
Recent development of allyl–allyl cross-coupling and its application in natural product synthesis
V.
Ravichandiran
*ab and
Anupam
Jana
 *a
*a
aDepartment of Pharmaceutical Analysis, National Institute of Pharmaceutical Education and Research, Hajipur, Bihar – 844102, India
bNational Institute of Pharmaceutical Education and Research, Kolkata, 168, Maniktala Main Road, Kolkata – 700054, India. E-mail: directorniperkolkata@gmail.com; anupamjana2@gmail.com
First published on 15th October 2022
Abstract
The allyl–allyl cross-coupling reaction is one of the most important cross-coupling reactions as it provides a practical synthetic route for the direct construction of 1,5-dienes, which are abundant in terpenes, and are significant building blocks in chemical synthesis. The transition metal-catalyzed cross-coupling has the power to generate synthetically important 1,5-dienes from an allylic nucleophile and allylic electrophile. Catalysts derived from different metals, including Pd, Ni, Cu, Ir etc. are extensively studied and show excellent regio- and enantioselective control. Moreover, this strategy has been successfully applied in stereoselective total syntheses of a number of complex natural products. Major developments have been observed over the past decade in the allyl–allyl cross-coupling strategy, and the application of a new tool for regio-and enantioselective C–C bond formation to form the 1,5-diene has revolutionized synthetic chemistry. The present Short Review summarises the most significant advancements that have provided access to a wide variety of branched/and linear 1,5 dienes of synthetic and pharmaceutical importance during the past decade. To the best of our knowledge, this is the first review on the allyl–allyl cross-coupling reaction.
 V. Ravichandiran | Dr V. Ravichandiran was born in Tamilnadu, India. He finished his PhD studies in Pharmaceutical Sciences in the year 2006 from TN Dr MGR Medical University, Chennai, India. Currently, he is working as Director at NIPER-Kolkata, India and NIPER-Hajipur. He is a member of the Scientific Advisory Board in the Central Council for Research in Ayurvedic Sciences (CCRAS), Ministry of AYUSH, Govt. of India. He has 27 years of professional and research experience in the field of pharmacy, drug discovery, and natural products. |
 Anupam Jana | Dr Anupam Jana was born in Tarakeswar, India. He received his MSc degree in Organic Chemistry from the University of Calcutta, India. In 2015, He obtained his PhD under the supervision of Professor Subrata Ghosh at the Indian Association for the Cultivation of Science (IACS), Kolkata, working on the total synthesis of natural products. In 2015, He moved to Europe for postdoctoral research at the University of Warsaw, Poland with Professor Karol Grela. He is currently working as a scientist in the National Institute of Pharmaceutical Education and Research (NIPER), Hajipur, India. His research activity is focused on the total synthesis of bioactive natural products, and the synthesis and application of organometallic complexes in organic synthesis. |
1. Introduction
Metal-catalyzed C–C bond formation reactions are among the most important tools in organic synthesis.1 During the last few decades, a large number of reports have been published, and the scope and utility of this transformation has been greatly extended.2–7 The prominence of “the development of the palladium catalyzed cross-coupling reaction in organic synthesis” was recognized by the award of the Nobel Prize for Chemistry jointly to Richard Heck, Akira Suzuki and Ei-ichi Neghishi in 2010.8 Among the different cross-coupling reactions, allylic substitution has attracted major attention due to the versatility and flexibility of the protocol.9–12 This method allows the introduction of alkyl, aryl, vinyl, alkynyl allenyl etc. fragments with high enantioselectivity using organometallic nucleophiles.13–18 Although great efforts have been made for the rapid development of this reaction, allyl–allyl cross-coupling reactions using an allyl metal reagent is a rather under-represented area. The major problem in metal-catalyzed allyl coupling reactions is the tendency of allyl–metal intermediates to undergo β-hydride elimination. Secondly, allyl–allyl coupling suffers from homocoupling and unwanted regioselectivities, where such disadvantages may result in low yields and limited scope. Therefore, the stability of allyl metal reagents and the difficulty of controlling the regioselectivity of the product may have hampered the investigation of an effective catalytic system as well as the reaction conditions to enable this reaction with a high stereoselectivity. Despite these drawbacks, among the frontiers of organic synthesis has been a quest for the development of allyl–allyl coupling between allyl nucleophiles and allyl electrophiles under the influence of transition-metal catalysts as this is a powerful method for providing direct access to chiral 1,5-dienes, which are abundant in naturally occurring terpenes,19–24 and are versatile synthetic intermediates because of the usefulness of having two alkene functionalities for further transformations. Since being first reported by Morken and co-workers25 in 2010, asymmetric allyl–allyl cross-coupling has become a burgeoning field of research in chemical synthesis. In this asymmetric allylic allylation, a new stereogenic center is created by the attack of a carbon nucleophile on a prochiral substrate. Catalysts derived from different metals, including Pd, Ni, Cu, Ir etc., were explored and showed excellent regio- and enantioselective control. Surprisingly, there is a scarcity of any comprehensive reviews on the allyl–allyl cross-coupling reaction, and its synthetic applications. Therefore, we summarize the development of catalysts, ligands and synthetic applications of allyl–allyl coupling reactions from 2008 to the most recent results.2. Mechanism
There are three different mechanisms of allyl–allyl cross-coupling proposed to rationalise the product formation presented Fig. 1. Professor Barry M. Trost26 and Professor John K. Stille27 independently postulated the reaction where an outer-sphere attack of the allyl nucleophile at a metal–π-allyl intermediate is involved through an SE2′ mechanism, leading to the formation of a single major isomer, rather than a distribution of regioisomers formed through an inner-sphere mechanism that involved transmetalation followed by a reductive elimination sequence. The generation of a linear 1,5-diene via inner-sphere reductive elimination from a bis-η3-allyl–palladium complex was described by Schwartz and Goliaszewski in 1984.28 Most importantly, there is a chance of forming two different 1,5-dienes via inner-sphere reductive elimination when the allyl groups are different (see Fig. 2).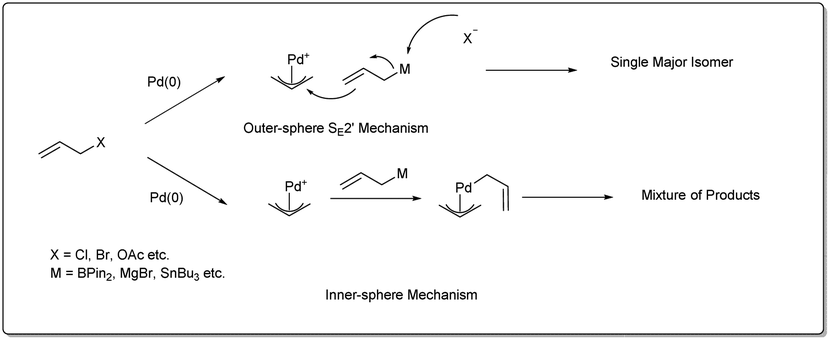 | ||
| Fig. 1 Outer-sphere and inner-sphere mechanisms. | ||
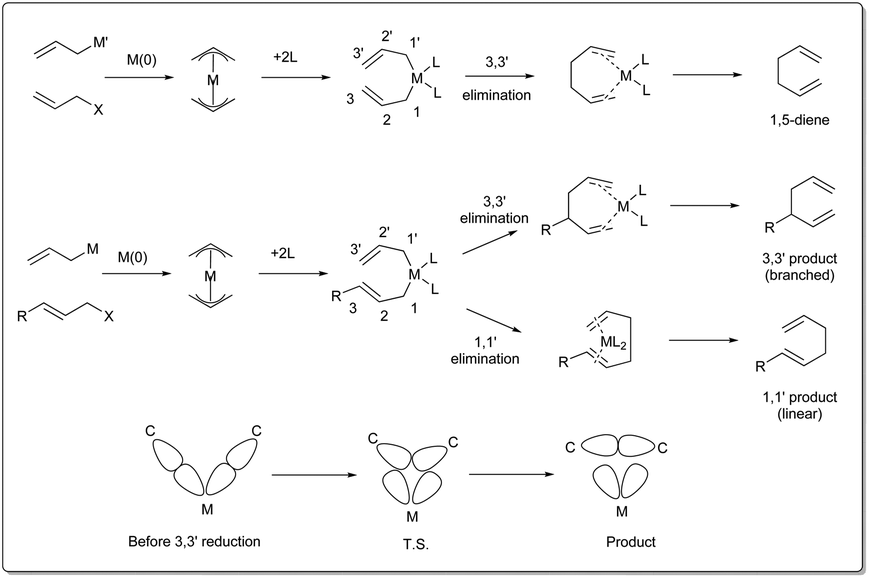 | ||
| Fig. 2 3,3′ vs. 1,1′ elimination. | ||
3. Metal-catalyzed allyl–allyl coupling
3.1. Palladium-catalyzed coupling
Over the 40 years of history on cross-coupling reactions, palladium metal catalysts have been those most commonly explored. Enormous success has been achieved in the synthesis of complex and functionalized molecules by using cross-coupling. Similarly, over the last decade of exploration of allyl–allyl cross-coupling reactions, the palladium-catalyzed cross-coupling has been the one most frequently investigated.In 2009, Kobayashi and co-workers disclosed29 Pd-catalyzed intermolecular Suzuki-type sp3–sp3 allyl–allyl cross-coupling between allyl carbonates and allyl boronates under mild conditions (Scheme 1). The reaction was carried out in the presence of 2 mol% of Pd(PPh3)4 in ethyl acetate at room temperature and provided the linear 1,5-diene 4 as a major product. Various carbonates, substituted at the α-, β-, or γ-positions, relative to the leaving group, were found to be compatible with the reaction conditions. They mentioned that aromatic substrates (R1 = Ar) generally led to linear products with excellent selectivities. However, nitrile-substituted aromatic substrates and α,β-unsaturated aliphatic esters provided predominately branched dienes. Additionally, they also demonstrated that certain substrates (like 1c, 1d, 2cetc.) gave worse results due to a β-hydride elimination associated with intermediary allyl–Pd species, which can be overcome by employing a commercially available Ni(PPh3)4 catalyst.
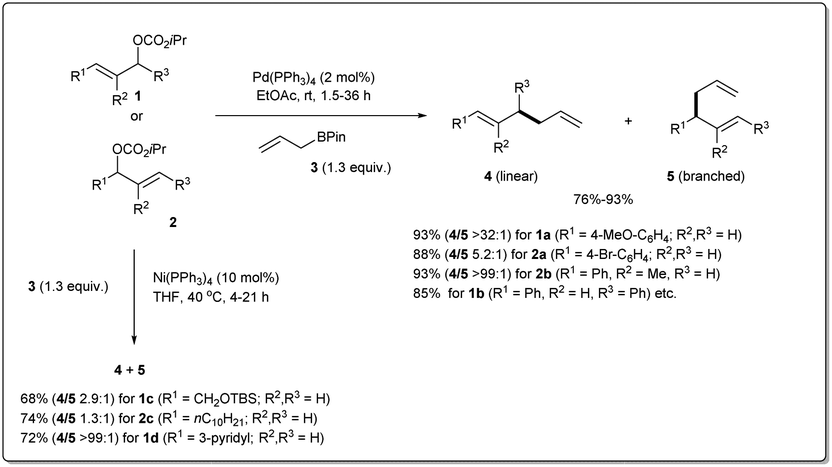 | ||
| Scheme 1 Pd-catalyzed allyl–allyl cross-coupling between allyl carbonates and allyl boronates. | ||
In 2010, Morken uncovered25 a highly regio- and enantioselective Pd-catalyzed cross-coupling of allylic carbonates and allylB(pin) to produce chiral branched 1,5 dienes (Scheme 2). In their report, they explained that the formation of the chiral, branched 3,3′ elimination product (branched diene 6) relative to the linear 1,1′ elimination product (linear diene 7) is dependent on the bite angle of the ligand where a small-bite-angle ligand favours a branched substitution product (see Table 1) while a small-bite-angle ligand disfavours the 1,1′ elimination relative to the 3,3′ by increasing the C1–C1′ separation. The higher bite angle of a bidentate ligand decreases the ∠CPdC and pushes the two carbon atoms closer together, and this accelerates C–C bond formation and subsequent reductive elimination.30 They found that 2,2′-bis(difurylphosphino)-6,6′-dimethoxybiphenyl31 (A in Scheme 2) provides the allyl–allyl cross-coupling product with high regio- and enantioselectivity with aromatic allylic carbonates. However, this ligand showed a low yield for the aliphatic substrate and the problematic low yield was overcome by applying the ligand (R,R)-QuinoxP* (B).32 The inner-sphere reductive elimination was validated by isotope-labeling experiments. Notably, in another report, they explained33 the role of the ligand on branched/linear selectivity in palladium-catalyzed allyl–allyl cross-couplings.
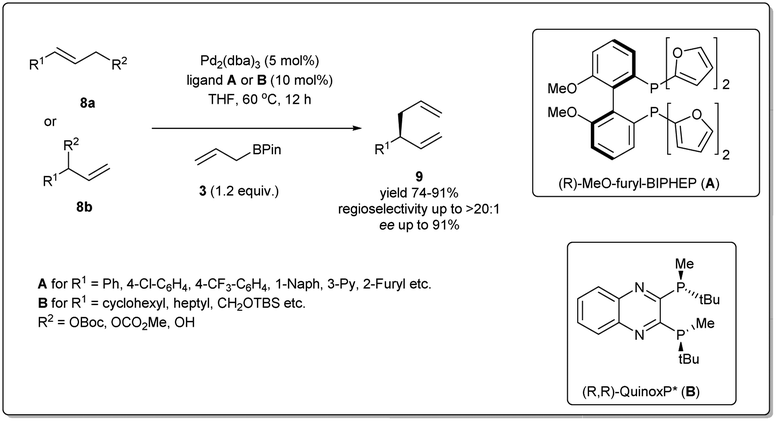 | ||
| Scheme 2 Pd-catalyzed enantioselective allyl–allyl cross-coupling. | ||
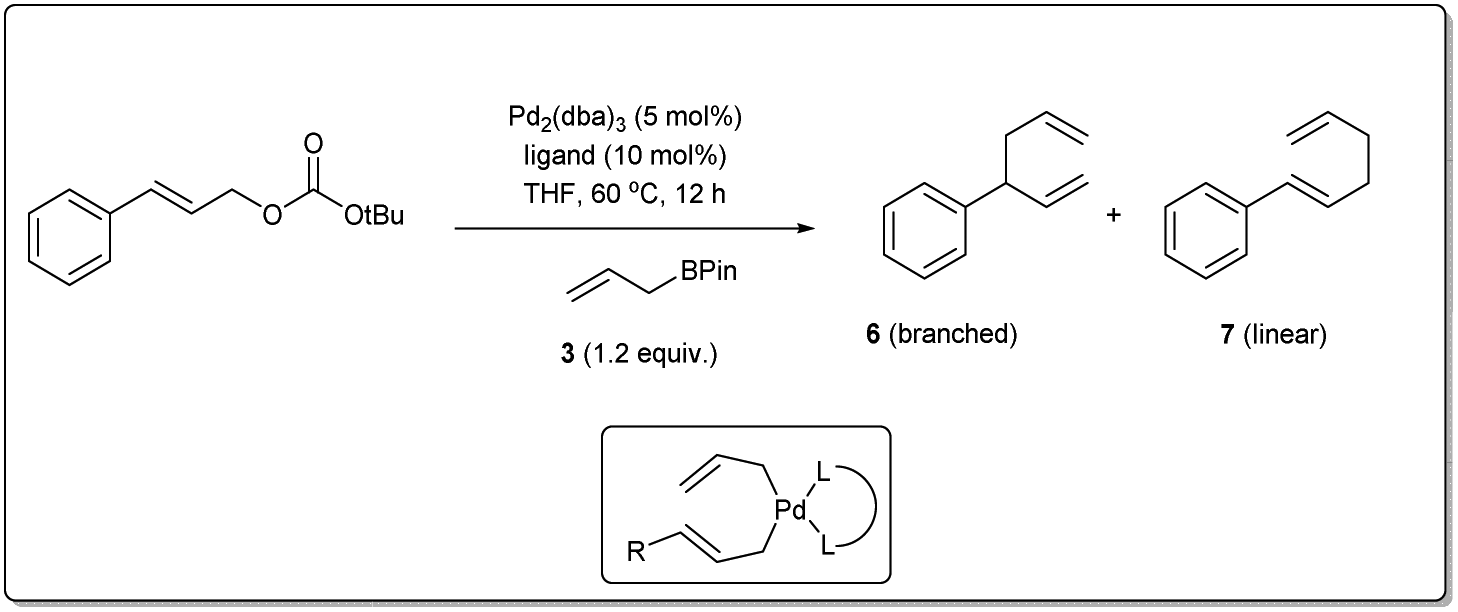
In 2011, they applied the above methodology for highly regioselective and enantioselective installations of all-carbon quaternary centers from racemic allylic carbonates and allyl boronates (Scheme 3).35 The reaction proceeds via a 3,3′-reductive elimination of bis-(η1-allyl)palladium intermediates in the presence of 2 mol% Pd2(dba)3 and 4 mol% of ligand A at 60 °C. The process is amenable to a wide range of aromatic and aliphatic tertiary allylic carbonates including oxygen and halogen-substituted substrates. Most importantly, this enantioselective reaction can be applied to the creation of hindered quaternary carbon centers. In 2014, they demonstrated36 the application of allyl–allyl cross-couplings to generate 1,5-dienes 11a with tertiary centers adjacent to all-carbon quaternary centers (Scheme 3). A number of congested boronates 3a were coupled with different cinnamyl chlorides 10a with high enantioselectivity and moderate diastereoselectivity.
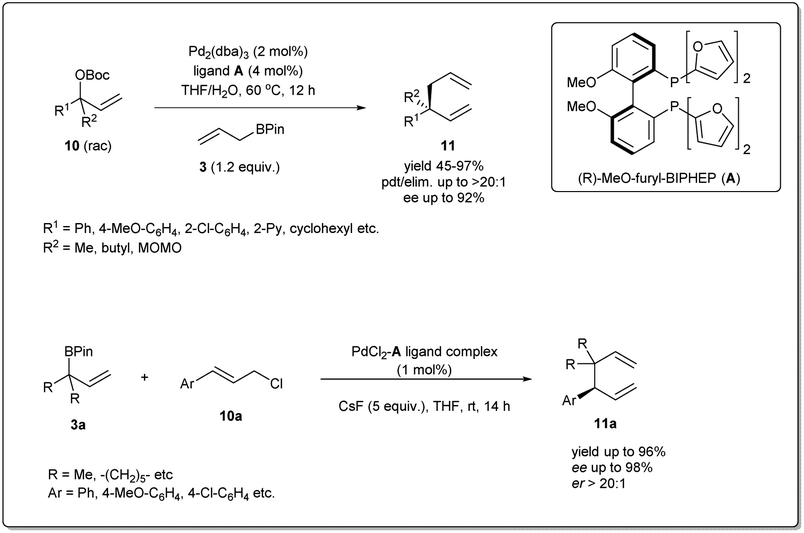 | ||
| Scheme 3 Construction of all-carbon quaternary centers by Pd-catalyzed allyl–allyl cross-coupling. | ||
In 2011, they reported37 a highly effective palladium-catalyzed allyl–allyl cross-coupling for the creation of vicinal stereocenters from prochiral allyl boronates and prochiral allyl chlorides (Scheme 4). A number of γ-substituted allyl boronates and allyl electrophiles were investigated in cross-coupling reactions in the presence of 2.5 mol% of Pd2(dba)3 and 5 mol% of ligand (A or B), and excellent levels of diastereoselectivity were observed in the products bearing adjacent tertiary stereocenters. Significantly, the allyl–allyl coupling reaction occurs in a 3,3′ mode, which enables coupling of two hindered tertiary centers with a high level of diastereocontrol and enantiocontrol, though not in the less hindered 1,1′ mode. Computational studies by Echavarren38 established that cross-coupling reactions occur through a chair-like structure (Scheme 4). The η1-allyl ligands are tilted to minimize interaction with the furyl rings present in the ligand, which makes one chair form to be more favored over the other conformer and results in high asymmetric induction.
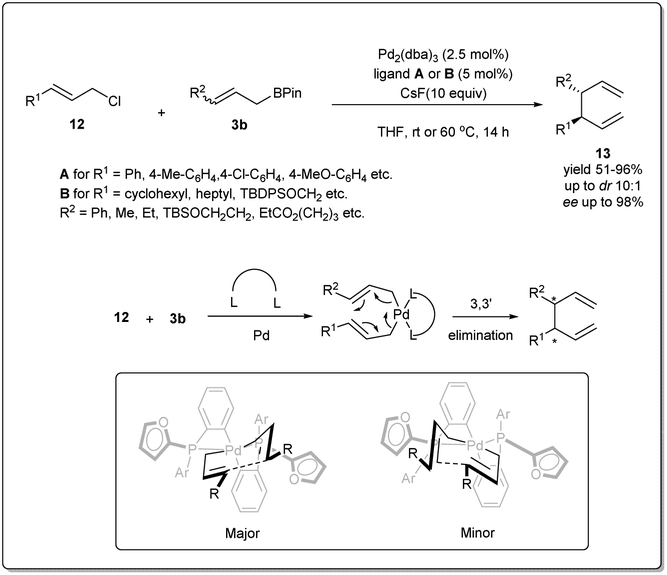 | ||
| Scheme 4 Pd-catalyzed diastereo- and enantioselective allyl–allyl cross-coupling. | ||
In 2013, they demonstrated39 a highly effective catalytic enantioselective allyl–allyl cross-coupling that offered versatile borylated chiral 1,5-hexadienes, which can be applied in a number of ways to construct functionalized chiral building blocks for asymmetric synthesis (Scheme 5). Similar to previous cases, the most important feature of this reaction is that the reaction proceeds via a stereoselective 3,3′-reductive elimination, which provides a borylated branched 1,5-diene as the predominant product. They found that excellent reactivity and enantioselectivity could be achieved when cinnamyl chloride 17a (or 17b) was treated with bis(boryl) nucleophile 3b in the presence of [(allyl)PdCl]2 and (R)-methoxyfurylbiphep (A) as the catalyst system. With these mentioned conditions, borylated 1,5-diene 18 was isolated in up to 79% yield and 98% enantiomeric excess. However, for aliphatic substrates, ligand B was applied to obtain the best results.
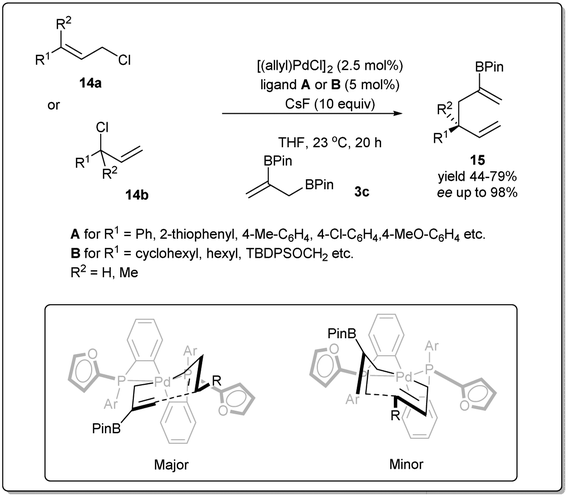 | ||
| Scheme 5 Asymmetric allyl–allyl cross-coupling of allyl boronate 3c and allylic chlorides. | ||
In 2014, they investigated40 the Pd-catalyzed allyl–allyl cross-coupling reaction using internal allyl electrophiles, which allowed the rapid construction of substituted 1,5-dienes including those with all carbon quaternary centers with high regio- and enantioselectivity (Scheme 6). Notably, an important feature of the cross-coupling reactions is that Pd is generally unable to migrate from one allyl-face to the other. This feature omits the requirement for chiral ligands and the enantioselectivity of the reaction depends on the starting electrophiles. Different acyclic, aliphatic substrates, including those undergoing a sterically demanding substitution, were well tolerated, resulting in products with good to excellent yields. Importantly, the method can be used to synthesize molecules with minimal steric differentiation with excellent enantiomeric excess.
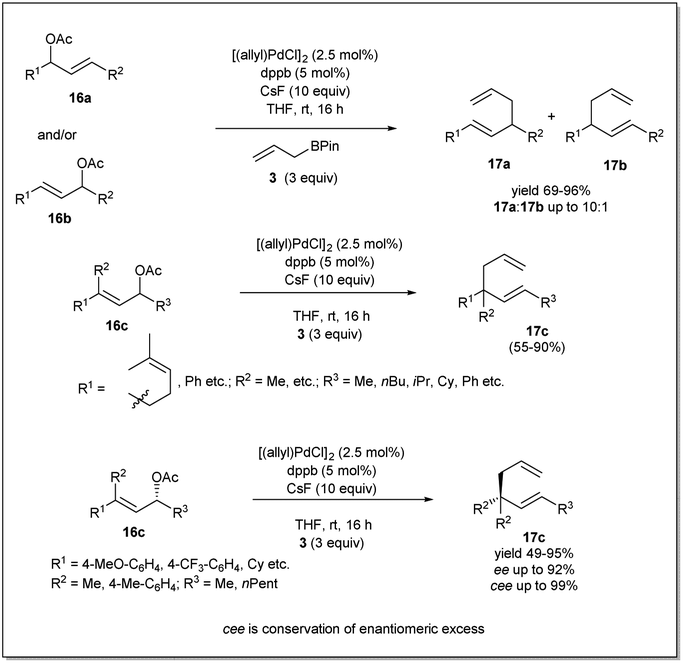 | ||
| Scheme 6 Allyl–allyl cross-coupling of secondary allyl acetates. | ||
In 2016, Ding et al. reported41 a highly efficient asymmetric allyl–allyl cross-coupling reaction of racemic Morita–Baylis–Hillman adducts and allyl boronates (Scheme 7). They used 1.25 mol% of a spiroketal-based bis(phosphine) ligand (SKP) and 0.5 mol% of palladium complex [Pd(C3H5)Cl]2 as the catalyst, and obtained highly functionalized chiral 1,5-dienes in good yields with high regioselectivities, and excellent enantioselectivities (95–99% ee). This powerful transformation afforded a range of functionalized 1,5-dienes, which provided versatile scaffolds for a number of complex molecules, including some polycyclic lactones and biologically important polycyclic lactams.
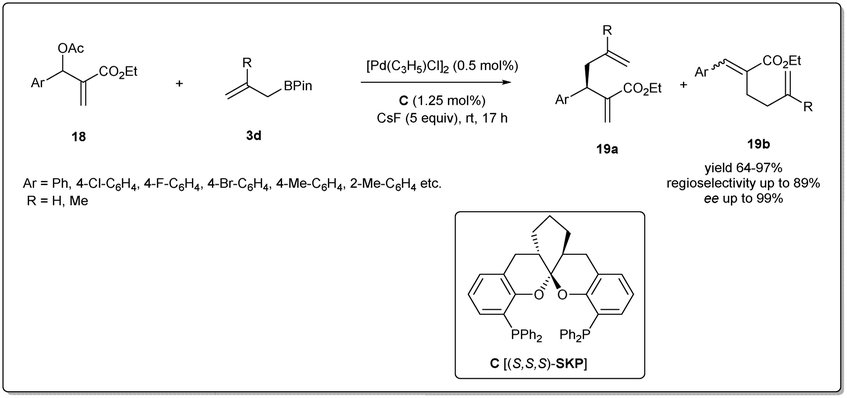 | ||
| Scheme 7 Enantioselective allyl–allyl cross-coupling reaction of racemic Morita–Baylis–Hillman adducts and allyl boronates. | ||
In 2021, Song and coworkers demonstrated42 a Pd-catalyzed C-glycosylation by allyl–allyl cross-coupling where Achmatowicz rearrangement products act as donors and methylcoumarins as acceptors under mild conditions (Scheme 8). The reaction proceeds smoothly in the presence of 5 mol% of Pd(OAc)2 when various Achmatowicz rearrangement products 21 and methylcoumarins 20 were used as substrates to obtain the desired C-glycosides 22 in good yields (up to 74%) with excellent diastereomeric ratios (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). This methodology allows a general and practical route to construct biologically important C-glycosides with excellent regioselectivities and diastereoselectivities. Moreover, they applied this method to the stereodivergent enantioselecive synthesis of C-glycosides.
:1). This methodology allows a general and practical route to construct biologically important C-glycosides with excellent regioselectivities and diastereoselectivities. Moreover, they applied this method to the stereodivergent enantioselecive synthesis of C-glycosides.
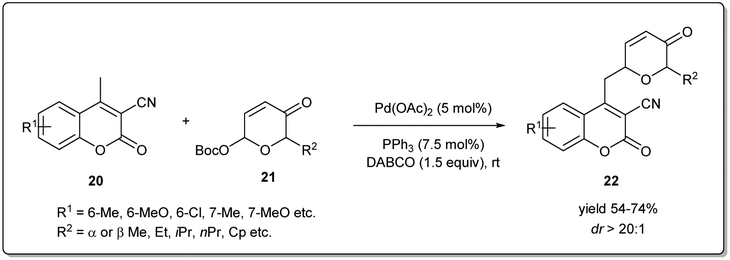 | ||
| Scheme 8 Pd-catalyzed C-glycosylation by allyl–allyl cross-coupling. | ||
In 2021 Huang et al. disclosed43 a unique palladium-catalyzed allyl–allyl reductive coupling of allylamines and allylic alcohols with H2 as the sole reductant under mild reaction conditions (Scheme 9). The reaction proceeds via heterolytic cleavage of the H–H bond by the in situ formed Pd–N or Pd–O bond from the allylic substrate and the cleavage of the C–N bond or C–O bond. A number of allylamines and allylic alcohols, as well as allylic ethers, respond to this allyl–allyl reductive coupling under 1 atm of hydrogen. Notably, kinetic studies of the reaction suggested that the dinuclear palladium species formed from self-transmetalation was involved in the catalytic cycle and the formation of the dinuclear palladium species was the rate-determining step.
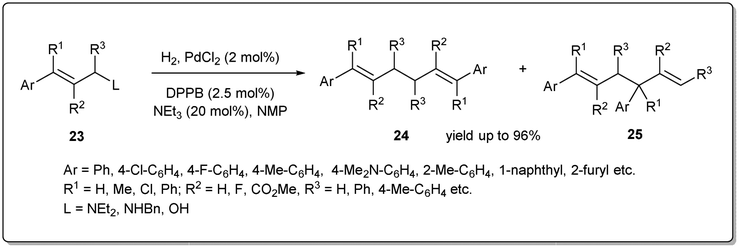 | ||
| Scheme 9 Palladium-catalyzed allyl–allyl reductive coupling. | ||
In 2022, Hall et al. synthesized44 optically enriched 2-allylated 3,4-dehydropiperidines in good yields using a palladium-catalyzed stereospecific allyl–allyl cross-coupling reaction between an enantioenriched piperidinyl allylic boronate and allylic carbonates (Scheme 10). The coupling reaction was carried out in the presence of 5 mol% Pd(OAc)2 and 10 mol% (p-CF3C6H4)3P as the ligand for 16 h to afford up to 76% product yield. Despite the possibility of the formation of four regioisomers from the coupling of two unsymmetrical allyl fragments, the procedure resulted in linear 2-allylated 3,4-dehydropiperidines exclusively (>98:2 γ-linear selectivity) with enantiospecificity up to 99%. They proposed that the coupling reaction proceeds via an enantiofacially controlled double allylic rearrangement mechanism, which explains the large preference for the γ-linear regioisomer (S)-28 over the three other possible regioisomers. It provides a useful alternative for preparing chiral piperidine derivatives for applications in drug discovery.
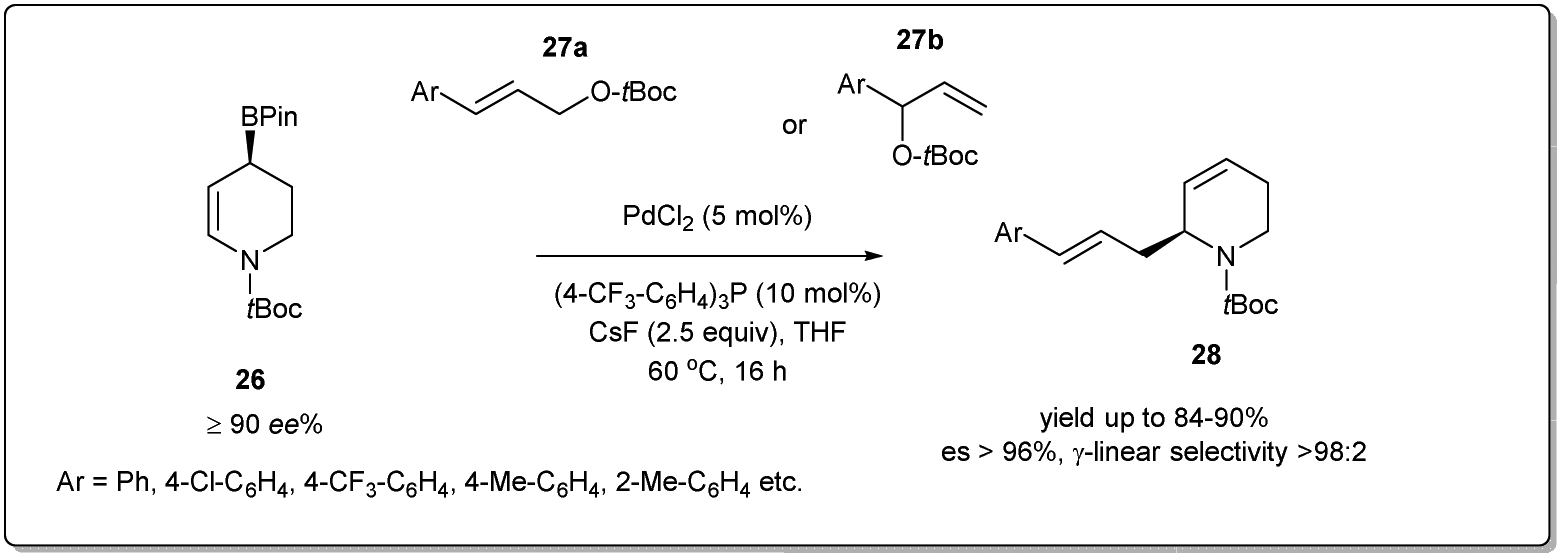 | ||
| Scheme 10 Allyl–allyl cross-coupling piperidinyl allylic boronate and allylic carbonates. | ||
3.2. Iridium-catalyzed reactions
Since the first reports of iridium-catalyzed allylic substitution reactions,9,45 iridium-based catalysts were found to be very effective for allylic substitution reactions. In the last decade, there has been an increasing trend in iridium-catalyzed allylic substitutions, as Ir catalysts normally provide branched products with good regio- and enantioselectivities.9 Hence, a number of allyl–allylations were documented, which will be discussed in this part of the review.In 2014, Carreira et al. described46 a new Ir-catalyzed, enantioselective allyl–allyl cross-coupling reaction between racemic secondary alcohols and simple alkenes to construct chiral 1,5-dienes in good yields and excellent enantioselectivities (Scheme 11). The atom economical method is catalyzed by an Ir–phosphine complex and the allyl–Ir intermediate is sufficiently electrophilic to engage olefins in the substitution. Hence, the process does not require any stoichiometric allyl–metal reagents to activate the allylic alcohol partner. The reaction was carried out in the presence of 4 mol% Ir catalyst, and 16 mol% D and 50 mol% unsymmetrical sulfonimide E as Brønsted acid activators. Various alcohols and alkenes were investigated to check the scope and universality of this allyl–alkene coupling protocol.
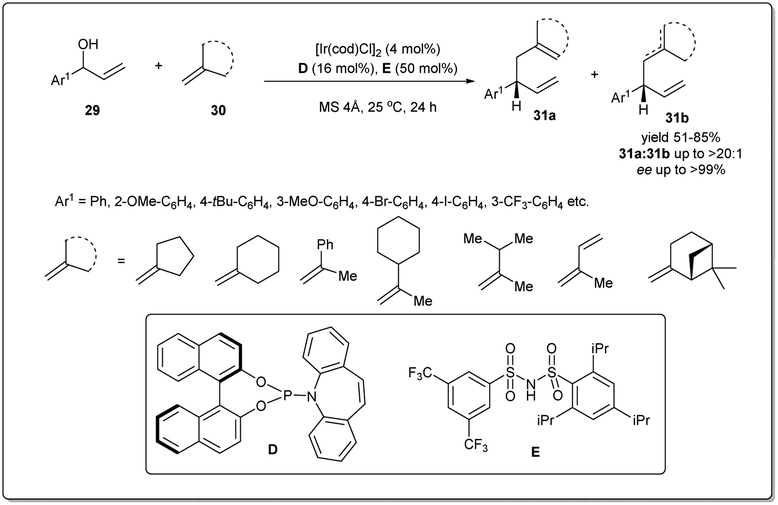 | ||
| Scheme 11 Ir-catalyzed enantioselective allyl–allyl cross-coupling reaction. | ||
In 2014, Carreira and co-workers reported47 an operationally convenient Ir-catalyzed enantioselective allyl substitution reaction of unactivated racemic secondary allylic alcohols by employing readily available and bench-stable allylsilanes to provide optically active 1,5-dienes with excellent regio- and enantioselectivity, and high functional group tolerance (Scheme 12). After a lot of screening, they proposed that vinyl carbinol 32 underwent a smooth reaction with allylsilane 33 in the presence of the chiral Ir/D catalyst and diphenyl phosphate in dioxane to give dienes 34a and 34b in up to 99% ee with the branched product (34a) predominant. Notably, the promoter for the activation of allylic alcohols in intermolecular allylic substitution reactions, Sc(OTf)3, showed complete conversion, leading to product with high stereoselectivity and regioselectivity compared to other Lewis acids as well as Brønsted acids. Different functionalised naphthyl and phenyl vinyl carbinols 32 and allylsilanes were employed in this reaction and underwent the described allyl–allylsilane cross-coupling with very high enantioselectivty and regioselectivity.
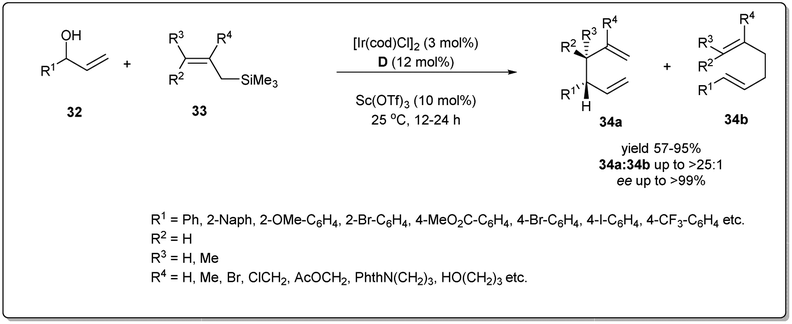 | ||
| Scheme 12 [Ir(cod)Cl]2-catalyzed enantioselective allyl–allyl cross-coupling reaction. | ||
In 2015, Zhang and co-workers disclosed48 an Ir-catalyzed allyl–allyl cross-coupling reaction between allylic carbonates and 1,3-diarylpropenes under mild conditions to give linear 1,5-dienes regioselectively in excellent yields (Scheme 13). Very weakly acidic (E)-1,3-diarylpropenes were used directly as allyl pro-nucleophiles. To explore the utility, the coupling reaction was studied using different types of arylpropenes and tert-butyl cinnamyl carbonates in the presence of 1.5 equivalents of NaHMDS, 2.0 mol% of [Ir(cod)Cl]2 and 4.4 mol% of dppf as the catalyst in THF. To measure the activity of the catalyst, they conducted a study with tert-butyl cinnamyl 35a (0.4 mmol), (E)-1,3-diphenylpropene 36a (0.6 mmol), in the presence of 0.025 mol% [Ir(cod)Cl]2 (S/Ir = 2000; S/Ir = substrate/iridium) and obtained 79% product yield with more than 99% regioselectivity. To probe the mechanism of the cross-coupling reaction, they carried out a reaction between (E)-1,3-di(β-naphthyl)propene 36b and tert-butyl cis-(5-phenyl-2-cyclohexenyl)carbonate 35b and observed only trans-isomers of 37c in 35% isolated yield (Scheme 13). Formation of a trans-isomer advocates that the coupling reactions proceeded via an inner-sphere coordination of the hard 1,3-diarylallyl nucleophile with the Ir catalyst, followed by reductive elimination to give the product.
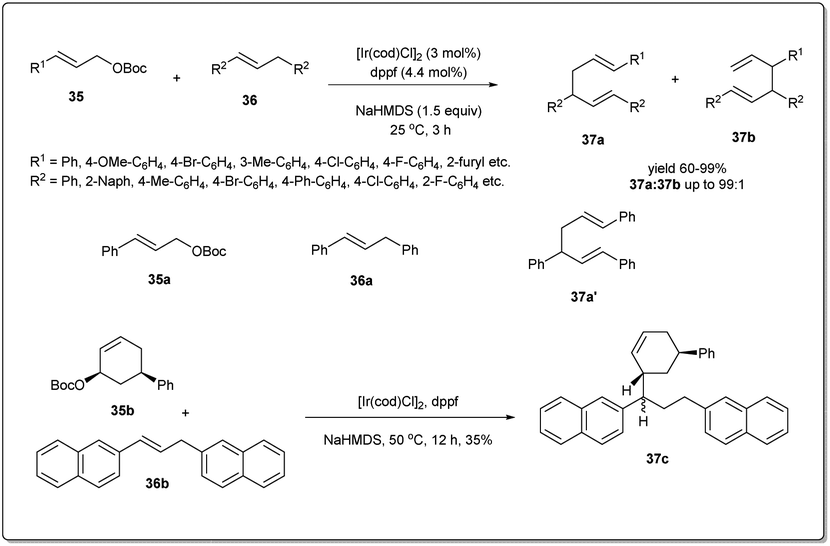 | ||
| Scheme 13 Ir-catalyzed allyl–allyl cross-coupling reaction between carbonates and 1,3-diarylpropenes. | ||
In 2018, Yang and co-workers demonstrated49 a highly efficient iridium-catalyzed allyl–allyl cross-coupling reaction between allylboronic esters and unactivated racemic secondary allylic alcohols to provide chiral branched 1,5-dienes (Scheme 14). The good functional group compatibility with high regio- and stereoselectivity are also major features of this method. Different 2-naphthyl- and phenyl-vinyl carbinols smoothly underwent the coupling giving excellent isolated product yields in the presence of 4 mol% Ir-catalyst, 16 mol% D and 10 mol% Zn(OTf)2 in DCE.
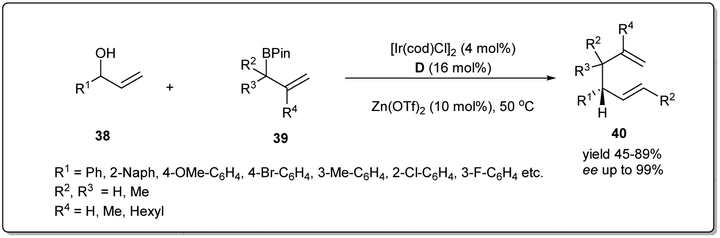 | ||
| Scheme 14 Cross-coupling reaction between allyl boronic esters and racemic secondary allylic alcohols. | ||
3.3. Gold-catalyzed coupling
In 2008, Echavarren et al. reported50 an unprecedented gold-catalyzed intramolecular allyl–allyl coupling of allyl acetates with allyl stannanes (Scheme 15). The mechanism of the cross-coupling reaction begins with the oxidative addition of gold into the allyl acetate bond. Then, transmetalation followed by an allyl–allyl reductive elimination leads to the formation of the desired product. A variety of dienes were used to assess the coupling reaction and excellent yields were observed. The coupling reaction proceeds in the presence of 3 mol% of catalyst F in DCE.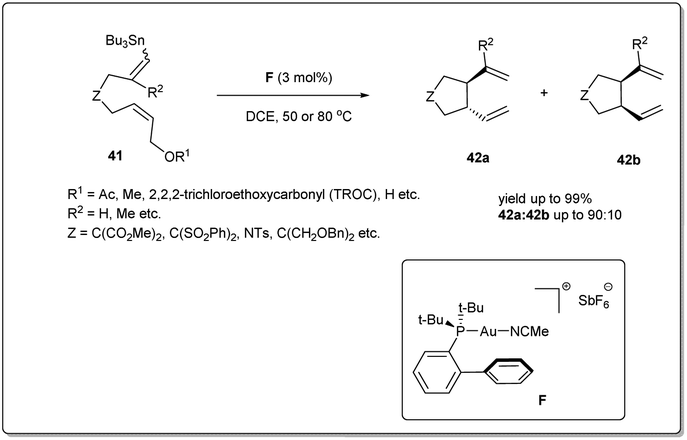 | ||
| Scheme 15 Gold-catalyzed allyl–allyl cross-coupling reaction. | ||
3.4. Copper-catalyzed coupling
The earth-abundant metal of copper, with its relatively low toxicity and high tunability, is one of the most studied metals in the area of catalysis. Copper-catalyzed allylic substitution is a powerful Csp3–Csp3 bond-forming reaction for syntheses of synthons for biologically active natural products. Due to their low cost, high compatibility with different functional groups, and resultant products with high regio- and enantioselectivities, copper catalysts have helped this transformation become a very efficient tool for the enantioselective introduction of alkyl, aryl, allenyl, vinyl, or alkynyl fragments in the construction of stereogenic centers using nonstabilized organometallic nucleophiles.13–18 However, copper-catalyzed asymmetric allylic allylation with control of enantioselectivity faces a major challenge as allylcopper species can exist in a delocalized contact η3 π-allyl ion-pair structure.51–53The first example of copper-catalyzed enantioselective allyl–allyl cross-coupling dates back to 2013 when Feringa reported54 the synthesis of chiral 1,5-dienes using a Cu(I)-phosphoramidite-based catalytic system (Scheme 16). The enantioselective allyl–allyl cross-coupling between an allyl Grignard reagent and allyl bromides resulted in a large variety of chiral 1,5-dienes in good yields with high enantioselectivity. At first, they initiated their study with different phosphoramidite-based ligands and observed that G showed the best result. However, a Cu salt with non-coordinating counteranions, (CuOTf)2·C6H6, increased the regioselectivity and showed high enantioselectivity. A wide range of allylic electrophiles bearing different functionalities, including protected alcohols, amines, alkenes, and acetals, were tested, showing excellent enantioselectivities in almost all cases. Importantly, this method provided a number of chiral building blocks (45a) for natural product syntheses, e.g.45c, an intermediate in the synthesis of (+)-sabinene,55 a monoterpene broadly distributed in essential oils from plants.
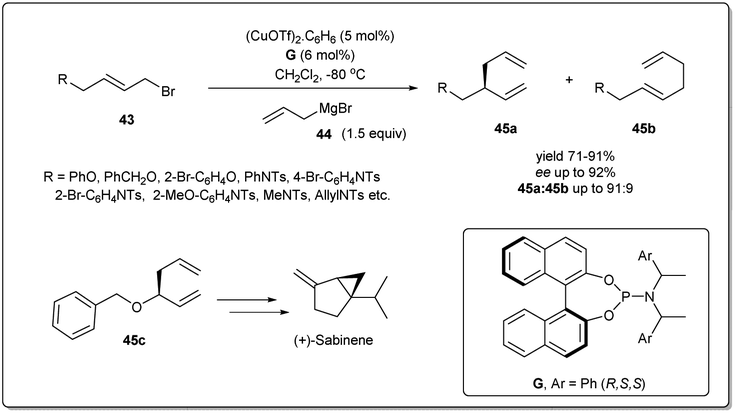 | ||
| Scheme 16 Cu(I)-phosphoramidite-catalyzed enantioselective allyl–allyl cross-coupling. | ||
Since the isolation and full characterization of a free N-heterocyclic carbene (NHC),56 catalysis mediated by NHC-based metal complexes has emerged as a powerful method for the construction of the carbon–carbon bond.57–64 Nevertheless, Cu–NHC-catalyzed enantioselective reactions had been underdeveloped until Sawamura and coworkers reported65 regio- and enantioselective allyl–allyl coupling between substituted allyllic boronates and phosphate (Scheme 17). Remarkably, the limitation of regioselectivity faced by Feringa and co-workers was solved when they reported copper-catalyzed enantioselective allyl–allyl cross-coupling between (Z)-allylic phosphates and allyl boronates using phenol/NHC chiral ligands. In their studies, they displayed that even a simple Cu/NHC (SIMes) complex gave exclusive γ-selectivity (γ/α > 99:1). During an investigation with various chiral ring-saturated NHC ligands bearing a 2-hydroxyphenyl group, excellent enantioselectivity and exceptional regioselectivity were obtained from the ligand H. Several leaving groups were effective, but phosphate provided higher enantioselectivity and γ-selectivity. In addition to a leaving group, different bases were employed and KOMe provided the highest γ-regioselectivity and enantioselectivity. Sawamura's conditions can be applied to a range of Z-allylic phosphates with different aliphatic γ-substituents as well as various functional groups, including 1,3-benzodioxole, THP ether, benzyl ether, silyl ether, pivalate, carbamate and p-toluenesulfonate groups. However, when allyl boronate derivatives with hexyl, benzyl, and phenyl groups at the β-position were subjected to this reaction, the corresponding chiral 1,5-hexadienes with enantiomeric excesses over 80%, with moderate γ-selectivities, were provided. Importantly, the geometry of the electrophiles had a strong influence on both the regioselectivity and enantioselectivity.
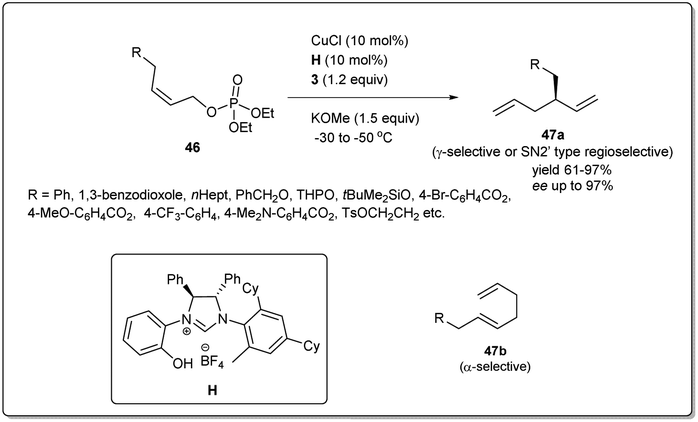 | ||
| Scheme 17 Copper–NHC-catalyzed allyl–allyl cross-coupling. | ||
After successfully establishing the strategy delineated in Scheme 17 for Cu–NHC-catalyzed enantioselective allyl–allyl cross-coupling reactions using a simple allyl boronate 3, they focussed on the construction of a substituted 1,5-diene employing β-substituted allyl boronate derivatives 48 (Scheme 18).66 Under similar conditions mentioned above, different substituted allyl boronates were subjected to a cross-coupling reaction with phosphonate 49 and good to excellent yields with high regio- and enantioselectivity were observed.
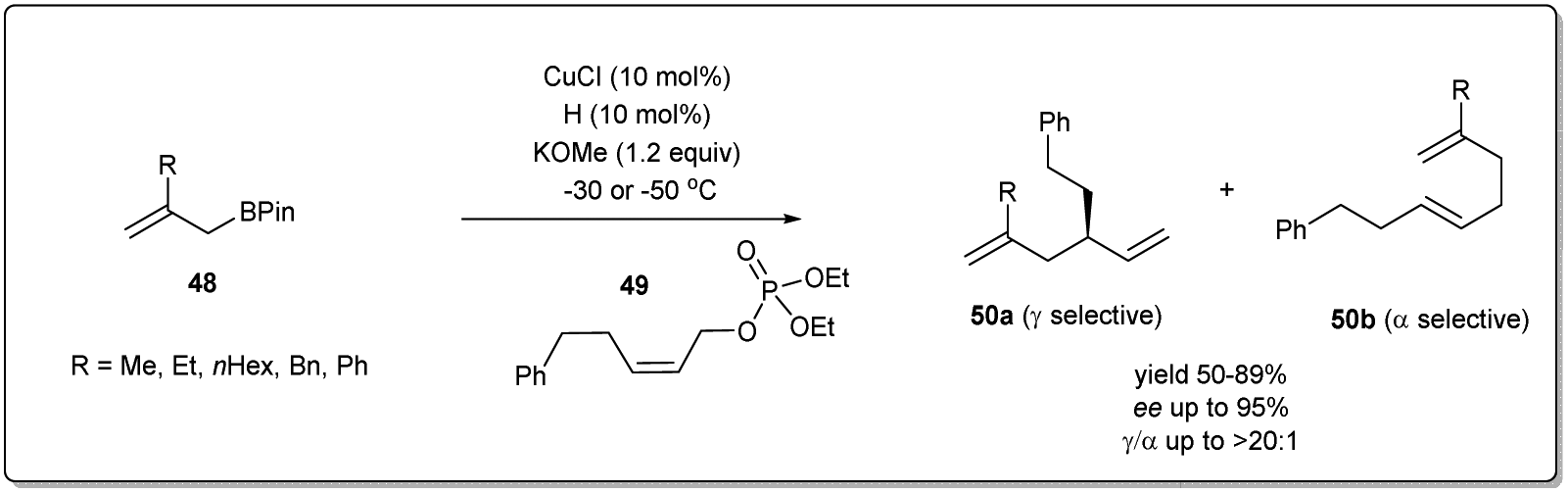 | ||
| Scheme 18 Scope with respect to the allylic boronates. | ||
In 2014, borylative asymmetric allyl–allyl coupling in the presence of an N-heterocyclic carbene-based copper catalyst using allenes, B2(pin)2, and allyl phosphates had been developed by Hoveyda et al. (Scheme 19).67 When allenes were treated with B2(pin)2 in the presence of in situ generated copper–NHC, the allyl copper intermediate bearing a boryl functionality at the β-position is produced catalytically, and reacts with allyl phosphate. Different NHC and phosphine ligands were screened to identify the optimised conditions for the reaction. Surprisingly, the NHC–Cu complex showed excellent site selectivity and enantioselectivity whereas phosphine complexes were ineffective. The authors mentioned that phenylglycinol-derived air-stable imidazolinium salt I exhibited the best results. Different functional groups, such as an alkyne, an amine, or an amide containing allenes, were employed and a variety of chiral trisubstituted alkenyl–B(pin) products with complete Z selectivity were obtained with excellent stereo- and regioselectivities. The boryl functionality is very useful for further derivatization. Notably, Pd-catalyzed cross-coupling of alkenyl–B(pin) with aryl bromides resulted in the complete retention of stereochemistry.
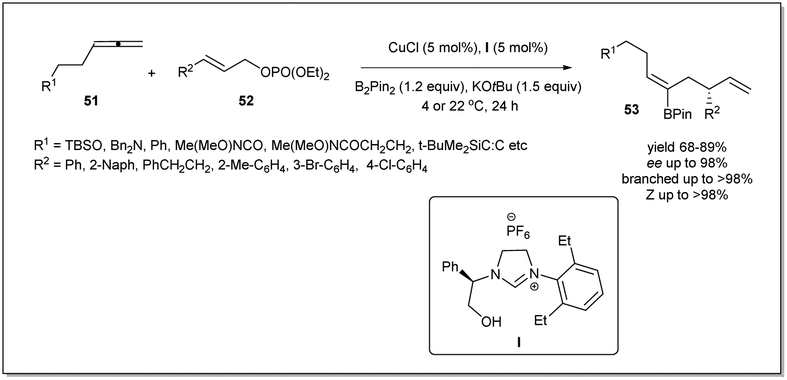 | ||
| Scheme 19 Borylative asymmetric allyl–allyl coupling. | ||
Highly stereo- and regioselective borylative allyl–allyl cross-coupling using allenes, B2(pin)2, and allyl phosphates was also reported by Tsuji et al. (Scheme 20)68 in the presence of a copper catalyst bearing an N-heterocyclic carbene ligand J. Different boryl-substituted 1,5-diene derivatives were synthesized from diversely substituted allenes and allyl phosphates in good to high yields with high regioselectivity and Z selectivity. The borylation proceeds via β-boryl (Z)-σ-allyl copper species, which reacted with (Z)-55 to afford (Z)-56.
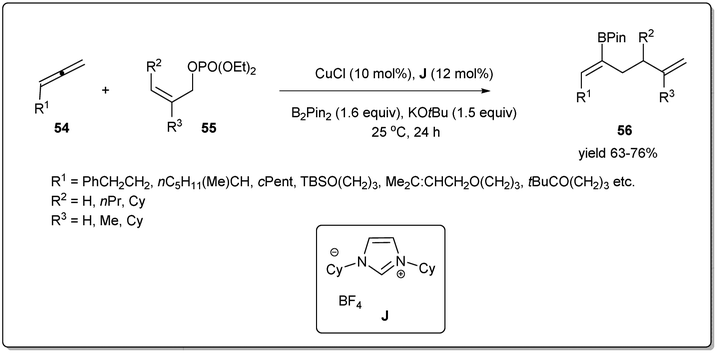 | ||
| Scheme 20 CuCl-catalyzed borylative allyl–allyl coupling. | ||
Copper-catalyzed site-specific and enantioselective reductive allyl–allyl coupling reactions of allenes with allylic phosphates to afford 1,5-dienes without the use of any pre-formed allylic metals were reported for the first time by Xiong and co-workers (Scheme 21).69 It was found that ligand K exhibited excellent reactivity and enantioselectivity. In the presence of the in situ generated (NHC)CuH, allenes bearing aliphatic, alkoxyl, halogen, –CF3etc. substituents on the aryl rings, showed high levels of reactivities and excellent enantioselectivities. Furthermore, haloaryl and heteroaromatic ring-containing allylic phosphates and β-ethyl-, propyl- or H-substituted allylic phosphates also facilitated the reaction providing chiral 1,5-dienes. Notably, a 1,1-disubstituted allene was also tolerated in this transformation; however, an alkyl-substituted allylic phosphate was not a suitable substrate for this reductive cross-coupling reaction. Most importantly, the study was further expanded to provide a facile route to access chiral 1,5-dienes bearing more hindered all-carbon quaternary stereocenters, which are broadly distributed in many important biologically active molecules and natural products. After optimising the reaction conditions, a series of quaternary carbon center-containing chiral 1,5-dienes were synthesized with exclusive regioselectivity and excellent enantioselectivity. A possible mechanism of the reductive cross-coupling was described in the report.
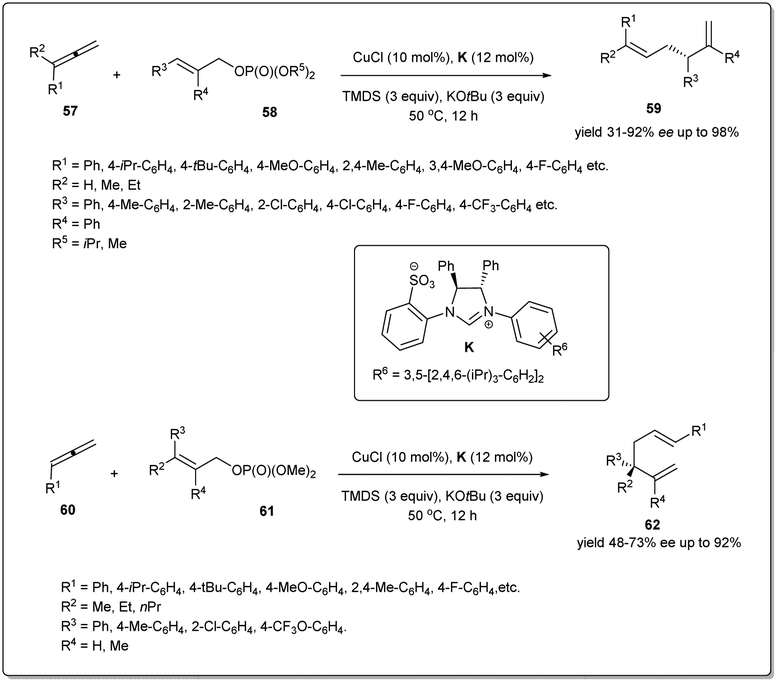 | ||
| Scheme 21 Synthesis of tertiary/quaternary stereogenic center-containing 1,5-dienes. | ||
3.5. Nickel-catalyzed coupling
Recent attention to allyl–allyl cross-coupling has been focused on nickel-based catalysts as nickel is less expensive and more earth abundant. A number of efforts focussing on nickel-catalyzed regioselective allyl–allyl cross-coupling reactions have been made to obtain 1,5-dienes.In 2011, Kobayashi demonstrated70 a regiospecific allyl–allyl coupling reaction between various allyl alcohols and allyl boronates under nickel catalysis (Scheme 22). Significant features of this method include the earth-abundant catalyst, good reactivity, exclusive regioselectivity and good functional group tolerance. The use of moderately Lewis acidic boron reagents along with a nickel(0)–phosphine catalyst helps facile C–O bond activation. The reactions proceed smoothly under mild conditions with excellent linear- and γ-selectivity. Importantly, this method can be applied to the selective preparation of vinyl silanes, which makes the method significant to synthetic chemists.
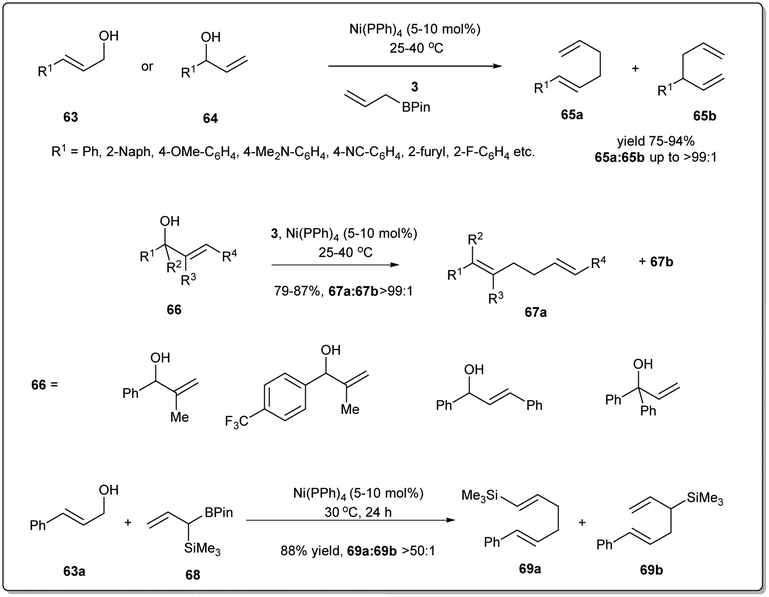 | ||
| Scheme 22 Nickel-phosphine-catalyzed allyl–allyl coupling reaction. | ||
Chen developed71 a hydroallylative reaction to form 1,5-dienes from 1,3-dienes and allyl boronates in the presence of a nickel catalyst (Scheme 23). The protocol is highly regiospecific and is amenable to a variety of 1,3-dienes, as well as congested substrates. In the presence of 5 mol% of Ni(COD)2 and 10 mol% of phosphine ligand, this coupling reaction exhibits high reactivity with good functional group tolerance.
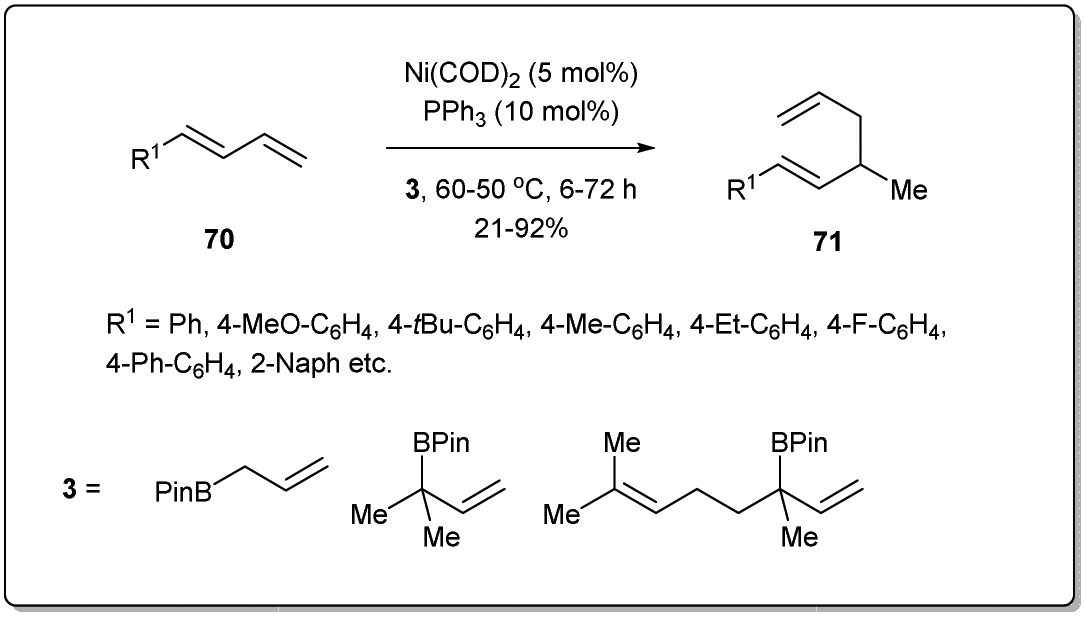 | ||
| Scheme 23 Hydroallylative reaction between 1,3-dienes and allyl boronates. | ||
In 2020, Liu uncovered72 a nickel-catalyzed allyl–allyl cross-coupling reaction via sequential borylation of allyl alcohol followed by a cross-coupling reaction of the resulting allyl boronate with another allyl alcohol (Scheme 24). They employed a number of different alcohols, including linear and branched allyl alcohols, and primary and secondary allyl alcohols, with desired 1,5-dienes afforded in moderate yields with good linear selectivity. The reaction proceeds in the presence of 5 mol% of Ni(COD)2, 10 mol% of PPh3 and 2 equivalents of boron reagent. The intermediate allyl boronate was formed under very simple conditions without the presence of a ligand and was confirmed by NMR.
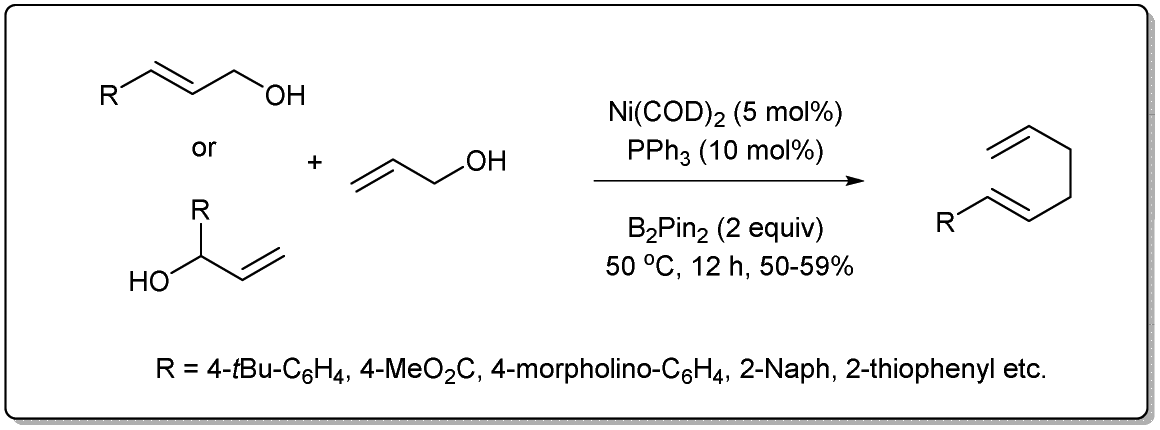 | ||
| Scheme 24 Sequential borylation followed by cross-coupling reaction. | ||
4. Metal-free coupling
Although transition metal catalysts showed a high level of regio- and stereoselectivity, the high cost and complicated purification steps required, due to the presence of heavy transition-metal impurities, limit their application. Therefore, a few attempts have been made to explore metal-free allyl–allyl cross-coupling reactions.In 2012, Liu et al. developed73 a new metal-free method of allylic allylation of acetates using a Brønsted acid catalyst (Scheme 25). The operationally simple, highly efficient metal-free intermolecular Csp3–Csp3 allyl–allyl cross-coupling reaction between allyl acetates and allyltrimethylsilanes proceeds under remarkably mild reaction conditions in the presence of catalytic triflimide to form 1,5-dienes. They screened the scope and limitation of the reaction with a variety of allylic acetates and allyltrimethylsilanes and observed smooth conversions to the desired 1,5-dienes in moderate to good yields and regioselectivities. Notably, only 0.5 mol% of cheap and well-known Tf2NH is required to carry out the reaction and the environmentally benign reagent, acetic acid, is the only waste produced during the reaction.
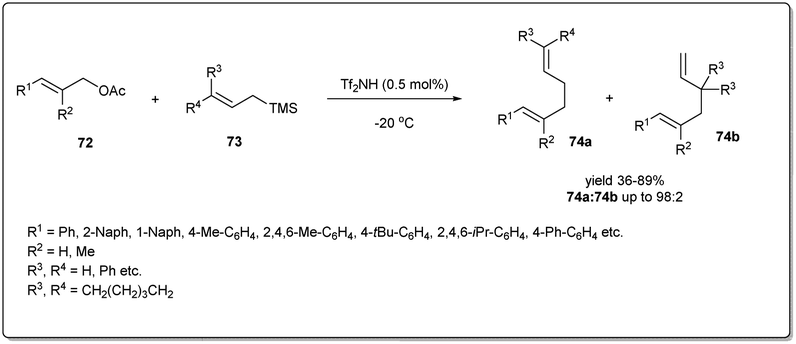 | ||
| Scheme 25 Tf2NH-catalyzed metal-free method of allyl–allyl coupling. | ||
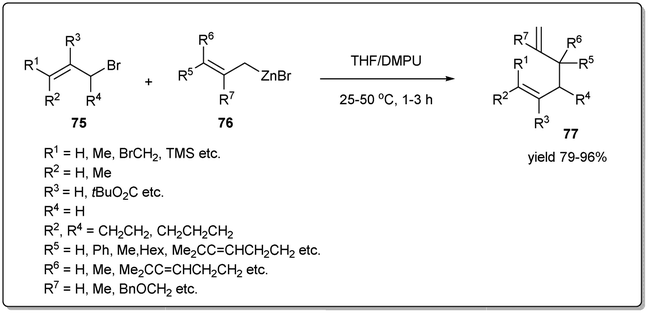
In 2016, Knochel uncovered74 metal-free SN2-type substitutions of allylic bromides by allylic zinc halides in the presence of a 1:1 mixture of THF and DMPU providing 1,5-dienes regioselectively (Scheme 26). The coupling reaction is carried out at rt for 1–3 h in the presence of 1.2 equivalents of an organozinc reagent, and several functional groups, including ester, nitrile etc., are tolerated under the reaction conditions. Importantly, unsymmetrical allylic zinc reagents react almost exclusively through the most branched side of the allylic system, furnishing exclusively γ,α′-allyl–allyl cross-coupling products. Notably, the stereochemistry of the double bond in the allylic halide is retained during the coupling reaction.
 | ||
| Scheme 26 DMPU-mediated allyl–allyl coupling. | ||
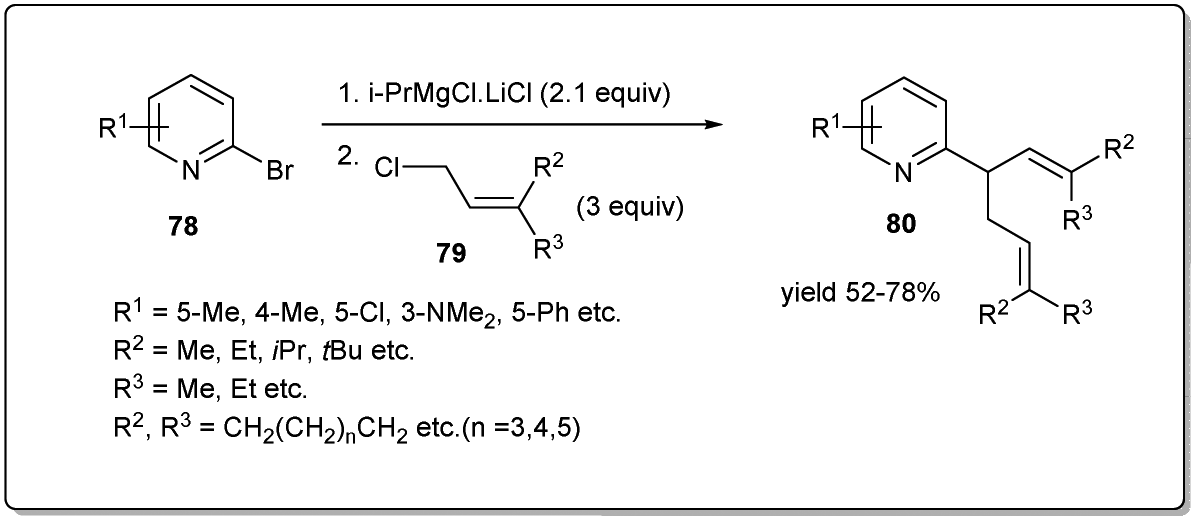
In 2017, Zhang and coworkers demonstrated75 a one-pot domino allylation reaction of 2-pyridinyl Grignard reagents with polysubstituted allyl chlorides (Scheme 27). The tandem pyridinyl–allyl–allyl cross-coupling reaction provides a regioselective synthesis of pyridinyl substituted 1,5-diene derivatives. They proposed that the reaction proceeded through the coupling of substituted allyl chloride to 2-PyMgX, which was generated from 2-bromopyridine and i-PrMgCl·LiCl.
 | ||
| Scheme 27 One-pot domino allylation reaction of 2-pyridinyl Grignard reagents with allyl chlorides. | ||
5. Allyl–allyl cross-coupling reactions in total syntheses
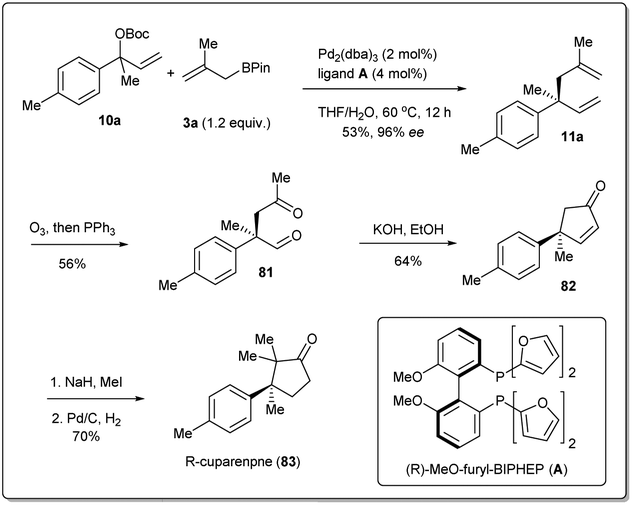
Total synthesis of natural products is one of the most important areas of organic synthesis.76,77 Despite massive achievements, the development of efficient routes for the scalable synthesis of complex natural products remains a challenging task. The allyl–allyl coupling reaction is one of the most useful methods for the synthesis of natural products as it provides versatile synthetic intermediates in 1,5-dienes, which can be used for further transformations. Therefore, a number of total syntheses have been attempted by different groups, which will be described in this section.In 2011, Morken et al. applied35 their highly regioselective and enantioselective allyl–allyl cross-coupling reaction to the synthesis of (R)-cuparenone (Scheme 28),78,79 which is the shortest catalytic asymmetric synthesis of this molecule. They chose one of the products, 11a, as the starting material for the total synthesis and this was converted to diketo compound 12 by ozonolysis. An intramolecular aldol reaction was followed by methylation and hydrogenation, and the desired product (R)-cuparenone was obtained.
 | ||
| Scheme 28 Synthesis of (R)-cuparenone. | ||
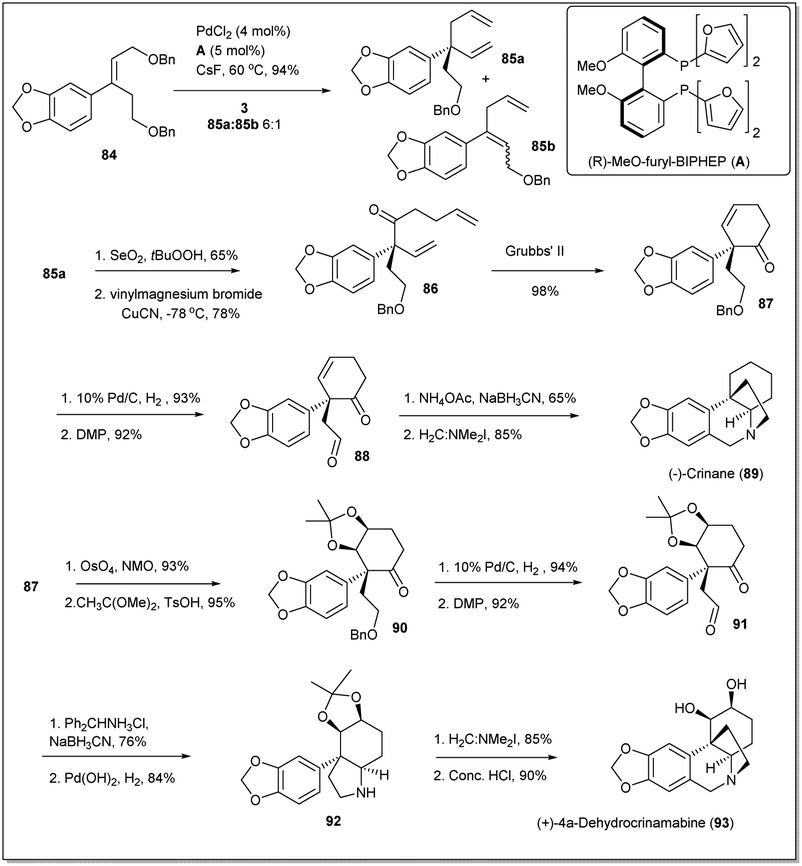
In 2017, Wang and co-workers demonstrated80 a new strategy highlighting a Pd-catalyzed asymmetric allyl–allyl cross-coupling reaction for the synthesis of the cis-3a-arylhydroindole unit with a quaternary carbon center in asymmetric syntheses of crinane alkaloids (Scheme 29). Initially, as the starting material for the cross-coupling reaction, compound 84 was readily synthesized from 1,3-benzodioxole. Though, the coupling reaction was carried out in the presence of different Pd sources and ligands, the best yield was obtained when PdCl2 and ligand A were used. Then, the cross-coupling product 85a was converted to RCM precursor 86 on treatment with selenium dioxide followed by a vinyl Grignard reagent. The diene 86 was subjected to ring closing metathesis in the presence of the Grubbs 2nd generation catalyst (G-II) and the metathesis product 87 was obtained in 98% yield. Then, palladium-catalyzed hydrogenation and DMP oxidation of the cyclohexanone 87 gave γ-keto aldehyde 88 in excellent yield. Finally, (−)-crinane was synthesized through reductive amination of diketo compound 88 followed by a Pictet–Spengler reaction.81 After that, they achieved the first asymmetric total synthesis of (+)-4a-dehydroxycrinamabine from the product 87. A two-step method involving sequential dihydroxylation of 87 with OsO4 followed by acetonide protection proceeded to obtain product in excellent yield. Hydrogenation followed by oxidation of the resulting alcohol gave compound 91, which was subjected to reductive amination with diphenylmethylamine hydrochloride followed by N-deprotection to afford compound 92. Then, addition of the Eschenmoser salt to 92 and deprotection of the acetonide unit gave the final desired product of (+)-4a-dehydroxycrinamabine. Importantly, the method offers an another important route for the syntheses of crinane derivatives, including other amaryllidaceae natural products.
 | ||
| Scheme 29 Asymmetric synthesis of crinane and (+)-4a-dehydroxycrinamabine. | ||
To demonstrate the synthetic utility and efficiency of Cu-catalyzed asymmetric allylic allylation, in 2013, Feringa and co-workers54 attempted a larger scale (5 mmol, 1.5 g) production of the martinelline alkaloid chromene derivative core82,83 from the allyl–allyl cross-coupling product 94 (Scheme 30). Allyl bromide 43a was used as the substrate for the coupling reaction and 5 mol% copper triflate was used as catalyst. Allyl–allyl coupling product 94 was converted to the 6-member exocyclic ring 95 by using a Heck reaction. Ozonolysis followed by reductive amination gave product 99.
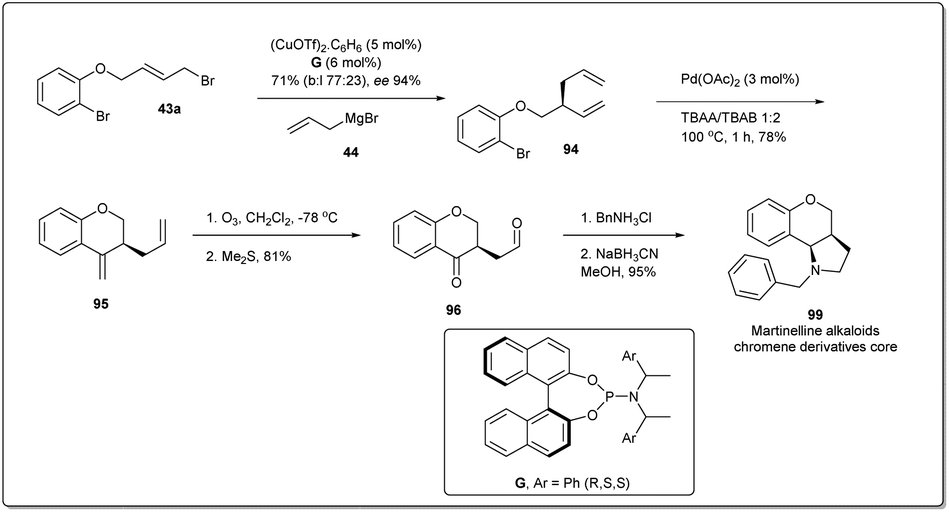 | ||
| Scheme 30 Enantioselective synthesis of a martinelline alkaloid chromene derivative core. | ||
In 2014, rottnestol was synthesized by Hoveyda et al.67 from allyl–allyl cross-coupling product 102, which was constructed from allene derivative 100 and phosphonate 101 (Scheme 31). At first, cross-coupling product 102 was transformed to vinyl iodide 104, which was further subjected to a Suzuki–Miyaura reaction with prebuilt boronate unit 105 in the presence of Pd(dppf)Cl2. Enantiomerically pure rottnestol in gram scale was obtained in 21.5% overall yield after acid treatment of the coupling product.
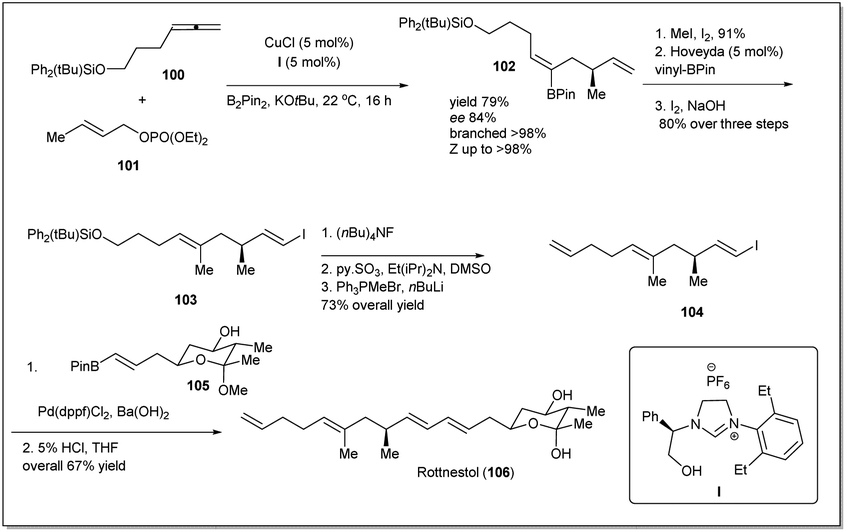 | ||
| Scheme 31 Enantioselective synthesis of rottnestol. | ||
Similarly, Cu–NHC-catalyzed borylative enantioselective allyl–allyl cross-coupling product 108 was used for the synthesis of anti-tumour agent herboxidiene (Scheme 32). At first, alkenyl–B(pin) 108 was converted into vinyl boronate 109, which was treated with a prebuilt vinyl iodide 110 unit in the presence of a phosphine–Pd-catalyst to obtain a Suzuki cross-coupling product. After three steps, 1.03 g of herboxidiene was obtained in 5.5% overall yield, which is more efficient than other previously reported syntheses.
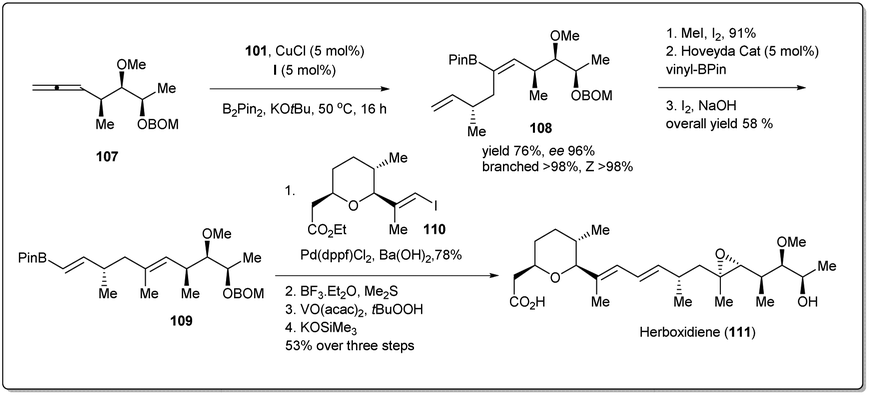 | ||
| Scheme 32 Enantioselective gram-scale synthesis of herboxidiene. | ||
In 2014, Carreira et al. applied46 the new Ir-catalyzed, enantioselective allyl–allyl cross-coupling reaction between secondary alcohols and alkenes to a concise synthesis of γ-secretase modulator JNJ-4041867784,85 to demonstrate the synthetic utility of their reaction (Scheme 33). At first, a suitable alcohol 112 and an alkene 113 were subjected to an allyl–allyl cross-coupling reaction to obtain the desired 1,5-diene 114. Then, chemoselective reduction of the disubstituted alkene, followed by oxidative cleavage of another terminal olefin provided JNJ-40418677 (115).
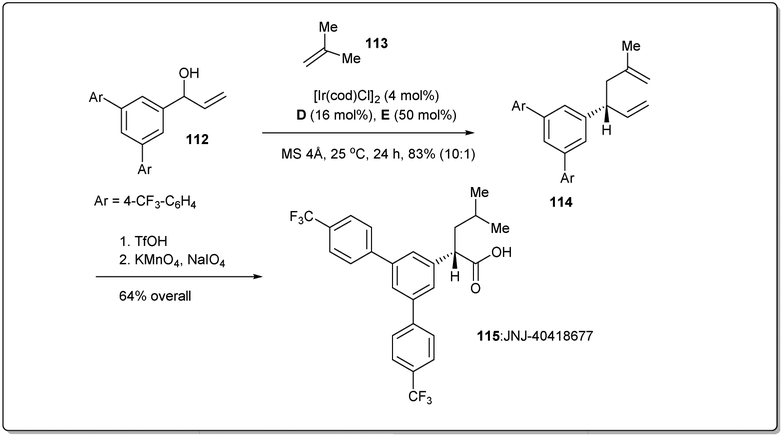 | ||
| Scheme 33 Synthesis of JNJ-40418677. | ||
In 2014, Carreira and co-workers demonstrated47 the importance of the allyl–allyl cross-coupling reaction through a concise enantioselective synthesis of the pyrethroid insecticide protrifenbute (Scheme 34).86 Notably, the cross-coupling reaction was also conducted on a larger scale (1.7 g, 10 mmol of 116) with excellent results. Selective hydroboration of 1,5-diene 117 and subsequent Suzuki coupling gave the product 119. Next, treatment of 119 with zinc carbenoid resulted in the desired product 120.
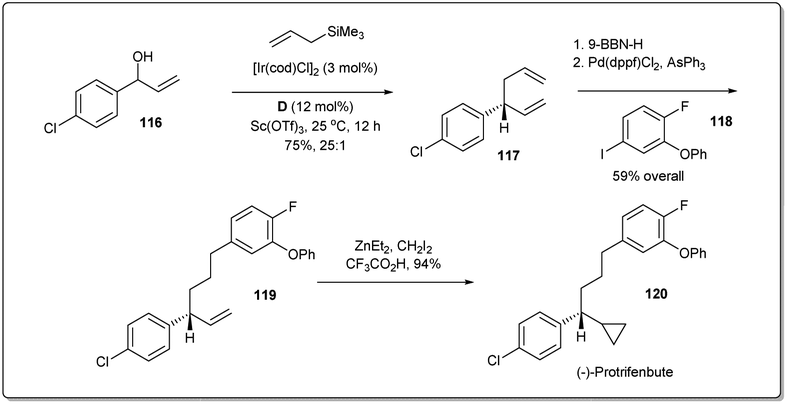 | ||
| Scheme 34 Synthesis of (−)-protrifenbute. | ||
In 2018, Yang and co-workers illustrated49 in a concise catalytic asymmetric synthesis of (−)-preclamol,87 a potent dopaminergic drug, the application of an iridium-catalyzed allyl–allyl cross-coupling reaction between allylboronic ester 3 and racemic secondary allylic alcohol 121 (Scheme 35). The 1,5-diene 122 was then transformed to diol 123 through selective hydroboration–oxidation and oxidative cleavage followed by reductive workup. The advanced intermediate 123 could be further converted into (−)-preclamol with a known three-step sequence.87
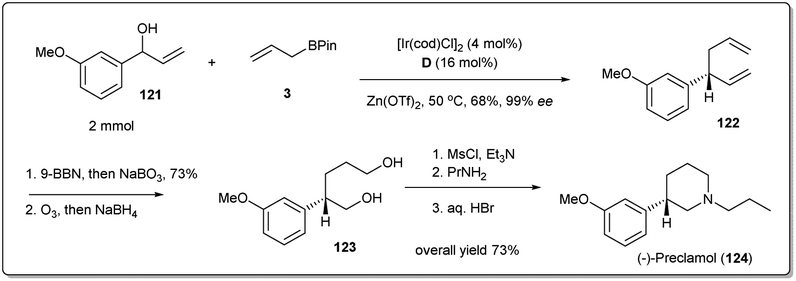 | ||
| Scheme 35 Synthesis of (−)-preclamol. | ||
In 2016, Ding et al. applied41 their palladium-catalyzed allyl–allyl cross-coupling reaction to access the antidepressant drug (−)-paroxetine with high optical purities. At first, racemic Morita–Baylis–Hillman adducts 18 were coupled in the presence of a 0.5 mol% palladium source and 1.25 mol% of ligand C to afford product 125 in 95% enantiomeric excess. Then, after selective epoxidation of an electron-rich double bond followed by oxidative cleavage, compound 126 was formed in 68% overall yield. Reductive amination followed by cyclisation afforded compound 127, which was reduced to primary alcohol 128 in 83% yield. Compound 128 is the key intermediate for the synthesis of (−)-paroxetine and can be converted to the target molecule by following the reported method (Scheme 36).88
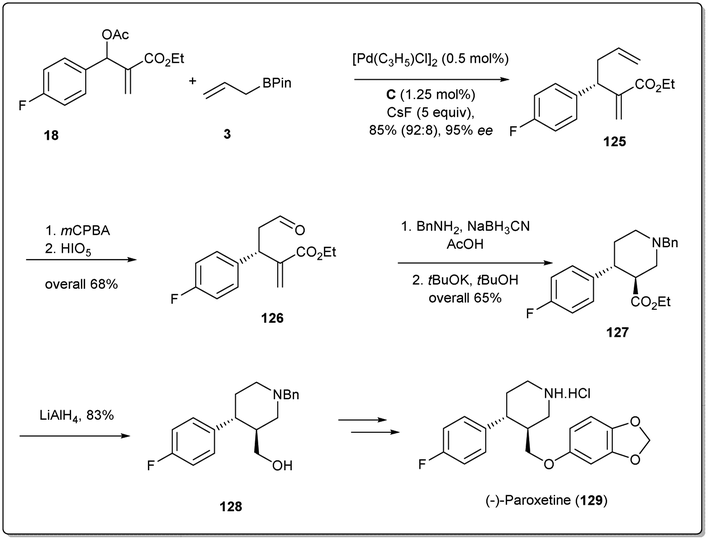 | ||
| Scheme 36 Synthesis of paroxetine. | ||
6. Conclusion and future outlook
Allyl–allyl cross-coupling provides a powerful tool to synthesize compounds containing the 1,5-diene unit. The major breakthroughs have been realized over the past decade in an allyl–allyl cross-coupling strategy, and the application of the new tool for regio- and enantioselective C–C bond formation to form the 1,5-diene has revolutionized synthetic organic chemistry. Thorough mechanistic investigations into reactivity, regio- and enantioselectivity have enhanced the recognition and application of the cross-coupling reaction. Moreover, asymmetric allyl–allyl cross-coupling reactions have been successfully applied in stereoselective total syntheses of a number of complex chiral natural products, providing evidence of the effectiveness of this approach in controlling both regio- and enantioselectivities. From the above discussion, it is evident that the most efficient and most common catalyst for this method is a Pd-based catalyst. However, a number of efforts have extended the scope of the reaction to many other metal catalysts like copper, iridium, nickel etc. The Cu catalysts could be considered as alternatives to the expensive Pd catalysts, as copper is a cheap coinage metal. Although different organometallic reagents have been employed in the cross-coupling reaction, at the present, the most studied allylmetal reagents are allyl boronates. The tendency of allyl–metal intermediates to undergo β-hydride elimination and homocoupling is the main problem in the allyl–allyl cross-coupling reaction. However, the challenges in controlling reaction regio- and enantioselectivity have been addressed and resolved by different groups, and the reaction has proved its power through applications in the synthesis of natural products and medicinally relevant chemicals.Author contributions
Anupam Jana: writing – original draft, writing – review and editing; V. Ravichandiran: writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge generous financial support from the National Institute of Pharmaceutical Education and Research (NIPER)-Hajipur, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India.References
- F. de Meijere and A. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Second Edition, Wiley- VCH, New York, 2008 Search PubMed.
- N. Miyaura and A. Suzuki, Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- F. X. Felpin and S. Sengupta, Biaryl Synthesis with Arenediazonium Salts: Cross-Coupling, CH-Arylation and Annulation Reactions, Chem. Soc. Rev., 2019, 48, 1150–1193 RSC.
- Y. Nakao and T. Hiyama, Silicon-Based Cross-Coupling Reaction: An Environmentally Benign Version, Chem. Soc. Rev., 2011, 40, 4893–4901 RSC.
- M. M. Heravi, V. Zadsirjan, P. Hajiabbasi and H. Hamidi, Advances in Kumada–Tamao–Corriu cross-coupling reaction: an update, Springer Vienna, 2019, vol. 150 Search PubMed.
- C. Cordovilla, C. Bartolomé, J. M. Martínez-Ilarduya and P. Espinet, The Stille Reaction, 38 Years Later, ACS Catal., 2015, 5, 3040–3053 CrossRef CAS.
- R. Jana, T. P. Pathak and M. S. Sigman, Advances in Transition Metal (Pd, Ni, Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-Organometallics as Reaction Partners, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed.
- Nobel Prize Chem. 2010. NobelPrize.org. Nobel Prize Outreach AB 2022. Thu. 19 May 2022. < https//www.nobelprize.org/prizes/chemistry/2010/summary/>.
- Q. Cheng, H. Tu, C. Zheng, J. Qu, G. Helmchen and S. You, Iridium-Catalyzed Asymmetric Allylic Substitution Reactions, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS PubMed.
- A. Alexakis, J. E. Ba, N. Krause, O. Pa and M. Die, Enantioselective Copper-Catalyzed Conjugate Addition and Allylic Substitution Reactions, Chem. Rev., 2008, 108, 2796–2823 CrossRef CAS PubMed.
- S. R. Harutyunyan, T. Den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Catalytic Asymmetric Conjugate Addition and Allylic Alkylation with Grignard Reagents, Chem. Rev., 2008, 108, 2824–2852 CrossRef CAS PubMed.
- B. M. Trost and D. L. Van Vranken, Asymmetric Transition Metal-Catalyzed Allylic Alkylations, Chem. Rev., 1996, 96, 395–422 CrossRef CAS.
- K. B. Selim, Y. Matsumoto, K. Yamada and K. Tomioka, Efficient Chiral N-Heterocyclic Carbene/Copper(I) -Catalyzed Asymmetric Allylic Arylation with Aryl Grignard Reagents, Angew. Chem., 2009, 121, 8889–8891 CrossRef.
- Y. Shido, M. Yoshida, M. Tanabe, H. Ohmiya and M. Sawamura, Copper-Catalyzed Enantioselective Allylic Substitution with Alkylboranes, J. Am. Chem. Soc., 2012, 134, 18573–18576 CrossRef CAS PubMed.
- F. Gao, Y. Lee, K. Mandai and A. H. Hoveyda, Quaternary Carbon Stereogenic Centers through Copper-Catalyzed Enantioselective Allylic Substitutions with Readily Accessible Aryl- or Heteroaryllithium Reagents and Aluminum Chlorides, Angew. Chem., Int. Ed., 2010, 49, 8370–8374 CrossRef CAS PubMed.
- F. Gao, J. L. Carr and A. H. Hoveyda, Copper-Catalyzed Enantioselective Allylic Substitution with Readily Accessible Carbonyl- and Acetal-Containing Vinylboron Reagents, Angew. Chem., 2012, 124, 6717–6721 CrossRef.
- B. Jung and A. H. Hoveyda, Site- and Enantioselective Formation of Allene-Bearing Tertiary or Quaternary Carbon Stereogenic Centers through NHC-Cu-Catalyzed Allylic Substitution, J. Am. Chem. Soc., 2012, 134, 1490–1493 CrossRef CAS PubMed.
- J. Dabrowski, F. Gao and A. H. Hoveyda, Enantioselective Synthesis of Alkyne-Substituted Quaternary Carbon, J. Am. Chem. Soc., 2011, 2, 4778–4781 CrossRef PubMed.
- E. Breitmaier, Terpenes, Flavors, Fragrances, Pharmaca, Pheromones, Wiley-VCH, Weinheim, 2006 Search PubMed.
- Medicinal Natural Products: A Biosynthetic Approach, ed. P. M. Dewick, Wiley, Chichester, 2002 Search PubMed.
- Y. Nakamura and H. Yamamoto, Handbook of Organopalladium Chemistry for Organic Synthesis: Vol. 2, Wiley-Interscience, West Lafayette, 2002 Search PubMed.
- J. A. Feducia, M. R. Gagne, C. Hill and N. Carolina, Reversibility Effects on the Stereoselectivity of Pt(II) -Mediated Cascade Poly-Ene Cyclizations Multicyclic Steroid-like Structures Stands as One of the Crowning, J. Am. Chem. Soc., 2008, 130, 592–599 CrossRef CAS PubMed.
- A. Cope, R. J. Felix, D. Weber, O. Gutierrez, D. J. Tantillo and M. R. Gagne, A Gold-Catalysed Enantioselective Cope Rearrangement of Achiral 1,5-Dienes, Nat. Chem., 2012, 4, 405–409 CrossRef PubMed.
- Y. Zhao, S. Chng and T. Loh, Lewis Acid-Promoted Intermolecular Acetal-Initiated Cationic Polyene Cyclizations, J. Am. Chem. Soc., 2007, 129, 492–493 CrossRef CAS PubMed.
- P. Zhang, L. A. Brozek and J. P. Morken, Pd-Catalyzed Enantioselective Allyl-Allyl Cross-Coupling, J. Am. Chem. Soc., 2010, 132, 10686–10688 CrossRef CAS PubMed.
- H. M. Trost, Allylstannanes as Electrofugal Partners in Allylic Alkylation, Tetrahedron Lett., 1980, 21, 2595–2598 CrossRef.
- J. Godschalx and J. K. Stille, Catalyzed Cross-Coupling of Allyl Bromides with Allyl Tin Reagents, Tetrahedron Lett., 1980, 21, 2599–2602 CrossRef CAS.
- A. Goliaszewski and J. Schwartz, Carbon-Carbon Bond Formation by Induced Elimination from Unsymmetrically Substituted (AllylXallyl′)Opalladium Complexes, J. Am. Chem. Soc., 1984, 106, 5028–5030 CrossRef CAS.
- E. F. Flegeau, U. Schneider and S. Kobayashi, Palladium(0) versus Nickel(0) Catalysis in Selective Functional- Grouptolerant Sp3-Sp3 Carbon-Carbon Bond Formations, Chem. – Eur. J., 2009, 15, 12247–12254 CrossRef CAS PubMed.
- J. E. Marcone and K. G. Moloy, Kinetic Study of Reductive Elimination from the Complexes (Diphosphine) Pd (R)(CN), J. Am. Chem. Soc., 1998, 120, 8527–8528 CrossRef CAS.
- E. A. Broger, J. Foricher, B. Heiser and R. Schmid, U.S. Patent5274125, 1993 Search PubMed.
- T. Imamoto, K. Sugita and K. Yoshida, An Air-Stable P-Chiral Phosphine Ligand for Highly Enantioselective Transition-Metal-Catalyzed Reactions, J. Am. Chem. Soc., 2005, 127, 11934–11935 CrossRef CAS PubMed.
- M. J. Ardolino and J. P. Morken, Branched/Linear Selectivity in Palladium-Catalyzed Allyl-Allyl Cross-Couplings: The Role of Ligands, Tetrahedron, 2015, 71, 6409–6413 CrossRef CAS.
- P. W. N. M. Van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Ligand Bite Angle Effects in Metal-Catalyzed C-C Bond Formation, Chem. Rev., 2000, 100, 2741–2769 CrossRef CAS PubMed.
- P. Zhang, H. Le, R. E. Kyne and J. P. Morken, Enantioselective Construction of All-Carbon Quaternary Centers by Branch-Selective Pd-Catalyzed Allyl–Allyl Cross-Coupling, J. Am. Chem. Soc., 2011, 133, 9716–9719 CrossRef CAS PubMed.
- M. J. Ardolino and J. P. Morken, Congested C−C Bonds by Pd-Catalyzed Enantioselective Allyl−Allyl Cross-Coupling, a Mechanism-Guided Solution, J. Am. Chem. Soc., 2014, 136, 7092–7100 CrossRef CAS PubMed.
- L. A. Brozek, M. J. Ardolino and J. P. Morken, Diastereocontrol in Asymmetric Allyl-Allyl Cross-Coupling: Stereocontrolled Reaction of Prochiral Allylboronates with Prochiral Allyl Chlorides, J. Am. Chem. Soc., 2011, 133, 16778–16781 CrossRef CAS PubMed.
- D. J. Cárdenas and A. M. Echavarren, Mechanistic aspects of C–C bond formation involving allylpalladium complexes: the role of computational studies, New J. Chem., 2004, 28, 338–347 RSC.
- H. Le, R. E. Kyne, L. A. Brozek and J. P. Morken, Catalytic Enantioselective Allyl-Allyl Cross-Coupling with a Borylated Allylboronate, Org. Lett., 2013, 15, 1432–1435 CrossRef CAS PubMed.
- H. Le, A. Batten and J. P. Morken, Catalytic Stereospecific Allyl-Allyl Cross-Coupling of Internal Allyl Electrophiles with AllylB(Pin), Org. Lett., 2014, 16, 2096–2099 CrossRef CAS PubMed.
- X. Wang, X. Wang, Z. Han, Z. Wang and K. Ding, Palladium-Catalyzed Asymmetric Allylic Allylation of Racemic Morita–Baylis–Hillman Adducts, Angew. Chem., Int. Ed., 2017, 56, 1116–1119 CrossRef CAS PubMed.
- J. Li, N. Zheng, X. Duan, R. Li and W. Song, Palladium-Catalyzed Regioselective and Diastereoselective C-Glycosylation by Allyl-Allyl Coupling, Adv. Synth. Catal., 2021, 363, 846–850 CrossRef CAS.
- X. Zhou, G. Zhang, R. Huang and H. Huang, Palladium-Catalyzed Allyl−Allyl Reductive Coupling of Allylamines or Allylic Alcohols with H2 as Sole Reductant, Org. Lett., 2021, 23, 365–369 CrossRef CAS PubMed.
- M. Estaitie and D. G. Hall, Regiocontrolled synthesis of enantioenriched 2-substituted dehydropiperidines by stereospecific allyl–allyl cross-coupling of a chiral allylic boronate, Chem. Commun., 2022, 58, 1370–1373 RSC.
- R. Takeuchi and M. Kashio, Highly Selective Allylic Alkylation with a Carbon Nucleophile at the More Substituted Allylic Terminus Catalyzed by an Iridium Complex: An Efficient Method for Constructing Quaternary Carbon Centers, Angew. Chem., Int. Ed. Engl., 1997, 36, 263–265 CrossRef CAS.
- J. Y. Hamilton, D. Sarlah and E. M. Carreira, Iridium-Catalyzed Enantioselective Allyl-Alkene Coupling, J. Am. Chem. Soc., 2014, 136, 3006–3009 CrossRef CAS PubMed.
- J. Y. Hamilton, N. Hauser, D. Sarlah and E. M. Carreira, Iridium-Catalyzed Enantioselective Allyl-Allylsilane Cross-Coupling, Angew. Chem., Int. Ed., 2014, 53, 10759–10762 CrossRef CAS PubMed.
- Q. Yuan, K. Yao, D. Liu and W. Zhang, Iridium-Catalyzed Allyl-Allyl Cross-Coupling of Allylic Carbonates with (E)-1,3-Diarylpropenes, Chem. Commun., 2015, 51, 11834–11836 RSC.
- Y. Zheng, B. B. Yue, K. Wei and Y. R. Yang, Iridium-Catalyzed Enantioselective Allyl-Allyl Cross-Coupling of Racemic Allylic Alcohols with Allylboronates, Org. Lett., 2018, 20, 8035–8038 CrossRef CAS PubMed.
- S. Porcel, V. López-Carrillo, C. García-Yebra and A. M. Echavarren, Gold-Catalyzed Allyl-Allyl Coupling, Angew. Chem., Int. Ed., 2008, 47, 1883–1886 CrossRef CAS PubMed.
- M. Yamanaka, S. Kato and E. Nakamura, Mechanism and Regioselectivity of Reductive Elimination of π-Allylcopper(III) Intermediates, J. Am. Chem. Soc., 2004, 126, 6287–6293 CrossRef CAS PubMed.
- E. R. Bartholomew, S. H. Bertz, S. Cope, M. Murphy and C. A. Ogle, Preparation of σ- and π-Allylcopper(III) Intermediates in SN2 and SN2′ Reactions of Organocuprate(I) Reagents with Allylic Substrates, J. Am. Chem. Soc., 2008, 130, 11244–11245 CrossRef CAS PubMed.
- A. S. E. Karlström and J. Bäckvall, Experimental Evidence Supporting a CuIII Intermediate in Cross-Coupling Reactions of Allylic Esters with Diallylcuprate Species, Chem. – Eur. J., 2001, 7, 1981–1989 CrossRef.
- V. Hornillos, M. Pérez, M. Fañanás-Mastral and B. L. Feringa, Copper-Catalyzed Enantioselective Allyl − Allyl Cross-Coupling, J. Am. Chem. Soc., 2013, 135, 2140–2143 CrossRef CAS PubMed.
- H. Urabe, K. Suzuki and F. Sato, Intramolecular Cyclization of 2,7- or 2,8-Bis-Unsaturated Esters Mediated by (H2-Propene)Ti(O-i-Pr)2. Facile Construction of Mono- and Bicyclic Skeletons with Stereoselective Introduction of a Side Chain. A Synthesis of d-Sabinene, J. Am. Chem. Soc., 1997, 119, 10014–10027 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, A Stable Crystalline Carbene, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- Science of Synthesis, in N-Heterocyclic Carbenes in Catalytic Organic Synthesis 1, ed. S. Nolan, Georg Thieme Verlag KG, 2017 Search PubMed.
- A. Jana and K. Grela, Mild Functionalization of Tetraoxane Derivatives via Olefin Metathesis: Compatibility of Ruthenium Alkylidene Catalysts with Peroxides, Org. Lett., 2017, 19, 520–523 CrossRef CAS PubMed.
- A. Jana, K. Misztal, A. Zak and K. Grela, Synthesis of Selectively Substituted or Deuterated Indenes via Sequential Pd and Ru Catalysis, J. Org. Chem., 2017, 82, 4226–4234 CrossRef CAS PubMed.
- A. Jana, D. Trzybiński, K. Woźniak and K. Grela, Well-Defined Chiral Copper NHC Complex in the Asymmetric Conjugated β-Borylation and One-Pot Metathesis-Asymmetric β-Borylation Reactions, Chem. – Eur. J., 2018, 24, 891–897 CrossRef CAS.
- A. Jana and K. Grela, Forged and Fashioned for Faithfulness - Ruthenium Olefin Metathesis Catalysts Bearing Ammonium Tags, Chem. Commun., 2017, 54, 122–139 RSC.
- S. Planer, A. Jana and K. Grela, Ethyl Lactate: A Green Solvent for Olefin Metathesis, ChemSusChem, 2019, 12, 4655–4661 CrossRef CAS PubMed.
- A. Jana, G. K. Zieliński, S. Czarnocka-Śniadała, K. Grudzień, D. Podwysocka, M. Szulc, A. Kajetanowicz and K. Grela, Synthesis of Substituted β-Functionalised Styrenes by Microwave-Assisted Olefin Cross-Metathesis and Scalable Synthesis of Apremilast, ChemCatChem, 2019, 11, 5808–5813 CrossRef CAS.
- A. Jana, V. Ravichandiran and S. P. Swain, Application of Organometallic Catalysts for the Synthesis of o-Tolyl Benzonitrile, a Key Starting Material for Sartans, New J. Chem., 2021, 45, 17753–17771 RSC.
- Y. Yasuda, H. Ohmiya and M. Sawamura, Copper-Catalyzed Enantioselective Allyl–Allyl Coupling between Allylic Boronates and Phosphates with a Phenol/N-Heterocyclic Carbene Chiral Ligand, Angew. Chem., Int. Ed., 2016, 55, 10816–10820 CrossRef CAS PubMed.
- Y. Yasuda, H. Ohmiya and M. Sawamura, Copper-Catalyzed Enantioselective Coupling between Allyl-Boronates and Phosphates Using a Phenol-Carbene Chiral Ligand: Asymmetric Synthesis of Chiral Branched 1,5-Dienes, Synthesis, 2018, 50, 2235–2246 CrossRef CAS.
- F. Meng, K. P. McGrath and A. H. Hoveyda, Multifunctional Organoboron Compounds for Scalable Natural Product Synthesis, Nature, 2014, 513, 367–374 CrossRef CAS PubMed.
- K. Semba, N. Bessho, T. Fujihara, J. Terao and Y. Tsuji, Copper-Catalyzed Borylative Allyl-Allyl Coupling Reaction, Angew. Chem., Int. Ed., 2014, 53, 9007–9011 CrossRef CAS PubMed.
- G. Xu, B. Fu, H. Zhao, Y. Li, G. Zhang, Y. Wang, T. Xiong and Q. Zhang, Enantioselective and Site-Specific Copper-Catalyzed Reductive Allyl-Allyl Cross-Coupling of Allenes, Chem. Sci., 2019, 10, 1802–1806 RSC.
- A. Jiménez-Aquino, E. Ferrer Flegeau, U. Schneider and S. Kobayashi, Catalytic Intermolecular Allyl-Allyl Cross-Couplings between Alcohols and Boronates, Chem. Commun., 2011, 47, 9456–9458 RSC.
- D. W. Ji, G. C. He, W. S. Zhang, C. Y. Zhao, Y. C. Hu and Q. A. Chen, Nickel-Catalyzed Allyl-Allyl Coupling Reactions between 1,3-Dienes and Allylboronates, Chem. Commun., 2020, 56, 7431–7434 RSC.
- Y. Gan, H. Hu and Y. Liu, Nickel-Catalyzed Homo- And Cross-Coupling of Allyl Alcohols via Allyl Boronates, Org. Lett., 2020, 22, 4418–4423 CrossRef CAS PubMed.
- F. Ding, R. William, F. Wang and X. W. Liu, Triflimide-Catalyzed Allyl–Allyl Cross-Coupling: A Metal-Free Allylic Alkylation, Chem. Commun., 2012, 48, 8709–8711 RSC.
- M. Ellwart, I. S. Makarov, F. Achrainer, H. Zipse and P. Knochel, Regioselective Transition-Metal-Free Allyl–Allyl Cross-Couplings, Angew. Chem., Int. Ed., 2016, 55, 10502–10506 CrossRef CAS PubMed.
- L. Zhang, Z. Chen, H. Li, W. Yin, J. Xu, M. Miao and H. Ren, Transition Metal-Free Tandem Pyridinyl–Allyl–Allyl Cross-Coupling Reaction, Synth. Commun., 2017, 47, 1668–1675 CrossRef CAS.
- P. S. Baran, Natural Product Total Synthesis: As Exciting as Ever and Here to Stay, J. Am. Chem. Soc., 2018, 140, 4751–4755 CrossRef CAS PubMed.
- L. Li, Z. Chen, X. Zhang and Y. Jia, Divergent Strategy in Natural Product Total Synthesis, Chem. Rev., 2018, 118, 3752–3832 CrossRef CAS PubMed.
- A. I. Meyers and B. A. Lefker, Chiral Bicyclic Lactams for Asymmetric Synthesis of Quaternary Carbons. The Total Synthesis of (-)-a-Cuparenone, J. Org. Chem., 1986, 51, 1541–1544 CrossRef CAS.
- A. Natarajan, D. Ng, Z. Yang and M. A. Garcia-Garibay, Parallel Syntheses of (+)- and (-)-α-Cuparenone by Radical Combination in Crystalline Solids, Angew. Chem., Int. Ed., 2007, 46, 6485–6487 CrossRef CAS PubMed.
- Y. R. Gao, D. Y. Wang and Y. Q. Wang, Asymmetric Syntheses of Amaryllidaceae Alkaloids (-)-Crinane and (+)-4a-Dehydroxycrinamabine, Org. Lett., 2017, 19, 3516–3519 CrossRef CAS PubMed.
- L. Li, Q. Yang, Y. Wang and Y. Jia, Catalytic Asymmetric Total Synthesis of (-)-Galanthamine and (-)-Lycoramine, Angew. Chem., Int. Ed., 2015, 54, 6255–6259 CrossRef CAS PubMed.
- S. Ikeda, M. Shibuya and Y. Iwabuchi, Asymmetric Total Synthesis of Martinelline and Martinellic Acid, Chem. Commun., 2007, 504–506 RSC.
- N. Arumugam, R. Raghunathan, A. I. Almansour and U. Karama, An Efficient Synthesis of Highly Functionalized Novel Chromeno[4,3-b] Pyrroles and Indolizino[6,7-b]Indoles as Potent Antimicrobial and Antioxidant Agents, Bioorg. Med. Chem. Lett., 2012, 22, 1375–1379 CrossRef CAS PubMed.
- C. Y. Ho, WO2009052341A1, 2009.
- A. R. B. Van Broeck, J.-M. Chen, G. Tréton, M. Desmidt, C. Hopf, N. Ramsden, E. Karran and M. Mercken, Chronic Treatment with a Novel G-Secretase Modulator, JNJ-40418677, Inhibits Amyloid Plaque Formation in a Mouse Model of Alzheimer's Disease, Br. J. Pharmacol., 2011, 163, 375–389 CrossRef PubMed.
- T. G. Cullen, P. E. P. Brust, G. A. Meier and J. F. Engel, US5223536A, 1993.
- J. Hu, Y. Lu, Y. Li and J. Zhou, Highly Active Catalysts of Bisphosphine Oxides for Asymmetric Heck Reaction, Chem. Commun., 2013, 49, 9425–9427 RSC.
- D. A. Devalankar, P. U. Karabal and A. Sudalai, Optically Pure γ-Butyrolactones and Epoxy Esters via Two Stereocentered HKR of 3-Substituted Epoxy Esters: A Formal Synthesis of (-)-Paroxetine, Ro 67-8867 and (+)-Eldanolide, Org. Biomol. Chem., 2013, 11, 1280–1285 RSC.
| This journal is © the Partner Organisations 2023 |