
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStop and restart of polycondensation reactions of highly reactive sol–gel precursors for nanoscale surface molding†
Norihiro
Mizoshita
 * and
Yuri
Yamada
* and
Yuri
Yamada
Toyota Central R&D Labs., Inc., Nagakute, Aichi 480-1192, Japan. E-mail: nmizoshita@mosk.tytlabs.co.jp
First published on 13th March 2023
Abstract
Sol–gel processes have been widely used for synthesizing functional inorganic and organic–inorganic hybrid materials. However, it has been challenging to apply mechanical nano-processing to sol–gel materials due to the rapid polycondensation and curing of concentrated precursors. In this study, a versatile polycondensation process for sol–gel precursors with intrinsic high reactivity has been developed by combining acid-catalyzed transesterification of metal alkoxides with addition of basic amine compounds. The reaction mixtures prepared using naphthalimide-derived alkoxysilanes result in long-term stable unhardened films after the removal of solvents, and their curing can be thermally restarted at the desired timing. Using this reaction method, nanoimprinted porous films have been obtained reproducibly from various organosilane precursors as well as some metal tetraalkoxides. The present approach would be applicable to a wide range of sol–gel reactions involving metal alkoxide species.
1. Introduction
Materials synthesis based on sol–gel chemistry is one of the most important approaches to the fabrication of covalently crosslinked robust and chemically stable materials.1,2 Preparation of metal oxide particles and films from metal alkoxides (M(OR)n; M = Si, Ti, etc.) and synthesis of organic–inorganic hybrid materials from organic-functionalized alkoxysilane precursors (X[Si(OR)3]n, X: organic groups) have widely prevailed for various applications, such as electronic and optical devices,3,4 solid catalysts5 and functional coatings.6 Introduction of nanostructures into metal oxides has also attracted attention for enhancing optical and electronic properties,7–9 heat confinement effects10,11 and catalytic activities.12Tuning the reactivity of metal alkoxide species is the key to the sol–gel process. Alkoxysilane compounds with moderate reactivity easily afford stable sol solutions that are available for synthesizing various silica-based materials.13 On the other hand, it is challenging to control sol–gel reactions of highly reactive precursors, such as organosilanes exhibiting strong intermolecular interactions and low solubility due to large aromatic moieties and/or hydrogen-bonding groups,14–16 and metal alkoxides with center metals except for silicon.17–19 Their high reactivities lead to prompt hardening of the framework, which is problematic for the morphological control of products, including uniform thin film coatings, ordered porous materials and periodic surface nanostructures, unless semi-solid substances are generated from precursor mixtures.20,21 Reaction systems that are less reactive during film formation or surface molding and can shift to a highly reactive state at the desired timing are greatly attractive toward opening up new possibilities for traditional sol–gel materials.22–24
In the present study, we show that sol–gel reactions of highly reactive organosilane precursors can be stopped and restarted by acid-catalyzed transesterification of metal alkoxide groups and subsequent addition/removal of amine compounds (Fig. 1). Recently we developed naphthalimide-based aromatic organosilane precursors (1) and applied their sol–gel films to nanoimprinting.25,26 The nanoimprinted porous organosilica films were proven to function successfully as analytical substrates in laser desorption/ionization mass spectrometry.25,26 However, the nanoimprinting of the sol–gel films was successful only under limited conditions. For 1c, the reaction conditions of the sol solution had to be carefully adjusted according to the hydrolysis and condensation behavior of the precursor.25 For hydrogen-bonding 1a and 1b (1b forms a diol group after acid-catalyzed deprotection of the acetal group), nanoimprinting was unsuccessful under conventional acidic sol–gel conditions. In the conventional process where reactivity is controlled based on hydrolytic conditions, highly reactive Si–OH species are generated at a high concentration, which makes curing control difficult because the polycondensation of the concentrated Si–OH species proceeds quickly. In this study, we achieve stop and progress of the curing of organosilica films by tuning moisture-sensitivity of precursor/catalyst mixtures prior to hydrolysis. Here, triisopropoxysilyl groups are introduced into the organosilane precursors to prevent undesired polycondensation during the synthesis and purification of the precursors. We utilize transesterification of the alkoxysilyl groups to tune the reactivity of the reaction mixtures without forming concentrated Si–OH species. This makes it possible to prepare nanoimprinted films reproducibly from highly reactive precursors. The reaction mechanism is investigated and discussed, focusing on the study of 1. Moreover, we extend the present reaction method to other metal alkoxides and demonstrate nanoscale surface molding of ceramic films.
 | ||
| Fig. 1 (a) Sol–gel polycondensation of 1 using different reaction methods. (b) Photographs of the spin-coated films prepared from 1a: (A) 1a-SG film immediately after spin-coating; (B) 1 min after spin-coating (rubbed with cotton wool applicator); (C) a film prepared by spin-coating of the transesterified reaction mixture; (D) 30 min after spin-coating (rubbed with cotton wool applicator); (E) 1a-TE/A film obtained by heating at 80 °C for 3 min (rubbed with cotton wool applicator). (c) AFM images of the nanoimprinted films: 1a-SG (mold I), 1a-TE/A (mold II), 1b-TE/A (mold I) and 1b-TE/A (mold III). Mold I: nanopillar array; diameter: 145 nm; height: 250 nm; pitch: 250 nm. Mold II: nanopillar array; diameter: 270 nm; height: 200 nm; pitch: 460 nm. Mold III: line-and-space; line width: 70 nm; height: 150 nm; pitch: 140 nm. | ||
2. Results and discussion
2.1. Reaction control and surface molding of organosilica films
Polycondensation of 1a and 1b proceeded rapidly in the spin-coated films and even in the sol solutions prepared by the conventional acidic sol–gel method (Fig. 1a, denoted as SG method). For example, sol solutions of 1a and 1b in ethanol containing a small amount of hydrochloric acid (0.02 M) formed precipitates of the polycondensed materials in 2 and 1 h, respectively. Homogeneous sol–gel films (1a-SG, 1b-SG) could be obtained by spin-coating the sol solutions reacted for 30 min. However, as shown in Fig. 1b (A → B), the films were hardened less than 1 min after the spin-coating.Transesterification of alkoxysilyl groups was used to suppress the reactivity of the precursors without forming the silanol-rich state. The transesterification approach is effective to tune the reactivity, solubility and crystallinity of the alkoxysilane compounds.27–30 In order to increase the fluidity of the films after spin-coating, transesterification of 1a and 1b (90 mg) was first carried out using 2-methoxyethanol having a flexible oxyethylene unit (2-ME, 1.0 mL; boiling point: 124 °C) and p-toluenesulfonic acid monohydrate (TsOH·H2O, 2 mg, 0.01 mmol) as an acid catalyst. The transesterification reaction using excess 2-ME at 130 °C for 15–60 min gave stable precursor solutions, which formed no precipitates for more than 24 h at room temperature. However, when spin-coated films were formed using the transesterified precursor solutions (denoted as TE method), unexpectedly, the films (1a-TE, 1b-TE) were cured rapidly and no fluid state was induced. TsOH likely functioned as a strong acid catalyst in the spin-coated films and promoted the polycondensation of the precursors under the influence of trace amounts of water contained in the catalyst, solvents and air.
We discovered that temporary stop and heat-driven restart of the sol–gel polycondensation reactions could be achieved by adding basic amine compounds to the transesterified reaction mixtures (Fig. 1a, denoted as TE/A method). Triethylamine (10 μL, 0.07 mmol) was added to the reaction mixtures (1.0 mL) to suppress the acidity of TsOH. Moisture from the reagents or the atmosphere was sufficient to cause curing of the films, so no additional water was required. Fig. 1b shows photographs of the spin-coated films prepared using 1a. The spin-coating of the reaction mixture containing transesterified 1a, TsOH and triethylamine resulted in an unhardened fluid film (Fig. 1b, C). The film was easily removed by rubbing with a cotton wool applicator, even 30 min after the preparation (Fig. 1b, D). The fluid state was maintained for more than 20 h at room temperature (humidity: 50–60%). Interestingly, after heating the film at 80–100 °C for 1–3 min, the film (1a-TE/A) hardened rapidly and became resistant to the surface rubbing (Fig. 1b, E). Thus, the TE/A method realizes the formation of long-term stable fluid films from highly reactive sol–gel precursors and the rapid curing of the films at the desired timing. Since the polycondensation in the TE/A method is affected by atmospheric moisture, the fluid state could be maintained for 2–3 days in low humidity (ca. 30%) environments. Storage of the precursor films under reduced pressure further suppressed the generation of silanol species, thus retaining the fluid state.
The stop and restart behavior of the sol–gel reaction was applied to the nanoimprinting of 1a-TE/A and 1b-TE/A films (Fig. 1c). As mentioned above, nanoimprinting of the 1a-SG and 1b-SG films was unsuccessful. When the polymer mold with a nanopillar array (height: 250 nm) was pressed into the 1a-SG and 1b-SG films just after spin-coating, only distorted and shallow hollows (depth: 15–20 nm) were formed on the surface due to their rapid curing (Fig. 1c). In contrast, the use of the TE/A method enabled the nanoimprinting of the organosilica films. Vertically oriented nanopore arrays with different structural periodicities (pitch: 460 nm or 250 nm) and a line-and-space pattern (pitch: 140 nm) were clearly formed on the 1a-TE/A and 1b-TE/A films by the structural transfer from the polymer molds (Fig. 1c). The depth of the nanopatterns (holes: 170–200 nm; line-and-space: 110 nm) was 75–85% of the height of the surface patterns of the molds, whereas the in-plane shrinkage was relatively suppressed probably due to constraints by the substrate. This volume shrinkage was due to the elimination of 2-ME from the transesterified precursors and the subsequent dehydration condensation.
2.2. Reaction mechanism in TE/A method
The reaction process in the TE/A method was examined using 29Si NMR measurements. Fig. 2a shows solid-state 29Si magic-angle-spinning (MAS) NMR spectra of the organosilicas prepared from 1 using the SG and TE/A methods. NMR signals corresponding to condensed Tn species (Tn: –Si(OSi)n(OH)3–n) were observed at −49 (T1), −59 (T2), and −68 (T3) ppm. 1a-SG exhibited a strong signal corresponding to T2 species, whereas the framework of 1a-TE/A mainly consisted of T1 species. Similar 29Si MAS NMR spectra were obtained for organosilicas prepared from 1b and 1c. The degrees of condensation were calculated to be 61, 55 and 57% for 1a-SG, 1b-SG and 1c-SG, and 44, 43 and 48% for 1a-TE/A, 1b-TE/A and 1c-TE/A, respectively (Fig. S5, ESI†). These results suggest that the polycondensation of 1 is slightly suppressed in the TE/A method. Incorporation of sulfonic acids or amines into the organosilica framework cannot be ruled out, but the NMR spectra do not indicate stable formation of such species (Fig. 2 and Fig. S4, ESI†). The reasons for the low degree of condensation will be discussed later. | ||
| Fig. 2 (a) 29Si MAS NMR spectra of the organosilicas prepared from 1 using SG and TE/A methods. (b) 29Si NMR spectra of 1c in 2-ME (A), the transesterified sol mixture of 1c (B), and the fluid film exposed to the air for 3 h (redissolved in 2-ME) (C). (c) 29Si MAS NMR spectrum of 1c-TE. (d) 29Si MAS NMR spectrum of 1c-SG/A. | ||
The introduction of bulky and flexible –Si(OCH2CH2OCH3)3 (–Si(ME)3) groups surrounding the large organic moiety by transesterification of 1 with 2-ME leads to the great enhancement of solubility and hydrolytic reactivity of the precursors. The spectral analysis in solutions was performed mainly for 1c having a simple alkyl substituent. 29Si NMR measurements indicate that monomeric species (T0) with –Si(ME)3 and –Si(ME)2(OiPr) groups are present as the main components in the precursor solution, with a small amount (∼35%) of T1 species mixed in (Fig. 2b, A → B and Scheme 1, A → B). If the precursor is concentrated without adding amine compounds, TsOH acts as a strong acid catalyst, and hydrolysis and polycondensation proceed (Scheme 1, C). Fig. 2c shows that 1c-TE forms a well-crosslinked framework and its degree of condensation is 67%. When triethylamine in excess to TsOH is added to the sol mixtures, the formation of the salt (HN+Et3·TsO−) suppresses the high catalytic activity of TsOH (Scheme 1, D). It has also been confirmed that polycondensation of 1 is not promoted in the presence of triethylamine under the present spin-coating conditions. There are almost no M–OH species in the reaction mixtures prepared by the TE/A method; therefore, the polycondensation reaction does not proceed easily after the evaporation of the solvents. When the reaction mixture was exposed to the atmosphere for 3 h after the evaporation of solvent, the T0-rich unhardened state was maintained, although a slight increase in chemical species containing Si–OH groups was observed probably due to the influence of moisture (Fig. 2b, C). The amount of water is insufficient to hydrolyze all –Si(ME)3 groups, which may lead to the formation of the T1-rich framework after curing. While transesterification of alkoxysilane compounds results not in a single precursor component but in precursor mixtures, their sol–gel polycondensation affords homogeneous films probably due to the formation of a T1-rich organosilica framework.
 | ||
| Scheme 1 Reaction process of transesterified organosilane precursors (ME = –OCH2CH2OCH3). | ||
The heat-driven polycondensation in the TE/A method was confirmed to be catalyzed by the salt HN+Et3·TsO− (Scheme 1, D → E). When the spin-coated fluid films are heated, excessive triethylamine evaporates and the salt remains. We have checked that the salt HN+Et3·TsO− does not decompose upon heating at 100 °C for 2 h (Fig. S13, ESI†). It has also been confirmed that this salt exhibits moderate acidity (pH = 3–4) in a 5 wt% aqueous solution (Table S1, ESI†). In other words, the acidic salt acts as a catalyst to restart the polycondensation reaction of the transesterified precursors. The introduction of –Si(ME)3 groups into the precursors probably increased the compatibility with the salt. The catalytic activity of the salt for –Si(ME)3 species was verified by observing the polycondensation behavior of Me–Si(ME)3 in the presence of the salt and/or triethylamine (Table S2, ESI†). It is reasonable that the degree of condensation of 1-TE/A became lower than those obtained by the SG and TE methods including strong acid catalysts (HCl or TsOH). The degree of condensation can be increased to the same extent by post-treatments such as exposure to hydrochloric acid vapor after curing (Fig. S9, ESI†). Compared to the SG method using silanol-rich precursor mixtures, film formation by the TE/A method delays the timing of curing because the reaction proceeds with exposure to the ambient moisture. However, the degree of condensation of the product was strongly dependent on the catalytic species rather than the amount of water used or atmospheric exposure time. The steric effect of the bulky organosilane precursors may hinder the polycondensation reaction. As a reference sample, triethylamine was added to a sol solution prepared by the conventional SG method (denoted as SG/A method). The acid catalyst (HCl) was neutralized and the degree of condensation of the spin-coated film was decreased to 52% (Fig. 2d). However, no fluid film was obtained because the reaction mixture contained a high concentration of reactive Si–OH species and the curing reaction occurred immediately after the evaporation of solvents. These results indicate that the stop and restart of the sol–gel reaction are realized by adopting the appropriate procedures to control the subsequent steps as shown in Scheme 1. The effects of acid and base catalysts on reaction kinetics and microstructures of products have been investigated in conventional hydrolysis and polycondensation processes.31 In the present study, addition of acid/base mixed catalysts to organosilane precursors prior to hydrolysis led to the construction of a new processing method of sol–gel thin films.
Moreover, we used amines with different basicities (triethylamine (pKa: 10.7), imidazole (pKa: 7.0) and pyridine (pKa: 5.3)) in the TE/A method and the curing times of transesterified 1a were measured at room temperature to confirm the effect of basic compounds for suppressing the catalytic activity of the acids (Table S3, ESI†). The curing times were longer when stronger bases were used, which indicates that the presence of a strong base is necessary to stop the hardening of the films.
2.3. Versatility of TE/A method
We confirmed that nanoscale surface molding through the TE/A method could be achieved for other organosilica films prepared from highly reactive precursors with four silyl groups and larger π-conjugated moieties such as naphthalimide dimer (2)29 and perylene bisimide (3)30 (Fig. 3). The nanoimprinted films (2-TE/A and 3-TE/A) have vertically oriented nanopore arrays with the structural periodicity of 250 nm and the depth of ca. 180–190 nm, which are almost the same as those formed for 1-TE/A films using mold I. For these precursors, nanoimprinting using the SG method was not achievable due to their rapid curing after the evaporation of solvents (Fig. 3, 2-SG and 3-SG). The TE/A method is applicable not only to precursors with isopropoxysilyl groups but also to general organosilane precursors with ethoxysilyl groups. Moreover, it was found that the stop and restart behavior of the polycondensation of alkoxysilane precursors could be induced using different reagents with similar roles, e.g., 2-ethoxyethanol, trifluoroacetic acid and dipropylamine, instead of 2-ME, TsOH and NEt3, respectively (Fig. S8, ESI†). These results suggest the great potential of the TE/A method for nano-molding of a wide range of organosilica hybrid materials. The combination of 2-ME, TsOH and NEt3 is experimentally preferred, because acids should be less volatile during transesterification at high temperatures, and amines and alcohols should have appropriate volatility during film formation. | ||
| Fig. 3 (a) Chemical structure of 2 and AFM images of nanoimprinted 2-TE/A and 2-SG films (prepared using mold I). (b) Chemical structure of 3 and AFM images of nanoimprinted 3-TE/A and 3-SG films (prepared using mold I). | ||
We further expected that the present approach could be effective for sol–gel reactions using other metal alkoxide precursors. The TE/A method was applied to the synthesis of nanoimprinted TiO2, ZrO2 and GeO2 films (TiO2-TE/A, ZrO2-TE/A and GeO2-TE/A) from Ti(OBu)4, Zr(OiPr)4 and Ge(OiPr)4, respectively. These metal tetraalkoxides are more reactive than tetraalkoxysilanes. When the SG method was applied to these metal tetraalkoxides, the sol mixtures resulted in precipitates of polycondensed materials immediately after the addition of hydrochloric acid. Up to now, mechanical surface nano-processing of inorganic sol–gel films had only been reported in limited cases, using metal complexes with bulky ligands and assistance by photoreactive organic components.32–34 In contrast, nanoimprinting of the ceramic films was successfully achieved by applying the TE/A method. Fig. 4a–d shows the AFM images of the nanoimprinted metal oxide films prepared using polymer molds with 460 nm-pitch nanopillar or nanohole patterns. Nanohole and nanopillar arrays were formed by the structural transfer from the polymer molds. It was possible to induce curing at the desired timing by heating and atmospheric exposure of the precursor thin films composed of the metal alkoxides and the mixed catalysts, which led to nanoimprinting with good reproducibility. The heights of the patterned nanostructures are ca. 55–75% of those of the surface structures of the molds due to volume shrinkage of the frameworks. After calcination at 550 °C, further volume shrinkage of the ceramic frameworks was observed, but the nanoscale periodic patterns were retained for all the films (Fig. 4e, f and Fig. S14, ESI†), accompanied by the crystallization of each framework (Fig. 4g).35–37
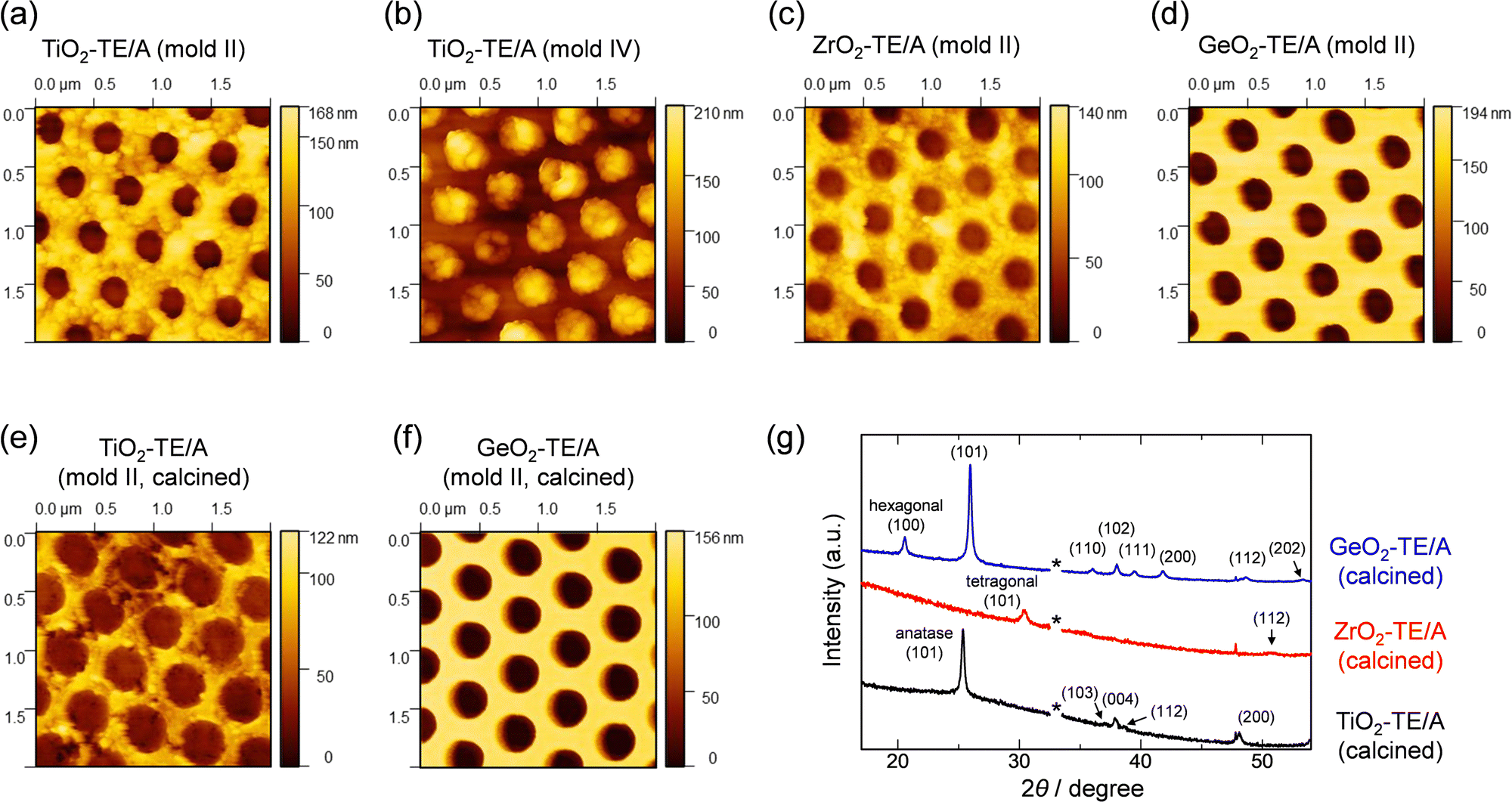 | ||
| Fig. 4 AFM images of the nanoimprinted metal oxides using the TE/A method: (a) TiO2-TE/A (mold II); (b) TiO2-TE/A (mold IV); (c) ZrO2-TE/A (mold II); (d) GeO2-TE/A (mold II); (e) TiO2-TE/A (mold II) after calcination at 550 °C; (f) GeO2-TE/A (mold II) after calcination at 550 °C. (g) X-ray diffraction patterns of TiO2-TE/A, ZrO2-TE/A, and GeO2-TE/A after calcination at 550 °C (the asterisk indicates the position of an X-ray diffraction peak derived from the Si substrate). Mold IV: nanohole array; diameter: 240 nm; height: 200 nm; pitch: 460 nm. | ||
3. Conclusions
We established a reaction method to temporarily stop and restart the polycondensation of sol–gel precursors with intrinsic high reactivity. Using this simple method combining acid-catalyzed transesterification with addition of basic amine compounds, nanoimprinting could be performed for various sol–gel materials with high reproducibility. Formation of nanostructures via stabilized fluid precursor films has great potential for large-scale production such as roll-to-roll processes. We expect that our approach will open a new path to synthesizing various organic–inorganic hybrid materials and nanostructured ceramics, and to enhancing their optical, electronic and catalytic functionalities. Application to other metal oxides and composite materials containing multiple metals is currently underway.Author contributions
N. M.: supervision, conceptualization, methodology, investigation, validation, data curation, visualization, writing – original draft preparation. Y. Y.: methodology, investigation, validation, data curation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Mr Kenichi Yagi and Ms Miku Maeyama (Toyota Central R&D Labs., Inc.) for 29Si NMR measurements of solution samples. The authors are also grateful to Dr Yasutomo Goto, Mr Masakazu Murase and Dr Masaru Ogawa (Toyota Central R&D Labs., Inc.) for helpful discussions.Notes and references
- A. E. Danks, S. R. Hall and Z. Schnepp, The evolution of ‘sol–gel’ chemistry as a technique for materials synthesis, Mater. Horiz., 2016, 3, 91–112 RSC.
- K. Deshmukh, T. Kovářík, T. Křenek, D. Docheva, T. Stich and J. Pola, Recent advances and future perspectives of sol–gel derived porous bioactive glasses: a review, RSC Adv., 2020, 10, 33782–33835 RSC.
- R. Chen and L. Lan, Solution-processed metal-oxide thin-film transistors: a review of recent developments, Nanotechnology, 2019, 30, 312001 CrossRef CAS PubMed.
- D. Chen, Anti-reflection (AR) coatings made by sol–gel processes: a review, Sol. Energy Mater. Sol. Cells, 2001, 68, 313–336 CrossRef CAS.
- S. Esposito, Traditional sol–gel chemistry as a powerful tool for the preparation of supported metal and metal oxide catalysts, Materials, 2019, 12, 668 CrossRef CAS PubMed.
- R. B. Figueira, C. J. R. Silva and E. V. Pereira, Organic–inorganic hybrid sol–gel coatings for metal corrosion protection: a review of recent progress, J. Coat. Technol., 2015, 12, 1–35 CrossRef CAS.
- W. K. Tan, H. Muto, G. Kawamura, Z. Lockman and A. Matsuda, Nanomaterial fabrication through the modification of sol–gel derived coatings, Nanomaterials, 2021, 11, 181 CrossRef CAS PubMed.
- K. Davis, R. Yarbrough, M. Froeschle, J. White and H. Rathnayake, Band gap engineered zinc oxide nanostructures via a sol–gel synthesis of solvent driven shape-controlled crystal growth, RSC Adv., 2019, 9, 14638–14648 RSC.
- P. P. Conti, E. Scopel, E. R. Leite and C. J. Dalmaschio, Nanostructure morphology influences in electrical properties of titanium dioxide thin films, J. Mater. Res., 2020, 35, 3012–3020 CrossRef CAS.
- K. Feddi, M. Kria, M. El-Yadri, F. C. Fobasso Mbognou, G. Long, A. Tiutiunnyk, L. M. Pérez, D. Laroze and E. Feddi, Geometrical confinement effects on fundamental thermal properties of rutile and anatase TiO2 cylindrical and tubular nanostructures, Phys. Scr., 2020, 95, 105706 CrossRef CAS.
- Y. Yamada, M. Murase, K. Yatsugi and N. Mizoshita, Laser desorption/ionization mass spectrometry using TiO2 nanopillar array substrates with tunable surface roughness and wettability, ACS Appl. Nano Mater., 2021, 4, 13884–13895 CrossRef CAS.
- P. Sudarsanam, A. Köckritz, H. Atia, M. H. Amin and A. Brückner, Synergistic nanostructured MnOx/TiO2 catalyst for highly selective synthesis of aromatic imines, ChemCatChem, 2021, 13, 1990–1997 CrossRef CAS.
- R. Ciriminna, A. Fidalgo, V. Pandarus, F. Beland, L. M. Ilharco and M. Pagliaro, The sol–gel route to advanced silica-based materials and recent applications, Chem. Rev., 2013, 113, 6592–6620 CrossRef CAS PubMed.
- O. J. Dautel, G. Wantz, R. Almairac, D. Flot, L. Hirsch, J.-P. Lere-Porte, J.-P. Parneix, F. Serein-Spirau, L. Vignau and J. J. E. Moreau, Nanostructuration of phenylenevinylenediimide-bridged silsesquioxane: from electroluminescent molecular J-aggregates to photoresponsive polymeric H-aggregates, J. Am. Chem. Soc., 2006, 128, 4892–4901 CrossRef CAS PubMed.
- J. J. E. Moreau, B. P. Pichon, M. Wong Chi Man, C. Bied, H. Pritzkow, J.-L. Bantignies, P. Dieudonn and J.-L. Sauvajol, A better understanding of the self-structuration of bridged silsesquioxanes, Angew. Chem., Int. Ed., 2004, 43, 203–206 CrossRef CAS PubMed.
- Y. Guo, A. Mylonakis, Z. Zhang, P. I. Lelkes, K. Levon, S. Li, Q. Feng and Y. Wei, Oligoaniline-contained electroactive silsesquioxane precursor for synthesizing novel siliceous materials, Macromolecules, 2007, 40, 2721–2729 CrossRef CAS.
- D. C. Bradley, Metal alkoxides as precursors for electronic and ceramic materials, Chem. Rev., 1989, 89, 1317–1322 CrossRef CAS.
- E. P. Turevskaya, M. I. Yanovskaya and N. Y. Turova, Preparation of oxide materials from metal alkoxides, Inorg. Mater., 2000, 36, 330–341 CrossRef.
- N. Pinna and M. Niederberger, Surfactant-free nonaqueous synthesis of metal oxide nanostructures, Angew. Chem., Int. Ed., 2008, 47, 5292–5304 CrossRef CAS PubMed.
- T. Kaneda, D. Hirose, T. Miyasako, P. T. Tue, Y. Murakami, S. Kohara, J. Li, T. Mitani, E. Tokumitsu and T. Shimoda, Rheology printing for metal-oxide patterns and devices, J. Mater. Chem. C, 2014, 2, 40–49 RSC.
- S.-H. Shin, S.-Y. Choi, M. H. Lee and J. Nah, High-performance piezoelectric nanogenerators via imprinted sol−gel BaTiO3 nanopillar array, ACS Appl. Mater. Interfaces, 2017, 9, 41099–41103 CrossRef CAS PubMed.
- Y.-H. Kim, J.-S. Heo, T.-H. Kim, S. Park, M.-H. Yoon, J. Kim, M. S. Oh, G.-R. Yi, Y.-Y. Noh and S. K. Park, Flexible metal-oxide devices made by room temperature photochemical activation of sol–gel films, Nature, 2012, 489, 128–132 CrossRef CAS PubMed.
- J.-W. Jo, J. Kim, K.-T. Kim, J.-G. Kang, M.-G. Kim, K.-H. Kim, H. Ko, Y.-H. Kim and S. K. Park, Highly stable and imperceptible electronics utilizing photoactivated heterogeneous sol-gel metal–oxide dielectrics and semiconductors, Adv. Mater., 2015, 27, 1182–1188 CrossRef CAS PubMed.
- N. Martín-Arbella, I. Bretos, R. Jiménez, M. L. Calzada and R. Sirera, Photoactivation of sol–gel precursors for the low-temperature preparation of PbTiO3 ferroelectric thin films, J. Am. Ceram. Soc., 2011, 94, 396–403 CrossRef.
- N. Mizoshita, Y. Yamada, M. Murase, Y. Goto and S. Inagaki, Direct nanoimprinting of nanoporous organosilica films consisting of covalently crosslinked photofunctional frameworks, Nanoscale, 2020, 12, 21146–21154 RSC.
- N. Mizoshita, Y. Yamada, M. Murase, Y. Goto and S. Inagaki, Nanoporous substrates with molecular-level perfluoroalkyl/alkylamide surface for laser desorption/ionization mass spectrometry of small proteins, ACS Appl. Mater. Interfaces, 2022, 14, 3716–3725 CrossRef CAS PubMed.
- S. Hartmann, D. Brandhuber and N. Hüsing, Glycol-modified silanes: novel possibilities for the synthesis of hierarchically organized (hybrid) porous materials, Acc. Chem. Res., 2007, 40, 885–894 CrossRef CAS PubMed.
- M. Weinberger, S. Puchegger, T. Fröschl, F. Babonneau, H. Peterlik and N. Hüsing, Sol–gel processing of a glycolated cyclic organosilane and its pyrolysis to silicon oxycarbide monoliths with multiscale porosity and large surface areas, Chem. Mater., 2010, 22, 1509–1520 CrossRef CAS.
- N. Mizoshita, M. Murase, Y. Yamada and Y. Goto, Nanostructured organosilicas constructed by homopolycondensation of a transesterified bulky precursor and their potential in laser desorption/ionization, Mater. Chem. Front., 2021, 5, 851–861 RSC.
- N. Mizoshita, T. Tani, H. Shinokubo and S. Inagaki, Mesoporous organosilica hybrids consisting of silica-wrapped π–π stacking columns, Angew. Chem., Int. Ed., 2012, 51, 1156–1160 CrossRef CAS PubMed.
- C. J. Brinker, Hydrolysis and condensation of silicates: effects on structure, J. Non-Cryst. Solids, 1988, 100, 31–50 CrossRef CAS.
- F. Back, M. Bockmeyer, E. Rudigier-Voigt and P. Löbmann, Hybrid polymer sol–gel material for UV-nanoimprint: microstructure and thermal densification, J. Sol-Gel Sci. Technol., 2013, 66, 73–83 CrossRef CAS.
- H.-H. Park, X. Zhang, S.-W. Lee, K.-D. Kim, D.-G. Choi, J.-H. Choi, J. Lee, E.-S. Lee, H.-H. Park, R. H. Hill and J.-H. Jeong, Facile nanopatterning of zirconium dioxide films via direct ultraviolet-assisted nanoimprint lithography, J. Mater. Chem., 2011, 21, 657–662 RSC.
- S. S. Dinachali, M. S. M. Saifullah, R. Ganesan, E. S. Thian and C. He, A universal scheme for patterning of oxides via thermal nanoimprint lithography, Adv. Funct. Mater., 2013, 23, 2201–2211 CrossRef CAS.
- J. He, Y. Du, Y. Bai, J. An, X. Cai, Y. Chen, P. Wang, X. Yang and Q. Feng, Facile formation of anatase/rutile TiO2 nanocomposites with enhanced photocatalytic activity, Molecules, 2019, 24, 2996 CrossRef CAS PubMed.
- Y.-F. Lin and F.-L. Liang, Synthesis of a ZrO2/carbon aerogel composite with tetragonal ZrO2 structures assisted by the formation of phenol formaldehyde resin, CrystEngComm, 2015, 17, 678–685 RSC.
- Y. Sun, W. Xu, X. Fu, Z. Sun, J. Wang, J. Zhang, D. Rosenbach, R. Qi, K. Jiang, C. Jing, Z. Hu, X. Ma and J. Chu, Evaluation of lattice dynamics, infrared optical properties and visible emissions of hexagonal GeO2 films prepared by liquid phase deposition, J. Mater. Chem. C, 2017, 5, 12792–12799 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization of organosilica films, properties of catalysts and supplementary data on metal oxide films. See DOI: https://doi.org/10.1039/d3qm00070b |
| This journal is © the Partner Organisations 2023 |