
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of VIVO–8-hydroxyquinoline species with RNase A: the effect of metal ligands in the protein adduct stabilization†
Giarita
Ferraro
a,
Luigi
Vitale
a,
Giuseppe
Sciortino
 b,
Federico
Pisanu
c,
Eugenio
Garribba
*c and
Antonello
Merlino
*a
b,
Federico
Pisanu
c,
Eugenio
Garribba
*c and
Antonello
Merlino
*a
aDepartment of Chemical Sciences, University of Naples Federico II, I-80126 Napoli, Italy. E-mail: antonello.merlino@unina.it
bInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, 43007 Tarragona, Spain
cDipartimento di Medicina, Chirurgia e Farmacia, Università di Sassari, Viale San Pietro, I-07100 Sassari, Italy. E-mail: garribba@uniss.it
First published on 11th July 2023
Abstract
Herein, we studied the interaction of bovine pancreatic ribonuclease (RNase A) with [VIVO(8-HQ)2] – a promising antitrypanosomal, antituberculosis, and antitumor vanadium complex (VC) formed by 8-hydroxyquinolinato (8-HQ), reaching IC50 values lower than cisplatin in HCT116 and A2780 cancer lines (0.99 and 0.96 μM vs. 15.0 and 3.4 μM) and the minimum inhibitory concentration (MIC) of 3.7 μM against strains of Mycobacterium tuberculosis. The system was investigated by spectroscopic (UV-vis absorption spectroscopy, circular dichroism, and electron paramagnetic resonance), computational (docking and DFT calculations), and X-ray crystallographic multi-technique approach. Crystallographic results at pH 5.1 demonstrate that upon interaction with the protein, one of the 8-HQ ligands is replaced by a water molecule and by Glu111 side-chain, resulting in the inhibition of the RNase A activity. EPR data confirm the formation of this adduct, with VIV coordinated by 8-HQ and by the couples (Glu-COO−; H2O) at pH 5.1, (His-N; H2O) at pH 7.4, and (His-N; OH−) or (Ser/Thr-O−, H2O) at pH 8.4. Computational data unveil the reasons behind the ligand exchange, not observed for similar complexes like [VIVO(picolinato)2] and not predicted by thermodynamic considerations. Overall, our results show that the VC–protein binding is defined not only by the thermodynamic stability of VC but also by other factors, such as the structure and steric requirements of the metal ligands, with important implications in the drug delivery strategies.
Introduction
The applications of vanadium in materials science, catalysis, biology, pharmacy, and medicine have been discussed over the last few years and constitute a field of extensive research.1–3 A growing interest in vanadium complexes (VCs) concerns their possible use in medicine as antibacterial,4 antiparasitic, but mainly as antidiabetic5–7 and anticancer1,8–10 agents.Among the VCs with pharmacological properties, species with stoichiometry VIVOL2 were studied with L being a bidentate and monoanionic ligand. Among them, the bis-chelated complexes formed by maltolato (malt), 1,2-dimethyl-3-hydroxy-4(1H)-pyridinonato (dhp), picolinato (pic), and acetylacetonato (acac) are worthy of being cited.1,11,12 Recently, several molecules with ascertained biological activity were proposed as metal ligands for new potential drugs. Among them, 8-hydroxyquinolines (8-HQ), found in many biologically active natural products, have emerged as a privileged skeleton for new drugs;13 such ligands belong to the series of metabolites of tryptophan that participate in the regulation of many cells of the immune system and are involved in many immune-mediated diseases and disorders.14,15 Transition metal complexes with 8-HQ and its derivatives were recently designed and tested, both in vitro and in vivo, against several tumour cell lines.16–19 Focusing our attention on vanadium, VCs of 8-HQ-based ligands were tested as potential antitrypanosomal, antituberculosis, and antitumor agents.20–24 The biological effectiveness depends on the nature of the substituents on the 8-HQ moiety, and in some cases, IC50 values smaller than 1 μM were found, for example, in the case of the VIVO complex formed by 5,7-dichloro-8-quinolinato against Trypanosoma cruzi.22 Similarly, the anticancer potential is rather promising and the IC50 value against the cisplatin sensitive/resistant ovarian A2780 cell line of the homoleptic species of VVO with 8-HQ and of the mixed species of VIVO with 8-HQ and picolinate or salicylideneglycinato is in the range of 5–14 μM,20 while that of VIVO complexes formed by 8-hydroxyquinoline hydrazones is <6.3 μM on malignant melanoma (A-375) cancer cells,24 all these complexes being more active than cisplatin on the same cells (22.4 and 11.2 μM on A2780 and A-375, respectively).20,24
The binding of VCs with proteins plays a crucial role in the processes of transport in organisms and action mechanism.25,26 Over the last years, a growing number of research studies discussed the interaction with small proteins, such as hen egg white lysozyme (HEWL),27,28 myoglobin,29 ubiquitin,28,30 cytochrome c,31 or large proteins like human serum transferrin in the apo or holo form,32 human serum albumin,33 hemoglobin,32 and immunoglobulin G.32 The interaction depends on several factors: the structure of VIVOL2 complex in aqueous solution, its thermodynamic stability, the presence of accessible residues on the protein structure, the stabilization of the adduct by secondary interactions, such as hydrogen bonds or van der Waals contacts,25,34 making the description of these systems a demanding, but stimulating, challenge for the inorganic and bioinorganic chemists. However, despite the importance of the binding VC–protein, also with regard to the design of new drugs and their industrial development, only few X-ray diffraction (XRD) studies on single crystals are available for VIVO–L–protein adducts up to now: those formed by VIVO(pic)2 fragment with HEWL35 and bovine pancreatic ribonuclease (RNase A),36 and by VIVO(bipy/phen)2+ (bipy = 2,2′-bipyridine, phen = 1,10-phenanthroline),37 VIVO(malt)+/VIVO(malt)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38 and VIVO(empp)+/VIVO(empp)2 (empp = 1-methyl-2-ethyl-3-hydroxy-4(1H)-pyridinonato)39 with HEWL. Depending on the experimental conditions, both covalent and non-covalent interactions were revealed, with Asp, Asn, and Glu residues being involved in the first type of binding and Asp, Asn, Glu, Gln, Cys, Lys, and Arg in the second one.35–38 To the best of our knowledge, no other structures based on VIV–L moiety were deposited in Protein Data Bank (PDB).40
38 and VIVO(empp)+/VIVO(empp)2 (empp = 1-methyl-2-ethyl-3-hydroxy-4(1H)-pyridinonato)39 with HEWL. Depending on the experimental conditions, both covalent and non-covalent interactions were revealed, with Asp, Asn, and Glu residues being involved in the first type of binding and Asp, Asn, Glu, Gln, Cys, Lys, and Arg in the second one.35–38 To the best of our knowledge, no other structures based on VIV–L moiety were deposited in Protein Data Bank (PDB).40
Here, we studied the interaction of [VIVO(8-HQ)2] with RNase A through a combined application of experimental and computational techniques. The major biochemical function of RNases is the catalytic scission of RNA,41,42 but recent results suggested that RNA is implicated in many human diseases, and for this reason, RNases are considered as potential targets in the drug action. In addition, RNase A has also been frequently used as a model protein in metalation studies and structures with several metallodrugs,43,44 including cisplatin, carboplatin, oxaliplatin,45,46 Ru-,47 Rh-,48 Pd-,49 Ir-,50 and Au-based drugs,51 are known. Recently, the metalation of arsenoplatin-1, a dual pharmacophore anticancer agent, has been presented.52 In this study, the binding of [VIVO(8-HQ)2] to RNase A has been examined at the solid state by XRD studies and in aqueous solution by collecting spectroscopic data through UV-vis absorption spectroscopy, circular dichroism (CD), and electron paramagnetic resonance (EPR). The results have been rationalized by in silico methods, such as docking and density functional theory (DFT). The aims of the study are: (i) the characterization of the possible VC–RNase A adducts; (ii) the comparison between the different behaviour with RNase A of the two structurally similar complexes, [VIVO(pic)2(H2O)] and [VIVO(8-HQ)2(H2O)] (derived in aqueous solution from the hydrolysis of the square pyramidal species [VIVO(8-HQ)2]), which – in principle – could undergo the replacement of the equatorial water ligand by a residue side-chain; (iii) the role of the protein in the ligand exchange of [VIVOL2] complexes (it will be demonstrated here that RNase A cleaves the coordination of one of the two chelating ligands in [VIVO(8-HQ)2]); (iv) finally, the role of the metal ligand in the stabilization of the adducts formed with a protein that – in turn – may be related to the transport in biological fluids and mode of action of the vanadium-based potential drugs.
Our results disclose that the interaction of VCs with proteins can lead to non-trivial speciation schemes and, in some cases, not predictable with thermodynamic considerations based on the behavior of solutions containing only the specific VC.
Experimental and computational section
Materials
Water was deionised prior to use through the purification system Millipore MilliQ Academic or purchased from Sigma-Aldrich (LC-MS grade). VIVOSO4·3H2O, 8-hydroxyquinoline, and 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), sodium phosphate, sodium citrate, sodium acetate, NaOH, and H2SO4 were Sigma-Aldrich products of the highest grade available and used as received. The complex [VIVO(8-HQ)2] was synthesised as reported in the literature.53 RNase A was purchased from Sigma-Aldrich (type XIIA) and used without further purification.UV-vis absorption spectroscopy and circular dichroism
The stability of [VIVO(8-HQ)2] has been studied by UV-vis absorption spectroscopy, using a Varian Cary 5000 UV-vis-NIR spectrophotometer, recording the spectra of the systems containing only the VC (50 μM) and containing RNase A plus VC (molar ratio 1:3 and metal concentration of 50 μM). The spectra were collected at room temperature in 10 mM sodium citrate buffer (pH 5.1) and in 10 mM sodium phosphate buffer (pH 7.4) in the range 240–700 nm as a function of time. The following parameters were used: data pitch 1.0 nm, bandwidth 2.0 nm, scan speed 600 nm min−1, and quartz cuvette with 1.0 cm path length.
Circular dichroism (CD) spectra were recorded with a Jasco J-715 spectropolarimeter equipped with a Peltier temperature controller. Far-UV CD measurements were carried out at a protein concentration of 7.3 μM in 10 mM sodium citrate buffer (pH 5.1) and 10 mM sodium phosphate buffer (pH 7.4) at 20 °C, using a cell with an optical path length of 0.1 cm. The instrumental conditions were as follows: response time 2 s, scan rate 50 nm min−1, 2.0 nm bandwidth, and 1.0 nm data pitch. The spectra were the average of three scans.
The thermal stability was determined by recording the CD signal at 222 nm in the temperature mode in the range 20–95 °C, with a scan rate of 1.0 °C min−1. The melting temperatures (Tm) were determined through analysis of the first derivative of the unfolding curves.
EPR spectroscopy
The solutions for EPR measurements were prepared by dissolving VIVOSO4, 8-HQ, and RNase A in ultra-pure water to get a VIVO2+ concentration of 0.8 mM and a VIVO2+:8-HQ:RNase A molar ratio of 1:1:1 and 1:2:1. The benchmark spectra of [VIVO(8-HQ)(H2O)2]+ were obtained with a V concentration of 1.0 mM at pH 4.0 using a VIVO2+:8-HQ ratio of 1:1 and at pH 3.0 using a VIVO2+:8-HQ ratio of 1:2. Argon was bubbled through the solutions to ensure the oxygen absence. 0.1 M HEPES buffer was added to the solutions. The pH was adjusted to the desired value with NaOH or H2SO4 diluted solutions. Under these experimental conditions, the oxidation of VIV to VV is negligible, as proven by the examination of the relative intensity of EPR (and UV-vis absorption spectra) as a function of the time (0–4 h).
Anisotropic EPR spectra were recorded at 120 K on aqueous solutions with an X-band Varian E-9 spectrometer equipped with a variable temperature unit. The microwave frequency was 9.15–9.16 GHz, microwave power 20 mW, time constant 0.5 s, modulation frequency 100 kHz, and modulation amplitude 0.4 mT. The spectra were immediately measured after the samples were transferred into the EPR tubes. In the text, only the high-field part of the EPR spectra is shown because it is more sensitive than the low-field region to the identity of the equatorial donors and the amount of the several species in solution.54,55
Crystallization, X-ray diffraction data collection, structure, and refinement
Metal-free RNase A crystals were grown by hanging drop vapor diffusion method mixing equal volumes of protein (22 mg mL−1) and reservoir solutions (22% PEG4K, 10 mM sodium citrate, pH 5.1) at 20 °C. Metal-free protein crystals were then exposed to solid aliquots of [VIVO(8-HQ)2] to obtain the adduct. These crystals were then captured in silicon loops and cryoprotected using a solution containing the reservoir and 25% glycerol after 50 soaking days. Cryoprotected crystals were then flash-frozen in nitrogen and shipped to Elettra synchrotron, Trieste, Italy.X-ray diffraction data were collected at 100 K at XRD2 beamline. Data were indexed, integrated, and scaled using the Global Phasing AutoPROC pipeline.56 Data collection statistics are reported in Table S1 of the ESI.† The structure of the adduct was solved by molecular replacement with Phaser implemented in the CCP4 suite,57 using the structure of metal-free RNase A deposited in the PDB under the accession code 1JVT (chain A)58 as starting model. The structures were refined using Refmac;59 manual interventions on the models based on observation of the electron density map were carried out using Coot.60 The electron density map of the protein chain is rather well defined, with the exclusion of residues 17–21 and 18–20 of chains A and B, respectively. The location of the V atom is confirmed by inspection of the anomalous difference electron density map. The final model of the adduct, which presents 2305 atoms and an average B-factor of 30.52 Å2, refines to an R-factor of 0.174 and R-free of 0.222. Refinement statistics are reported in Table S1.† The final structure of the adduct was validated using the PDB validation server and deposited in the PDB under the accession code 7QWH (https://www.rcsb.org/structure/7QWH).
Quantum and docking calculations
All the DFT calculations were carried out with Gaussian 16 (revision B.01).61 The geometry of VIVO species and harmonic frequencies were computed at the level of theory B3P86/6-311g(d,p) using the SMD model for water.62,63 This guarantees a good degree of accuracy in the structure optimization of first-row transition metal complexes64,65 and, particularly, of VCs.66–68Docking calculations were carried out through GOLD 5.8 software69 on the X-ray structure available in the PDB of free RNase A (PDB code: 1JVT58). The PDB structure was cleaned, removing all the small molecules and crystallographic water, and hydrogen atoms were added with the UCSF Chimera program.70 Residue side-chain protonation state was computed by means of the PROPKA algorithm.71
The DFT optimized structure of cis-[VIVO(8-HQ)2(H2O)] was preliminarily treated by replacing the equatorial leaving H2O ligand with a dummy hydrogen atom according to what was recently established.27,72
To identify the possible RNase A binding sites for the VIVO species, relative Solvent Excluded Surface (SES) calculations73 were preliminarily performed, focusing on the most exposed potential coordinating residues. The docking simulations were carried out by constructing in the region of interest an evaluation sphere of 12 Å. Side-chain flexibility was taken into account, considering the GOLD implemented rotamer libraries.74 Genetic algorithm (GA) parameters have been set to 50 GA runs and a minimum of 100000 operations. The other parameters of GA were set to default.
The scoring (Fitness of GoldScore) was evaluated by applying the modified version of GoldScore scoring function, which was validated in previously published papers.27,72 The best solutions (binding poses) were evaluated through three main criteria: (i) the mean (Fmean) and the highest value (Fmax) of the scoring (Fitness of GoldScore) associated with each pose; (ii) the population of the cluster containing the best pose; (iii) the position in the Fitness ranking of the computed cluster.
The refinement of the VC–protein adducts found by dockings was performed by cutting out the region with [VIVO(8-HQ)2] and [VIVO(8-HQ)(H2O)]+ fragments and neighbour interacting amino acid side-chains within a radius of 5 Å. The extracted clusters were treated at full DFT theory level for all atoms, completing the valence and freezing the backbone atoms to simulate the protein constraints.75 The geometry optimizations were performed using B3LYP/6-31g(d,p) level of theory including D3 Grimme's correction for dispersion for the high level (BS1). The final potential energies were obtained by single point calculations on the optimized geometries using the extended basis set BS2, consisting of the triple-ζ def2-TZVP for the main group element and the quadruple-ζ def2-QZVP set for vanadium.76
Ribonucleolytic activity assays
RNase A catalytic activity was qualitatively measured according to the Kunitz method77 by monitoring cleavage of yeast RNA via UV-vis absorption spectroscopy in the absence and presence of VC in 0.050 M sodium acetate (pH 5.0) at room temperature. The VC–protein adduct was obtained by incubating RNase A with [VIVO(8-HQ)2] for 3, 24, and 72 h using 1:0.5, 1:5, 1:10, and 1:30 protein-to-metal molar ratios. The experiments were carried out in triplicate with RNA concentration of 0.5 mg mL−1 and RNase A concentration of 7.3 μM.
Results and discussion
Behaviour of VIVO2+/8-HQ system in aqueous solution
Bis-chelated oxidovanadium(IV) complexes can give in aqueous solution a series of transformations that can affect their transport and interaction with biomolecules.25,34,78 In particular, they can change the number and coordination geometry upon hydrolysis with the loss of one or both the ligands. In the case of 8-hydroxyquinoline, the square pyramidal solid complex [VIVO(8-HQ)2] transforms into a mixture of octahedral cis-[VIVO(8-HQ)2(H2O)] isomers with an equatorial-equatorial and an equatorial-axial arrangement of the two ligands, a water molecule in cis to the V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, and two quinoline-N or two phenolato-O in the equatorial plane,53 as illustrated in Scheme 1. The formation of cis-octahedral [VIVO(8-HQ)2(H2O)] species is confirmed by the X-ray structures of [VVO2(8-HQ)2]− or [VVO(O-iPr)(8-HQ)2],79,80 in which the second oxido ligand or the isopropoxido anion, in cis position to the VVO bond, derive from the oxidation or replacement of the water molecule of the corresponding VIVO complex. Depending on the experimental conditions, such as pH, V concentration, and molar ratio, cis-[VIVO(8-HQ)2(H2O)] may give the 1:1 species [VIVO(8-HQ)(H2O)2]+.53
O bond, and two quinoline-N or two phenolato-O in the equatorial plane,53 as illustrated in Scheme 1. The formation of cis-octahedral [VIVO(8-HQ)2(H2O)] species is confirmed by the X-ray structures of [VVO2(8-HQ)2]− or [VVO(O-iPr)(8-HQ)2],79,80 in which the second oxido ligand or the isopropoxido anion, in cis position to the VVO bond, derive from the oxidation or replacement of the water molecule of the corresponding VIVO complex. Depending on the experimental conditions, such as pH, V concentration, and molar ratio, cis-[VIVO(8-HQ)2(H2O)] may give the 1:1 species [VIVO(8-HQ)(H2O)2]+.53
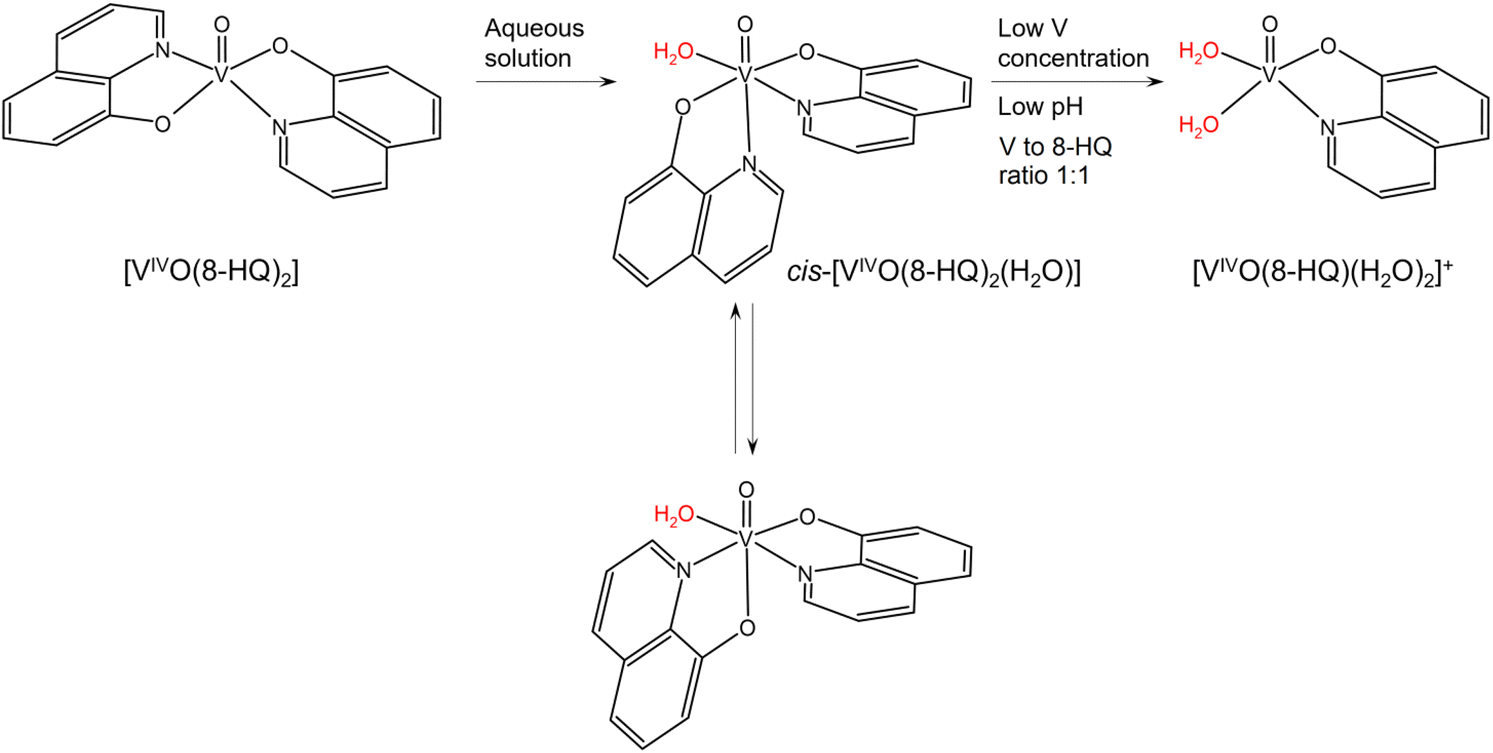 | ||
| Scheme 1 Possible transformation of [VIVO(8-HQ)2] in aqueous solution. The water molecules replaceable by protein residue side-chains are indicated in red. | ||
UV-vis absorption spectroscopy
UV-vis absorption spectroscopy was used to obtain the first indications on the behaviour of [VIVO(8-HQ)2] in 10 mM sodium phosphate buffer at pH 7.4 and in 10 mM sodium citrate buffer at pH 5.1 in the absence and presence of RNase A (Fig. S1†). The investigated experimental conditions were chosen to get information on the potential interaction between VC and protein at neutral pH and under the experimental conditions used to obtain crystals of RNase A in the presence of VC (vide infra). Spectra collected as a function of time reveal significant differences between the behaviour of the systems without and with the protein, both at pH 5.1 and 7.4, indicating an interaction of VIVO–8-HQ fragment(s) with RNase A (cfr. panels A and C in Fig. S1† with those B and D); moreover, the comparison of the absorptions at pH 5.1 and 7.4 show that they are different, suggesting the formation of various adducts when pH increases. The spectra do not change for at least 7 days, and this means that the adduct(s) formed with RNase A are stable at room temperature and pH 5.1 and 7.4 and does(do) not undergo any transformations such as a ligand replacement and/or oxidation.EPR spectroscopy
EPR spectra were recorded at 120 K as a function of pH in the systems containing [VIVO(8-HQ)2] or [VIVO(8-HQ)(H2O)2]+ plus RNase A. The high-field region of the spectra is represented in Fig. 1, which is divided into two parts: part A on the left shows the systems containing [VIVO(8-HQ)2] and RNase A (VIVO2+:8-HQ:RNase A molar ratio 1:2:1), while part B on the right shows the systems containing [VIVO(8-HQ)(H2O)2]+ and RNase A (VIVO2+:8-HQ:RNase A ratio 1:1:1).
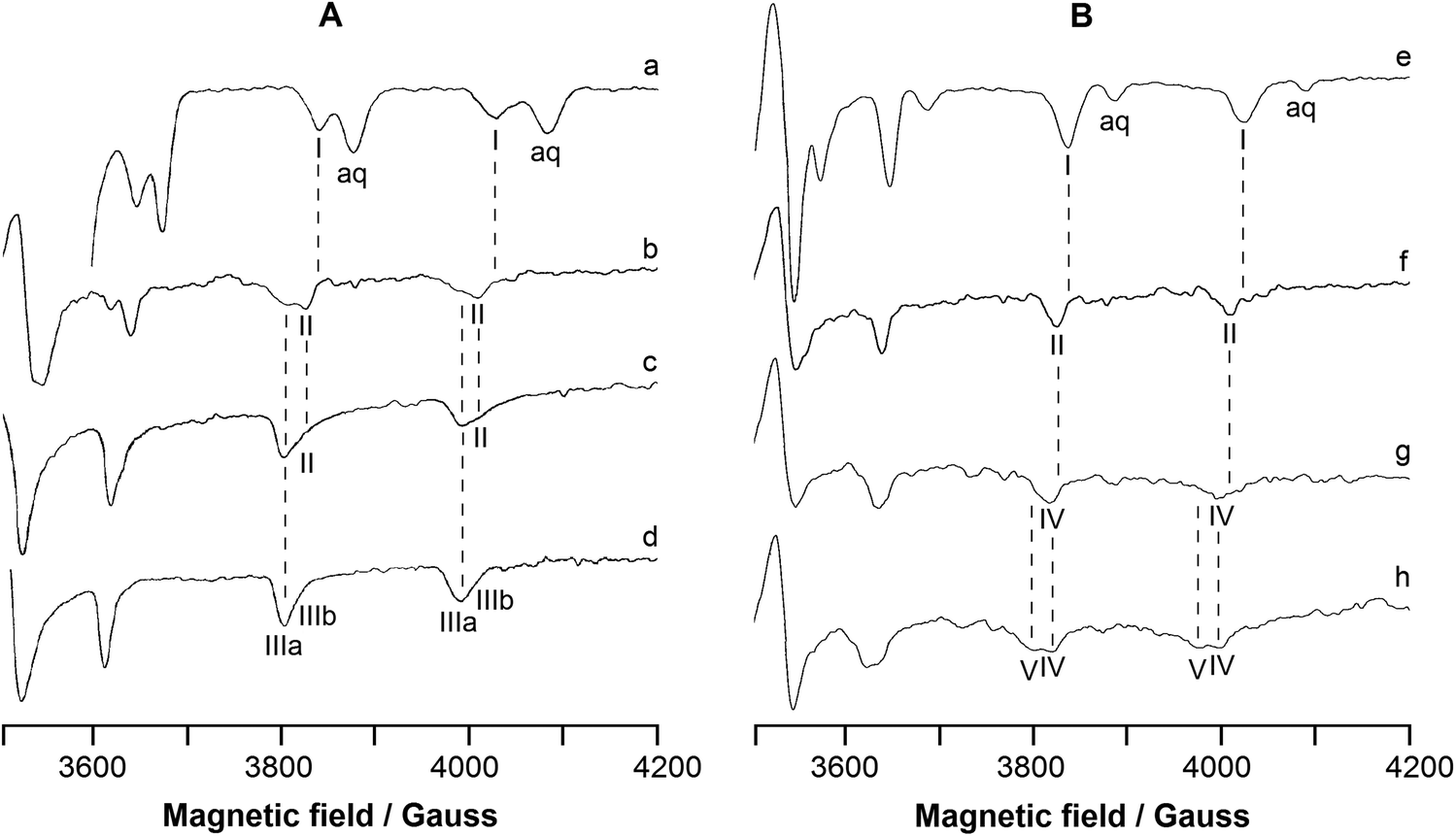 | ||
| Fig. 1 High-field region of anisotropic X-band EPR spectra at 120 K in an aqueous solution containing the systems: (A) [VIVO(8-HQ)2] and RNase A (molar ratio VIVO2+:8-HQ:RNase A 1:2:x with x = 0–1) and (B) [VIVO(8-HQ)(H2O)2]+ and RNase A (molar ratio VIVO2+:8-HQ:RNase A 1:1:y with y = 0–1). Traces a–h show the spectra recorded with: (a) VIVO2+:8-HQ:RNase A 1:2:0, pH 3.0; (b) VIVO2+:8-HQ:RNase A 1:2:1, pH 5.1; (c) VIVO2+:8-HQ:RNase A 1:2:1, pH 6.4; (d) VIVO2+:8-HQ:RNase A 1:2:1, pH 7.4; (e) VIVO2+:8-HQ:RNase A 1:1:0, pH 4.0; (f) VIVO2+:8-HQ:RNase A 1:1:1, pH 5.1; (g) VIVO2+:8-HQ:RNase A 1:1:1, pH 7.4; (h) VIVO2+:8-HQ:RNase A 1:1:1, pH 8.4. V concentration is 1.0 mM in traces a and e, and 0.8 mM in traces b–d and f–h. I indicates the MI = 5/2, 7/2 resonances of [VIVO(8-HQ)(H2O)2]+, II of the adduct [VIVO(8-HQ)(H2O)]+–RNase A with Asp/Glu-COO− coordination, IIIa and IIIb of the two isomers cis-[VIVO(8-HQ)2(H2O)], IV of the adduct [VIVO(8-HQ)(H2O)]+–RNase A with His-N coordination, and V of the adduct [VIVO(8-HQ)(OH)]–RNase A with His-N coordination or [VIVO(8-HQ)(H2O)]+–RNase A with Ser/Thr-O− coordination, while aq denotes the MI = 7/2 resonances of the aqua ion [VIVO(H2O)5]2+. The positions of the MI = 5/2, 7/2 resonances of I, II, IIIa, IIIb, IV, and V species are also indicated by the dotted lines. | ||
In Fig. 1A, the trace a shows the spectrum of the hydrolyzed form of [VIVO(8-HQ)2], i.e., [VIVO(8-HQ)(H2O)2]2+ taken as a benchmark, measured in the binary system VIVO2+:8-HQ 1:2 at pH as low as 3.0 (the MI = 5/2, 7/2 resonances are indicated by I); its spin Hamiltonian parameters along the z axis are gz = 1.942 and Az = 170.3 × 10−4 cm−1, in agreement with what was reported in the literature.53 The value of Az(51V) estimated with the “additivity relationship”, the empirical rule that allows predicting the hyperfine coupling constant along z axis from the contribution of the four donors in the equatorial plane of VIVO2+ ion,81,82 is 170.2 × 10−4 cm−1. The spectra of the ternary system VIVO2+:8-HQ:RNase A with a molar ratio of 1:2:1 are represented in traces b–d. At pH 5.1, the same used for XRD determination (vide infra), the spectrum is characterized by gz = 1.944 and Az = 168.2 × 10−4 cm−1 (II in Fig. 1); the decrease of Az compared to [VIVO(8-HQ)(H2O)2]2+ suggests that equatorial water is replaced by a stronger ligand that, at this pH, may be a carboxylate of an Asp or Glu residue. Notably, for a species with equatorial coordination [(quinoline-N, phenolato-O); Asp/Glu-COO; H2O], Az estimated by the “additivity relationship” is 166.7 × 10−4 cm−1. The adduct II may be indicated with [VIVO(8-HQ)(H2O)]+–RNase A and coexists in solution with the two isomers of the bis-chelated species cis-[VIVO(8-HQ)2(H2O)] (indicated with IIIa and IIIb, see also Scheme 1). The spin Hamiltonian parameters for IIIa and IIIb are gz = 1.943, Az = 165.6 × 10−4 cm−1 and gz = 1.946 and Az = 162.8 × 10−4 cm−1, which agree well with the data previously reported.53 With increasing pH, [VIVO(8-HQ)(H2O)]+–RNase A survives up to pH 6.4, while the relative amount of cis-[VIVO(8-HQ)2(H2O)], favoured at these metal to ligand ratios and pH values, becomes larger. At pH 7.4, only IIIa and IIIb are detected in solution.
Trace e of Fig. 1B shows the benchmark spectrum of [VIVO(8-HQ)(H2O)2]+ recorded in the system containing only VIVO2+:8-HQ 1:1: at pH 4.0. The spectra recorded in the system VIVO2+:8-HQ:RNase A 1:1:1 are presented in traces f–h. At pH 5.1 (trace f), the adduct [VIVO(8-HQ)(H2O)]+–RNase A (II) is the major species in solution and this suggests that the binding of a COO− group is favoured under these experimental conditions. When pH is brought close to the physiological value (trace g), the spectrum changes and a new adduct with gz = 1.945 and Az = 165.9 × 10−4 cm−1 is revealed (IV in Fig. 1). As observed for the system [VIVO(pic)2(H2O)]/RNase A,36 this variation could be ascribed to the coordination of a His-N, deprotonated at this pH, that replaces Asp/Glu-COO−; for this species, with equatorial coordination [(quinoline-N, phenolato-O); His-N; H2O], Az estimated by the “additivity relationship” is 164.7 × 10−4 cm−1. The change in the EPR parameters can be explained considering that at pH 5.1, histidine residues are still protonated and cannot interact with VIV, whereas around pH 7.4, they undergo deprotonation of the imidazolium group and His-N can replace the carboxylate group of Asp/Glu. Notably, at these conditions (VIVO2+ concentration 0.8 mM, VIVO2+:8-HQ 1:1 and pH 7.4), the percentage amounts of [VIVO(8-HQ)(H2O)2]+ and [VIVO(8-HQ)2(H2O)] in solution – predicted from the thermodynamic stability constants53 – are ca. 0% and 48% (Table 1); in other words, the 1:1 VIVO–8-HQ complex should not exist in solution. Instead, IV is the only revealed species and no resonances attributable to [VIVO(8-HQ)2(H2O)] (IIIa and IIIb in the traces c and d) are detected; this suggests that the interaction with RNase A stabilizes the [VIVO(8-HQ)(H2O)]+ moiety, as XRD technique and computational methods allowed us to confirm (vide infra).
| VIVO2+:8-HQ ratio |
pH | [VIVO(H2O)5]2+ | [VIVO(8-HQ)(H2O)2]+ | [VIVO(8-HQ)2(H2O)] | Hydroxido speciesb |
|---|---|---|---|---|---|
| a Percentages calculated using the stability data for VIVO complexes reported in ref. 53. b Hydroxido species are [VIVO(OH)]+, [(VIVO)2(OH)2]2+, [(VIVO)2(OH)5]−, [VIVO(OH)3]−, and the hydroxido complex formed by 8-HQ, [VIVO(8-HQ)2(OH)]−. | |||||
| 5.1 | 0.0 | 5.1 | 94.9 | 0.0 | |
| 1:2 |
6.4 | 0.0 | 0.8 | 98.7 | 0.4 |
| 7.4 | 0.0 | 0.0 | 95.3 | 4.7 | |
| 5.1 | 10.4 | 59.2 | 20.3 | 10.0 | |
| 1:1 |
7.4 | 0.0 | 0.2 | 47.7 | 52.1 |
| 8.4 | 0.0 | 0.0 | 34.7 | 65.3 | |
Finally, when pH is increased to 8.4, the appearance of a further species (V in Fig. 1), which coexists with IV, is observed. Two options are compatible with the experimental spin Hamiltonian parameters, gz = 1.947 and Az = 161.2 × 10−4 cm−1: the deprotonation of a water molecule in IV to give a hydroxido complex with the donor set [(quinoline-N, phenolato-O); His-N; OH−] or the interaction of the moiety [VIVO(8-HQ)(H2O)]+ with a side-chain of a residue stronger than histidine, like Ser or Thr, which would undergo deprotonation at this pH value to give the coordination mode [(quinoline-N, phenolato-O); Ser/Thr-O−; H2O]. It is worth noticing that this third adduct V exists even though the percentage of [VIVO(8-HQ)(H2O)2]+ predicted in solution by thermodynamics is zero (Table 1); as pointed out above, the reasons for the increased stability of [VIVO(8-HQ)(H2O)]+ fragment are assessed by docking and DFT calculations.
A plausible structure of the three adducts revealed by EPR spectroscopy (indicated with II, IV and V in Fig. 1) is shown in Scheme S1.†
Circular dichroism: effect of the VC binding on protein structure and VC–protein adduct stability
To evaluate the effect of the VC binding on the structure of RNase A, far-UV circular dichroism spectra of free RNase A and the protein in the presence of [VIVO(8-HQ)2] with a molar ratio of 1:0.5, 1:2, and 1:3 have been collected in 10 mM sodium phosphate buffer at pH 7.4 (i.e., the physiological pH) and at pH 5.1 (i.e., the value used in the crystallization experiments) in 10 mM sodium citrate buffer (Fig. S2A and S2B†). CD spectra suggest that the protein does not experience any change in secondary structure content in solution upon the VC binding, retaining its overall structure.
Since it has been demonstrated that the metalation of proteins can alter their thermal stability,43,83 CD spectra have also been used to evaluate the stability of the VC–protein adduct obtained by incubating the protein with [VIVO(8-HQ)2] with a molar ratio of 1:2 and 1:3. Experimental data collected in 10 mM sodium phosphate buffer at pH 7.4 and in 10 mM sodium citrate buffer at pH 5.1 after 24 h from incubation reveal that the adducts are stable and that the binding does not alter at all the thermal stability of RNase A (Table 2). Indeed, melting temperatures (Tm) of RNase A are 64 ± 1 °C and 66 ± 1 °C in sodium phosphate buffer and in sodium citrate buffer, respectively, and similar Tm values were obtained for the adducts prepared upon 24 h incubation of the protein with VC. This result is in agreement with what was observed for the adduct formed upon the reaction of RNase A with [VIVO(pic)2(H2O)].36
| T m (°C) in sodium citratea | T m (°C) in sodium phosphateb | |
|---|---|---|
| a Value measured in 10 mM sodium citrate buffer at pH 5.1. b Value measured in 10 mM sodium phosphate at pH 7.4. | ||
| Free RNase A | 66 ± 1 °C | 64 ± 1 °C |
| RNase A upon 24 h incubation with [VIVO(8-HQ)2] using a 1:2 protein to metal molar ratio |
65 ± 1 °C | 64 ± 1 °C |
| RNase A upon 24 h incubation with [VIVO(8-HQ)2] using a 1:3 protein to metal molar ratio |
66 ± 1 °C | 64 ± 1 °C |
X-ray structure
Soaking of monoclinic crystals of RNase A containing two molecules in the asymmetric unit (chain A and B hereafter) with an excess of [VIVO(8-HQ)2] leads to the formation of crystals of the VC–protein adduct (Fig. 2A). In the absence of the protein, the VC formed by 8-hydroxyquinolinato is stable under the experimental conditions used for the soaking experiments as demonstrated by the analysis of UV-vis absorption spectra of [VIVO(8-HQ)2] collected as function of time (Fig. S2C†). Crystals of the adduct diffract X-rays at 1.57 Å resolution. Data collection and refinement statistics for the structure are reported in Table S1.† The position of the V fragment was unambiguously identified by inspection of 2Fo–Fc, Fo–Fc, and anomalous difference electron density maps. The V fragment binds to the side-chain of Glu111 in chain A, while chain B remains unaltered, as occurs in the adduct formed upon reaction of the protein with [VIVO(pic)2(H2O)].36 Rmds of Cα atoms of the two RNase A chains in the asymmetric unit, which can be used as a good estimate for the comparison between the structure of the adduct and that of the metal-free protein, is as low as 0.44 Å. RNase A retains its characteristic structure consisting of two antiparallel β-sheets that form a cationic cleft that binds RNA (see ref. 84). The cleft contains subsites (B1, B2, and B3) that interact specifically with bases and subsites (P0, P1, and P2) that interact with phosphoryl groups. In the V binding site, which is located in the B2 subsite,85 V adopts a slightly distorted square pyramidal geometry, with one of the two original 8-HQ ligands that is replaced by a solvent molecule (water) and by the OE1 of Glu111, giving a [VIVO(8-HQ)(H2O)]+–RNase A adduct (Fig. 2B). The 8-HQ ligand is held in its position by stacking interactions with the His119 side-chain. Oxygens from the V coordination sphere are hydrogen bonded to N atoms of the side-chains of Gln69 and Asn71 and to water molecules.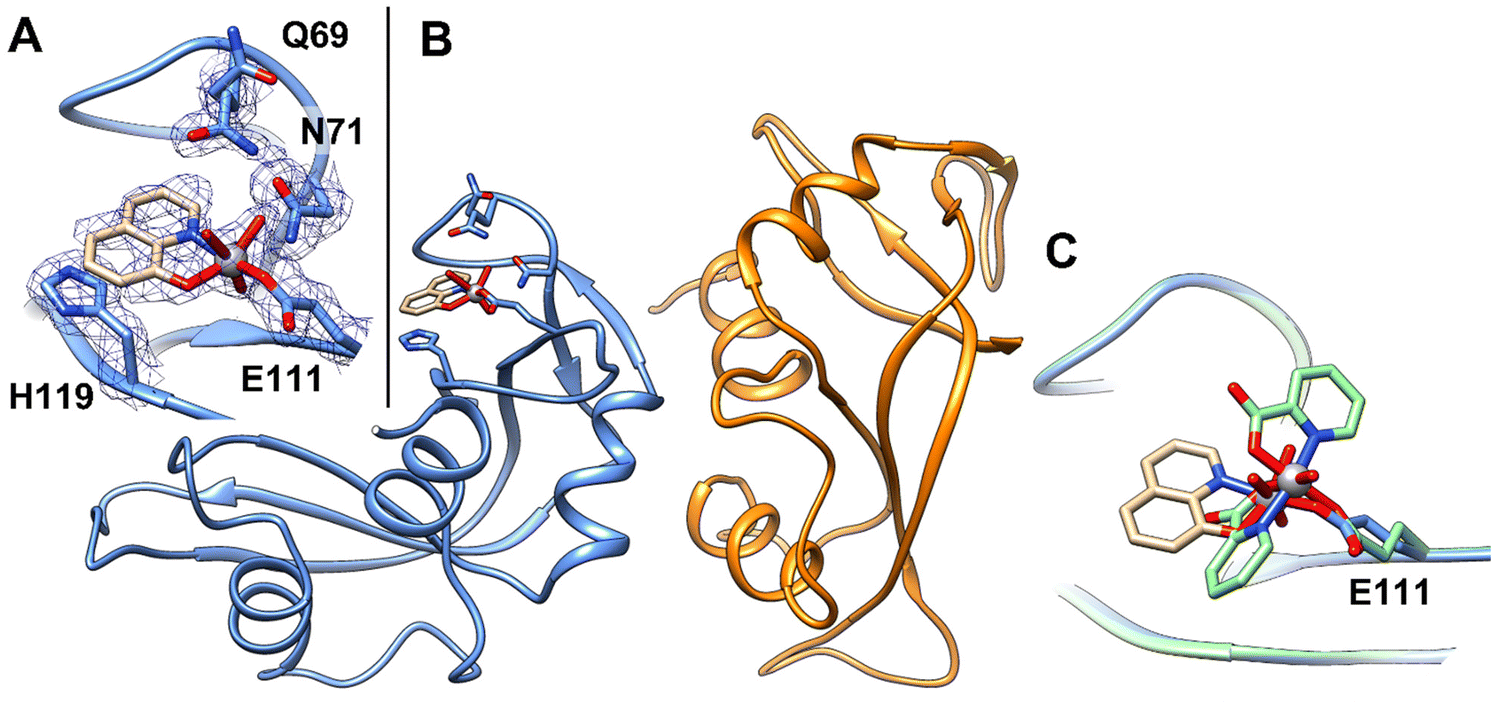 | ||
| Fig. 2 (A) Detail of the V binding site on RNase A structure. 2Fo–Fc electron density maps are contoured at the 1.0σ level (in blue). (B) Overall structure of the adduct formed upon reaction of [VIVO(8-HQ)(H2O)]+ with RNase A (PDB code: 7QWH (https://www.rcsb.org/structure/7QWH)). The two molecules in the asymmetric unit are reported in blue and orange, respectively. (C) Superimposition of the structures of the adducts formed upon reaction of RNase A with [VIVO(8-HQ)2] (tan) and with [VIVO(pic)2(H2O)] (green). | ||
The VIVO(oxido) bond was found at a length of 1.66 Å, which is not far from the expected value (1.57–1.65 Å);86 the V–O(8-HQ) and V–N(8-HQ) bond lengths are 1.78 and 1.97 Å, respectively. The VIV–OE1(Glu111) bond length is 2.06 Å, while the distance of V from water O atom is 1.94 Å. The VC refines with occupancy equal to 0.70 and B-factors within the range 23.67–33.73 Å2.
The V binding site and the identity of the V fragment identified in the structure have been further verified using a second data collection on an additional crystal of the adduct obtained under the same experimental conditions (data not shown).
The comparison between the structures of the adducts [VIVO(8-HQ)(H2O)]+–RNase A (here solved) and [VIVO(pic)2]–RNase,36 obtained under the same experimental conditions, reveals significant differences: (i) the structural superposition shows that the V moieties bind to the side-chain of Glu111 with a different orientation of the equatorial-equatorial chelating ligand (Fig. 2C); (ii) in the present study, [VIVO(8-HQ)(H2O)]+–RNase A is formed with the mono-chelated VIVO(8-HQ)+ fragment, while with picolinato the adduct [VIVO(pic)2]–RNase A is formed with the bis-chelated moiety VIVO(pic)2. The results of the two research works (this study and ref. 36) appear to be in contrast with the observation of the relationship between the thermodynamic stability of VIVO fragments and the formation of the adducts with proteins. Indeed, it has been recently proposed that the composition of VC–protein adducts generally follows the stability of the VIVO species in solution.87 For example, with HEWL, the VCs of pic and dhp form [VIVOL2]–HEWL adducts,35,87 while those of malt and acac yield [VIVOL]+–HEWL adducts;87,88 this is in agreement with their speciation profiles in solution, being [VIVOL2(H2O)] the most abundant complex when L = pic, dhp, and [VIVOL(H2O)2]+ the major species when L = malt, acac. Notably, in the present system, the adduct [VIVO(8-HQ)(H2O)]+–RNase A is formed, even though the 1:2 complex [VO(8-HQ)2(H2O)] is the most abundant species in solution. In fact, at pH 5.1 – i.e., the value in the soaking experiments where the adducts of RNase A with VIVO(pic)2 and VIVO(8-HQ)+ fragments were crystallized – the percent amount of VIVO(pic)(H2O)2+ and VIVO(pic)2 is 7.8% and 89.7% with a V concentration of 1 mM (in agreement with the experimental adduct [VIVO(pic)2]–RNase A reported recently36), while it is 4.5% and 95.4% for VIVO(8-HQ)(H2O)2+ and VIVO(8-HQ)2, namely in contrast with the X-ray structure presented in this study (it must be pointed out that these values were calculated using the thermodynamic stability constants determined for the complexes of the VIVO2+ ion with pic and 8-HQ ligands in refs. 53 and 89 see Table S2† for the percent amounts with V concentrations in the range 10 μM–1 mM). These conclusions indicate that thermodynamic stability is not the only factor governing the formation of the protein adducts, but – presumably – other variables play an important role. The computational study allowed us to rationalize this apparent contradiction (vide infra).
Computational studies
The novelty of the revealed transformation led us to undertake a multiscale computational analysis of the binding process of [VIVO(8-HQ)2] to RNase A. Our strategy consisted of: (i) the prediction of the binding of the cis-VIVO(8-HQ)2 moiety to the active site of protein by molecular docking and (ii) from the docking-predicted structure, the analysis of the 8-HQ ligand exchange that entails the formation of the final adduct [VIVO(8-HQ)(H2O)]+–RNase A, with the coordination of Glu111 residue, through full-DFT cluster calculations.As a result of our first docking assay, among the four isomers of cis-[VIVO(8-HQ)2(H2O)], i.e., the species existing in solution (see Scheme 1), those with OC-6-42 and OC-6-44 arrangement (N-donor in the axial position, Scheme S2†) display higher scoring values than those with OC-6-24 and OC-6-23 (O-donor in the axial position, Scheme S2†). This is due to an extra stabilization coming from a hydrogen bond with the Asn71 side-chain (Table S3†). It is important to note that the best docking-predicted adduct for OC-6-42 displays the equatorial 8-HQ in the same arrangement as the reported X-ray structure (Fig. S3†) and, for this reason, it has been selected for the following refinement studies. The full-DFT optimization of the docking predicted structure led to a barrierless isomerization from the initial six-coordinated octahedral [VIVO(8-HQ)2]–RNase A adduct (cis-VIVO(8-HQ-κ2O,N)2(Glu111-O), with two equatorial O-donors and one N-donor from 8-HQ plus an additional O-donor from Glu111) to the five-coordinated VIVO(8-HQ-κ2O,N)(8-HQ-κO)(Glu111-O) (intermediate I, Fig. 3A). This is stabilized by the π-stacking interaction between the amide side-chains of Asn67 and Gln69 and 8-HQ aromatic rings, dragging the axial N-donor and the metal centre about 2.8 Å apart and leading to the breaking of the V–N coordinative bond. Having a less extended conjugated system, picolinato ligand in [VIVO(pic)2]–RNase A does not interact with Gln69, thus the V–N bond is not weakened and remains stable.36 Furthermore, 8-HQ (pKa of OH group 9.6153) is more easily protonated than pic (pKa of COOH group <189), thus favouring the release of one ligand molecule under the acidic conditions used in the crystallization experiments. From intermediate I, a water molecule can enter the first coordination sphere of the metal, displacing the equatorial phenolato-O donor that migrates in the axial position forming the six-coordinated cis-[VIVO(8-HQ-κ2O,N)(8-HQ-κO)(Glu111-O)(H2O)] (Fig. 3B); the activation energy barrier associated with this step, 15.3 kcal mol−1, is compatible with a fast process at room temperature. The final species, [VIVO(8-HQ-κ2O,N)(H2O)(Glu111-O)], falling at −13.4 kcal mol−1, is obtained by protonation and consequent release of the axial 8-HQ along with a concomitant flip of Asn71 in an exergonic process driving the reaction (Fig. 3C). This final DFT-characterized adduct perfectly matches the solid-state structure obtained by X-ray crystallography (see Fig. S4†).
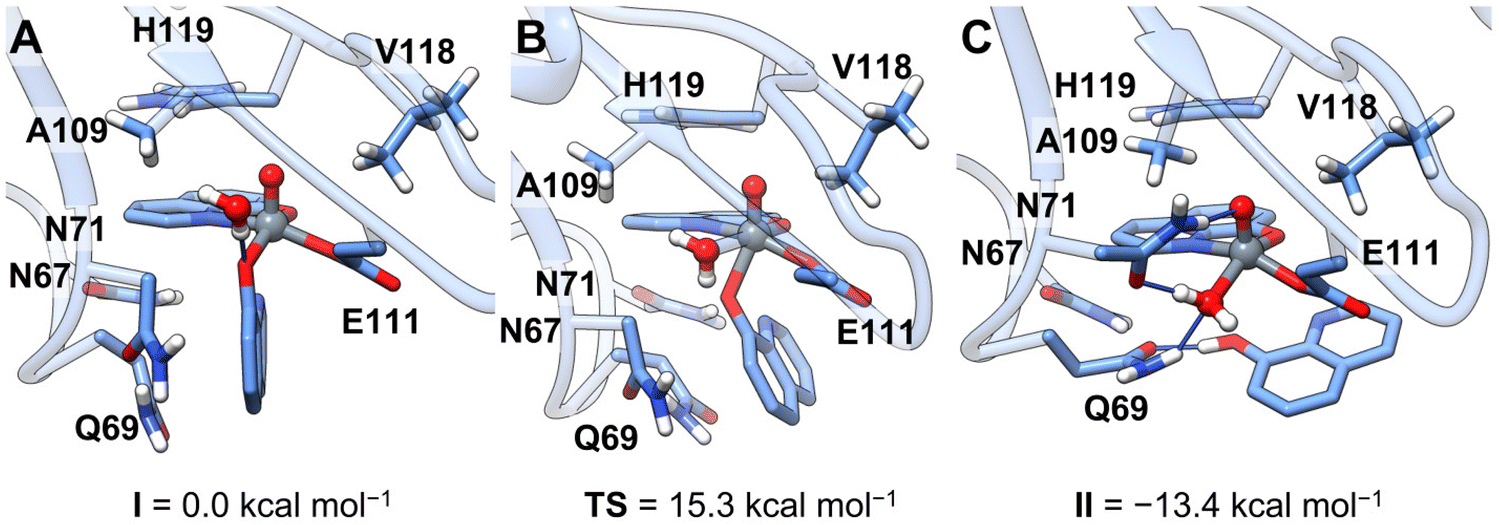 | ||
| Fig. 3 Optimized geometries (solid sticks) over the X-ray structure of RNase A (transparent ribbons) of (A) best docking solution for the OC-6-42 isomer, with an additional water molecule (intermediate I); (B) transition state for equatorial water coordination and concomitant 8-HQ migration to the axial position, TS; (C) final adduct upon protonation and release of the axial 8-HQ ligand, II. Hydrogen bonds are depicted as narrower blue sticks. | ||
The coordinated 8-HQ ligand is stabilized by π-stacking with the His119 side-chain and is involved in a hydrogen bond network with the N-atoms of the side-chains of Gln69 and Asn71 and water molecules. The VIVO(oxido atom) bond distance is 1.59 Å, in the expected range,86 while the V–O(8-HQ) and V–N(8-HQ) bond lengths are 1.95 and 2.13 Å, respectively. Finally, the VIV–OE1 for Glu111 and VIV–OH2 distances are 1.96 and 2.05 Å.
For the sake of completeness, additional docking explorations were carried out to characterize the species inferred by EPR data at pH 7.4 (see Fig. 1), namely the binding of the moiety [VIVO(8-HQ)(H2O)]+ by donors other than carboxylates, allowing us to identify His105 and His119 as putative coordinating residues at physiological pH; notably, and in contrast with what was observed for [VIVO(malt)2],38 the relative orientation of the [VIVO(8-HQ)(H2O)(Glu111-O)] adduct hinders the binding of a second [VIVO(8-HQ)(H2O)]+ moiety to His119 (Fig. 4). Moreover, as commented before, this last residue is involved in π-stacking stabilization of the [VIVO(8-HQ)(H2O)(Glu111-O)] adduct, having as a consequence that His105 is the most likely candidate in the VIV binding.
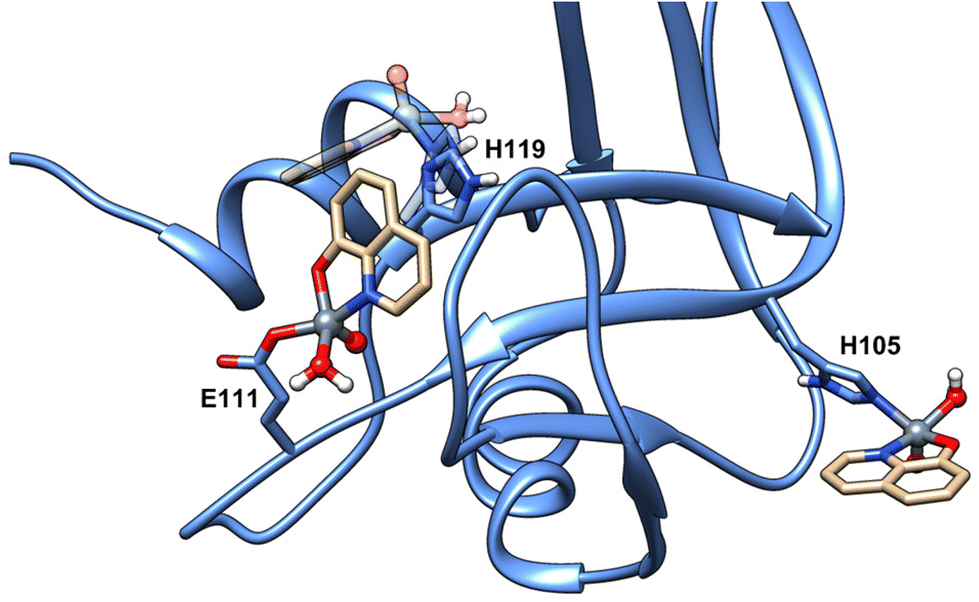 | ||
| Fig. 4 Docking predicted binding of the moiety [VIVO(8-HQ)(H2O)]+ to the residues His105 and His119. The overlap of the binding sites with Glu111 and His119 residues can be observed. The predicted adduct with His119 is shown in transparency. | ||
The change in the EPR spectra at pH 8.4 could be explained – as pointed out above – with the deprotonation of the equatorial water molecule or the replacement of water by Ser or Thr side-chains. Considering – as seen above – that the histidine coordination is restricted to His105 residue, the hydroxido complex would have the formula [VIVO(8-HQ)(OH)(His105-N)]. The interaction with Ser-O− and Thr-O− could also be possible due to at least 17 surface residues, which could bind the metal fragment to form adducts with coordination [VIVO(8-HQ)(H2O)(Ser/Thr-O−)]. Notably, these two possibilities (water deprotonation or Ser binding) were invoked to explain the decrease of Az from pH 4.2 to 8.4 of the reduced vanadium bromoperoxidase (VBrPO) with VIV in the active site.32,90
Ribonucleolytic activity assays
Since the V-containing moiety binds RNase A in the B2 subsite, it can be expected that the protein can lose its ability to degrade RNA in the presence of the VC formed by 8-HQ. To verify this hypothesis, the catalytic activity of the adduct formed upon reaction of [VIVO(8-HQ)2] with RNase A has been investigated using the Kunitz assay.77 As expected, the experimental data, reported in Fig. S5,† reveal that the presence of the VC alters the ribonuclease activity of the protein, although the latter is not completely lost even when incubated for 72 h with a 1:30 RNase A to VC molar ratio. A comparable behaviour was observed for the [VIVO(pic)2]–RNase A adduct,36 and for the species formed upon reaction of RNase A with Ru-,47 Pd-,49 Au-,51 and Pt-based drugs.91
Conclusions
VCs could be employed as therapeutic agents in a wide range of medical conditions, mainly as antiparasitic, antidiabetic, and anticancer potential drugs.8 In their path in the organism from the administration to the target cells, they can interact with proteins,25 and the formation of VC–protein adducts may be relevant in the transport of putative V drugs, in cellular uptake and mechanism of action. A solid knowledge of the type and structure of the VC–protein adducts and of the factors that favour and stabilize their formation is therefore essential to boost the design of new biologically active VCs at the industrial level.Here, we have studied the interaction of [VIVO(8-HQ)2] – a promising antitrypanosomal, antituberculosis, and antitumor VC formed by 8-hydroxyquinolinato ligand – with RNase A by a combination of biophysical techniques, including X-ray crystallography. The X-ray structure of the adduct obtained at pH 5.1 reveals that: (i) the VC binds RNase A without altering the overall conformation and thermal stability of the protein; (ii) the [VIVO(8-HQ)(H2O)]+ fragment binds to the protein, after the release of one anionic 8-HQ ligand; (iii) the metal centre is anchored to the side-chain of Glu111, the same residue involved in the interaction with [VIVO(pic)2(H2O)] recently published;36 (iv) the binding of [VIVO(8-HQ)(H2O)2]+ affects the catalytic activity of RNase A by obstructing the B2 subsite.
Notably, [VIVO(pic)2(H2O)] and [VIVO(8-HQ)2] form two different types of adducts. While [VIVO(pic)2(H2O)] gives the species [VIVO(pic)2]–RNase A upon the replacement of water by the carboxylate group of Glu111, [VIVO(8-HQ)2] gives [VIVO(8-HQ)(H2O)]+–RNase A, although the thermodynamic stability should favour the binding of [VIVO(8-HQ)2] between pH 5.0 and 7.4. The computational results highlight that the adduct [VIVO(8-HQ)(H2O)]+–RNase A derives from an exergonic transformation of [VIVO(8-HQ)2]–RNase A (i.e., a species similar to [VIVO(pic)2]–RNase A) with a low energy barrier, driven by the interaction with Gln69 residue within the binding pocket, which stabilizes the intermediate and the transition state via a π-stacking interaction.
In conclusion, our data confirm that VC–protein binding is a complex process, influenced not only by the thermodynamic stability of VC but also by other factors such as the concentration in solution, the presence of accessible residues on the protein structure, the nature and steric requirements of the metal ligand, and the stabilization of the adduct by secondary forces like hydrogen bonds and van der Waals contacts. All these factors should be taken into account in the prediction of the interaction with potential targets and, considering that in some cases, the reaction of VCs with proteins leads to the formation of adducts with high anti-proliferative activity,92 in the design and development of new potential vanadium-based drugs.
Conflicts of interest
There are no conflicts to declare.Author contributions
The manuscript was written through the contributions of all authors, who contributed equally. All authors have given approval to the final version of the manuscript.Acknowledgements
This work was supported by Fondazione di Sardegna (grant FdSGarribba2017). A. M. and G. F. thank N. Demitri for his help with X-ray diffraction data collection at Elettra synchrotron. G. S. also thanks Spanish MINECO’ Juan de la Cierva program, FJC2019-039135-I.References
- D. Rehder, Perspectives for vanadium in health issues, Future Med. Chem., 2016, 8, 325–338 CrossRef CAS PubMed.
- A. Ścibior, Ł. Pietrzyk, Z. Plewa and A. Skiba, Vanadium: Risks and possible benefits in the light of a comprehensive overview of its pharmacotoxicological mechanisms and multi-applications with a summary of further research trends, J. Trace Elem. Med. Biol., 2020, 61, 126508 CrossRef PubMed.
- M. Aureliano, N. I. Gumerova, G. Sciortino, E. Garribba, A. Rompel and D. C. Crans, Polyoxovanadates with emerging biomedical activities, Coord. Chem. Rev., 2021, 447, 214143 CrossRef CAS.
- B. Bueloni, D. Sanna, E. Garribba, G. R. Castro, I. E. León and G. A. Islan, Design of nalidixic acid-vanadium complex loaded into chitosan hybrid nanoparticles as smart strategy to inhibit bacterial growth and quorum sensing, Int. J. Biol. Macromol., 2020, 161, 1568–1580 CrossRef CAS PubMed.
- D. C. Crans, L. Henry, G. Cardiff and B. I. Posner, in Essential Metals in Medicine: Therapeutic Use and Toxicity of Metal Ions in the Clinic, ed. P. L. Carver, De Gruyter GmbH, Berlin, 2019, pp. 203–230 Search PubMed.
- K. H. Thompson, J. Lichter, C. LeBel, M. C. Scaife, J. H. McNeill and C. Orvig, Vanadium treatment of type 2 diabetes: A view to the future, J. Inorg. Biochem., 2009, 103, 554–558 CrossRef CAS PubMed.
- S. Treviño, A. Díaz, E. Sánchez-Lara, B. L. Sanchez-Gaytan, J. M. Perez-Aguilar and E. González-Vergara, Vanadium in Biological Action: Chemical, Pharmacological Aspects, and Metabolic Implications in Diabetes Mellitus, Biol. Trace Elem. Res., 2019, 188, 68–98 CrossRef PubMed.
- J. Costa Pessoa, S. Etcheverry and D. Gambino, Vanadium compounds in medicine, Coord. Chem. Rev., 2015, 301–302, 24–48 CrossRef PubMed.
- E. Kioseoglou, S. Petanidis, C. Gabriel and A. Salifoglou, The chemistry and biology of vanadium compounds in cancer therapeutics, Coord. Chem. Rev., 2015, 301–302, 87–105 CrossRef CAS.
- C. Amante, A. L. De Sousa-Coelho and M. Aureliano, Vanadium and Melanoma: A Systematic Review, Metals, 2021, 11, 828 CrossRef CAS.
- D. Rehder, The potentiality of vanadium in medicinal applications, Inorg. Chim. Acta, 2020, 504, 119445 CrossRef CAS.
- D. Sanna and E. Garribba, Pharmacologically Active Vanadium Species: Distribution in Biological Media and Interaction with Molecular Targets, Curr. Med. Chem., 2021, 28, 7339–7384 CrossRef CAS PubMed.
- V. Oliveri and G. Vecchio, 8-Hydroxyquinolines in medicinal chemistry: A structural perspective, Eur. J. Med. Chem., 2016, 120, 252–274 CrossRef CAS PubMed.
- K. Walczak, E. Langner, K. Szalast, A. Makuch-Kocka, P. Pożarowski and T. Plech, A Tryptophan Metabolite, 8-Hydroxyquinaldic Acid, Exerts Antiproliferative and Anti-Migratory Effects on Colorectal Cancer Cells, Molecules, 2020, 25, 1655 CrossRef CAS PubMed.
- H.-L. Zhang, A.-H. Zhang, J.-H. Miao, H. Sun, G.-L. Yan, F.-F. Wu and X.-J. Wang, Targeting regulation of tryptophan metabolism for colorectal cancer therapy: a systematic review, RSC Adv., 2019, 9, 3072–3080 RSC.
- A. Casado-Sánchez, C. Martín-Santos, J. M. Padrón, R. Mas-Ballesté, C. Navarro-Ranninger, J. Alemán and S. Cabrera, Effect of electronic and steric properties of 8-substituted quinolines in gold(III) complexes: Synthesis, electrochemistry, stability, interactions and antiproliferative studies, J. Inorg. Biochem., 2017, 174, 111–118 CrossRef PubMed.
- D. Rogolino, A. Cavazzoni, A. Gatti, M. Tegoni, G. Pelosi, V. Verdolino, C. Fumarola, D. Cretella, P. G. Petronini and M. Carcelli, Anti-proliferative effects of copper(II) complexes with hydroxyquinoline-thiosemicarbazone ligands, Eur. J. Med. Chem., 2017, 128, 140–153 CrossRef CAS PubMed.
- C. P. Matos, Y. Yildizhan, Z. Adiguzel, F. R. Pavan, D. L. Campos, J. Costa Pessoa, L. P. Ferreira, A. I. Tomaz, I. Correia and C. Acilan, New ternary iron(III) aminobisphenolate hydroxyquinoline complexes as potential therapeutic agents, Dalton Trans., 2019, 48, 8702–8716 RSC.
- N. Ribeiro, I. Bulut, B. Sergi, V. Pósa, G. Spengler, G. Sciortino, V. André, L. P. Ferreira, T. Biver, V. Ugone, E. Garribba, J. Costa Pessoa, É. A. Enyedy, C. Acilan and I. Correia, Promising anticancer agents based on 8-hydroxyquinoline hydrazone copper(II) complexes, Front. Chem., 2023, 11, 1106349 CrossRef CAS PubMed.
- I. Correia, P. Adão, S. Roy, M. Wahba, C. Matos, M. R. Maurya, F. Marques, F. R. Pavan, C. Q. F. Leite, F. Avecilla and J. Costa Pessoa, Hydroxyquinoline derived vanadium(IV and V) and copper(II) complexes as potential anti-tuberculosis and anti-tumor agents, J. Inorg. Biochem., 2014, 141, 83–93 CrossRef CAS PubMed.
- G. Scalese, I. Machado, C. Fontana, G. Risi, G. Salinas, L. Pérez-Díaz and D. Gambino, New heteroleptic oxidovanadium(V) complexes: synthesis, characterization and biological evaluation as potential agents against Trypanosoma cruzi, J. Biol. Inorg. Chem., 2018, 23, 1265–1281 CrossRef CAS PubMed.
- G. Scalese, I. Machado, I. Correia, J. Costa Pessoa, L. Bilbao, L. Pérez-Diaz and D. Gambino, Exploring oxidovanadium(IV) homoleptic complexes with 8-hydroxyquinoline derivatives as prospective antitrypanosomal agents, New J. Chem., 2019, 43, 17756–17773 RSC.
- K. Choroba, L. R. Raposo, J. Palion-Gazda, E. Malicka, K. Erfurt, B. Machura and A. R. Fernandes, In vitro antiproliferative effect of vanadium complexes bearing 8-hydroxyquinoline-based ligands – the substituent effect, Dalton Trans., 2020, 49, 6596–6606 RSC.
- N. Ribeiro, I. Bulut, V. Pósa, B. Sergi, G. Sciortino, J. Costa Pessoa, L. B. Maia, V. Ugone, E. Garribba, É. A. Enyedy, C. Acilan and I. Correia, Solution chemical properties and anticancer potential of 8-hydroxyquinoline hydrazones and their oxidovanadium(IV) complexes, J. Inorg. Biochem., 2022, 235, 111932 CrossRef CAS PubMed.
- J. Costa Pessoa, M. F. A. Santos, I. Correia, D. Sanna, G. Sciortino and E. Garribba, Binding of vanadium ions and complexes to proteins and enzymes in aqueous solution, Coord. Chem. Rev., 2021, 449, 214192 CrossRef.
- M. Aureliano, N. I. Gumerova, G. Sciortino, E. Garribba, C. C. McLauchlan, A. Rompel and D. C. Crans, Polyoxidovanadates’ interactions with proteins: An overview, Coord. Chem. Rev., 2022, 454, 214344 CrossRef CAS.
- G. Sciortino, D. Sanna, V. Ugone, G. Micera, A. Lledós, J.-D. Maréchal and E. Garribba, Elucidation of Binding Site and Chiral Specificity of Oxidovanadium Drugs with Lysozyme through Theoretical Calculations, Inorg. Chem., 2017, 56, 12938–12951 CrossRef CAS PubMed.
- G. Sciortino, D. Sanna, V. Ugone, J.-D. Maréchal, M. Alemany-Chavarria and E. Garribba, Effect of secondary interactions, steric hindrance and electric charge on the interaction of VIVO species with proteins, New J. Chem., 2019, 43, 17647–17660 RSC.
- G. Sciortino, D. Sanna, V. Ugone, J. D. Marechal and E. Garribba, Integrated ESI-MS/EPR/computational characterization of the binding of metal species to proteins: vanadium drug-myoglobin application, Inorg. Chem. Front., 2019, 6, 1561–1578 RSC.
- V. Ugone, D. Sanna, G. Sciortino, J. D. Marechal and E. Garribba, Interaction of Vanadium(IV) Species with Ubiquitin: A Combined Instrumental and Computational Approach, Inorg. Chem., 2019, 58, 8064–8078 CrossRef CAS PubMed.
- V. Ugone, D. Sanna, S. Ruggiu, G. Sciortino and E. Garribba, Covalent and non-covalent binding in vanadium–protein adducts, Inorg. Chem. Front., 2021, 8, 1189–1196 RSC.
- G. Sciortino and E. Garribba, The binding modes of VIVO2+ ions in blood proteins and enzymes, Chem. Commun., 2020, 56, 12218–12221 RSC.
- G. Sciortino, D. Sanna, G. Lubinu, J. D. Marechal and E. Garribba, Unveiling VIVO2+ Binding Modes to Human Serum Albumins by an Integrated Spectroscopic-Computational Approach, Chem. – Eur. J., 2020, 26, 11316–11326 CrossRef CAS PubMed.
- G. Sciortino, J.-D. Maréchal and E. Garribba, Integrated experimental/computational approaches to characterize the systems formed by vanadium with proteins and enzymes, Inorg. Chem. Front., 2021, 8, 1951–1974 RSC.
- M. F. A. Santos, I. Correia, A. R. Oliveira, E. Garribba, J. Costa Pessoa and T. Santos-Silva, Vanadium Complexes as Prospective Therapeutics: Structural Characterization of a VIV Lysozyme Adduct, Eur. J. Inorg. Chem., 2014, 2014, 3293–3297 CrossRef CAS.
- G. Ferraro, N. Demitri, L. Vitale, G. Sciortino, D. Sanna, V. Ugone, E. Garribba and A. Merlino, Spectroscopic/Computational Characterization and the X-ray Structure of the Adduct of the VIVO–Picolinato Complex with RNase A, Inorg. Chem., 2021, 60, 19098–19109 CrossRef CAS PubMed.
- M. F. A. Santos, G. Sciortino, I. Correia, A. C. P. Fernandes, T. Santos-Silva, F. Pisanu, E. Garribba and J. Costa Pessoa, Binding of VIVO2+, VIVOL, VIVOL2 and VVO2L Moieties to Proteins: X-ray/Theoretical Characterization and Biological Implications, Chem. – Eur. J., 2022, 28, e202200105 CrossRef CAS PubMed.
- G. Ferraro, M. Paolillo, G. Sciortino, E. Garribba and A. Merlino, Multiple and Variable Binding of Pharmacologically Active Bis(maltolato)oxidovanadium(IV) to Lysozyme, Inorg. Chem., 2022, 61, 16458–16467 CrossRef CAS PubMed.
- G. Ferraro, M. Paolillo, G. Sciortino, F. Pisanu, E. Garribba and A. Merlino, Implications of Protein Interaction in the Speciation of Potential VIVO–Pyridinone Drugs, Inorg. Chem., 2023, 62, 8407–8417 CrossRef CAS PubMed.
- H. Berman, K. Henrick and H. Nakamura, Announcing the worldwide Protein Data Bank, Nat. Struct. Mol. Biol., 2003, 10, 980–980 CrossRef CAS PubMed.
- S. Sorrentino, The eight human “canonical” ribonucleases: Molecular diversity, catalytic properties, and special biological actions of the enzyme proteins, FEBS Lett., 2010, 584, 2194–2200 CrossRef CAS PubMed.
- K. D. Dyer and H. F. Rosenberg, The RNase a superfamily: Generation of diversity and innate host defense, Mol. Diversity, 2006, 10, 585–597 CrossRef CAS PubMed.
- A. Merlino, Recent advances in protein metalation: structural studies, Chem. Commun., 2021, 57, 1295–1307 RSC.
- L. Messori and A. Merlino, Protein metalation by metal-based drugs: X-ray crystallography and mass spectrometry studies, Chem. Commun., 2017, 53, 11622–11633 RSC.
- L. Messori and A. Merlino, Cisplatin Binding to Proteins: Molecular Structure of the Ribonuclease A Adduct, Inorg. Chem., 2014, 53, 3929–3931 CrossRef CAS PubMed.
- L. Messori, T. Marzo and A. Merlino, Interactions of carboplatin and oxaliplatin with proteins: Insights from X-ray structures and mass spectrometry studies of their ribonuclease A adducts, J. Inorg. Biochem., 2015, 153, 136–142 CrossRef CAS PubMed.
- J. Hildebrandt, H. Görls, N. Häfner, G. Ferraro, M. Dürst, I. B. Runnebaum, W. Weigand and A. Merlino, Unusual mode of protein binding by a cytotoxic π-arene ruthenium(II) piano-stool compound containing an O,S-chelating ligand, Dalton Trans., 2016, 45, 12283–12287 RSC.
- G. Ferraro, A. Pratesi, L. Messori and A. Merlino, Protein interactions of dirhodium tetraacetate: a structural study, Dalton Trans., 2020, 49, 2412–2416 RSC.
- G. Ferraro, A. M. Mansour and A. Merlino, Exploring the interactions between model proteins and Pd(II) or Pt(II) compounds bearing charged N,N-pyridylbenzimidazole bidentate ligands by X-ray crystallography, Dalton Trans., 2018, 47, 10130–10138 RSC.
- M. Caterino, A. A. Petruk, A. Vergara, G. Ferraro, D. Marasco, F. Doctorovich, D. A. Estrin and A. Merlino, Mapping the protein-binding sites for iridium(III)-based CO-releasing molecules, Dalton Trans., 2016, 45, 12206–12214 RSC.
- L. Messori, F. Scaletti, L. Massai, M. A. Cinellu, I. Russo Krauss, G. di Martino, A. Vergara, L. Paduano and A. Merlino, Interactions of gold-based drugs with proteins: crystal structure of the adduct formed between ribonuclease A and a cytotoxic gold(III) compound, Metallomics, 2014, 6, 233–236 CrossRef CAS PubMed.
- Đ. Miodragović, A. Merlino, E. P. Swindell, A. Bogachkov, R. W. Ahn, S. Abuhadba, G. Ferraro, T. Marzo, A. P. Mazar, L. Messori and T. V. O'Halloran, Arsenoplatin-1 Is a Dual Pharmacophore Anticancer Agent, J. Am. Chem. Soc., 2019, 141, 6453–6457 CrossRef PubMed.
- E. Garribba, G. Micera, D. Sanna and E. Lodyga-Chruscinska, Oxovanadium(IV) complexes of quinoline derivatives, Inorg. Chim. Acta, 2003, 348, 97–106 CrossRef CAS.
- B. D. Liboiron, in High Resolution EPR, ed. G. Hanson and L. Berliner, Springer New York, New York, NY, 2010, vol. 28, pp. 507–549 Search PubMed.
- D. Sanna, L. Biro, P. Buglyo, G. Micera and E. Garribba, Biotransformation of BMOV in the presence of blood serum proteins, Metallomics, 2012, 4, 33–36 CrossRef CAS PubMed.
- C. Vonrhein, C. Flensburg, P. Keller, A. Sharff, O. Smart, W. Paciorek, T. Womack and G. Bricogne, Data processing and analysis with the autoPROC toolbox, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 293–302 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Overview of the CCP4 suite and current developments, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed.
- L. Vitagliano, A. Merlino, A. Zagari and L. Mazzarella, Reversible substrate-induced domain motions in ribonuclease A, Proteins, 2002, 46, 97–104 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, REFMAC5 for the refinement of macromolecular crystal structures, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Features and development of Coot, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- I. Tolbatov and A. Marrone, Computational strategies to model the interaction and the reactivity of biologically-relevant transition metal complexes, Inorg. Chim. Acta, 2022, 530, 120686 CrossRef CAS.
- M. Bühl, C. Reimann, D. A. Pantazis, T. Bredow and F. Neese, Geometries of Third-Row Transition-Metal Complexes from Density-Functional Theory, J. Chem. Theory Comput., 2008, 4, 1449–1459 CrossRef PubMed.
- I. Tolbatov, A. Marrone, C. Coletti and N. Re, Computational Studies of Au(I) and Au(III) Anticancer MetalLodrugs: A Survey, Molecules, 2021, 26, 7600 CrossRef CAS PubMed.
- E. Lodyga-Chruscinska, D. Sanna, E. Garribba and G. Micera, Potentiometric, spectroscopic, electrochemical and DFT characterization of oxovanadium(IV) complexes formed by citrate and tartrates in aqueous solution at high ligand to metal molar ratios: the effects of the trigonal bipyramidal distortion in bis-chelated species and biological implications, Dalton Trans., 2008, 2008, 4903–4916 RSC.
- G. Micera and E. Garribba, Application of DFT Methods in the Study of VIVO2+–Peptide Interactions, Eur. J. Inorg. Chem., 2010, 2010, 4697–4710 CrossRef.
- G. Micera and E. Garribba, The effect of the functional, basis set, and solvent in the simulation of the geometry and spectroscopic properties of VIVO2+ complexes chemical and biological applications, Int. J. Quantum Chem., 2012, 112, 2486–2498 CrossRef CAS.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, Development and validation of a genetic algorithm for flexible docking1, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera-A visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- M. H. M. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- G. Sciortino, E. Garribba and J.-D. Maréchal, Validation and Applications of Protein–Ligand Docking Approaches Improved for Metalloligands with Multiple Vacant Sites, Inorg. Chem., 2019, 58, 294–306 CrossRef CAS PubMed.
- M. Connolly, Analytical molecular surface calculation, J. Appl. Crystallogr., 1983, 16, 548–558 CrossRef CAS.
- S. C. Lovell, J. M. Word, J. S. Richardson and D. C. Richardson, The penultimate rotamer library, Proteins, 2000, 40, 389–408 CrossRef CAS.
- P. E. M. Siegbahn and F. Himo, The quantum chemical cluster approach for modeling enzyme reactions, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 323–336 CAS.
- G. Sciortino, M. Aureliano and E. Garribba, Rationalizing the Decavanadate(V) and Oxidovanadium(IV) Binding to G-Actin and the Competition with Decaniobate(V) and ATP, Inorg. Chem., 2021, 60, 334–344 CrossRef CAS PubMed.
- M. Kunitz, A spectrophotometric method for the measurement of ribonuclease activity, J. Biol. Chem., 1946, 164, 563–568 CrossRef CAS PubMed.
- J. Costa Pessoa and I. Correia, Misinterpretations in Evaluating Interactions of Vanadium Complexes with Proteins and Other Biological Targets, Inorganics, 2021, 9, 17 CrossRef.
- C. P. Rao, A. Sreedhara, P. Venkateswara Rao, M. Bindu Verghese, K. Rissanen, E. Kolehmainen, N. K. Lokanath, M. A. Sridhar and J. Sashidhara Prasad, Syntheses, structure, reactivity and species recognition studies of oxo-vanadium(V) and -molybdenum(VI) complexes, J. Chem. Soc., Dalton Trans., 1998, 1998, 2383–2394 RSC.
- W. R. Scheidt, Crystallographic study of the structural trans effect. Molecular structure of oxoisopropoxobis(8-hydroxyquinolinato)vanadium(V), Inorg. Chem., 1973, 12, 1758–1761 CrossRef CAS.
- D. N. Chasteen, in Biological Magnetic Resonance, ed. L. J. J. Berliner and J. Reuben, Plenum Press, New York, 1981, vol. 3, pp. 53–119 Search PubMed.
- T. S. Smith II, R. LoBrutto and V. L. Pecoraro, Paramagnetic spectroscopy of vanadyl complexes and its applications to biological systems, Coord. Chem. Rev., 2002, 228, 1–18 CrossRef.
- M. P. Sullivan, M. Groessl, S. M. Meier, R. L. Kingston, D. C. Goldstone and C. G. Hartinger, The metalation of hen egg white lysozyme impacts protein stability as shown by ion mobility mass spectrometry, differential scanning calorimetry, and X-ray crystallography, Chem. Commun., 2017, 53, 4246–4249 RSC.
- R. T. Raines and A. Ribonuclease, Chem. Rev., 1998, 98, 1045–1066 CrossRef CAS PubMed.
- M. V. Nogués, M. Vilanova and C. M. Cuchillo, Bovine pancreatic ribonuclease A as a model of an enzyme with multiple substrate binding sites, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1995, 1253, 16–24 CrossRef PubMed.
- L. F. Vilas Boas and J. Costa Pessoa, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon Press, Oxford, 1987, vol. 3, pp. 453–583 Search PubMed.
- V. Ugone, D. Sanna, G. Sciortino, D. C. Crans and E. Garribba, ESI-MS Study of the Interaction of Potential Oxidovanadium(IV) Drugs and Amavadin with Model Proteins, Inorg. Chem., 2020, 59, 9739–9755 CrossRef CAS PubMed.
- G. Sciortino, V. Ugone, D. Sanna, G. Lubinu, S. Ruggiu, J.-D. Maréchal and E. Garribba, Biospeciation of Potential Vanadium Drugs of Acetylacetonate in the Presence of Proteins, Front. Chem., 2020, 8, 345 CrossRef CAS PubMed.
- E. Kiss, K. Petrohan, D. Sanna, E. Garribba, G. Micera and T. Kiss, Solution speciation and spectral studies on oxovanadium(IV) complexes of pyridinecarboxylic acids, Polyhedron, 2000, 19, 55–61 CrossRef CAS.
- E. De Boer, K. Boon and R. Wever, Electron paramagnetic resonance studies on conformational states and metal ion exchange properties of vanadium bromoperoxidase, Biochemistry, 1988, 27, 1629–1635 CrossRef CAS.
- D. Picone, F. Donnarumma, G. Ferraro, G. Gotte, A. Fagagnini, G. Butera, M. Donadelli and A. Merlino, A comparison study on RNase A oligomerization induced by cisplatin, carboplatin and oxaliplatin, J. Inorg. Biochem., 2017, 173, 105–112 CrossRef CAS PubMed.
- E. Griffin, A. Levina and P. A. Lay, Vanadium(V) tris-3,5-di-tert-butylcatecholato complex: Links between speciation and anti-proliferative activity in human pancreatic cancer cells, J. Inorg. Biochem., 2019, 201, 110815 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Data collection and refinement statistics (Table S1), distribution of the species formed in the systems VIVO2+:pic and VIVO2+:8-HQ at pH 5.1 (Table S2), docking solutions for the binding of VIVO2+–8-HQ species with RNase A (Table S3), UV-Vis and far-UV CD spectra of [VIVO(8-HQ)2] at pH 5.1 and 7.4 (Fig. S1 and S2), superimposition of the docking- and DFT-predicted structures of [VIVO(8-HQ)(H2O)]+–RNase A with the crystallographic adduct (Fig. S3 and S4), hydrolysis of yeast RNA at pH 5.0 (Fig. S5), possible complexation of [VIVO(8-HQ)(H2O)2]+ and RNase A as a function of pH (Scheme S1) and isomers of cis-[VIVO(8-HQ)2(H2O)] in aqueous solution (Scheme S2). See DOI: https://doi.org/10.1039/d3qi01023f |
| This journal is © the Partner Organisations 2023 |