
Recent advances in carbon-resistant anodes for solid oxide fuel cells
Wei
Zhang
,
Jialu
Wei
,
Fusheng
Yin
and
Chunwen
Sun
 *
*
School of Chemical and Environmental Engineering, China University of Mining & Technology (Beijing), Beijing 100083, P. R. China. E-mail: csun@cumtb.edu.cn
First published on 27th February 2023
Abstract
Solid oxide fuel cells (SOFCs) can efficiently satisfy the power supply demand at any time and place in an environmentally friendly manner. However, considering the commercial application of SOFCs, the deposition of carbon on conventional SOFC anodes (nickel-based anodes) during their operation with hydrocarbon fuels, which leads to a degradation in the cell performance, needs to be well addressed. In this review, we discuss the carbon deposition process in SOFCs, carbon detection methods, and strategies to solve anode carburization, with a primary focus on alternative anode materials. Specifically, the coking mechanism, carbon-resistant strategies, and research development of bimetallic-cermet materials are reviewed in detail. In addition, the principle of in situ nanoparticle exsolution from perovskite materials and factors affecting the growth of dissolved nanoparticles are introduced, and the application of in situ exsolution in anti-carbon anodes is also discussed. Furthermore, we present the carbon deposition phenomenon in SOFC anodes and alternative anode design ideas in terms of simulation, calculation, and reaction kinetic models. Finally, we also give future research directions, especially proposing the potential application of single-atom based anode catalysts in hydrocarbon-fueled SOFCs.
 Wei Zhang | Wei Zhang is currently a PhD candidate under the supervision of Prof. Chunwen Sun at China University of Mining and Technology (Beijing). He received his BS Degree in Applied Chemistry from Shandong University of Technology. His research interests include solid oxide fuel cells operating at intermediate and low temperatures, mainly focusing on designing anodes of bimetallic-cermet materials, in situ exsolution of perovskite materials and single-atom catalysts. |
 Jialu Wei | Jialu Wei is currently pursuing her MS Degree under the supervision of Prof. Chunwen Sun at China University of Mining and Technology (Beijing). She received her BS Degree in Applied Chemistry from China University of Mining and Technology (Beijing). Her research interests are mainly focused on developing anode materials for solid oxide fuel cells. |
 Fusheng Yin | Fusheng Yin is currently pursuing his MS Degree under the supervision of Prof. Chunwen Sun and Dr Zhijun Zhang at China University of Mining and Technology (Beijing). He received his BS Degree in Chemical Engineering and Technology from Jiangsu University. His research interests are first principles computational chemistry and all-solid-state batteries. |
 Chunwen Sun | Prof. Chunwen Sun received his PhD in Condensed Matter Physics from the Institute of Physics (IOP), Chinese Academy of Sciences (CAS) in 2006. He is a Full Professor and Group Leader of Energy Storage Materials and Devices at the School of Chemical and Environmental Engineering at China University of Mining & Technology (Beijing). His research interests include lithium-ion batteries, all-solid-state batteries, solid oxide fuel cells and electrocatalysis. He has published 150 peer-reviewed papers with citations of more than 14 |
1. Introduction
The sustainable development of modern society is hindered by the issues of energy shortage and environmental pollution. Therefore, solving or alleviating these problems has become a notable research direction. In this case, the development of renewable energy, the improvement of storage technologies and advanced energy conversion are essential to solve the above-mentioned challenges.1–4Fuel cells (FCs) are electrochemical energy conversion devices that directly convert chemical energy into electrical energy,5 which are highly efficient compared to other conventional energy conversion devices.6,7 Further, FCs can operate in the opposite mode, that is, they can also convert electrical energy generated by modern renewable technologies such as solar and wind energy back into chemical energy.8 Therefore, FCs are a promising technology in the clean utilization of energy. FCs are desirable all-solid-state structures, i.e., there are no moving parts, and thus there is no risk of leakage in FCs. Consequently, these systems are expected to possess high reliability and long life. Moreover, unlike regular batteries, FCs allow arbitrary scaling between power (determined by fuel cell size) and capacity (determined by fuel storage size). Thus, based on their significant advantages, various types of FCs with different electrolyte materials have been developed.5,6 Among them, proton-exchange membrane fuel cells (PEMFCs) and solid oxide fuel cells (SOFCs) have received significant attention owing to their relatively higher efficiency. Nevertheless, PEMFCs only use precious metals anode (e.g., Pt) and to prevent poisoning of Pt catalysts by CO, high-purity (∼99.99%) H2 is required. All these restrictions greatly increase the production cost. In FCs, the chemical energy that is not converted into electricity will partially be converted into heat, but the high operating temperature of SOFCs (typically between 600–1000 °C) is different from that in other types of FCs, and hence the waste heat can be well used in SOFCs. Therefore, some of the energy of SOFCs can be recovered by introducing the reaction products into the steam turbine. SOFC/gas turbine hybrid systems provide combined heat and power with efficiencies of up to 90%.6,7 Given that oxides and base metals are sufficiently active under high operating temperature, the use of precious metals in the electrodes can be avoided. Briefly, SOFCs have several advantages over PEMFCs, including relatively inexpensive materials (e.g., metal-ceramic and metal supported),9–13 high fuel flexibility (e.g., hydrocarbon, syngas, solid carbon, and ammonia)14–21 and very high efficiency.
However, although SOFCs exhibit excellent electrocatalytic properties when hydrogen is employed as the fuel, it is difficult to store and transport H2 due to its low volumetric energy density. Alternatively, as mentioned above, SOFCs offer excellent fuel flexibility and can use hydrocarbon fuels at high operating temperatures. By avoiding the energy losses associated with external reformers, directing the electrochemical oxidation of CH4 to generate electricity can significantly improve the efficiency (5–7%)22 and accelerate the applications of SOFCs in transportation and power distribution equipment.5,23 Presently, a well-developed methane-rich natural gas infrastructure is available, which can be readily and easily used in residential or office buildings. Therefore, hydrocarbon fuels are considered a potential alternative to hydrogen fuels in SOFCs. However, due to the deposition of carbon caused by the incomplete oxidation of hydrocarbons and sulfur poisoning of the catalyst caused by common pollutants in the fuel, the catalytic activity of the most investigated anode materials of SOFCs, i.e., Ni–YSZ, is dramatically affected during operation with hydrocarbon fuels.10,24–28
To achieve the large-scale commercial applications of SOFCs, many studies have been devoted to investigating carbon-resistant anodes, and some breakthroughs have been achieved in this regard. In this review, we explain the causes and types of carbon deposits in hydrocarbon-fueled SOFCs and summarize the relevant characterization methods. In particular, the methods for reducing carbon deposition on the anode based on thermodynamics and reaction kinetics are extensively reviewed, and the recent advances in carbon-resistant anodes are summarized in detail according to three aspects, as follows: (1) bimetallic-cermet materials, (2) ceramic materials, and (3) anode reforming layer materials. Furthermore, the outlook for some exciting approaches is discussed to provide some suggestions to broaden the research vision.
1.1 Working principle of SOFCs
An SOFC consists of an oxygen electrode (cathode), a fuel electrode (anode) and a dense ionic conductor (solid electrolyte) sandwiched between them, as well as an external circuit connecting the anode and cathode to collect electric energy, as shown in Fig. 1. During the working process, the electrodes do not directly participate in the reaction, they only act as a catalyst and provide ion and electron transport. If hydrocarbons are used as fuel gas for SOFCs, and assuming that the fuel is completely oxidized during operation, then the following reactions will occur spontaneously at the cathode and anode, as follows: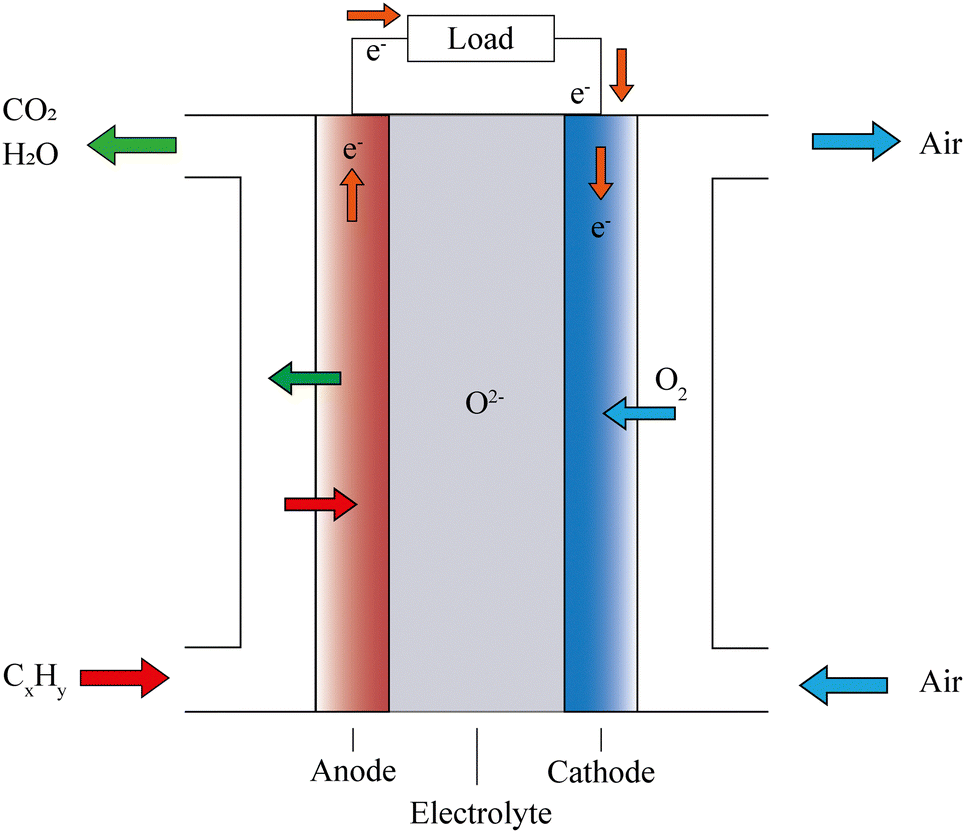 | ||
| Fig. 1 Schematic diagram of a hydrocarbon-fueled SOFC. | ||
Cathode reaction:
| O2 + 4e− → 2O2− | (1) |
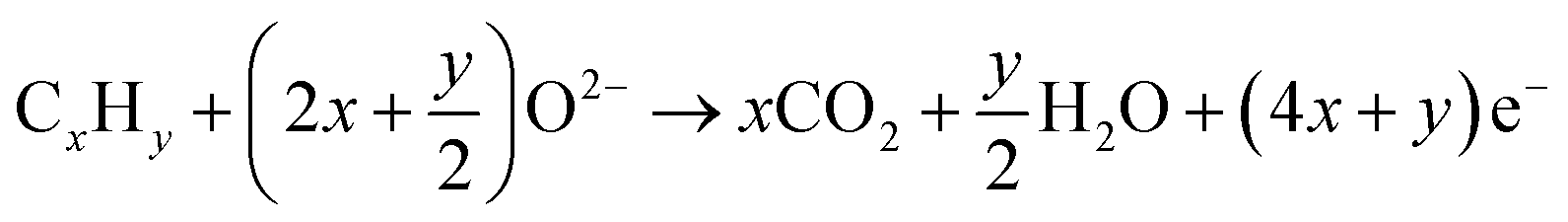 | (2) |
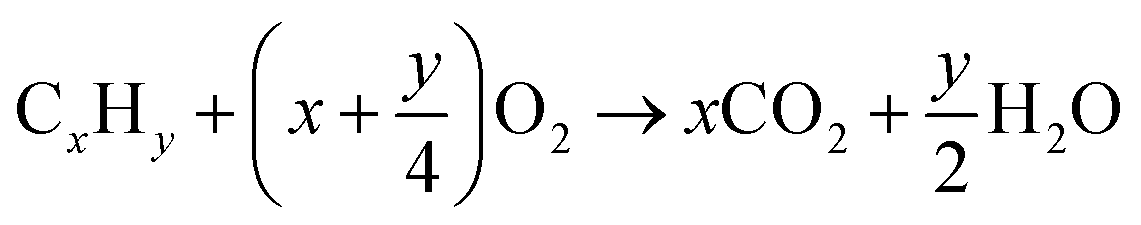 | (3) |
The cathode is in contact with oxygen, and molecular O2 is reduced to oxide anions (O2−) using electrons from the anode. Subsequently, the O2− ions travel through the electrolyte and enter the anode, where they oxidize fuels to produce water and carbon dioxide, finally releasing electrons at the anode. The resulting electrons pass through the external circuit and return back to the cathode. When fuel and oxidizer are abundant, the SOFC can continue to supply power to the external circuit.
The ideal cathode electrode is usually a hybrid conductor that conducts ions and electrons. This material should have high oxidation resistance and high catalytic activity in the cathode environment, which can easily dissociate molecular oxygen into O2− ions. The most commonly used material for the cathode electrode in SOFCs is Sr-doped LaMnO3 (LSM), but it exhibits relatively poor electrical performance at temperatures below 800 °C because there are fewer oxygen vacancies. Consequently, some alternative cathode materials have been developed,29 including La0.8Sr0.2CoO3 (LSC),30 La0.8Sr0.2FeO3 (LSF),31 La0.8Sr0.2Co0.2Fe0.8O3 (LSCF),32 Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF),33 PrBa0.5Sr0.5Co1.5Fe0.5O5+δ(PBSCF),34 Ba1−xPrxCo1−yFeyO3−δ(BPCF),35 Sr0.95Ce0.05CoO3−δ(SCCO),36 and Ba0.9Co0.7Fe0.2Nb0.1O3−δ(BCFN).37
There are two charged substances in the electrochemical reaction of SOFCs, i.e., electrons and ions. In the case of electrons, their transfer from the region they produced to the region they consumed is relatively easy. In contrast, for ions, their transport is much more difficult, mainly because they are much larger and heavier than electrons, and thus an electrolyte is necessary to provide a path for ions to transport. Ions move through the electrolyte via a “jump” mechanism, specifically, ions “jump” from one position to another in the lattice of the electrolyte material and the jumping process only occurs in places such as vacancies and gap-filling ions, which is prompted by lattice defects. Therefore, the conductivity of the electrolyte is controlled by the defects in the lattice. Generally, the intrinsic defect concentration of the electrolyte is low, and thus the non-intrinsic defect concentration is introduced in the electrolyte lattice by doping. The most commonly used electrolyte material is yttria-stabilized zirconia (YSZ), which shows excellent stability in both highly reducing and highly oxidizing environments, but it has low ionic conductivity. Therefore, some other doped oxides, such as scandia-stabilized zirconia (ScSZ), gadolinium-doped ceria (GDC) and samarium-doped ceria (SDC), are being investigated because they have higher ionic conductivities and good compatibilities with fuel electrode materials.38
The ionic conductivity of the electrolyte is also closely related to the temperature, and thus the appropriate operating temperature of SOFCs depends on whether sufficient ionic conductivity is achieved in the electrolyte at this temperature.38 Here, even in the case of excellent electrolytes, their ionic conductivity is usually 4–8 orders of magnitude lower than the electronic conductivity of metals, and hence the area-specific resistance (ASR) of an SOFC is mainly determined by the resistance of the electrolyte. Consequently, ion transport can cause significant resistance loss, which can degrade the performance of the SOFC. Thus, to attenuate this effect, the electrolyte should technically be as thin as possible to shorten the ion conduction pathway. There are many commonly used methods to prepare thin electrolytes, such as tape casting,39,40 spin coating,41 wet powder spraying42 and dip coating.43–45 Among the processes employed for the fabrication of thin films, magnetron sputtering46 and pulsed laser deposition47 have emerged as unique technologies to obtain oxide films by selecting appropriate process parameters. Furthermore, the electrolyte must be dense enough to separate the oxygen and fuel, while being thin, and it must be an electronic insulator. In addition, given that the partial pressure of O2 varies from ∼1 atm at the cathode to ∼10−20 atm or less at the anode, the electrolyte needs to have appropriate mechanical strength.48 When SOFCs operate at high temperatures, their materials will expand, producing cracks. Therefore, each component of SOFCs needs to possess a similar thermal expansion coefficient (TEC) to prevent the formation of cracks, and their TEC compatibilities are usually ensured by mixing with the electrolyte material.
Anode materials usually have high conductivity, good catalytic activity, and suitable adsorption and desorption of reactants. The ability of the anode layer to catalyze the oxidation of fuels has always been the focus of SOFC research. To maximize the reaction surface area, anodes with porous micro/nano-structure need to be prepared to achieve close contact between the gas (gas phase) and electronic (electron-conducting phase) and oxygen ion (ion-conducting phase), and these reaction sites are called the three-phase boundary (TPB). The TPB concept is an important guide for the optimization of anode and cathode microstructures. It should be noted that this simplified description of the TPB does not consider the fact that some oxides, which are often added to the anode, have mixed electronic and ionic conductivity (MEIC), where there addition of a MEIC oxide will expand the TPB.
Optimizing the thickness of the anode catalyst layer requires a fine balance between mass transport and catalytic activity, given that thinner layers facilitate better gas diffusion and catalyst utilization, whereas thicker layers contain a great catalyst load and provide more TPB regions. The most commonly used material for the anode electrode in SOFCs is a nickel and YSZ composite (Ni–YSZ), which is a ceramic and metal mixture. Nickel provides electronic conductivity and catalytic activity, while YSZ provides ionic conductivity, thermal expansion compatibility comparable to the electrolyte, and maintains the high porosity and large surface area of the anode structure.49 However, in addition to the above-mentioned carbon deposition problems in Ni-based anodes, cell breakage caused by the sintering of nickel particles during high-temperature operation of SOFCs and significant volume changes during re-oxidation process will lead to the interruption of the partial electron conduction network and failure of the catalytic active sites in the anode. Furthermore, carbon deposition, nickel agglomeration and poor redox cycling all block the reaction sites at the TPB, and consequently the performance of Ni-based anode SOFCs degrades.50,51 However, thus far, no anode catalyst has been able to match the competitive price of the Ni cermet anode with good catalytic performance and long-term stability. Fortunately, there numerous studies have been conducted to address the above-mentioned problems, where among them, the development of carbon-resistant anode catalysts is one of the most promising methods to achieve high SOFC efficiency.24 Carbon-resistant anode materials will be described in detail later.
The performance of fuel cells can be summarized by a current–voltage characteristic diagram (I–V), where it is critical to maintain high voltages under high current loads of fuel cells. Unfortunately, due to the unavoidable losses, the actual voltage output of fuel cells is always lower than that predicted by thermodynamic theory. In general, there are three main voltage losses, which can be expressed as follows:
| V = Ethermo − ηact − ηohmic − ηconc | (4) |
The activation loss is the overpotential required to exceed the half reaction energy barrier. At low operation temperature (e.g., PEMFCs), the output voltage drops sharply due to activation loss. However, at high operating temperatures in SOFCs, this problem does not exist. The main factors that affect the voltage drop in SOFCs are ohmic loss and concentration loss.52
1.2 Challenges in hydrocarbon-fueled SOFCs
As mentioned above, the high operating temperature of SOFCs is actually in the temperature zone where the direct internal reforming (DIR) reaction of hydrocarbon occurs, thus opening up the possibility of using hydrocarbon as fuel. Hydrocarbon fuels undergo a variety of basic reactions under the operating conditions of SOFCs, and the elementary reactions are usually divided into the following reactions:14,53–57methane steam reforming:
| CH4 + H2O → CO + 3H2 | (5) |
| CH4 + CO2 → 2CO + 2H2 | (6) |
| 2CH4 + O2 + CO2 → 3H2 + 3CO + H2O | (7) |
 | (8) |
| CH4 + 2O2 → CO2 + 2H2O | (9) |
| CO + H2O → CO2 + H2 | (10) |
Basically, during methane-fueled SOFC operation, three types of reactions are considered as sources of carbon deposition, as follows:58
Methane cracking:
| CH4 → C + 2H2, ΔH = +19 kJ mol−1 | (11) |
| CO + H2 → C + H2O, ΔH = −131 kJ mol−1 | (12) |
| 2CO → C + CO2, ΔH = −172 kJ mol−1 | (13) |
Carbon can be formed by the thermal cracking of hydrocarbons or the decomposition of CO.59 Given that the Gibbs free energy of the Boudouard reaction is the largest, it is generally considered to be the main carbon-forming reaction in research. When any one or more of these reactions occur, the following carbon deposition formation processes will appear. A schematic of the process on the anode catalyst (e.g., Ni metal) is shown in Fig. 2, consisting of the following steps:13,60 in stage 1, at the beginning of any one of the above-mentions reactions, tiny amounts of carbon begin to be deposited on the active catalytic site, but it does not cover the entire anode surface. In this process, the current density of the SOFC increases slightly, and hence the deposited carbon can improve the conductivity of the anode. In stage 2, the carbon deposited on the active catalytic site is interconnected into layers, which will cover the entire anode surface. Considering that the deposited carbon is porous, fuel gases are still allowed to pass through and react on the anode surface, but the slowly formed coverage of deposited carbon can reduce the reaction TPB region and hinder the reaction rate. Therefore, at this stage, the current and voltage will decrease as time progress. In stage 3, the deposited carbon forms a thick layer on the anode and prevents fuel gas from penetrating into the TBP region to react, severely impeding the reaction rate. Consequently, with time, the output power of the SOFC will slowly change to zero.
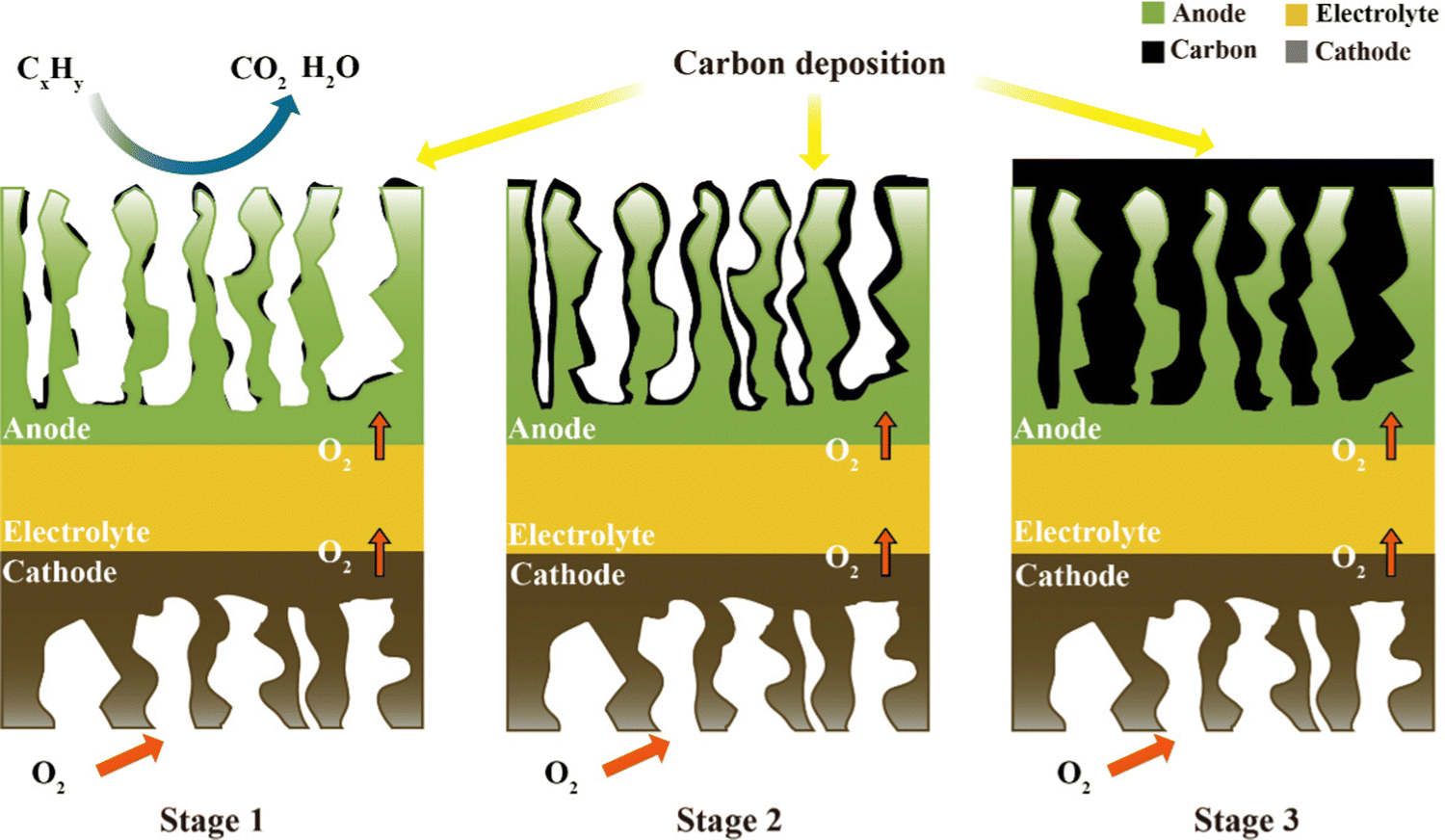 | ||
| Fig. 2 Schematic of the carbon deposition processes. | ||
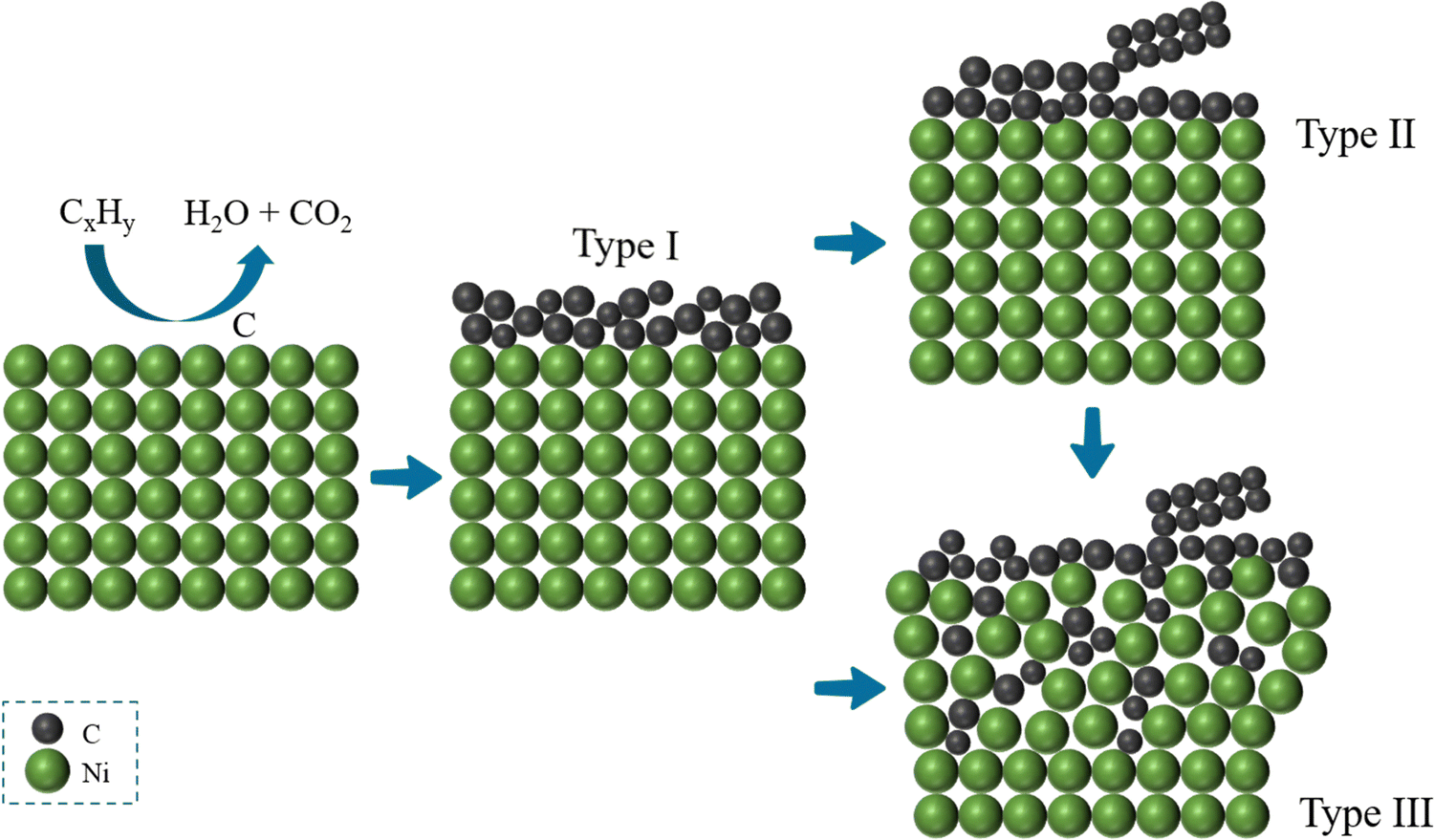
In the process of carbon deposition, generally three types of carbon species exist14,61,62 including amorphous carbon (Type I), crystalline graphite form (Type II) and injected carbon (Type III). As illustrated in Fig. 3, if the carbon cannot be removed from oxidized form, Type I carbon first appears on the surface of the catalyst active sites. The high-resolution transmission electron microscopy image (HR-TEM) of Type I carbon in the TEM grid is shown in Fig. 4(a).63 As the reaction proceeds, a large amount of carbon continues to be added to the active sites, and the Type I carbon will go through two different pathways,64 as follows: (1) Type I carbon automatically undergoes a graphitization reaction upward to become Type II carbon, depending on the various catalysts and Type II carbon will behave as varying forms of carbon materials, such as carbon flakes, carbon fibers and carbon nanotubes. The scanning electron microscopy (SEM) images of Type II carbon are shown in Fig. 4(b) and (c), where the deposited carbon appears as carbon nanofibers at the interface between Ni and the oxide particles on the anode, and Fig. 4(c) is a magnified image of Fig. 4(b) by ten times.65 (2) Type I carbon injects downward into metal crystalline structure, causing the structure to form Type III carbon (metal carbides), resulting in morphological changes in the metal anode structure, causing it to expand and peel off.66 As shown in Fig. 4(d), the SEM image of the cross-section of the anode demonstrates that a large number of carbon species grew inside the cell. Also, this is further proven in Fig. 4(e) and (f), where the size of the Ni particles is in the nanometer range and separated from the zirconia by a carbon layer, which confirms that Type III carbon was formed.67 In addition, Type II carbon can be directly transformed into Type III carbon, as shown in Fig. 4(g), where type II carbon is slowly converted to Type III carbon, and some of the Ni particles were stripped off the anode after 7 h.68 Given that Type III carbon is an irreversible structure and cannot be removed in the operating environment of the SOFC, this type of carbon species is extremely destructive to the catalytic performance and structure of the anode.
 | ||
| Fig. 3 Schematic of three types of carbon species. | ||
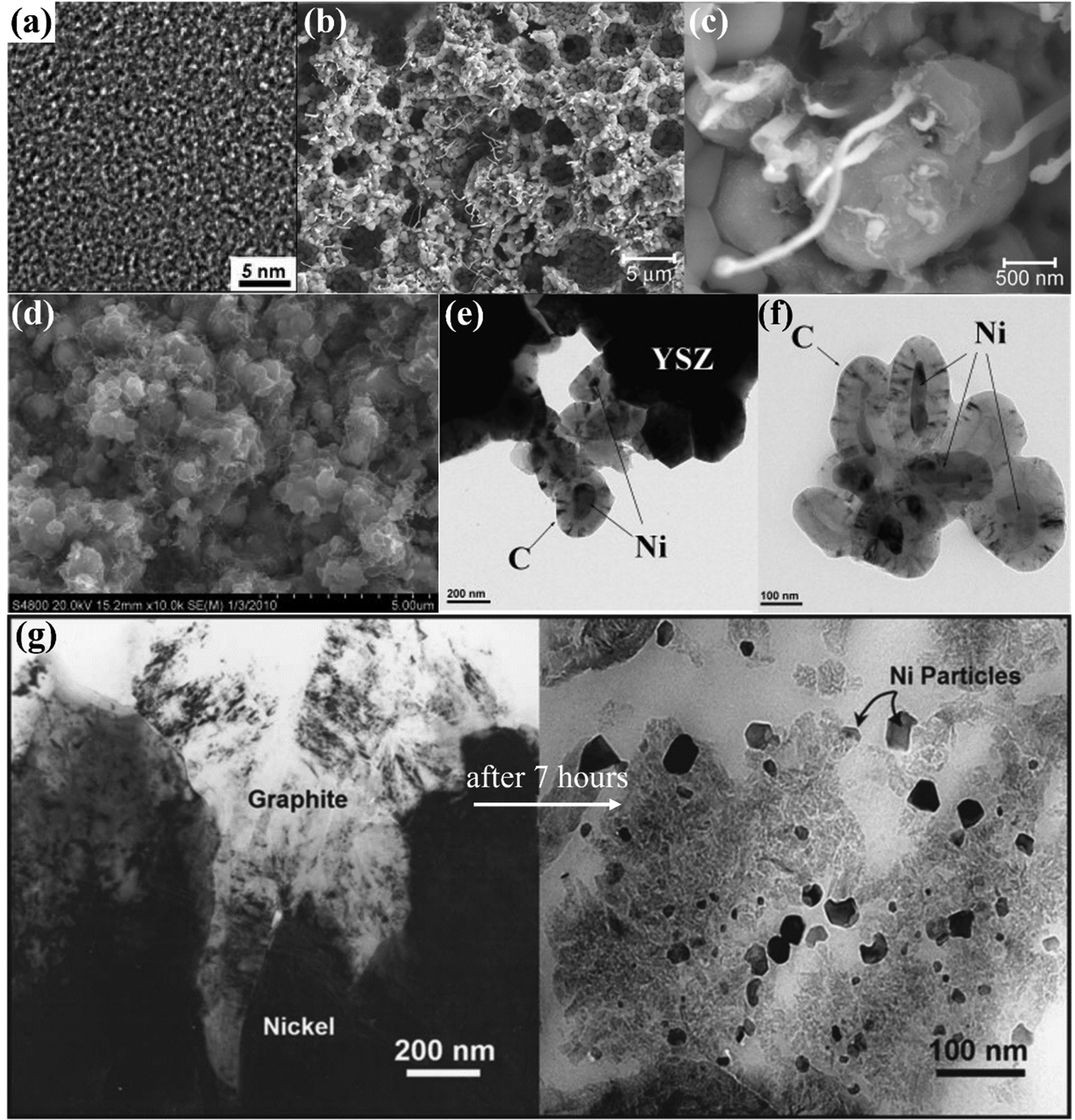 | ||
| Fig. 4 Characterization of different types of carbon species. (a) HR-TEM image of Type I carbon. (Reprinted from ref. 63 with permission from the American Chemical Society.) (b and c) SEM images of Type II carbon. (Reprinted from ref. 65 with permission from Elsevier Ltd.) (d–f) SEM and TEM images of Type III carbon. (Reprinted from ref. 67 with permission from Elsevier Ltd.) (g) TEM images of the formation process with Type III carbon. (Reprinted from ref. 68 with permission from The Electrochemical Society Ltd.) | ||
Compared with Type II and Type III carbon, Type I carbon can be more easily removed by oxygen ions, and hence a better carbon tolerance anode catalyst should allow carbon species to form Type I than the others.54,69
1.3 Characterization of carbon deposition
In the design of a favorable carbon-resistant anode, the carbon deposition phenomena must be studied in detail by using suitable characterization methods. In this case, SEM is the most direct method for characterization, clearly showing the morphological structure of the carbon deposition existing on the surface of the anode. Additionally, when combined with energy dispersive X-ray spectroscopy (EDS), the presence of carbon that is not visible to the naked eye (e.g., Type III carbon) can also be detected. However, whether it can be detected depends on the depth of the electron beams of EDS and the depth at which carbon deposition is present at the anode, thus it is best to scan the cross-section of the SOFC when performing SEM-EDS characterization.Type III carbon can cause volume expansion of the metal layer and change the lattice parameters, resulting in an increase in the crystal plane spacing of the material, and this X-ray diffraction (XRD) can be used to detect the presence of Type III carbon. Bayer et al. used an ionized magnetron sputter deposition system to deposit metastable Ni3C nanocomposite films in an amorphous carbon matrix and studied the changes in their composition and structure at both low annealing (300 °C) and high annealing (800 °C) temperatures. It can be seen from Fig. 5(a) that in the annealing process, the Ni3C phase slowly changed to a metallic Ni phase. In addition, the shift in the Ni3C diffraction peak to the right as the Ni diffraction peak also proved that the crystal plane spacing of the Ni metal decreased due to the decomposition of Ni3C.70 XRD can also be used to detect the existence of Type II carbon with slower and smaller scanning steps, but a detailed carbon database is required to match the detection data.71
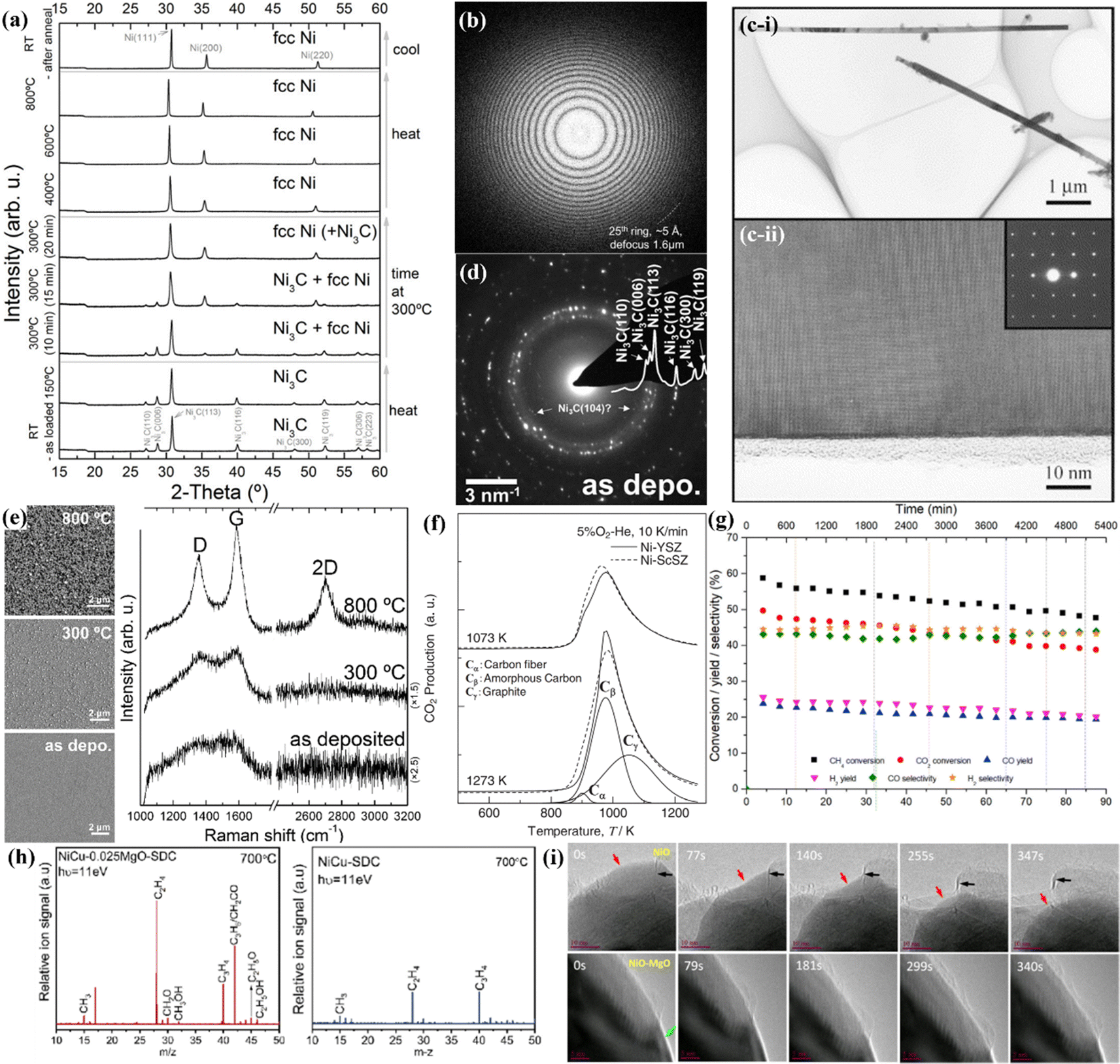 | ||
| Fig. 5 Characterization methods for carbon deposits. (a) XRD patterns showing the modification in the diffraction peak of Ni3C nanocomposite films in an amorphous carbon matrix with temperature and time. (Reprinted from ref. 70 with permission from the American Chemical Society.) (b) Thorn-ring characteristics in HRTEM-SAED of amorphous carbon. (Reprinted from ref. 72 with permission from MYJoVE Corporation.) (c and d) Lattice pattern characteristics in HRTEM-SAED of crystalline carbon. (Reprinted from ref. 73 with permission from Elsevier Ltd and from ref. 70 with permission from the American Chemical Society.) (e) Raman spectra showing that nickel can catalyze the graphitization of amorphous carbon with an increase in the annealing temperature. (Reprinted from ref. 70 with permission from the American Chemical Society.) (f) Peak deconvolution of TPO curve can confirm the presence of amorphous carbon, carbon fiber and graphite. (Reprinted from ref. 76 with permission from the American Chemical Society.) (g) In situ FTIR spectra quantifying the composition of outlet gases from the SOFC. (Reprinted from ref. 77 with permission from Elsevier Ltd.) (h) SVUV-PIMS spectra of gas-phase components for humid CH4 reacting over NiCu–0.025MgO–SDC and NiCu–SDC catalysts. (Reprinted from ref. 78 with permission from Elsevier Ltd.) (i) In situ TEM showing the sustainable graphite layer deposition phenomenon on NiO and MgO-modified NiO catalysts. (Reprinted from ref. 79 with permission from the American Chemical Society.). | ||
HR-TEM and selected area electron diffraction (SAED) can determine the presence of all three types of deposited carbon. HRTEM-SAED can use electron diffraction analysis through selected microregions to identify the crystal structure of the material, which allows the detection of the type and crystallinity of the material. Amorphous materials (Type I carbon) will exhibit a halo pattern, as shown in Fig. 5(b), while amorphous carbon shows Thorn-ring characteristics in the HRTEM-SAED image.72 Alternatively, crystalline materials (Type II and Type III carbon) show a spot pattern in the SAED image. As illustrated in Fig. 5(c-i), carbon nanotubes were observed under low-magnification HR-TEM, and in the close-up image in Fig. 5(c-ii), the carbon nanotubes are highly crystalline; moreover, the insert picture in Fig. 5(c-ii) displays the lattice pattern of the crystalline structure of the carbon nanotube (Type II carbon).73 In addition, in the study by Bayer et al., the HRTEM-SAED pattern demonstrated the successful deposition of Ni3C (Type III carbon) in the amorphous carbon matrix, as shown in Fig. 5(d).70 Further, by calibrating the diffraction spots and comparing them with the XRD standard card library, the detailed information of the crystalline materials can be obtained.63,73,74
Carbon species display two main peaks at 1360 cm−1 (D band) and 1575 cm−1 (G band) in Raman spectroscopy. The G band is often attributed to the presence of crystalline graphite (Type II carbon),whereas the D band is attributed to amorphous species (Type I carbon). According to the enhancement in the signals of the D band and the G band in the Raman spectra in Fig. 5(e), Bayer et al. found that nickel could catalyze the graphitization of amorphous carbon at a high annealing temperature of 800 °C.70 Also, the ratio of the intensity of the D and G band (Id/Ig) is used to identify the degree of graphitization in the carbon deposits, where the magnitude of this value reflects the ability of the anode to resist carbon deposition, given that the presence of type I carbon has the least impact on the anode performance among the carbon species.75
The majority of deposited carbon except Type III in the anode can be removed as gas via oxidation by oxygen.71 Therefore, temperature-programmed oxidation (TPO) can be used to semi-quantitatively detect the carbon content and type in the SOFC anode by fitting the test curve. As shown in Fig. 5(f), Sumi et al. performed peak deconvolution of the TPO curve of the Ni–ScSZ anode after exposure to 10% CH4-N2 for 1 h, and the results indicated that amorphous carbon (Type I carbon), carbon fiber and graphite (type II carbon) were all present in the anode. Furthermore, comparing the size of the different peak areas, it is possible to estimate the percentage of different carbon types, and the peak area of Cα indicates the presence of a small amount of carbon fiber.76
In situ detection can reflect the composition of the reaction intermediates during operation, which is of instructive significance for understanding the carbon deposition process and the anti-carbon deposition principle of the designed anode. Chlipała et al. described that in situ Fourier transform infrared spectroscopy (FTIR) can be employed to quantify the composition of outlet gases from SOFCs. Fig. 5(g) shows the outlet gas concentrations of CH4, CO2, CO, and H2 over time, and the yield and selectivity of CO and H2 could be calculated immediately given that the alterations in the concentration of individual gases can be monitored in real time by in situ FTIR. Furthermore, non-equilibrium analysis of in situ FTIR in a biogas fuel SOFC showed that methane steam reforming (MSR) was the most beneficial direct internal reforming reaction due to the presence of observable H2O in the reaction atmosphere, and CH4 cracking appeared to be the main reaction in the reaction, leading to carbon deposition, given that the carbon activity coefficient of this process was orders of magnitude higher than other reactions.77 Xie et al. used synchrotron-based vacuum ultraviolet photoionization mass spectrometry (SVUV-PIMS) to further explore the impact of MgO on CH4-involved reactions. As shown in Fig. 5(h), when measured at 700 °C and acquired at the photon energy of 11 eV, the SVUV-PIMS spectra demonstrated improved signals for C2H4 and C3H6 compared to the Ni–Cu catalyst without MgO, suggesting that the introduction of MgO improved the selectivity for C2+ hydrocarbons and inhibited CH4 cracking to carbon directly.78
Sun et al. studied the carbon formation mechanism on Ni-based catalysts using environmental transmission electron microscopy (ETEM) over a wide temperature range in combination with molecular dynamics simulations and density functional theory (DFT) calculations. In situ TEM observation performed in a C2H2/H2 atmosphere provided real-time evidence that Ni3C is an intermediate phase that decomposes to graphitic carbon and metallic Ni, leading to the formation of carbon. The mechanisms of acetylene decomposition and evolution of the carbon atom configuration were revealed by molecular dynamics simulations, which verified the experimental results. The modification of MgO on NiO could effectively decrease the formation of graphitic layers, and thus enhance the catalytic performance of NiO. The black arrows in Fig. 5(i) indicate the growth direction of the deposited graphite layer, and compared to NiO, the graphite layer in the MgO-modified NiO was basically unchanged with time and thinner in thickness. These results provide insight into the origin of the carbon deposition and developing effective approaches to mitigate it.79
Each characterization has its unique strengths and drawbacks, and thus in specific SOFC anode research, a variety of characterizations should be rationally used to analyze the carbon deposits in combination with the existing scientific research conditions.
1.4 Strategies to control deposition of carbon
To prevent carbon deposition, two general methods can be considered. One is based on thermodynamics, while the other by changing the reaction kinetics. From a thermodynamic point of view, the first strategy to control carbon deposition is to reduce the operating temperature of SOFCs. Sumi et al. reported that methane cracking reactions mainly occur above 800 °C58 and Steele found that at the lower operating temperature of SOFCs (below 750 °C), the carbon produced by CH4 cracking will not deposit on most oxides.80 Low temperatures are not conducive to carbon deposition in SOFCs, while the conductivity of oxygen ions in the electrolyte also decreases greatly, and thus this method is not ideal given that lowering the operating temperature will lead to a lower SOFC performance.According to the study by Sasaki et al. on balancing the C, H, and O contents in fuels and their relationship with carbon formation, carbon deposition will occur at equilibrium.81 Thus, to avoid using an SOFC operating environment in the carbon deposition zone, the second strategy from a thermodynamic point is to introduce O atoms (oxygen and carbon dioxide) in the hydrocarbon fuel or add steam to the fuel gas to further improve the O/C and H/C ratios in the fuel composition. Thus, mixture fuels avoid being in the carbon deposition region and inhibit carbon formation. Carbon-containing adsorbents are susceptible to oxidation by the chemically adsorbed oxygen species present on the surface of metals, and therefore when there is enough steam, the carbon can be removed at a faster rate. However, due to the dilution of the fuel by a large amount of steam and the increase in p(O2) in the fuel, the SOFC efficiency and the open-circuit voltage will decrease, and the rate of carbon deposition is proportional to the mass of the hydrocarbon fuel, i.e., hydrocarbons with longer chains require more steam. In addition, steam enhances the formation of Ni(OH)2, and excess steam may also cause thermal stress to damage the SOFC components. Ideally, the minimum amount of steam required should be generated in situ by an electrochemical reaction to avoid any additional loss in SOFC efficiency.
The reaction kinetic methods to avoid carbon deposition include increasing the carbon removal rate or reducing the carbon deposition rate by adjusting the anode catalyst composition. Based on DFT calculations, the anodic oxidation pathways of CH4 fuel in nickel-based materials are as follows: (1) CH4 begins adsorption on Ni and (2) the adsorbed CH4 decomposes into *CH3, *CH2, *CH and *H. However, for the adsorbed *H, it further migrates and overflows on the oxygen atoms in the electrolyte (Oelectrolyte) to form adsorbed *OH, and the adsorbed hydrogen further overflows on *OH to form adsorbed *H2O, which then desorbs to form H2O. However, for *CH, it will have two different oxidation pathways, depending on whether it occurs on Ni(111) or Ni(211). (3a) In the case of Ni(111), *CH continues to be oxidized to *CHO by spilling Oelectrolyte, (4a) *CHO is broken down into *CO and *H, and (5) in the case of *H, it follows a similar path as above, and *CO desorbs to CO or further oxidized to CO2 at high temperature. In contrast, for Ni(211), the step followed is the same as (3b), where *CH is decomposed into *C and *H. Also, *C will further become carbon deposits on adjacent Ni surfaces.82
Carbon-resistant SOFC anodes should be designed with the intrinsic adsorption capacity of CO2 and H2O. Thus, the anodes adsorb CO2, greatly improving the in situ CO2 reforming of methane or reacting with the carbon deposits at the anode to form gas-phase CO (C + CO2 → 2CO), thereby removing the deposited carbon. In the case of anodes that can adsorb H2O, the adsorbed *H2O is converted to *OH with the oxygen vacancy in the material  , while *OH can react with *C to form *CHO. Subsequently, *CHO follows similar steps as (4a) and (5). Both pathways will mitigate the carbon deposits typically encountered by nickel-based anodes in hydrocarbon fuels.83,84
, while *OH can react with *C to form *CHO. Subsequently, *CHO follows similar steps as (4a) and (5). Both pathways will mitigate the carbon deposits typically encountered by nickel-based anodes in hydrocarbon fuels.83,84
Given that nickel acts as an excellent dehydrogenation catalyst, this reaction manifests itself as coke formation when nickel-based anodes are used for hydrocarbon-fueled SOFC.85 As an alternative, non-nickel-based catalysts have been developed.
Most oxide ceramics do not catalyze the formation of carbon in the same way as Ni, and some oxide anodes of SOFCs can directly oxidize methane given they are inert to carbon formation. Steele et al. used Bi2O3–Pr6O11 as an anode and proved that the metal oxide anodes did not tend to catalyze CH4 into C2 compounds. Therefore, CH4 could be completely oxidized to CO2 and H2O, reducing the formation of coke.86 Some other non-nickel based ceramic material anodes were studied, such as Sr0.88Y0.08TiO3−δ forming yttria-stabilized zirconia (YST–YSZ),87 lanthanum-doped SrTiO3 (LaxSr1−xTiO3),88 La0.75Sr0.25Cr0.5Mn0.5O3 (LSCM),51 La4.0Sr8.0Ti11.0Mn0.5Ga0.5O37.5 (LSTGM)89 and Sr2MgMoO6−δ(SMMO).90 However, although the direct electrochemical oxidation of methane was achieved on the ceramic anode, the conductivity of the electrons was insufficient to give acceptable performance compared to the traditional Ni/YSZ anodes.
Metals commonly used as dehydrogenation catalysts and for the manufacturing of carbon nanotube, such as Ni, Co and Fe, will result in the deposition of carbon at the metal sites. Although the solubility of carbon in Cu, Ag and Au is lower than that of Ni, Fe, Co and will not result in the formation of substantial Type II or Type III carbon,91,92 some researchers believe that Cu is a poor catalyst for hydrocarbon hydrogenolysis, and it is this property that endows Cu cermet with excellent coking stability. However, this that leads to some limitations. Firstly, Cu does not exhibit an acceptable electrocatalytic performance compared to Ni; furthermore, it has a low melting point and sinters easily, resulting in relatively poor thermal stability and making it incompatible with many standard high-temperature SOFCs manufacturing technologies; finally, it is prone to interface reactions with other SOFC components. Accordingly, one way to improve the activity and stability of Cu-based anodes is to alloy Cu with a second metal with higher catalytic activity, and from another perspective, a way to avoid Ni-based anode coking is to alloy Ni with low-activity metals such as Fe and Cu to inhibit the carbon deposition rate. Consequently, the use of alloy metals seems to be a good approach to solve the trade-off between electrochemical properties and carbon coking tolerance.
2. The development of hydrocarbon-fueled anodes
The development of novel anodes focuses on carbon-resistant materials that meet the critical requirements of hydrocarbon-fueled SOFC. Based on the keyword “SOFC hydrocarbon anode” search in Scopus, the corresponding number of papers published annually from 1995 to 2022 showed an upward trend. Overall, several types of materials are employed in the study of SOFC anodes for hydrocarbon utilization, as follows: (1) bimetallic-cermet materials, (2) ceramic materials, and (3) anode reforming layer materials.2.1 Bimetallic-cermet materials
Cermet is a porous structure of ceramic-metallic composites. Excellent SOFC anodes should have mixed ionic-electronic conductors to catalyze electrochemical reactions, and therefore O2− conductive ceramic materials such as YSZ and CeO2 can be added to cermet. In addition, the cermet will help match the TEC of the anode with the electrolyte and control metal sintering.The bifunctional synergy of bimetallic catalysts can modify the electronic properties and surface structure of the electrode (minimizing Ni particles and increasing TPB length), and additional the alloy reduces the carbon–metal bonding energy, thus increasing the tendency of carbon to be oxidized by oxygen ions.93 It is well known that nickel, as a highly active dehydrogenation catalyst, can produce hydrogen and coke from hydrocarbon.85 In the presence of carbon, due to the interaction between the 2p electrons of carbon and the 3d electrons of nickel, they will lead to the formation of nickel carbides, which are difficult to remove. If another metal is added to form an Ni-based alloy, the d-band center of the Ni atom can be moved away from the Fermi level, thereby reducing the formation of nickel carbides kinetically.
Kim et al. and Lü et al. began to study Ni–Cu alloys around the same time. Kim et al. studied an Ni–Cu–CeO2–YSZ anode-supported SOFC for the direct oxidation of methane. Ni–Cu alloy composites divided into 20% Ni and 80% Cu were exposed to dry methane for 500 h, showing a significant increase in power density over time. The open-circuit voltage (OCV) was approximately 1.0 V in both H2 and CH4 at 1073 K, which is marginally lower than the theoretical OCV at this temperature, and the peak power density of the cell reached 440 mW cm−2 for H2 and 330 mW cm−2 for CH4. The experimental results of this work had many implications in the development of anodes in SOFCs. Firstly, a small amount of carbon can significantly lead to an obvious improvement in the performance of SOFCs, given it connects the isolated metal particles inside the anode, enhancing the electronic conductivity of the anode. This is the same as stage 1 of carbon deposition discussed earlier. Secondly, SOFC anodes based on Cu–Ni alloys have different properties compared to Ni-based cermet or Cu-based cermet, where the catalytic properties of an alloy are more than just the sum of the properties of its two separate metals. Overall, they demonstrated that a direct oxidation SOFC made of Cu–Ni alloy cermet was feasible for CH4 at 1073 K.94 Lü et al. investigated the performance of Ni0.7Cu0.3–YSZ/Ni0.3Cu0.7–YSZ cermet as an anode for SOFCs under hydrogen and city coal gas fuel, and the XRD results proved that the anode was a composite material. The OCV of the cell exceeded 1.1 V in H2, which was very close to the theoretical voltage, but the output power density was very low, i.e., only about 33–43 mW cm−2 at 900 °C. However, when changing the fuel from H2 to city coal gas, there was only a slight drop in the output current and power density, and the anode reacted quickly to the fuel conversion without significant decay. When pitch was present in the city coal gas, tar-like carbon was only deposited on the Al2O3 tube of the cell, not on the anode, and hence the Ni–Cu–YSZ anode displayed a self-cleaning function. However, they did not further test the life of these anodes.95
Song et al. prepared NiCu–CeO2-based anode materials for intermediate-temperature SOFCs using hydrocarbon fuels and investigated the promotion effect of Ni–Cu alloy on their electrochemical activity and stability. Specifically, they focused on the power density and short-term stability tests of the SOFCs with six different anodes in 97% CH4/3% H2O fuel at 700 °C. The peak power density of the cells with the Ni–CeO2, Cu–CeO2 and NiCu–CeO2 anodes was 60, 28 and 80 mW cm−2, while that of the cells with the Ni–Zr0.1Ce0.9O2−δ (ZDC), Cu–ZDC and NiCu–ZDC anodes was 92, 32 and 87 mW cm−2, respectively. Short-term stability tests for all the cells at 700 °C were conducted at a constant voltage of 0.6 V under 97% CH4/3% H2O. After the stability test, the carbon content on the surface of the Ni–CeO2, Cu–CeO2 and NiCu–CeO2 anodes was 15.62, 2.19 and 4.93 wt%, while that of the Ni–ZDC, Cu–ZDC and Ni–Cu–ZDC anodes was 10.72, 1.93 and 2.88 wt%, respectively. These experimental data show that compared with the pure CeO2-based anodes, Zr0.1Ce0.9O2−δ exhibited good thermal stability and activity. Moreover, the stability of Ni0.5Cu0.5–ZDC was comparable to that of Cu–ZDC, and its high activity is comparable to that of Ni–ZDC.96
In the study of Ni–Cu alloy anodes, it was found that the addition of a small amount of Cu also resulted in a slight improvement in the stability of an SOFC in hydrocarbon fuels. Akdeniz et al. altered the anode structure by impregnating Cu-nitrate solution and Ce-nitrate solution in the Ni–YSZ backbone to improve the carbon-resistance performance of the SOFC under direct methane fuel. By controlling the variables of molarity of metal-nitrate solution; the amount of impregnation in mL (Cu-nitrate: Ce-nitrate); the amount of pore former and anode functional layer composition, they determined the most appropriate proportions of Ni–Cu–CeO2–YSZ for the carbon-resistant anode. Specifically, the optimal dosages and molarities of copper-nitrate and cerium-nitrate solution were determined to be 3 mL–1 M and 2 mL–1 M, respectively. The Ni–Cu–CeO2–YSZ anode-optimized SOFC showed a voltage loss of only 6% after 5 h of operation in dry methane fuel, and the carbon resistance and lifetime of the cell were significantly higher than that of traditional nickel-based anodes.97
Ni–Cu alloy was also used in BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb) and Pr0.5Ba0.5MnO3−δ (PBMO) anodes. Li et al. prepared Cu–, Ni– and Ni–Cu–BZCYYb anodes by impregnating metal salt solutions in a BZCYYb scaffold. BZCYYb is not a good electronic conductor, and thus metal oxides were introduced to form a good electron path; however, BZCYYb can absorb H2O on its surface to increase the local H2O content for carbon removal. The impregnation of Ni or Ni–Cu significantly reduced the ohmic resistance and polarization resistance of the BZCYYb anodes when exposed to H2 or CH4, while Cu impregnation only reduced the ohmic resistance in H2 and CH4, and furthermore only the Ni–Cu–BZCYYb anode was resistant to carbon deposition in wet and dry CH4.98 Kim et al. also synthesized Ni–, Ni–Cu–PBMO anodes in a PBMO scaffold using the impregnation method, which were expected to provide synergistic effects due the high catalytic activity of nickel and the high carbon coking tolerance of the transition metals. Subsequently, at 700 °C in the C3H8 fuel test, it was observed by SEM that the Ni–Cu–PBMO anode had no carbon deposition compared to the Ni–PBMO anode, even though the peak power density decreased from 280 to 200 mW cm−2. In addition, they demonstrated through DFT calculations that the binding energy of the Ni–Cu alloy to C species was lower than that of Ni and calculations showed that the Ni–Cu–PBMO anodes had excellent tolerance to carbon deposition.99
A second alloy metal can be introduced in the anode through different methods, which will improve the performance of the SOFC in some ways. By using a microwave irradiation process, Islam et al. directly deposited Cu particles on the Ni–YSZ anode in just 15 without a calcination step. SEM analysis confirmed that the morphology of the deposited Cu particles was spherical and that the average size of the particles was less than 100 nm. The electrochemical performance of the electrolyte-supported Ni–Cu–YSZ anode was improved over time when operating at 1073 K in dry CH4 fuel at a current density of 20 mA cm−2, which was similar to the performance trend of the Ni–Cu–CeO2–YSZ anode prepared by Kim et al. using the impregnation method.94 In the study by Kim et al., the weight ratio of Cu to Ni was 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and the maximum power density (MPD) of the cell with the impregnated Ni–Cu–CeO2–YSZ anode in methane at 1073 K was 330 mW cm−2. Also, in the study by Islam et al., the weight ratio of Cu to Ni was only 0.06:1, but the microwave-irradiated Ni–Cu–YSZ anode had an MPD of merely 49 mW cm−2 in methane at 1073 K. The difference in power density was due to the difference in the electrolyte thickness (60 μm vs. 300 μm) and the presence of CeO2. Specifically, the microwave radiation process can be employed to prepare Ni–Cu–YSZ anodes in less time than other methods such as impregnation and electrodeposition.100
:1, and the maximum power density (MPD) of the cell with the impregnated Ni–Cu–CeO2–YSZ anode in methane at 1073 K was 330 mW cm−2. Also, in the study by Islam et al., the weight ratio of Cu to Ni was only 0.06:1, but the microwave-irradiated Ni–Cu–YSZ anode had an MPD of merely 49 mW cm−2 in methane at 1073 K. The difference in power density was due to the difference in the electrolyte thickness (60 μm vs. 300 μm) and the presence of CeO2. Specifically, the microwave radiation process can be employed to prepare Ni–Cu–YSZ anodes in less time than other methods such as impregnation and electrodeposition.100
Park et al. prepared an Ni–Cu–YSZ anode by plating Cu on a porous Ni–YSZ cermet anode using an aqueous copper sulfate solution. In methane fuel at 700 °C, the power density of the cell with the Ni–Cu–YSZ anode (240 mW cm−2) was slightly lower than that of the cell with the Ni–YSZ anode (280 mW cm−2), possibly due to the increase in anode resistance from 1.81 Ω cm2 to 1.93 cm2 by copper plating. However, the Ni–Cu–YSZ anode cell demonstrated a stable performance over 200 h, while the Ni–YSZ anode cell degraded dramatically within 21 h due to carbon deposition.101 Kumar et al. prepared an Ni0.9–Cu0.1–YSZ0.95–GDC0.05 anode by co-tape casting, followed by co-firing, where the advantage of this method is that it maintains the integrity of the anode-electrolyte interface. The optimized anode had an excellent power density of 436 mW cm−2 at 850 °C in methane fuel. Carburizing studies in methane showed that the Ni0.9–Cu0.1–YSZ0.95–GDC0.05 anode had a tendency to electrochemically oxidize deposited carbon and reduce carbon deposition by up to 50%. Therefore, this study showed that the new anode composition and the preparation route employed can serve as a precedent for the development of high-power density SOFCs for hydrocarbon fuels.102
Co has better oxidation resistance than Ni and does not exhibit corrosiveness at a high overpotential or high p(O2).103 Cho et al. synthesized Ni1−x–Cox–Ce0.8Gd0.2O1.9 (GDC) anodes using a glycine nitrate process. It was found that the polarization resistance in an H2 atmosphere increased with an increase in the Co content, whereas in a CH4 atmosphere the polarization resistance decreased with an increase in the Co content. In CH4 fuel at 800 °C, the MPDs of single cells of Ni–GDC and Ni0.8–Co0.2–GDC were 150 and 250 mW cm−2, respectively. Consequently, they considered that Ni–Co alloy reduced the electronic conductivity but still alleviated carbon deposition by inhibiting the formation of C–C bonds and/or reducing the thermodynamic driving force required for carbon nucleation.104 Nicharee et al. prepared Ni–YSZ and Ni–Co–YSZ by impregnating YSZ with nitrates of nickel and cobalt, followed by calcination and hydrogen reduction, and the introduced Co could be completely dissolved in the Ni lattice. Ni–Co alloys improve their electrochemical performance under CH4 fuel by reducing the resistance and anode overvoltage. The MPDs of the cells with the Ni–YSZ and Ni0.85–Co0.15–YSZ anodes were 94 and 136 mW cm−2 at 750 °C and at 20% v/v CH4 in He, respectively. The ohmic resistance and polarization resistance of the Ni–YSZ and Ni0.85–Co0.15–YSZ anodes were investigated at 750 °C for 60 h at a constant loaded current of 80 mA, and the ohmic resistance of Ni0.85–Co0.15–YSZ basically did not change, and the polarization resistance increased slightly, but the overall performance was more stable than Ni–YSZ. Therefore, Ni–Co alloy showed high efficiency in inhibiting carbon deposition and improved the electrochemical performance of SOFCs operating with CH4 fuel.105
Cesario et al. successfully synthesized Ni–, Ni–Co– and Ni–Cu-based alloys with GDC by using citric acid as a chelating agent through a one-step synthesis route and utilized them in the dry methane reforming (DMR) reaction of SOFC. XRD analysis combined with thermodynamic calculations revealed that the anode formed a complete solid solution of Ni–Co and Ni–Cu-based alloys. Surface area (SBET) measurements suggested that the Ni–Co–GDC samples had a higher surface area, which favored the expansion of the reaction TPB. In addition, according to the H2-TPR results, the Ni–Co–GDC anode exhibited higher reducing and stronger metal-ceramic interactions compared to the Ni–Cu–GDC anode. This tight Ni–Co cermet contact allowed better adsorption of CO2 on the anode and reduced carbon formation by inverse Boudouard reaction (CO2 + C ⇌ 2CO). Finally, the DRM results demonstrated that the Ni–Co–GDC anode showed higher CH4 and CO2 conversion than the Ni–Cu–GDC and Ni–GDC catalysts, which could mitigate carbon deposition on the surface of the anode.106
Alloying Fe with Ni was also studied as a viable option. Kan et al. synthesized an Ni0.9–Fe0.1–GDC anode using a solid-state reaction and the MPD of the cell reached 340 mW cm−2 at 650 °C in dry methane, while the cell with Ni-GDC had a maximum power of 300 mW cm−2. Subsequently, the long-term stability of the cell with the Ni–GDC and Ni–Fe–GDC anodes was evaluated at a constant current density of 0.2 A cm−2 in dry methane fuel, where the performance of the SOFCs with the Ni0.9–Fe0.1–GDC anode did not degrade for 50 h, while that with the Ni–GDC anode only operated for 12 h. The exit gas of SOFC was analyzed by gas chromatography during the long-term stability test, and the results showed that the Ni–GDC anode produced 41.1% H2, 19.6% CO, 8.1% CO2 and 22.3% H2O after 10 h. The high concentrations of H2 and CO indicated the incomplete combustion of methane (CH4 + O2− → CO + 2H2 + 2e−) and the ratio of C to H in the outlet stream was 0.88:4, which was lower than the expected ratio of 1:4 (CH4 + 4O2− → CO2 + 2H2O + 8e−). In contrast, the outlet stream of the Ni0.9–Fe0.1–GDC anode contained 33.2% H2, 11.2% CO, 19.5% CO2 and 27.8% H2O after 10 h, and the ratio of C to H in the outlet stream was 1:4, indicating that no carbon deposition occurred inside the SOFC. Due to the addition of Fe, the changes in the catalytic performance resulted in more complete oxidation of the methane fuel, which enhanced the long-term stability of the Ni–Fe–GDC catalysts.107 Kim et al. investigated the catalytic activity of Ni–, Ni–Cu–, Ni–Fe–PBMO anodes under C3H8 fuels. The Ni and Ni–Fe catalysts had the best electrochemical performance, while at 700 °C in C3H8 fuel, the MPDs of the cells with Ni–PBMO, Ni–Cu–PBMO and Ni–Fe–PBMO anodes were 280, 200 and 300 mW cm−2, respectively. However, the Ni–PBMO catalysts formed carbon fibers under dry C3H8 operating conditions, while the Ni–Cu–PBMO and Ni–Fe–PBMO catalysts exhibited excellent carbon deposition tolerance. Considering the catalytic activity and carbon deposition resistance, the Ni–Fe–PBMO catalyst is best-suited for SOFCs based on the YSZ electrolyte support.99
Lee et al. and Park et al. also prepared Ni–Fe alloys, but unlike other researchers, they added a layer of Ni–Fe alloy on top of the cermet anode layer, not within it. Specially, Lee et al. prepared a common Ni–YSZ anode, a dual-layered anode of Ni–Fe alloy layer and Ni–YSZ anode layer, and a hybrid double-anode (HB Ni–Fe) SOFC with a porous Ni–Fe alloy layer added to the dual-layered anode. In methane fuel tests in SOFCs, the cells with the Ni–YSZ and double- and triple-layer anodes exhibited current densities of 0.45, 0.59, and 0.66 A cm−2 at 0.8 V and 750 °C, respectively. In addition, according to the impedance analysis, the cell with Ni–Fe additional layers exhibited lower charge transfer resistance than that without additional layers, which may be due to the reduced coke formation because of the catalytic activity of the Ni–Fe layer in methane fuel. However, they also proposed that the morphology and process of the SOFC system could be further optimized.108 Park et al. did similar work to that of Lee et al. in the same year, and still concluded that SOFCs with an additional Ni–Fe layer in methane fuel had a greater current output and lower charge transfer resistance than that without the Ni–Fe layer, and the porosity and sintering temperature of the additional Ni–Fe layer need to be optimized.109
For alloy anode research, the morphology of the Ni–Fe alloy is also an influencing factor that affects the cell performance. In high-temperature environments, metal spherical particles often agglomerate, thus affecting the microstructure of the electrode and cell stability. Thus, to overcome this problem, Lee et al. employed electrospinning to make Ni–Fe alloy nanofiber anodes, which were compared to Ni–Fe spherical powder anodes. The XRD analysis showed that Ni and Fe formed a solid solution under reducing conditions, and the SEM image displayed that the Ni–Fe nanofibers retained a porous network with an average pore size of 30 μm even after high-temperature sintering. Furthermore, the cell with the Ni–Fe fiber anode exhibited an MPD of 1640 mW cm−2 at 1173 K in hydrogen fuel, while that with the Ni–Fe spherical powder anode had only 1450 mW cm−2. At all the operating temperatures, the polarization resistance of the Ni–Fe nanofiber anode was lower than that of the Ni–Fe powder anode. These results demonstrate that the highly porous structure of the Ni–Fe nanofiber anode structure can produce excellent charge transfer paths and enhance the gas diffusion performance, thereby enhancing the electrochemical performance and stability of the SOFC.110 Also, Although they did not test the SOFCs in hydrocarbon fuel, it is expected that the Ni–Fe nanofiber anode SOFCs can perform well in anti-carbon deposition research.
Among the transition metals, Mo is an ideal nickel-based alloy metal. Hua et al. reported the preparation of a novel Ni–Mo bimetallic alloy anode, which when fueled with CH4–50 ppm H2S, the MPD of the cell with the Ni–Mo–YSZ anode was 594 mW cm−2 at 800 °C, and this bimetallic catalyst also provided a consistently stable power output over a long period of 120 h, making it a promising anode catalyst with excellent coke resistance and thermal stability.111
Bkour et al. used an Ni–Mo–YSZ catalyst for the partial oxidation of isooctane, which was an alternative to the commonly used gasoline. The XRD results showed that the larger Mo atoms partially replaced Ni atoms to form Ni–Mo solid solutions. The Raman spectroscopy results also indicated that the presence of well-dispersed Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species on the Ni surface was an active site related to the high coke resistance of the Ni–Mo–YSZ catalyst. Further, they used the Ni–Mo–YSZ catalyst as a reforming layer for the traditional Ni–YSZ anode SOFC, with a maximum power output of 452 mW cm−2 at 750 °C in isooctane/air operation. In the 15 h performance stability test, the cell with the micro-reforming layer showed a lower degradation rate, while the cell without the reforming layer stopped working within 12 h. This improved stability suggests that the use of an Ni–Mo–YSZ reforming layer in isooctane/air operation mode has great benefits for the electrochemical performance of SOFCs. In addition, DFT-based calculations demonstrated that the d-band center of the Ni atoms located close to the Mo atoms shifted away from the Fermi level, thus reducing the electron interaction between carbon and nickel. Consequently, less carbon was adsorbed in the Ni–Mo system.112 Moreover, they previously studied the catalyst capacity of an Ni–Mo–CZ catalyst in isooctane, and the carbon conversion rate reached 100% and the H2 yield reached 75%. When Ni–Mo–CZ was used as the reforming layer on the traditional Ni–YSZ-based SOFC, it still showed an excellent voltage degradation rate of 4.8 mV h−1 after 12 h operation at 750 °C in isooctane fuel.113 Similarly, Hou et al. developed a dual-functional Ni–Mo–CZ as an anode for isooctane-fueled SOFCs. The MPD of the cell with the Ni–CZ anode was 70 mW cm−2 at 800 °C in isooctane/air, indicating its very poor anode activity, where the MPD tripled to 212 mW cm−2 after the addition of 5 wt% Mo to the Ni–CZ anode. Also, the SOFC with the Ni–5Mo–CZ anode could run steadily for 30 h in an isooctane/air mixture, which has great potential in SOFCs by using complex liquid isooctane fuels.114
O species on the Ni surface was an active site related to the high coke resistance of the Ni–Mo–YSZ catalyst. Further, they used the Ni–Mo–YSZ catalyst as a reforming layer for the traditional Ni–YSZ anode SOFC, with a maximum power output of 452 mW cm−2 at 750 °C in isooctane/air operation. In the 15 h performance stability test, the cell with the micro-reforming layer showed a lower degradation rate, while the cell without the reforming layer stopped working within 12 h. This improved stability suggests that the use of an Ni–Mo–YSZ reforming layer in isooctane/air operation mode has great benefits for the electrochemical performance of SOFCs. In addition, DFT-based calculations demonstrated that the d-band center of the Ni atoms located close to the Mo atoms shifted away from the Fermi level, thus reducing the electron interaction between carbon and nickel. Consequently, less carbon was adsorbed in the Ni–Mo system.112 Moreover, they previously studied the catalyst capacity of an Ni–Mo–CZ catalyst in isooctane, and the carbon conversion rate reached 100% and the H2 yield reached 75%. When Ni–Mo–CZ was used as the reforming layer on the traditional Ni–YSZ-based SOFC, it still showed an excellent voltage degradation rate of 4.8 mV h−1 after 12 h operation at 750 °C in isooctane fuel.113 Similarly, Hou et al. developed a dual-functional Ni–Mo–CZ as an anode for isooctane-fueled SOFCs. The MPD of the cell with the Ni–CZ anode was 70 mW cm−2 at 800 °C in isooctane/air, indicating its very poor anode activity, where the MPD tripled to 212 mW cm−2 after the addition of 5 wt% Mo to the Ni–CZ anode. Also, the SOFC with the Ni–5Mo–CZ anode could run steadily for 30 h in an isooctane/air mixture, which has great potential in SOFCs by using complex liquid isooctane fuels.114
Majewski et al. investigated the carbon tolerance of Ni–Mo alloys in steam reforming and dry reforming of methane combining five different ceramic materials, which were YSZ, GDC, CSZ, Al2O3 and TiO2. The choice of ceramic materials significantly affected the reaction kinetics of the Ni–Mo catalysts and the yield of ideal products for H2 and CO during CH4 reforming. Also, in the process of methane reforming, the presence of Mo2C was also detected in the Ni–Mo–YSZ and Ni–Mo–Al2O3 catalysts, which could improve the catalytic activity of the steam reforming reactions. In general, the catalytic performance of the Ni–Mo-combined ceramic catalysts decreased in the order of Al2O3 > YSZ > TiO2 > CSZ > GDC.115 These findings can guide the design of Ni–Mo bimetallic anodes for carbon-resistant SOFCs.
In addition to transition metals, Sn plays a major role in the research of hydrocarbon fuels. There are many reports in the literature on Ni–Sn alloys. Nikola et al. combined kinetic studies, isotope labeling experiments and DFT calculations to make outstanding contributions to the study of Ni–Sn alloy catalysts for hydrocarbon fuels. In particular, according to the results of DFT calculations, compared with Ni–YSZ, the Sn–Ni–YSZ catalysts (0.5–1 wt%, with respect to Ni) showed that the surface of the Ni–Sn alloy preferentially oxidized C atoms, promoting the formation of C–O bonds and further forming gas-phase CO and CO2, rather than forming C–C bonds. Further catalyst characterization studies showed that the presence of Ni–Sn–YSZ had a great effect on the reduction of carbon deposition for methane steam reforming, and X-ray photoelectron spectroscopy (XPS) and EDS studies indicated that Ni–Sn surface alloys were thermodynamically preferred structures at the limit of low Sn concentrations.116–118 Their study demonstrated that Ni–Sn–YSZ catalysts were more resistant to carbon deposition than Ni–YSZ catalysts in isooctane steam reforming.119
Kan et al. investigated the application of Ni–Sn–YSZ catalysts as anodes in methane-fueled SOFCs. an MPD of 390 mW cm−2 was obtained at 800 °C in methane fuel using the Sn-doped Ni–YSZ cell, which was equivalent to the results obtained with the Ni–YSZ cell (400 mW cm−2). Furthermore, in a stability test at 800 °C with a steady applied current density of 0.2 A cm−2, the Ni–YSZ anode cell stopped operating after 2.3 h, while the Ni–Sn–YSZ anode cell could run for 49.0 h. Moreover, graphitic carbon was deposited on the Ni–YSZ anode, while amorphous carbon was deposited on the Ni–Sn–YSZ anode. It was demonstrated that on the Sn-doped Ni–YSZ, the transition from amorphous carbon to graphite was hindered.120 Subsequently, they added a functional layer between the native anode and the electrolyte to obtain a high power density even at intermediate temperatures, and studied the performance of the SOFC with the (Ni–Sn–YSZ|Ni–Sn–YSZ|YSZ|LSM + YSZ) configuration in comparison to that with the (Ni–YSZ|Ni–Sn–YSZ|YSZ|LSM + YSZ) configuration in wet methane fuel. The cells with the added functional layer showed a high-power density output at an operating temperature of 650 °C in methane fuel, and the Ni–YSZ and Sn-doped Ni–YSZ functional layers displayed similar power densities (390 mW cm−2vs. 410 mW cm−2), while the Sn-doped Ni–YSZ showed enhanced long-term stability (27 h vs. 137 h). Similar to the results of their previous studies, this good stability was ascribed to the much lower amorphous carbon deposition rate. As mentioned above that amorphous carbon (Type I carbon) can be easily removed, they retested the long-term stability of the Sn-doped Ni/YSZ functional layer cells and removed the carbon deposits on the cell every 100 h by high-temperature oxidative calcination, and consequently the cells could run stably for 300 h with no observed degradation. The Sn content measured by energy dispersive X-ray spectroscopy (EDX) prior to stability testing was 5.2 wt% in the functional layer and 17.2 wt% in the native anode. After 300 h of operation, the change in Sn content was negligible. The above-mentioned results confirmed that Sn was stable on the Ni surface despite the many hours of operation.121
Jiang et al. separately synthesized 1 wt% Cu- and Sn-impregnated traditional Ni/YSZ anodes. In the biogas fuel test, the MPD of the cells with Ni–, Ni–Cu–, Ni–Sn–YSZ anodes was 101, 85 and 272 mW cm−2 at 750 °C. At a constant current density of 0.2 A cm−2 at 750 °C, it was found that the stability of the pure Ni–YSZ anode SOFC deteriorated rapidly, and the operation stopped after 19 h. Also, the voltage of Ni–Cu–YSZ dropped to 0.025 V at a fast rate after 48 h test, while the cell with the Ni–Sn–YSZ anode remained unchanged throughout the test period, and the voltage reduction rate was only 2.98 × 10−4 V h−1. Furthermore, they used SEM to characterize the anodes, and observe severe carbon deposition on the surface of the Ni–YSZ anode, whereas no observable fibrous carbon was found on the surface of the Ni–Sn–YSZ anode. After measuring the morphology of the anode surface, the elements on the anode surface were quantified in weight percentage through EDX, where the content of C element in the Ni–, Ni–Cu–, and Ni–Sn–YSZ anodes was 5.32, 3.61 and 4.5 wt%, respectively. Hence, the long-term stability of the SOFCs under biogas fuel could be significantly improved by adding 1 wt% Sn to the Ni/YSZ anode.122
Singh et al. prepared (1 wt%) Sn–Ni–YSZ anodes at a high sintering temperature (1450 °C) and evaluated their direct utilization of CH4 at 800 °C. They found that Sn enhanced the shrinkage of the Ni–YSZ anode during the sintering stage of the preparation process, and this shrinkage matched better with the electrolyte shrinkage, which may help to reduce the stresses during the fabrication process. However, Sn was not detected in the anode after the sintering stage, resulting in the presence of Sn having no significant effect on carbon formation on the Ni–YSZ anode.123 Farrell et al. showed that the carbon deposition catalyzed by Ni in ethanol fuel of the Ni–YSZ anode could be reduced by alloying nickel with a small amount of Sn. The Ni–Sn alloys had a lower tendency to activate carbon deposition formation compared to monometallic Ni and did not significantly reduce the reaction rate.124
For the same alloy, the properties of various ceramic materials will be different. Better results were observed when Ni–Sn alloys were used with GDC and SDC than YSZ. Yoon et al. synthesized (1.99 atom%) Sn–Ni–GDC anodes using the impregnation method. In terms of the electrochemical performance of CH4 fuel at 650 °C, the cells with Ni–GDC and Ni–Sn–GDC anodes showed MPDs of 370 and 470 mW cm−2 at 650 °C, respectively. Hydrophilic Sn doped on the surface of nickel was hydrated and hydroxylated by reaction with water generated from electrochemical oxidation of fuels, and the hydroxyl groups (*OH) contributed to the oxidation of the adjacent carbon and removed as CO or CO2. Also, the Ni–Sn alloy prevented the enlargement of Ni particles, which not only hindered the sintering of Ni metal, but also expanded the TPB of the anode, This enabled O2− to quickly reach the carbon deposition area and electrooxidize it, thereby converting it into carbon dioxide and releasing it from the anode. In addition, Sn: [Kr]4d105s25p2 with an external electron configuration analogous to C: [He]2s22p2 (two valence electrons close to stable s-orbital) hindered the formation of nickel carbide. Therefore, with this anti-carbon deposition advantage, the power generation time of the CH4-fueled SOFC with the Ni–Sn–GDC anode could last for more than 200 h without any performance degradation.125 Myung et al. developed Sn-doped nano-sized Ni and GDC conjugated on a core GDC nano-composite anode (n-SNGG), where this structure had more uniformly distributed Sn because Sn was doped on nanoscale NiO embedded in the core GDC. Due to its excellent micro-junction, the cell with the n-SNGG anode showed an MPD of 930 mW cm−2 at 650 °C in methane fuel and could operate stably for more than 40 h without attenuation in its performance.126 Yang et al. reported that in humidified CH4, the MPDs of the cell with (1 wt%) Sn–Ni–SDC anodes reached 640, 390, and 280 mW cm−2 at 800 °C, 750 °C, and 700 °C, respectively. Also, after running at 700 °C at a constant voltage of 0.8 V in wet CH4 for 230 h, the current density dropped by only 30%. High-angle annular dark-field scanning transmission electron microscopy (HAADF STEM) images showed that the addition of a small amount of Sn to the surface of Ni–SDC favored the formation of highly active Ni3Sn, which was one of the highly active catalysts for hydrocarbon oxidation. Moreover, during the preparation of the cell, they found that the key process of oxidizing impregnated SnCl2 to SnO2 at temperatures above 800 °C was essential.127 Li et al. studied the performance of single SOFCs (Ni–Sn–SDC|SDC|Ba0.5Sr0.5Co0.8Fe0.2O3−δ) in the temperature range of 600–700 °C. The cell with the (5 molar%) Sn–Ni–SDC anode exhibited an MPD of about 600 mW cm−2 at 700 °C with CH4 as fuel. Also, at a constant output current of 300 mA at 600 °C with CH4 as the fuel, its performance degraded by only 1.5% during 72 h of operation, while the Ni–SDC anode dropped by 3.7%. The addition of Sn significantly inhibited carbon deposition on the anode, thereby improving the stability of the single cells fueled by dry methane.128
Arifin et al.44 reported that the electrochemical performance of Ni–Sn–ScSZ in simulated biogas (CH4/CO2 = 2) was better than that of undoped Ni–ScSZ. When the fuel changed from hydrogen to biogas, an immediate decrease in OCV and MPD was observed, with a decrease of 86.5% and 33.3% for the cells with Ni–YSZ and Ni–Sn–ScSZ anodes, respectively. However, they also found higher levels of carbon deposition in Ni–Sn–ScSZ.129
Previous work suggested that a high Sn loading (Sn/Ni ratio >1 wt%) adversely affects the performance of SOFCs. Thus, the loading amount of Sn needs to be adjusted to a suitable ratio, which can maximize the catalytic performance of Sn without hindering the Ni active site and TPB.
By varying the amount of Sn impregnated in the functional layer, Kan et al. found that although a small amount of Sn significantly improved the cell performance, this enhancement decreased with an increase in the Sn content. This was because Sn is preferentially retained on the surface of the anode and the presence of excess Sn will occupy a high percentage of the surface-active sites, thus weakening the anode response.121 The study by Singh et al. showed that the electrochemical performance of SOFCs in H2 and CH4 decreased when the Sn content increased from 1% to 5%.130 Jang et al. investigated that the impregnation of 15 to 30 mg Sn on an anode with a surface area of 3.14 cm2 resulted in the highest power density, whereas 60 mg Sn reduced the power density. To better understand the observed phenomena, they also conducted model experiments. The modeling study showed that the oversupplied Sn agglomerated, resulting in fewer reaction sites, and therefore lower power generation.131
Troskialina et al. impregnated Sn with mass ratios of with 0.00, 0.20, 0.38, 0.66 and 1.02 wt% Sn/Ni in Ni–YSZ catalysts, respectively. Repeated cells tests showed that the electrochemical performance of the SOFC was very sensitive to the amount of Sn doped in the Ni/YSZ anode, and among them, the cell with an Sn/Ni loading of 0.38 wt% had the highest performance in both H2 and biogas fuels. According to the performance trend, it was predicted that when the Sn loading is > 1.02 wt%, the presence of Sn has a significant negative impact on the electrochemical oxidation of H2, which consequently affects the application in hydrocarbon fuels. This finding can explain the results reported by Singh et al.,130 because the cells studied by Singh et al. were under high Sn loads (1 and 5 wt%), and the anode was negatively affected by Sn on hydrogen electrochemical oxidation, resulting in the positive effect of Sn on the Ni–YSZ anode being ignored.
Similar to nickel, noble metals such as Ag, Pd, Ru, Rh and Pt are the commonly used metals in SOFCs. Although silver wires are commonly used as current collectors, silver itself is known to improve the performance of SOFCs.132 Wu et al. impregnated Ag with mass ratios of 0.00, 0.89, 1.59 and 2.48 wt% in Ni–YSZ catalysts, which were denoted as blank cell, cell A, B and C, respectively. For dry CH4, the MPDs of the cells at 750 °C were 542, 262, 307 and 271 mW cm−2, respectively. However, in the stability test, the blank cell could work for only 5 h, while the modified cells could run stably for longer time without performance degradation. The SEM results showed that for cell B, the carbon deposition was greatly reduced both inside and on the surface of the anode, and thus Ag greatly improved the carbon deposition resistance of the Ni–YSZ anodes fueled by dry CH4, which is mainly due to the fact that the deposited carbon could be easily separated by Ag into isolated pieces.133 Jiang et al. also found that doping the Ni–YSZ anode with 1 wt% Ag reduced the MPD of the cell when operating under the condition of 50% CH4–25% CO2–25% N2, but the long-term stability could be greatly improved.122 Wu et al. further improved the relevant works about introducing Ag in the Ni–YSZ anode by the electroless plating method, instead of using the impregnation method. Also, it was found that the MPD of the SOFCs with the Ni–Ag–YSZ anode increased whether in H2, dry CH4 or dry C2H6 fuel. In particular, the Ni–YSZ cell only operated for a few minutes when working in dry C2H6, while the Ni–Ag–YSZ cell could run for up to more than 24 h.134
Hibino et al. studied (0–10 wt%) Ni–Pd–SDC as an SOFC anode in a mixture of methane and air fuels between 450 °C and 550 °C. They found that methane was greatly activated on the anode by adding a small amount of Pd (0.145 mg cm−2), which significantly contributed to the oxidation of methane by oxygen to form hydrogen gas and carbon monoxide. This result led to a high MPD of 644 mW cm−2 at 550 °C when using an SDC electrolyte with a thickness of 0.15 mm.135 Babaei et al. investigated that the presence of Pd in Ni–GDC greatly promoted the electrooxidation of hydrocarbon, especially methanol and ethanol fuels. Specially, in the methanol oxidation reaction, the activation energy of the reaction decreased from 154 to 101 kJ mol−1 on a 0.15 mg cm−2 PdO-impregnated Ni–GDC anode. Also, they found that although carbon depositions were present in the working environment of all these fuels, no filament carbon fibers were formed in the reaction of methanol and ethanol compared to methane fuels. A possible reason for this was that the hydroxide group (i.e., OH) present in the molecular structure of the alcohols removed some of the carbon from the electrode surface.136
Modafferi et al. synthesized an Ni–Ru–GDC anode via the hydrothermal method and studied its steam reforming (SR) and auto-thermal reforming (ATR) of propane. At reaction temperatures above 700 °C, high propane conversion and syngas (H2 + CO) productivity were obtained. However, under SR conditions, the catalyst was inactivated due to the large deposition of filamentous carbon and amorphous carbon. In contrast, under ATR conditions, the formation of coke was completely inhibited because the presence of oxygen promoted the vaporization of the carbon residues.137 In addition, Boaro et al. drew the same conclusion for an Ni–Rh–GDC anode.138 Alternatively, Hibino et al. studied the use of Ni–Ru–GDC catalysts for SOFC anodes also, and when methane, ethane, and propane were used as the fuel, the cells with the Ni–Ru–GDC anode had MPDs of 750, 716, and 648 mW cm−2 at 600 °C, respectively. The high electrochemical performance was due to the fact that Ru could efficiently promote the reforming reactions of H2O and CO2 produced by the electrochemically oxidized hydrocarbon fuels. In addition, in the stability test of the constant current of 600 mA cm−2, the voltage of the cell did not change for almost 20 h.139
Hussain et al. studied the performance of the Ni–Pt–GDC–Sr0.94Ti0.9Nb0.1O3 (STN) composite anode at a low temperature (400–600 °C), and the SOFC showed a 0.10 Ωcm2 polarization resistance at 600 °C. The HR-TEM analysis displayed that nanocrystalline alloys of Ni–Pt were formed in the Ni–Pt–GDC electrocatalyst together with the coexistence of Ni and Pt nanoparticles as separate phases.140
Pd, Pt, Rh and Ru are actually classified as Pt group metals and have superior reforming catalytic properties compared to Ni, Fe and Co; therefore, Pt group metals were used as electrodes in the early SOFC studies. However, due to the high cost of noble metals, there are few reports on the use of noble metals in bimetallic-anode research, which also limits the use of commercial SOFCs in the future.
The acidic sites on the anode are known to facilitate hydrocarbon cracking reactions. Thus, it is expected that the formation of carbon can be inhibited by adding alkali metals or alkaline earth metal oxides (such as Li, Na, CaO, BaO, and MgO) to the Ni-based cermet anode to improve its alkalinity. Moreover, the excellent hydrophilicity and adsorption of CO2 by alkali metals or alkaline earth metal oxides can accelerate the removal rate of carbon depositions through in situ steam/CO2 reforming hydrocarbon fuels and the adsorption hydroxyl groups (*OH) oxidized with adjacent carbon.
Liu et al. developed a simple chemical Li/Na insertion method to prepare an Li/Na-modified Ni–SDC anode (Li–Ni–SDC, Na–Ni–SDC), and found that the MPDs of the cells with the Li- and Na-modified Ni–SDC anodes reached about 212 and 231 mW cm−2 at 800 °C for CH4 fuel, respectively, which are much higher than that of the original Ni–SDC anode SOFC (105 mW cm−2). Moreover, they showed better long-term stability in 70 h, which was attributed to the unique ability of the alkaline metals to absorb H2O and CO2 under the SOFC operating conditions.141
Takeguchi et al. found that the addition of CaO with strong basicity to Ni–YSZ cermet modified the metal Ni to a slightly cationic state, which was effective in inhibiting carbon deposition without reducing the reforming activity.24 Asamoto et al. also drew a comparable conclusion, namely, that the addition of CaO to the Ni–SDC anode slightly reduced the MPD; however, it enhanced the stability of the SOFC performance and inhibited carbon deposition on the anode. In methane fuel at 700 °C, the cell with the Ni–SDC anode exhibited an MPD of 40.3 mW cm−2, while that with the Ni–CaO–SDC anode had an MPD of about 35.6 mW cm−2. The reduced electrochemical performance of the SOFC was attributed to the dissolution of CaO in the ZrO2 or CeO2 lattice. However, after 30 min, there was 36.1 mg of carbon deposited on the Ni–SDC anode, compared to only 31.0 mg on Ni–CaO–SDC.142
La Rosa et al. investigated the effect of BaO doping in the Ni0.53Cu0.47–GDC anode, and for the cells doped or not doped with BaO, similar MPDs of 284 and 310 mW cm−2 were obtained in dry methane fuel at 750 °C, respectively. Alternatively, the study using XRD to characterize the cells after running in dry methane for 200 h showed that the BaO-doped cells did not have significant carbon deposition, while the undoped anodes displayed the presence of carbon deposits.143 Islam et al. conducted a more in-depth study by incorporating BaO in the SOFC Ni–YSZ anode via two different methods, i.e., impregnation and microwave irradiation. When the impregnation method was employed, BaO was stable on Ni, but diffused to YSZ and interacted with it to block O2− transport, resulting in morphological changes and volume expansion of YSZ. However, the selectively deposited BaO only on Ni by using microwave irradiation minimized the interaction between BaO and YSZ. Furthermore, the microwave-prepared anodes possessed comparable electrochemical performance with the impregnated anodes in CH4, but showed lower carbon accumulation. The increase in carbon tolerance was attributed to the discrete BaO/Ni interface, which preferentially adsorbed H2O, and subsequently gasified the adjacent carbon deposits.144 McIntyre et al. used in situ vibration Raman spectroscopy in their study and found that the infiltration of 1% BaO significantly reduced the carbon accumulated on the Ni–YSZ cermet anode in CH4 fuel at 730 °C. This can be attributed to the following reasons: (1) BaO occupied the catalytic site of Ni, slowing down the surface reaction at the anode and (2) the presence of BaO changed the binding energy of carbon and the Ni catalysts. They further studied the ability of different gas-phase reformers to remove carbon, and found that H2O was the most effective reformer for carbon removal, followed by O2, and then CO2. However, H2O and CO2 only partially oxidized the anode, while prolonged exposure to O2 completely oxidized nickel to nickel oxide.145 Itagaki et al. noticed that the addition of 0.2 wt% BaO to Ni-SDC anodes significantly reduced the carbon accumulated in CH4 fuel. This is because the addition of BaO further improved the dispersion of Ni particles, which increased the electrochemical reaction field of CH4.146
Hu believed that NiO in an NiO–MgO solid solution was more difficult to be reduced than pure NiO, which is mainly because MgO isolates NiO and inhibits the formation of Ni–Ni bonds during reduction, which contributes to the formation of very small Ni particles to inhibit carbon deposition. Also, due to the surface alkalinity of the NiO–MgO solid solution, it demonstrated excellent activity and selectivity for CO2 reforming of methane.147 Zanganeh et al. also considered that for NixMg1−xO solid-solution catalysts, the high dispersion of reduced nickel species, the alkalinity of the carrier surface, and the nickel-support interaction endowed the catalyst with very high resistance to carbon formation.148
Yang et al. obtained an excellent MPD of 714 mW cm−2 in methane fuel at 800 °C by impregnating 2.5 wt% MgO in an Ni–SDC porous anode, and the SOFC could run for 330 h with a power attenuation about 12.5%. Furthermore, through material characterization and theoretical calculations, they believed that the addition of a small amount of MgO to nickel cermet will increase the Lewis alkalinity of the anode, thereby enhancing the ability of the catalyst to chemically adsorb H2O and CO2 generated from SOFC operations, and hence reduce coking. Also, HAADF-STEM showed that small Ni particles were located on the surface of the MgO nanoplates.149 Xie et al. prepared a nickel-based anode modified with MgO nanolayers via the in situ reduction of an Ni0.9−xCu0.1MgxO solid solution, which could greatly eliminate sample inhomogeneity and additional costs due to the manufacturing methods. The MPD of the SOFC using the Ni–Cu–0.025MgO–SDC anode reached 670 mW cm−2 in methane fuel at 700 °C, which is about 10% higher than that using the Ni–Cu–SDC anode. Combined with synchrotron vacuum ultraviolet photoionization mass spectra, they deduced that MgO improved the selectivity for C2+ hydrocarbons and directly inhibited the cracking of CH4, and due to the high Lewis alkalinity of MgO, steam adsorption and dissociation were promoted, thereby producing more CO by indirect means.78
Yan et al. fabricated a Co–W–YSZ anode by infiltrating cobalt nitrate and ammonium metatungstate in YSZ, and found that the formed Co3W and Co7W6 intermetallic nanoparticles had high thermal stability up to 900 °C and showed good anti-coking in methane. Exposure of the Co–YSZ anode to CH4 at 800 °C for 5 h resulted in carbon deposition of 7 wt% on the SOFC, compared to less than 1.5 wt% on the Co–W–YSZ anode. The carbon removal mechanism of this anode was manifested by the formation of tungsten acid on the surface of the Co–W alloy during SOFC operation, which inhibited carbon deposition in methane.153
Kaur et al. explored the use of Cu–Fe alloy. They prepared Cu–Fe–CeO2–YSZ anodes with different Cu/Fe molar ratios (1:0, 3:1, and 1:1) by dipping and used them for SOFC research in n-C4H10 fuel. The XRD results showed the formation of cubic phases of Cu–Fe metals. With an increase in the Fe loading, an improvement the performance was observed, where the single cell with a Cu:Fe molar ratio of 1:1 reached an MPD of 240 mW cm−2 at 800 °C in n-C4H10. They found that the output power dropped only slightly over the operation for 50 h, and TGA and SEM showed that the carbon formed at the anode was non-graphite.154 Further, they used the Cu–Fe–CeO2–YSZ anode for CH4 fuel research, and the MPDs of the cell with the Cu–CeO2–YSZ and Cu–Fe–CeO2–YSZ anodes were 70 and 90 mW cm−2 in CH4 fuel at 800 °C, respectively, and the MPD increased with an increase in the Cu–Fe metal loading. During a 46 h stability test of the Cu–Fe–CeO2–YSZ anode, the performance only dropped from 125 mW cm−2 to 100 mW cm−2, which showed the good stability of the SOFC in methane.155Table 1 presents a summary some typical bimetallic-cermet anode materials and their electrochemical performances.
| No. | Anode | Temperature (°C) | Fuel | MPD (mW cm−2) | Long term (h) | Ref. |
|---|---|---|---|---|---|---|
| 1 | Ni–Cu–CeO2–YSZ | 800 | CH4 | 330 | 500+ | 94 |
| 2 | Ni–Cu–YSZ | 900 | City coal gas | 43 | — | 95 |
| 3 | Ni–Cu–ZDC | 700 | CH4 | 87 | 48+ | 96 |
| 4 | Ni–Cu–CeO2–YSZ | 700 | CH4 | 120 | 5+ | 97 |
| 5 | Ni–Cu–BZCYYb | 750 | CH4 | 106 | 130+ | 98 |
| 6 | Ni–Cu–PBMO | 700 | C3H8 | 200 | — | 99 |
| 7 | Ni–Cu–YSZ | 800 | CH4 | 49 | 25+ | 100 |
| 8 | Ni–Cu–YSZ | 700 | CH4 | 240 | 200+ | 101 |
| 9 | Ni–Cu–YSZ–GDC | 850 | CH4 | 436 | — | 102 |
| 10 | Ni–Co–GDC | 800 | CH4 | 250 | — | 104 |
| 11 | Ni–Co–YSZ | 750 | 20% v/v CH4 in He | 136 | 60+ | 105 |
| 12 | Ni–Fe–GDC | 650 | CH4 | 340 | 50+ | 107 |
| 13 | Ni–Fe–PBMO | 700 | C3H8 | 300 | — | 99 |
| 14 | Ni–Mo–YSZ | 800 | CH4–50 ppm H2S | 594 | 120+ | 111 |
| 15 | Ni–Mo–CZ | 800 | Isooctane | 212 | 30+ | 114 |
| 16 | Ni–Sn–YSZ | 800 | CH4 | 390 | 49+ | 120 |
| 17 | Ni–Sn–YSZ functional layer | 650 | CH4 | 410 | 137+ | 121 |
| 18 | Ni–Sn–YSZ | 750 | Biogas | 272 | 50+ | 122 |
| 19 | Ni–Sn–GDC | 650 | CH4 | 470 | 200+ | 125 |
| 20 | Sn doped Ni/GDC–GDC | 650 | CH4 | 930 | 40+ | 126 |
| 21 | Ni–Sn–SDC | 700 | CH4 | 280 | 230+ | 127 |
| 22 | Ni–Sn–SDC | 700 | CH4 | 600 | 72+ | 128 |
| 23 | Ni–Ag–YSZ | 750 | CH4 | 407 | 100+ | 133 |
| 24 | Ni–Ag–YSZ | 750 | CH4 | 251 | 24+ | 134 |
| 25 | Ni–Ag–YSZ | 750 | C2H6 | 379 | 24+ | 134 |
| 26 | Ni–Pd–SDC | 550 | CH4 | 644 | — | 135 |
| 27 | Ni–Ru–GDC | 600 | CH4 | 750 | 20+ | 139 |
| 28 | Ni–Ru–GDC | 600 | C2H6 | 716 | 20+ | 139 |
| 29 | Ni–Ru–GDC | 600 | C3H8 | 648 | 20+ | 139 |
| 30 | Ni–Li–SDC | 800 | CH4 | 212 | 70+ | 141 |
| 31 | Ni–Na–SDC | 800 | CH4 | 231 | 70+ | 141 |
| 32 | Ni–CaO–SDC | 700 | CH4 | 35.6 | 0.5+ | 142 |
| 33 | Ni–Cu–BaO–SDC | 750 | CH4 | 284 | 200+ | 143 |
| 34 | Ni–MgO–SDC | 800 | CH4 | 714 | 330+ | 149 |
| 35 | Ni–Cu–MgO–SDC | 700 | CH4 | 670 | — | 78 |
| 36 | Co–Cu–CeO2–YSZ | 800 | CH4 | 250 | 500+ | 150 |
| 37 | Cu–Fe–CeO2–YSZ | 800 | n-C4H10 | 240 | 50+ | 154 |
| 38 | Cu–Fe–CeO2–YSZ | 800 | CH4 | 90 | 46+ | 155 |
Considering the efficient catalytic ability of Ni on hydrocarbon fuels, the research on carbon-resistant alloys mainly focuses on Ni-based bimetallic cermet at present. For the second metal species in nickel-based alloys, this review mainly summarizes Cu, Co, Fe, Mo, Sn, Ag, Pd, and Ru metals, as well as alkaline earth metal oxides such as CaO, BaO, and MgO. It is worth noting that in the Ellingham diagram drawn according to the change in enthalpy of metal oxide formation at different temperatures, the metal alloys that can be reduced by hydrogen at the SOFC operating temperature are limited, but there are more types of metals that can be reduced by CO than hydrogen.156 Thus, future alloying studies should consider the synthesis of a wider variety of bimetallic cermets through non-hydrogen reduction pathways.
For the bimetallic-cermet preparation process, the producing method can not only affect the mass of metals and the fabrication time, but also determine the most important property, which is the cermet structure. In addition to the traditional alloy preparation methods, such as ball milling with firing and impregnation method, plating, electrodeposition, microwave irradiation process and co-tape casting can also be considered. In addition to alkane fuel, alcohol fuel should also be studied because the hydroxyl group in the molecular structure of alcohol fuels is beneficial to resist carbon deposition. However, compared with alkane fuels, the molecules in alcohol fuels require more oxygen ions to react, and thus higher requirements are demanded on the charge transfer ability of the anode materials.
For existing hydrogen reduction systems, the alloys of Mo, Sn, and Ru with Ni and the solid solution of MgO with Ni demonstrate better electrochemical performances, and thus these systems should be given more attention. Moreover, researchers have also established a relatively mature theoretical system for the excellent stability and carbon-resistant mechanism of the above-mentioned systems in hydrocarbon fuels. Briefly, the excellent stability and carbon-resistant mechanism of bimetallic cermet anode can be summarized as follows: (I) hydrophilic metal M and H2O generated by the electrochemical oxidation of fuel are conducive to methane steam reforming, and upon further hydration to hydroxylation reaction, the hydroxyl group (*OH) helps the oxidation of adjacent carbon, and is removed in the form of CO or CO2. (II) The outstanding adsorption of metal M to CO2 accelerates the in situ dry methane reforming and the combination of adsorbed CO2 with deposited carbon to form gas-phase CO. (III) The surface of the metal alloy preferentially oxidizes the C atom, promoting the formation of C–O bonds instead of forming C–C bonds. (IV) Carbon deposited on alloy-modified catalysts is more likely to form disordered amorphous carbon. (V) Ni-based alloys prevent an increase in Ni particles, thereby increasing the active sites and expanding the reaction TPB region. (VI) The external electron configuration of metal M is similar to C: [He]2s22p2 (two valence electrons close to stable s-orbitals), and hence the presence of Ni-based alloys hinders the formation of nickel carbide.
Although these bimetallic-cermet anodes show good potential for hydrocarbon catalytic performance, during the actual application environment of SOFCs, they will undergo multiple redox cycles and temperature alteration processes, which will reduce the mechanical strength of the bimetallic-cermet and even cause cracks in the cells. Alternatively, metals or alloys formed by exsolving from perovskite materials of the anode can improve the stability of the cell, which will be discussed in Section 2.2.
2.2 Ceramic materials
Ceramic oxide anodes, such as SrTiO3, La4.0Sr8.0Ti11.0Mn0.5Ga0.5O37.5, La0.75Sr0.25Cr0.5Mn0.5O3 and Sr2MgMoO6−δ mentioned above, are known for their slow carbon deposition, high temperature and redox stability. Unlike metals, they are not catalysts for C–H bond breaking, but contribute to the direct electrochemical oxidation of hydrocarbon fuels. However, the main disadvantages of oxide anodes are their low catalytic activity and electronic conductivity. Typically, they are doped or used for in situ exsolution of cations to form highly reactive metals or alloy nanoparticles to achieve the desired properties.Yun et al. studied the Ni–Sm0.2Ce0.8O2−δ (SDC) anode as an alternative anode in methane fuel and compared it to the traditional Ni–YSZ anode. The mixed ion and electron conductive properties of SDC under reduction conditions enlarged the reaction TPB, resulting in a 20–25% improvement in the performance of the SOFC with the Ni–SDC anode compared to that with the Ni–YSZ anode. In addition, the performance of the SDC-coated Ni–SDC anode under 0.1 A cm−2 at 850 °C for 180 h did not show any significant degradation, indicating that SDC caused the oxidation of methane and inhibited carbon deposition by electrochemically oxidizing the carbon produced from methane cracking on Ni. In contrast, Ni–SDC had lower performance when switching from H2 to CH4. This study, based on the most widely used SOFC Ni–YSZ anode demonstrated the advantages of CeO2-based materials in hydrocarbon fuels.160
Putna et al. investigated Rh supported on SDC as an anode, demonstrating that it was possible to oxidize CH4 electrochemically in an SOFC without prior steam reforming, but the power density of the cell was very poor (less than 7 mW cm−2 at 800 °C).161 Given that Cu–YSZ anodes, unless cerium dioxide is added, perform poorly in methane,162 Lu et al. did similar work to study the effects of adding CeO2 by testing Cu–SDC and Au–SDC anodes in n-butane fuels. The MPDs for these two cells made were 122 and 132 mW cm−2 at 650 °C with H2 fuel, respectively, and both around 50 mW cm−2 at 650 °C with n-butane fuel. These similar performances showed that the two metals were only electronic conductors and had no catalytic effect in the direct-oxidation. The addition of CeO2 suggested that it played an important role in improving the anode performance by increasing its catalytic activity or mixed ion conductivity.163
Gd-doped ceria (GDC) has also been studied by many researchers because of its excellent performance in various SOFC fuels. Marina et al. studied a Gd-doped ceria (GDC) anode with a 10–15 μm thick Au–GDC composite layer as the current-collector. It was found that the MPD of the cell was 178.5 mW cm−2 at 1000 °C with 4.9% CH4 + 2% H2 + 3% H2O + 90.1% N2 fuel. Moreover, after 1000 h of operation at 1000 °C with a low partial pressure of CH4 and H2O of 10 and 3 kPa, respectively, no carbon depositions were found on the GDC anode. The cell could last several rapid thermal cycles in the temperature range of 200–1000 °C and underwent a complete redox cycle without degradation.164 Wisniewski et al. applied Ir-loaded-Ce0.9Gd0.1O2−x (Ir–GDC) in CH4/CO2 ratios between 2 and 0.66 at 600–800 °C. They found that the Ir–GDC catalysts showed considerable stability for DRM, and no carbon formation was observed except under highly reductive conditions of CH4:CO2 = 2 at 800 °C.165 Han et al. studied the performance of Ni-GDC anodes in H2, CH4, C3H8, NH3 or simulated underground coal gasification (UCG) gases. The composition of the simulated UCG gas at room temperature was 66% H2 + 19% CO + 15% CH4. Among them, the MPD of the cells in different fuel gases was reduced in the order of H2 > simulated UCG gas > NH3 > CH4 > C3H8. The Ni-GDC anodes showed good stability in other fuel gases except for C3H8 after 18 h at the constant voltage of 0.6 V at 600 °C. Among them, in CH4 and C3H8 fuels, the coking results of the cells and the long-term stability were not as bad as predicted in the function of balance composition (moles%) with the fuel and the temperature between 100–900 °C. These results were attributed to two possible causes, where one was that the slow kinetics at 600 °C led to less pyrolysis of CH4 and C3H8, resulting in less coke formation. The other was due to the rapid migration of oxide ions. The relatively abundant supply of O2− reacted with the deposited solid carbon to help suppress carbon deposition at low operating temperatures.166
Using an Ni–YSZ/yttria-doped ceria (YDC) anode, Murray et al. pioneered the work of direct electrochemical oxidation of methane at low operating temperature (generated power densities of up to 370 mW cm−2 at 650 °C). The amount of carbon deposited increased with an increase in temperature when the temperature was higher than 700 °C, but at a given temperature, less carbon deposition occurred on Ni–YSZ–YDC than on the Ni–YSZ anode.16
Ahn et al. examined the effect on the SOFC anode performance of replacing CeO2 with a Ce0.6Zr0.4O2 (ZDC) solid solution in Cu–YSZ composites. Before Cu was added, CeO2 was calcined at 723 and 1273 K, respectively, resulting in a significant reduction in the anode performance with the MPD decreasing from 280 to 170 mW cm−2 in humidified (3% H2O) H2 fuel at 973 K. Conversely, the MPD of the cell decreased from 240 to 210 mW cm−2 when CeO2 was replaced with Ce0.6Zr0.4O2. Compared with CeO2, Ce0.6Zr0.4O2 improved the thermal stability of the anode, which was ascribed to the improved reducibility of the Ce0.6Zr0.4O2 solid solution. Although Cu–ZDC–YSZ has not been tested in hydrocarbon fuels, we expect that it will also demonstrate good thermal stability.167 Similar work was performed by Song et al. using 10% ZDC, which verifies our hypothesis. Compared with pure CeO2-based anodes, the Zr0.1Ce0.9O2−δ anode exhibited good thermal stability and activity in 97% CH4 + 3% H2O fuel at 700 °C. The MPD of the cell with the Cu–ZDC anode was 32 mW cm−1, while the cell with the Cu–CeO2 anode showed a peak power density of only 28 mW cm−2. Short-term stability tests for all the cells at 700 °C were conducted at a constant voltage of 0.6 V under 97% CH4/3% H2O. After the stability test, the carbon content on the anode surfaces of Cu–ZDC was 1.93 wt%, while that on Cu–CeO2 reached 2.19 wt%96.
A co-doped CeO2-based anode with Y and Yb was first reported by Zhou et al. for use in direct methane SOFCs. This novel ionic conductor, Ce0.8Y0.1Yb0.1O1.9, exhibited higher ionic conductivity between 400–750 °C than single-doped CeO2 (Ce0.8Y0.2O1.9 and Ce0.8Yb0.2O1.9). The two rare earth elements worked synergistically to improve the ionic conductivity and reforming or oxidation of hydrocarbon fuels. The MPD of the Ni–CYYb configuration cell was 94 mW cm−2 at 800 °C in dry CH4 fuel. After the test at constant current density of 200 mA cm−2 at 750 °C for 120 h, SEM–EDS characterization showed that there was essentially no carbon deposition on the anode. In addition, the stability of the Ni–CYYb SOFC anode in H2 contaminated with 5 ppm H2S was also studied, and they found that it could catalyze the conversion of H2S to SO2. All these results indicate the outstanding potential of this new material as an SOFC anode.168Table 2 presents a summary of some typical cerium-based anode materials and their electrochemical performances.
| No. | Anode | Temperature (°C) | Fuel | MPD (mW cm−2) | Long term (h) | Ref. |
|---|---|---|---|---|---|---|
| 1 | SDC-coated Ni–SDC | 850 | CH4 | 248 | 180+ | 160 |
| 2 | Rh–SDC | 800 | CH4 | 7 | — | 161 |
| 3 | Au–SDC | 650 | n-butane | 50 | — | 163 |
| 4 | Cu–SDC | 650 | n-butane | 50 | — | 163 |
| 5 | Au–GDC | 1000 | 4.9% CH4 + 2% H2 + 3% H2O + 90.1% N2 | 179 | 1000+ | 164 |
| 6 | Ni–GDC | 600 | 66% H2 + 19% CO+ 15% CH4 | 537 | 48+ | 166 |
| 7 | Ni–YSZ–YDC | 650 | CH4 | 370 | — | 16 |
| 8 | Cu–ZDC | 700 | CH4 | 32 | 48+ | 96 |
| 9 | Ni–CYYb | 800 | CH4 | 94 | 120+ | 168 |
The process of storing and releasing oxygen from cerium-based oxides is maintained by the Ce4+/Ce3+ redox couple and doping suitable metal ions in the cubic fluorite phase CeO2 can reduce the concentration of Ce4+, thereby increasing the oxygen vacancy concentration for excellent catalytic activity. The research on cerium-based oxide doping is currently mature, mainly including samarium-doped ceria (SDC), gadolinium-doped ceria (GDC), yttria–doped ceria (YDC), zirconia–doped ceria (ZDC), and Ce0.8Y0.1Yb0.1O1.9. For the ceramic composition of the anode, the bi-ceramic composition (e.g., Ni–YSZ–YDC) is beneficial for the expansion of the TPB when the structure of the cell is reasonable. Also, in addition to the common cerium-based materials, the above-mentioned BaZr0.1Ce0.7Y0.1Yb0.1O3−δ, Pr0.5Ba0.5MnO3−δ and in situ exsolution of perovskite materials are also popular research areas for ceramic materials and may have better properties than cerium-based oxides.
 | ||
| Fig. 6 (a) Unit cells of perovskite-type oxides and typical cations occupying the A and B sites.186 (Reprinted from ref. 186 with permission from Multidisciplinary Digital Publishing Institute.) (b) Comparison of heterogeneous electrodes manufactured by infiltration and exudation. (c) Exsolution of Ni at B-site on the surface of perovskite. Reproduced with permission.187 (Reprinted from ref. 187 with permission from the American Chemical Society.) (d) Average CH4 conversion rate of Ni/LSM n = 1 (prepared by exsolution), Ni/LSMO n = 1 (prepared by impregnation) and Ni/YSZ at an extended reaction time (reduction preprocessing condition: in diluted H2 for 4 h, T = 850 °C. (Reprinted from ref. 174 with permission from Elsevier Ltd.) (e) Schematic diagram of segregation at the A-site and its driving force in LaMnO3 perovskite system.183 (Reprinted from ref. 183 with permission from the American Chemical Society.). | ||
Different cations and defects will make perovskites exhibit a variety of properties due to their structure characteristics.169 The atoms on the surface of perovskites usually have lower coordination numbers than their inner atoms, and thus the perovskites have higher surface-free energies, which enable the rearrangement of their surface atoms and the separation of certain cations from the matrix to the surface to generate metal nanoparticles. Accordingly, the composition, microstructure of perovskite oxide, and the related properties of reforming, oxidation, and electrocatalysis of fuel will also be changed. When two different materials come into contact, a heterogeneous interface will be formed, and thus the exsolved metal nanoparticles and the perovskite body will become a metal-oxide heterogeneous structure.170 Heterostructures usually have a small band gap and their internal electric fields accelerate the ion diffusion dynamics, resulting in excellent electrical conductivity. In addition, compared with the weak interaction between the parent and nanoparticles produced by impregnation, the robust interaction between the parent phase and the exsolution particles enhances the structural stability of fuel cells under the working conditions,171 as shown in Fig. 6(b). Therefore, the construction of heterogeneous interfaces by in situ exsolution can optimize the catalytic performance of the materials. Over the years, in situ exsolution of perovskite has become an effective method to prepare highly active materials.
Usually, catalytic nanoparticles dispersed on the outer surface of porous materials are prepared via the physical vapor deposition method or chemical impregnation method and attached to the surface of the carrier material by a high-temperature calcination process. However, the impregnation method restricts the size, uniform dispersion, and firmness of the deposited species;172 in addition, they tend to aggregate at high temperatures because of the weak mutual effect between the nanoparticles and bulk phase, and their performance will inevitably degrade with the extension of the working time.173Fig. 6(d) shows the in situ exsolution material in La1.5Sr1.5Mn1.5Ni0.5O7±δ (Ni/LSM n = 1), the impregnated material Ni/La0.5Sr1.5MnO4±δ (Ni/LSMO) n = 1) and the average conversion of CH4 at 850 °C for the traditional anode Ni/YSZ constructed by Sebastián et al.174 The conversion rate of Ni after in situ exsolution was 9.71 ± 0.50 mol%, which was higher than that of the impregnated material (7.89 ± 0.20 mol%), and the hydrogen yield was also higher. After 30 h, the CH4 conversion rate of the impregnated material decreased significantly, which for Ni/YSZ was almost zero. However, the conversion rate of Ni/LSM did not change much in the exsolution system, and the researchers believed that this is due to the sintering or condensation of the impregnated particles, resulting in the uneven distribution of nickel on the material surface, reducing catalyst activity, and forming carbon deposits on the material surface. Recently, researchers have found a more advanced and effective way to dissolve nanoparticles, i.e., in situ exsolution. Compared with the impregnation and deposition methods, in situ exsolution is cheaper and is a simpler process; moreover, the nanoparticles constructed in this way do not easily agglomerate at high temperatures and have reversibility to avoid catalyst agglomeration through reoxidation because they are more evenly distributed and smaller. On account of the strong mutual action between the nanoparticles and matrix, the problem of particle coarsening is effectively alleviated, the life of the catalyst will be greatly improved,172 and the stability of the cell will also be better.
In general, the segregation of ABO3 perovskite oxide cations can be divided into A-site cations (such as Sr) and B-site cations, i.e., exsolution elements. A-site cation segregation is a common phenomenon in perovskite oxidation cathode,175 and Sr is commonly used as an A-site dopant, and thus it is usually called “Sr segregation”. B-site cationic segregation refers to the doping of transition metals or noble metals at the B-site, and the doped metals are subsequently induced to exsolve out of the lattice in situ as active nanoparticles in a reducing atmosphere.176 Cathode materials usually work in a strong oxidizing atmosphere at high temperatures, which promotes the separation of the A-site cations from the surface, while the B-site is more prone to exsolve in a reducing atmosphere. For instance, if the B sites of perovskite oxides are doped with Pd,177 Ru,178 Pt,179 Co,180 Ni,181 and other catalytic active noble metals or transition metals, they will exsolve from the perovskite skeleton structure to form nanoparticles in a high-temperature reduction atmosphere, embedding on the perovskite phase and forming a heterogeneous structure.175
Niania et al. proposed the mechanism of A-site cation (Sr) surface segregation in the La0.6Sr0.4Co0.2Fe0.8O3−δ perovskite material based on in situ surface characterization results, where Sr segregates on the exposed grain surface and forms a monolayer, and more strontium will form Sr-based particles over time. Strontium nucleates at the grain boundaries with an increasing segregation rate and the resulting defects can also be heterogeneous nucleation sites, and eventually strontium agglomerates due to surface migration.182 Lee et al. suggested that the foremost driving force of cation segregation in perovskite-based oxides is the elastic and electrostatic interactions between the dopants in the perovskite oxides and the surrounding lattice.183 These researchers demonstrated that the purpose of A-site dopant cation segregation was to decrease the elastic energy to the minimum, which is caused by the mismatching between the size of the dopant and the host cation, and decrease the electrostatic energy produced by the interaction between the surface and the dopant charged defects near the surface, as shown in Fig. 6(e). Neagu et al. observed the annealing of La0.43Ca0.37Ti0.94Ni0.06O3 at 900 °C under high vacuum and proposed the mechanism of the in situ exsolution of the B-cations (Ni) from the perovskite, as shown in Fig. 6(c). Sr segregation at the A-site is considered to be one of the important reasons for the chemical instability of the perovskite surface and the corresponding performance degradation of the SOC electrode,184 for example, the SrOx segregation layer on SrTi1−xFexO3(STF) could reduce the oxygen reduction kinetics of the electrode, and thus affect the cell performance.185 In contrast, the exsolution of the B-cations usually improves the electrochemical performance of the electrode.
The classical nucleation theory indicates that the main reason for exsolution and nucleation in the reduction process is the over-saturation of the cations in the main lattice. In the case of perovskites composed of AB1−xMxO3, the metal element M is more easily reduced to metal from cations and exsolved than other transition metals B. As mentioned earlier, the Gibbs free energy variation (ΔG) in the process from Mn+ to M is the dominating driving force for the exsolution. Gao et al. proposed that exsolution is the result of four physical processes, i.e., diffusion, reduction, nucleation, and growth, as shown in Fig. 7(a). The Ni2+ ions exsolved from the matrix to the surface, then were reduced to zero-valent Ni, and the reduced Ni became nanoparticles, which increased in size with time. After nucleation, the growth rate of the particle size slowed down, indicating that ΔG began to increase after reaching the minimum value.188Fig. 7(b) shows the ΔG of each metal element oxide reduced to metal in La0.4Sr0.4Sc0.9Ni0.1O3−δ. The ΔG > 0 of the La, Sr, and Sc metal oxides show that they are thermodynamically stable in the reduction conditions; nevertheless, the ΔG < 0 of Ni shows that it is easier to be reduced, which is conducive to exsolution.
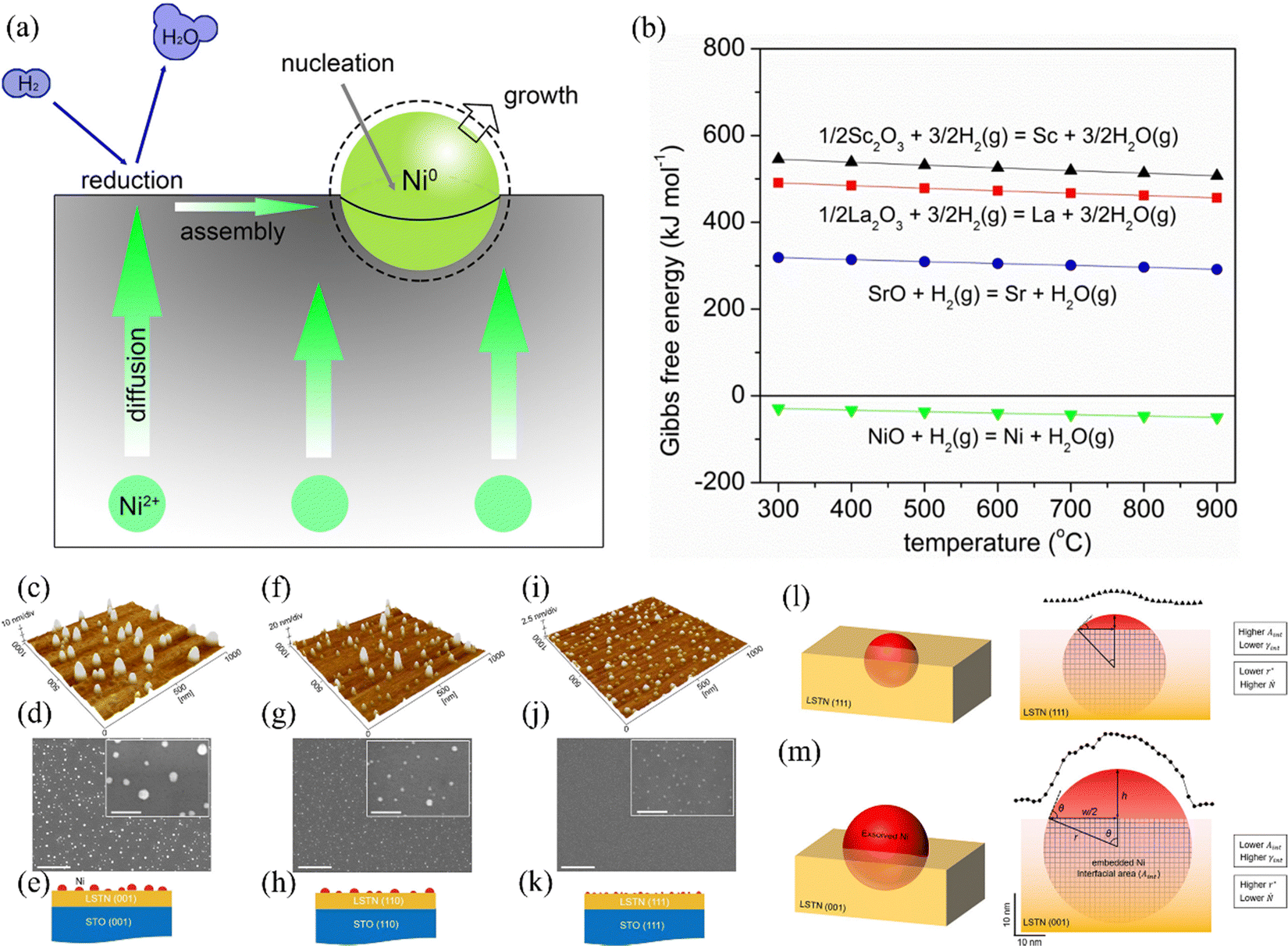 | ||
| Fig. 7 (a) Schematic diagram of the exsolution process. (b) Sc, La, Sr, and Ni oxides are reduced in H2 according to the Gibbs free energy of the corresponding metal. (Reprinted from ref. 188 with permission from Elsevier Ltd.) (c, d), (f, g) and (i, g) are (001)-oriented film, (110)-oriented film, and (111)-oriented LSTN film AFM and SEM top view images, respectively. The scales of (d, g and j) and their inset are 1 μm and 200 nm, respectively. (e, h and k) Surface schematic of an LSTN film exsolved with Ni nanoparticles. AFM imaging area = 1 μm2. The thickness of LSTN film is about 300 nm. AFM height and transverse diameter distribution of representative exsolved particles. (l) (111)-oriented films and (m) (001)-oriented films show that Ni particles exsolved from LSTN and embedded in the films, the lower normalized interface area in the (001)-oriented films mean a higher interface energy. (Reprinted from ref. 191 with permission from the American Chemical Society.). | ||
Oh et al. used the COMSOL Multiphysics software to calculate the elastic field and free energy in the exsolution process, indicating that the interaction between the surface free energy and strain energy is the driving force for exsolution.189 In addition, the surface and interface energy of the nucleation particles and the local strain field in the matrix may further affect the distribution of grain size. In the initial study of the surface effect, the researchers believed that the (110) face was most suitable for B-site cation exsolution.190 However, Kim et al. came to a different conclusion that other planes such as the (001) and (111) planes also exhibited strong exsolution tendencies.191 La0.2Sr0.7Ti0.9Ni0.1O3−δ (LSTN) films in the (001), (110), and (111) orientation were deposited on the corresponding single-crystal SrTiO3 substrates in the (001), (110), and (111) orientation using pulsed laser deposition (PLD). The exsolution of the nickel nanoparticles on the three oriented LSTN films is shown in Fig. 7(c)–(k). On the film in the (111) orientation, the largest number of particles (195 μm−2), the smallest particle size (20 ± 7 nm), the strongest embedding, and the shortest distance between particles were observed, which was the most uniform oriented film. The smaller average particle size indicates higher catalytic activity, and without a strong shielding effect or diffusion effect due to the large distance. Fig. 7(l) and (m) shows that the surface interface energy of the (111) orientation was the lowest, and because the normalized projected area of the partial (001)-oriented film connected to the LSTN matrix is smaller than the corresponding area of the (111)-oriented film, the interface energy (γint) of the (001) oriented film was higher. On account of the higher interface energy, the assembly nuclear energy barrier and critical nucleation size increased and the nucleation rate and particle population density decreased.
Early studies focused on perovskite oxides with a stoichiometric ratio of A/B = 1, which exsolved cations through the inherent reduction driving force once used by researchers to prepare “smart catalysts” for controlling automobile emissions.192 For example, Nishihata et al. found that the Pd-perovskite catalyst had higher activity than the traditional Pd–Al2O3 catalyst.193 The method for the preparation of this “smart catalyst” has been widely used in the preparation of in situ exsolution perovskite oxide SOFC anodes lately.194 However, the intrinsic reducibility of perovskite can only drive limited reducible cations to exsolution176 and the types of metal elements that can be exsolved in situ are limited to noble metals such as Ru, Pt, Pd, and Ni. Fig. 6(d) shows that the average CH4 conversion of Ni/YSZ was 14.00 ± 0.44 mol%, which was close to the reaction equilibrium value 15 mol%, while the average conversion of the exsolution was only 9.71 ± 0.50 mol%. Obviously, the electrocatalytic activity of the nano-catalyst exsolved by inherent reducibility is insufficient and its electrochemical performance is still limited compared with the traditional nickel-based catalyst. Thus, recently, to improve the catalytic activity of in situ exsolved perovskite, many researchers have focused on investigating A-site defects (i.e., perovskite oxides with stoichiometric ratio of A/B < 1) using cheap and stable transition metals to regulate the exsolution.195 The A-site defect increases the tendency of the B-site cation to exsolve from the lattice. Also, the exsolution restores the perovskite to a stable structure locally, making the whole structure “defect-free”, thus conforming to the chemometrics of ABO3. This mechanism can be explained by the point defect reaction proposed by Neagu et al.,172 as follows:
 | (14) |
 | (15) |
 | (16) |
 | (17) |
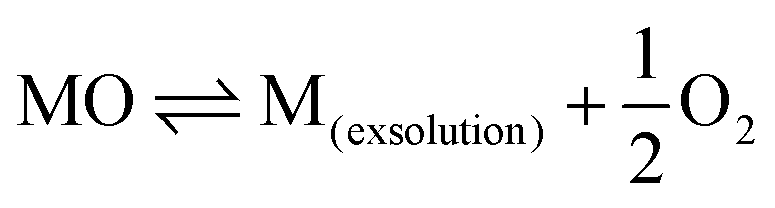 | (18) |
A large number of A-site defects  produced by doping will make formula (14) move to the left, limiting the number of inherent Schottky defects, thus reducing the number of
produced by doping will make formula (14) move to the left, limiting the number of inherent Schottky defects, thus reducing the number of  . Simultaneously, formula (15) moves to the right under a certain p(O2) to counteract the changes in formula (14), which promotes the reduction of lattice oxygen and the doped metal to
. Simultaneously, formula (15) moves to the right under a certain p(O2) to counteract the changes in formula (14), which promotes the reduction of lattice oxygen and the doped metal to 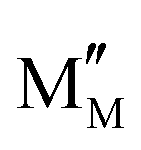 in the reducing gas. Afterward, the metal atoms
in the reducing gas. Afterward, the metal atoms 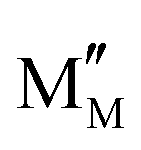 spontaneously detach from the oxide lattice, leaving cationic vacancies
spontaneously detach from the oxide lattice, leaving cationic vacancies  , as shown in formula (16). Another metal exsolution path is shown in formulas (17) and (18). The doped metal and lattice O generate MO, and then the metal will exsolve in the reduction process. The presence of vacancies destroys the stability of the perovskite lattice, partially causing the B-site cations to spontaneously exsolve to reconstruct the stoichiometry of the bulk phase. In addition, perovskites with A, O-site defects can be considered as B-site excess, which explains the tendency of B-site exsolution from another perspective. At the atomic scale, the process of removing oxygen from perovskites lacking A-site cations can be seen as moving the B-site dopant locally from the main perovskite skeleton to the outside to form an initial exsolution state, as shown in Fig. 8(a).172 The A-site-occupied unit cell remains unaffected, while the B site in the A-site missing unit cell is excess and will be discharged.
, as shown in formula (16). Another metal exsolution path is shown in formulas (17) and (18). The doped metal and lattice O generate MO, and then the metal will exsolve in the reduction process. The presence of vacancies destroys the stability of the perovskite lattice, partially causing the B-site cations to spontaneously exsolve to reconstruct the stoichiometry of the bulk phase. In addition, perovskites with A, O-site defects can be considered as B-site excess, which explains the tendency of B-site exsolution from another perspective. At the atomic scale, the process of removing oxygen from perovskites lacking A-site cations can be seen as moving the B-site dopant locally from the main perovskite skeleton to the outside to form an initial exsolution state, as shown in Fig. 8(a).172 The A-site-occupied unit cell remains unaffected, while the B site in the A-site missing unit cell is excess and will be discharged.
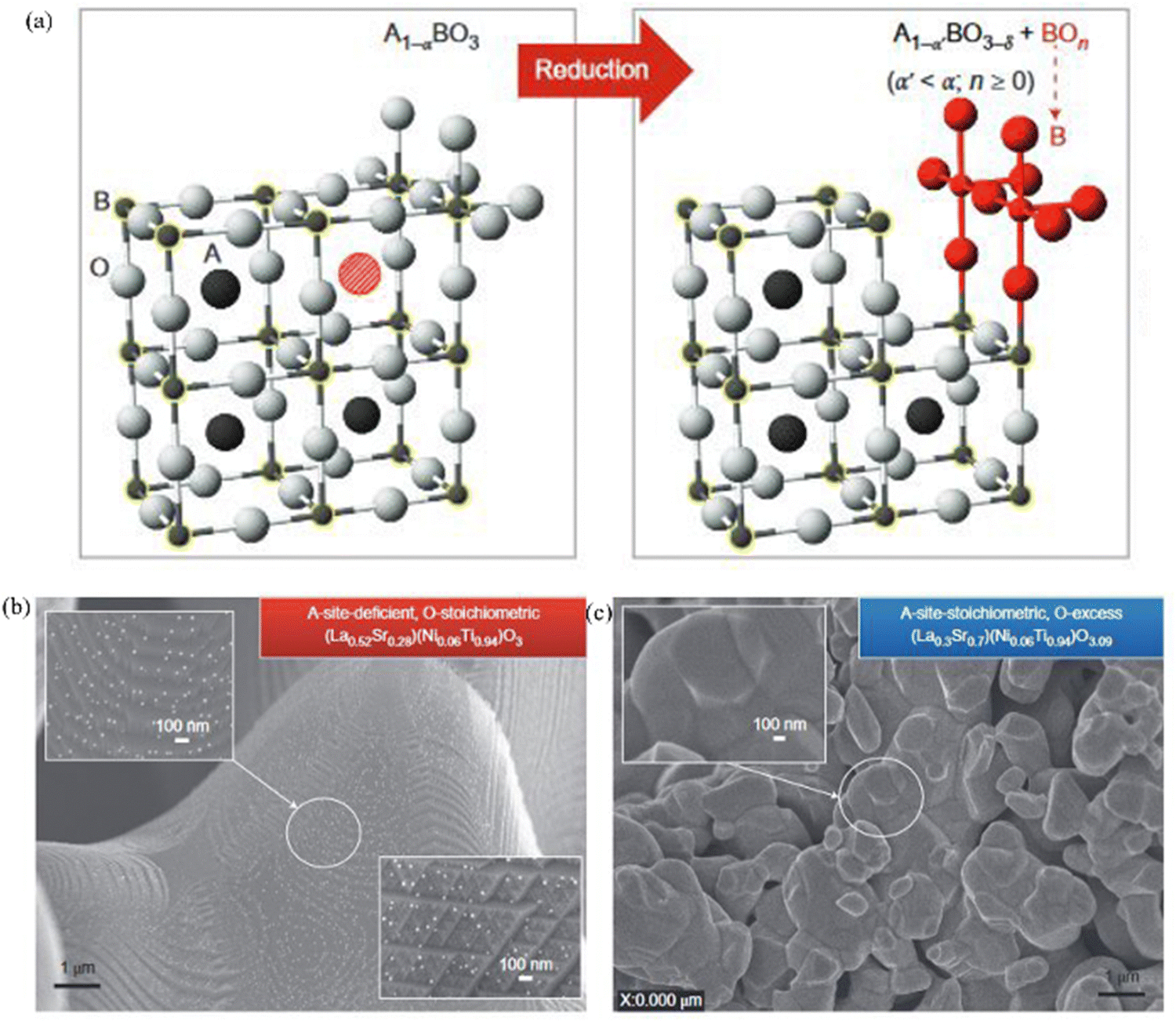 | ||
| Fig. 8 (a) Schematic diagram of B-site cation exsolution in A-site defect perovskite, where O-site is represented by silver balls; B site is represented by yellow highlighted gray balls; black big spheres represent A site; and red color indicates that VA is an A-site vacancy. By reducing the oxygen in the A-site defect unit, some B-sites are partially isolated from the perovskite matrix and enter the initial BOn exsolution, as showed in the red atom group in the right figure. The effect of non-stoichiometry in stoichiometry materials and A-site-deficient perovskite exsolution showed by SEM. (b) Oxygen stoichiometry and A-site-deficient La0.52Sr0.28Ni0.06Ti0.94O3 after reduction at 930 °C for 20 h, and Ni was exsolved in 5% H2/Ar. (c) La0.3Sr0.7Ni0.06Ti0.94O3.09 sample with excess stoichiometric O with A site stoichiometric did not exsolve after reduction at 930 °C for 20 h under 5% H2/Ar conditions. (Reprinted from ref. 172 with permission from Macmillan Publishers Ltd.) | ||
Neagu also studied the exsolution of Ni in two samples containing 6% Ni at the B-site with different oxygen vacancy concentrations at the A-site under the same reduction conditions.172 The A-site oxygen-deficient material La0.52Sr0.28Ni0.06Ti0.94O3 generated a large number of metal Ni nanoparticles, which exsolved to the perovskite surface, as shown in Fig. 8(b). In contrast, no particle growth was observed in La0.3Sr0.7Ni0.06Ti0.94O3.09, which is stoichiometric and oxygen-excess, as shown in Fig. 8(c).
This experiment showed that the B-site atoms had a greater tendency to undergo exsolution in A-site-deficient perovskites than that in oxygen-rich perovskites, and A-site defects help to form oxygen vacancies with high fluidity, an increase in which can improve the ionic conductivity.196 When the composition of the perovskite oxide is changed to A/B < 1, more cations can segregate on the surface of the perovskite bulk, which may be a more efficient method to construct perovskite structures with well-dispersed nanoparticles. Therefore, it is possible to control the surface modification of perovskites by constructing a non-stoichiometric system, which cannot be achieved by traditional-component perovskites.172 Sun et al. used the in situ exsolution method to prepare nickel-doped and strontium-doped lanthanum chromate (LSC) with A-site defects as SOFC anodes.176 The electrochemical performance of the perovskite anodes promoted by the A-site defects in the SOFC was evaluated for the first time and it was found that the A-site defects indeed facilitated the in situ exsolution of Ni on the nickel-doped anodes (La0.7Sr0.3)CrO3(LSCNi). The cell with the A-site-defect anode showed an MPD of 460 mW cm−2 in 5000 ppm H2S-H2, while that of the cell using the stoichiometric LSCNi anode was only 135 mW cm−2. Dueñas et al. compared the stoichiometric perovskite oxide La0.70Sr0.3Cr0.85Ni0.15O3−δ(L70SCrN) with 5% A-site deficient La0.65Sr0.3Cr0.85Ni0.15O3−δ(L65SCrN) and found that the latter had higher exsolution ability. The researchers calculated that the net oxygen consumption of L70SCrN was 0.27 (mol O mol−1), while the net oxygen consumption of L65SCrN was 0.36 (mol O mol−1).197 The lack of A site enhanced its reducibility, which is conducive to the exsolution of metal Ni. Zhu et al. prepared Sr0.95(Ti0.3Fe0.63Ni0.07)O3(STFN), which is A-site-deficient, by substituting Ni for Fe in the SrTi0.3Fe0.7O3(STF) perovskite anode.198 The MPD of the cell with the STFN anode that exsolved Ni–Fe nanoparticles was 1300 mW cm−2 at 850 °C, while the MPD of the cell with the STF anode was 1100 mW cm−2. In summary, researchers assume that the presence of A-site defects in perovskite means there is the driving force to trigger the external exsolution of the B-site cations, and the produced nanoparticles have a more uniform surface distribution and coverage with better catalytic performance.
In addition, some reports demonstrated that the particle size, coverage or distribution of in situ exsolution nanoparticles on the perovskite surface can be adjusted by changing factors such as the reduction temperature, working time, and number of oxygen vacancies. Kobsiriphat et al. found that the volume of B-site Ru nanoparticles increased with an increase in temperature, which exsolved from La0.8Sr0.2Cr1−xRuxO3−δ. After annealing in H2 at 600 °C, 700 °C and 800 °C for 15 min, the average diameter of the Ru nanoparticles increased from 1 nm at 600 °C (Fig. 9(a)) to 3–4 nm at 800 °C (Fig. 9(c)), and the grain density also decreased with an increase in annealing temperature.199 He et al. also found that temperature changes can adjust the exsolution of the nanostructures. These researchers doped cerium dioxide in titanate at 1400 °C and found that up to one-third of the doped cerium was exsolved from the A-site of La0.8Ce0.1Ni0.4Ti0.6O3−δ at 1300 °C to form a lamellar structure on the grain boundary, as shown in Fig. 9(d). When the temperature was lower than 1300 °C, cerium oxide cubes of different sizes appeared on the perovskite surface. Fig. 9(j) shows that when La0.8Ce0.1Ni0.4Ti0.6O3−δ was annealed at 1300–900 °C, the cubic exsolution tendency of CeO2 increased with temperature from the bottom to top, and the lower temperature could make more CeO2 cubic phase grow with a smaller size. When annealed at 1200 °C, the particle diameter was about 220 nm, and there was about one particle per μm2 on the perovskite surface (Fig. 9(e)). When the annealing temperature was reduced to 1100 °C, a large number of CeO2 particles was exsolved on the surface of titanate. Compared with 1200 °C, the ceria particles were smaller (130 nm) and their density was higher (about 9 particles per μm2), as shown in Fig. 9(f). Although the exsolution of the ceria cubes could be observed under scanning electron microscopy at 1100 °C, it was possible that XRD could not detect the diffraction peak of CeO2 because the amount of ceria exsolution at low temperature was lower than the detection limit (<1 wt%). Further lowering the annealing temperature resulted in a smaller cerium dioxide cubic phase, with particle sizes of about 55 and 40 nm at 1000 °C and 900 °C, and densities of about 20 and 10 grains per μm2, as shown in Fig. 9(g) and (h), respectively. Fig. 9(i) shows that no CeO2 cubes exsolved from the surface of the perovskite after annealing at 800 °C, possibly due to the insufficient thermal energy to support cerium cation diffusion at this low temperature. Therefore, the temperature is considered a key factor affecting the exsolution and separation of CeO2 in this perovskite material.173
 | ||
| Fig. 9 (a–c) TEM images of La0.8Sr0.2Cr1−xRuxO3−δ annealed in H2 at 600 °C, 700 °C, and 800 °C for 15 min, respectively. The average diameter of ruthenium nanoparticles increased from 1 nm at 600 °C to 3–4 nm at 800 °C, and the grain density decreased with an increase in the annealing temperature. (Reprinted from ref. 199 with permission from Elsevier Ltd.) (d)–(h) Backscattered electron micrograph of the surface of the La0.8Ce0.1Ni0.4Ti0.6O3−δ particle surface annealed in the air for 2 h. (d) 1300 °C, (e) 1200 °C, (f) 1100 °C, (g) 1000 °C, (h) 900 °C and (i) 800 °C. (j) Schematic showing the tendency of CeO2 cubic exsolution with an increase in temperature (from bottom to top). (Reprinted from ref. 173 with permission from Elsevier Ltd.) (k–m) In situ TEM images of PBMCo in 0.5 Pa H2, at 770–850 °C, with one image per 40 °C. (n) In situ XRD patterns of PBMCo at room temperature to 800 °C in 10% H2–N2. (o) I–V–P curves of the cell with PBMCo anode in the range of 800–900 °C in H2. (Reprinted from ref. 200 with permission from the American Chemical Society.) | ||
Sun et al. studied the in situ exsolution of Co nanoparticles from a double perovskite-structured crystal, Pr0.5Ba0.5MnOx(PBMCo). Fig. 9(k)–(m) show that there was no evidence of exsolution on the perovskite surface at 770 °C, and Co did not segregate until the temperature increased to 810 °C. When the temperature further increased to 850 °C, large particles appeared on the edge of the perovskite bulk, which adhered to it, and at a certain temperature, these exsolved particles became clearer than before. As shown in Fig. 9(n), the in situ TEM image is consistent with the results of in situ XRD, and a significant Co diffraction peak appeared at 800 °C, indicating that the exsolution of Co mainly occurred above 800 °C, and the exsolution rate was also high. These researchers also characterized the performance of the catalyst as an SOFC anode in H2, and Fig. 9(o) shows the I–V–P curves at different temperatures. As the test temperature increased, more and more Co nanoparticles exsolved, and their MPD also increased, which was 520 mW cm−2 at 800 °C and reached 1100 mW cm−2 at 900 °C.200
The running time also has an influence on the performance of the cell with an exsolution nanoparticle anode. Madsen et al. tested the electrochemical performance of the cell loaded with La0.8Sr0.2Cr0.82Ru0.18O3−δ–GDC as the anode at 800 °C and working in wet H2 over time. The MPD over 15 min was about 250 mW cm−2 and stabilized at 390 mW cm−2 over time to 100 h. The results were similar to the tests at other temperatures, where the MPD of the cell increased from 120 mW cm−2 at 15 min to 300 mW cm−2 at 100 h at 750 °C.201
Neagu et al. introduced the concept of oxygen-deficient δ by reducing perovskite (A1−αBO3) with the A-site deficient. δ not only means the number of oxygen vacancies  , but also indicates the degree of reduction, which means that the amount of oxygen removed per formula unit of perovskite is quantitatively equivalent to
, but also indicates the degree of reduction, which means that the amount of oxygen removed per formula unit of perovskite is quantitatively equivalent to  .172 It is also related to the number of reduced B-site ions
.172 It is also related to the number of reduced B-site ions 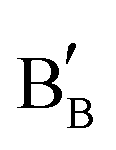 , which has a great influence on exsolution. The formation of defects can be expressed by the Kröger-Vink formula, as follows:
, which has a great influence on exsolution. The formation of defects can be expressed by the Kröger-Vink formula, as follows:
 | (19) |
Given the effects of δ on solubility and conductivity, Neagu et al. studied the changes in oxygen vacancies (δ) and conductivity (σ) with the quantity of Ga doping (x) after the reduction of La0.4Sr0.4Gx0.4GaxO3−x/2−δ (0 ≤ x ≤ 0.15), and proposed that the oxide ion mobility of the perovskite oxide n-type conductor bulk could be increased by increasing the number of oxygen vacancies, thereby improving the conductivity.169 It is known that both ion conduction and electron conduction contribute to the conductivity of the ion-electron mixed conductor. When the B-site cation is partially substituted with other low-valent elements (e.g., Ga element), oxide ion vacancies are formed to keep the electrical neutrality of the perovskite structure, as shown in formula (19), while the oxide ion conduction in most solid electrolytes is achieved by jumping from the lattice position to an adjacent vacancy (oxygen vacancy) due to the activation energy.202 Therefore, after replacing Ti4+ with Ga3+, the oxygen generated by compensation doping can support ion conduction together with the vacancies produced in the course of the reduction process. The most likely reason for the occurrence of electron conduction is that electrons jump from B(n−1)+ to Bn+ through oxygen bridges, where the higher the carrier (e−) concentration  and mobility μe−, the higher the conductivity σe−, as follows:
and mobility μe−, the higher the conductivity σe−, as follows:
 | (20) |
The experimental results showed that both oxygen vacancy δ and conductivity σ increased significantly with an increase in the Ga doping amount. It has been reported that the electronic conductivity of LSGM under reduction conditions is very small, and thus the reduction of Ga has no positive contribution to conductivity. The increase in conductivity is mainly affected by the increase in the concentration of oxygen vacancies, and the change in conductivity is largely determined by the concentration of oxygen vacancies. Fig. 14(b) and (c) show that the effect of the doping number of δ and σ has a similar trend. Under the condition of 1000 °C and 5% H2/Ar, the δ of the doped sample increased from 0.035 to 0.060. With the Ga doping amount gradually increasing, the conductivity increased initially, and then decreased, but the overall value is larger than that of undoped Ga samples. When the doping amount x = 0.05, the conductivity reached the maximum value of 50 S cm−1. According to formula (19), it can be seen that δ is proportional to the concentration of free electrons, while formula (20) shows that σ is proportional to the concentration of electrons, indicating that σ is proportional to δ, that is, under certain conditions, the conductivity will also increase with an increase in the concentration of oxygen vacancies. The number of oxygen vacancies increases with an increase in the doping amount x, but the conductivity increases first, and then decreases with an increase in x, showing that the increase of oxygen vacancies is not beneficial to redox activity all the time. In addition, for systems with α = 0.2 in the A-site stoichiometry (1 − α), when the value of δ is less than or approximately equal to 0.05, no or only a little exsolution was produced, as shown in Fig. 10(a). Given that δ depends on p(O2), temperature, and doped elements, adjusting these factors can almost control the B-site cations to exsolve from any A-site defect system after reduction.
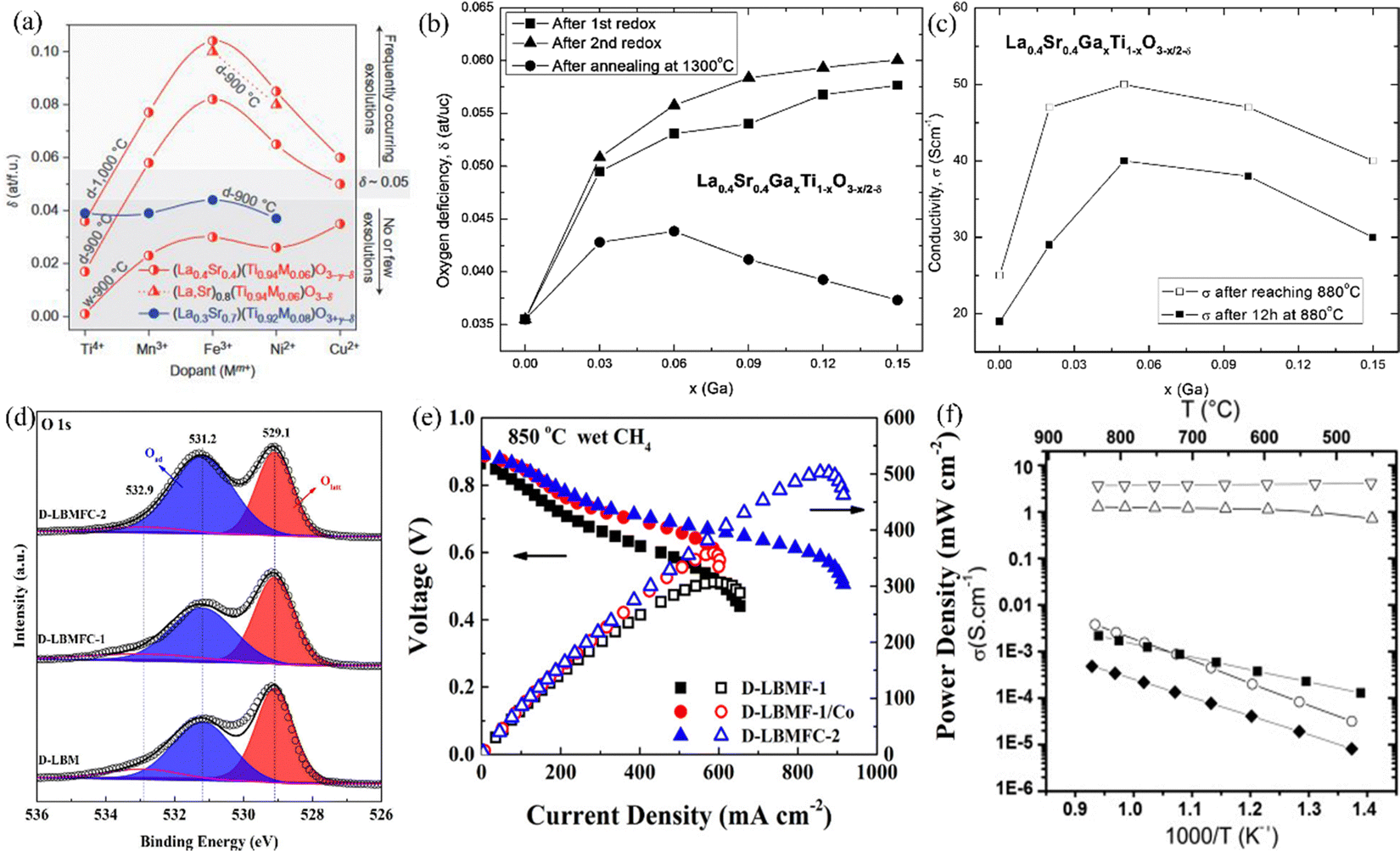 | ||
| Fig. 10 (a) Reduction degree δ (represented by oxygen atoms per formula unit of perovskite, at/f.u.) as a function of the dopant under different situations, indicating in the exsolution region with different temperatures, the degree of exsolution during reduction in a dry or wetted (d or w) reducing gas of 3% H2O or 5% H2O/Ar. (Reprinted from ref. 172 with permission from Macmillan Publishers Ltd.) (b) In different cases, the relationship between oxygen vacancy and Ga doping was measured by the increase in weight during oxidation after the first reduction (■), after the second reduction (▲), after annealing at 1300 °C (●). The sample was pellets with 60–65% density. The reduction step was in a flow of 5% H2/Ar at 1000 °C for 20 h. (c) Electrical conductivity (σ) as a function of Ga content (x) in 60–65% La0.4Sr0.4GaxTi1−xO3−x/2−δ pellets was measured and decreased in 5% H2/Ar at 1000 °C (20 h). (□) represents the conductivity of the sample after reaching the measurement temperature of 880 °C, and (■) represents the conductivity after 12 h at this temperature. (Reprinted from ref. 169 with permission from the American Chemical Society.) (d) High-resolution XPS spectra of O1s on the anode surfaces of D-LBM, D-LBMFC-1, and D-LBMFC-2, where O1s can be divided into three types: lattice oxygen Olatt (529.1 eV), adsorbed oxygen load Oad (531.2 eV) and adsorbed water (532.9 eV). (e) I–V–P curves of wet CH4-loaded single cells with different anodes. (Reprinted from ref. 192 with permission from the American Chemical Society.) (f) Relationship between conductivity and reciprocal temperature of La0.5Sr0.5Ti0.75Ni0.25O3: samples prepared in (■) air and in (◆) Ar/H2 (98/2); after 2 h reduction in Ar/H2(98/2) at 1200 °C. (△) was reduced at 1200 °C for 2 h, and then oxidized at 800 °C for 5 h in air. (○) was reduced at 800 °C for 48 h, and then tested in wet Ar/H2(98/2) (pH2O = 0.025 atm). (Reprinted from ref. 203 with permission from Elsevier Ltd.) | ||
La0.5Ba0.5MnO3−δ with co-doped Co–Fe was synthesized by Hou et al. using the Pechini method, and after reduction, the core–shell nanoparticles consisted of Co0.94Fe0.06 alloy exsolved from the surface, which significantly improved the catalytic performance of the anode. These researchers proposed that when the reduction temperature is lower, the oxygen is more unstable, resulting in the stronger redox ability of the sample and easier formation of oxygen vacancies. Surface oxygen is mainly adsorbed on the oxygen vacancies and easily desorbed at high temperatures. The XPS results of O1s (Fig. 10(d) and Table 3) showed that the content of lattice oxygen in the sample decreased, and the content of adsorbed oxygen increased, with the gradually increase in Fe and Co in the sample that had been reduced. The high content of adsorbed oxygen indicates that there is a large number of oxygen vacancies in the material, which could increase the high oxygen ionic conductivity. These researchers also tested the performance of the cells loaded with La0.5Ba0.5MnO3−δ (LBM), La0.5Ba0.5Mn0.9Fe0.05Co0.05O3−δ (LBMFC-1) and La0.5Ba0.5Mn0.8Fe0.1Co0.1O3−δ (LBMFC-2) at 850 °C using wet methane as the fuel. The MPD of the cell with the LBMFC-2 anode was 503 mW cm−2, as shown in Fig. 10(e). The addition of Co and Fe introduced more oxygen vacancies for perovskite, accelerating the anodic kinetics for electrocatalytic reactions.192 Arrivé et al. used La0.5Sr0.5Ti0.75Ni0.25O3 titanate as the anode of an SOFC, which could make Ni nanoparticles in situ exsolve after reduction. The conductivity of the sample tested under the SOFC anode conditions after reduction at 800 °C for 48 h (symbol ○ in Fig. 10(f)) was nearly an order of magnitude higher than that of the unreduced sample prepared under the same conditions (symbol ◆ in Fig. 10(f)). This is because the pre-reduced sample at 800 °C may have a higher oxygen vacancy concentration than the unreduced sample, resulting in higher ionic conductivity.203
| Anode | Lattice oxygen (%) | Adsorbed oxygen (%) | Adsorbed H2O (%) |
|---|---|---|---|
| D-LBM | 49.31 | 42.81 | 7.88 |
| D-LBMFC-1 | 47.32 | 44.46 | 8.22 |
| D-LBMFC-2 | 37.01 | 58.60 | 4.39 |
For most anodes loaded with Ni nanoparticles, it is still an inevitable problem that the Ni metal particles will catalyze the formation of carbon in a hydrocarbon-containing environment for a long time, but the well-distributed of metal particles on the catalyst can reduce the formation of coking, and therefore the perovskite oxide anode prepared by in situ exsolution is considered to have a better anti-coking performance.93 Oh et al. loaded Ni nanoparticles on La0.4Sr0.4TiO3 by in situ exsolution, nickel nitrate aqueous solution impregnation, and vapor deposition, respectively. Characterization methods such as atomic force microscopy were used to study the exsolution state of the Ni nanoparticles, and the anti-coking performance of the three samples was compared. It was revealed that the in situ exsolution metal particles embedded on the surface of perovskite were the main reason for the in situ exsolved metal particles having a better anti-coking performance.190Fig. 11(a) shows a TEM image of the exsolution particles at the (110) end. It can be seen that 30% of the Ni nanoparticles was submerged below the perovskite surface, and a clear comparison with the Ni loaded by impregnation and exsolution is shown in Fig. 11(c). The researchers etched the sample with concentrated HNO3, and it can be observed from Fig. 11(d) and (e) that the pits left were very similar to the density and degree of size distribution of the particles before, confirming that the nanoparticles were embedded in the perovskite surface. Fig. 11(f) shows the AFM image of the etched surface, indicating that they were indeed embedded deep in the perovskite. In contrast, the nanoparticles embedded in perovskites by traditional deposition methods were almost invisible on similar perovskites with A-site defects, indicating that the interaction between the deposited metal and the perovskite phase was much lower.
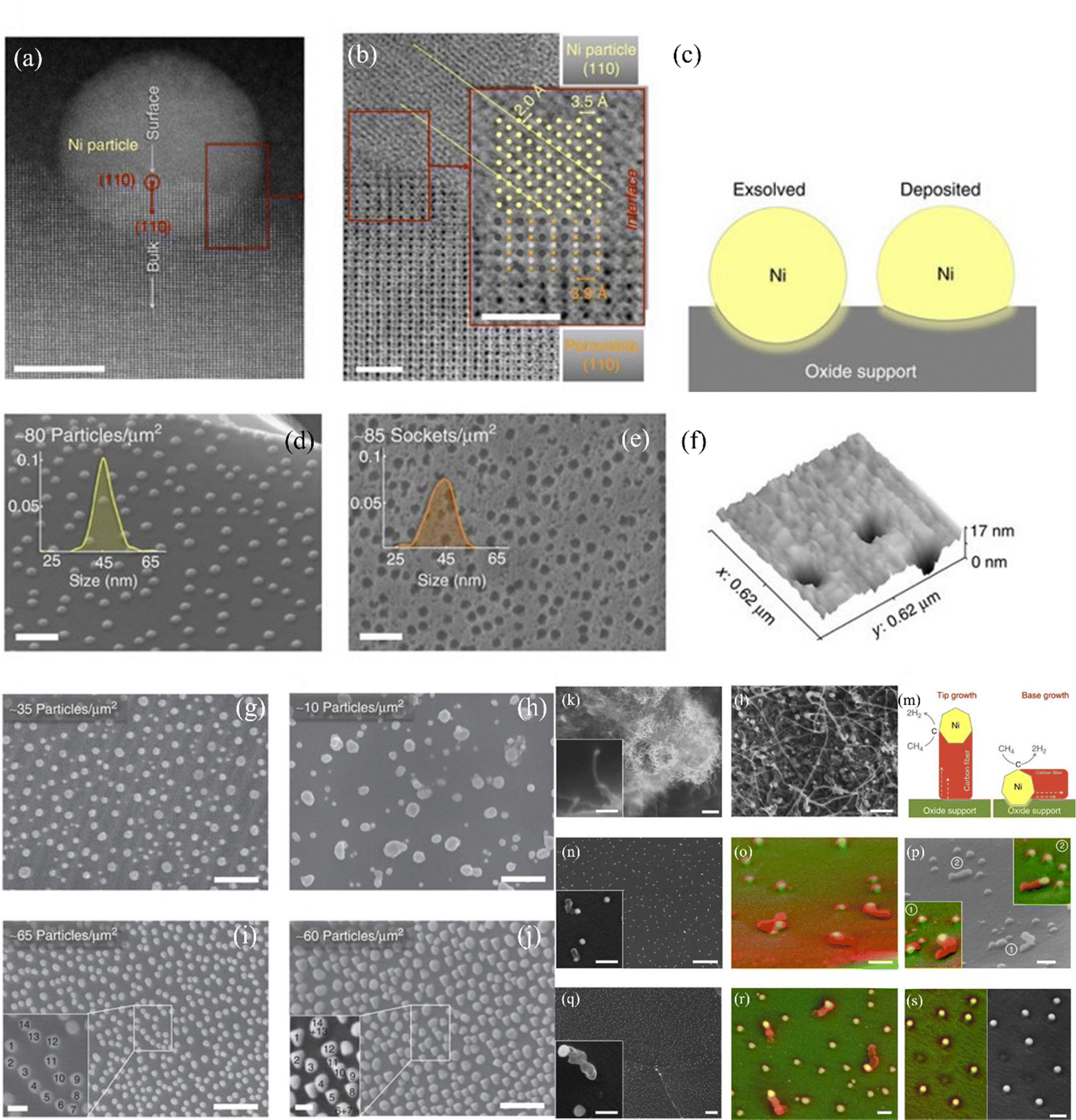 | ||
| Fig. 11 (a) TEM image of aged nickel particles dissolved on (110) as-grown surface (dark field) (3% H2O/5% H2/Ar, 930 °C, 60 h). (b) TEM image of the metal-perovskite interface (bright field). (c) Schematic diagram of the particle-substrate interface of deposited and dissolved nickel particles. (d) SEM image of particles before etching. (e) SEM image the particles after etching in HNO3, illustrated by histograms of particle and pore sizes determined by image analysis. (f) Three-dimensional atomic force microscopy image similar to a hole in (e). SEM images of vapor-deposition nickel particles from La0.4Sr0.4TiO3 (g) before aging and (h) after aging (H2, 650 °C, 24 h and 800 °C, 6 h, respectively). SEM images of Ni particles precipitated from La0.52Sr0.28Ni0.06Ti0.94O3 (5% H2/Ar, 900 °C, 12 h) before aging (i) and after aging (j) (5% H2/Ar, 900 °C, 70 h). (k) After coking test, Ni particles of about 20 nm were formed on La0.4Sr0.4TiO3, and the growth of carbon fibers was obvious. (l) Growth of considerable carbon fibers from Ni particles (30–100 nm) grown on La0.4Sr0.4TiO3 by vapor deposition. (m) Diagram of the probable carbon fiber growth mechanism based on the literature. (n) After coking test, exsolution of La0.52Sr0.28Ni0.06Ti0.94O3 (5% H2, 880 °C, 6 h) to form Ni particles of about 25 nm and only limited carbon fibers grew. (o) Pseudo-color micrographs depicting the side details of the sample in (n). (p) Side-view micrographs and pseudo-color micrograph detail illustrations of different areas in material (n). (q) After coking, about 60 nm Ni particles were formed on La0.52Sr0.28Ni0.06Ti0.94O3 (5% H2/Ar, 1000 °C, 6 h) by exsolution and only limited coking occurred. (r) Sample and (q) pseudo-color top-view micrograph. (s) Pseudo-color photographs of different parts of g and matching secondary electron micrographs show the empty pits and the particles next to them. The coking test was performed at 800 °C in 20% CH4/H2 for 4 h. In the pseudo-color micrograph, green represents perovskite, red represents carbon, and yellow represents nickel metal.190 (Reprinted from ref. 190 with permission from Macmillan Publishers Ltd.) | ||
Compared with Ni particles just deposited on the perovskite surface, the in situ exsolution Ni particles showed a relatively low tendency of agglomeration and coking. Fig. 11(g)–(j) compare the thermal stability of the deposited particles with the exsolution particles with the same size. The former rapidly agglomerated at 800 °C or lower temperatures, as shown in Fig. 11(g) and (h), while the latter remained stable at 900 °C. The number of Ni particles loaded on the in situ exsolution sample was about twice that of the deposited sample, as shown in Fig. 11(j). To test the anti-coking stability, the samples were placed in 20% CH4/H2 at 800 °C for about 4 h. The Ni particles (20 nm) prepared by the impregnation method severely coked and produced carbon fibers, as shown in Fig. 11(k). The Ni particles with a size of 30–100 nm prepared by vapor deposition also produced a large number of micron-sized carbon fibers, as shown in Fig. 11(l). As shown in Fig. 11(n) and (q), the in situ exsolution Ni metal particles of the same size had very little carbon fiber growth because the robust mutual effect between the nanoparticles and the perovskite support prevented carbon fiber growth and particle removal. The growth mechanism of the carbon fibers in the case of in situ exsolution can be explained as the Ni particles adhered to the substrate, and the carbon fibers could only grow on the substrate next to the Ni particles simultaneously, as shown in Fig. 11(m), but the particles occasionally lifted, as showed in region 1 in Fig. 11(p).
Managutti et al. prepared a perovskite in situ exsolution system with Ni-doped (Pr0.5Ba0.5)1−x/2Mn1−x/2Nix/2O3−δ (S-PBMNx) as an SOFC anode, where x = 0, 0.05, 0.1 and 0.2. The CH4 conversions of PBMN0 and PBMN0.05 (R-PBMN0 and R-PBMN0.05) were less than 1% after reduction and in situ exsolution of Ni nanoparticles, while R-PBMN0.2 with the highest Ni stoichiometry had the best performance in DRM, and the CH4 and CO2 conversions were 11% and 32%, respectively. Moreover, R-PBMN0.2 had almost no carbon deposition (just 0.017 g gcat−1 h−1) after continuous operation for 5 h at 800 °C. Researchers have also found that bimetallic alloy exsolution anodes of SOFCs may have better performance than single metal exsolution anodes of SOFCs. Iron has been widely studied as a metal promoter because of its ability to effectively enhance the catalytic performance.204 Sun et al. prepared A-site-deficient LaSrCrO3 (A-LSC) as an anode material doped with Ni, Fe, and Ni-Fe, respectively. By introducing A-site defects under a reducing atmosphere, the in situ exsolution method promoted the uniform distribution of nano-Ni, Fe, and Ni–Fe alloys. The Ni–Fe composites obviously changed the reduction mode of Ni and Fe oxides, and adding Fe changed the reduction mode of the material. Fe and Ni may exsolve on (La0.7Sr0.3)(Cr0.85Ni0.12Fe0.03)O3−x (LSCNi–Fe) firstly, and then form alloys. The presence of the alloy promotes the generation of oxygen vacancies, which is conducive to the exsolution of Ni and Fe. The original reduction behavior of Ni includes two stages, i.e., Ni3+ reduction to Ni2+, and then Ni2+ reduction to metal Ni. The reduction behavior of Fe includes three stages, i.e., Fe3+ reduction to Fe2+, Fe2+ reduction to metallic Fe, and Fe3+ reduction to metallic Fe. The performance tests of the cell showed that compared with the cell with the single metal anode catalyst, that with the bimetallic anode catalyst of nano-Ni–Fe alloy with high catalytic activity exhibited a significantly improved electrochemical performance in 5000 ppm H2S/syngas and the cell was tolerant to carbon deposition, with an MPD of 560 mW cm−2. This was higher than the 450 mW cm−2 of (La0.7Sr0.3)(Cr0.85Ni0.15)O3−x and 360 mW cm−2 of (La0.7Sr0.3)(Cr0.85Fe0.15)O3−x, which were loaded with Ni and Fe, respectively. Based on DFT calculations, the researchers claimed that Ni–Fe alloys can clear away carbon deposition on the electrocatalyst surface, unlike the individual metal Ni, which promotes the formation of C–C bonds, and this conclusion is consistent with the experimental results, indicating that the addition of Fe significantly promotes the removal of carbon deposition.205
Ma et al. studied Ni-doped La0.6Sr0.4FeO3−δ (LSFN), which could exsolve Ni–Fe alloy nanoparticles, as an anode for hydrocarbon-fueled SOFCs. Fig. 12(a)–(c) show the high-resolution TEM images of the LSFN powder after calcination and reduction for 2 h under a humid H2 atmosphere at 750 °C, where it can be seen that small nanoparticles with an average diameter of about 20 nm were uniformly dispersed on the sample surface. Also, the particles in Fig. 12(b) have clear lattice fringes, proving that the nanoparticles were single crystals. As shown in Fig. 12(d) and (e), the LSFN symmetric cell operated in wet C3H8 and CH4 at 750 °C, with an open circuit voltage of 1.18 and 1.0 V and MPDs of 400 and 230 mW cm−2, but the MPDs of the LSF symmetric cell were just 80 and 240 mW cm−2, respectively. Fig. 12(f) shows that LSFN had good long-term stability for hydrocarbon fuel oxidation compared with LSF. After more than 36 runs, only a slight power reduction and a small amplitude fluctuation were observed. It showed excellent stability in wet C3H8 fuel at 750 °C, while the output current of LSF showed a large fluctuation trend.206 Tang et al. prepared an anode material, i.e., (La0.4Sr0.4)(Ti0.85Ru0.07Ni0.08)O3−δ (L0.4STRN), with good catalytic performance and stability by doping Ru in La0.4Sr0.4Ti0.85Ni0.15O3−δ (L0.4STN). It was found that the doped Ru could avoid the agglomeration of the exsolution Ni nanoparticles, generating a large number of defects in the perovskite, and thus increasing the amount of oxygen vacancies. The researchers also prepared single cells loaded with L0.4STN and L0.4STRN (R-L0.4STN and R-L0.4STRN) after reduction. The MPD of the R-L0.4STN|SDC|LSGM|LSCF cell was 445 mW cm−2, using wet CH4 as the fuel at 800 °C, which was better than that of the R-L0.4STN cell of 202 mW cm−2. In the stability test, the cell with R-L0.4STRN retained a voltage of about 0.78 V, while that of the R-L0.4STN cell decreased significantly at a rate of 0.379% h−1. The cell with R-L0.4STRN exhibited excellent carbon deposition resistance, which may be due to the synergistic effect of the well-distributed Ni nanoparticles and doped Ru.207
 | ||
| Fig. 12 (a) Low-magnification and (b and c) high-magnification TEM images of La0.6Sr0.4Fe0.9Ni0.1O3−δ powder after reduction. (Inset: Fast Fourier transform pattern of selected region.) (d and e) I–V–P curves of cells with different anodes: LSFN (d) and LSF (e). (f) Current density of the cell with different anodes tested at a constant voltage of 0.7 V in different electrodes and humidified C3H8 as a function of time. (Reprinted from ref. 206 with permission from Elsevier Ltd.) | ||
Although many studies have constructed heterostructures via exsolution to improve the catalytic ability of perovskites, most of the exsolution species are limited to reducible metal cations, such as nickel, cobalt, iron and noble metals. He et al. reported an improved exsolution method for the co-exsolution of active oxides and metal nanoparticles from a perovskite material, i.e., La0.8Ce0.1Ni0.4Ti0.6O3−δ (LCeNT), and constructed a metal cerium titanate heterostructure, CeO2–Ni@LCeNT, with stable activity to enhance the catalytic performance. After the air annealing treatment of LCeNT, CeO2 cubes grew on the support, and its morphology could be adjusted by changing the annealing temperature, while the nickel nanoparticles continued to exsolve during the subsequent reduction process. The MPD of the cell with the CeO2-Ni@LCeNT anode reached 642 mW cm−2 at 900 °C in CH4 (3% H2O). When the stability test was carried out in wet CH4 at 850 °C to evaluate its carbon deposition resistance, it was found that the current density of the cell decreased slightly before the conversion of CH4 reached equilibrium after running in CH4 (3% H2O) for about 1 h. In the subsequent 19 h running time, the current density stability did not deteriorate significantly. When the working time was delayed to 105 h with the same cell, the current density of the cell was maintained at about 1 A cm−2, which proved that the CeO2–Ni@LCeNT cell had outstanding stability. The SEM and STEM tests showed that there was no fibrous carbon on the anode, confirming that CeO2–Ni@LCeNT has an excellent anti-coking performance. The in situ exsolution of CeO2 and Ni nanoparticles embedded in the LCeNT substrate to form a strong particle–substrate interaction and a strong structure is the main reason for the excellent anti-coking performance.173Table 4 presents a summary of some typical in situ exsolution perovskite materials and their electrochemical performances.
| No. | Anode | Temperature (°C) | Fuel | MPD (mW cm−2) | Long term (h) | Ref. |
|---|---|---|---|---|---|---|
| 1 | (La0.7Sr0.3)(Cr0.85Ni0.12Fe0.03)O3−x | 850 | 5000 ppm H2S-syngas | 400 | — | 205 |
| 2 | Ni-doped La0.6Sr0.4FeO3−δ | 750 | C3H8 | 400 | 35+ | 206 |
| 3 | Ni-doped La0.6Sr0.4FeO3−δ | 750 | CH4 | 230 | — | 206 |
| 4 | La0.8Sr0.2FeO3 | 750 | C3H8 | 240 | 30+ | 206 |
| 5 | La0.8Sr0.2FeO3 | 750 | CH4 | 80 | — | 206 |
| 6 | (La0.4Sr0.4)(Ti0.85Ru0.07Ni0.08)O3−δ | 650 | CH4 | ∼100 | 25+ | 207 |
| 7 | (La0.4Sr0.4)(Ti0.85Ru0.15)O3−δ | 800 | CH4 | 202 | — | 207 |
| 8 | La0.8Ce0.1Ni0.4Ti0.6O3−δ | 900 | CH4 | 306 | — | 173 |
| 9 | Pr0.5Ba0.5MnOx | 900 | Syngas | 900 | — | 200 |
| 10 | La0.5Ba0.5Mn0.8Fe0.1Co0.1O3−δ | 850 | CH4 | 503 | 200+ | 192 |
| 11 | La0.5Ba0.5MnO3−δ | 850 | CH4 | 226 | — | 192 |
In summary, we discussed the in-depth mechanism of in situ exsolution of perovskite materials and their application advantages in SOFC anodes. Due to the higher surface free energy of perovskites and the interaction with strain energy, transition metal or noble metal doped at the B-site of perovskite can be exsolved in situ out of the lattice as active nanoparticles in a reducing atmosphere. Although the impregnation method is the traditional and effective method for constructing metal nanoparticles on the surface of the anode, it requires multiple cycle processes, and the amount of metal loading is restricted by the volume of open pores in the anode. Moreover, the metal nanoparticles formed by the impregnation technique are unstable at high temperatures because they are in an unrestricted environment. In contrast, the metal nanoparticles formed by in situ exsolution are more uniformly distributed and smaller than that formed by the impregnation method, and the precipitated metal particles are semi-embedded in the perovskite matrix, resulting in strong particle–substrate interactions. Hence, the in situ exsolved nanoparticles are not easy to agglomerate at high temperatures. Moreover, for hydrocarbon SOFCs, the Ni particles on the anode prepared by the impregnation method are severely coked and produce a large number of carbon fibers, and even if Ni particles with a particle size of 30–100 nm are prepared by the vapor deposition technique, they also produce a large number of micron-sized carbon fibers, while the Ni metal particles of the same size exsolved in situ have only a small amount of carbon fiber. This is attributed to the powerful interaction between the exsolved nanoparticles and the perovskite matrix, which prevents the growth of carbon fibers and metal nanoparticle lifting.190
The early research about the stoichiometric ratio of perovskite oxides mainly focused on A/B = 1, where the exsolution of the B-site cations relied on the inherent reduction driving force. In contrast, A-site-deficient perovskites (i.e., perovskite oxides with stoichiometric ratio of A/B < 1) increase the tendency of the B-site cations to exsolve from the lattice with a wider distribution and better coverage on the surface of perovskites, given that the exsolution restores the perovskite oxides to a stable structure locally, making the stoichiometry of the perovskite conform to ABO3. In addition, A-site-deficient perovskites contribute to the formation of highly mobile oxygen vacancies. There are many intrinsic and extrinsic factors that affect the nucleation and growth of exsolved nanoparticles. The intrinsic factors include surface properties (surface and interfacial energy of nucleated particles), local strain fields in the perovskite matrix, A-site defects and concentration of oxygen vacancies. External factors include processing time and temperature. Thus, by exploring the above-mentioned influencing factors, we can further understand the mechanism of in situ exsolution of perovskite to achieve more artificial regulation to improve the electrochemical performance of the exsolution system.
The development of in situ exsolution is restricted by the fact that most exsolved species are limited to reducible metals such as Ni, Co, Fe and precious metals, limiting the selection of active species. Thus, increasing the category of species from in situ exsolution is the focus of future research. Also, many researchers have focused on the application of SOFC anodes with in situ exsolution for H2 fuel instead of hydrocarbon fuels, but the anti-carbon deposition advantages of the mosaic structure constructed by in situ exsolution cannot be ignored, and thus more studies should be carried out on hydrocarbon fuels in the future. Furthermore, most of the operating temperatures of cells are mainly at 750 °C or even higher, and the construction of in situ exsolution systems at a lower temperature remains to be solved.
Bimetallic alloys may have better properties than single metals for in situ exsolution metal component systems, and thus the study of bimetallic-cermet may provide more guidance for the in situ exsolution of perovskite materials. In addition, among the various matrix materials, the cell with a Pr0.5Ba0.5MnOx anode exhibited an outstanding performance, which may deserve more attention and exploration. We believe that the excellent heterojunction structure will greatly improve the catalytic performance and stability of hydrocarbon SOFCs.
2.3 Anode reforming layer materials
In addition to directly changing the composition of the fuel cell anode, considering the cost, simplified system, anti-coking, and selectivity of materials, the assembly of a catalytic reforming layer with high catalytic ability for hydrocarbon fuels on the cell maybe an effective way to improve the anti-carbon deposition stability of direct hydrocarbon fuel SOFCs.Noble metals have good catalytic performance for the reforming reaction. Wang et al. proved that a 3–7 wt% Ru–Al2O3 composite had great catalytic ability for the partial oxidation of methane and CO2/H2O reforming reaction. H2-TPR and TEM results showed that there was a robust interaction between RuOx and Al2O3 in the catalysts, which may be why the 3 wt% Ru–Al2O3 catalyst exhibited good catalytic stability. In addition, the carbon deposited on Ru–Al2O3 was less than that deposited on Ni–Al2O3, indicating that it is easier to eliminate the potential carbon deposited on Ru–Al2O3 catalyst.208
By adding an Ru–CeO2 catalyst layer to Ni–YSZ, Zhan et al. compared the cell performance test results with the test results of the conventional Ni–YSZ anode-supported fuel cell using a propane-air mixture. Fig. 13(a) shows the temperature dependence of propane conversion in the cell operated in 10.7% C3H8-air fuel with an Ru–CeO2 catalyst layer on the YSZ anode. The propane conversion increased from 20% at 450 °C to 80% at 500 °C, and Ru–CeO2 could catalyze the C3H8 partial oxidation at T > 500 °C. The conversion of propane on the Ni–YSZ anode reached 80% at 650 °C, which is 150 °C higher than that of the Ru–CeO2 catalyst. This indicates that the Ru–CeO2 layer is a more effective low-temperature catalyst than Ni–YSZ for propane partial oxidation.209 Generally, it is believed that the electrochemical reaction in SOFCs usually occurs in the TPB. Thus, to promote gas diffusion, prolong the length of the TPB and form a continuous ion electron conduction path, Sun et al. prepared a fuel cell with a flower-like mesoporous cerium oxide microsphere/ruthenium nanoparticle catalyst layer by loading Ru on CeO2 using the impregnation-reduction method. As shown in Fig. 13(b) and (c), most of the CeO2 particles in the flower-like mesoporous CeO2–Ru microspheres were monodispersed spherical particles with a hollow structure. It can also be seen in the high-magnification SEM and TEM image in Fig. 13(d) and (e), respectively, that many petal-shaped nanosheets formed an open porous flower-like structure, and the microspheres were hollow structures, which facilitated the diffusion of the fuel. At 600 °C, the MPD of the cell was 654 mW cm−2, which was much higher than the of the cell without a catalyst layer of 81 mW cm−2, as shown in Fig. 13(f).210 Similarly, Sun et al. synthesized a Pd layer consisting of 3D nanoflowers with a diameter of 1.0–2.0 μm as an anodic catalytic reforming layer on an Ni–YSZ anode by galvanic replacement reaction. The MPDs of the Pd–Ni–YSZ single cell with catalytic reforming layer and single cell with just the Ni–YSZ anode were 196 and 153 mW cm−2, respectively, when ethanol was used as the fuel, indicating that the Pd catalytic reforming layer increased the MPD by 43 mW cm−2. Furthermore, the voltage of the cell with the catalytic reforming layer decreased significantly within 59 h, while the cell loaded with the Ni–YSZ anode had a sharp drop in voltage after 6 h, which was because the Pd nanoparticles could act as a barrier layer to prevent diffusion, and thus significantly suppress carbon deposition on the nickel-based anode surface.211
 | ||
| Fig. 13 (a) Relationship between propane conversion and temperature of Ni–YSZ anode-supported Ru–CeO2 catalyst layer with 10.7% C3H8–18.7% O2–70.6% Ar as the fuel. Single-cell Ru–CeO2|PSZ|Ni–YSZ|YSZ|LSCF-GDC, LSCF. (Reprinted from ref. 209 with permission from Elsevier Ltd.) (b and d) SEM images and (c and e) TEM images of the flower-like mesoporous CeO2 microspheres. (f) Voltage and power density versus current density for the anode-supported SOFCs with and without catalyst layers, tested in 5% iso-octan–9% air–3% H2O–83% CO2 at 100 mL min−1 in the anode and ambient air in the cathode at 600 and °C. (Reprinted from ref. 210 with permission from Elsevier Ltd.) | ||
Wang et al. prepared mesoporous cerium oxide Ce0.8Sm0.22O1.9 (SDC) with a high specific surface area as the catalytic layer of CH4-fueled SOFCs. After calcination at 700 °C for 3 h, the SDC powder still had a mesoporous structure with a high surface area of 77 m2 g−1 and a large pore volume of 0.2276 cm3g−1, which is conducive to free gas diffusion. After impregnation of the mesoporous SDC catalyst layer with Ru, the supported cell exhibited an excellent MPD of about 462 mW cm−2 at 650 °C.212 Zhao et al. applied a reforming layer composed of Ni loaded on La-doped ceria such as La2Ce2O7 (LDC) and La1.95Sm0.05Ce2O7 (LSDC) on an Ni–Ce0.8Sm0.2O2−x anode. In wet CH4 at 650 °C, the MPD of the conventional cells was only 580 ± 20 mW cm−2, but after the addition of the Ni-LDC and LSDC layers, the MPD of both increased to 699 ± 20 mW cm−2 and 639 ± 20 mW cm−2, respectively. In the stability test at 650 °C and 0.2 A cm−2 in wet methane, the voltage of the conventional cell decreased significantly within 10 h, while the cell with any one of the catalyst layers was stable for 26 h.213
When used in monometallic catalysts, the catalytic activity of metals such as iron, cobalt, and copper is relatively low, but the addition of other metals to form new substances or bimetallic catalysts may result in catalysts with good performance, and among them, cobalt is the most appropriate metal for changing nickel-based catalysts because the electronic interaction between nickel and cobalt contribute to improving the dispersion of metal alloys and carbon deposition resistance.214
Due to the low reforming performance of Cu-based catalysts, a second phase catalyst needs to be added when hydrocarbons are fed. Therefore, Jin et al. synthesized spinel oxide Cu1.3Mn1.7O4 as an anode inner reforming layer for an Ni–SDC anode-supported SOFC for direct operation in methane, and after the in situ reduction of CH4, well dispersed nano-Cu metal mesh was obtained in the MnO matrix. The MPDs of the cells reached 242 and 311 mW cm−2 at 650 °C and 700 °C, respectively. The power output of the cell increased by about 30% compared with the cell without an internal reforming layer under the same test conditions. For the cells with a Cu1.3Mn1.7O4 internal reforming layer, the cell voltage became more stable for 40 h. In contrast, the cell without a Cu1.3Mn1.7O4 catalyst layer rapidly failed after working for 16 h. In the case of the CuB2O4 (B = Fe, Mn, Cr, Ga, Al, etc.) spinel oxides after reduction, the stability of the spinel oxides, metal oxides and the strong interaction between the metal oxides and Cu contributed to the outstanding catalyst durability.215
Because RuxCo1−x supported on a CoO catalyst has high activity for dissociating H2O/CO2 and Ni has catalytic activity for CH4, Chen et al. dispersed Ru and Ni on CeO2 to form a catalyst. It is expected that Ru and Ni can catalyze H2O and CH4, respectively, and thus the coupling of the two dissociated substances can play a role in the synergistic activation of H2O and CH4. Based on this, these researchers prepared a Ce0.90Ni0.05Ru0.05O2 (CNR) reforming catalyst supported on a BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb) anode. The single cell composed of the of PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) cathode and SDC electrolyte ran directly on nearly dry (containing only 3.5% H2O) methane at 500 °C, and its MPD was 370 mW cm−2. This is obviously higher than the 142 mW cm−2 reported as the highest value for a proton ceramic fuel cell fueled with steam-rich CH4 and 78% higher than the fuel cell without catalyst layer, and the cell showed no signs of coking after about 550 h continuous operation.216 However, catalyst materials doped with noble metals such as Ru,217 Pd, and Pt218 are limited by their high cost, and thus cannot be applied widely.
Therefore, researchers began to avoid using noble metals, such as preparing nanostructured materials with other metals or adding non-noble metal elements in the anode catalytic reforming layer. Kim et al. innovatively coated a cerium oxide layer modified with metal nano-catalysts by cathodic electrochemical deposition (CELD). The first CELD was performed in 0.05 M Ce + Sm nitrate solution with 5% Sm content to obtain the SDC layer. A cathodic potential of 0.65 V per saturated calomel electrode (SCE) was applied to the nickel electrode to reduce the NO3 anions or dissolved O2 to produce OH−, with solid (Ce, Sm)(OH)3 on the electrode surface. The low deposition rate of 0.65 V per SCE could produce a more nanostructured cerium oxide morphology, ensuring that the catalytic base had the best electrocatalytic activity. Through drying or heat treatment in air, these deposits were oxidized to obtain an oxide coating, which became a petal-like SDC nanostructure composed of high-density, randomly oriented nanosheets. Ni(OH)2 was obtained by the second CELD in the same way, and finally the deposited layer was in situ transformed into SDC nanostructures and nickel metal particles were dispersed on it in a reduced H2 atmosphere. The catalytic performance of the SDC nanostructures modified by 12 at% Ni nanoparticles was tested by gas chromatography (GC) at 550–700 °C for CH4 steam reforming (3% H2O/CH4). The results were expressed as the ratio of generated H2 concentration to the theoretically predicted H2 concentration. The ratios of the anodes after two-step CELD at two temperatures reached 54.1% and 79.2%, while that of the uncoated samples was 3.5% and 12.4%, respectively. The bare nickel catalyst layer had a higher EA value of 113.25 kJ mol−1, while that of the CELD anode layer was just 35.75 kJ mol−1, which was lower than the EA value of the typical Ni–CeO2 (63–88 kJ mol−1), indicating that the reforming layer significantly promoted CH4 steam reforming. Also, the cell loaded with the reforming layer could operate stably at 650 °C for 90 h.219 Hua et al. introduced Ni–Sn bimetallic nanoparticles in the traditional nickel-based anode to form a porous and flexible nickel foam-loaded Ni–Sn/Al2O3 nanocluster reforming catalytic layer. The MPD of the cell was as high as 0.946 W cm−2 at 850 °C in a CH4–CO2–200 ppm H2S atmosphere. In CH4–CO2 containing 200 ppm H2S, the conversion of CH4 was still as high as 95.6%. The output voltage of the cell with the conventional Ni–YSZ anode without this catalytic reforming layer decreased continuously during the 48 h stability test, while the CH4 conversion and CO selectivity of the cell loaded with this reforming layer remained at about 95% during the 48 h test.220
Zhao et al. deposited Ni–Mo–Ce0.5Zr0.5O2−δ (Ni–Mo–CZ) on the top of a conventional Ni–YSZ anode as an internal catalytic reforming layer. The Ni–Mo–CZ layer effectively increased the gasoline conversion and increased the H2 and CO yields of Ni–YSZ at 750 °C. The gasoline conversion of the Ni–YSZ anode without the Ni–Mo–CZ catalyst was just 40%, and the yields of H2 and CO were 5% and 8%, respectively. When the nanoscale Ni–Mo–CZ catalyst was added, the gasoline conversion rate increased from 40% to 57%, and the H2 and CO yield increased to 17% and 22%, respectively. However, the thicker catalyst layer resulted in higher mechanical stress after sintering, and thus only a limited amount of catalyst (about 20 mg) could be loaded on the traditional button cell, which prevents the improvement in the cell performance. Therefore, the researchers produced a tubular single-cell device to load a larger amount of catalyst (about 80 mg), and the gasoline conversion rate increased to 78%, the CO yield increased to 55%, and the H2 yield reached 40%. In addition, the MPD of the tubular single cell without the Ni–Mo–CZ catalyst was 144 mW cm−2, which increased to 254 mW cm−2 by filling with catalyst. The single cell without the catalyst was damaged after running for 6 h, while the stability of the button cell with the catalyst layer decreased after running for about 10 h. However, even after 15 h operation, the tubular cell with catalyst did not show a significant performance degradation, which was 42% longer than the button cell life. Thus, the above-mentioned test results show that the cell with Ni–Mo–CZ had a good catalytic reforming performance using gasoline as fuel, and for tubular single cell, one of the advantages is that more catalyst can be added to the reforming layer, further improving the cell performance.221Table 5 presents a summary of some typical anode reforming layer materials and their electrochemical performances.
| No. | Catalyst layer | Temperature (°C) | Fuel | MPD (mW cm−2) | Long term (h) | Ref. |
|---|---|---|---|---|---|---|
| 1 | Ru–Al2O3 | 750 | CH4:O2 = 4:1 |
581 | 6+ | 208 |
| 2 | Ru–Al2O3 | 750 | CH4:H2O = 2:1 |
489 | — | 208 |
| 3 | Ru–Al2O3 | 750 | CH4:CO2 = 2:1 |
478 | — | 208 |
| 4 | Ru–CeO2 | 750 | 10.7% C3H8 +18.7% O2 + 70.6% Ar | 480 | — | 209 |
| 5 | Flowerlike CeO2–Ru | 600 | 5% isooctane + 9% air + 3% H2O + 83% CO2 | 654 | — | 210 |
| 6 | 3D flower-like Pd | 750 | Ethanol | 196 | — | 211 |
| 7 | Ru–SDC | 650 | CH4 | 462 | — | 212 |
| 8 | Ni–La2Ce2O7 | 650 | CH4 | 699 | 26+ | 213 |
| 9 | Ni–La1.95Sm0.05Ce2O7 | 650 | CH4 | 639 | 26+ | 213 |
| 10 | Cu1.3Mn1.7O4 | 650 | CH4 | 242 | 60+ | 215 |
| 11 | Ce0.90Ni0.05Ru0.05O2 | 500 | CH4 | 370 | 380+ | 216 |
| 12 | Ni NP–SDC | 650 | CH4 | — | 6+ | 219 |
| 13 | Ni–Sn–Al2O3 | 850 | 200 ppm H2S –CH4 + CO2 | 946 | 48+ | 220 |
| 14 | Ni–Mo–CZ | 750 | Gasoline/air | 254 | 15+ | 221 |
In summary, the addition of a reforming layer with high catalytic activity has been demonstrated to be an effective way to improve the performance and long-term stability of hydrocarbon-based SOFCs. To prepare the reforming layer, in addition to finding a catalyst that can efficiently catalyze the reforming reaction, it is also necessary to optimize the microstructure and solve the gas diffusion problem caused by the reforming layer, such as preparing the catalyst in a porous form, which is an important direction to overcome the shortcomings of the reforming layer. Furthermore, it is noteworthy to match the thermal expansion coefficient of the reforming layer with the anode material, where an inappropriate thermal expansion coefficient will cause the reforming layer to fall off from the cell in the high-temperature operating environment.
3. The kinetic anode models
Considering the possibility of competing or co-existing reforming reactions on SOFC anodes, studying the mechanism of reforming hydrocarbon fuel is the first step in realizing the direct application of hydrocarbon fuel. Therefore, the modeling research based on the first-principles method is conducive to the design of SOFCs suitable for various fuels before the actual experiments, as well as the interpretation of experimental phenomena, guiding the subsequent experiments to a certain extent. In this part, we summarize some of the reaction mechanisms that have been discovered and give an in-depth description of the causes and solutions of anode carbon deposition.Yang et al. calculated the process by DFT that the adsorbed *H2O is converted to *OH, while *OH can react with the *C near the Ni/BaO interface to form *CHO, which is then electrochemically oxidized at the TPB. It was found that water associated with the Ni/BaO interface plays a critical role in increasing the tolerance to coking and inactivation. They introduced Ni, YSZ and BaO powder samples into wet argon (3 vol% H2O), respectively, and found that the weight of the Ni and YSZ powders remained almost unchanged based on the thermogravimetric analysis results, but a large degree of weight alteration was observed in the BaO powder test, indicating the good water absorption of BaO. Similarly, after introducing Ni–BaO samples into wet argon, peaks around 1600 cm−1 were observed in the Raman spectrum, corresponding to the bending mode of water, which also confirmed the water absorption capacity of BaO. Furthermore, the cell with or without the BaO-modified anode was introduced into dry C3H8 fuel gas at a constant current density of 500 mA cm−2 at 750 °C, and it was found that the voltage of the cell without the BaO-modified anode fell to 0 V in 0.5 h, while the cell with the BaO-modified anode was stable at 0.8 V for more than 100 h. This suggested that water-mediated carbon removal reactions were more advantageous at the Ni/BaO interface. Also, it was found that water dissociation occurred at BaO and carbon generation occurred at the Ni site of Ni/BaO boundary. The results showed that the performance and properties of resistance to carbon deposition were largely dependent on the direct involvement of the Ni/BaO interface.222
Based on the principle that noble metal reactions increase the activity of water–gas shift reactions and formaldehyde oxidation at low temperature, Yang et al. chose the relatively stable Ni(111) surface to model and used the VASP software to perform ab initio calculations. Combined with the PAW method and CI-NEB method, the dissociation adsorption of H2O on MgO, and the reaction between surface diffusion and carbon deposition of intermediate materials at the Ni/MgO interface were calculated by simplifying the reaction mechanism. The results of thermogravimetric analysis and temperature-programmed desorption of CO2 measurements indicated that the MgO nanoparticles on the surface of Ni ceramics were beneficial to capture H2O and CO2, significantly increased the number of *OH substances for Ni stably, and thus enhanced the coke resistance. The cell with the MgO-modified anode could operate in CH4 (3 v% H2O) fuel at 800 °C for more than 325 h. Also, this principle can be directly applied to various fields such as direct methanol fuel cells and other catalytic reactions.149
Ahmed et al. used mathematical models of finite size to analyze the temperature, current, voltage and internal distribution of the components in SOFC anodes, considering the three reactions, i.e., water–gas shift, methane steam reforming and reverse methanation (RMTN). Based on the fact that Xu et al. found that the activation energy of RMTN is very close to the activation energy of methane disappearance,223 they believed RMTN should be considered in addition to the water–gas shift reaction when kinetic rate expressions are used to simulate the methane steam reforming reaction because the lack of simulation calculations about RMTN reaction leads to prediction bias for characteristic variables such as current density of SOFC systems. This study found that from the perspective of SOFC degradation, a fast kinetic rate did not necessarily lead to the optimal temperature distribution and current density; on the contrary, the use of an anode with a slightly lower kinetic rate could improve the operational stability.224
The reactive force field molecular dynamics simulation (ReaxFF-MD) model can correctly describe a series of complex physicochemical processes in the TPB of SOFCs, and Duin et al. used ReaxFF-MD to study the transport performance of the protons in Y-doped BaZrO3 and oxygen ions in YSZ, and verified the feasibility of the model in SOFC applications through experiments.225,226 Also, Lu et al. used ReaxFF-MD to clearly display the process of coke formation on the anode and simulated the number of C and O atoms in Ni crystals over time, as shown in Fig. 14, explaining the increase in the amount of methane in the early stages of methanol and methane absorption. The results showed that alkyl groups are more likely to stick to the surfaces during adsorption. Initially, the C in Ni(100) provides the driving force, causing changes in the formation of new carbon cluster phases in the later stage, resulting in Ni(100) damage. Finally, more and more C is absorbed on the surface, leading to the minimization of the reforming process. Thus, doping the surface of nickel with other elements can increase the steric hindrance of the carbon adsorption process.227
Kim et al. used FIB-SEM tomography and finite element analysis to perform three-dimensional reconstruction of reduced and re-oxidized Ni–YSZ anode functional layers, analyzed their morphology and stress distribution, and explained the very common stack fault phenomenon. As showed in Fig. 15, by virtually heating at 500–900 °C to obtain the thermal stress curves, they analyzed the thermal stress distribution of the re-oxidized anodes. According to the simulation results, it can be seen that the thermal stress is concentrated at the NiO–YSZ interface, and the stress at the NiO interface is higher than that at the YSZ interface. This means that there is a high probability of rupture in the NiO region.228
 | ||
| Fig. 14 Process of coke formation on the anode and change in the number of C and O atoms with time in the Ni crystal. (Reprinted from ref. 227 with permission from Elsevier Ltd.) | ||
 | ||
| Fig. 15 Stress distribution diagram under different heating conditions. (Reprinted from ref. 228 with permission from Elsevier Ltd.) | ||
Guo et al. found that an all porous solid oxide fuel cell (AP-SOFC) exhibited excellent resistance to carbon deposition in experimental tests,229,230 and further Xu et al. developed a 2D model of AP-SOFC to study the effects of key parameters such as O/C ratio and electrochemical reaction rate on carbon deposition in SOFCs. They found that the process of diffusion of O2− from the cathode to the anode improved the O/C ratio of the anode, which can effectively inhibit methane coking and carbon deposition. In addition, the data obtained through design experiments showed that in different CH4 molar fraction ratios of fuel gases, the CH4 conversion efficiency of the anode-supported AP-SOFC was above 97%, and the outlet syngas was abundant, thus, AP-SOFCs can co-generate electricity and syngas without carbon deposition. The anode support of AP-SOFC can obtain a greater MPD than the electrolyte support (1607 vs. 146 mW cm−2), and for the same cell, the fuel composition and fuel flow rate affect the O/C ratio of the anode surface, which in turn affects the electrochemical performance.231
Combined with Raman spectroscopy, Bkour et al. found that well-dispersed MoOx substances, especially MoO bonds, on Ni nanoparticles can promote the coke resistance and catalyst stability of Ni–Mo–YSZ catalyst samples. Calculations based on DFT showed that compared with Ni–YSZ catalysts, the Mo-doped Ni surface sites on the YSZ support are thermodynamically feasible and can enhance the carbon tolerance by increasing the activation barriers of C–H bond cleavage and C–C coupling reactions, as show in Fig. 16. In addition, for the projected density of states (PDOS) of the Ni 3d orbitals in Ni(111) with Mo, the d-band center of the Ni atom is located near the Mo atom away from the Fermi level, thus reducing the electron interaction between carbon and nickel. Therefore, less carbon will be adsorbed in the Ni–Mo–YSZ catalyst system.112 The DFT calculation result of the Ni–Mo–YSZ catalyst was similar to the study of the Ni–Sn–YSZ catalyst,116 where the activation energy barrier of C–O was much lower than that of C–C on the surface of the Ni–Sn alloy calculated by DFT, and thus the C atom will be preferentially oxidized rather than form carbon deposits.
 | ||
| Fig. 16 In Ni–Mo/YSZ and Ni/YSZ model systems (A) transition states of C–C coupling paths and (B) their corresponding configurations of initial state (IS), transition state (TS) and final state (FS) in partial representation. The purple, silver, red, brown, light green and dark green spheres represent Mo, Ni, O, C, Zr, and Y atoms, respectively. (Reprinted from ref. 112 with permission from Elsevier Ltd.) | ||
Audasso et al. proposed a two-dimensional modeling tool to simulate the performance of SOFCs under direct internal methane reforming and introduced a coefficient matrix (σ) to represent the distribution of active sites. The same method was also used to determine the optimized catalyst distributions. For the DIR process, the code SIMFC (SIMulation of Fuel Cell) considered two different approaches proposed in the literature, i.e., thermodynamic equilibrium and reaction kinetics. The SOFC crosscurrent configuration supported by a single cell anode was also considered, and (i) how to simulate the effect of reforming catalyst inactivation on SOFC performance and (ii) how local control could be used to detect optimized catalyst distributions to provide minimal thermal stress to the cell. In addition, the σ coefficient matrix was appropriately adjusted to obtain the optimal distribution that is easily achievable in the manufacturing process. Studies found that steep temperature distributions lead to higher mechanical stress, which leads to creep formation and ultimately leads to the degradation of the overall anode.232 This model is reliable for guiding SOFC direct internal reforming studies, and because of the simplicity of the calculation, it can be integrated into more complex simulation software such as Aspen Plus.
Su et al. synthesized H2-reduced Sr2ZnMoO6 (R-SZMO) as an SOFC anode material. Due to the inherent anion-Frenkel defect pairs in the material, it showed good catalytic capacity and coking resistance. According to DFT calculations, from the perspective of local structure, with the help of the electroactive O site on the optimal SZMO(110) surface, the optimal adsorption of key intermediates methyl, methylene and formaldehyde was located near O and Mo sites, showing excellent catalytic capacity for methane. Meanwhile, high methane catalytic capacity triggers high energy output and carbon deposition resistance. When CH4 was used as the fuel gas, compared with other typical double perovskite anode materials (Sr2CoMoO6, Sr2MgMoO6, and La0.75Sr0.25Cr0.5Mn0.5O3), the cell with the Sr2ZnMoO6 anode exhibited the highest output power and could operate stably in CH4 for 110 h without significant degradation.233
Overall, molecular dynamics, finite element simulation, supplementary mathematical models and other simulation methods are comprehensively used to simulate and calculate the causes and solutions of anode carbon deposits in SOFCs. They lay a good theoretical foundation for further understanding the anode reaction kinetics and the design of carbon-resistant anodes. In the future, the reaction mechanism and reaction kinetics of hydrocarbon fuels on carbon-resistant anodes should be studied in depth, and the theoretical calculation or simulation model can be verified by corresponding experiments under certain conditions.
4. Future anode research direction
4.1 Multilayer anode
It should be noted that the electrochemical reactions in high-performance electrodes mainly occur in the range of about 10 μm from the electrolyte, and consequently the anode can be divided into two layers, i.e., a thinner functional layer for electrocatalysis and a thicker conductive layer for current harvesting. Consequently, by considering the desired characteristics of each layer separately, the catalytic performance of the anode can be maximized.61Ye et al. successfully prepared a dual-layer structure containing a Cu–CeO2–YSZ support anode and Ni–ScSZ functional anode by acid leaching of nickel and wet impregnation method. In the case of ethanol fuels, each ethanol molecule needed to react with six times the oxygen ions compared to each hydrogen molecule, which meant more charge transfer was required. Therefore, the performance of the Cu–CeO2–ScSZ composite anode mainly depended on the conductivity and connectivity of Cu with CeO2 particles in the porous matrix for ethanol fuels. The anode with a greater copper loading provided further conductive paths for electrons as well as connected additional highly efficient CeO2 catalytic active reaction sites. The additional anode functional layer significantly improved the effective reaction region because the limited inherently porosity of the original single-layer anode inhibited the total loading of copper and cerium dioxide. The MPDs of the Cu–CeO2–YSZ|Ni–ScSZ|ScSZ|PCM single cell reached 604 and 408 mW cm−2 in hydrogen and ethanol steam at 800 °C, which were higher than 372 and 222 mW cm−2 of the Cu–CeO2–ScSZ anode in their previous work, respectively.234 In addition, they found that the double-layer SOFC anode could stably output power density for more than 50 h in ethanol steam in 0.7 V at 800 °C. Also, the cell did not exhibit the phenomenon of layer-to-layer peeling in the 50 h test environment at high temperature given that this dual-layer anode structure was fabricated by multi-layer tape casting and co-sintering processes. However, they also emphasized that the long-term stability of the anode in hydrocarbon fuels still needs further study. The great electrochemical performance improvement proved the possibility of enhancing the properties of Cu–CeO2 anodes by optimizing the microstructure of dual-layer anodes.235
Hao et al. prepared an La0.2Sr0.7TiO3–Ni–YSZ functional gradient anode (FGA) support SOFC using the co-tape casting method and sintered it using the field-assisted sintering technique (FAST). Specially, LST–Ni–YSZ FGA was a composite anode that possessed a variable anode component content and porosity from the anode to the electrolyte. They confirmed that the FGA structure was acquired and maintained excellent stability after the FAST process through SEM images, successfully avoiding the deformation and delamination that occur after conventional sintering. This SOFC anode exhibited the MPD of 600 mW cm−2 at 750 °C in methane fuel, and after 70 h of operation, no carbon deposits were detected through Raman spectroscopy. The authors emphasized that unlike the traditional sintering processes, distortion and delamination between the layers of the anode could be avoided due to the use of FAST technology to sinter LST–Ni–YSZ FGA. Thus, the phenomenon of interlayer stripping did not occur during the 70 h test.236
Wang et al. prepared an Ni–Fe/Ni–YSZ dual-layered anode by using the tape casting/lamination/sintering method. The electrochemical impedance measurement and relaxation time distribution analysis indicated that the cell with a double-layer straight hole path allowed rapid gas phase transport, thereby reducing concentration polarization, and also decreasing activation polarization by enhancing the accessibility of the electrochemical reaction sites. Furthermore, after eight redox cycles, the dual-layered anode support SOFC maintained its structure, while the Ni–YSZ single-layer anode support SOFC was damaged after one redox cycle. The results demonstrated that the dual-layered anode exhibited good long-term stability, and they also noticed that for the same cell, according to the cross-sectional SEM/EDX images, the re-oxidation stage involved the separation of the alloy phase during a redox cycle.237
The construction of multilayer anodes contributes to the diffusion and reforming reaction of hydrocarbon fuel, which can realize a greater loading of active catalytic component and appropriate porosity. However, it is worth noting that multilayer anodes may undergo interlayer stripping during cell testing, Therefore, this problem needs to be considered when constructing multilayer anodes.
4.2 Bimetallic-perovskite
The different carbon deposition resistances observed between different nickel cermets indicate the importance of the anode ceramic composition, and the TPB region of the different ceramics is also important in terms of carbon deposition resistance.The novel A-site layered PBMO exhibited a superior performance and remarkable stability under the reducing conditions of SOFCs. Sengodan et al. annealed Pr0.5Ba0.5MnO3−δin situ at 800 °C under a hydrogen atmosphere to make layered perovskite PrBaMn2O5+δ (PBMO) and used it as an SOFC anode for the first time. They further impregnated the Co0.5Fe0.5 catalyst solution in the PBMO skeleton, and the Co–Fe–PBMO anode SOFC delivered MPDs of 1770, 1320 and 570 mW cm−2 in humidified H2, C3H8 and CH4 at 850 °C, respectively. They also systematically studied that when the pure H2 was changed to H2 containing 30 ppm H2S, the cells with layered PBMO with Co–Fe catalyst did not show significant degradation of the voltage during 50 h of operation, indicating that the layered PBMO anode had superior sulfur resistance. In addition, the Co–Fe–PBMO anode cell was subjected to a constant current load of 0.2 A cm−2 at 700 °C and operated in C3H8 for more than 500 h without significant voltage degradation.238 Li et al. infiltrated Pr0.5Ba0.5MnO3−δ nanoparticles in the YSZ backbone, and then subjected it to redox cycles to form nanoparticle-based alloy Co–Mo–PBMO5–YSZ anodes. Also, when tested in simulated biogas, the Co–Mo–PBMO5–YSZ anode cells exhibited an MPD of 1100 mW cm−2, while that of the Co–YSZ anode cell was only 800 mW cm−2. Moreover, the Co–Mo–PBMO5–YSZ anode cell showed no performance degradation in the 30 h stability test, while the Co–YSZ cell manifested a poor performance due to coking.239 These experimental results showed that the layered perovskite PBMO anode exhibited good redox stability and excellent resistance to coking in hydrocarbon fuels and sulfur pollution, which established new design principles for the optimization and stabilization of nanoparticle-based catalyst SOFC.
The excellent catalytic activity and anti-sintering of alloy-ceramic catalysts are closely related to the strong interaction between the alloy and support. We believe that bimetallic materials combining multiple types of perovskites may be the focus of future research.
4.3 Application of single-atom catalyst anode in intermediate-temperature SOFCs
Designing single-atom catalysts (SACs) by managing the catalytic sites at the atomic scale can result in high-quality activity and minimize the use of metals, and SACs have unique selectivity compared to nanoparticle catalysts due to the lack of collection sites. The presence of high surface energies in the unsaturated and/or low-coordination active sites of SACs can lower the energy barrier and enhance the charge transfer.240–242Tang et al. reported the synergistic effect of two groups of single-atom sites (Ni1 and Ru1) anchored on the surface of CeO2 nanorods, which were highly active for reforming methane with CO2 to generate syngas (CO + H2). By comparing the three groups of catalysts of Ce0.95Ni0.025Ru0.025O2, Ce0.95Ru0.05O2 and Ce0.95Ni0.05O2, they found that Ce0.95Ni0.025Ru0.025O2 catalysts had the lowest apparent activation barrier and the highest CO and H2 conversion rates. Computational studies had found that the synergistic effect of Ni1 and Ru1 sites was manifested in the complementary functions of (1) Ni1 (activated CH4) and Ru1 (activated CO2), and (2) Ni1 site activated CH4 and generated H atoms, and then coupled H atoms at Ru1 sites to form H2. And they also found that the single-atom sites anchored on the surface of CeO2 remained monodisperse during catalysis up to 600 °C, which could basically meet the application of single-atom catalysts in hydrocarbon SOFC.243
Similarly, Wu et al. successfully modulated oxygen vacancies (OV) at different concentrations of CeO2 by replacing the Ce4+ cations with smaller cations M (M = Mg, Co, Zn). Also, Ni1/MCeO2 SAC was used for methane dry reforming reactions, which maintained a highly active state in the catalytic processes up to 800 °C. The Ni SACs exhibited extremely high activity in the activation of C–H bonds; however, carbon deposited on Ni caused their inactivation, but the in situ adsorbed oxygen (Oad) species adjacent to the Ni SACs were efficient reactants to eliminate carbon deposition, which could react with the deposited carbon to form gas-phase CO and CO2, thereby removing the carbon deposits. Also, Oad species were typically generated on the oxygen vacancies (OVs), specifically, OV activated CO2 to produce CO and Oad species, and thus the issue of carbon deposition can be solved by building enough OV around the Ni SACs. During the methane dry reforming reaction, carbon deposition decreased by 50% as the OV concentration increased from 21.9% to 30.8%. The synergistic combination of the effective C–H activation function of the Ni SAC and the CO2 activation function of OV led to a highly active and efficient carbon removal process for methane conversion in the OV–SAC-catalyzed system.244
For fuel cells, SACs with high ORR performance are mainly used in proton exchange membrane fuel cells (PEMFC) and anion exchange membrane fuel cells (AEMFC). However, few researchers have applied single-atom catalysts in SOFCs, which is mainly because their high operating temperature leads to severe single-atom aggregation.245 However, it is worth noting that reducible oxide supports, such as Fe2O3, CeO2, and TiO2, are particularly suitable for stabilizing single atoms due to the strong covalent metal support interactions (CMSIs), and high-temperature thermally stable single-atom catalysts can be obtained through this interaction.246 Li et al. linked SACs with reversible proton-conducting solid oxide cells (R-PSOCs) and selectively anchored Pt atoms to position B in Pr4Ni3O10+δ by a CMSI-based mechanism. The obtained Pt single-atom catalyst could withstand a high temperature of 700 °C and was highly active against oxygen reduction and oxygen evolution after 10% H2O–air and 10% H2O–10% CO2-air for 100 h, respectively. They also assembled an Ni–BZCYYb1711|BZCYYb1711|Pt–Pr4Ni3O10+δ cell, and its electrochemical performance was improved by almost 100% compared with Ni–BZCYYb1711|BZCYYb1711| Pr4Ni3O10+δ, and this catalyst preparation method is suitable for industrial-scale production.247
Therefore, we believe that single-atom anode SOFC catalysts based on the CMSI mechanism and combined with a large number of mature single-atom catalyst systems in hydrocarbon research will have broad prospects and commercial value in the future.
5. Conclusions and perspectives
In summary, the process of carbon deposition with the classification of carbon species, carbon detection methods, and strategies to solve anode carburizing of SOFCs were discussed. The use of alternative anodes is more promising than other strategies such as reducing the SOFC operating temperature and adjusting the O/C and H/C ratios of fuels. Therefore, a comprehensive overview of several decades of development and recent trends of carbon-resistant anode materials were provided, which were classified as follows: (1) bimetallic-cermet materials, (2) ceramic materials, and (3) anode reforming layer. Also, we briefly summarized the theoretical calculations and simulation models of anodes for hydrocarbon SOFCs, aiming to promote the research and development of alternative anodes. It can be seen that the commercialization of hydrocarbon SOFCs has slowed down due to many major technical challenges, where among them, the carbon deposition generated at the anodes during the operation in hydrocarbon fuel seriously decreases the catalytic ability and durability of the anodes, and damages the structure of the entire cell. Thus, to overcome these challenges to obtain efficient and stable carbon-resistant anodes, several research directions are proposed, as follows:(1) A large number of studies proved that bimetallic-cermet or solid solution of metal-alkaline earth metal oxide ceramic anodes have excellent electrochemical performances and long-term stability in hydrocarbon fuels. Future cermet anode research should consider the synthesis of more types with bimetallic alloys and solid solution of metal-alkaline metal oxides by non-hydrogen reduction pathways, such as reduction by carbon or carbon monoxide, given that hydrogen limits the reduction of some metal oxides.
(2) The catalytic activity and anti-coking of bimetallic-cermet are related to the interaction between the alloy and carrier ceramics, and for the ceramic components, the following two directions can be considered: (I) constructing bi-ceramic components (e.g., Ni–YSZ–YDC) and (II) using or synthesizing ceramics that can change their structure under a high-temperature reducing atmosphere, then forming a stable structure containing high conductivity and substantial oxygen vacancies (e.g., Pr0.5Ba0.5MnO3−δ).
(3) The heterojunction structure anode formed by in situ exsolution in perovskite materials will greatly improve the catalytic performance and stability of hydrocarbon SOFCs. (I) A-site-deficient perovskites (i.e., perovskite oxides with stoichiometric ratio of A/B < 1) increase the tendency of the B-site cations to exsolve from the lattice with wider distribution and better coverage on the surface of the perovskite, which is a more efficient method for constructing heterojunction structure anodes. (II) Moreover, combined with sufficient bimetallic-cermet research, the study of alloy metal nanoparticles with high catalytic activity in situ exsolved from perovskite materials should be valued.
(4) In addition to improving the alternative anode material composition, the anode microstructure should be optimized to obtain a greater loading of active catalytic component and appropriate porosity. The construction of a multilayer anodes contributes to the diffusion and reforming reaction of hydrocarbon fuel.
(5) The comprehensive use of molecular dynamics, finite element simulation, supplementary mathematical models and other simulation methods is very important to understand the reaction process of hydrocarbon fuel on the catalyst and provide theoretical guides for the lifecycle assessment and large-scale fabrication of SOFCs. Hence, advanced in situ characterization methods integrated with theoretical calculation and other novel material design strategies such as single-atom design ideas are helpful to effectively optimize the composition and microstructure of carbon-resistant anodes.
(6) Combined with the practical application environment of hydrocarbon-based fuel cells, the research on alternative anodes should be optimized and integrated with the current advanced SOFC cathodes and electrolytes. Also, in the actual long-term operation of the cells, the atmosphere of the anodes will change from single CH4 to CH4, CO, CO2, H2, H2O and other gas mixing systems, and hence it is more meaningful to study the effects of the fuel composition.
From the perspective of global energy strategy, it is imperative to vigorously promote the development of the natural gas industry. Presently, a well-developed methane-rich natural gas infrastructure is available, which can be readily and easily employed in residential or office buildings, significantly reducing the additional cost of using hydrocarbon-fuel SOFCs, accelerating the application of SOFCs in transportation and power distribution equipment. The above-mentioned research directions can promote the process of direct electrooxidation of hydrocarbon fuels to form CO2 and H2O or the process of indirect oxidation of syngas (CO + H2) through reforming reaction to form CO2 and H2O, and play an essential role in improving the electrochemical performance, durability and cost-effectiveness of SOFCs. The optimized anode structures also make it easy to extend them to various cell structures, such as planar, tubular and honeycomb types, thereby speeding up the commercial application of hydrocarbon-fueled SOFCs.
To further facilitate the application of SOFCs, in addition to accelerating the development of multi-fuel compatible anodes, it is also necessary: (I) to develop efficient and green methods for the fabrication of SOFCs, e.g., 3D printing combined with theoretical model optimization. (II) Developing hybrid power systems based on SOFCs to improve their energy efficiency and economic viability, e.g., integrated coal gasification fuel cell combined cycle (IGFC), given that some of the heat energy of SOFCs can be recovered by introducing the reaction products into the steam turbine, where SOFC/gas turbine hybrid systems provide combined heat and power with efficiencies of up to 90%. (III) Reducing the operating temperature of SOFCs. Reducing the working temperature can improve the rapid start-up of the cell, alleviate the mismatch of the thermal expansion coefficient of different material systems and reduce the heat loss of the cell stack. Although the core influencing factor of the intermediate-to-low temperature SOFC is the ionic conductivity of the electrolyte, anode materials with high catalytic activity at intermediate-to-low temperature, matching electrolytes with high ionic conductivity and perovskite-based cathode materials with high activity and redox reaction stability will be a more suitable cell system. It is expected that hydrocarbon-fueled SOFCs will be widely used in stationary power generation and automobile applications.
Abbreviations
| FCs | Fuel cells |
| PEMFCs | Proton-exchange membrane fuel cells |
| SOFCs | Solid oxide fuel cells |
| LSM | Sr-doped LaMnO3 |
| LSC | La0.8Sr0.2CoO3 |
| LSF | La0.8Sr0.2FeO3 |
| LSCF | La0.8Sr0.2Co0.2Fe0.8O3 |
| BSCF | Ba0.5Sr0.5Co0.8Fe0.2O3−δ |
| PBSCF | PrBa0.5Sr0.5Co1.5Fe0.5O5+δ |
| BPCF | Ba1−xPrxCo1−yFeyO3−δ |
| SCCO | Sr0.95Ce0.05CoO3−δ |
| BCFN | Ba0.9Co0.7Fe0.2Nb0.1O3−δ |
| YSZ | Yttria-stabilized zirconia |
| ScSZ | Scandia-stabilized zirconia |
| GDC | Gadolinium-doped ceria |
| SDC | Samaria- doped ceria |
| ASR | Area specific resistance |
| TEC | Thermal expansion coefficient |
| TPB | Three-phase boundary |
| MEIC | Mixed electronic and ionic conductivity |
| DIR | Direct internal reforming |
| HR-TEM | High-resolution transmission electron microscopy image |
| SEM | Scanning electron microscope |
| EDS | Energy dispersive X-ray spectroscopy |
| XRD | X-ray diffraction |
| SAED | Selected area electron diffraction |
| TPO | Temperature programmed oxidation |
| FTIR | Fourier transform infrared spectroscopy |
| MSR | Methane steam reforming |
| SVUV-PIMS | Synchrotron-based vacuum ultraviolet photoionization mass spectrometry |
| ETEM | Environmental transmission electron microscopy |
| DFT | Density functional theory |
| YST–YSZ | Sr0.88Y0.08TiO3−δ formed yttria-stabilized zirconia |
| LSCM | La0.75Sr0.25Cr0.5Mn0.5O3 |
| LSTGM | La4.0Sr8.0Ti11.0Mn0.5Ga0.5O37.5 |
| SMMO | Sr2MgMoO6−δ |
| OCV | Open-circuit voltage |
| ZDC | Zr0.1Ce0.9O2−δ |
| BZCYYb | BaZr0.1Ce0.7Y0.1Yb0.1O3−δ |
| PBMO | Pr0.5Ba0.5MnO3−δ |
| MPD | Maximum power density |
| DRM | Dry methane reforming |
| CZ | Ce0.5Zr0.5O2 |
| CSZ | Ceria-stabilized zirconia |
| XPS | X-ray photoelectron spectroscopy |
| EDX | Energy dispersive X-ray spectroscopy |
| n-SNGG | Sn-doped Ni/gadolinium-doped ceria–gadolinium-doped ceria |
| HAADF STEM | High-angle annular dark-field scanning transmission electron microscopy |
| SR | Steam reforming |
| ATR | Auto-thermal reforming |
| STN | Sr0.94Ti0.9Nb0.1O3 |
| UCG | Underground coal gasification |
| YDC | Yttria-doped ceria |
| CYYb | Ce0.8Y0.1Yb0.1O1.9 |
| LSMO | La0.5Sr1.5MnO4±δ |
| STF | SrTi1−xFexO3 |
| LSTN | La0.2Sr0.7Ti0.9Ni0.1O3−δ |
| LSC | Strontium-doped lanthanum chromate |
| LSCNi | Nickel-doped (La0.7Sr0.3)CrO3 |
| L70SCrN | La0.70Sr0.3Cr0.85Ni0.15O3−δ |
| L65SCrN | La0.65Sr0.3Cr0.85Ni0.15O3−δ |
| STFN | Sr0.95(Ti0.3Fe0.63Ni0.07)O3 |
| PBMCo | Pr0.5Ba0.5MnOx |
| LBM | La0.5Ba0.5MnO3−δ |
| LBMFC-1 | La0.5Ba0.5Mn0.9Fe0.05Co0.05O3−δ |
| LBMFC–2 | La0.5Ba0.5Mn0.8Fe0.1Co0.1O3−δ |
| PBMNx | (Pr0.5Ba0.5)1−x/2Mn1−x/2Nix/2O3−δ |
| A-LSC | A-site-deficient LaSrCrO3 |
| LSCNi–Fe | (La0.7Sr0.3)(Cr0.85Ni0.12Fe0.03)O3−x |
| LSFN | Ni-doped La0.6Sr0.4FeO3−δ |
| L0.4STRN | (La0.4Sr0.4)(Ti0.85Ru0.07Ni0.08)O3−δ |
| L0.4STN | L0.4STN La0.4Sr0.4Ti0.85Ni0.15O3−δ |
| LSGM | Lanthanum gallate with strontium doping |
| LCeNT | La0.8Ce0.1Ni0.4Ti0.6O3−δ |
| GDC–PBCC | Gd0.2Ce0.8O1.9–PrBa0.8Ca0.2Co2O5+δ |
| LDC | La2Ce2O7 |
| LSDC | La1.95Sm0.05Ce2O7 |
| CNR | Ce0.90Ni0.05Ru0.05O2 |
| CELD | Cathodic electrochemical deposition |
| SCE | Saturated calomel electrode |
| GC | Gas chromatography |
| NiMo–CZ | NiMo–Ce0.5Zr0.5O2−δ |
| PAW | Projector-augmented wave |
| CI-NEB | Climbing image-nudged elastic band |
| RMTN | Reverse methanation |
| ReaxFF-MD | Reactive force field molecular dynamics simulation |
| FIB-SEM | Focused ion beam-scanning electron microscope |
| AP-SOFC | All porous solid oxide fuel cell |
| PDOS | Projected density of states |
| IS | Initial state |
| TS | Transition state |
| FS | Final state |
| SIMFC | SIMulation of fuel cell |
| R-SZMO | H2-reduced Sr2ZnMoO6 |
| FGA | Functional gradient anode |
| FAST | Field assisted sintering technique |
| SACs | Single-atom catalysts |
| AEMFC | Anion-exchange membrane fuel cells |
| CMSIs | Covalent metal support interactions |
| R-PSOCs | Reversible proton-conducting solid oxide cells |
Latin symbols
| I | Current |
| V | Voltage |
| p | Gas pressure |
| ΔG | Gibbs free energy variation |
| γ int | Interface energy |
| P | Cell power |
| δ | Oxygen vacancy |
| σ | Conductivity |
| e− | Carrier concentration |
| μ | Mobility |
Subscript
| E thermo | Thermodynamic predicted voltage |
| η act | Activation loss |
| η ohmic | Ohmic loss |
| η conc | Concentration loss |
| Id | Intensity of D band |
| Ig | Intensity of G band |
| S BET | Surface area |
| gcat | The mass of catalyst |
| Oad | Adsorbed oxygen |
| OVs | Oxygen vacancies |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by 21C Innovation Laboratory, Contemporary Amperex Technology Ltd by project No. 21C-OP-202212, Foundation of Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, the Foundation of State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering (Grant No. 2022-K15), China University of Mining & Technology (Beijing), Beijing National Laboratory for Condensed Matter Physics, and the National Natural Science Foundation of China (No. 51172275, 51672029 and 51372271).References
- S. R. Bull, Renewable energy today and tomorrow, Proc. IEEE, 2001, 89, 1216–1226 CrossRef.
- J. P. Barton and D. G. Infield, Energy storage and its use with tntermittent renewable energy, IEEE Trans. Energy Convers., 2004, 19, 441–448 CrossRef.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- J. S. Sawyer, Man-made carbon dioxide and the “greenhouse” Effect, Nature, 1972, 239, 23–26 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Materials for fuel-cell technologies, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- R. O'Hayre, S.-W. Cha, W. Colella and F. B. Prinz, Fuel Cell Fundamentals-Chapter 1: Introduction, Fuel Cell Fundamentals, Wiley Online Library, 2016, pp. 1–24 Search PubMed.
- A. L. Dicks and D. A. J. Rand., Fuel Cell Systems Explained-Introducing Fuel Cells, Fuel Cell Systems Explained, 2018, pp. 1–26 Search PubMed.
- M. A. Laguna-Bercero, Recent advances in high temperature electrolysis using solid oxide fuel cells: A review, J. Power Sources, 2012, 203, 4–16 CrossRef CAS.
- C. Sun and U. Stimming, Recent anode advances in solid oxide fuel cells, J. Power Sources, 2007, 171, 247–260 CrossRef CAS.
- A. J. Jacobson, Materials for solid oxide fuel cells, Chem. Mater., 2009, 22, 660–674 CrossRef.
- M. A. Hassan, O. B. Mamat and M. Mehdi, Review: Influence of alloy addition and spinel coatings on Cr-based metallic interconnects of solid oxide fuel cells, Int. J. Hydrogen Energy, 2020, 45, 25191–25209 CrossRef CAS.
- M. Dewa, W. Yu, N. Dale, A. M. Hussain, M. G. Norton and S. Ha, Recent progress in integration of reforming catalyst on metal-supported SOFC for hydrocarbon and logistic fuels, Int. J. Hydrogen Energy, 2021, 46, 33523–33540 CrossRef CAS.
- H. A. Shabri, M. H. D. Othman, M. A. Mohamed, T. A. Kurniawan and S. M. Jamil, Recent progress in metal-ceramic anode of solid oxide fuel cell for direct hydrocarbon fuel utilization: A review, Fuel Process. Technol., 2021, 212, 106626 CrossRef CAS.
- S. Senthil Kumar and S. T. Aruna, Hydrocarbon compatible SOFC anode catalysts and their syntheses: A review, Sustainable Chem., 2021, 2, 707–763 CrossRef CAS.
- V. Marcantonio, L. Del Zotto, J. P. Ouweltjes and E. Bocci, Main issues of the impact of tar, H2S, HCl and alkali metal from biomass-gasification derived syngas on the SOFC anode and the related gas cleaning technologies for feeding a SOFC system: A review, Int. J. Hydrogen Energy, 2022, 47, 517–539 CrossRef CAS.
- E. P. Murray, T. Tsai and S. A. Barnett, A direct-methane fuel cell with a ceria-based anode, Nature, 1999, 400, 649–651 CrossRef CAS.
- F. Yu, T. Han, Z. Wang, Y. Xie, Y. Wu, Y. Jin, N. Yang, J. Xiao and S. Kawi, Recent progress in direct carbon solid oxide fuel cell: Advanced anode catalysts, diversified carbon fuels, and heat management, Int. J. Hydrogen Energy, 2021, 46, 4283–4300 CrossRef CAS.
- P. Zhang, Z. Yang, Y. Jin, C. Liu, Z. Lei, F. Chen and S. Peng, Progress report on the catalyst layers for hydrocarbon-fueled SOFCs, Int. J. Hydrogen Energy, 2021, 46, 39369–39386 CrossRef CAS.
- L. Fan, C. e Li, L. van Biert, S.-H. Zhou, A. N. Tabish, A. Mokhov, P. V. Aravind and W. Cai, Advances on methane reforming in solid oxide fuel cells, Renewable Sustainable Energy Rev., 2022, 166, 112646 CrossRef CAS.
- W. Wang, C. Su, Y. Wu, R. Ran and Z. Shao, Progress in solid oxide fuel cells with nickel-based anodes operating on methane and related fuels, Chem. Rev., 2013, 113, 8104–8151 CrossRef CAS PubMed.
- S. S. Rathore, S. Biswas, D. Fini, A. P. Kulkarni and S. Giddey, Direct ammonia solid-oxide fuel cells: A review of progress and prospects, Int. J. Hydrogen Energy, 2021, 46, 35365–35384 CrossRef CAS.
- W. Winkler and H. Lorenz, The design of stationary and mobile solid oxide fuel cell-gas turbine systems, J. Power Sources, 2002, 105, 222–227 CrossRef CAS.
- B. C. H. Steele, Running on natural gas, Nature, 1999, 400, 619–621 CrossRef CAS.
- T. Takeguchi, Y. Kani, T. Yano, R. Kikuchi, K. Eguchi, K. Tsujimoto, Y. Uchida, A. Ueno, K. Omoshiki and M. Aizawa, Study on steam reforming of CH4 and C2 hydrocarbons and carbon deposition on Ni–YSZ cermets, J. Power Sources, 2002, 112, 588–595 CrossRef CAS.
- A. Atkinson, S. Barnett, R. J. Gorte, J. T. S. Irvine, A. J. McEvoy, M. Mogensen, S. C. Singhal and J. Vohs, Advanced anodes for high-temperature fuel cells, Nat. Mater., 2004, 3, 17–27 CrossRef CAS PubMed.
- M. Flytzani-Stephanopoulos, M. Sakbodin and Z. Wang, Regenerative adsorption and removal of H2S from hot fuel gas streams by rare earth oxides, Science, 2006, 312, 1508–1510 CrossRef CAS PubMed.
- Z. Tao, M. Fu and Y. Liu, A mini-review of carbon-resistant anode materials for solid oxide fuel cells, Sustainable Energy Fuels, 2021, 5, 5420–5430 RSC.
- S. Zarabi Golkhatmi, M. I. Asghar and P. D. Lund, A review on solid oxide fuel cell durability: Latest progress, mechanisms, and study tools, Renewable Sustainable Energy Rev., 2022, 161, 112339 CrossRef CAS.
- C. Sun and R. Hui, and J. Roller, Cathode materials for solid oxide fuel cells: a review, J. Solid State Electrochem., 2009, 14, 1125–1144 CrossRef.
- W. S. Wang, Y. Y. Huang, S. W. Jung, J. M. Vohs and R. J. Gorte, A comparison of LSM, LSF, and LSCo for solid oxide electrolyzer anodes, J. Electrochem. Soc., 2006, 153, A2066–A2070 CrossRef CAS.
- M. A. Laguna-Bercero, J. A. Kilner and S. J. Skinner, Development of oxygen electrodes for reversible solid oxide fuel cells with scandia stabilized zirconia electrolytes, Solid State Ionics, 2011, 192, 501–504 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- W. Zhang, B. Yu and J. Xu, Investigation of single SOEC with BSCF anode and SDC barrier layer, Int. J. Hydrogen Energy, 2012, 37, 837–842 CrossRef CAS.
- L. Jiang, T. Wei, R. Zeng, W.-X. Zhang and Y.-H. Huang, Thermal and electrochemical properties of PrBa0.5Sr0.5Co2−xFexO5+δ (x = 0.5, 1.0, 1.5) cathode materials for solid-oxide fuel cells, J Power Sources, 2013, 232, 279–285 CrossRef CAS.
- R. Hui, C. Sun, S. Yick, C. Decès-Petit, X. Zhang, R. Maric and D. Ghosh, Ba1−xPrxCo1−yFeyO3−δ as cathode materials for low temperature solid oxide fuel cells, Electrochim. Acta, 2010, 55, 4772–4775 CrossRef CAS.
- W. Yang, T. Hong, S. Li, Z. Ma, C. Sun, C. Xia and L. Chen, Perovskite Sr1−xCexCoO3−δ (0.05 ≤ x ≤ 0.15) as superior cathodes for intermediate temperature solid oxide fuel cells, ACS Appl. Mater. Interfaces, 2013, 5, 1143–1148 CrossRef CAS PubMed.
- Y. Gong, C. Sun, Q. A. Huang, J. A. Alonso, M. T. Fernandez-Diaz and L. Chen, Dynamic octahedral breathing in oxygen-deficient Ba0.9Co0.7Fe0.2Nb0.1O3−δ Perovskite Performing as a Cathode in Intermediate-Temperature SOFC, Inorg. Chem., 2016, 55, 3091–3097 CrossRef CAS PubMed.
- D. J. Brett, A. Atkinson, N. P. Brandon and S. J. Skinner, Intermediate temperature solid oxide fuel cells, Chem. Soc. Rev., 2008, 37, 1568–1578 RSC.
- H. Moon, S. Kim, S. Hyun and H. Kim, Development of IT-SOFC unit cells with anode-supported thin electrolytes via tape casting and co-firing, Int. J. Hydrogen Energy, 2008, 33, 1758–1768 CrossRef CAS.
- L. Zhang, S. P. Jiang, W. Wang and Y. Zhang, NiO/YSZ, anode-supported, thin-electrolyte, solid oxide fuel cells fabricated by gel casting, J. Power Sources, 2007, 170, 55–60 CrossRef CAS.
- X. Xu, C. Xia, S. Huang and D. Peng, YSZ thin films deposited by spin-coating for IT-SOFCs, Ceram. Int., 2005, 31, 1061–1064 CrossRef CAS.
- W. Zhou, H. Shi, R. Ran, R. Cai, Z. Shao and W. Jin, Fabrication of an anode-supported yttria-stabilized zirconia thin film for solid-oxide fuel cells via wet powder spraying, J. Power Sources, 2008, 184, 229–237 CrossRef CAS.
- J.-H. Kim, R.-H. Song, K.-S. Song, S.-H. Hyun, D.-R. Shin and H. Yokokawa, Fabrication and characteristics of anode-supported flat-tube solid oxide fuel cell, J. Power Sources, 2003, 122, 138–143 CrossRef CAS.
- Y. Du and N. M. Sammes, Fabrication and properties of anode-supported tubular solid oxide fuel cells, J. Power Sources, 2004, 136, 66–71 CrossRef CAS.
- S. D. Kim, S. H. Hyun, J. Moon, J.-H. Kim and R. H. Song, Fabrication and characterization of anode-supported electrolyte thin films for intermediate temperature solid oxide fuel cells, J. Power Sources, 2005, 139, 67–72 CrossRef CAS.
- Y. Yang, Y. Zhang and M. Yan, A review on the preparation of thin-film YSZ electrolyte of SOFCs by magnetron sputtering technology, Sep. Purif. Technol., 2022, 298, 121627 CrossRef CAS.
- X.-F. Ding, J.-Y. Shen, X.-J. Gao and J. Wang, Enhanced electrochemical properties of Sm0.2Ce0.8O1.9 film for SOFC electrolyte fabricated by pulsed laser deposition, Rare Metals, 2014, 40, 1294–1299 CrossRef.
- S. McIntosh and R. J. Gorte, Direct hydrocarbon solid oxide fuel cells, Chem. Rev., 2004, 104, 4845–4865 CrossRef CAS PubMed.
- I. Sreedhar, B. Agarwal, P. Goyal and S. A. Singh, Recent advances in material and performance aspects of solid oxide fuel cells, J. Electroanal. Chem., 2019, 848, 113315 CrossRef CAS.
- D. Simwonis, F. Tietz and D. Stover, Nickel coarsening in annealed Ni/8YSZ anode substrates for solid oxide fuel cells - In memoriam to Professor H. Tagawa, Solid State Ionics, 2000, 132, 241–251 CrossRef CAS.
- S. Tao and J. T. Irvine, A redox-stable efficient anode for solid-oxide fuel cells, Nat. Mater., 2003, 2, 320–323 CrossRef CAS PubMed.
- J. B. Young, Thermofluid modeling of fuel cells, Annu. Rev. Fluid Mech., 2007, 39, 193–215 CrossRef.
- L. S. Skutina, A. I. Vylkov, I. N. Bainov, K. A. Chistyakov, D. K. Kuznetsov, O. B. Pavlenko and D. A. Medvedev, Catalytic properties of Sr2Ni0.75Mg0.25MoO6–δ based composites for application in hydrocarbon-fuelled solid oxide fuel cells, Int. J. Hydrogen Energy, 2021, 46, 16899–16906 CrossRef CAS.
- S. A. Saadabadi, B. Illathukandy and P. V. Aravind, Direct internal methane reforming in biogas fuelled solid oxide fuel cell; the influence of operating parameters, Energy Sci. Eng., 2021, 9, 1232–1248 CrossRef CAS.
- A. Abdulrasheed, A. A. Jalil, Y. Gambo, M. Ibrahim, H. U. Hambali and M. Y. Shahul, Hamid, A review on catalyst development for dry reforming of methane to syngas: Recent advances, Renewable Sustainable Energy Rev., 2019, 108, 175–193 CrossRef CAS.
- L. Cai, T. He, Y. Xiang and Y. Guan, Study on the reaction pathways of steam methane reforming for H2 production, Energy, 2020, 207, 118296 CrossRef CAS.
- L. Bobrova, D. Andreev, E. Ivanov, N. Mezentseva, M. Simonov, L. Makarshin, A. Gribovskii and V. Sadykov, Water–gas shift reaction over Ni/CeO2 Catalysts, Catalysts, 2017, 7, 310 CrossRef.
- H. Sumi, Y.-H. Lee, H. Muroyama, T. Matsui, M. Kamijo, S. Mimuro, M. Yamanaka, Y. Nakajima and K. Eguchi, Effect of carbon deposition by carbon monoxide disproportionation on electrochemical characteristics at low temperature operation for solid oxide fuel cells, J. Power Sources, 2011, 196, 4451–4457 CrossRef CAS.
- A. L. Dicks, Advances in catalysts for internal reforming in high temperature fuel cells, J. Power Sources, 1998, 71, 111–122 CrossRef CAS.
- S. McIntosh, J. M. Vohs and R. J. Gorte, Role of hydrocarbon deposits in the enhanced performance of direct-oxidation SOFCs, J. Electrochem. Soc., 2003, 150, A470–A476 CrossRef CAS.
- M. D. Gross, J. M. Vohs and R. J. Gorte, Recent progress in SOFC anodes for direct utilization of hydrocarbons, J. Mater. Chem., 2007, 17, 3071–3077 RSC.
- C. M. Chun and T. A. Ramanarayanan, Mechanism and control of carbon deposition on high temperature alloys, J. Electrochem. Soc., 2007, 154, C465–C471 CrossRef CAS.
- D. Lozano-Castello, D. Cazorla-Amoros, A. Linares-Solano, K. Oshida, T. Miyazaki, Y. J. Kim, T. Hayashi and M. Endo, Comparative characterization study of microporous carbons by HRTEM image analysis and gas adsorption, J. Phys. Chem. B, 2005, 109, 15032–15036 CrossRef CAS PubMed.
- T. Nakamura, N. Ohmura, K. Yahiro and K. Amezawa, Structural changes of Ni/YSZ cermet for solid oxide fuel cells in hydrocarbon gas containing atmospheres, ECS Trans., 2013, 57, 1539–1544 CrossRef.
- H. Sumi, T. Yamaguchi, K. Hamamoto, T. Suzuki and Y. Fujishiro, Impact of direct butane microtubular solid oxide fuel cells, J. Power Sources, 2012, 220, 74–78 CrossRef CAS.
- D. Lee, J. Myung, J. Tan, S.-H. Hyun, J. T. S. Irvine, J. Kim and J. Moon, Direct methane solid oxide fuel cells based on catalytic partial oxidation enabling complete coking tolerance of Ni-based anodes, J. Power Sources, 2017, 345, 30–40 CrossRef CAS.
- T. Chen, W. G. Wang, H. Miao, T. Li and C. Xu, Evaluation of carbon deposition behavior on the nickel/yttrium-stabilized zirconia anode-supported fuel cell fueled with simulated syngas, J. Power Sources, 2011, 196, 2461–2468 CrossRef CAS.
- C. M. Chun and T. A. Ramanarayanan, Mechanism and control of carbon deposition on high temperature alloys, J. Electrochem. Soc., 2007, 154, 9 Search PubMed.
- X. Li, K. Blinn, Y. Fang, M. Liu, M. A. Mahmoud, S. Cheng, L. A. Bottomley, M. El-Sayed and M. Liu, Application of surface enhanced Raman spectroscopy to the study of SOFC electrode surfaces, Phys. Chem. Chem. Phys., 2012, 14, 5919–5923 RSC.
- B. C. Bayer, D. A. Bosworth, F. B. Michaelis, R. Blume, G. Habler, R. Abart, R. S. Weatherup, P. R. Kidambi, J. J. Baumberg, A. Knop-Gericke, R. Schloegl, C. Baehtz, Z. H. Barber, J. C. Meyer and S. Hofmann, In situ observations of phase transitions in metastable nickel (carbide)/carbon nanocomposites, J. Phys. Chem. C, 2016, 120, 22571–22584 CrossRef CAS PubMed.
- B. Novosel, M. Marinsek and J. Macek, Deactivation of Ni–YSZ material in dry methane and oxidation of various forms of deposited carbon, J. Fuel Cell Sci. Technol., 2012, 9, 061003 CrossRef.
- M. Rames, Y. Yu and G. Ren, Optimized negative staining: a high-throughput protocol for examining small and asymmetric protein structure by electron microscopy, J. Vis. Exp., 2014, 90, e51087 Search PubMed.
- H. Nishino, R. Nishida, T. Matsui, N. Kawase and I. Mochida, Growth of amorphous carbon nanotube from poly(tetrafluoroethylene) and ferrous chloride, Carbon, 2003, 41, 2819–2823 CrossRef CAS.
- Y. Chen, Y. Zhang, Y. Lin, Z. Yang, D. Su, M. Han and F. Chen, Direct-methane solid oxide fuel cells with hierarchically porous Ni-based anode deposited with nanocatalyst layer, Nano Energy, 2014, 10, 1–9 CrossRef CAS.
- A. Cuesta, P. Dhamelincourt, J. Laureyns, A. Martínez-Alonso and J. M. D. Tascón, Raman microprobe studies on carbon materials, Carbon, 1994, 32, 1523–1532 CrossRef CAS.
- H. Sumi, H. Shimada, T. Yamaguchi, K. Hamamoto, T. Suzuki and Y. Fujishiro, Development of microtubular solid oxide fuel cells using hydrocarbon fuels, Advances in Solid Oxide Fuel Cells and Electronic Ceramics, 2015, pp. 93–104 Search PubMed.
- M. Chlipała, P. Błaszczak, S. F. Wang, P. Jasiński and B. Bochentyn, In situ study of a composition of outlet gases from biogas fuelled solid oxide fuel cell performed by the Fourier Transform Infrared Spectroscopy, Int. J. Hydrogen Energy, 2019, 44, 13864–13874 CrossRef.
- Y. Xie, N. Shi, X. Hu, M. Liu, Y. Yang, D. Huan, Y. Pan, R. Peng and C. Xia, Novel in-situ MgO nano-layer decorated carbon-tolerant anode for solid oxide fuel cells, Int. J. Hydrogen Energy, 2020, 45, 11791–11801 CrossRef CAS.
- C. Sun, R. Su, J. Chen, L. Lu and P. Guan, Carbon formation mechanism of C2H2 in Ni-based catalysts revealed by in situ electron microscopy and molecular dynamics simulations, ACS Omega, 2019, 4, 8413–8420 CrossRef CAS PubMed.
- B. Steele, Appraisal of Ce1−yGdyO2−y/2 electrolytes for IT-SOFC operation at 500 °C, Solid State Ionics, 2000, 129, 95–110 CrossRef CAS.
- K. Sasaki and Y. Teraoka, Equilibria in fuel cell gases - II. The C-H-O ternary diagrams, J. Electrochem. Soc., 2003, 150, 885–888 CrossRef.
- N. Galea, D. Knapp and T. Ziegler, Density functional theory studies of methane dissociation on anode catalysts in solid-oxide fuel cells: Suggestions for coke reduction, J. Catal., 2007, 247, 20–33 CrossRef CAS.
- E. Fabbri, D. Pergolesi and E. Traversa, Materials challenges toward proton-conducting oxide fuel cells: a critical review, Chem. Soc. Rev., 2010, 39, 4355–4369 RSC.
- H. Zhu, R. J. Kee, V. M. Janardhanan, O. Deutschmann and D. G. Goodwin, Modeling elementary heterogeneous chemistry and electrochemistry in Solid-Oxide Fuel Cells, J. Electrochem. Soc., 2005, 152, 12 Search PubMed.
- S. Ahmed, A. Aitani, F. Rahman, A. Al-Dawood and F. Al-Muhaish, Decomposition of hydrocarbons to hydrogen and carbon, Appl. Catal., A, 2009, 359, 1–24 CrossRef CAS.
- B. C. H. Steele, I. Kelly, H. Middleton and R. Rudkin, Oxidation of methane in solid state electrochemical reactors, Solid State Ionics, 1988, 28–30, 1547–1552 CrossRef.
- H. He, Characterization of YSZ-YST composites for SOFC anodes, Solid State Ionics, 2004, 175, 171–176 CrossRef CAS.
- O. A. Marina, N. L. Canfield and J. W. Stevenson, Thermal, electrical, and electrocatalytical properties of lanthanum-doped strontium titanate, Solid State Ionics, 2002, 149, 21–28 CrossRef CAS.
- J. C. Ruiz-Morales, J. Canales-Vazquez, C. Savaniu, D. Marrero-Lopez, W. Zhou and J. T. Irvine, Disruption of extended defects in solid oxide fuel cell anodes for methane oxidation, Nature, 2006, 439, 568–571 CrossRef CAS PubMed.
- Y. H. Huang, R. I. Dass, Z. L. Xing and J. B. Goodenough, Double perovskites as anode materials for solid-oxide fuel cells, Science, 2006, 312, 254–257 CrossRef CAS PubMed.
- M. L. Toebes, J. H. Bitter, A. J. van Dillen and K. P. de Jong, Impact of the structure and reactivity of nickel particles on the catalytic growth of carbon nanofibers, Catal. Today, 2002, 76, 33–42 CrossRef CAS.
- R. T. K. Baker and J. J. Chludzinski, Jr., In-situ electron microscopy studies of the behavior of supported ruthenium particles. 2. Carbon deposition from catalyzed decomposition of acetylene, J. Phys. Chem., 1986, 90, 4734–4738 CrossRef CAS.
- R. Shiozaki, A. G. Andersen, T. Hayakawa, S. Hamakawa, K. Suzuki, M. Shimizu and K. Takehira, Partial oxidation of methane over a Ni/BaTiO3 catalyst prepared by solid phase crystallization, J. Chem. Soc., Faraday Trans., 1997, 93, 3235–3242 RSC.
- H. Kim, C. Lu, W. L. Worrell, J. M. Vohs and R. J. Gorte, Cu-ni cermet anodes for direct oxidation of methane in Solid-Oxide Fuel Cells, J. Electrochem. Soc., 2002, 149, 3 CrossRef.
- Z. Lü, L. Pei, T.-M. He, X.-Q. Huang, Z.-G. Liu, Y. Ji, X.-H. Zhao and W.-H. Su, Study on new copper-containing SOFC anode materials, J. Alloys Compd., 2002, 334, 299–303 CrossRef.
- S. Song, M. Han, J. Zhang and H. Fan, NiCu–Zr0.1Ce0.9O2−δ anode materials for intermediate temperature solid oxide fuel cells using hydrocarbon fuels, J. Power Sources, 2013, 233, 62–68 CrossRef CAS.
- Y. Akdeniz, B. Timurkutluk and C. Timurkutluk, Development of anodes for direct oxidation of methane fuel in solid oxide fuel cells, Int. J. Hydrogen Energy, 2016, 41, 10021–10029 CrossRef CAS.
- M. Li, B. Hua, J. Pu, B. Chi and L. Jian, Electrochemical performance and carbon deposition resistance of M-BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (M=Pd, Cu, Ni or NiCu) anodes for solid oxide fuel cells, Sci. Rep., 2015, 5, 7667 CrossRef CAS PubMed.
- S. Kim, C. Kim, J. H. Lee, J. Shin, T.-H. Lim and G. Kim, Tailoring Ni-based catalyst by alloying with transition metals (M = Ni, Co, Cu, and Fe) for direct hydrocarbon utilization of energy conversion devices, Electrochim. Acta, 2017, 225, 399–406 CrossRef CAS.
- S. Islam and J. M. Hill, Preparation of Cu–Ni/YSZ solid oxide fuel cell anodes using microwave irradiation, J. Power Sources, 2011, 196, 5091–5094 CrossRef CAS.
- E. W. Park, H. Moon, M.-S. Park and S. H. Hyun, Fabrication and characterization of Cu–Ni–YSZ SOFC anodes for direct use of methane via Cu-electroplating, Int. J. Hydrogen Energy, 2009, 34, 5537–5545 CrossRef CAS.
- S. Senthil Kumar, V. Jayaram and S. T. Aruna, Co-fired anode-supported solid oxide fuel cell for internal reforming of hydrocarbon fuel, Energy, Ecol. Environ., 2020, 6, 55–68 CrossRef.
- A. Ringuedé, D. Bronine and J. R. Frade, Ni1−xCox/YSZ cermet anodes for solid oxide fuel cells, Electrochim. Acta, 2002, 48, 437–442 CrossRef.
- C.-K. Cho, B.-H. Choi and K.-T. Lee, Effect of Co alloying on the electrochemical performance of Ni–Ce0.8Gd0.2O1.9 anodes for hydrocarbon-fueled solid oxide fuel cells, J. Alloys Compd., 2012, 541, 433–439 CrossRef CAS.
- W. Nicharee, S. Chaianansutcharit and K. Sato, Electrochemical performance and stability of Ni1−xCox-based cermet anode for direct methane-fuelled solid oxide fuel cells, MATEC Web Conf., 2017, 130, 03005 CrossRef.
- M. R. Cesario, G. S. Souza, F. J. A. Loureiro, A. J. M. Araújo, J. P. F. Grilo, S. Aouad, H. L. Tidahy, D. A. Macedo, D. P. Fagg, C. Gennequin and E. Abi-Aad, Synthesis of Co–Ni and Cu–Ni based-catalysts for dry reforming of methane as potential components for SOFC anodes, Ceram. Int., 2021, 47, 33191–33201 CrossRef CAS.
- H. Kan and H. Lee, Enhanced stability of Ni–Fe/GDC solid oxide fuel cell anodes for dry methane fuel, Catal. Commun., 2010, 12, 36–39 CrossRef CAS.
- S. H. Lee and H. Kim, Dual layered anode support for the internal reforming of methane for solid oxide fuel cells, Ceram. Int., 2014, 40, 5959–5966 CrossRef CAS.
- Y. M. Park and H. Kim, An additional layer in an anode support for internal reforming of methane for solid oxide fuel cells, Int. J. Hydrogen Energy, 2014, 39, 16513–16523 CrossRef CAS.
- S. Lee, J. H. Park, K. T. Lee and Y.-W. Ju, Anodic properties of Ni-Fe bimetallic nanofiber for solid oxide fuel cell using LaGaO3 electrolyte, J. Alloys Compd., 2021, 875, 159911 CrossRef CAS.
- B. Hua, M. Li, Y.-Q. Zhang, J. Chen, Y.-F. Sun, N. Yan, J. Li and J.-L. Luo, Facile synthesis of highly active and robust Ni–Mo bimetallic electrocatalyst for hydrocarbon oxidation in Solid Oxide Fuel Cells, ACS Energy Lett., 2016, 1, 225–230 CrossRef CAS.
- Q. Bkour, F. Che, K.-M. Lee, C. Zhou, N. Akter, J. A. Boscoboinik, K. Zhao, J. T. Gray, S. R. Saunders, M. Grant Norton, J.-S. McEwen, T. Kim and S. Ha, Enhancing the partial oxidation of gasoline with Mo-doped Ni catalysts for SOFC applications: An integrated experimental and DFT study, Appl. Catal., B, 2020, 266, 118626 CrossRef CAS.
- Q. Bkour, K. Zhao, L. Scudiero, D. J. Han, C. W. Yoon, O. G. Marin-Flores, M. G. Norton and S. Ha, Synthesis and performance of ceria-zirconia supported Ni–Mo nanoparticles for partial oxidation of isooctane, Appl. Catal., B, 2017, 212, 97–105 CrossRef CAS.
- X. Hou, K. Zhao, O. A. Marina, M. Grant Norton and S. Ha, NiMo-ceria-zirconia-based anode for solid oxide fuel cells operating on gasoline surrogate, Appl. Catal., B, 2019, 242, 31–39 CrossRef CAS.
- A. J. Majewski, S. K. Singh, N. K. Labhasetwar and R. Steinberger-Wilckens, Nickel–molybdenum catalysts for combined solid oxide fuel cell internal steam and dry reforming, Chem. Eng. Sci., 2021, 232, 116341 CrossRef CAS.
- E. Nikolla, J. Schwank and S. Linic, Promotion of the long-term stability of reforming Ni catalysts by surface alloying, J. Catal., 2007, 250, 85–93 CrossRef CAS.
- E. Nikolla, J. Schwank and S. Linic, Comparative study of the kinetics of methane steam reforming on supported Ni and Sn/Ni alloy catalysts: The impact of the formation of Ni alloy on chemistry, J. Catal., 2009, 263, 220–227 CrossRef CAS.
- E. Nikolla, J. Schwank and S. Linic, Direct electrochemical oxidation of hydrocarbon fuels on SOFCs: improved carbon tolerance of Ni alloy anodes, J. Electrochem. Soc., 2009, 156, 11 CrossRef.
- E. Nikolla, J. Schwank and S. Linic, Hydrocarbon steam reforming on Ni alloys at solid oxide fuel cell operating conditions, Catal. Today, 2008, 136, 243–248 CrossRef CAS.
- H. Kan, S. H. Hyun, Y.-G. Shul and H. Lee, Improved solid oxide fuel cell anodes for the direct utilization of methane using Sn-doped Ni/YSZ catalysts, Catal. Commun., 2009, 11, 180–183 CrossRef CAS.
- H. Kan and H. Lee, Sn-doped Ni/YSZ anode catalysts with enhanced carbon deposition resistance for an intermediate temperature SOFC, Appl. Catal., B, 2010, 97, 108–114 CrossRef CAS.
- Z. Jiang, N. A. Arifin, P. Mardle and R. Steinberger-Wilckens, Electrochemical performance and carbon resistance comparison between tin, copper and silver-doped nickel/yttria-stabilized zirconia anodes SOFCs operated with biogas, J. Electrochem. Soc., 2019, 166, F393–F398 CrossRef CAS.
- A. Singh and J. M. Hill, Evaluation of Sn-modified Ni/YSZ SOFC anodes for the direct utilization of methane, ECS Trans., 2011, 35, 1397–1406 CrossRef CAS.
- B. Farrell and S. Linic, Direct electrochemical oxidation of ethanol on SOFCs: Improved carbon tolerance of Ni anode by alloying, Appl. Catal., B, 2016, 183, 386–393 CrossRef CAS.
- D. Yoon and A. Manthiram, Hydrocarbon-fueled solid oxide fuel cells with surface-modified, hydroxylated Sn/Ni–Ce0.8Gd0.2O1.9 heterogeneous catalyst anode, J. Mater. Chem. A, 2014, 2, 17041–17046 RSC.
- J.-h Myung, S.-D. Kim, T. H. Shin, D. Lee, J. T. S. Irvine, J. Moon and S.-H. Hyun, Nano-composite structural Ni–Sn alloy anodes for high performance and durability of direct methane-fueled SOFCs, J. Mater. Chem. A, 2015, 3, 13801–13806 RSC.
- Q. Yang, J. Chen, C. Sun and L. Chen, Direct operation of methane fueled solid oxide fuel cells with Ni cermet anode via Sn modification, Int. J. Hydrogen Energy, 2016, 41, 11391–11398 CrossRef CAS.
- P. Li, Z. Wang, X. Yao, N. Hou, L. Fan, T. Gan, Y. Zhao, Y. Li and J. W. Schwank, Effect of Sn addition on improving the stability of Ni–Ce0.8Sm0.2O1.9 anode material for solid oxide fuel cells fed with dry CH4, Catal. Today, 2019, 330, 209–216 CrossRef CAS.
- N. A. Arifin, A. H. Shamsuddin and R. Steinberger-Wilckens, Evaluating the drop of electrochemical performance of Ni/YSZ and Ni/ScSZ solid oxide fuel cells operated with dry biogas, Int. J. Energy Res., 2020, 45, 6405–6417 CrossRef.
- A. Singh and J. M. Hill, Carbon tolerance, electrochemical performance and stability of solid oxide fuel cells with Ni/yttria stabilized zirconia anodes impregnated with Sn and operated with methane, J. Power Sources, 2012, 214, 185–194 CrossRef CAS.
- H. Jang, J. Eom, H. Ju and J. Lee, Ameliorated performance in a direct carbon fuel cell using Sn mediator on Ni–YSZ anode surface, Catal. Today, 2016, 260, 158–164 CrossRef CAS.
- A. Cantos-Gómez, R. Ruiz-Bustos and J. van Duijn, Ag as an alternative for Ni in direct hydrocarbon SOFC anodes, Fuel Cells, 2011, 11, 140–143 CrossRef.
- X. Wu, X. Zhou, Y. Tian, X. Kong, J. Zhang, W. Zuo, X. Ye and K. Sun, Preparation and electrochemical performance of silver impregnated Ni–YSZ anode for solid oxide fuel cell in dry methane, Int. J. Hydrogen Energy, 2015, 40, 16484–16493 CrossRef CAS.
- X. Wu, Y. Tian, J. Zhang, W. Zuo, X. Kong, J. Wang, K. Sun and X. Zhou, Enhanced electrochemical performance and carbon anti-coking ability of solid oxide fuel cells with silver modified nickel-yttrium stabilized zirconia anode by electroless plating, J. Power Sources, 2016, 301, 143–150 CrossRef CAS.
- T. Hibino, A. Hashimoto, M. Yano, M. Suzuki, S.-I. Yoshida and M. Sano, High performance anodes for SOFCs operating in methane-air mixture at reduced temperatures, J. Electrochem. Soc., 2001, 149, A133–A136 CrossRef.
- A. Babaei, L. Zhang, E. Liu and S. P. Jiang, Performance and carbon deposition over Pd nanoparticle catalyst promoted Ni/GDC anode of SOFCs in methane, methanol and ethanol fuels, Int. J. Hydrogen Energy, 2012, 37, 15301–15310 CrossRef CAS.
- V. Modafferi, G. Panzera, V. Baglio, F. Frusteri and P. L. Antonucci, Propane reforming on Ni–Ru/GDC catalyst: H2 production for IT-SOFCs under SR and ATR conditions, Appl. Catal., A, 2008, 334, 1–9 CrossRef CAS.
- M. Boaro, V. Modafferi, A. Pappacena, J. Llorca, V. Baglio, F. Frusteri, P. Frontera, A. Trovarelli and P. L. Antonucci, Comparison between Ni–Rh/gadolinia doped ceria catalysts in reforming of propane for anode implementations in intermediate solid oxide fuel cells, J. Power Sources, 2010, 195, 649–661 CrossRef CAS.
- T. Hibino, A. Hashimoto, M. Yano, M. Suzuki and M. Sano, Ru-catalyzed anode materials for direct hydrocarbon SOFCs, Electrochim. Acta, 2003, 48, 2531–2537 CrossRef CAS.
- A. M. Hussain, J. V. T. Høgh, W. Zhang and N. Bonanos, Efficient ceramic anodes infiltrated with binary and ternary electrocatalysts for SOFCs operating at low temperatures, J. Power Sources, 2012, 216, 308–313 CrossRef CAS.
- L. Liu, Q. Yang, W. Yang, X. Qi, C. Sun and L. Chen, Li/Na modified Ni-SDC anode for methane-fueled Solid Oxide Fuel Cells, ECS Trans., 2015, 68, 1403–1409 CrossRef CAS.
- M. Asamoto, S. Miyake, K. Sugihara and H. Yahiro, Improvement of Ni/SDC anode by alkaline earth metal oxide addition for direct methane–solid oxide fuel cells, Electrochem. Commun., 2009, 11, 1508–1511 CrossRef CAS.
- D. La Rosa, A. Sin, M. L. Faro, G. Monforte, V. Antonucci and A. S. Aricò, Mitigation of carbon deposits formation in intermediate temperature solid oxide fuel cells fed with dry methane by anode doping with barium, J. Power Sources, 2009, 193, 160–164 CrossRef CAS.
- S. Islam and J. M. Hill, Barium oxide promoted Ni/YSZ solid-oxide fuel cells for direct utilization of methane, J. Mater. Chem. A, 2014, 2, 1922–1929 RSC.
- M. D. McIntyre, J. D. Kirtley, A. Singh, S. Islam, J. M. Hill and R. A. Walker, Comparing in situ carbon tolerances of Sn-infiltrated and BaO-infiltrated Ni–YSZ cermet anodes in Solid Oxide Fuel Cells exposed to methane, J. Phys. Chem. C, 2015, 119, 7637–7647 CrossRef CAS.
- Y. Itagaki, S. Yamaguchi and H. Yahiro, Anodic performance of BaO-added Ni/SDC for solid oxide fuel cell fed with dry CH4, Front. Energy Res., 2021, 9, 652239 CrossRef.
- Y. H. Hu, Solid-solution catalysts for CO2 reforming of methane, Catal. Today, 2009, 148, 206–211 CrossRef CAS.
- R. Zanganeh, M. Rezaei and A. Zamaniyan, Dry reforming of methane to synthesis gas on NiO–MgO nanocrystalline solid solution catalysts, Int. J. Hydrogen Energy, 2013, 38, 3012–3018 CrossRef CAS.
- Q. Yang, F. Chai, C. Ma, C. Sun, S. Shi and L. Chen, Enhanced coking tolerance of a MgO-modified Ni cermet anode for hydrocarbon fueled solid oxide fuel cells, J. Mater. Chem. A, 2016, 4, 18031–18036 RSC.
- S.-I. Lee, K. Ahn, J. M. Vohs and R. J. Gorte, Cu-Co bimetallic anodes for direct utilization of methane in SOFCs, Electrochem. Solid-State Lett., 2005, 8, 1 Search PubMed.
- M. D. Gross, J. M. Vohs and R. J. Gorte, A study of thermal stability and methane tolerance of Cu-based SOFC anodes with electrodeposited Co, Electrochim. Acta, 2007, 52, 1951–1957 CrossRef CAS.
- A. Fuerte, R. X. Valenzuela, M. J. Escudero and L. Daza, Effect of cobalt incorporation in copper-ceria based anodes for hydrocarbon utilisation in Intermediate Temperature Solid Oxide Fuel Cells, J. Power Sources, 2011, 196, 4324–4331 CrossRef CAS.
- N. Yan, J. Pandey, Y. Zeng, B. S. Amirkhiz, B. Hua, N. J. Geels, J.-L. Luo and G. Rothenberg, Developing a thermal- and coking-resistant cobalt–tungsten bimetallic anode catalyst for Solid Oxide Fuel Cells, ACS Catal., 2016, 6, 4630–4634 CrossRef CAS.
- G. Kaur and S. Basu, Performance studies of copper–iron/ceria–yttria stabilized zirconia anode for electro-oxidation of butane in solid oxide fuel cells, J. Power Sources, 2013, 241, 783–790 CrossRef CAS.
- G. Kaur and S. Basu, Physical characterization and electrochemical performance of copper-iron-ceria-YSZ anode-based SOFCs in H2 and methane fuels, Int. J. Energy Res., 2015, 39, 1345–1354 CrossRef CAS.
- M. Hasegawa, Ellingham Diagram, Treatise Process Metallurgy, 2014, pp. 507–516 Search PubMed.
- C. Sun, H. Li and L. Chen, Nanostructured ceria-based materials: synthesis, properties, and applications, Energy Environ. Sci., 2012, 5, 8475–8505 RSC.
- N. V. Skorodumova, S. I. Simak, B. I. Lundqvist, I. A. Abrikosov and B. Johansson, Quantum origin of the oxygen storage capability of ceria, Phys. Rev. Lett., 2002, 89, 166601 CrossRef CAS PubMed.
- C. Sun, J. Sun, G. Xiao, H. Zhang, X. Qiu, H. Li and L. Chen, Mesoscale organization of nearly monodisperse flowerlike ceria microspheres, J. Phys. Chem. B, 2006, 110, 13445–13452 CrossRef CAS PubMed.
- J. W. Yun, S. P. Yoon, H. S. Kim, J. Han and S. W. Nam, Effect of Sm0.2Ce0.8O1.9 on the carbon coking in Ni-based anodes for solid oxide fuel cells running on methane fuel, Int. J. Hydrogen Energy, 2012, 37, 4356–4366 CrossRef CAS.
- E. S. Putna, J. Stubenrauch, J. M. Vohs and R. J. Gorte, Ceria-based anodes for the direct oxidation of methane in Solid Oxide Fuel Cells, Langmuir, 1995, 11, 4832–4837 CrossRef CAS.
- S. Park, R. Craciun, J. M. Vohs and R. J. Gorte, Direct oxidation of hydrocarbons in a Solid Oxide Fuel Cell: I. methane oxidation, J. Electrochem. Soc., 1999, 146, 3603–3605 CrossRef CAS.
- C. Lu, W. L. Worrell, J. M. Vohs and R. J. Gorte, A comparison of Cu-ceria-SDC and Au-ceria-SDC composites for SOFC anodes, J. Electrochem. Soc., 2003, 150, A1357–A1359 CrossRef CAS.
- O. Marina, A solid oxide fuel cell with a gadolinia-doped ceria anode: preparation and performance, Solid State Ionics, 1999, 123, 199–208 CrossRef CAS.
- M. Wisniewski, A. Boréave and P. Gélin, Catalytic CO2 reforming of methane over Ir/Ce0.9Gd0.1O2−x, Catal. Commun., 2005, 6, 596–600 CrossRef CAS.
- M.-F. Han, J. Xiong, T. Zhu and S. C. Singhal, Performance of Solid Oxide Fuel Cells on H2, NH3 and hydrocarbon fuels, ECS Trans., 2013, 50, 163 CrossRef.
- K. Ahn, H. He, J. M. Vohs and R. J. Gorte, Enhanced thermal stability of SOFC anodes made with CeO2-ZrO2 Solutions, Electrochem. Solid-State Lett., 2005, 8, A414–A417 CrossRef CAS.
- X. Zhou, J. Zhen, L. Liu, X. Li, N. Zhang and K. Sun, Enhanced sulfur and carbon coking tolerance of novel co-doped ceria based anode for solid oxide fuel cells, J. Power Sources, 2012, 201, 128–135 CrossRef CAS.
- D. Neagu and J. T. S. Irvine, Enhancing electronic conductivity in strontium titanates through correlated A and B-Site doping, Chem. Mater., 2011, 23, 1607–1617 CrossRef CAS.
- C. Zhao, Y. Li, W. Zhang, Y. Zheng, X. Lou, B. Yu, J. Chen, Y. Chen, M. Liu and J. Wang, Heterointerface engineering for enhancing the electrochemical performance of solid oxide cells, Energy Environ. Sci., 2020, 13, 53–85 RSC.
- Y. Li, J. Zhang, Q. Chen, X. Xia and M. Chen, Emerging of heterostructure materials in energy storage: A review, Adv. Mater., 2021, 33, e2100855 CrossRef PubMed.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Menard and J. T. Irvine, In situ growth of nanoparticles through control of non-stoichiometry, Nat. Chem., 2013, 5, 916–923 CrossRef CAS PubMed.
- S. He, M. Li, J. Hui and X. Yue, In-situ construction of ceria-metal/titanate heterostructure with controllable architectures for efficient fuel electrochemical conversion, Appl. Catal., B, 2021, 298, 120588 CrossRef CAS.
- S. Vecino-Mantilla, P. Simon, M. Huvé, G. Gauthier and P. Gauthier-Maradei, Methane steam reforming in water-deficient conditions on a new Ni-exsolved Ruddlesden-Popper manganite: Coke formation and H2S poisoning, Int. J. Hydrogen Energy, 2020, 45, 27145–27159 CrossRef CAS.
- Y. Li, W. Zhang, Y. Zheng, J. Chen, B. Yu, Y. Chen and M. Liu, Controlling cation segregation in perovskite-based electrodes for high electro-catalytic activity and durability, Chem. Soc. Rev., 2017, 46, 6345–6378 RSC.
- Y. Sun, J. Li, Y. Zeng, B. S. Amirkhiz, M. Wang, Y. Behnamian and J. Luo, A-site deficient perovskite: the parent for in situ exsolution of highly active, regenerable nano-particles as SOFC anodes, J. Mater. Chem. A, 2015, 3, 11048–11056 RSC.
- A. Marcucci, F. Zurlo, I. N. Sora, E. Placidi, S. Casciardi, S. Licoccia and E. Di Bartolomeo, A redox stable Pd-doped perovskite for SOFC applications, J. Mater. Chem. A, 2019, 7, 5344–5352 RSC.
- N. K. Monteiro, F. B. Noronha, L. O. O. da Costa, M. Linardi and F. C. Fonseca, A direct ethanol anode for solid oxide fuel cell based on a chromite-manganite with catalytic ruthenium nanoparticles, Int. J. Hydrogen Energy, 2012, 37, 9816–9829 CrossRef CAS.
- H. Tanaka, M. Uenishi, M. Taniguchi, I. Tan, K. Narita, M. Kimura, K. Kaneko, Y. Nishihata and J. I. Mizuki, The intelligent catalyst having the self-regenerative function of Pd, Rh and Pt for automotive emissions control, Catal. Today, 2006, 117, 321–328 CrossRef CAS.
- M. van den Bossche and S. McIntosh, Pulse reactor studies to assess the potential of La0.75Sr0.25Cr0.5Mn0.4X0.1O3−δ(X = Co, Fe, Mn, Ni, V) as direct hydrocarbon Solid Oxide Fuel Cell anodes, Chem. Mater., 2010, 22, 5856–5865 CrossRef CAS.
- B. H. Park and G. M. Choi, Ex-solution of Ni nanoparticles in a La0.2Sr0.8Ti1−xNixO3−δ alternative anode for solid oxide fuel cell, Solid State Ionics, 2014, 262, 345–348 CrossRef CAS.
- M. Niania, R. Podor, T. B. Britton, C. Li, S. J. Cooper, N. Svetkov, S. Skinner and J. Kilner, In situ study of strontium segregation in La0.6Sr0.4Co0.2Fe0.8O3−δ in ambient atmospheres using high-temperature environmental scanning electron microscopy, J. Mater. Chem. A, 2018, 6, 14120–14135 RSC.
- W. Lee, J. W. Han, Y. Chen, Z. Cai and B. Yildiz, Cation size mismatch and charge interactions drive dopant segregation at the surfaces of manganite perovskites, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS PubMed.
- B. Koo, K. Kim, J. K. Kim, H. Kwon, J. W. Han and W. Jung, Sr segregation in perovskite oxides: why it happens and how it exists, Joule, 2018, 2, 1476–1499 CrossRef CAS.
- Y. Chen, W. Jung, Z. Cai, J. J. Kim, H. L. Tuller and B. Yildiz, Impact of Sr segregation on the electronic structure and oxygen reduction activity of SrTi1−xFexO3 surfaces, Energy Environ. Sci., 2012, 5, 7979–7988 RSC.
- E. N. Sgourou, Y. Panayiotatos, K. Davazoglou, A. L. Solovjov, R. V. Vovk and A. Chroneos, Self-diffusion in perovskite and perovskite related oxides: insights from modelling, Appl. Sci., 2020, 10, 2286 CrossRef CAS.
- D. Neagu, V. Kyriakou, I. L. Roiban, M. Aouine, C. Tang, A. Caravaca, K. Kousi, I. Schreur-Piet, I. S. Metcalfe, P. Vernoux, M. C. M. van de Sanden and M. N. Tsampas, In situ observation of nanoparticle exsolution from perovskite oxides: from atomic scale mechanistic insight to nanostructure tailoring, ACS Nano, 2019, 13, 12996–13005 CrossRef CAS PubMed.
- Y. Gao, D. Chen, M. Saccoccio, Z. Lu and F. Ciucci, From material design to mechanism study: Nanoscale Ni exsolution on a highly active A-site deficient anode material for solid oxide fuel cells, Nano Energy, 2016, 27, 499–508 CrossRef CAS.
- T. S. Oh, E. K. Rahani, D. Neagu, J. T. Irvine, V. B. Shenoy, R. J. Gorte and J. M. Vohs, Evidence and model for strain-driven release of metal nanocatalysts from perovskites during exsolution, J. Phys. Chem. Lett., 2015, 6, 5106–5110 CrossRef CAS PubMed.
- D. Neagu, T. S. Oh, D. N. Miller, H. Menard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. S. Irvine, Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution, Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- K. J. Kim, H. Han, T. Defferriere, D. Yoon, S. Na, S. J. Kim, A. M. Dayaghi, J. Son, T. S. Oh, H. M. Jang and G. M. Choi, Facet-dependent in situ growth of nanoparticles in epitaxial thin films: the role of interfacial energy, J. Am. Chem. Soc., 2019, 141, 7509–7517 CrossRef CAS PubMed.
- N. Hou, T. Yao, P. Li, X. Yao, T. Gan, L. Fan, J. Wang, X. Zhi, Y. Zhao and Y. Li, A-site ordered double perovskite with in situ exsolved core-shell nanoparticles as anode for Solid Oxide Fuel Cells, ACS Appl. Mater. Interfaces, 2019, 11, 6995–7005 CrossRef CAS PubMed.
- Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Self-regeneration of a Pd-perovskite catalyst for automotive emissions control, Nature, 2002, 418, 164–167 CrossRef CAS PubMed.
- T. Cao, O. Kwon, R. J. Gorte and J. M. Vohs, Metal exsolution to enhance the catalytic activity of electrodes in Solid Oxide Fuel Cells, Nanomaterials, 2020, 10, 2445 CrossRef CAS PubMed.
- Z. Li, M. Li and Z. Zhu, Perovskite cathode materials for low-temperature Solid Oxide Fuel Cells: fundamentals to optimization, Electrochem. Energy Rev., 2021, 5, 263–311 CrossRef.
- Z. Du, H. Zhao, S. Yi, Q. Xia, Y. Gong, Y. Zhang, X. Cheng, Y. Li, L. Gu and K. Świerczek, High-performance anode material Sr2FeMo0.65Ni0.35O6−δ with in situ exsolved nanoparticle catalyst, ACS Nano, 2016, 10, 8660–8669 CrossRef CAS PubMed.
- D.-M. Amaya-Dueñas, G. Chen, A. Weidenkaff, N. Sata, F. Han, I. Biswas, R. Costa and K. A. Friedrich, A-site deficient chromite with in situ Ni exsolution as a fuel electrode for solid oxide cells (SOCs), J. Mater. Chem. A, 2021, 9, 5685–5701 RSC.
- T. Zhu, H. E. Troiani, L. V. Mogni, M. Han and S. A. Barnett, Ni-substituted Sr(Ti,Fe)O3 SOFC anodes: achieving high performance via metal alloy nanoparticle exsolution, Joule, 2018, 2, 478–496 CrossRef CAS.
- W. Kobsiriphat, B. D. Madsen, Y. Wang, L. D. Marks and S. A. Barnett, La0.8Sr0.2Cr1−xRuxO3−δ–Gd0.1Ce0.9O1.95 solid oxide fuel cell anodes: Ru precipitation and electrochemical performance, Solid State Ionics, 2009, 180, 257–264 CrossRef CAS.
- Y. F. Sun, Y. Q. Zhang, J. Chen, J. H. Li, Y. T. Zhu, Y. M. Zeng, B. S. Amirkhiz, J. Li, B. Hua and J. L. Luo, New opportunity for in situ exsolution of metallic nanoparticles on perovskite parent, Nano Lett., 2016, 16, 5303–5309 CrossRef CAS PubMed.
- B. D. Madsen, W. Kobsiriphat, Y. Wang, L. D. Marks and S. A. Barnett, Nucleation of nanometer-scale electrocatalyst particles in solid oxide fuel cell anodes, J. Power Sources, 2007, 166, 64–67 CrossRef CAS.
- C. Sun, J. A. Alonso and J. Bian, Recent advances in perovskite-type oxides for energy conversion and storage applications, Adv. Energy Mater., 2020, 11, 2000459 CrossRef.
- C. Arrivé, T. Delahaye, O. Joubert and G. Gauthier, Exsolution of nickel nanoparticles at the surface of a conducting titanate as potential hydrogen electrode material for solid oxide electrochemical cells, J. Power Sources, 2013, 223, 341–348 CrossRef.
- P. B. Managutti, S. Tymen, X. Liu, O. Hernandez, C. Prestipino, A. Le Gal La Salle, S. Paul, L. Jalowiecki-Duhamel, V. Dorcet, A. Billard, P. Briois and M. Bahout, Exsolution of Ni nanoparticles from A-Site-deficient layered double perovskites for dry reforming of methane and as an anode material for a Solid Oxide Fuel Cell, ACS Appl. Mater. Interfaces, 2021, 13, 35719–35728 CrossRef CAS PubMed.
- Y. F. Sun, J. H. Li, L. Cui, B. Hua, S. H. Cui, J. Li and J. L. Luo, A-site-deficiency facilitated in situ growth of bimetallic Ni-Fe nano-alloys: a novel coking-tolerant fuel cell anode catalyst, Nanoscale, 2015, 7, 11173–11181 RSC.
- Z. Ma, C. Sun, C. Ma, H. Wu, Z. Zhan and L. Chen, Ni doped La0.6Sr0.4 FeO3−δ symmetrical electrode for solid oxide fuel cells, Chin. J. Catal., 2016, 37, 1347–1353 CrossRef CAS.
- Y. Tang, H. Wang, R. Wang, Q. Liu, Z. Yan, L. Xu and X. Liu, Synergistically promoting coking resistance of a La0.4Sr0.4Ti0.85Ni0.15O3−δ anode by Ru-doping-induced active twin defects and highly dispersed Ni nanoparticles, ACS Appl. Mater. Interfaces, 2022, 14, 44002–44014 CrossRef CAS PubMed.
- W. Wang, R. Ran and Z. Shao, Combustion-synthesized ru–Al2O3 composites as anode catalyst layer of a solid oxide fuel cell operating on methane, Int. J. Hydrogen Energy, 2011, 36, 755–764 CrossRef CAS.
- Z. Zhan and S. Barnett, Use of a catalyst layer for propane partial oxidation in solid oxide fuel cells, Solid State Ionics, 2005, 176, 871–879 CrossRef CAS.
- C. Sun, Z. Xie, C. Xia, H. Li and L. Chen, Investigations of mesoporous CeO2–Ru as a reforming catalyst layer for solid oxide fuel cells, Electrochem. Commun., 2006, 8, 833–838 CrossRef CAS.
- L.-l Sun, L.-l Liu, L.-h Luo, Y.-f Wu, J.-j Shi, L. Cheng, X. Xu and Y.-m Guo, Facile synthesis of flower-like Pd catalyst for direct ethanol solid oxide fuel cell, J. Fuel Chem. Technol., 2016, 44, 607–612 CrossRef CAS.
- K. Wang, R. Ran and Z. Shao, Methane-fueled IT-SOFCs with facile in situ inorganic templating synthesized mesoporous Sm0.2Ce0.8O1.9 as catalytic layer, J. Power Sources, 2007, 170, 251–258 CrossRef CAS.
- J. Zhao, X. Xu, W. Zhou, I. Blakey, S. Liu and Z. Zhu, Proton-conducting La-doped ceria-based internal reforming layer for direct methane Solid Oxide Fuel Cells, ACS Appl. Mater. Interfaces, 2017, 9, 33758–33765 CrossRef CAS PubMed.
- P. Qiu, S. Sun, X. Yang, F. Chen, C. Xiong, L. Jia and J. Li, A review on anode on-cell catalyst reforming layer for direct methane solid oxide fuel cells, Int. J. Hydrogen Energy, 2021, 46, 25208–25224 CrossRef CAS.
- C. Jin, C. Yang, F. Zhao, A. Coffin and F. Chen, Direct-methane solid oxide fuel cells with Cu1.3Mn1.7O4 spinel internal reforming layer, Electrochem. Commun., 2010, 12, 1450–1452 CrossRef CAS.
- Y. Chen, B. deGlee, Y. Tang, Z. Wang, B. Zhao, Y. Wei, L. Zhang, S. Yoo, K. Pei, J. H. Kim, Y. Ding, P. Hu, F. F. Tao and M. Liu, A robust fuel cell operated on nearly dry methane at 500 °C enabled by synergistic thermal catalysis and electrocatalysis, Nat. Energy, 2018, 3, 1042–1050 CrossRef CAS.
- Z. Zhan and S. A. Barnett, An octane-fueled solid oxide fuel cell, Science, 2005, 308, 844–847 CrossRef CAS PubMed.
- S. Futamura, A. Muramoto, Y. Tachikawa, J. Matsuda, S. M. Lyth, Y. Shiratori, S. Taniguchi and K. Sasaki, SOFC anodes impregnated with noble metal catalyst nanoparticles for high fuel utilization, Int. J. Hydrogen Energy, 2019, 44, 8502–8518 CrossRef CAS.
- J. Kim, Y. Choi, D.-K. Lim, J. Yoo, H. G. Seo, S. Kim, S. Kim and W. Jung, Electrochemically plated nickel-decorated ceria nanostructures for direct hydrocarbon solid oxide fuel cell electrodes, J. Mater. Chem. A, 2022, 10, 20886–20895 RSC.
- B. Hua, M. Li, Y.-F. Sun, Y.-Q. Zhang, N. Yan, J. Chen, J. Li, T. Etsell, P. Sarkar and J.-L. Luo, Biogas to syngas: flexible on-cell micro-reformer and NiSn bimetallic nanoparticle implanted solid oxide fuel cells for efficient energy conversion, J. Mater. Chem. A, 2016, 4, 4603–4609 RSC.
- K. Zhao, X. Hou, M. G. Norton and S. Ha, Application of a NiMo–Ce0.5Zr0.5O2−δ catalyst for solid oxide fuel cells running on gasoline, J. Power Sources, 2019, 435, 226732 CrossRef CAS.
- L. Yang, Y. Choi, W. Qin, H. Chen, K. Blinn, M. Liu, P. Liu, J. Bai, T. A. Tyson and M. Liu, Promotion of water-mediated carbon removal by nanostructured barium oxide/nickel interfaces in solid oxide fuel cells, Nat. Commun., 2011, 2, 357 CrossRef PubMed.
- J. Xu and G. F. Froment, Methane steam reforming, methanation and water-gas shift: I. Intrinsic kinetics, AIChE J., 1989, 35, 88–96 CrossRef CAS.
- K. Ahmed and K. Föger, Analysis of equilibrium and kinetic models of internal reforming on solid oxide fuel cell anodes: Effect on voltage, current and temperature distribution, J. Power Sources, 2017, 343, 83–93 CrossRef CAS.
- A. C. van Duin, B. V. Merinov, S. S. Han, C. O. Dorso and W. A. Goddard, 3rd, ReaxFF reactive force field for the Y-doped BaZrO3 proton conductor with applications to diffusion rates for multigranular systems, J. Phys. Chem. A, 2008, 112, 11414–11422 CrossRef CAS PubMed.
- A. C. T. van Duin, B. V. Merinov, S. S. Jang and W. A. Goddard, ReaxFF reactive force field for solid oxide fuel cell systems with application to oxygen ion transport in yttria-stabilized zirconia, J. Phys. Chem. A, 2008, 112(14), 3133–3140 CrossRef CAS PubMed.
- H. Lu, D. Hua, T. Iqabl, X. Zhang, G. Li and D. Zhang, Molecular dynamics simulations of the coke formation progress on the nickel-based anode of solid oxide fuel cells, Int. Commun. Heat Mass Transfer, 2018, 91, 40–47 CrossRef CAS.
- J. W. Kim, K. Bae, H. J. Kim, J.-W. Son, N. Kim, S. Stenfelt, F. B. Prinz and J. H. Shim, Three-dimensional thermal stress analysis of the re-oxidized Ni–YSZ anode functional layer in solid oxide fuel cells, J. Alloys Compd., 2018, 752, 148–154 CrossRef CAS.
- Y. Guo, M. Bessaa, S. Aguado, M. C. Steil, D. Rembelski, M. Rieu, J.-P. Viricelle, N. Benameur, C. Guizard, C. Tardivat, P. Vernoux and D. Farrusseng, An all porous solid oxide fuel cell (SOFC): a bridging technology between dual and single chamber SOFCs, Energy Environ. Sci., 2013, 6, 2119–2123 RSC.
- Y. M. Guo, G. Largiller, C. Guizard, C. Tardivat and D. Farrusseng, Coke-free operation of an all porous solid oxide fuel cell (AP-SOFC) used as an O2 supply device, J. Mater. Chem. A, 2015, 3, 2684–2689 RSC.
- H. Xu, B. Chen, P. Tan, W. Cai, W. He, D. Farrusseng and M. Ni, Modeling of all porous solid oxide fuel cells, Appl. Energy, 2018, 219, 105–113 CrossRef CAS.
- E. Audasso, F. R. Bianchi and B. Bosio, 2D simulation for CH4 internal reforming-SOFCs: an approach to study performance degradation and optimization, Energies, 2020, 13, 4116 CrossRef CAS.
- Y. Su, T. Wei, Y. Li, B. Yin, Y. Huan, D. Dong, X. Hu and B. Huang, A highly active CH4 catalyst correlated with solid oxide fuel cell anode performance, J. Mater. Chem. A, 2021, 9, 5067–5074 RSC.
- X.-F. Ye, B. Huang, S. R. Wang, Z. R. Wang, L. Xiong and T. L. Wen, Preparation and performance of a Cu–CeO2–ScSZ composite anode for SOFCs running on ethanol fuel, J. Power Sources, 2007, 164, 203–209 CrossRef CAS.
- X.-F. Ye, S. R. Wang, Q. Hu, J. Y. Chen, T. L. Wen and Z. Y. Wen, Improvement of Cu–CeO2 anodes for SOFCs running on ethanol fuels, Solid State Ionics, 2009, 180, 276–281 CrossRef CAS.
- X. Hao, D. Han, J. Wang, Y. Liu, D. Rooney, W. Sun, J. Qiao, Z. Wang and K. Sun, Co-tape casting fabrication, field assistant sintering and evaluation of a coke resistant La0.2Sr0.7TiO3–Ni/YSZ functional gradient anode supported solid oxide fuel cell, Int. J. Hydrogen Energy, 2015, 40, 12790–12797 CrossRef CAS.
- M. Wang, N. Li, Z. Wang, C. Chen and Z. Zhan, Electrochemical performance and redox stability of solid oxide fuel cells supported on dual-layered anodes of Ni–YSZ cermet and Ni–Fe alloy, Int. J. Hydrogen Energy, 2022, 47, 5453–5461 CrossRef.
- S. Sengodan, S. Choi, A. Jun, T. H. Shin, Y. W. Ju, H. Y. Jeong, J. Shin, J. T. Irvine and G. Kim, Layered oxygen-deficient double perovskite as an efficient and stable anode for direct hydrocarbon solid oxide fuel cells, Nat. Mater., 2015, 14, 205–209 CrossRef CAS PubMed.
- M. Li, B. Hua, Y. Zeng, B. Shalchi Amirkhiz and J.-L. Luo, Thermally stable and coking resistant CoMo alloy-based catalysts as fuel electrodes for solid oxide electrochemical cells, J. Mater. Chem. A, 2018, 6, 15377–15385 RSC.
- R. Lang, T. Li, D. Matsumura, S. Miao, Y. Ren, Y. T. Cui, Y. Tan, B. Qiao, L. Li, A. Wang, X. Wang and T. Zhang, Hydroformylation of olefins by a rhodium single-atom catalyst with activity comparable to RhCl(PPh3)3, Angew. Chem., Int. Ed., 2016, 55, 16054–16058 CrossRef CAS PubMed.
- Y. Pan, S. Liu, K. Sun, X. Chen, B. Wang, K. Wu, X. Cao, W. C. Cheong, R. Shen, A. Han, Z. Chen, L. Zheng, J. Luo, Y. Lin, Y. Liu, D. Wang, Q. Peng, Q. Zhang, C. Chen and Y. Li, A bimetallic Zn/Fe polyphthalocyanine-derived single-atom Fe-N4 catalytic site: a superior trifunctional catalyst for overall water splitting and Zn-air batteries, Angew. Chem., Int. Ed., 2018, 57, 8614–8618 CrossRef CAS PubMed.
- J. Han, J. Bian and C. Sun, Recent advances in single-atom electrocatalysts for oxygen reduction reaction, Research, 2020, 2020, 9512763 CAS.
- Y. Tang, Y. Wei, Z. Wang, S. Zhang, Y. Li, L. Nguyen, Y. Li, Y. Zhou, W. Shen, F. F. Tao and P. Hu, Synergy of single-atom Ni1 and Ru1 sites on CeO2 for dry reforming of CH4, J. Am. Chem. Soc., 2019, 141, 7283–7293 CrossRef CAS PubMed.
- J. Wu, J. Gao, S. Lian, J. Li, K. Sun, S. Zhao, Y. D. Kim, Y. Ren, M. Zhang, Q. Liu, Z. Liu and Z. Peng, Engineering the oxygen vacancies enables Ni single-atom catalyst for stable and efficient C-H activation, Appl. Catal., B, 2022, 314, 121516 CrossRef CAS.
- C. T. Campbell, The energetics of supported metal nanoparticles: relationships to sintering rates and catalytic activity, Acc. Chem. Res., 2013, 46, 1712–1719 CrossRef CAS PubMed.
- K. Liu, X. Zhao, G. Ren, T. Yang, Y. Ren, A. F. Lee, Y. Su, X. Pan, J. Zhang, Z. Chen, J. Yang, X. Liu, T. Zhou, W. Xi, J. Luo, C. Zeng, H. Matsumoto, W. Liu, Q. Jiang, K. Wilson, A. Wang, B. Qiao, W. Li and T. Zhang, Strong metal-support interaction promoted scalable production of thermally stable single-atom catalysts, Nat. Commun., 2020, 11, 1263 CrossRef CAS PubMed.
- X. Li, Z. Chen, Y. Yang, D. Huan, H. Su, K. Zhu, N. Shi, Z. Qi, X. Zheng, H. Pan, Z. Zhan, C. Xia, R. Peng, S. Wei and Y. Lu, Highly stable and efficient Pt single-atom catalyst for reversible proton-conducting solid oxide cells, Appl. Catal., B, 2022, 316, 121627 CrossRef CAS.
| This journal is © the Partner Organisations 2023 |