC.
- T. J. Fulton, A. Y. Chen, M. D. Bartberger and B. M. Stoltz, Enantioselective total synthesis of (-)-myrifabral A and B, Chem. Sci., 2020, 11, 10802–10806 RSC.
- B. E. Maryanoff, H. C. Zhang, J. H. Cohen, I. J. Turchi and C. A. Maryanoff, Cyclizations of N-acyliminium ions, Chem. Rev., 2004, 104, 1431–1628 CrossRef CAS PubMed.
- R. B. Othman, R. Affani, M.-J. Tranchant, S. Antoniotti, V. Dalla and E. Duñach, N-Acyliminium ion chemistry: highly efficient and versatile carbon-carbon bond formation by nucleophilic substitution of hydroxy groups catalyzed by Sn(NTf2)4, Angew. Chem., Int. Ed., 2010, 49, 776–780 CrossRef PubMed.
- B. Touati, A. El Bouakher, M. S. Azizi, C. Taillier, R. B. Othman, M. Trabelsi-Ayadi, S. Antoniotti, E. Duñach and V. Dalla, Atom-Economic Catalytic Direct Substitution of N,O-Acetals with Simple Ketones, Eur. J. Org. Chem., 2017, 4445–4460 CrossRef CAS.
- Z. Andrade, C. Kleber and R. A. Matos, Niobium Pentachloride-mediatedNucleophilic Additions to CyclicN-AcyliminiumIons, Synlett, 2003, 1189–1191 CrossRef.
- E. L. Eliel and O. Hofer, Conformational analysis. XXVII. Solvent effects in conformational equilibriums of heterosubstituted 1,3-dioxanes, J. Am. Chem. Soc., 1973, 95, 8041–8045 CrossRef CAS.
- A. Pal, N. D. Koduri, Z. Wang, E. L. Quiroz, A. Chong, M. Vuong, N. Rajagopal, M. Nguyen, K. P. Roberts and S. R. Hussaini, Copper-catalyzed chemoselective cross-coupling reaction of thioamides and α-diazocarbonyl compounds: Synthesis of enaminones, Tetrahedron Lett., 2017, 58, 586–589 CrossRef CAS.
- N. D. Koduri, H. Scott, B. Hileman, J. D. Cox, M. Coffin, L. Glicksberg and S. R. Hussaini, Ruthenium catalyzed synthesis of enaminones, Org. Lett., 2012, 14, 440–443 CrossRef CAS.
- J. P. Michael, A. S. Parsons and R. Hunter, Synthesis of two pyrrolidine alkaloids, peripentadenine and dinorperipentadenine, Tetrahedron Lett., 1989, 30, 4879–4880 CrossRef CAS.
- B. A. D. Neto, A. A. M. Lapis, A. B. Bernd and D. Russowsky, Studies on the Eschenmoser coupling reaction and insights on its mechanism. Application in the synthesis of Norallosedamine and other alkaloids, Tetrahedron, 2009, 65, 2484–2496 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2169386. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qo01460b |
| This journal is © the Partner Organisations 2023 |