
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tumor microenvironment responsive nanocarriers for efficient antisense DNA delivery and enhanced chemodynamic therapy†
Gowtham
Raj
a,
Vasudev
D. S.
a,
Nikhil Dev
Narendradev
 b,
Viswa Kalyan Kumar
Dommeti
a,
Saurabh
Shriwas
a,
P. M.
Ajay Sekhar
a,
Leah Susan
Jacob
b,
S. Murty
Srinivasula
b and
Reji
Varghese
*a
b,
Viswa Kalyan Kumar
Dommeti
a,
Saurabh
Shriwas
a,
P. M.
Ajay Sekhar
a,
Leah Susan
Jacob
b,
S. Murty
Srinivasula
b and
Reji
Varghese
*a
aSchool of Chemistry, Indian Institute of Science Education and Research (IISER), Thiruvananthapuram, Trivandrum-695551, Kerala, India. E-mail: reji@iisertvm.ac.in
bSchool of Biology, Indian Institute of Science Education and Research (IISER), Thiruvananthapuram, Trivandrum-695551, Kerala, India
First published on 20th March 2023
Abstract
Cancer is undoubtedly one of the major threats to mankind. Herein, we demonstrate a supramolecular approach for the design of tumour microenvironment sensitive nanoparticles composed of artemisinin, glutathione-responsive and redox-active ferrocene and antisense DNA via a single-step assembly. Assembly between the antisense DNA and ferrocene derivative results in the noncovalent tethering of the latter along the backbone of antisense DNA and leads to the formation of the corresponding supramolecular amphiphile. The addition of hydrophobic artemisinin during the assembly of the supramolecular amphiphile permits the encapsulation of artemisinin and leads to the formation of glutathione-responsive NPs composed of antisense DNA, ferrocene derivative and artemisinin. The most remarkable feature of our design strategy is the glutathione-triggered cascade reactions of the NPs inside the cancer cell in a cooperative fashion. Cleavage of disulfide bonds of the NPs by the overexpressed glutathione in the tumour microenvironment leads to the following cascade chemodynamic therapy reactions: (i) glutathione-triggered uncaging of the ferrocene derivative to release amino ferrocene, which then undergoes reaction with endogenous hydrogen peroxide to produce ˙OH radicals and Fe3+ and (ii) the in situ reduction of Fe3+ by glutathione to form Fe2+ and the subsequent Fe2+-triggered cleavage of the peroxide bond of artemisinin to form carbon-centered free radicals. Furthermore, disassembly of NPs releases antisense DNA, which efficiently downregulates the translation of Bcl-2 mRNA and thereby causes cell apoptosis. Excellent anticancer efficacy is demonstrated by using HeLa cells as a representative example and the enhanced therapeutic efficacy of our approach is attributed to the co-operative cascade CDT and antisense therapy.
Introduction
Cancer is undoubtedly one of the major causes of death to mankind. Recent years have witnessed the emergence of chemodynamic therapy (CDT) as a powerful strategy for the treatment of cancer.1,2 Chemodynamic therapy relies on the generation of highly cytotoxic reactive oxygen species (ROS) through Fenton or Fenton-like reactions by exploring the endogenous H2O2 present inside the cancer cells.3–5 Typically, CDT involves the delivery of a suitable Fenton catalyst into the cancer cells, which catalyses the conversion of endogenous H2O2 present inside the cells to the cytotoxic ROS that causes oxidative damage to DNA, proteins and lipids, which eventually leads to cell death. Even though the concentration of endogenous H2O2 inside the cancer cells (50–100 μM) is significantly higher when compared to healthy cells, it is still insufficient to perform an efficient Fenton reaction and hence traditional CDT suffers from poor therapeutic efficacy. One way to address this challenge is through the direct delivery of H2O2 and Fenton catalyst to the cancer cell by using a nanocarrier. Alternatively, the intratumoral H2O2 level can be boosted by delivering H2O2 generating natural enzymes such as glucose oxidase,6,7 or nicotinamide adenine dinucleotide phosphate oxidase.8,9 Though significant improvements have been achieved by using these strategies, the therapeutic efficacy of these approaches is not impressive for clinical applications. Therefore, the development of novel strategies that permit the elevation of the intratumoral H2O2 level is extremely important for effective CDT.10–16To overcome the above-mentioned challenges, an ideal strategy would be the design of nanoagents that generate ROS in an H2O2 independent pathway in addition to the hydroxyl radicals generated by conventional CDT so that the overall efficacy of CDT can be significantly improved. This is typically achieved by delivering any peroxide linkage containing molecule into the cancer cell, which undergoes peroxide cleavage in the presence of metal ions (Fe2+) and increases the radical species inside the cells and leads to cell apoptosis.17 Artemisinin (ART), a popular antimalarial drug, is an organic molecule having an endoperoxide bridge and has recently been identified as a potential anticancer agent.18 Though various factors are responsible for its anticancer activity, the generation of cytotoxic carbon-centered free radicals by Fe2+-catalysed cleavage of the peroxide bond of ART is proven to be the major cause of its activity.19–21 This suggests that ART could be a potential candidate as a peroxide containing molecule that can generate cytotoxic radical species in Fe2+-catalysed reaction and hence would be a promising molecule for CDT. Two major hurdles that limit the scope of ART for CDT applications are (i) poor water solubility of ART and (ii) lack of control over the delivery of ART and the metal ions. The poor water solubility of ART can be addressed to a large extent by the use of a suitable nanocarrier as the delivery vehicle, but the controlled delivery of ART and metal ions into the cancer cell remains challenging. Furthermore, the lack of adequate concentration of Fe2+ inside the cancer cell is another obstacle for the therapeutic efficacy of CDT. Hence, the design of nanocarriers that permit the tumour microenvironment-triggered co-delivery of ART and Fe2+ in a controlled fashion would be the ideal approach for an efficient CDT.
Nanostructures derived from DNA have received enormous attention in recent years as a stimuli-responsive delivery vehicle for various biomedical applications.22–25 This is mainly due to their remarkable structural and functional characteristics including (i) excellent biocompatibility, (ii) stimuli responsive nature, (iii) enhanced cell permeability and (iv) high resistance to enzymatic degradation.26–32 In addition, an anionic phosphodiester backbone or the nucleobases of DNA can act as a ligand for the coordination of various metal ions, which has been extensively applied for the crafting of DNA-metal condensates and for the development of various anticancer drugs.33 Metal bound DNA NPs have also been explored as responsive nanoagents for CDT, which showed enhanced generation of ROS.34,35 Since this approach involves the direct binding of metal ions onto the anionic backbone of DNA by means of strong electrostatic interactions, these systems lack stimuli-responsiveness for the disassembly of NPs inside the cancer cell. Moreover, this approach may damage the functional properties of DNA by the reaction with the in situ generated radicals and potentially reduce the therapeutic efficacy of functional DNA.
Herein, we report a novel supramolecular strategy for the design of tumour microenvironment sensitive DNA nanoparticles (NPs) composed of ART, glutathione (GSH) responsive and redox-active ferrocene (1) and antisense DNA (DNA1) via a single-step assembly, guided mainly by electrostatic interactions. The redox-active ferrocene 1 is functionalized with an ammonium group at the end of the alkyl chain through a GSH responsive disulfide bond. Assembly between the antisense DNA (DNA1) and 1 results in the noncovalent tethering of 1 along the anionic backbone of DNA1 and leads to the formation of supramolecular amphiphile, DNA1@1. The addition of hydrophobic ART during the assembly of DNA1@1 permits the encapsulation of ART inside the hydrophobic pockets of DNA1@1 and leads to the formation of GSH-responsive DNA1@1-ART NPs. The presence of a disulfide bond in the NPs not only permits their GSH-triggered disassembly inside the cancer cell, but also helps to deplete the GSH concentration, which is advantageous for efficient CDT. In addition, a disulfide bond keeps the ferrocene in a caged state (prodrug) and the GSH-triggered uncaging of ferrocene leads to the generation of ROS, which makes DNA less susceptible to oxidative damage caused by the in situ radicals.36,37 The most remarkable feature of our design strategy is the GSH-triggered cascade reactions of the NPs inside the cancer cell in a cooperative fashion for synergistic cancer therapy. Cleavage of disulfide bonds of DNA1@1-ART NPs by the overexpressed GSH in the tumour microenvironment leads to the following cascade CDT reactions: (i) GSH-triggered uncaging of 1 releases amino ferrocene, which then undergoes reaction with endogenous H2O2 to produce ˙OH radicals and Fe3+ and (ii) the in situ reduction of Fe3+ by GSH to form Fe2+ and the subsequent Fe2+-triggered cleavage of the peroxide bond of ART to form carbon-centered free radicals (H2O2 independent pathway). Furthermore, disassembly of DNA1@1-ART NPs releases DNA1, which is an 18 mer oligonucleotide complementary to the first six codons of Bcl-2 (anti-apoptotic) mRNA, which efficiently downregulates the translation of Bcl-2 mRNA (anti-apoptotic) and thereby causes cell apoptosis. Excellent anticancer efficacy is demonstrated by using HeLa cells as a representative example and the enhanced therapeutic efficacy of our approach is attributed to the cooperative cascade CDT and antisense therapy (Scheme 1).
 | ||
| Scheme 1 Schematic representation depicting the formation of a DNA1@1 supramolecular amphiphile by the noncovalent tethering of 1 along DNA1 and its subsequent amphiphilicity-driven self-assembly in the presence of ART to the formation of DNA1@1-ART NPs. The cellular internalization of DNA1@1-ART NPs followed by the GSH-triggered cascade reactions leading to the release of antisense DNA (DNA1), ˙OH and carbon-centered radicals through Fenton and Fenton-like reaction for synergistic cancer therapy are also shown. | ||
Results and discussion
Synthesis of cationic ferrocene probe 1 was achieved through multistep organic reactions and the details are provided in the ESI.†DNA1 is an 18-mer antisense DNA (G3139), which was synthesized on a DNA synthesizer using standard protocols. Sequences of all DNAs used in our study are tabulated in Table 1. In a typical procedure, synthesis of DNA1@1 and DNA1@1-ART NPs was achieved by annealing a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of cationic ferrocene probe 1 (2 μM) and DNA1 (0.1 μM) in deionized water at 90 °C for 10 minutes followed by slow cooling to room temperature at a cooling rate of 5 °C min−1. The molar ratio of 20:1 was used in our studies after confirming that the NP formation was complete only at this molar ratio. A molar ratio of 1 less than 20 always leads to incomplete formation of the NPs (Fig. S1, ESI†). During the annealing process, ART was added to the solution at 60 °C for the synthesis of DNA1@1-ART NPs. After aging this solution for an additional 12 h, DNA1@1 or DNA1@1-ART aggregated species were centrifuged using a molecular weight cut-off filter at a speed of 4000 rpm for 10 min in order to remove any unbound ART or 1 from the solution. Atomic force (AFM) and transmission electron (TEM) microscopic analyses revealed the formation of spherical NPs for DNA1@1 aggregates (Fig. 1a and b), respectively. Nanoparticle formation was further supported by confocal laser scanning microscopic (CLSM) analyses by using Cy3.5 labelled DNA1 (DNA2), which showed the formation of fluorescent NPs (green, DNA2@1) (Fig. 1c). Dynamic light scattering (DLS) analyses of DNA1@1 aggregates confirmed the formation of spherical NPs in solution with an average diameter of ∼150 nm (Fig. 1d). It is to be noted that though 1 is amphiphilic in nature, self-assembly of 1 in the absence of DNA1 resulted only in the formation of ill-defined aggregates, disclosing the unique role of DNA as a template in controlling the morphology of the aggregated species (Fig. S1 and S2, ESI†). The encapsulation of ART was confirmed by comparing the absorption spectrum of free ART with the corresponding DNA1@1-ART NPs. A strong absorption band centred at 290 nm was observed for free ART (5 μM in DMSO) containing NaOH in water (50 mM). Whereas, no noticeable absorption was observed for the filtrate obtained after centrifuging DNA1@1-ART NPs using a molecular weight cut off filter, clearly indicating the efficient encapsulation of ART inside the NPs (Fig. 1e). The loading efficiency of ART was calculated to be 80% (Fig. S3, ESI†). Moreover, no significant change in the size of the NPs was observed before and after ART encapsulation (Fig. S5, ESI†). Energy-dispersive X-ray spectroscopy (EDS) analyses of DNA1@1 NPs revealed the presence of iron and sulfur from 1, and phosphorous from DNA1 (Fig. S4a, ESI†). The zeta potential measurement of DNA1@1 NPs showed a value of −1.46 mV, whereas DNA1 and 1 exhibited values of −9.72 and +9.64 mV, respectively. The almost neutral zeta potential value for DNA1@1 is indicative of the formation of DNA1@1 NPs (Fig. 1f). The X-ray photoelectron spectroscopy (XPS) analyses of DNA1@1 NPs revealed peaks at 710.0 and 723.6 eV, which is due to the presence of Fe(II) 2p3/2 and Fe(II) 2p1/2 ions, respectively (Fig. S4b, ESI†). Based on these results, the formation of NPs can be envisaged to be the initial formation of supramolecular amphiphile DNA1@1 by the non-covalent tethering of 1 along the anionic backbone of DNA1 and the subsequent amphiphilicity-driven self-assembly of DNA1@1 leads to the formation of DNA1@1 NPs and in the presence of hydrophobic ART self-assembly results in the formation of ART encapsulated NPs (DNA1@1-ART NPs).
:1 molar ratio of cationic ferrocene probe 1 (2 μM) and DNA1 (0.1 μM) in deionized water at 90 °C for 10 minutes followed by slow cooling to room temperature at a cooling rate of 5 °C min−1. The molar ratio of 20:1 was used in our studies after confirming that the NP formation was complete only at this molar ratio. A molar ratio of 1 less than 20 always leads to incomplete formation of the NPs (Fig. S1, ESI†). During the annealing process, ART was added to the solution at 60 °C for the synthesis of DNA1@1-ART NPs. After aging this solution for an additional 12 h, DNA1@1 or DNA1@1-ART aggregated species were centrifuged using a molecular weight cut-off filter at a speed of 4000 rpm for 10 min in order to remove any unbound ART or 1 from the solution. Atomic force (AFM) and transmission electron (TEM) microscopic analyses revealed the formation of spherical NPs for DNA1@1 aggregates (Fig. 1a and b), respectively. Nanoparticle formation was further supported by confocal laser scanning microscopic (CLSM) analyses by using Cy3.5 labelled DNA1 (DNA2), which showed the formation of fluorescent NPs (green, DNA2@1) (Fig. 1c). Dynamic light scattering (DLS) analyses of DNA1@1 aggregates confirmed the formation of spherical NPs in solution with an average diameter of ∼150 nm (Fig. 1d). It is to be noted that though 1 is amphiphilic in nature, self-assembly of 1 in the absence of DNA1 resulted only in the formation of ill-defined aggregates, disclosing the unique role of DNA as a template in controlling the morphology of the aggregated species (Fig. S1 and S2, ESI†). The encapsulation of ART was confirmed by comparing the absorption spectrum of free ART with the corresponding DNA1@1-ART NPs. A strong absorption band centred at 290 nm was observed for free ART (5 μM in DMSO) containing NaOH in water (50 mM). Whereas, no noticeable absorption was observed for the filtrate obtained after centrifuging DNA1@1-ART NPs using a molecular weight cut off filter, clearly indicating the efficient encapsulation of ART inside the NPs (Fig. 1e). The loading efficiency of ART was calculated to be 80% (Fig. S3, ESI†). Moreover, no significant change in the size of the NPs was observed before and after ART encapsulation (Fig. S5, ESI†). Energy-dispersive X-ray spectroscopy (EDS) analyses of DNA1@1 NPs revealed the presence of iron and sulfur from 1, and phosphorous from DNA1 (Fig. S4a, ESI†). The zeta potential measurement of DNA1@1 NPs showed a value of −1.46 mV, whereas DNA1 and 1 exhibited values of −9.72 and +9.64 mV, respectively. The almost neutral zeta potential value for DNA1@1 is indicative of the formation of DNA1@1 NPs (Fig. 1f). The X-ray photoelectron spectroscopy (XPS) analyses of DNA1@1 NPs revealed peaks at 710.0 and 723.6 eV, which is due to the presence of Fe(II) 2p3/2 and Fe(II) 2p1/2 ions, respectively (Fig. S4b, ESI†). Based on these results, the formation of NPs can be envisaged to be the initial formation of supramolecular amphiphile DNA1@1 by the non-covalent tethering of 1 along the anionic backbone of DNA1 and the subsequent amphiphilicity-driven self-assembly of DNA1@1 leads to the formation of DNA1@1 NPs and in the presence of hydrophobic ART self-assembly results in the formation of ART encapsulated NPs (DNA1@1-ART NPs).
| DNA | Sequence (5′→3′) |
|---|---|
| DNA1 | TCT CCC AGC GTG CGC CAT |
| DNA2 | Cy3.5-TCT CCC AGC GTG CGC CAT |
| DNA3 | TCT CCC AGG CTG GCC CAT |
 | ||
| Fig. 1 (a) AFM and (b) TEM images of DNA1@1 NPs. (c) CLSM image of DNA2@1 NPs and (d) DLS analysis of DNA1@1 NPs. (e) Comparison of absorption spectra of free ART and DNA1@1-ART NPs. (f) Comparison of zeta potential values of 1, DNA1 and DNA1@1 NPs. Experiments were performed in triplicate and repeated three times with similar results. | ||
After confirming the nanoparticle formation for DNA1@1 and DNA1@1-ART, we initially studied the ˙OH radical generation by DNA1@1 NPs. This was investigated by using a methylene blue degradation assay by monitoring the absorption changes of the dye at 665 nm. As expected, when H2O2 (10 mM) alone was added to the mixture of DNA1@1 NPs (100 μM) and methylene blue (15 μM), no decrease of absorbance of the dye at 665 nm was observed, indicating the lack of generation of ˙OH radicals (Fig. 2a). As designed, this clearly suggests that GSH-triggered cleavage of the disulfide bond of 1 is the first and triggering step for the production of ˙OH. Accordingly, significant decreases in absorbance of the dye at 665 nm were observed when DNA1@1 NPs were treated with H2O2 (100 μM) and GSH (0.5 mM). This can be attributed to the initial cleavage of the disulfide bond of the prodrug 1 by the reaction with GSH, leading to the release of reactive amino ferrocene, which in-turn undergoes redox reaction with H2O2 and produces ˙OH. This causes the degradation of methylene blue and decreases the absorbance of the dye at 665 nm. A time-dependent methylene blue degradation assay showed that 35% degradation of the dye occurs at ∼6 h of incubation (Fig. S5, ESI†). These observations are in full accordance with our design. The catalytic activity of the NPs was found to increase with the decrease of the pH and the optimum pH was found to be 4.5 (Fig. S9, ESI†). Furthermore, the catalytic activity of the DNA1@1 NPs showed dose-dependent enhancement in a methylene blue degradation assay with respect to an increase in H2O2 concentration (Fig. S10, ESI†). DNA1@1 NPs also showed enhanced catalytic activity at high intracellular GSH concentration of ∼0.5 mM when compared to low extracellular GSH concentration of ∼2 μM (Fig. S11, ESI†). The formation of ˙OH was further confirmed by electron paramagnetic resonance (EPR) analyses of GSH and H2O2-treated DNA1@1 NPs at pH 5.5, which exhibited the characteristic 1:2:2:1 spectrum of ˙OH with hyperfine coupling constants of aN = 14.9 G and aH = 14.9 G, as reported (Fig. 2b).38 Furthermore, the free Fe3+ ions formed from amino ferrocene, one of the products of the Fenton reaction, were detected using a chromogenic reaction with 2,2′-bipyridine (bipy) (300 μM). This was achieved by reducing Fe3+ to Fe2+ by using sodium dithionate, and the in situ formed Fe2+ was then trapped by complexing with bipy, which forms a red coloured [Fe(bipy)3]2+ complex. Accordingly, red coloured [Fe(bipy)3]2+ complex formation was observed for GSH and H2O2 treated-DNA1@1 NPs, whereas no complex formation was observed for H2O2 alone treated-DNA1@1 NPs (Fig. 2c). The release efficiency of Fe3+ was calculated to be 42% for GSH and H2O2 treated-DNA1@1 NPs, whereas only negligible release of Fe3+ (4%) was observed for H2O2 alone treated-DNA1@1 NPs (Fig. 2d). This further supports our hypothesis that GSH-triggered disassembly of DNA1@1 NPs is indeed responsible for the Fenton reaction and the subsequent release of Fe3+. In addition, GSH-triggered disassembly of DNA1@1 NPs was further characterized by monitoring the release of DNA1 (Fig. S12, ESI†). As expected, no gel electrophoretic mobility was observed for DNA1@1 NPs. Interestingly, GSH-treated DNA1@1 NPs showed a band with electrophoretic mobility comparable to free DNA1, which can be attributed to the DNA1 released from the NPs after its disassembly. The decreased mobility observed can be attributed to the electrostatic association of the ammonium salt of ethyl thiol (obtained after GSH reaction of 1) onto the DNA1 backbone.
 | ||
| Fig. 2 (a) Comparison of the absorption spectra of MB in the presence of different stimuli such as H2O2 (blue graph), H2O2 + DNA1@1 NPs (purple graph) and H2O2 + GSH + DNA1@1 NPs (black graph). (b) EPR spectrum of DNA1@1 NPs in the presence of 100 mM DMPO, 8 mM H2O2 and 0.5 mM GSH in dibasic sodium phosphate-citric acid buffer. (c) Reaction scheme depicting the formation of a red coloured [Fe(bipy)3]2+ complex and the corresponding visual colour changes of the DNA1@1 NPs solution with the addition of H2O2 and H2O2 in the presence of GSH. (d) Comparison of quantification of Fe3+ release from DNA1@1 NPs with addition of H2O2 and H2O2 in the presence of GSH. Experiments were performed in triplicate and repeated three times with similar results. | ||
We then investigated the reactivity of the probe 1 towards GSH, which was studied by monitoring the reaction of GSH with 5,5′-dithiobis[2-nitrobenzoic acid] (DTNB) in the presence and absence of 1. Reaction of GSH with DTNB is known to produce the chromogenic molecule, 2-nitro-5-thiobenzoic acid with distinct spectral features. The absorption maximum of DTNB undergoes bathochromic shift from 320 to 410 nm upon reaction with GSH. Accordingly, a decrease of the absorption band at 320 nm with the concomitant increase of the absorption band at 410 nm was observed with the addition of GSH to DTNB and was attributed to the formation of 2-nitro-5-thiobenzoic acid (Fig. 3a). In a separate experiment, GSH was incubated with probe 1 for 6 h, centrifuged and the supernatant was mixed with DTNB and the absorption spectrum of the supernatant was recorded. A comparison of the absorption spectra of the supernatant with the corresponding solution without 1 revealed a significant reduction of the intensity of the peak of 2-nitro-5-thiobenzoic acid at 400 nm. This clearly indicates that 1 undergoes an efficient reaction with GSH and as a consequence of this reaction, no significant amount of GSH was available for the subsequent reaction with DTNB. This clearly shows the high reactivity of 1 towards GSH as designed.
 | ||
| Fig. 3 (a) Comparison of the UV-vis absorption spectra of 1 (black trace), DTNB (blue), GSH (dark blue), DTNB and 1 (green), DTNB and GSH (grey) and DTNB, GSH and 1 (red). (b) Time-dependent absorption spectra of DPBF in the presence of Fe2+ and ART. | ||
After confirming the H2O2-mediated formation of ˙OH and GSH-directed cleavage of the disulfide bond of 1, we then investigated the generation of carbon centered radical species via a H2O2-independent pathway, that is, by the reaction between ART and Fe2+. The formation of carbon centered radical species was monitored by using 1,3-diphenylisobenzofuran (DPBF) as the probe. The dye, DPBF, has its characteristic absorption band centered at 410 nm, and the oxidation of the dye by the in situ generated carbon centered radical species causes the decrease of the characteristic absorption band at 410 nm. This is typically used for the characterization of radical species in solution.39 For this purpose, ART was treated with Fe2+ (FeCl2·4H2O) in the presence of DPBF in ethanol and the absorption of DPBF at 410 nm was monitored with time. As shown in Fig. 3b, a gradual decrease in the absorption of DPBF was observed with time, indicating the oxidation of DPBF by the reaction with the in situ generated carbon centered radical species. As expected, no decrease in the absorption of DPBF was observed when it was treated just with ART (Fig. S6, ESI†). This clearly proves our hypothesis that Fenton-like reaction between ART and Fe2+ efficiently generates radical species in an H2O2 independent pathway for the synergetic CDT.
After the characterization of NPs and its responsive behaviours with respect to various stimuli, we then investigated the internalization of the NPs into cancerous cells. For this purpose, HeLa cells were taken as a representative cell line and were incubated with DNA2@1 NPs for 6 h. Control experiments were also performed with free DNA2 as well. CLSM images (Fig. 4a) showed strong fluorescence for DNA2@1 NP-treated cell lines (red fluorescence) when compared to the corresponding free DNA2-treated cells (Fig. 4b). This validates the efficient internalization of DNA2@1 NPs into HeLa cells, whereas no internalization was observed for DNA2-treated cells, as expected. Furthermore, time-dependent CLSM analyses showed that the internalization of DNA2@1 NPs into the HeLa cells occurs within 3 h of incubation (Fig. S7, ESI†). On the other hand, no internalization was observed for DNA2-treated cells even after 6 h of incubation (Fig. 4b). These observations were further quantified using fluorescence activated cell sorting (FACS) analyses (Fig. 4c and d). In support of CLSM observations, high mean fluorescence intensity (MFI) shift was associated with DNA2@1-treated HeLa cells (24450) when compared with the MFI shift (265) of DNA2-treated cells. Collectively, CLSM and FACS analyses conclude the efficient internalization of DNA2@1 NPs into the HeLa cells and the NPs are mainly distributed in the cytoplasm.
 | ||
| Fig. 4 (a) CLSM images of DNA2@1 NPs-treated HeLa cells and (b) the corresponding FACS analysis after 6 h of incubation. (c) Time-dependent CLSM images of DNA2-treated HeLa cells and (d) the corresponding FACS analyses. The incubation time for FACS is 6 h. | ||
Because DNA2@1 NPs contain disulfide bonds (from 1), the internalization of the NPs may occur through a thiol-mediated uptake pathway. To shed more light on the internalization pathway of the NPs, we next investigated whether DNA2@1 NPs enter into the HeLa cells through the endocytotic pathway or thiol-mediated uptake. Towards this end, HeLa cells were initially treated with endocytosis inhibitor, methyl-β-cyclodextrin (MβCD, 50 μM), which is known to efficiently block the endocytosis pathway. Interestingly, endocytosis inhibitor MβCD showed remarkable influence on the cellular uptake efficiency of DNA2@1 NPs (red) and no significant uptake of DNA2@1 NPs was observed as evident from the CLSM analyses (Fig. 5a). On the other hand, 5,5′-dithiobis-2-nitrobenzoic acid (DTNB), which is a known exofacial thiol blocking agent, had no notable effect on the uptake of DNA2@1 NPs. These results rule out the possibility of thiol-mediated uptake of the NPs and DNA2@1 NPs enter into the HeLa cell solely through the endocytosis pathway. In support of this, the uptake efficiency of DNA2@1 NPs was found to be significantly reduced when the cells were pre-treated at 4 °C, as endocytosis is a temperature-dependent process. These observations were further quantified using FACS analyses, which revealed mean fluorescence intensity (MFI) shifts of 2800, 2604, 1844 and 177 for untreated cells, DTNB-treated cells, MβCD-treated cells and pre-treated cells at 4 °C, respectively (Fig. S15, ESI†). In order to get better insight into the location of the NPs after their cellular entry, various colocalization experiments were performed. For this, lysosomes of HeLa cells were stained with Lysotracker deep red (LysoTracker, red fluorescence). Merged CLSM analyses obviously revealed no colocalization of green fluorescence of DNA2@1 NPs and the red fluorescence of LysoTracker, suggesting that the NPs escape from the lysosomes, as reported in similar examples (Fig. 5b). The extent of colocalization of DNA2@1 NPs and lysosome was quantified by using Pearson's correlation coefficient and the value was found to be 0.035. This value is significantly lower compared to the threshold value (>0.5) required for a correlation. These results conclude that DNA2@1 NPs enter the cancer cell through endocytosis, undergo lysosomal escape and are distributed mainly in the cytosol.
 | ||
| Fig. 5 (a) CLSM images of DNA2@1 NP-treated HeLa cells (red fluorescence) in the presence of different inhibitors; DTNB (4.8 mM), MβCD (50 μM), and after pre-treatment at 4 °C. The nucleus of the cells was stained with Hoechst (blue fluorescence) to monitor the colocalization of DNA2@1 NPs. (b) CLSM images of DNA2@1 NP-treated HeLa cells with lysosomes stained with Lysotracker deep red. (c) CLSM images of DNA1@1-ART NP-treated HeLa cells with the nucleus (green fluorescence) and lysosomes (red fluorescence) stained with Acridine Orange. (d) CLSM images of DNA2@1 NP-treated HeLa cells (green fluorescence) with the mitochondria stained with MitoTracker (red fluorescence). (e) CLSM images of DNA2@1 NP-treated HeLa cells with TMRM stain. | ||
It is known that the lysosomal compartment of cancer cells is highly sensitive to oxidative damage caused by the radical species. After the cellular entry, the NPs undergo disassembly due to the presence of GSH and H2O2 followed by the in situ generation of ROS and carbon centered radical species, which can potentially damage the lysosome membrane. In order to check whether any damage occurs to the lysosome membrane due to the disassembly of the NPs (DNA1@1-ART NPs), an Acridine Orange staining assay was carried out. For this, HeLa cells were initially stained with Acridine Orange and the red fluorescence of Acridine Orange was monitored using CLSM, which is an indicator of acidic regions inside the cells including lysosomes. If the lysosomal membrane is damaged or the lysosome itself is being disrupted, an increase in pH inside the lysosome and hence a decrease in fluorescence intensity was expected. At the same time, Acridine Orange is also known to stain the nucleus of the cells (green fluorescence). CLSM analyses of DNA1@1-ART NP-treated HeLa cells did not show any decrease in the fluorescence intensity when compared to the untreated control cells, suggesting that no lysosomal damage is caused by the NPs (Fig. 5c). This was further confirmed by using FACS analyses, which showed nearly the same MFI shifts of 65708 and 64344 for the control and DNA1@1-ART NP-treated cells, respectively (Fig. S16, ESI†). This is in accordance with our earlier observation that DNA2@1 NPs undergo lysosomal escape. Interestingly, colocalization studies of mitochondria with MitoTracker deep red (red fluorescence) revealed excellent colocalization of the green fluorescence of DNA2@1 NPs and the red fluorescence of MitoTracker deep red. This can be attributed to the disassembly of DNA2@1 NPs inside the cytoplasm and the subsequent localization of DNA2 (antisense DNA, G3139) in the mitochondria due to its high affinity for mitochondria (Fig. 5d). In addition, Cy3.5 is also known to have high affinity for mitochondria.40 However, the binding of DNA2 to the mitochondria did not cause any damage to the mitochondria or its membrane, which was confirmed using a tetramethylrhodamine methyl ester perchlorate (TMRM) assay.
TMRM exhibits orange fluorescence when accumulated in negatively charged polarized mitochondria. When the mitochondrial membrane potential collapses due to apoptosis or metabolically stressed cells, TMRM reagent is dispersed throughout the cell cytosol, and the fluorescence intensity drops down dramatically. Interestingly, DNA2@1 NP-treated HeLa cells did not show any decrease in the fluorescence from mitochondria when compared to untreated control cells, suggesting that no mitochondrial damage has occurred (Fig. 5e). This was further verified through FACS analyses, which revealed similar MFI shifts of 2894 and 2777 for the control and DNA1@1-ART NP-treated cells, respectively (Fig. S17, ESI†). Collectively these experiments conclude that after the lysosomal escape of DNA2@1 NPs, they immediately undergo GSH-triggered disassembly, and release DNA2 and Fe2+ at the cytosol. Because of the mitochondria affinity and also due to the presence of Cy3.5, the antisense DNA (DNA2) gets accumulated at the mitochondria.
We next studied the generation of radical species by DNA1@1 and DNA1@1-ART NPs inside the HeLa cells by using 2,7-dichlorofluorescein-diacetate (DCFH-DA) as the marker for radical species, which produces green fluorescent 2,7-dichlorofluorescein (DCF) upon radical detection. As shown in Fig. 6a, DNA1@1-ART NP-treated HeLa cells showed obviously stronger DCF green fluorescence than DNA1@1 or ART-treated cells, indicating higher radical (both ˙OH and carbon cantered radicals) generation for DNA1@1-ART NP-treated cancer cells through Fenton and Fenton-like reactions, as explained previously. The corresponding FACS analyses showed MFI shifts of 473, 506, 2882 and 16691 for the control, ART, DNA1@1 NPs and DNA1@1-ART NPs, respectively (Fig. S18, ESI†). This is in full agreement with the CLSM results. To further validate that the radical generation indeed occurs via Fe2+-catalysed Fenton reactions, the cells were pre-incubated with a Fe2+ binding agent, 2,2′-bipyridyl and then treated with DNA1@1-ART NPs. As expected, a significant decrease in green fluorescence of DCF was observed when compared to the corresponding bipyridyl-untreated cells (FACS revealed MFI value of 626, Fig. S19, ESI†), which indicates that the intracellular radical production indeed occurs through the Fe2+-catalysed cascade Fenton reactions (Fig. 6b).
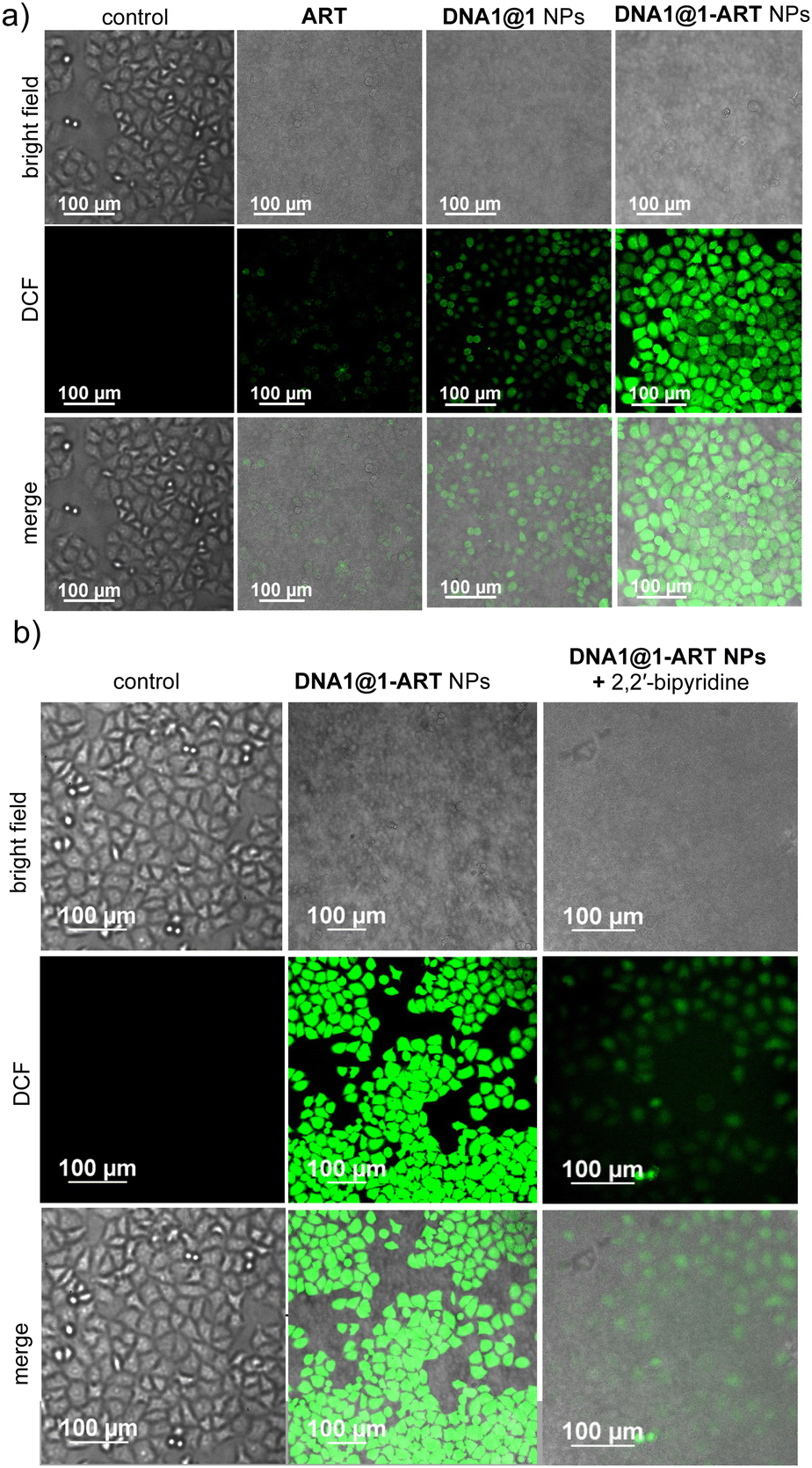 | ||
| Fig. 6 (a) CLSM images of ART, DNA1@1 and DNA1@1-ART NP-treated HeLa cells stained with DCFH-DA. (b) CLSM images of DCFH-DA stained and DNA1@1-ART NP-treated HeLa cells in the presence of Fe2+ chelator, 2,2′-bipyridyl. | ||
The cytotoxicity of DNA1@1-ART NPs was evaluated using the MTT assay on HeLa cell lines. Before investigating the cytotoxicity of the DNA1@1-ART NPs, the cytotoxicity of its individual components was studied. Initially, HeLa cell lines were incubated with free DNA1 for 24 h with varying concentrations in the range of 50–1000 nM. Though DNA1 is an antisense DNA and can potentially induce cell apoptosis, no significant cell death was observed in this case, and the cell viability remained more than 80% in the entire concentration range (Fig. 7a). This might be due to the cell impermeable nature of free oligonucleotides and hence DNA1 is unable to induce any cell apoptosis. Subsequently, ART of different concentrations was incubated with HeLa cells for 24 h to assess the cytotoxicity of free ART. In this case as well, cell viability remains more than 60% even at a higher concentration of 250 μM, and this is very much expected due to the poor water solubility of ART in the physiological media (Fig. 7b). On the other hand, incubation of HeLa cells with DNA1@1-ART NPs showed reasonable cytotoxicity with a cell death of more than 75% (Fig. 7c). Though commendable cytotoxicity was achieved, the relatively reduced efficacy of the system can be attributed to the large size of the NPs, which hinders the efficient cellular internalization of the NPs. This suggests that the efficacy can be improved by reducing the size of the NPs. In order to validate the role of DNA1 in the overall cytotoxicity of DNA1@1-ART NPs, we synthesized two other control NPs, DNA3@1-ART and DNA3@1 NPs. In these cases, DNA3 is a scrambled DNA with no therapeutic value. MTT analyses of both DNA3@1-ART and DNA3@1 NPs revealed reduced cytotoxicity when compared to the corresponding DNA1@1-ART NPs. These results clearly support our hypothesis that the excellent cytotoxicity of DNA1@1-ART NPs is due to the combined effect of Fenton reactions and the gene therapeutic effect of DNA1. A comparison of the cytotoxicity of free ART with the corresponding DNA1@1-ART NPs revealed enhanced cytotoxicity for the latter in a dose-dependent manner (Fig. 7d). The reduced therapeutic efficacy of free ART must be due to the poor water solubility of ART and its water solubility is significantly improved when encapsulated inside DNA1@1 NPs and hence exhibits increased therapeutic efficacy. The potential of DNA as a template to carry various metal ions for Fenton-like reaction for CDT application has been well demonstrated by several research groups.31–33 In these approaches, typically metal ions are released by the degradation of DNA and hence the therapeutic value of DNA is completely lost in these cases. On the other hand, in our approach, GSH-triggered cleavage of the disulfide bond of 1 causes the disassembly as well as activation of metal ions from their prodrug state to the active state, and hence the damage caused to DNA is minimized. In order to validate this hypothesis, NPs comprising Fe2+ and G3139 (DNA1@Fe2+) were synthesized by annealing of Fe2+ and G3139 in 1:50 molar ratio by following a literature procedure and characterized using various microscopic techniques.31 Comparison of cell viability of DNA1@Fe2+ with DNA1@1 NPs revealed enhanced cytotoxicity for DNA1@1 NPs (Fig. S20, ESI†).
 | ||
| Fig. 7 Cell viability assay of HeLa cells (a) with varying concentrations of DNA1 and (b) ART. (c) Comparison of cell viability of HeLa cells treated with DNA3, DNA1, DNA3@1 NPs, DNA3@1-ART NPs and DNA1@1-ART NPs for 24 h incubation. (d) Comparison of cell viability of HeLa cells treated with different concentrations of ART for free ART and DNA1@1-ART NPs. | ||
Finally, the cytotoxicity of DNA1@1-ART NPs was directly visualized by calcein-AM/ethidium homodimer-1 co-staining assay. For this purpose, HeLa cells were treated with DNA1@1-ART NPs and then stained with calcein-AM and ethidium homodimer-1 dyes for 24 h. Calcein-AM stains viable cells and gives green fluorescence, whereas ethidium homodimer-1 stains dead cells and gives red fluorescence. As shown in Fig. 8a, untreated HeLa cells (control) showed only calcein-AM staining and no ethidium homodimer-1 staining, indicating that all the cells are alive, as expected. On the other hand, notable reduction in calcein-AM staining and significant dead cell staining by ethidium homodimer-1 were observed for DNA1@1-ART NP-treated cells. These results are unambiguous to demonstrate the effective killing of cancer cells by DNA1@1-ART NPs and are in full agreement with MTT analyses. Subsequently, an annexin V-FITC (AV)/propidium iodide (PI) assay was performed to gather better insight into the mechanism of cell death. Annexin V is a specific marker for apoptosis, which binds specifically to the exposed phosphatidylserine (PS) on the cell surface in the early-stage of apoptosis, and PI stains the nuclear chromatin during the late stage of apoptosis. As shown in Fig. 8b, DNA1@1-ART NP-treated cells exhibited strong green fluorescence from annexin V-FITC at the cell membrane and red fluorescence of PI from the nuclei indicating the late-stage apoptosis. These results suggest that the mechanism of cell death is apoptosis and is not following the necrosis pathway. This is further confirmed through the FACS analyses. For this, HeLa cells were treated with DNA1@1-ART NPs for 24 h and analysed by flow cytometry after staining with annexin V-FITC (AV)/propidium iodide (PI). As shown in Fig. S21 (ESI†), the number of apoptotic cells increased to 86.56% (including early apoptosis and late apoptosis). These results unequivocally conclude that the mechanism of cell death is indeed due to the apoptosis mechanism and not due to the necrotic pathway.
 | ||
| Fig. 8 (a) Calcein-AM/ethidium homodimer-1 live/dead cell assay of DNA1@1-ART NP-treated HeLa cells. (b) Annexin V-FITC/propidium iodide assay of DNA1@1-ART NP-treated HeLa cells. | ||
Conclusions
In summary, we have demonstrated a supramolecular strategy for the crafting of tumour microenvironment sensitive NPs composed of ART, ferrocene derivative 1 (GSH and redox-active) and DNA1 (antisense DNA) via a single-step assembly process. The most remarkable feature of our design strategy is the GSH-triggered cascade reactions of the NPs inside the cancer cell in a cooperative fashion for synergistic cancer therapy. Cleavage of disulfide bonds of the NPs by the overexpressed GSH in the tumour microenvironment leads to the following cascade CDT reactions: (i) GSH-triggered uncaging of 1 releases amino ferrocene, which then undergoes reaction with endogenous H2O2 to produce ˙OH radicals and Fe3+ and (ii) the in situ reduction of Fe3+ by GSH to form Fe2+ and the subsequent Fe2+-triggered cleavage of the peroxide bond of ART to form carbon-centered free radicals (H2O2 independent pathway). Furthermore, disassembly of the NPs releases DNA1, which downregulates the translation of Bcl-2 mRNA and thereby causes cell apoptosis. The enhanced therapeutic efficacy of our approach is attributed to the cooperative cascade CDT and antisense therapy. We hope the simple yet efficient supramolecular approach demonstrated here may encourage other researchers to design stimuli-responsive multifunctional nanosystems for combination cancer therapy.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from DBT (BT/PR30172/NNT/28/1593/-2018) is gratefully acknowledged. The help of Sarika Mohan S. is acknowledged for the FACS analyses.References
- Z. Tang, Y. Liu, M. He and W. Bu, Chemodynamic Therapy: Tumour Microenvironment-Mediated Fenton and Fenton-like Reactions, Angew. Chem., Int. Ed., 2019, 58, 946–956 CrossRef CAS PubMed.
- E. Hwang and H. S. Jung, Metal–organic complex-based chemodynamic therapy agents for cancer therapy, Chem. Commun., 2020, 56, 8332–8341 RSC.
- C. Cao, X. Wang, N. Yang, X. Song and X. Dong, Recent advances of cancer chemodynamic therapy based on Fenton/Fenton-like chemistry, Chem. Sci., 2022, 13, 863–889 RSC.
- H. Lin, Y. Chen and J. Shi, Nanoparticle-triggered in situ catalytic chemical reactions for tumour-specific therapy, Chem. Soc. Rev., 2018, 47, 1938–1958 RSC.
- Y. Li, W. Xiu, K. Yang, Q. Wen, L. Yuwen, Z. Luo, X. Liu, D. Yang, X. Xie and L. Wang, A multifunctional Fenton nanoagent for microenvironment-selective anti-biofilm and anti-inflammatory therapy, Mater. Horizons, 2021, 8, 1264–1271 RSC.
- J. Tan, H. Li, X. Hu, R. Abdullah, S. Xie, L. Zhang, M. Zhao, Q. Luo, Y. Li, Z. Sun, Q. Yuan and W. Tan, Size-Tunable Assemblies Based on Ferrocene-Containing DNA Polymers for Spatially Uniform Penetration, Chem, 2019, 5, 1775–1792 CAS.
- X. Li, Q. Zhou, A. A.-W. M. M. Japir, D. Dutta, N. Lu and Z. Ge, Protein-Delivering Nanocomplexes with Fenton Reaction-Triggered Cargo Release to Boost Cancer Immunotherapy, ACS Nano, 2022, 16, 14982–14999 CrossRef CAS PubMed.
- P. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang, Z. Gu and J. Lin, Enhanced Cisplatin Chemotherapy by Iron Oxide Nanocarrier-Mediated Generation of Highly Toxic Reactive Oxygen Species, Nano Lett., 2017, 17, 928–937 CrossRef CAS PubMed.
- Q. Chen, J. Zhou, Z. Chen, Q. Luo, J. Xu and G. Song, Tumor-Specific Expansion of Oxidative Stress by Glutathione Depletion and Use of a Fenton Nanoagent for Enhanced Chemodynamic Therapy, ACS Appl. Mater. Interfaces, 2019, 11, 30551–30565 CrossRef CAS PubMed.
- L.-S. Lin, J. Song, L. Song, K. Ke, Y. Liu, Z. Zhou, Z. Shen, J. Li, Z. Yang, W. Tang, G. Niu, H.-H. Yang and X. Chen, Simultaneous Fenton-like Ion Delivery and Glutathione Depletion by MnO2-Based Nanoagent to Enhance Chemodynamic Therapy, Angew. Chem., Int. Ed., 2018, 57, 4902–4906 CrossRef CAS PubMed.
- J. Fu, Y. Shao, L. Wang and Y. Zhu, Lysosome-controlled efficient ROS overproduction against cancer cells with a high pH-responsive catalytic nanosystem, Nanoscale, 2015, 7, 7275–7283 RSC.
- Y. He, A. del Valle, Y. Qian and Y.-F. Huang, Near infrared light-mediated enhancement of reactive oxygen species generation through electron transfer from graphene oxide to iron hydroxide/oxide, Nanoscale, 2017, 9, 1559–1566 RSC.
- G. Lan, K. Ni, Z. Xu, S. S. Veroneau, Y. Song and W. Lin, Nanoscale Metal-Organic Framework Overcomes Hypoxia for Photodynamic Therapy Primed Cancer Immunotherapy, J. Am. Chem. Soc., 2018, 140, 5670–5673 CrossRef CAS PubMed.
- X. Zhong, X. Wang, L. Cheng, Y. Tang, G. Zhan, F. Gong, R. Zhang, J. Hu, Z. Liu and X. Yang, GSH-Depleted PtCu3 Nanocages for Chemodynamic- Enhanced Sonodynamic Cancer Therapy, Adv. Funct. Mater., 2020, 30, 1907954 CrossRef CAS.
- L. V. Nair, S. S. Nazeer, R. S. Jayasree and A. Ajayaghosh, Fluorescence Imaging Assisted Photodynamic Therapy Using Photosensitizer-Linked Gold Quantum Clusters, ACS Nano, 2015, 9, 5825–5832 CrossRef CAS PubMed.
- C. Tong, T. Liu, V. Saez Talens, W. E. M. Noteborn, T. H. Sharp, M. M. R. M. Hendrix, I. K. Voets, C. L. Mummery, V. V. Orlova and R. E. Kieltyka, Squaramide-Based Supramolecular Materials for Three-Dimensional Cell Culture of Human Induced Pluripotent Stem Cells and Their Derivatives, Biomacromolecules, 2018, 19, 1091–1099 CrossRef CAS PubMed.
- W. Xuan, Y. Xia, T. Li, L. Wang, Y. Liu and W. Tan, Molecular Self-Assembly of Bioorthogonal Aptamer-Prodrug Conjugate Micelles for Hydrogen Peroxide and pH-Independent Cancer Chemodynamic Therapy, J. Am. Chem. Soc., 2020, 142, 937–944 CrossRef CAS PubMed.
- P. Ji, H. Huang, S. Yuan, L. Wang, S. Wang, Y. Chen, N. Feng, H. Veroniaina, Z. Wu, Z. Wu and X. Qi, ROS-Mediated Apoptosis and Anticancer Effect Achieved by Artesunate and Auxiliary Fe(II) Released from Ferriferous Oxide-Containing Recombinant Apoferritin, Adv. Healthc. Mater., 2019, 8, 1900911 CrossRef CAS PubMed.
- X. Wan, H. Zhong, W. Pan, Y. Li, Y. Chen, N. Li and B. Tang, Programmed Release of Dihydroartemisinin for Synergistic Cancer Therapy Using a CaCO3 Mineralized Metal–Organic Framework, Angew. Chem., Int. Ed., 2019, 58, 14134–14139 CrossRef CAS PubMed.
- L. Shao, T. Hu, X. Fan, X. Wu, F. Zhou, B. Chen, S. Tan, H. Xu, A. Pan, S. Liang and Y. He, Intelligent Nanoplatform with Multi Therapeutic Modalities for Synergistic Cancer Therapy, ACS Appl. Mater. Interfaces, 2022, 14, 13122–13135 CrossRef CAS PubMed.
- Y. Luo, X. Sun, L. Huang, J. Yan, B.-Y. Yu and J. Tian, Artemisinin-Based Smart Nanomedicines with Self-Supply of Ferrous Ion to Enhance Oxidative Stress for Specific and Efficient Cancer Treatment, ACS Appl. Mater. Interfaces, 2019, 11, 29490–29497 CrossRef CAS PubMed.
- H. Pei, N. Lu, Y. Wen, S. Song, Y. Liu, H. Yan and C. Fan, A DNA Nanostructure-based Biomolecular Probe Carrier Platform for Electrochemical Biosensing, Adv. Mater., 2010, 22, 4754–4758 CrossRef CAS PubMed.
- A. T. Veetil, K. Chakraborty, K. Xiao, M. R. Minter, S. S. Sisodia and Y. Krishnan, Cell-targetable DNA nanocapsules for spatiotemporal release of caged bioactive small molecules, Nat. Nanotechnol., 2017, 12, 1183–1189 CrossRef CAS PubMed.
- S. Walia, V. Morya, A. Gangrade, S. Naskar, A. Guduru Teja, S. Dalvi, P. K. Maiti, C. Ghoroi and D. Bhatia, Designer DNA Hydrogels Stimulate 3D Cell Invasion by Enhanced Receptor Expression and Membrane Endocytosis, ACS Biomater. Sci. Eng., 2021, 7, 5933–5942 CrossRef CAS PubMed.
- S. Kadekar, G. N. Nawale, V. K. Rangasami, V. Le Joncour, P. Laakkonen, J. Hilborn, O. P. Varghese and O. P. Oommen, Redox responsive Pluronic micelle mediated delivery of functional siRNA: a modular nano-assembly for targeted delivery, Biomater. Sci., 2021, 9, 3939–3944 RSC.
- H. V. P. Thelu, S. Atchimnaidu, D. Perumal, K. S. Harikrishnan, S. Vijayan and R. Varghese, Self-Assembly of an Aptamer-Decorated, DNA–Protein Hybrid Nanogel: A Biocompatible Nanocarrier for Targeted Cancer Therapy, ACS Appl. Bio Mater., 2019, 2, 5227–5234 CrossRef CAS PubMed.
- S. Atchimnaidu, D. Perumal, K. S. Harikrishanan, H. V. P. Thelu and R. Varghese, Phototheranostic DNA micelles from the self-assembly of DNA-BODIPY amphiphiles for the thermal ablation of cancer cells, Nanoscale, 2020, 12, 11858–11862 RSC.
- D. Bousmail, L. Amrein, J. J. Fakhoury, H. H. Fakih, J. C. C. Hsu, L. Panasci and H. F. Sleiman, Chem. Sci., 2017, 8, 6218–6229 RSC.
- J. Liu, L. Song, S. Liu, Q. Jiang, Q. Liu, N. Li, Z.-G. Wang and B. Ding, A DNA-Based Nanocarrier for Efficient Gene Delivery and Combined Cancer Therapy, Nano Lett., 2018, 18, 3328–3334 CrossRef CAS PubMed.
- H. Shi, X. Ye, X. He, K. Wang, W. Cui, D. He, D. Li and X. Jia, Au@Ag/Au nanoparticles assembled with activatable aptamer probes as smart “nano-doctors” for image-guided cancer thermotherapy, Nanoscale, 2014, 6, 8754–8761 RSC.
- Y. Yang, W. Zhu, L. Feng, Y. Chao, X. Yi, Z. Dong, K. Yang, W. Tan, Z. Liu and M. Chen, G-Quadruplex-Based Nanoscale Coordination Polymers to Modulate Tumor Hypoxia and Achieve Nuclear-Targeted Drug Delivery for Enhanced Photodynamic Therapy, Nano Lett., 2018, 18, 6867–6875 CrossRef CAS PubMed.
- Q. Zhang, Q. Jiang, N. Li, L. Dai, Q. Liu, L. Song, J. Wang, Y. Li, J. Tian, B. Ding and Y. Du, DNA origami as an in vivo drug delivery vehicle for cancer therapy, ACS Nano, 2014, 8, 6633–6643 CrossRef CAS PubMed.
- B. Liu, F. Hu, J. Zhang, C. Wang and L. Li, A Biomimetic Coordination Nanoplatform for Controlled Encapsulation and Delivery of Drug–Gene Combinations, Angew. Chem., Int. Ed., 2019, 58, 8804–8808 CrossRef CAS PubMed.
- L. Lin, J. Yu, H. Lu, Z. Wei, Z. Chao, Z. Wang, W. Wu, H. Jiang and L. Tian, Mn–DNA coordination of nanoparticles for efficient chemodynamic therapy, Chem. Commun., 2021, 57, 1734–1737 RSC.
- C. Liu, Y. Chen, J. Zhao, Y. Wang, Y. Shao, Z. Gu, L. Li and Y. Zhao, Self-Assembly of Copper–DNAzyme Nanohybrids for Dual-Catalytic Tumor Therapy, Angew. Chem., Int. Ed., 2021, 60, 14324–14328 CrossRef CAS PubMed.
- H. Hagen, P. Marzenell, E. Jentzsch, F. Wenz, M. R. Veldwijk and A. Mokhir, Aminoferrocene-based prodrugs activated by reactive oxygen species, J. Med. Chem., 2012, 55, 924–934 CrossRef CAS PubMed.
- S. Lei, J. Chen, K. Zeng, M. Wang and X. Ge, Visual dual chemodynamic/photothermal therapeutic nanoplatform based on superoxide dismutase plus Prussian blue, Nano Res., 2019, 12, 1071–1082 CrossRef CAS.
- S. E. Lehman, A. S. Morris, P. S. Mueller, A. K. Salem, V. H. Grassian and S. C. Larsen, Silica nanoparticle-generated ROS as a predictor of cellular toxicity: mechanistic insights and safety by design, Environ. Sci.: Nano, 2016, 3, 56–66 RSC.
- H. Hou, X. Huang, G. Wei, F. Xu, Y. Wang and S. Zhou, Fenton Reaction-Assisted Photodynamic Therapy for Cancer with Multifunctional Magnetic Nanoparticles, ACS Appl. Mater. Interfaces, 2019, 11, 29579–29592 CrossRef CAS PubMed.
- J. Liu, L. Yang, C. Xue, G. Huang, S. Chen, J. Zheng and R. Yang, Reductase and Light Programmatical Gated DNA Nanodevice for Spatiotemporally Controlled Imaging of Biomolecules in Subcellular Organelles under Hypoxic Conditions, ACS Appl. Mater. Interfaces, 2021, 13, 33894–33904 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qm00073g |
| This journal is © the Partner Organisations 2023 |