DOI:
10.1039/D3QI01081C
(Research Article)
Inorg. Chem. Front., 2023,
10, 5678-5685
Chalcogen atom abstraction from NCE− (E = O, S, Se) and i-Pr2S by the excited state of a luminescent tricyano osmium(VI) nitride†
Received
9th June 2023
, Accepted 7th August 2023
First published on 16th August 2023
Introduction
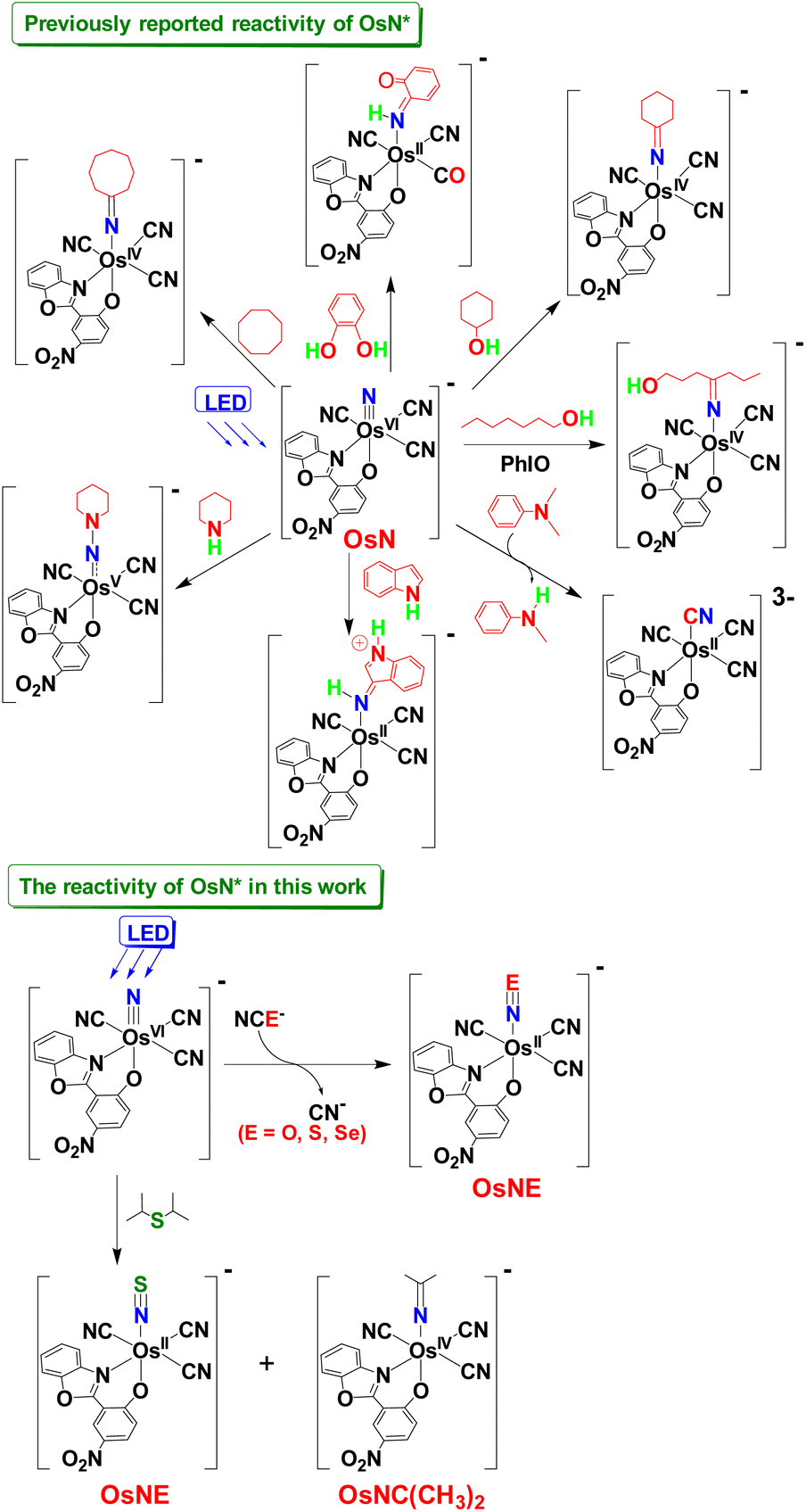
Metal nitrido complexes (M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) have been proposed as key intermediates in N2 fixation; they are also potentially useful reagents for the nitrogenation of various organic substrates.1–5 A number of electrophilic nitrido complexes have been reported recently. For instance, cis- and trans-[OsVI(N)(tpy)Cl2]+ (tpy = 2,2′:6′,2′′-terpyridine) have been shown to exhibit novel electrophilic properties. A variety of reagents such as phosphines, amines, cyanide, azide, arylboranes, amine N-oxides, alkenes, and benzenethiols have been reported to react with the osmium nitrido complexes.6 Higher reactivity is found for RuN and FeN, such as [Ru(N)(salchda)(MeOH)]+ (salchda = N,N′-bis(salicylidene)-o-cyclohexyldiamine dianion)7 and [PhB(RIm)3FeN] (Im = imidazol, R = tBu, Mes, and iPr2).8 Although these nitrido complexes exhibit novel electrophilic properties and react readily with a variety of nucleophiles, their reactivity is still relatively limited compared to analogous metal-oxo (M
N) have been proposed as key intermediates in N2 fixation; they are also potentially useful reagents for the nitrogenation of various organic substrates.1–5 A number of electrophilic nitrido complexes have been reported recently. For instance, cis- and trans-[OsVI(N)(tpy)Cl2]+ (tpy = 2,2′:6′,2′′-terpyridine) have been shown to exhibit novel electrophilic properties. A variety of reagents such as phosphines, amines, cyanide, azide, arylboranes, amine N-oxides, alkenes, and benzenethiols have been reported to react with the osmium nitrido complexes.6 Higher reactivity is found for RuN and FeN, such as [Ru(N)(salchda)(MeOH)]+ (salchda = N,N′-bis(salicylidene)-o-cyclohexyldiamine dianion)7 and [PhB(RIm)3FeN] (Im = imidazol, R = tBu, Mes, and iPr2).8 Although these nitrido complexes exhibit novel electrophilic properties and react readily with a variety of nucleophiles, their reactivity is still relatively limited compared to analogous metal-oxo (M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) species.9
O) species.9
In search of more reactive MN species that are comparable to MO, we recently started to investigate the reactivity of MN in their excited states. Accordingly, a highly luminescent Os(VI) nitrido complex, [OsVI(N)(L)(CN)3]− (OsN, HL = 2-(2-hydroxy-5-nitrophenyl)benzoxazole) with long-lived LMCT excited state has been prepared.10 This species is highly reactive in the excited state (OsN*) due to its nitridyl [OsN˙] character. Indeed, upon irradiation with visible light (λ > 460 nm), OsN* readily activates the strong C–H bonds of alkanes and arenes,11 undergoes oxidative N-dealkylation of various tertiary amines12 and C–O bond cleavage of dihydroxybenzene,13 exhibits formal N atom transfer to aliphatic secondary amines14 and ring-nitrogenation of aromatic amines.15 Recently, we have also found that OsN* could activate both α- and δ-C–H bonds of alcohols in the presence of PhIO, due to the formation of the highly potent oxidant PhIO+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 (Fig. 1).
16 (Fig. 1).
 |
| | Fig. 1 (a) Reported reactivity of OsN* towards various substrates; (b) reaction of OsN* with NCE− and i-Pr2S in this work. | |
We report herein that OsN* readily undergoes unprecedented chalcogen atom abstraction from the stable inorganic anions NCE− (N = O, S, Se), as well as from organic sulphide such as i-Pr2S.
Results and discussion
Upon irradiation of a solution of OsN in CH2Cl2 containing 10 equiv. of (PPh4)NCSe with blue LED (λ > 460 nm) for 24 h, the bright yellow solution turned pale-yellow. Electrospray ionization mass spectrometry (ESI/MS, −ve mode) of the resulting solution exhibits a new peak at m/z 617, which is assigned to [Os(NSe)(L)(CN)3]− (OsNSe). Similarly, ESI/MS for the photoreaction of OsN with 10 equiv. of (PPh4)NCS for 48 h shows a major peak at m/z 571, which is assigned to [Os(NS)(L)(CN)3]− (OsNS) (Fig. 2 and S1†). When the reactions were carried out on a preparative scale, (PPh4)[Os(NE)(L)(CN)3] (OsNE, E = Se and S) were isolated as light yellow crystalline solids with 45% and 52% yields, respectively. ESI/MS (−ve mode) of these photoreaction solutions also show a small peak at m/z = 26 due to the formation of CN−. M-NE complexes have been prepared from the reaction of metal nitrides with S8 or elemental Se.17–19 However, to the best of our knowledge, S/Se atom transfer from NCE− anions, which involves cleavage of strong CE bonds, has not been reported. The photoreaction of OsN with (PPh4)NCO has also been investigated; however, its reaction rate is much slower, and the yield of (PPh4)[Os(NO)(L)(CN)3] (OsNO)20,21 is only ∼10%, which is probably due to the stronger CO bond than CS and CSe bonds in NCE−. O atom transfer to metal nitride usually occurs with oxidants such as Me3NO.17a When 15N-labelled Os15N was used, ESI/MS shows that the parent OsNE ions all increases by one mass unit, indicating that the N atoms in OsNE are from the nitrido ligand rather than from NCE−.
 |
| | Fig. 2 ESI/MS of the photoreaction of OsN with 10 equiv. of (PPh4)NCS for 24 h showing a new product peak at m/z 571 (OsNS). | |
Both OsNS and OsNSe are stable for >2 weeks in the solid state or in various solvents at room temperature. However, upon exposure to air for more than two months, these complexes were partially converted into OsNO (m/z 555). Attempts were also made to synthesize the tellurium analog (OsNTe) from the photoreaction of OsN and NCTe−. However, no products could be isolated, presumably due to the instability of the [Os(NTe)(L)(CN)3]− species. Also, no reaction of OsN* with elemental tellurium in various solvents was observed, presumably due to the poor solubility of Te.
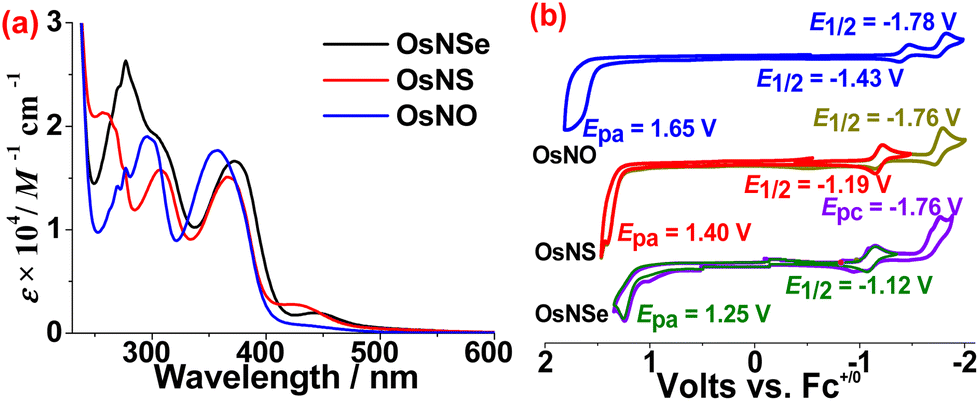
The IR spectrum of OsNSe shows strong v(CN) stretches at 2148, 2133 cm−1 and v(NSe) stretch at 1136 cm−1. Similar v(CN) stretches in OsNS and OsNO are also found at 2149, 2133 cm−1 and 2150, 2139 cm−1, respectively; while the v(NS) and v(NO) stretches occur at 1291 cm−1 and 1849 cm−1, respectively. The ratio of v(NO) to v(NS) stretching frequencies in OsNO and OsNS is 1.432, a typical value for structurally similar NO and NS compounds.22 The UV/vis spectra of these compounds show strong absorption bands due to ligand centered π–π* transitions below 400 nm, with molar extinction coefficients (ε) on the order of 104 M−1 cm−1 (Fig. 3a). OsNO also shows a weak absorption band tailing down to the visible region, while for OsNS and OsNSe there is a well-defined absorption band in the visible region; these are tentatively assigned to the O^N ligand to metal (Os) charge transfer (LMCT) (Fig. S2 and S3†). All compounds are diamagnetic, as evidenced by their sharp proton signals in the normal range in their 1H NMR spectra (Fig. S4–S6†).
 |
| | Fig. 3 (a) UV/vis spectra of OsNE in CH3CN; (b) CV of OsNE in CH3CN containing 0.1 M [nBu4N](PF6) with a scan rate of 0.1 V s−1. | |
Cyclic voltammetry (CV) of OsNE was conducted in 0.1 M [nBu4N](PF6) CH3CN solution. As shown in Fig. 3b, these compounds all show two reduction waves. The first reduction potentials E1/2 are in the range of −1.12 to −1.43 V (vs. Fc+/0), which are dependent on the coordinated NE+ ligand with the order of NSe (−1.12 V) > NS (−1.19 V) > NO (−1.43 V). Thus, the first reduction waves are tentatively assigned to the ligand-centered NE+/0 reduction. On the other hand, the second reduction waves have very similar potentials for these complexes, hence they are assigned to the reduction of the O^N ligand. OsNE also exhibits an irreversible wave with Epa of OsNO (1.65 V) > OsNS (1.40 V) > OsNSe (1.25 V), in line with the decreasing π-accepting ability of these chalcogenonitrosyl ligands on going from O to Se. Thus, these oxidation waves should be metal centered, since the OsII center is more stabilized by the stronger π-accepting NO ligand.
Molecular structures
The molecular structures of OsNE have been determined by X-ray crystallography and selected bond parameters are listed in Table 1. As shown in Fig. 4, the coordination geometries of the metal centers are similar to that of OsN. The Os centers are all 6-coordinated by three CN− in a meridional configuration, a bidentate O^N ligand and a chalcogenonitrosyl ligand. The Os–N4 bond length in the complexes are similar; 1.727(5) Å in OsNO, 1.767(5) Å in OsNS and 1.749(3) Å in OsNSe, indicating that they have double bond character. These bond lengths follow the order: Os–NO < Os–NSe < Os–NS. This trend is also found in the iridium chalcogenonitrosyl series [Ir(NE){N(CHCHPtBu2)2}][PF6].23 The Os1–N4–E bonds are close to linear; 174.1(4)°, 177.8(4)°, 175.4(2)°, respectively, for E = O, S, Se. The N–E bond lengths are 1.206(6) Å, 1.507(5) Å, and 1.675(3) Å, respectively, for E = O, S, Se, which are very close to the sum of the covalent radii of the double bonds; N–O = 1.17 Å; N–S = 1.54 Å; N–Se = 1.67 Å. The Os–O1(phenoxy) bond lengths are comparable to the value of 2.024(2) Å in (PPh4)[Os(NH3)(L)(CN)3],13 indicating the absence of the strong trans influence for these chalcogenonitrosyl ligands.
 |
| | Fig. 4 The structures of the anions OsNO (a), OsNS (b), and OsNSe (c). | |
Table 1 Selected bond parameters (Å, °) for OsNE
| |
OsNO
|
OsNS
|
OsNSe
|
| Os1–N4 |
1.727(5) |
1.767(5) |
1.749(3) |
| Os1–C2 |
2.082(7) |
2.084(8) |
2.069(4) |
| Os1–C3 |
2.041(5) |
2.037(6) |
2.020(4) |
| Os1–C1 |
2.091(7) |
2.083(8) |
2.066(4) |
| Os1–N5 |
2.129(4) |
2.117(5) |
2.127(3) |
| Os1–O1 |
2.022(3) |
2.034(4) |
2.037(2) |
| N4–E |
1.206(6) |
1.507(5) |
1.675(3) |
| Os1–N4–E |
174.1(4) |
177.8(4) |
175.4(2) |
| O1–Os1–N4 |
174.4(2) |
177.1(2) |
175.3(1) |
In our previous work, we showed that OsN* readily undergoes an initial one-electron oxidation of various substrates,12–16 hence it is reasonable to propose that the present photoreactions proceed via an initial 1e− transfer (ET) from NCE− to OsN* to generate OsVN and the NCE˙ radical; this is followed by rapid recombination of the two species to afford the unstable intermediate [OsIV(L)(CN)3(N-ECN)]2−, which then undergoes spontaneous E–CN bond cleavage to produce OsNE and CN− (Fig. 5). The estimated reduction potentials (E0) for NCE˙/NCE− are 1.27 V, 1.63 V, and 2.15 V for E = Se, S, and O, respectively.24 The observed reaction rates are inversely dependent on the E° values, consistent with ET being involved in the rate-determining step.
 |
| | Fig. 5 Proposed mechanism for the reaction of OsN* with NCE−. | |
Reaction of OsN* with diisopropyl sulfide
The above results indicate that OsN* readily abstracts various chalcogen atoms from NCE−. A similar reaction occurs between OsN* and organic sulphide such as diisopropyl sulfide (i-Pr2S). Upon irradiation of a solution of OsN with 300 equiv. of i-Pr2S in CH2Cl2 for 4 h, ESI/MS shows two new product peaks at m/z 571 and 581 (Fig. 6), which are assigned to OsNS and [OsIV(L)(CN)3(NC(CH3)2)]− (Os–NC(CH3)2),12 respectively (Fig. 7). The reaction was also followed by UV/vis spectroscopy, which shows that the absorption band due to OsN gradually decreases, while those of the products gradually increase with time (Fig. S7†). When the reaction was carried out on a preparative scale, the two species were isolated with a molar ratio of ∼1:2.
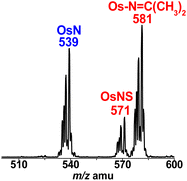 |
| | Fig. 6 ESI/MS of the photoreaction of OsN with excess i-Pr2S for 24 h. | |
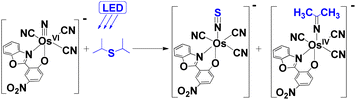 |
| | Fig. 7 The photoreaction of OsN with i-Pr2S. | |
Oxidative desulfurization is an important process for the removal of sulfur from liquid fuels. In this process, sulfur-containing compounds are converted to their corresponding sulfones/sulfoxides using various oxidants.25 To the best of our knowledge, direct S atom abstraction from S-containing substrates has yet to be reported. The proposed mechanism for the reaction of OsN* with i-Pr2S is shown in Fig. 8. The first step is ET from i-Pr2S to OsN* to produce OsVN and the radical cation i-Pr2S˙+, this is followed by their combination to give the Os(IV) species (I). I then undergoes an internal 2e− transfer to give an Os(II) species (II). This is followed by rapid oxidation of II by two 2OsN*, resulting C–S bond cleavage and the formation of OsVN, OsNS, and CH3CH+CH3. CH3CH+CH3 then adds to the nitrido ligand of OsVN to form the Os(V) species III, which further undergoes an internal 1e− oxidative dehydrogenation to afford Os–NC(CH3)2.12
 |
| | Fig. 8 Proposed mechanism for the reaction of OsN* with i-Pr2S. | |
We have carried out DFT calculations on the reaction of OsN* + NCS− and OsN* + i-Pr2S (Fig. S9† and Fig. 9), which support our proposed mechanisms. As shown in Fig. 9, the reaction is downhill in energy after the initial combination of OsVN and the radical cation i-Pr2S˙+ to give the Os(IV) species. The Os(IV) species then undergoes an internal 2e− transfer to give an Os(II) species (3INT2) and this is followed by rapid oxidation of 3INT2 by two 2OsVIN, resulting in C–S bond cleavage and the formation of OsVN, OsNS, and CH3CH+CH3.
 |
| | Fig. 9 Gibbs free energy profile for reaction of OsN* with i-Pr2S at the B3LYP-D3(BJ)/def2-TZVPD level. | |
Dechalcogenation of OsNE
Dechalcogenation of OsNS and OsNSe occur readily by using PPh3. As shown in Fig. 10a, ESI/MS of OsNS with 100 equiv. of PPh3 for 6 h at 313 K shows a new product peak at m/z 801, which is tentatively assigned to [OsIV(L)(CN)3(NPPh3)]− (OsIVNPPh3).26 A minor peak also occurs at m/z 539, which is due to OsN, suggesting that OsN may be an intermediate that further reacts with excess PPh3 to give OsIVNPPh3. The reaction rate of OsNSe with PPh3 is much faster; as shown in Fig. 10b, the ESI/MS for the reaction of OsNSe with 5 equiv. of PPh3 for 0.5 h at 298 K shows two major product peaks at m/z 539 and 801, while the parent peak at m/z 617 disappears completely.
 |
| | Fig. 10 (a) ESI/MS for OsNS with 100 equiv. of PPh3 in CH2Cl2 at 313 K for 6 h; (b) ESI/MS for OsNSe with 5 equiv. of PPh3 in CH2Cl2 at 298 K for 0.5 h. | |
The reaction of OsNE (E = S, Se) with PPh3 can be represented by the following equation.
| | | [OsII(NE)(L)(CN)3]− + PPh3 = [OsVI(N)(L)(CN)3]− + EPPh3 | (1) |
| | | [OsVI(N)(L)(CN)3]− + PPh3 = [OsIV(NPPh3)(L)(CN)3]− | (2) |
When the reactions were carried out on a preparative scale, SPPh3 and SePPh3 were formed and could be extracted with Et2O with >80% yield (Fig. 11). Although for both OsNS and OsNSe, the Os(IV) phosphineiminato complex [OsIV(L)(CN)3(NPPh3)]− (OsIVNPPh3, m/z 801) could be observed by ESI/MS, the osmium product that was actually isolated (after chromatography) is the osmium(III) complex [OsIII(L)(CN)3(NHPPh3)]− with PPh4+ counter ion in ∼70% yield (OsIIINHPPh3). ESI/MS (−ve mode) of OsIIINHPPh3 in MeOH shows a predominant parent anion peak at m/z 802, which is one mass unit higher than OsIVNPPh3. Presumably OsIVNPPh3 is reduced to OsIIINHPPh3 during the work-up. The IR spectrum of OsIIINHPPh3 shows v(CN) stretches at 2113 and 2084 cm−1, and v(N–H) stretches at 3276 cm−1. It has a room-temperature magnetic moment of 1.91μB (Gouy method, solid sample), consistent with its formulation as a low-spin d5 OsIII compound. The CV of OsIIINHPPh3 in CH3CN containing 0.1 M [nBu4N](PF6) shows a quasi-reversible Os(III/II) couple at E1/2 = −1.27 V (vs. Fc+/0) (Fig. S8†). There is also a broad wave at ∼0.17 V, which is tentatively assigned to OsIVNPPh3/OsIIINHPPh3.
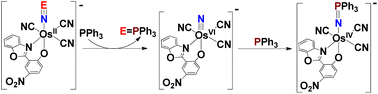 |
| | Fig. 11 Chalcogen abstraction by PPh3 from OsNE (E = S, Se). | |
Oxidative dechalcogenation of OsNSe and OsNS also occurs by using PhIO (Fig. 12), which is much more rapid than with PPh3. Upon mixing OsNS or OsNSe with 5 equiv. of PhIO for 0.5 h in CH3CN, ESI/MS (−ve mode) for both solutions show a predominant peak at m/z 539. When the reactions were carried out on a preparative scale, OsN could be isolated with >90% yield, indicating that OsNSe and OsNS were almost quantitatively converted to OsN by PhIO. Moreover, ESI/MS (−ve mode) for the OsNS + PhIO solution also shows a minor peak at m/z 81, possibly due to HSO3−. On the other hand, for the reaction of OsNSe with PhIO, a grey precipitate was observed, which is presumably SeO2. However, no reaction of OsNO with PhIO was observed, probably due to its much stronger N–O bond.
 |
| | Fig. 12 (a) ESI/MS of OsNSe and (b) after addition of 5 equiv. PhIO for 0.5 h in CH3CN; (c) ESI/MS of OsNS and (d) after addition of 5 equiv. PhIO for 0.5 h in CH3CN. | |
Conclusions
We have demonstrated that OsN* readily undergoes unprecedented chalcogen atom abstraction from NCE− and i-Pr2S; such reaction has not been observed even by metal oxo species. We propose that these reactions occur by initial one-electron oxidation of the substrates by OsN*. The resulting OsNE products readily undergoes dechalcogenation by using PPh3 or PhIO. Notably PhIO regenerates OsN from OsNE, which suggests that it is possible to construct a catalytic cycle for dechalcogenation of substrates based on OsN/PhIO/visible light.
Experimental
(PPh4)[Os(NSe)(L)(CN)3] (OsNSe)
Ten Pyrex tubes (15 × 2 cm) each containing OsN (5 mg, 5.7 μmol) and (PPh4)NCSe (12 mg, 27 μmol) in CH2Cl2 were irradiated with blue LED light (λ > 460 nm) for 96 h, whereby the light-yellow solutions became pale yellow. The solvent was removed by a rotary evaporator and the residue was dissolved in a minimum amount of CH2Cl2 and then loaded onto a silica gel column. The pale yellow band was eluted by CH2Cl2/acetone (v:v, 5:1). (PPh4)[Os(NSe)(L)(CN)3] was obtained as a yellow microcrystalline solid. Yield: 25 mg, 45%. Light yellow crystals were obtained from the slow diffusion of diethyl ether into a CH2Cl2 solution of OsNSe. Selected IR (KBr disc, cm−1): v(CN) 2148 and 2133; v(NSe) 1136; ESI/MS (−ve mode): m/z 617 ([M]−); UV/vis (CH3CN): λmax [nm] (ε [mol−1 dm3 cm−1]): 256 (21320), 277sh (15950), 307 (15770), 367 (15080), 427sh (2750). 1H NMR (400 MHz, CDCl3): δ 8.89 (d, J = 2.9 Hz, 1H, Ar–H), 8.29–8.23 (m, 1H, Ar–H), 7.95–7.89 (m, 5H, Ar–H and PPh4–H), 7.80 (td, J = 7.8, 3.6 Hz, 8H, PPh4–H), 7.75–7.71 (m, 1H, Ar–H), 7.70–7.62 (m, 8H, PPh4–H), 7.53 (dt, J = 10.9, 3.7 Hz, 2H, Ar–H), 6.52 (d, J = 9.3 Hz, 1H, Ar–H). Calcd (%) for C40H27N6O4OsPSe: C, 50.26; H, 2.85; N, 8.79; found: C, 50.28; H, 2.81; N, 8.76.
(PPh4)[Os(NS)(L)(CN)3] (OsNS)
The synthesis of OsNS is similar to that of OsNSe except (PPh4)NCS (23 mg, 58 μmol) was used instead. Yield for OsNS: 27 mg, 52%. Selected IR Selected IR (KBr disc, cm−1): v(CN) 2149 and 2133; v(NS) 1291; ESI/MS (−ve mode): m/z 571 ([M]−); UV/vis (CH3CN): λmax [nm] (ε [mol−1 dm3 cm−1]): 270sh (23710), 277 (26320), 302sh (19240), 374 (16600), 447 (1940). 1H NMR (400 MHz, CDCl3): δ 8.90 (d, J = 2.9 Hz, 1H, Ar–H), 8.15–8.11 (m, 1H, Ar–H), 7.95–7.89 (m, 5H, Ar–H and PPh4–H), 7.80 (td, J = 7.8, 3.6 Hz, 8H, PPh4–H), 7.75–7.70 (m, 1H, Ar–H), 7.70–7.62 (m, 8H, PPh4–H), 7.56–7.49 (m, 2H, Ar–H), 6.51 (d, J = 9.3 Hz, 1H, Ar–H). Calcd (%) for C40H27N6O4OsPS: C, 52.86; H, 2.99; N, 9.25; found: C, 52.82; H, 3.02; N, 9.27.
(PPh4)[Os(NO)(L)(CN)3] (OsNO)
The synthesis of OsNO is similar to that of OsNSe except (PPh4)NCO (22 mg, 58 μmol) was used instead. Yield for OsNO: 5 mg, 10%. The OsNO also could be obtained from oxidation of the guanidine precursor (OsG) by excess m-chloroperbenzoic acid (m-cpba) in CH3CN with about 60% yield.20,21 Selected IR (KBr disc, cm−1): v(CN) 2150 and 2139; v(NO) 1849; ESI/MS (−ve mode): m/z 555 ([M]−); UV/vis (CH3CN): λmax [nm] (ε [mol−1 dm3 cm−1]): 270 (14210), 277 (15950), 295 (18980), 357 (17660), 435sh (760). 1H NMR (400 MHz, CDCl3): δ 8.92 (s, 1H, Ar–H), 8.03 (d, J = 9.5 Hz, 1H, Ar–H), 7.92 (t, J = 7.4 Hz, 4H, PPh4–H), 7.80 (d, J = 7.8 Hz, 8H, PPh4–H), 7.73 (t, J = 7.4 Hz, 2H, Ar–H), 7.65 (dd, J = 12.8, 7.9 Hz, 8H, PPh4–H), 7.58–7.50 (m, 2H, Ar–H), 6.70 (d, J = 9.3 Hz, 1H, Ar–H). Calcd (%) for C40H27N6O5OsP: C, 53.81; H, 3.05; N, 9.41; found: C, 53.76; H, 3.02; N, 9.44.
Dechalcogenation of OsNE
(PPh4)[OsIII(L)(CN)3(NHPPh3)] (OsNHPPh3). OsNSe (20 mg, 20.8 μmol) and PPh3 (55 mg, 0.2 mmol) were dissolved in CH2Cl2 and stirred at room temperature for 6 h to give a dark red solution. The solvent was removed by a rotary evaporator and the residue was dissolved in a minimum amount of CH2Cl2 and then loaded onto a silica gel column. The red band was eluted by CH2Cl2/MeOH (v:v, 10:1). OsNHPPh3 was isolated as a red microcrystalline solid. Yield: 16.7 mg, 70%. Selected IR (KBr disc, cm−1): v(CN) 2113 and 2084; v(N–H) 3276; ESI/MS (−ve mode): m/z 802 ([M]−); μeff = 1.91μB; UV/vis (CH3CN): λmax [nm] (ε [mol−1 dm3 cm−1]): 263sh (15180), 268(15790), 275(15760), 290sh (14800), 311(14790), 392 (12810), 440sh (10580), 550sh (970). Calcd (%) for C58H43N6O4OsP2: C, 61.10; H, 3.80; N, 7.37; found: C, 61.13; H, 3.78; N, 7.39.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21771026), the Excellent Discipline Cultivation Project by JHUN (2023XKZ038), the Natural Science Foundation of Jingzhou Science and Technology Bureau (2022CC54-05) and “Laboratory for Synthetic Chemistry and Chemical Biology” under the Health@InnoHK Program launched by Innovation and Technology Commission, The Government of Hong Kong Special Administrative Region of the People's Republic of China. KCL and TCL also acknowledge financial support from a NSFC_RGC Joint Research Scheme (N_CityU111/20).
References
- R. A. Eikey and M. M. Abu-Omar, Nitrido and Imido Transition Metal Complexes of Groups 6–8, Coord. Chem. Rev., 2003, 243, 83–124 CrossRef CAS.
- J. F. Berry, Terminal Nitrido and Imido Complexes of the Late Transition Metals., Comments Inorg. Chem., 2009, 30, 28–66 CrossRef CAS.
- S. J. K. Forrest, B. Schluschaß, E. Y. Yuzik-Klimova and S. Schneider, Nitrogen Fixation via Splitting into Nitrido Complexes, Chem. Rev., 2021, 121, 6522–6587 CrossRef CAS PubMed.
- J. M. Smith, Reactive Transition Metal Nitride Complexes, Prog. Inorg. Chem., 2014, 58, 417–470 CAS.
-
(a) M. N. Cosio and D. C. Powers, Prospects and challenges for nitrogen-atom transfer catalysis, Nat. Rev. Chem., 2023, 7, 424–438 CrossRef CAS PubMed;
(b) T. Itabashi, K. Arashiba, A. Egi, H. Tanaka, K. Sugiyama, S. Suginome, S. Kuriyama, K. Yoshizawa and Y. Nishibayashi, Direct synthesis of cyanate anion from dinitrogen catalysed by molybdenum complexes bearing pincer-type ligand, Nat. Commun., 2022, 13, 6161 CrossRef CAS PubMed;
(c) M. Fritz, S. Rupp, C. I. Kiene, S. Kisan, J. Telser, C. Würtele, V. Krewald and S. Schneider, Photoelectrochemical Conversion of Dinitrogen to Benzonitrile: Selectivity Control by Electrophile- versus Proton-Coupled Electron Transfer, Angew. Chem., Int. Ed., 2022, 61, e202205922 CrossRef CAS PubMed;
(d) J. Song, Q. Liao, X. Hong, L. Jin and N. Mézailles, Conversion of Dinitrogen into Nitrile: Cross-Metathesis of N2-Derived Molybdenum Nitride with Alkynes, Angew. Chem., Int. Ed., 2021, 60, 12242–12247 CrossRef CAS PubMed.
-
(a) T. J. Meyer and M. H. V. Huynh, The Remarkable Reactivity of High Oxidation State Ruthenium and Osmium Polypyridyl Complexes, Inorg. Chem., 2003, 42, 8140–8160 CrossRef CAS PubMed;
(b) A. G. Maestri, K. S. Cherry, J. J. Toboni and S. N. Brown, [4+1] Cycloadditions of Cyclohexadienes with Osmium Nitrides, J. Am. Chem. Soc., 2001, 123, 7459–7460 CrossRef CAS PubMed;
(c) M. H. V. Huynh, T. J. Meyer, M. A. Hiskey and D. L. Jameson, Os(II)−Nitrosyl and Os(II)−Dinitrogen Complexes from Reactions between Os(VI)−Nitrido and Hydroxylamines and Methoxylamines, J. Am. Chem. Soc., 2004, 126, 3608–3615 CrossRef CAS PubMed;
(d) P. Q. Kelly, A. S. Filatov and M. D. Levin, A Synthetic Cycle for Heteroarene Synthesis by Nitride Insertion, Angew. Chem., Int. Ed., 2022, 61, e202213041 CrossRef CAS PubMed;
(e) H.-X. Wang, L. Wu, B. Zheng, L. Du, W.-P. To, C.-H. Ko, D. L. Phillips and C.-M. Che, C−H Activation by an Iron-Nitrido Bis-Pocket Porphyrin Species, Angew. Chem., Int. Ed., 2021, 60, 4796–4803 CrossRef CAS PubMed;
(f) D. Martelino, S. Mahato, W. VandeVen, N. M. Hein, R. M. Clarke, G. A. MacNeil, F. Thomas and T. Storr, Chromium Nitride Umpolung Tuned by the Locus of Oxidation, J. Am. Chem. Soc., 2022, 144, 11594–11607 CrossRef CAS PubMed;
(g) H. Shi, H. K. Lee, Y. Pan, K.-C. Lau, S.-M. Yiu, W. W. Y. Lam, W.-L. Man and T.-C. Lau, Structure and Reactivity of a Manganese(VI) Nitrido Complex Bearing a Tetraamido Macrocyclic Ligand, J. Am. Chem. Soc., 2021, 143, 15863–15872 CrossRef CAS PubMed.
-
(a) W.-L. Man, W. W. Y. Lam and T.-C. Lau, Reactivity of Nitrido Complexes of Ruthenium(VI), Osmium(VI), and Manganese(V) Bearing Schiff Base and Simple Anionic Ligands, Acc. Chem. Res., 2014, 47, 427–439 CrossRef CAS PubMed, and ref therein.
(b) W.-L. Man, J. Xie, P.-K. Lo, W. W. Y. Lam, S.-M. Yiu, K.-C. Lau and T.-C. Lau, Functionalization of Alkynes by a (Salen)ruthenium(VI) Nitrido Complex, Angew. Chem., Int. Ed., 2014, 53, 8463–8466 CrossRef CAS PubMed;
(c) J. Xie, W.-L. Man, C.-Y. Wong, X. Chang, C.-M. Che and T.-C. Lau, Four-Electron Oxidation of Phenols to p-Benzoquinone Imines by a (Salen)ruthenium(VI) Nitrido Complex, J. Am. Chem. Soc., 2016, 138, 5817–5820 CrossRef CAS PubMed.
-
(a) S. B. Muñoz III, W.-T. Lee, D. A. Dickie, J. J. Scepaniak, D. Subedi, M. Pink, M. D. Johnson and J. M. Smith, Styrene Aziridination by Iron(IV) Nitrides, Angew. Chem., Int. Ed., 2015, 54, 10600–10603 CrossRef PubMed;
(b) J. L. Martinez, H.-J. Lin, W.-T. Lee, M. Pink, C.-H. Chen, X. Gao, D. A. Dickie and J. M. Smith, Cyanide Ligand Assembly by Carbon Atom Transfer to an Iron Nitride, J. Am. Chem. Soc., 2017, 139, 14037–14040 CrossRef CAS PubMed;
(c) D. W. Crandell, S. B. Muñoz, J. M. Smith and M.-H. Baik, Mechanistic study of styrene aziridination by iron(IV) nitrides, Chem. Sci., 2018, 9, 8542–8552 RSC;
(d) G. E. Cutsail III, B. W. Stein, D. Subedi, J. M. Smith, M. L. Kirk and B. M. Hoffman, EPR, ENDOR, and Electronic Structure Studies of the Jahn–Teller Distortion in an FeV Nitride, J. Am. Chem. Soc., 2014, 136, 12323–12336 CrossRef PubMed;
(e) J. J. Scepaniak, C. S. Vogel, M. M. Khusniyarov, F. W. Heinemann, K. Meyer and J. M. Smith, Synthesis, Structure, and Reactivity of an Iron(V) Nitride, Science, 2011, 331, 1049–1052 CrossRef CAS PubMed;
(f) J. J. Scepaniak, R. P. Bontchev, D. L. Johnson and J. M. Smith, Snapshots of Complete Nitrogen Atom Transfer from an Iron(IV) Nitrido Complex, Angew. Chem., Int. Ed., 2011, 50, 6630–6633 CrossRef CAS PubMed;
(g) J. J. Scepaniak, M. D. Fulton, R. P. Bontchev, E. N. Duesler, M. L. Kirk and J. M. Smith, Structural and Spectroscopic Characterization of an Electrophilic Iron Nitrido Complex, J. Am. Chem. Soc., 2008, 130, 10515–10517 CrossRef CAS PubMed.
-
(a) B. Meunier, S. P. de Visser and S. Shaik, Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed;
(b) I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Structure and Chemistry of Cytochrome P450, Chem. Rev., 2005, 105, 2253–2278 CrossRef CAS PubMed;
(c) S. Fosshat, S. D. M. Siddhiaratchi, C. L. Baumberger, V. R. Ortiz, F. R. Fronczek and M. B. Chambers, Light-Initiated C–H Activation via Net Hydrogen Atom Transfer to a Molybdenum(VI) Dioxo, J. Am. Chem. Soc., 2022, 144, 20472–20483 CrossRef CAS PubMed;
(d) Y. Cao, J. A. Valdez-Moreira, S. Hay, J. M. Smith and S. P. de Visser, Reactivity Differences of Trigonal Pyramidal Nonheme Iron(IV)-Oxo and Iron(III)-Oxo Complexes: Experiment and Theory, Chem. – Eur. J., 2023, 29, e202300271 CrossRef CAS PubMed;
(e) J. A. Valdez-Moreira, D. M. Beagan, H. Yang, J. Telser, B. M. Hoffman, M. Pink, V. Carta and J. M. Smith, Hydrocarbon Oxidation by an Exposed, Multiply Bonded Iron(III) Oxo Complex, ACS Cent. Sci., 2021, 7, 1751–1755 CrossRef CAS PubMed.
- L. J. Luo, Q. Q. Su, S. C. Cheng, J. Xiang, W. L. Man, W. M. Shu, M. H. Zeng, S. M. Yiu, C. C. Ko and T. C. Lau, Tunable Luminescent Properties of Tricyanoosmium Nitrido Complexes Bearing a Chelating O^N Ligand, Inorg. Chem., 2020, 59, 4406–4413 CrossRef CAS PubMed.
- J. Xiang, X.-X. Jin, Q.-Q. Su, S.-C. Cheng, C.-C. Ko, W.-L. Man, M. Xue, L. Wu, C.-M. Che and T.-C. Lau, Photochemical Nitrogenation of Alkanes and Arenes by a Strongly Luminescent Osmium(VI) Nitrido complex, Commun. Chem., 2019, 2, 40 CrossRef.
- J. Xiang, M. Peng, Y. Pan, L. J. Luo, S. C. Cheng, X. X. Jin, S. M. Yiu, W. L. Man, C. C. Ko, K. C. Lau and T. C. Lau, Visible Light-Induced Oxidative N-dealkylation of Alkylamines by a Luminescent Osmium(VI) Nitrido Complex, Chem. Sci., 2021, 12, 14494–14498 RSC.
- J. Xiang, J. Zhu, M. Zhou, L. L. Liu, L. X. Wang, M. Peng, B. S. Hou, S. M. Yiu, W. P. To, C. M. Che, K. C. Lau and T. C. Lau, Oxidative C-O bond Cleavage of Dihydroxybenzenes and Conversion of Coordinated Cyanide to Carbon Monoxide Using a Luminescent Os(VI) Cyanonitrido Complex, Chem. Commun., 2022, 58, 7988–7991 RSC.
- Q. Q. Su, K. Fan, X. D. Huang, J. Xiang, S. C. Cheng, C. C. Ko, L. M. Zheng, M. Kurmoo and T. C. Lau, Field-induced Slow Magnetic Relaxation in Low-spin S=1/2 Mononuclear Osmium(V) Complexes, Dalton Trans., 2020, 49, 4084–4092 RSC.
- L.-L. Liu, L.-X. Wang, M. Peng, J. Xiang, H. Yang, S.-M. Yiu and T.-C. Lau, Ring Nitrogenation of Aromatic Amines by the Excited State of an Osmium(VI) Nitrido Complex, Inorg. Chem., 2023, 62, 1447–1454 CrossRef CAS PubMed.
- J. Xiang, Y. Pan, L.-L. Liu, L.-X. Wang, H. Yang, S.-C. Cheng, S.-M. Yiu, C.-F. Leung, C.-C. Ko, K.-C. Lau and T.-C. Lau, Visible Light-Induced Oxidation of Alcohols by a Luminescent Osmium(VI) Nitrido Complex: Evidence for the Generation of PhIO+ as a Highly Active Oxidant in the Presence of PhIO, J. Am. Chem. Soc., 2023, 145, 9129–9135 CrossRef CAS PubMed.
-
(a) T. J. Crevier, S. Lovell and J. M. Mayer, Chalcogen Atom Transfer to a Metal Nitrido. The First Transition Metal Selenonitrosyl Complex, J. Am. Chem. Soc., 1998, 120, 6607–6608 CrossRef CAS;
(b) B. L. Tran, R. Thompson, S. Ghosh, X. Gao, C. H. Chen, M. H. Baik and D. J. Mindiola, A Four-Coordinate Thionitrosyl Complex of Vanadium, Chem. Commun., 2013, 49, 2768–2770 RSC.
-
(a) M. G. Scheibel, I. Klopsch, H. Wolf, P. Stollberg, D. Stalke and S. Schneider, Thionitrosyl- and Selenonitrosyliridium Complexes, Eur. J. Inorg. Chem., 2013, 2013, 3836–3839 CrossRef CAS;
(b) H. Y. Ng, W. M. Cheung, E. K. Huang, K. L. Wong, H. H.-Y. Sung, I. D. Williams and W. H. Leung, Ruthenium Chalcogenonitrosyl and Bridged Nitrido Complexes Containing Chelating Sulfur and Oxygen Ligands, Dalton Trans., 2015, 44, 18459–18468 RSC;
(c) H. Y. Ng, N. M. Lam, M. Yang, X. Y. Yi, I. D. Williams and W. H. Leung, Ruthenium(VI) Nitrido Complexes with a Sterically Bulky Bidentate Schiff Base Ligand, Inorg. Chim. Acta, 2013, 394, 171–175 CrossRef CAS.
-
(a) A. Wu, A. Dehestani, E. Saganic, T. J. Crevier, W. Kaminsky, D. E. Cohen and J. M. Mayer, Reactions of Tp–Os Nitrido Complexes with the Nucleophiles Hydroxide and Thiosulfate, Inorg. Chim. Acta, 2006, 359, 2842–2849 CrossRef CAS;
(b) B. W. S. Kolthammer and P. Legzdins, Preparation of Dicarbonyl (eta. 5-cyclopentadienyl) Thionitrosylchromium(I). The First Organometallic Thionitrosyl Complex, J. Am. Chem. Soc., 1978, 100, 2247–2248 CrossRef CAS.
-
(a) J. Xiang, W. L. Man, S. M. Yiu, S. M. Peng and T. C. Lau, Reaction of an Osmium(VI) Nitrido Complex with Cyanide: Formation
and Reactivity of an Osmium(III) Hydrogen Cyanamide Complex, Chem. – Eur. J., 2011, 17, 13044–13051 CrossRef CAS PubMed;
(b) J. Xiang, Q. Wang, S. M. Yiu, W. L. Man, H. K. Kwong and T. C. Lau, Aerobic Oxidation of an Osmium(III) N-Hydroxyguanidine Complex to Give Nitric Oxide, Inorg. Chem., 2016, 55, 5056–5061 CrossRef CAS PubMed.
-
(a) J. Xiang, Q. Wang, S. M. Yiu and T. C. Lau, Dual Pathways in the Oxidation of an Osmium(III) Guanidine Complex. Formation of Osmium(VI) Nitrido and Osmium Nitrosyl Complex, Inorg. Chem., 2017, 56, 2022–2028 CrossRef CAS PubMed;
(b) J. Xiang, Q.-Q. Su, L.-J. Luo and T.-C. Lau, Synthesis and reactivity of an osmium(III) aminoguanidine complex, Dalton Trans., 2019, 48, 11404–11410 RSC.
-
(a) J. W. Dethlefsen, E. D. Hedegård, R. D. Rimmer, P. C. Ford and A. Døssing, Flash and Continuous Photolysis Studies of the Thionitrosyl Complex Cr(CH3CN)5(N S)2+ and the Nitric Oxide Analogs: Reactions of Nitrogen Monosulfide in Solution, Inorg. Chem., 2009, 48, 231–238 CrossRef CAS PubMed;
(b) J. R. Dethlefsen, A. Døssing and E. D. Hedegård, Electron Paramagnetic Resonance Studies of Nitrosyl and Thionitrosyl and Density Functional Theory Studies of Nitrido, Nitrosyl, Thionitrosyl, and Selenonitrosyl Complexes of Chromium, Inorg. Chem., 2010, 49, 8769–8778 CrossRef CAS PubMed.
- M. G. Scheibel, B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Closed-shell and Open-shell Square-planar Iridium Nitrido Complexes, Nat. Chem., 2012, 4, 552–558 CrossRef CAS PubMed.
- D. M. Stanbury, Reduction potentials involving inorganic free radicals in aqueous solution, Adv. Inorg. Chem., 1989, 33, 69–138 CrossRef CAS.
- F. Boshagh, M. Rahmani, K. Rostami and M. Yousefifar, Key Factors Affecting the Development of Oxidative Desulfurization of Liquid Fuels: A Critical Review, Energy Fuels, 2022, 36, 98–132 CrossRef CAS.
- T.-W. Wong, T.-C. Lau and W.-T. Wong, Osmium(VI) Nitrido and Osmium(IV) Phosphoraniminato Complexes Containing Schiff Base ligands, Inorg. Chem., 1999, 38, 6181–6186 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, crystal data, ESI/MS, and 1H NMR. CCDC 2267408–2267410. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi01081c |
| ‡ These two authors contributed equally. |
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Ji-Yan
Liu
a,
Kai-Chung
Lau
*a,
Ji-Yan
Liu
a,
Kai-Chung
Lau