
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular luminescent Pt(II) tweezers: aggregation studies and 1O2 production†
Guillermo
Romo-Islas
ab,
Rosa M.
Gomila
 c,
Antonio
Frontera
*c and
Laura
Rodríguez
*ab
c,
Antonio
Frontera
*c and
Laura
Rodríguez
*ab
aDepartament de Química Inorgànica i Orgànica, Secció de Química Inorgànica, Universitat de Barcelona, Martí i Franquès 1-11, E-08028 Barcelona, Spain. E-mail: laurarodriguezr@ub.edu
bInstitut de Nanociència i Nanotecnologia (IN2UB). Universitat de Barcelona, 08028 Barcelona, Spain
cDepartament de Química, Universitat de les Illes Balears, 07122 Palma de Mallorca, Spain. E-mail: toni.frontera@uib.es
First published on 27th July 2023
Abstract
Four series of Pt(II) tweezer compounds have been designed and synthesized in order to explore their ability to form supramolecular assemblies with luminescence ability. The compounds present the general chemical structure (diphos)Pt(N^N^N), where diphos is a diphosphane that differs in the flexibility and chain length (diphos = bis(diphenylphosphanyl)methane, dppm; 1,2-bis(diphenylphosphanyl)ethane, dppe and 1,2-bis(diphenylphosphanyl)benzene, dppbz) and confers the tweezer structure to the complexes. The four series are defined as a function of the substituents of the N^N^N ligands with different electronic properties (hydrogen vs. chloride) and bulkiness (p-tolyl vs. CF3). We can identify the presence of intra- and intermolecular Pt⋯Pt, Pt⋯π and π⋯π interactions based on the spectroscopic absorption and emission profiles and supported by DFT calculations giving rise to aggregates with different stabilities. The aggregation process has been carefully analyzed both in solution and in the solid state. The resulting emission properties are affected by aggregation, exemplified by the phosphorescence emission enhancement in the absence of the chloride substituent.
Introduction
The supramolecular chemistry of molecules and the resulting supramolecular functional materials are built upon a deep knowledge of the various noncovalent inter- and intramolecular forces. They are involved in the construction of high-order hierarchical structures through self-assembly processes that encompass the rational previous design of synthetic building blocks. It is highly relevant to consider the interplay of all kinds of possible weak interactions, both intra- and intermolecular contacts, and those with the surrounding microenvironment (e.g. possible solvation effect or formation of ion pairs). The deep knowledge of these weak interactions arises from complementary studies on different fields of research such as organic and inorganic chemistry, coordination chemistry, theoretical studies of non-covalent interactions, molecular recognition and molecular devices among others.1–18Regarding this, the use of transition metals (TM) as individual molecules or building blocks is particularly interesting due to their versatility. This strategy offers diverse control in the coordination geometry since TM can present a wide variety of coordination numbers that will drive the resulting geometries. Additionally, TM derivatives are of great interest from the photophysical point of view since those bearing a heavy metal center can promote efficient intersystem crossing (ISC) and populate the triplet excited state giving rise to phosphorescent emitters or energy transfer processes.19–22 When they are involved in the energy transfer processes, they can act as photosensitizers (PS) when the transfer of energy occurs from the molecules (PS) to the molecular oxygen to form the reactive singlet oxygen species, 1O2.23,24 The production of 1O2 is a research area that has seen increasing investigation in the last few years due to the wide variety of applications in fields such as organic synthesis of fine chemicals, destruction of contaminants (environmental issues) and photodynamic therapy.25–28
Among the different TM complexes, square planar d8 platinum derivatives are extensively explored due to their capability to establish metal⋯metal weak contacts (in addition to other non-covalent interactions).25–27 They are also well known for their rich photophysical properties and thus, they are interesting candidates to be used in the construction of luminescent supramolecular structures. On the other hand, the planar geometry of these complexes plays a key role in the possible formation of supramolecular aggregates affecting their spectroscopic properties due to a maximization of the orbital interactions between two π-systems and then enriching their photophysical and photochemical properties.28 In fact, the different degrees of aggregation modulate the emission wavelength (color), intensity, emission lifetime and quantum yields.13,29 The formation of these aggregates may affect their potential applications in different fields such as molecular recognition (sensing) and optoelectronic devices or even a combination of both by the design of optoelectronic architectures used as sensors where the detection process may depend on the weak interactions of the supramolecular assemblies.29
Molecular tweezers are one class of supramolecular structures that deserve investigation in different fields such as molecular recognition and selectivity by the fine tuning of the tweezer-cavity, flexibility and weak interactions with specific guest molecules.30–33 A tweezer molecule is characterized by the presence of two flat moieties (generally aromatic and identical) that are separated by a spacer with different rigidities and lengths. In this regard, diphosphanes are ideal candidates to be used as spacer units. They can easily coordinate to platinum(II) centers and thus, are adequate for the design and synthesis of supramolecular luminescent platinum tweezers. The choice of the particular diphosphane will confer the requisite rigidity, flexibility and aromaticity to the molecule. In the case of having an alkyl bridge between the two phosphorus atoms, the global aromaticity is broken and the tweezer acts as two monodentate phosphane systems. These supramolecular diphosphane platinum tweezers are relevant due to their chelating properties, the analysis of the establishment of intra- and intermolecular contacts, the understanding of the supramolecular assemblies and the resulting photophysical properties.
Taking all this into consideration, in this work, we have designed four series of diphosphane platinum tweezers that differ in the diphosphane bridging ligand. The platinum environment contains a N^N^N tridentate ligand with different substituents that also play an important role in the resulting supramolecular assemblies and observed emission. DFT calculations of the modelled systems disclose the existence of metallophilic interactions in all dinuclear complexes and different degrees of π-stacking of the N^N^N tridentate ligands. Moreover, the relative ability of self-aggregation has been investigated for one of the diphosphane bridging ligands, evidencing that the substituents on the N^N^N tridentate ligand strongly influence the aggregation.
Results and discussion
Synthesis and characterization
The synthesis of bis-(3-(substituted)-1H-1,2,4-triazol-5-yl)pyridine (N^N^N ligands H2L1–H2L4) was carried out following a similar procedure previously reported in the literature for H2L3.34 That is, the corresponding dicyanopyridine and hydrazine hydrate compounds were reacted in methanol at room temperature to generate the hydrazonamide intermediate A. Then, p-tolyl chloride (for H2L1 and H2L2, Scheme 1 above) or trifluoroacetic acid (for H2L3 and H2L4, Scheme 1 below) and ethylene glycol were added at 180 °C for 4 h in order to favor the cyclization of the molecule and to obtain the desired ligands (Scheme 1). | ||
| Scheme 1 Synthesis of the H2L1–H2L4 ligands. | ||
The four ligands differ in two different substituents: (i) the X group at the para position of the central pyridyl ring, which can be a H or a Cl atom (with different electronic density characteristics) and (ii) the substituent at the triazole rings, being p-tolyl or CF3, with different steric hindrance and electron-donating or electron-withdrawing characteristics. These chemical modifications have a direct impact on the resulting color of the powders, being white in the case of H2L1 and H2L3 (X = H) and brown and yellow in the case of H2L2 and H2L4 respectively (X = Cl).
The 1H NMR spectra of the four ligands show 0.16 and 0.09 ppm downfield shifts of the central pyridyl protons respectively for the ligands H2L1 and H2L3 compared to the protons of the 2,6-pyridinedicarbonitrile precursor. This shift is larger (2.46 and 0.45 ppm for the ligands H2L2 and H2L4 respectively) when a chloride substituent is present in this aromatic group. The IR spectra of all compounds show two well-defined signals between 3423–3410 cm−1 and 1694–1611 cm−1 that correspond to the N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibrations. The molecular [M + H]+ peaks recorded in the mass was the final evidence of the correct formation of the desired products.
N stretching vibrations. The molecular [M + H]+ peaks recorded in the mass was the final evidence of the correct formation of the desired products.
H2L1–H2L4 were reacted with the previously synthesized [PtCl2(DMSO)2] precursor in the presence of N,N-diisopropylethylamine (DIPEA) as a base to obtain the desired [Pt(DMSO)(L)] products in high yields (Scheme 2).35
 | ||
| Scheme 2 Synthesis of the [Pt(DMSO)(L)] compounds. | ||
The 1H NMR spectra of the four Pt(II) compounds show a signal centered at 2.70 ppm that corresponds to the protons of the methyl groups from DMSO coordinated to the Pt(II) center. The pyridyl protons display a downfield shift for the compounds [Pt(DMSO)L1] (0.02 ppm), [Pt(DMSO)L2] (1.24 ppm), and [Pt(DMSO)L3] (0.21 ppm). In contrast, the pyridyl protons of [Pt(DMSO)L4] are 0.38 ppm upfield shifted with respect to H2L4. This effect could be due to the two different electron-withdrawing groups (–Cl and –CF3) of the N^N^N ligand. The corresponding IR spectra present the additional SO stretching vibration band centered between 1023–1018 cm−1. These signals are shifted at lower energies compared to the corresponding SO of the free DMSO at 1044 cm−1.
Finally, the [Pt(DMSO)(L)] compounds were reacted with three different diphosphane ligands (bis(diphenylphosphino)methane, dppm, bis(diphenylphosphino)ethane, dppe, and bis(diphenylphosphino)benzene, dppbz) in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry, under an inert atmosphere, to yield the desired final Pt(II) tweezer compounds as shown in Scheme 3. The three different diphosphanes differ in the chain length between the two phosphorus atoms and their flexibility.
:1 stoichiometry, under an inert atmosphere, to yield the desired final Pt(II) tweezer compounds as shown in Scheme 3. The three different diphosphanes differ in the chain length between the two phosphorus atoms and their flexibility.
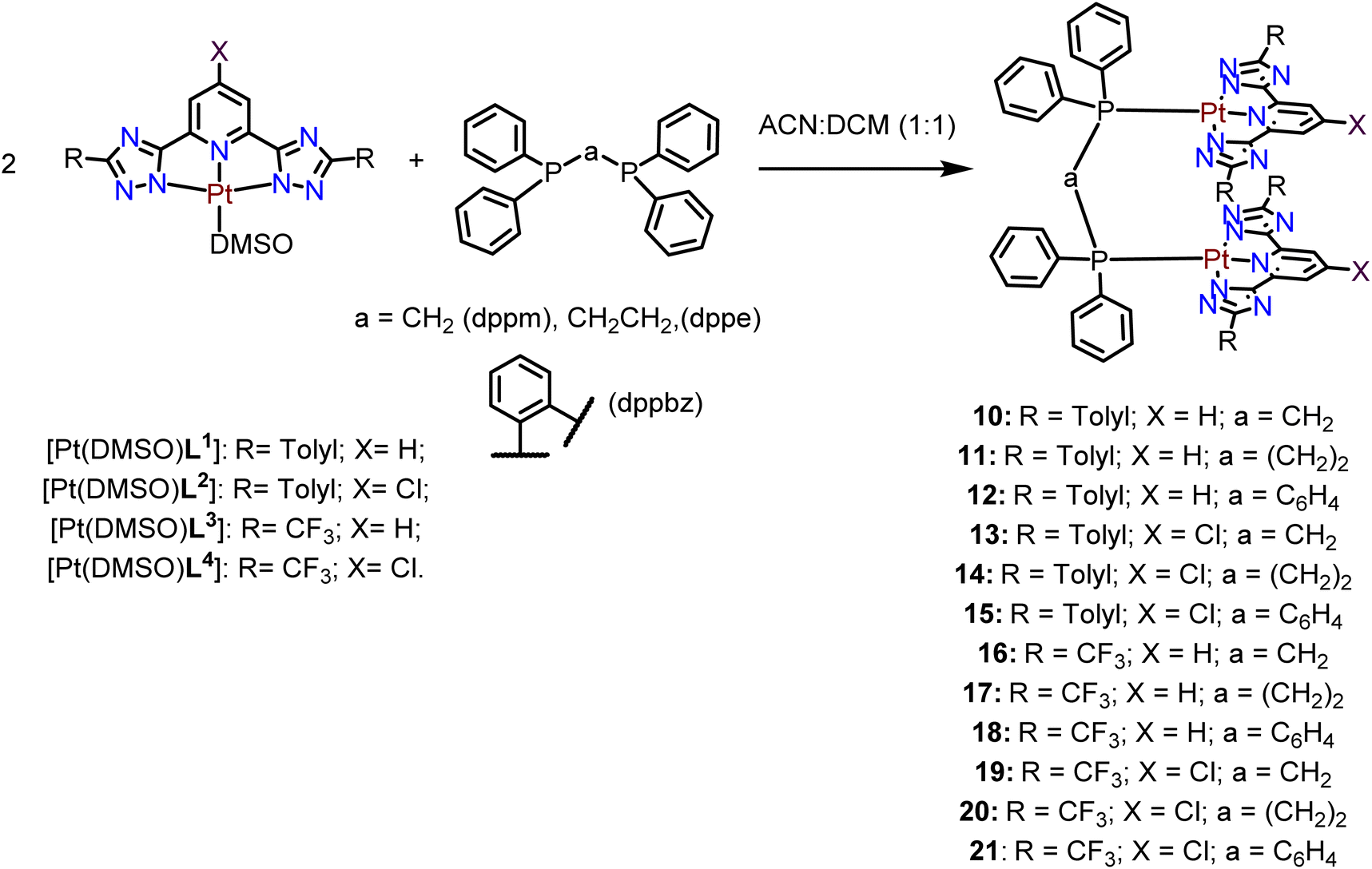 | ||
| Scheme 3 Synthesis of the Pt(II) tweezers. | ||
The 31P{1H} NMR spectra of the Pt(II) tweezers show, in all cases, an ca. 40–60 ppm downfield shift for the tweezers with the dppm and dppbz derivatives. These signals are flanked by platinum satellites, being a direct proof of the successful coordination of the diphosphanes. The 31P{1H} NMR spectra of dppe derivatives with the L1 and L3 ligands and X = H (compounds 11 and 17) present two different peaks that may be ascribed to cis/trans isomerization in the media due to the two different JPt–P coupling constants, 1173 and 1830 Hz, in agreement with cis and trans Pt(II) isomers containing diphosphane ligands, respectively.36,37 Additionally, the 1H NMR spectra of the compounds display the corresponding protons of the bridging diphosphane ligands. Several attempts to grow single crystals suitable for X-ray diffraction were unsuccessful. Thus, in order to investigate the most stable conformations of these compounds, DFT optimization was performed using the RI-BP86-D3/def2-TZVP level of theory, which is a good trade-off between the size of the systems and the accuracy of the results. The minimum energy geometries for compounds 10–12 are given in Fig. 1 and the rest in Fig. S43–S45.† The complexes display the expected face-to-face tweezer shape with slightly different Pt⋯Pt distances, ranging from 3.151 Å in 11 to 3.442 Å in 10. The main difference is the rotation of both tridentate ligands, one with respect to the other. That is, in compound 10 the two methyl groups of the stacked tolyl moieties point to the same direction (relative angle ∼0°), in 11 they are rotated ∼46° and in compound 12 they are rotated ∼64° (see Fig. 1, green arrows and angles). These differences can be related to the flexibility of the ligand and additional fine tuning of the final geometry due to the combination of metallophilic and π-stacking interactions.
 | ||
| Fig. 1 (a–c) Ball and stick representation of the minimum energy geometries using two orientations calculated for compounds 10–12 at the RI-BP86-D3/def2-TZVP level of theory. The Pt⋯Pt distances are also indicated. | ||
Photophysical characterization
The absorption and emission spectra of the Pt(II) tweezers were recorded in 10−5 M acetonitrile solutions at room temperature (Table 1). The absorption spectra of the compounds display broad bands centered between 270 and 350 nm and assigned to intraligand (1IL) and metal-to-ligand charge transfer (1MLCT) according to the literature (Fig. S46 and 47†).38–42 The broadening of these bands can be ascribed to some stable intra- and intermolecular weak interactions as predicted by DFT calculations due to the presence of Pt⋯Pt intramolecular contacts and in the computed stable dimers (see below). Chloride N^N^N substituted derivatives display an ca. 5 nm blue shift of the lowest energy absorption band which has been attributed to a lower coordination capacity in this ligand due to a decrease in the energy of the occupied d orbitals of the metal.43| Complex | A (ε, × 103 cm−1 m−1) | λ max Em (nm) | Φ fl (air-eq./N2 sat) | Φ phos (air-eq./N2 sat) | τ (air-eq./N2 sat, μs) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Corresponding to the highest energy emission band. The full values with their corresponding amplitudes are shown in Fig. S2.† | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 263 (59.38), 310 (30.97), 340 (16.58) | 570 | —/— | 0.01/0.09 | 0.96/4.02 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 255 (61.32), 267 (54.42), 315 (26.34) | 588 | —/— | 0.03/0.06 | 0.61/3.32 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 265 (45.44), 310 (22.37), 340 (11.56) | 586 | —/— | 0.02/0.09 | 0.71/2.41 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | 264 (25.85), 276 (19.33), 304 (10.54) | 435 | 0.01/0.01 | —/— | 3.7 × 10−3/4.7 × 10−3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | 273 (40.46), 310 (16.63) | 433 | 0.01/0.01 | —/— | 5.0 × 10−3/5.3 × 10−3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | 268 (46.37), 282 (34.97), 307 (14.62) | 432 | 0.01/0.01 | —/— | 5.8 × 10−3/4.7 × 10−3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 | 288 (21.37), 300 (19.33), 330 (7.35) | 365 | 0.01/0.01 | —/— | 4.4 × 10−3/8.3 × 10−3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 17 | 255 (37.22), 275 (26.55), 305 (18.77), 345 (8.47) | 358, 466, 500 | 0.01/0.01 | 0.01/0.02 | 5.6 × 10−3/7.1 × 10−3a |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 18 | 264 (48.92), 303 (27.10) | 364, 466, 497 | 0.01/0.01 | 0.01/0.02 | 2.2 × 10−3/2.3 × 10−3a |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 19 | 271 (27.67), 305 (12.95) | 356 | 0.01/0.01 | —/— | 1.4 × 10−3/1.4 × 10−3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 20 | 255 (37.97), 306 (13.09) | 361 | 0.01/0.01 | —/— | 1.4 × 10−3/1.5 × 10−3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 21 | 267 (34.68), 310 (13.96) | 365 | 0.01/0.01 | —/— | 1.5 × 10−3/1.6 × 10−3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
A new band at longer wavelengths (around 420 nm) has also been recorded in the excitation spectra of the p-tolyl derivatives 10–15 (Fig. S48†) and is absent in the corresponding uncoordinated H2L ligands (see the example in Fig. S48† for compound 10 and its precursors). This new band can be ascribed to the presence of aggregates in the ground state that are most favored for the p-tolyl derivatives since the spherical steric hindrance of the CF3 group hinders the formation of intermolecular contacts as has been supported by theoretical calculations (see below) and previous literature data.34,38
To further support the assignments of the bands, TD-DFT calculations were performed for the complexes with the most flexible diphosphane (dppe) and the smallest ligands L3 and L4 for computational economy. We computed the first 20 excited states using the CAM-B3LYP/def2-TZVP level of theory for compounds 17 and 20, and the results are presented in Fig. 2 and S49.† The lowest lying band corresponds to S0 → S3 excitation (λexc = 351 nm, f = 0.151) that is composed of two transitions (HOMO−1 → LUMO and HOMO → LUMO) with quite different contributions of 80 and 13%, respectively (other minor contributions are not shown for simplicity). The theoretical λ agrees well with the experimental one (see Table 1).
 | ||
| Fig. 2 Representation of the MOs involved in the S0 → S3 excitation of compound 17, with indication of the theoretical λexc, oscillator strength and relative contribution of each transition. | ||
The HOMO−1 is basically composed of both dz2 orbitals of Pt ions with a small contribution of the π-systems of the tridentate ligand. The HOMO is very similar to the HOMO−1 also dominated by the dz2 atomic orbitals of Pt(II). In contrast, the LUMO is localized basically at the aromatic rings of L3 disclosing the metal → ligand charge transfer. The TD-DFT analysis of compound 20, which incorporates chlorine in the pyridine ring, is similar in the sense that the band corresponds to S0 → S3 excitation involving the same transitions (HOMO−1 → LUMO and HOMO → LUMO). However, the relative contributions are opposite, with HOMO → LUMO being the dominant one (68%). Another significant difference is related to the composition of the LUMO−1, which is located at the aromatic rings. Therefore, the excitation can be defined as a mixture of ILCT and MLCT in 20. Since the only difference between the compositions of compounds 17 and 20 is the chlorine substituent in the pyridine ring, it can be concluded that this apparently innocent atom has a significant influence on the charge transfer.
The emission spectra of the compounds were recorded upon excitation with the samples at the lowest band recorded in the excitation spectra (Fig. 3 and S50–S51†). Different profiles can be observed in the different series of compounds. While pure room temperature phosphorescence (RTP) emission bands centered around 580 nm were recorded for compounds 10–12 (with X = H and the p-tolyl substituent), fluorescence emission governs the radiative deactivation processes of the other series of complexes. This could be ascribed to the lower steric hindrance of the compounds that is translated in a more favored intersystem crossing process. The planarity of the systems is expected due to the presence of the smaller hydrogen atom compared to the chlorine at the central pyridyl ring and to the more planar p-tolyl group instead of the more spherical CF3 at the triazole units of the N^N^N ligand. In this way, the two arms of the tweezer can more efficiently interact in solution, being in closer proximity and favoring the Pt⋯Pt interactions and thus, the intersystem crossing due to a higher (reinforced) heavy atom effect. On the other hand, it can be observed that the presence of the chloride substituent in the N^N^N tridentate ligand (L2 and L4) induces only fluorescence emission since some phosphorescence emission can also be detected for L3 derivatives (Fig. 3B and D). This could be related to the different orbital composition of the absorption band commented above.
 | ||
| Fig. 3 Emission spectra of the compounds in N2-saturated 1 × 10−5 M acetonitrile solutions (λexc = 365 for A and B and 330 nm for C and D). * Indicates residual of the Raman band. | ||
The observed emissions have been attributed to metal perturbed 3IL transition in the case of 13–15 (considering the emission energies and lifetimes and due to the vibronically structural shape41) and to 3MMLCT (due to the presence of intermolecular contacts, that is, aggregates)34 for the broad phosphorescence bands of 10–12 transitions and metal perturbed 3IL for the vibronically structured phosphorescence bands in 16–18 as described in the literature for similar N^N^N compounds and in agreement with the recorded values of decay times.34,38–40,42,44–46
Taking this into consideration, we can say that (i) the insertion of the chlorine atom at the para position of the pyridyl rings plays an important role in the emissive properties of the tweezers favoring fluorescence emission and (ii) the presence of the CF3 substituents does not favor the intermolecular contacts probably due to the spherical bulkiness of this group.
Some preliminary assays were performed to analyze the potential role of our tweezer compounds as sensors of polyaromatic hydrocarbon (PAH) guest molecules, which are well known as contaminants. These experiments have been performed considering the luminescence properties of the compounds that can be modified as a result of a sensing process. Thus, the addition of increasing amounts of either naphthalene or phenanthrene guests into an acetonitrile solution of 11 induces quenching of the room temperature phosphorescence emission band (see Fig. S52A and S52B†). We can expect similar host:guest interactions for both PAHs tested (in agreement with similar quenching effects) with association constant values, Kass, of 2 and 1.5 × 104 M−1 for the interaction with naphthalene and phenanthrene respectively. Hence, the Pt⋯Pt weak interactions are being modified due to the inclusion of the PAH within the tweezer structure. This opens new applications for our molecules that deserve to be explored in detail in future investigations.
Some additional information regarding the emission processes of these compounds can be retrieved from the calculated radiative and non-radiative rate constants (kr and knr) and from the corresponding luminescence quantum yields and lifetimes (Table 1 and Fig. S56–S79†). It can be observed in Table S1† that in all cases the non-deactivation pathways (knr values) are much more favored being in general two orders of magnitude larger than the corresponding kr. The increase in the phosphorescence emission intensity in compounds 10–12 upon deoxygenation is due to a decrease in the non-radiative deactivation pathways while radiative deactivation processes are not substantially affected. This is due to an increase of both emission quantum yields and lifetimes. On the other hand, tolyl derivatives with the Cl atom in the structure of ligands 13–15, and CF3 derivatives 16–21 seem to favor both the radiative and non-radiative deactivation channels with kr and knr values that are at least two orders of magnitude larger than those for the p-tolyl substituted compounds.
The emission spectra of the compounds were also recorded in the solid state. It can be observed that, in this case, only the compounds that contain the unsubstituted N^N^N tridentate ligands (X = H) are emissive (compounds 10–12 and 16–18). The emission of the p-tolyl derivatives 10–12 is quite similar to that recorded in solution (Fig. 4 left vs.Fig. 3A) with broad phosphorescence emission around 550 nm. On the other hand, the recorded emission of 16–18 in the solid state is dominated by the IL phosphorescence and a new red-shifted band around 600 nm appears due to the formation of aggregates (Fig. 4 right vs.Fig. 3B). This fact also supports the formation of aggregates both in solution and in the solid state for compounds 10–12 that are favored for 16–18 in the solid state producing this red-shift of their emission spectra (Table 2).
 | ||
| Fig. 4 Emission spectra of 10–12 and 16–18 in the solid state upon excitation the samples at the lower energy excitation band (λexc = 365 nm). | ||
| Complex | λ max Em (nm) | Φ | τ (μs) | k r (× 105 s−1) | k nr (× 105 s−1) |
|---|---|---|---|---|---|
| 10 | 558 | 0.06 | 4.59 | 0.13 | 2.05 |
| 11 | 567 | 0.02 | 1.61 | 0.12 | 6.09 |
| 12 | 549 | 0.05 | 2.52 | 0.20 | 3.77 |
| 16 | 467, 495, 530 | 0.02 | 0.65 | 0.30 | 15.1 |
| 17 | 479, 506, 594 | 0.04 | 1.37 | 0.29 | 7.01 |
| 18 | 458, 487, 521, 600 | 0.07 | 2.49 | 0.28 | 3.73 |
1H and 31P{1H} NMR experiments were performed for compounds 13 and 14 at different concentrations in order to retrieve experimental information about the intermolecular aggregation processes. These two compounds were chosen since, in agreement with the DFT calculations, we should expect a face-to-face (for 13) or tilted (for 14) conformation of the molecules that would be reflected in more shifted chemical shifts in the first case (dppm derivative) than in the second (dppe derivative). Nevertheless, no significant shifts have been recorded in any of the cases (see Fig. S77–S80†). Thus, we can expect that the formation of aggregates happens at lower concentrations (experimental conditions required for absorption and emission spectra being lower than those required for NMR). We must point out that these experiments were performed in DMSO-d6 due to solubility issues, and we cannot completely exclude different behaviours in acetonitrile, though the dielectric constant of both solvents is quite similar (37.5 and 46.7 for acetonitrile and DMSO, respectively).
Aggregation studies
The higher probability on the formation of intermolecular contacts leading to supramolecular assemblies for the p-tolyl compounds has been investigated by DFT calculations, with the dppe derivatives as a model. The dimerization energies in the case of compounds 11 and 14 (p-tolyl derivatives) are significantly more negative (favorable) than those of compounds 17 and 20 (CF3 derivatives), as shown in Fig. 5 and S81–583,† thus confirming the stronger ability of tolyl derivatives to aggregate. These dimers present Pt⋯π intermolecular contacts in addition to the π-stacking interactions of the aromatic ligands and the metallophilic Pt⋯Pt intramolecular contacts. The geometric features of the assemblies are also gathered in Table 3, showing similar π-stacking distances (both intra- and intermolecular). The Pt⋯π distance in compound 14 is the shortest one that also corresponds to the strongest interaction energy. It is worthy to comment that the interaction energies (calculated using acetonitrile as solvent) summarized in Table 3 are very large. In this sense, it should be emphasized that entropic effects are not considered in these calculations, so it is expected that the free energies of formation are less negative. Entropic effects are expected to be similar in all dimers; therefore the free energy differences among the compounds are also expected to be similar to the differences in the binding energies. The energy cost for the desolvation of the monomers to form the dimers is taken into consideration in the calculations, using the COSMO solvation continuum model as described in the theoretical methods. | ||
| Fig. 5 Two views of the optimized dimer of compound 11, with indication of the dimerization energy at the RI-BP86-D3(COSMO)/def2-TZVP level of theory in acetonitrile. Distances in Å. The π-stacking ones are calculated from the PtN3P mean plane of one molecule to the Pt atoms of the adjacent molecule or moiety. | ||
| Compound | ΔE | d intra (π⋯π) | d inter (π⋯π) | d inter (Pt⋯π) |
|---|---|---|---|---|
| 11 | −30.11 | 3.2 | 3.2 | 3.4 |
| 14 | −36.65 | 3.2 | 3.1 | 3.2 |
| 17 | −19.06 | 3.2 | 3.1 | 3.4 |
| 20 | −26.15 | 3.2 | 3.1 | 3.4 |
The UV-visible absorption at the highest energy band recorded for the four dppe derivatives (11, 14, 17 and 20) in acetonitrile display a sigmoidal profile as a function of temperature in agreement with an isodesmic mechanism of aggregation (see Fig. S84–S87†). The fitting of the data47 allowed us to calculate the corresponding ΔH energies of −26, −46, −23 and −25 kJ mol−1. These energies follow the same trend retrieved from the DFT studies and are much lower than the values calculated for other Pt(N^N^N) reported in the literature47 that were studied in water. Nevertheless, the calculated ΔH values support that aggregation happens earlier in acetonitrile although with a weaker energy (as expected) than in the presence of water.
The presence of the Pt(II) centers together with the N^N^N planar ligands and the DFT calculations predictions encouraged us to analyze in more detail the ability of the compounds to aggregate in solution and how it can affect their resulting photophysical properties. This study has been carried out by the preparation of several samples at the same concentration but with different acetonitrile:water mixtures ranging from 100% acetonitrile to 99% water. The results are observed to be dependent on the N^N^N ligand but not on the diphosphane. Hence, from now on, the dppe derivatives are exemplified in Fig. 6 and 7 in order to keep the discussion simple and understandable.
 | ||
| Fig. 6 Emission spectra of 11 (A), 14 (B), 17 (C), and 20 (D) compounds at different increasing amounts of water in the air-equibrated 1 × 10−5 M solutions (λexc = 350 nm). * Indicates residual of the Raman band. | ||
 | ||
| Fig. 7 Emission spectra of compounds 11 (A), 14 (B), 17 (C), and 20 (D) compounds at different % of water in air-equibrated 1 × 10−5 M acetonitrile:water solutions after 2 days (λexc = 350 nm). * Indicates residual of the Raman band. | ||
The corresponding absorption spectra of the samples show, in all cases, an increase on the baseline at higher water contents, in agreement with the formation of aggregated structures (Fig. S88 and 89†).48 The emission data are compatible with the previous data recorded in the solid state and in solution (Fig. 6). Starting from the non-emissive compounds in the solid state (13–15 and 19–21, containing the chlorine substituent at the N^N^N tridentate ligand), we can observe that the emission of the acetonitrile solutions of these compounds is quenched when increasing the water contents and thus, when the aggregation is much more favored, analogous to the behavior recorded in the solid state (Fig. 6B and D). On the other hand, the profile of the emission observed in the samples of compounds 10–12 and 16–18, containing the unsubstituted N^N^N tridentate ligand, when increasing the water contents in the solutions (more favored aggregation process) shows the same profile previously recorded in the solid state (see Fig. 6A and C).
Variable temperature absorption studies were also carried out in acetonitrile/water (25:75) for our systems. The resulting spectra display a reduction of the absorption baseline dispersion in agreement with the possible dissolution of the aggregates making the corresponding analysis of the aggregation mechanism not accurate.
The recorded emission quantum yields and lifetimes measured for the more aggregated emissive samples (compounds 10–12 and 16–18 at higher water contents) evidenced a higher emission efficiency for the samples with higher water contents (75 or 99%). This is compatible with a more favored radiative deactivation processes (larger kr values at larger % water, Table S3†) and less favored non-radiative competitive deactivation processes (smaller knr values at larger % water, Table S3†) thanks to longer emission decay times (Table 4).
| Compound | % water | Φ | τ (μs) | Compound | % water | Φ | τ (μs) |
|---|---|---|---|---|---|---|---|
| 10 | 99 | 0.05 | 1.20 | 16 | 99 | 0.02 | 0.33 |
| 75 | 0.04 | 1.14 | 75 | 0.02 | 0.26 | ||
| 50 | 0.03 | 0.51 | 50 | 0.01 | 0.01 | ||
| 11 | 99 | 0.03 | 1.14 | 17 | 99 | 0.02 | 0.20 |
| 75 | 0.03 | 1.15 | 75 | 0.02 | 0.08 | ||
| 50 | 0.02 | 0.82 | 50 | 0.02 | 0.01 | ||
| 12 | 99 | 0.03 | 0.85 | 18 | 99 | 0.01 | 0.60 |
| 75 | 0.03 | 0.72 | 75 | 0.02 | 0.42 | ||
| 50 | 0.02 | 0.50 | 50 | 0.02 | 0.01 |
The stability of the aggregated systems with time was examined by recording the corresponding emission spectra in all the different solutions after two days (see Fig. 7 and S91–S92†). In particular, the emission of compounds 10–12 and 16–18 gives rise to a blue-shifted band in the solutions with a lower amount of water, after two days (Fig. 7A and C). This band does not correspond to the emission previously recorded in acetonitrile solution. Thus, this new emission band may be ascribed to a possible new aggregate more stable thermodynamically, that emits around 450 nm. On the other hand, the emission spectra of the chlorinated derivatives (compounds 13–15 and 19–21) are not affected by time and thus, they would achieve more easily the thermodynamically stable species. This explanation agrees well with the larger dimerization energies observed for the chlorinated systems with respect to the non-chlorinated ones (see Table 3, complexes 17 and 20).
At this point, we decided to synthesize compound [Pt(dppm)2]Cl2 in order to discard the possible decomposition of our systems with time giving rise to the formation of more stable [Pt(diphenylphosphane)2]2+ species instead of the two suggested different aggregated structures.49 The synthesis of this compound was carried out according to the literature49 by the reaction of [Pt(COD)2Cl2] with two equivalents of dppm in CHCl3 for 2 hours at room temperature.
We observed that [Pt(dppm)2]Cl2 is not emissive under the same conditions used for the characterization of the Pt(II) tweezers. Thus, the observed emission with time (new emission at ca. 450 nm) cannot be a result of the formation of this decomposition product.
Additionally, 1H and 31P{1H} NMR spectra of the compounds 16 and [Pt(dppm)2]Cl2 were also recorded during the same periods of time (0 to 2 days, Fig. S93 and 94†). The 31P{1H} NMR spectrum of 16 is ca. 60 ppm downfield shifted with respect to the corresponding signal of [Pt(dppm)2]Cl2. Additionally, the chemical shifts recorded in the 1H NMR spectra of the compounds display a completely different profile in both cases. The different concentrations required for NMR and emission spectra analysis do not let us compare the results over time under the same conditions, but we can affirm that the resulting aggregates formed with time should not correspond to the possible formation of [Pt(dppm)2]Cl2 as a decomposition product supporting the formation of a new aggregated structure.
Optical microscopy was performed to analyze the effect of time on the shape of the different aggregates of the compounds. Selected results obtained for compound 16 in fresh samples and after 3 days are presented. Very large structures of a few micrometers can be detected in the resulting dried solutions at 50% water but with a completely different morphology depending on the conditions. While spherical aggregates can be detected in fresh samples, they tend to form some kind of needles with time (Fig. 8).
 | ||
| Fig. 8 Microscopical images obtained of the comparison of aggregates of compound 16 in a fresh 50% H2O:ACN solution (left) and after 3 days (right). | ||
1H and 31P{1H} NMR spectra of compounds 13 and 14 were also recorded at different DMSO-d6:D2O compositions (see Fig. S95–S98†). We can observe that the singlet corresponding to the pyridyl central protons of the N^N^N ligand immediately disappears in the presence of 25% of water in the medium. This indicates that is the part of the molecule more affected in the aggregation process, in agreement with the DFT predictions (see above, Fig. 5). Additionally, the protons from the tolyl group are also ca. 0.2 ppm downfield shifted when the aggregation is more favored (that is, in the presence of D2O) while the protons of the phosphine rings disappear due to their lower solubility in the more polar medium, as previously observed for other types of compounds that aggregate.11 There is not a significant shift on the 31P{1H} NMR data pointing to the more important role of the N^N^N ligand in the formation of intermolecular contacts than the diphosphane.
Theoretical studies
To further rationalize the behavior of the photophysical properties of the dppe-based complexes upon addition of incremental amounts of water, we have studied how the energies of the HOMO−1, HOMO and LUMO and the HOMO–LUMO gaps change depending on the solvent used in the calculations and the monomeric or dimeric form. Table 5 summarizes the results, showing that the HOMO–LUMO gaps are quite similar in both solvents. Therefore, the changes observed in the emission spectra commented above are likely due to aggregation processes instead of solvatochromic effects. Both the (HOMO−1)–LUMO and HOMO–LUMO gaps are reduced in the dimers with respect to the monomers. The reduction of the gaps varies from 0.06 to 0.27 eV in the dimers with respect to the monomers. Larger reductions can be expected for higher aggregates (oligomers). This reduction of the gaps agrees well with the larger emission quantum yields observed experimentally in the solid state and the aggregates in comparison with the values in air-equilibrated solutions since the most efficient population of the excited states is expected.| Compound | HOMO−1 | HOMO | LUMO | Gap (HOMO−1)–LUMO | Gap HOMO–LUMO |
|---|---|---|---|---|---|
| Acetonitrile | |||||
| (11)2 | −4.838 | −4.729 | −3.048 | 1.790 | 1.681 |
| 11 | −5.039 | −5.035 | −3.182 | 1.857 | 1.852 |
| (14)2 | −5.020 | −4.922 | −3.291 | 1.729 | 1.632 |
| 14 | −5.147 | −5.135 | −3.325 | 1.822 | 1.809 |
| (17)2 | −5.715 | −5.673 | −3.621 | 2.094 | 2.052 |
| 17 | −5.808 | −5.642 | −3.484 | 2.324 | 2.157 |
| (20)2 | −5.803 | −5.787 | −3.751 | 2.052 | 2.035 |
| 20 | −5.937 | −5.737 | −3.621 | 2.316 | 2.116 |
| Water | |||||
| (11)2 | −4.841 | −4.7325 | −3.052 | 1.789 | 1.680 |
| 11 | −5.046 | −5.041 | −3.188 | 1.859 | 1.854 |
| (14)2 | −5.026 | −4.928 | −3.298 | 1.728 | 1.630 |
| 14 | −5.154 | −5.141 | −3.331 | 1.823 | 1.810 |
| (17)2 | −5.713 | −5.671 | −3.620 | 2.093 | 2.051 |
| 17 | −5.807 | −5.638 | −3.481 | 2.326 | 2.158 |
| (20)2 | −5.802 | −5.786 | −3.752 | 2.050 | 2.033 |
| 20 | −5.935 | −5.732 | −3.618 | 2.317 | 2.114 |
Singlet oxygen production
Pt(II) compounds are widely studied regarding their potential biological activity as anticancer drugs. Photodynamic therapy is recently gaining increasing attention due to the biological activity of metal complexes. In this way, platinum compounds have been observed to act as photosensitizers to produce singlet oxygen, 1O2, being suitable candidates to be carefully analyzed in this field or in other applications such as photocatalysis, or even degradation of contaminants.50,51Dinuclear metal complexes and aggregates may be interesting to be explored in this field due to a possible cooperative process. For this reason, the 1O2 production quantum yields of 10–21 have been measured in this work and the obtained results are displayed in Table 6 and Fig. S99–S110.†
| Complex | Complex | ||
|---|---|---|---|
| p-Tolyl derivatives | Φ Δ (%) | CF3 derivatives | Φ Δ (%) |
| 10 | 23 | 16 | 27 |
| 11 | 36 | 17 | 36 |
| 12 | 17 | 18 | 26 |
| 13 | 16 | 19 | 12 |
| 14 | 31 | 20 | 36 |
| 15 | 18 | 21 | 47 |
The results show that the compounds present in general good results as photosensitizers (PS) to induce the production of singlet oxygen in solution. The obtained values are in the top-range of the previously reported in the literature.22,52–54 Interestingly, and as a difference with what we have seen in the photophysical behaviour, the diphosphane linker plays a direct role on the photosensitizing process. We observe that, in general, the compounds that contain the dppe linker (11, 14 and 18) present the highest values of singlet oxygen production compared to their respective dppm and dppbz derivatives. In the case of 10–12 this can be correlated with the lowest phosphorescence quantum yield value recorded for 11 due to the more efficient electron and energy transfer process. The exception is the series 19–21 where the highest value is observed for the dppbz tweezer (compound 21) where similar kET values would be expected in both dppe and dppbz derivatives. This hypothesis is supported by the application of the formula  for the compounds 10–12 where pure room temperature phosphorescence emission is recorded.22 The calculated values are 230, 600 and 255 × 105 s−1 respectively, in agreement with the more efficient energy transfer process for dppe derivatives.
for the compounds 10–12 where pure room temperature phosphorescence emission is recorded.22 The calculated values are 230, 600 and 255 × 105 s−1 respectively, in agreement with the more efficient energy transfer process for dppe derivatives.
On the other hand, the substituents at the triazole groups do not affect substantially this process.
Conclusions
The emissions recorded for the four series of (diphos)Pt(N^N^N) tweezer compounds present different profiles that are affected by the stability of the resulting intra- and intermolecular self-aggregation processes. DFT theoretical studies support the establishment of Pt⋯π intermolecular contacts in addition to the π-stacking interactions of the aromatic ligands and the metallophilic Pt⋯Pt intramolecular contacts. The substituent at the para position of the central pyridyl ring of the N^N^N ligand affects the resulting aggregation, being more stable for those compounds having a hydrogen atom instead of a chlorine atom. This results in MMLCT transitions and phosphorescence emissions for these compounds while MLCT and IL emissions can be associated with the transitions of the rest of the compounds. The HOMO–LUMO gaps computationally calculated for the dimers of all tweezer compounds display lower values compared to the monomer gaps, in agreement with larger emission quantum yields recorded experimentally.The aggregation process can be more favored in acetonitrile:water mixtures where the more aggregated samples present an analogous luminescence behavior compared to that recorded in the solid state.
The compounds have also been tested as singlet oxygen photosensitizers and, in general, the dppe derivatives display larger values of singlet oxygen quantum yields, in agreement with a more favored energy transfer process.
Experimental section
General information
All air- and moisture-sensitive manipulations were carried out by standard Schlenk techniques under a nitrogen atmosphere. Solvents were purchased from commercial sources and dried by distillation under a nitrogen atmosphere. Reagents 2,6-pyridinedicarbonitrile, 4-chloropyridine-2,6-carbonitrile, N,N-diisopropylethylamine (DIPEA), bis(diphenylphosphino)methane (dppm), bis(diphenylphosphino)ethane (dppe), bis(diphenylphosphino)benzene (dppbz), hydrazine hydrate, p-toluoyl chloride, trifluoroacetic acid were purchased from commercial sources and used without further purification. Precursor [PtCl2(DMSO)2] was prepared from the reaction of K2[PtCl4] with dimethylsulfoxide (DMSO).55Infrared spectra have been recorded on an FT-IR 520 Nicolet spectrophotometer. 1H NMR (δ(TMS) = 0.0 ppm), 19F NMR (δ(CFCl3) = 0.0 ppm), and 31P{1H} NMR (δ(85% H3PO4) = 0.0 ppm) spectra have been obtained on Bruker 400 and Bruker 500 (Universitat de Barcelona) instruments. Electrospray mass spectra (+) have been recorded on a Fisons VG Quatro spectrometer (Universitat de Barcelona). Absorption spectra were obtained in a 5 mm or 10 mm quartz cuvette in acetonitrile on a Cary 5000 UV–Vis–NIR or Shimadzu UV-2450 spectrophotometer. The emission spectra of the compounds in solution were obtained in a fluorescence quartz cuvette of 5 mm or 10 mm path length, using a Horiba-Jobin-Yvon Nanolog spectrofluorimeter.
Singlet oxygen quantum yields measurements
Room-temperature singlet oxygen phosphorescence was detected at 1270 nm with a Horiba-Jobin-Yvon SPEX Nanolog spectrofluorimeter (Universitat de Barcelona) using the DSS-IGA020L detector. The use of a Schott RG 1000 filter was essential to eliminate from the infrared signal all the first harmonic contribution of sensitizer emission in the region below 850 nm. The singlet oxygen formation quantum yield was then determined by direct measurement of the phosphorescence at 1270 nm following irradiation of the aerated aqueous solution of the samples. The samples were adjusted to an absorption nearly to 1 concerning the excitation wavelength to increase the sensitivity of the detection. 1H-Phenal-1-one in dichloromethane was used as standard reference, applying eqn (1). | (1) |
Theoretical calculations
The geometries of the complexes and dimeric assemblies were fully optimized without symmetry constrains at the RI-PB86-D3(COSMO)/def2-TZVP level of theory56–58 by means of the Turbomole 7.2 program.59 The level of theory has been validated by comparing the geometries of available X-ray structures of related compounds60 with those from the DFT optimization (see Fig. S111, ESI†). Moreover, this level of theory has been recently used by us to explain the aggregation of Au-complexes.61 The binding energies were computed by subtracting the sum of the energies of the monomers to the energy of the dimer. Solvent effects were taken into consideration using the COSMO-RS method62 as implemented in Turbomole program.59 The TD-DFT calculations were computed using 30 excited states at the CAM-B3LYP/def2-TZVP level of theory63 using ORCA 5.0 program.64Host![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :guest titrations
:guest titrations
The titrations were performed by the addition of subsequent aliquots of 9 × 10−4 M solutions of PAH (naphthalene or phenanthrene) into a 6 × 10−5 M solution of 11. Absorption and emission spectra were measured after each addition. The association constants of the 11 with the PAHs were retrieved from the fit of the fluorimetric titration data with the general equation reported in ref. 65.
Synthesis and characterization
N), 1472 (CC), 1422 (CH3), 1000 (C–N).
N), 1494 (CC), 1423 (CH3), 1022 (C–N), 747 (C–Cl).
N), 1458 (CC), 1138 (CF3), 1022 (C–N).
N), 1459 (CC), 1147 (CF3), 1022 (C–N), 809 (C–Cl).
N), 1467 (CC), 1436 (CH3), 1018 (SO), 929 (C–N).
N), 1496 (CC), 1385 (CH3), 1023 (SO), 916 (C–N), 746 (C–Cl).
N), 1476 (CC), 1125 (CF3), 1022 (SO).
N), 1476 (CC), 1129 (CF3), 1022 (SO), 742 (C–Cl).
N), 1462 (CC), 1432 (–CH3), 1000 (P–Car).
N), 1472 (CC), 1441 (–CH3), 1000 (P–Car).
N), 1483 (CC), 1432 (–CH3), 997 (P–Car).
N), 1508 (CC), 1441 (–CH3), 996 (P–Car), 738 (C–Cl).
N), 1494 (CC), 1432 (–CH3), 1018 (P–Car), 818 (C–Cl).
N), 1503 (CC), 1436 (–CH3), 1022 (P–Car), 831 (C–Cl).
N), 1462 (CC), 1130 (CF3), 989 (P–Car).
N), 1490 (CC), 1138 (CF3), 1000 (P–Car).
N), 1485 (CC), 1129 (CF3), 996 (P–Car).
N), 1436 (CC), 1098 (CF3), 996 (P–Car), 742 (C–Cl).
N), 1436 (CC), 1103 (CF3), 996 (P–Car), 711 (C–Cl).
N), 1432 (CC), 1103 (CF3), 1000 (P–Car), 689 (C–Cl).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Ministerio de Ciencia e Innovación of Spain (Projects PID2019-104121GB-I00 and PID2020-115637GB-I00). Authors would also like to acknowledge International Research Network Hetero-elements and Coordination Chemistry: from Concepts to Applications (HC3A) and Spanish network Materiales Supramoleculares Funcionales (RED2018-102331-T). G. R.-I. acknowledges Fundación Carolina and Secretaria de Relaciones Exteriores de México for the Ph.D. Scholarship. A. F. is grateful to the Alexander von Humboldt Foundation for the J. C. Mutis Award.References
- L. Escobar and P. Ballester, Molecular Recognition in Water Using Macrocyclic Synthetic Receptors, Chem. Rev., 2021, 121, 2445–2514 CrossRef CAS PubMed.
- C. Huang, A. Ciesielski and P. Samorì, Molecular Springs: Integration of Complex Dynamic Architectures into Functional Devices, Angew. Chem., Int. Ed., 2020, 59, 7319–7330 CrossRef CAS PubMed.
- M. Baroncini, L. Casimiro, C. de Vet, J. Groppi, S. Silvi and A. Credi, Making and Operating Molecular Machines: A Multidisciplinary Challenge, ChemistryOpen, 2018, 7, 169–179 CrossRef CAS PubMed.
- L. Martínez-Crespo, J. L. Sun-Wang, P. Ferreira, C. F. M. Mirabella, G. Aragay and P. Ballester, Influence of the Insertion Method of Aryl-Extended Calix[4]Pyrroles into Liposomal Membranes on Their Properties as Anion Carriers, Chem. – Eur. J., 2019, 25, 4775–4781 CrossRef PubMed.
- D. M. Ivanov, N. A. Bokach, V. Y. Kukushkin and A. Frontera, Metal Centers as Nucleophiles: Oxymoron of Halogen Bond-Involving Crystal Engineering, Chem. – Eur. J., 2022, 28, e2021031 Search PubMed.
- M. Benito, A. Frontera and E. Molins, Cocrystallization of Antifungal Compounds Mediated by Halogen Bonding, Cryst. Growth Des., 2023, 23, 2932–2940 CrossRef CAS PubMed.
- A. Jozeliūnaitė, T. Javorskis, V. Vaitkevičius, V. Klimavičius and E. Orentas, Fully Supramolecular Chiral Hydrogen-Bonded Molecular Tweezer, J. Am. Chem. Soc., 2022, 144, 8231–8241 CrossRef PubMed.
- A. F. Sierra, D. Hernández-Alonso, M. A. Romero, J. A. González-Delgado, U. Pischel and P. Ballester, Optical Supramolecular Sensing of Creatinine, J. Am. Chem. Soc., 2020, 142, 4276–4284 CrossRef CAS PubMed.
- C. Zhang, F. Wang, R. S. Patil, C. L. Barnes, T. Li and J. L. Atwood, Hierarchical Self-Assembly of Supramolecular Coordination Polymers Using Giant Metal-Organic Nanocapsules as Building Blocks, Chem. – Eur. J., 2018, 24, 14335–14340 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Functional Supramolecular Polymers, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- A. Pinto, C. Roma-Rodrigues, J. S. Ward, R. Puttreddy, K. Rissanen, P. V. Baptista, A. R. Fernandes, J. C. Lima and L. Rodríguez, Aggregation versus Biological Activity in Gold(I) Complexes. An Unexplored Concept, Inorg. Chem., 2021, 60, 18753–18763 CrossRef CAS PubMed.
- A. Pinto, J. Sonet, R. M. Gomila, A. Frontera, J. C. Lima and L. Rodríguez, Supramolecular Gold(I) Vesicles: An in-Depth Study of Their Aggregation Process, Inorg. Chem. Front., 2022, 9, 6047–6060 RSC.
- A. Lázaro, R. Bosque, J. S. Ward, K. Rissanen, M. Crespo and L. Rodríguez, Toward Near-Infrared Emission in Pt(II)-Cyclometallated Compounds: From Excimers’ Formation to Aggregation-Induced Emission, Inorg. Chem., 2023, 62, 2000–2012 CrossRef PubMed.
- V. W.-W. Yam, A. K.-W. Chan and E. Y.-H. Hong, Charge-Transfer Processes in Metal Complexes Enable Luminescence and Memory Functions, Nat. Rev. Chem., 2020, 4, 528–541 CrossRef CAS.
- J.-M. Lehn, Toward Self-Organization and Complex Matter, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed.
- M. H. Y. Chan and V. W. W. Yam, Toward the Design and Construction of Supramolecular Functional Molecular Materials Based on Metal-Metal Interactions, J. Am. Chem. Soc., 2022, 144, 22805–22825 CrossRef CAS PubMed.
- E. Aguiló, A. J. Moro, R. Gavara, I. Alfonso, Y. Pérez, F. Zaccaria, C. F. Guerra, M. Malfois, C. Baucells, M. Ferrer, J. C. Lima and L. Rodríguez, Reversible Self-Assembly of Water-Soluble Gold(I) Complexes, Inorg. Chem., 2018, 57, 1017–1028 CrossRef PubMed.
- A. K. W. Chan and V. W. W. Yam, Precise Modulation of Molecular Building Blocks from Tweezers to Rectangles for Recognition and Stimuli-Responsive Processes, Acc. Chem. Res., 2018, 51, 3041–3051 CrossRef CAS PubMed.
- H. S. Kim, J. Y. Lee, S. Shin, W. Jeong, S. H. Lee, S. Kim, J. Lee, M. C. Suh and S. Yoo, Enhancement of Reverse Intersystem Crossing in Charge-Transfer Molecule through Internal Heavy Atom Effect, Adv. Funct. Mater., 2021, 31, 2104646 CrossRef CAS.
- C. Tang, P. Hu, E. Ma, M. Huang and Q. Zheng, Heavy Atom Enhanced Generation of Singlet Oxygen in Novel Indenofluorene-Based Two-Photon Absorbing Chromophores for Photodynamic Therapy, Dyes Pigm., 2015, 117, 7–15 CrossRef CAS.
- A. Rodriguez-Serrano, V. Rai-Constapel, M. C. Daza, M. Doerr and C. M. Marian, Internal Heavy Atom Effects in Phenothiazinium Dyes: Enhancement of Intersystem Crossing via Vibronic Spin-Orbit Coupling, Phys. Chem. Chem. Phys., 2015, 17, 11350–11358 RSC.
- A. Lázaro, C. Cunha, R. Bosque, J. Pina, J. S. Ward, K. N. Truong, K. Rissanen, J. C. Lima, M. Crespo, J. S, S. De Melo and L. Rodríguez, Room-Temperature Phosphorescence and Efficient Singlet Oxygen Production by Cyclometalated Pt(II) Complexes with Aromatic Alkynyl Ligands, Inorg. Chem., 2020, 59, 8220–8230 CrossRef PubMed.
- P. R. Ogilby, Singlet Oxygen: There Is Indeed Something New under the Sun, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- M. Parasram and V. Gevorgyan, Visible Light-Induced Transition Metal-Catalyzed Transformations: Beyond Conventional Photosensitizers, Chem. Soc. Rev., 2017, 46, 6227–6240 RSC.
- A. Aliprandi, D. Genovese, M. Mauro and L. De Cola, Recent Advances in Phosphorescent Pt(II) Complexes Featuring Metallophilic Interactions: Properties and Applications, Chem. Lett., 2015, 44, 1152–1169 CrossRef CAS.
- G. Moreno-Alcántar, A. Aliprandi, R. Rouquette, L. Pesce, K. Wurst, C. Perego, P. Brüggeller, G. M. Pavan and L. De Cola, Solvent-Driven Supramolecular Wrapping of Self-Assembled Structures, Angew. Chem., Int. Ed., 2021, 60, 5407–5413 CrossRef PubMed.
- P. Picchetti, G. Moreno-Alcántar, L. Talamini, A. Mourgout, A. Aliprandi and L. De Cola, Smart Nanocages as a Tool for Controlling Supramolecular Aggregation, J. Am. Chem. Soc., 2021, 143, 7681–7687 CrossRef CAS PubMed.
- Y. Chen, K. Li, W. Lu, S. S. Y. Chui, C. W. Ma and C. M. Che, Photoresponsive Supramolecular Organometallic Nanosheets Induced by Pt II⋯PtII and C-H⋯π Interactions, Angew. Chem., Int. Ed., 2009, 48, 9909–9913 CrossRef CAS PubMed.
- D. K. Bhowmick, L. Stegemann, M. Bartsch, N. K. Allampally, C. A. Strassert and H. Zacharias, Controlled 2D-Confinement of Phosphorescent Pt(II) Complexes on Quartz and 6H-SiC(0001) Surfaces, J. Phys. Chem. C, 2015, 119, 5551–5561 CrossRef CAS.
- B. Doistau, A. Tron, S. A. Denisov, G. Jonusauskas, N. D. McClenaghan, G. Gontard, V. Marvaud, B. Hasenknopf and G. Vives, Terpy(Pt-Salphen) 2 Switchable Luminescent Molecular Tweezers, Chem. – Eur. J., 2014, 20, 15799–15807 CrossRef CAS PubMed.
- Z. Li, Y. Han, Z. Gao and F. Wang, Supramolecular Engineering of Discrete Pt(II)⋯Pt(II) Interactions for Visible-Light Photocatalysis, ACS Catal., 2017, 7, 4676–4681 CrossRef CAS.
- M. Liu, Y. Han, H. Zhong, X. Zhang and F. Wang, Supramolecular Chirogenesis Induced by Platinum(II) Tweezers with Excellent Environmental Tolerance, Angew. Chem., Int. Ed., 2021, 60, 3498–3503 CrossRef CAS PubMed.
- Y. Tanaka, K. Man-Chung Wong and V. Wing-Wah Yam, Phosphorescent molecular tweezers based on alkynylplatinum(II) terpyridine system: turning on of NIR emission via heterologous Pt⋯M interactions (M = PtII, PdII, AuIII and AuI), Chem. Sci., 2012, 3, 1185–1191 RSC.
- D. Septiadi, A. Aliprandi, M. Mauro and L. De Cola, Bio-Imaging with Neutral Luminescent Pt(II) Complexes Showing Metal⋯Metal Interactions, RSC Adv., 2014, 4, 25709–25718 RSC.
- S. Sinn, F. Biedermann and L. De Cola, Platinum Complex Assemblies as Luminescent Probes and Tags for Drugs and Toxins in Water, Chem. – Eur. J., 2017, 23, 1965–1971 CrossRef CAS PubMed.
- M. Ferrer, A. Pedrosa, L. Rodríguez, O. Rossell and M. Vilaseca, New Insights into the Factors That Govern the Square/Triangle Equilibria of Pd(II) and Pt(II) Supramolecules. Unexpected Participation of a Mononuclear Species in the Equilibrium, Inorg. Chem., 2010, 49, 9438–9449 CrossRef CAS PubMed.
- S. A. Al-Jibori, A. S. S. Al-Zaubai, M. Y. Mohammed and T. A. K. Al-Allaf, Mixed Ligand Palladium(II) and Platinum(II) Complexes of Tertiary Diphosphines and Benz-1,3-Imidazoline-2-Thione, Benz-1,3-Oxazoline-2-Thione or Benz-1,3-Thiazoline-2-Thione, Transition Met. Chem., 2007, 32, 281–286 CrossRef CAS.
- S. Chakraborty, A. Aliprandi and L. De Cola, Multinuclear PtII Complexes: Why Three Is Better Than Two to Enhance Photophysical Properties, Chem. – Eur. J., 2020, 26, 11007–11012 CrossRef CAS PubMed.
- S. C. Gangadharappa, I. Maisuls, I. P. Salto, S. Niemann, V. Bachtin, F. C. Herrmann and C. A. Strassert, Intermolecular Interactions and Self-Assembly in Pt(II) Complex-Nanoclay Hybrids as Luminescent Reporters for Spectrally Resolved PLIM, J. Phys. Chem. C, 2021, 125, 5739–5747 CrossRef.
- S. C. Gangadharappa, I. Maisuls, D. A. Schwab, J. Kösters, N. L. Doltsinis and C. A. Strassert, Compensation of Hybridization Defects in Phosphorescent Complexes with Pnictogen-Based Ligands - A Structural, Photophysical, and Theoretical Case-Study with Predictive Character, J. Am. Chem. Soc., 2020, 142, 21353–21367 CrossRef CAS PubMed.
- M. Mydlak, M. Mauro, F. Polo, M. Felicetti, J. Leonhardt, G. Diener, L. De Cola and C. A. Strassert, Controlling Aggregation in Highly Emissive Pt(II) Complexes Bearing Tridentate Dianionic N^N^N Ligands. Synthesis, Photophysics, and Electroluminescence, Chem. Mater., 2011, 23, 3659–3667 CrossRef CAS.
- L. Stegemann, J. Sanning, C. G. Daniliuc and C. A. Strassert, Influence of the Monodentate Ancillary Ligand on the Photophysical Properties of Pt(II) Complexes Bearing a Symmetric Dianionic Tridentate Luminophore, Z. Naturforsch., B: Chem. Sci., 2016, 71, 1087–1093 CrossRef CAS.
- V. W. W. Yam and K. M. C. Wong, Luminescent Metal Complexes of d6, d8 and d10 Transition Metal Centres, Chem. Commun., 2011, 47, 11579–11592 RSC.
- F. K.-W. Kong, A. K.-W. Chan, M. Ng, K.-H. Low and V. W.-W. Yam, Construction of Discrete Pentanuclear Platinum(II) Stacks with Extended Metal–Metal Interactions by Using Phosphorescent Platinum(II) Tweezers, Angew. Chem., 2017, 129, 15299–15303 CrossRef.
- C. Y. Sun, W. P. To, F. F. Hung, X. L. Wang, Z. M. Su and C. M. Che, Metal–organic framework composites with luminescent pincer platinum(II) complexes: 3MMLCT emission and photoinduced dehydrogenation catalysis, Chem. Sci., 2018, 9, 2357–2364 RSC.
- Q. Wan, W.-P. To, C. Yang and C.-M. Che, The Metal–Metal-to-Ligand Charge Transfer Excited State and Supramolecular Polymerization of Luminescent Pincer PdII–Isocyanide Complexes, Angew. Chem., 2018, 130, 3143–3147 CrossRef.
- X. Zheng, M. Ho-Yeung Chan, A. Kwun-Wa Chan, S. Cao, M. Ng, F. Kit Sheong, C. Li, E. Chrisana Goonetilleke, W. Wai Yan Lam, T.-C. Lau, X. Huang and V. W.-W. Yam, Elucidation of the key role of Pt⋯Pt interactions in the directional self-assembly of platinum(II) complexes, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2116543119 CrossRef CAS PubMed.
- A. Pinto, N. Svahn, J. C. Lima and L. Rodríguez, Aggregation Induced Emission of Gold(I) Complexes in Water or Water Mixtures, Dalton Trans., 2017, 46, 11125–11139 RSC.
- G. K. Anderson, J. A. Davies and D. J. Schoeck, A 31P{1H} NMR Study of the Reactions of [PtCl2L2] (L = RCN; L2 = 1,5-Cyclooctadiene) Complexes with Bidentate Ligands. The Effects of Solubility on Product Distribution, Inorg. Chim. Acta, 1983, 76, 251–252 CrossRef.
- H. Kim, W. Kim, Y. MacKeyev, G. S. Lee, H. J. Kim, T. Tachikawa, S. Hong, S. Lee, J. Kim, L. J. Wilson, T. Majima, P. J. J. Alvarez, W. Choi and J. Lee, Selective Oxidative Degradation of Organic Pollutants by Singlet Oxygen-Mediated Photosensitization: Tin Porphyrin versus C60 Aminofullerene Systems, Environ. Sci. Technol., 2012, 46, 9606–9613 CrossRef CAS PubMed.
- I. D. Rettig and T. M. McCormick, Enrolling reactive oxygen species in photon-to-chemical energy conversion: fundamentals, technological advances, and applications, Adv. Phys.: X, 2021, 6 Search PubMed.
- S. Goswami, R. W. Winkel and K. S. Schanze, Photophysics and Nonlinear Absorption of Gold(I) and Platinum(II) Donor-Acceptor-Donor Chromophores, Inorg. Chem., 2015, 54, 10007–10014 CrossRef CAS PubMed.
- I. Toubia, C. Nguyen, S. Diring, L. M. A. Ali, L. Larue, R. Aoun, C. Frochot, M. Gary-Bobo, M. Kobeissi and F. Odobel, Synthesis and Anticancer Activity of Gold Porphyrin Linked to Malonate Diamine Platinum Complexes, Inorg. Chem., 2019, 58, 12395–12406 CrossRef CAS PubMed.
- M. Yang, J. Deng, D. Guo, J. Zhang, L. Yang and F. Wu, A Folate-Conjugated Platinum Porphyrin Complex as a New Cancer-Targeting Photosensitizer for Photodynamic Therapy, Org. Biomol. Chem., 2019, 17, 5367–5374 RSC.
- J. H. Price, A. N. Williamson, R. F. Schramm and B. B. Wayland, Palladium(II) and Platinum(II) Alkyl Sulfoxide Complexes. Examples of Sulfur-Bonded, Mixed Sulfur- and Oxygen-Bonded, and Totally Oxygen-Bonded Complexes, Inorg. Chem., 1972, 11, 1280–1284 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- R. Ahlrichs, M. Bar, M. H. Iser, H. Horn and C. Ktjlmel, Electronic Structure calculations on workstation computers: the program system Turbomole, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- J. Sanning, P. R. Ewen, L. Stegemann, J. Schmidt, C. G. Daniliuc, T. Koch, N. L. Doltsinis, D. Wegner and C. A. Strassert, Scanning-Tunneling-Spectroscopy-Directed Design of Tailored Deep-Blue Emitters, Angew. Chem., Int. Ed., 2015, 54, 786–791 CrossRef CAS PubMed.
- A. Pinto, A. Llanos, R. M. Gomila, A. Frontera and L. Rodríguez, Ligand and Gold(I) Fluorescein–AIEgens as Photosensitizers in Solution and Doped Polymers, Inorg. Chem., 2023, 62, 7131–7140 CrossRef CAS PubMed.
- A. Klamt, C. Moya and J. Palomar, A Comprehensive Comparison of the IEFPCM and SS(V)PE Continuum Solvation Methods with the COSMO Approach, J. Chem. Theory Comput., 2015, 11, 4220–4225 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- F. Neese, Software update: The ORCA program system—Version 5.0, WIRES Comput. Mol. Sci., 2002, 12, e1606 Search PubMed.
- P. Cudic, M. Zinic, V. Tomisic, V. Simeon, J.-P. Vigneron and J.-M. Lehn, Binding of Nucleotides in Water by Phenanthridinium Bis(intercaland) Receptor Molecules, J. Chem. Soc., Chem. Commun., 1995, 1073–1075 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 31P and 19F NMR spectra for all complexes. ESI-TOF spectra for all complexes. Absorption and emission spectra for all compounds in solution (air-equilibrated and N2-saturated samples), in the solid state and in acetonitrile/water mixtures. Emission lifetime data of all compounds. Computed minimum energy geometries using two orientations calculated for compounds. Representation of the MOs involved in the S0 → S3 excitation of compound 20. Calculated dimer assemblies for compounds 14, 17 and 20. See DOI: https://doi.org/10.1039/d3qi01176c |
| This journal is © the Partner Organisations 2023 |