
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tricky but repeatable synthetic approach to branched, multifunctional silsesquioxane dendrimer derivatives†
Aleksandra
Mrzygłód
 ab,
Rafał
Januszewski
ab,
Julia
Duszczak
ab,
Michał
Dutkiewicz
c,
Maciej
Kubicki
a and
Beata
Dudziec
*ab
ab,
Rafał
Januszewski
ab,
Julia
Duszczak
ab,
Michał
Dutkiewicz
c,
Maciej
Kubicki
a and
Beata
Dudziec
*ab
aFaculty of Chemistry, Adam Mickiewicz University in Poznan, Uniwersytetu Pozańskiego 8, 61-614 Poznan, Poland. E-mail: beata.dudziec@gmail.com
bCentre for Advanced Technologies, Adam Mickiewicz University in Poznan, Uniwersytetu Poznańskiego 10, 61-614 Poznan, Poland
cAdam Mickiewicz University Foundation, Poznan Science and Technology Park, Rubież 46, 61-612 Poznan, Poland
First published on 10th July 2023
Abstract
Dendrimers are a wide group of chemical compounds that are still being studied extensively. New dendrimer cores are sought to improve their physicochemical properties. We present the syntheses of silsesquioxane dendrimers with different cores, from mono-T8, octa-T8 silsesquioxane to di- and tetrafunctional double-decker silsesquioxanes. These compounds were obtained by a sequence of hydrosilylation and reduction reactions in a one-pot protocol. As part of the research, the location of reactive Si–H and Si–Vi bonds on the selectivity of the hydrosilylation process was verified, as well as two reducing agents LiAlH4 and Red-Al®, on the reduction process. In addition, the reactivity of the obtained new hydrogen derivatives of these SQs was tested in the process of hydrosilylation with selected olefins and in a repetitive one-pot route to increase the generation of these dendritic systems.
Introduction
Dendrimers are specific macromolecular compounds of precisely defined, often spherical three-dimensional structure, built of a multifunctional core with respectively attached branching units (arms). Their important aspect is the architecture which is constituted of three parts: the core – placed in its interior, repeating units forming tree-like branched structure and external surface functional groups. Various types of connections are used in the chemistry of dendrimers, e.g. the polyamidoamines, polyamides, poly(L-lysine), organophosphorus or organosilicon compounds, i.e. silanes, carbosilanes, siloxanes, carbosiloxanes.1–3 Systems bearing phosphorus or silicon branching points have recently emerged within this family of dendrimers.4With regards to carbosilane dendrimers, two synthetic paths to their formation can be distinguished. The first method applies hydrosilylation reactions, e.g. tetravinylsilane hydrosilylated with dichloromethylsilane to obtain Si–Cl bonds. The Grignard reaction follows, which usually forms a compound possessing unsaturated carbon–carbon – silicon bonds, e.g. Si–vinyl, Si–allyl.5–9 The second protocol is based on the reduction of the Si–Cl bond to introduce Si–H.10 The compounds resulting from both these methods contain reactive Si–Vi or Si–H, susceptible to further transformation, increasing dendrimer's generation with more branching units. For carbosilanes, the procedure employing Grignards reagents is more popular.
The properties that determine dendrimers’ subsequent application depend on, i.e. their size and type of surface/end groups. There are reports on the investigation of pure carbosilane dendrimers series (G3–G8 generations) vs. their hybrid carbosilane–siloxane analogues (G4, G6, G7 generations).11,12 Interestingly, there is a correlation between the Tg and the type of functional groups in the surface layer but not in terms of dendrimer generation. On the contrary, the high molecular weight of dendrimers is responsive to specific interactions between molecules with the increasing generation and it is manifested by increases in melt viscosity and an effective liquid–solid transition. As for the surface groups, in addition to standard silane groups, the hydroxyl, imidazole, sulfonate, and carboxylate phosphonium groups enabling anchoring metal atoms are interesting.13–17 Considering organosilicon-based dendrimers, carbosilanes are one of particularly important systems due to their properties that influence further application. They have been used in diverse areas of chemistry or medicine, e.g. in catalysis, anticancer therapy, immunotherapy and drug/gene delivery.18–22 The cores of such systems can be constructed of silanes, cyclosiloxanes, 1,3,5-trisilacyclohexane, as well as more extensive structures, such as silsesquioxanes.3,23,24
The latter, i.e. silsesquioxanes (SQs), are hybrid compounds characterized by the presence of a rigid, inorganic Si–O–Si core and organic substituents attached to silicon atoms of this core. Their general formula, i.e. (RSiO1.5)n, where R stands for organic functional group and n reflects the number of repetitions of this unit from 6 to 18,25 does not give insight into the spatial architecture of these systems. They may constitute random resins or more defined ladder, open- or closed cage, and recently also double-decker silsesquioxanes (DDSQ).26–28 In the literature, there is limited information on the use of T8-type SQ dendrimer cores with a particular emphasis on octa-functionalized SQs due to theirs spherical architecture.24 The advantage of the surface area of octa-T8 SQs with diverse organic functionalities is the possibility of increasing their compatibility with organic materials. The use of this system makes it possible to obtain a large number of functional groups with a small number of synthetic steps.29,30 It should also be mentioned that SQs are present in dendrimers not only cores but also as repeat units (known as hyperbranched polymers based on SQs)31 or SQs as surface functionalities.32 However, the reports on the use of monofunctional T8-type SQ as the core of specific dendrons are limited.30,33,34 The research on the use of DDSQ as cores of dendrimers is even less explored.35 In general, the formation of SQ-based dendrimers is based on a classical approach concerning divergent and convergent methodology.24 The advantage of divergent protocols for SQs containing dendrimers is due to the multiplicity of reactive groups at SQs, their diversity which enables a wide range of chemistry tools and also on 3D SQ structure that diminish potential steric crowding of the end moieties. However, there are also few reports of the use of convergent routes.36 Due to diversity in the functional groups at SQs inorganic core, their modification is tunably varying from amidation, Michael addition, and thiol–ene reaction to metathesis or hydrosilylation, often employing reaction sequences.24 In the case of carbosilane arms at SQ cores, they may be classically modified via hydrosilylation of the followed by Grignard reaction.37 Reports on the use subsequent Si–Cl reduction to Si–H are very scarce.38 Resulting products may possess phosphine, ferrocene surface groups, which made them possible to be applied as potential catalysts, i.e. in hydroformylation, hydrocarbonylation and methoxy-carbonylation reactions, as platinum electrode modifiers, respectively.37,39–44 In general, the application of silsesquioxane dendrimer hybrid systems is comprehensive, ranging from catalysis, liquid crystals to photoactive hybrid systems used as probes, e.g. in MRI, photodynamic therapy or drug delivery.24
Here, we present studies on the elaboration of a synthetic protocol leading to the formation of silsesquioxane dendritic systems with different types of cores ranging from T8 to closed- and open-cage DDSQ systems. These compounds possess a various number of carbosilane functionalities, varying from one, two, four and eight, respectively. The synthetic protocol is based on the hydrosilylation reaction and the subsequent reduction performed in a one-pot repetitive reaction sequence. Within the scope of the studies, two reducing agents, i.e. LiAlH4 and Red-Al®, were verified. They turned out to be efficient with the respective type of silsesquioxane core. As a result, a series of SQs-based dendrimers of G2 generation possessing from 4 to 32 reactive Si–H end groups was effectively obtained. The presence of end Si–H groups opens possibilities for further functionalization/modification via i.e. consecutive hydrosilylation with, e.g. olefins (tests with allyl glycidyl ether, 1-octene), as well as the possibility to further increase of dendrimers generation (Scheme 1). This is the first comprehensive synthetic report describing the evaluation of the nature of SQ core and the type of chlorosilane on the efficiency of reactions and regioselectivity in the synthesis of silsesquioxane-based dendrimers.
 | ||
| Scheme 1 Synthetic route to obtain dendritic systems via hydrosilylation followed by reduction. | ||
Results and discussion
Synthesis of the first generation of carbosilane dendrimers with silsesquioxane cores
Hydrosilylation of C–C multiple bonds is still one of the major catalytic routes for functionalization of organosilicon compounds and is also common in industry. This process is characterized by its atom-efficiency, broad substrate scope, and widespread application.45,46 However, the reaction may not proceed with complete selectivity control and two main products may be obtained, i.e. α or β due to the Markovnikov rule. Moreover, the possibility for other side processes, i.e. dehydrogenative silylation, isomerization or hydrogenation may also be observed.47In general, for olefin hydrosilylation β-regioselectivity is preferred especially in terms of dendrimer formation. It is favoured, e.g. due to steric hindrance of the surface groups and their further availability. The reaction selectivity may be influenced by various factors, e.g. a suitable catalyst, and reaction temperature, but also by the location of Si–H and olefin (Si–Vi) reactive bonds in reactants. Therefore, it was decided to verify the location of the Si–H moiety, whether in the silsesquioxane structure or in the olefin, i.e. unsaturated chloroorganosilicon component, susceptible to further modification.
A model reaction was selected with the hydrogen derivative iBu7T8-OSiH, (synthesized according to the literature data)48 and dichloromethylvinylsilane (Cl2MeSiVi) to be tested in Pt-mediated hydrosilylation (Scheme 2, PATH A). In this way, the reaction could be monitored easily by FT-IR via analysis of changes in the area of the bands at ca. ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 2100 and 900 cm−1 corresponding to the stretching vibrations of Si–H bond, located in the SQs reagent. The reaction was carried out in the presence of Karstedt's catalyst ([Pt2(dvds)3]) for 24 h at 95 °C.49 After reaction completion, which was manifested by complete disappearance of the Si–H band in the FT-IR spectrum, the solvent and unreacted chlorosilane were evaporated. The crude product containing two Si–Cl moieties was subjected to consecutive reduction to form Si–H groups testing lithium aluminium hydride (LiAlH4) as a convenient reducing agent (Scheme 2).
= 2100 and 900 cm−1 corresponding to the stretching vibrations of Si–H bond, located in the SQs reagent. The reaction was carried out in the presence of Karstedt's catalyst ([Pt2(dvds)3]) for 24 h at 95 °C.49 After reaction completion, which was manifested by complete disappearance of the Si–H band in the FT-IR spectrum, the solvent and unreacted chlorosilane were evaporated. The crude product containing two Si–Cl moieties was subjected to consecutive reduction to form Si–H groups testing lithium aluminium hydride (LiAlH4) as a convenient reducing agent (Scheme 2).
 | ||
| Scheme 2 Possible selectivity of hydrosilylation reaction using mono-T8 SQ with different location of the Si–H functionality resulting in the formation of two products G1-1iBuOSi-2H-β and G1-1iBuOSi-2H-α. | ||
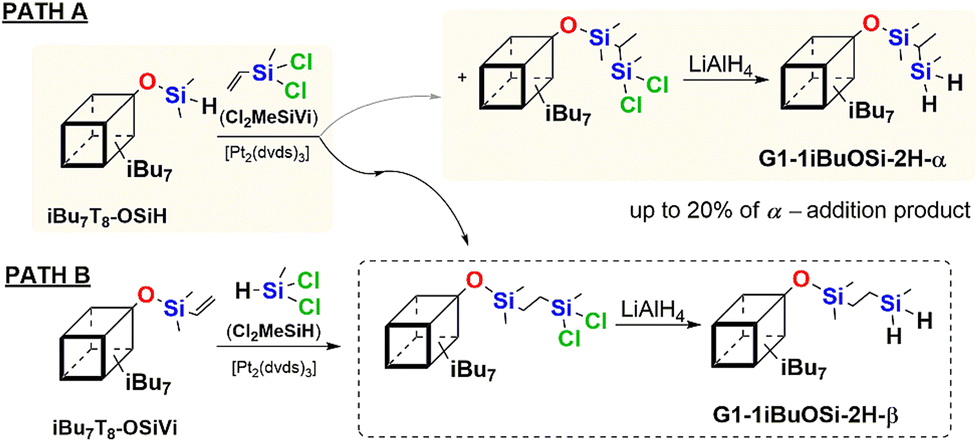
However, after the isolation of the product resulting from PATH A, the 29Si NMR analysis showed the presence of doubled peaks in the area of all types of silicon atoms present in the postulated compound containing two Si–H moieties. These resonance lines, e.g. 11.69, 11.44 (M-unit Si), −29.08, −30.21 (Si–H), −66.89, −67.03, −67.07, −67.10, −67.74, −67.87, −67.90 (T-unit Si), and −109.64 and −109.93 (Q-unit Si) ppm indicated the presence of a mixture of two products, i.e. β and α obtained in a 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 ratio, abbreviated as G1-1iBuOSi-2H-β and G1-1iBuOSi-2H-α, respectively (Fig. 1a). 13C NMR spectra revealed the presence of resonance lines at 2.37 and 11.80 ppm corresponding to –CH2– moieties of the β isomer (G1-1iBuOSi-2H-β) and 4.53 and 9.83 ppm correlating to
:20 ratio, abbreviated as G1-1iBuOSi-2H-β and G1-1iBuOSi-2H-α, respectively (Fig. 1a). 13C NMR spectra revealed the presence of resonance lines at 2.37 and 11.80 ppm corresponding to –CH2– moieties of the β isomer (G1-1iBuOSi-2H-β) and 4.53 and 9.83 ppm correlating to ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH and –CH3 units in α isomer (G1-1iBuOSi-2H-α). Also verified by DEPT-135 (see ESI, Fig. S50†).
CH and –CH3 units in α isomer (G1-1iBuOSi-2H-α). Also verified by DEPT-135 (see ESI, Fig. S50†).
 | ||
| Fig. 1 A selected range of stacked 29Si NMR spectra of (a) a post reaction mixture (PATH A) confirming the presence of G1-1iBu-OSi-2H-β and G1-1iBu-2H-α, (b) regioselective formation (PATH B) of G1-1iBu-2H-β product. | ||
As the hydrosilylation reaction resulted in formation β and α addition products some literature discusses the impact of the location of Si–H moiety (at the SQs core or olefin) on reaction selectivity, we decided to verify this.50,51 For this reason, an analogous hydrosilylation and reduction protocols were designed (PATH B) but varying in the structure of reagents, i.e.iBu7T8-OSiVi and dichloromethylsilane (Cl2MeSiH) were used as substrates. Interestingly, preservation of almost all reaction conditions, with the sole change in the location of the Si–H group, from silsesquioxane to chlorosilane, enabled the regioselective formation of G1-1iBuOSi-2H-β system, confirmed by NMR analyses. It may be seen, especially in the 29Si NMR spectrum the presence of peaks corresponding to respective silicon atoms, e.g. 11.44 (M-unit Si), −29.08 (Si–H), −67.07, −67.10, −67.87, −67.90 (T-unit Si), and −109.64 (Q-unit Si) ppm (Fig. 1b). The selectivity of this reaction may be a derivative of two factors, i.e. steric bulkiness of SQs core but also the electronic effect of substituents (i.e. Cl vs. O) in reactive Si–H and Si–HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 groups. It affects the type of olefin insertion into the Pt–H bond of active catalyst occurring as either 1,2- or 2,1-insertion that results in the formation of β or α isomer respectively. Also, it was reported that the presence of electron-withdrawing atom(s) increases the reactivity of Si–H moiety.51 This is in accordance with the Chalk–Harrod's mechanism.47,63
CH2 groups. It affects the type of olefin insertion into the Pt–H bond of active catalyst occurring as either 1,2- or 2,1-insertion that results in the formation of β or α isomer respectively. Also, it was reported that the presence of electron-withdrawing atom(s) increases the reactivity of Si–H moiety.51 This is in accordance with the Chalk–Harrod's mechanism.47,63
The obtained results facilitated the selection of the optimal location of the Si–H and Si–Vi bonds in applied reactants. This also helped to develop an efficient protocol for the synthesis of dendritic systems using different types of silsesquioxane cores, i.e. di- and tetra-DDSQ as well as octa-T8. As the usage of silsesquioxanes possessing reactive Si–Vi moieties affects the final regioselectivity of the β-addition product, it was decided to apply two compounds direct linked to the vinyl group to T8 core, without the –OSi(Me2)– unit between them. For this purpose, we omitted the β symbol in the abbreviations of products (unless needed) in further parts of the manuscript. Additionally, the iBu7T8-Vi is obtained in fewer steps of the synthetic path.52 For the proper exploration of the sequence of hydrosilylation (1.) and reduction reactions (2.), the iBu7T8-Vi was used as a model reagent to react with dichloromethylsilane (Cl2MeSiH) in order to optimize reaction conditions and enable the formation of G1-1iBu-2H. The first of the reaction sequence was hydrosilylation, which was carried out using Karstedt's catalyst ([Pt2(dvds)3]) at 95 °C for 24 h with stoichiometry [iBu7T8-Vi]:[Cl2MeSiH]:([Pt2(dvds)3]) = 1:2:1 × 10−4. Two-fold excess of Cl2MeSiH enabled >99% conversion of iBu7T8-Vi. After completion of the reaction, the solvent and unreacted chlorosilane were evaporated under reduced pressure and crude dichloro product as an intermediate product (G1-1iBu-2Cl) was dried and the subsequent step of the PATH B was maintained. The reduction was performed in toluene, and 1.2 equiv. of reducing agent per one reactive Si–Cl group was added and the reaction was conducted at room temperature for 20 h. In this case, LiAlH4 was used. When the reaction was finished, the by-products, such as lithium chloride and aluminium chloride, were removed by a syringe filter (0.2 μm). The product (G1-1iBu-2H) was obtained in 89% isolate yield. An analogue synthetic approach was applied for other monosubstituted T8 derivatives with iBu groups (G0-1iBu-1H, G2-2iBu-4H). The comparison of FT-IR spectra of iBu7T8-Vi and G1-1iBu-2H showed apparent proof of the differences in their structure. The disappearance of stretching vibration bands deriving from C–H and CC bonds at ca. = 3050 and = 1600 cm−1, was noted respectively, with the simultaneous presence of Si–H stretching vibration bands in the range of ca. = 2100 and 900 cm−1 (Fig. S53 in ESI†).
The double-decker silsesquioxane is specific construction of cage silsesquioxanes. It is characterized by two decks of cyclosiloxane rings that are stacked one above the another and connected by two oxygen bridges with phenyl groups at Si core atoms. They may occur in the form of di- and tetrafunctional analogues.53,54 As octafunctional T8 SQs are applied as cores for dendrimers and the reports on DDSQ-based systems resembling dumbbell-shaped dendrimers are very rare in the literature.35
For this, we applied the developed sequence of hydrosilylation reaction (1.) with subsequent reduction (2.) using LiAlH4 that was verified in terms of di- and tetrafunctional DDSQ, e.g. to obtain G0-2D-2H. The 1H NMR spectrum of crude product pointed to the disappearance of resonance lines from vinyl groups at ca. 5.90–6.20 ppm. However, as there was no resonance line deriving from Si–H bond which supposed to be present at ca. 3.50–3.70 ppm, there was no evidence on the successful completion of Si–Cl to Si–H reduction. The solution for this was to replace the reducing agent from LiAlH4 to sodium bis(2-methoxyethoxy) aluminum hydride (Red-Al®). Red-Al® possesses two active reactive hydrogen atoms in its structure and is considered a milder reducing agent.55 In the procedure elaborated to yield G0-2D-2H the step Si–Cl to Si–H reduction (2.) was performed using 0.6 fold excess of Red-Al® per one reactive Si–Cl group in the DDSQ.
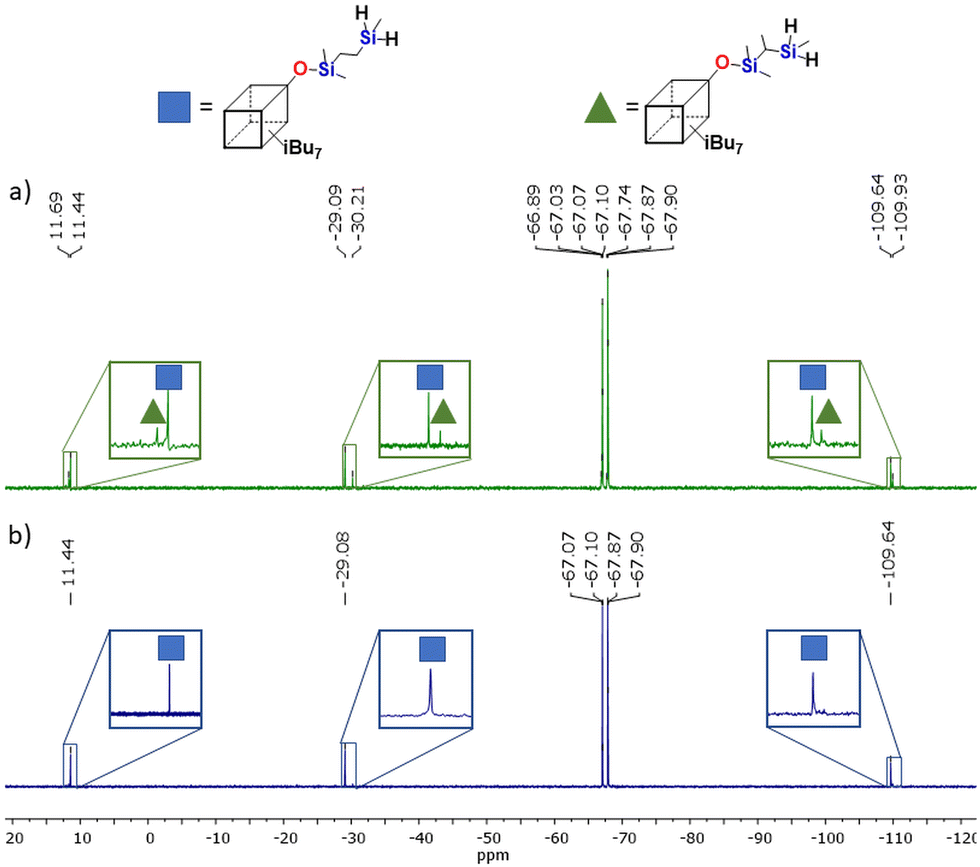
Despite the fact that reduction of Si–Cl to Si–H is a well-known reaction, it has never been verified in situ, so we decided to study this process using real-time FT-IR spectroscopy. The conducted experiment using G1-4D-8Cl (to gain G1-4D-8H), revealed that the consumption of Si–Cl with simultaneous formation of Si–H bonds proceeded extremely fast (Scheme 3). Specifically, it occurred immediately after the addition of Red-Al® which was evident from the appearance of a new band in the range of 880–970 cm−1 (Fig. 2a). We also observed a notable increase in the process temperature, from approximately 23 °C to 26 °C (Fig. 2b). However, the reaction was performed on a small scale, indicating that, for larger-scale synthesis, the addition of the reducing agent to the reaction mixture along with reaction time should be carefully controlled due to the exothermic nature of the process and possible diffusion issues.
 | ||
| Scheme 3 Synthetic path to obtain G1-4D-8Hvia hydrosilylation followed by reduction. | ||
 | ||
| Fig. 2 FT-IR in situ spectra of G1-4D-8Cl reduction using Red-Al® to gain G1-4D-8H: (a) 3D illustrations of the Si–H bond formation recorded in real time, (b) plot of Si–H peak area formation vs. temperature in real time of reaction. | ||
Another problem to be solved was the isolation of the pure G0-2D-2H product with good yield. In general, the purification of SQ-based compounds can be performed using various techniques such as column chromatography, precipitation, extraction or their combination. Herein, at first, the crude G0-2D-2H product was precipitated in acetonitrile (MeCN). This is a good solvent in which Red-Al® is soluble, so could be easily decantated. G0-2D-2H was properly separated from the impurities by washing with DCM to dissolve the G0-2D-2H. After decantation and evaporation of DCM, pure G0-2D-2H product was obtained in 64% yield. This isolation procedure was described as Method I and is depicted in Fig. 3 and in Table 1. Interestingly, we found that using a higher excess of reducing agent (i.e. 1.5 equiv. of Red-Al®) did not improve the product yield, despite being tested for its impact.
 | ||
| Fig. 3 Method I for isolating products from a reaction mixture from a reaction in which the reducing agent was Red-Al® for phenyl derivatives of DDSQ and T8. | ||
| G0-2D-2H | G1-2D-4H | G0-4D-4H | G1-4D-8H | |
|---|---|---|---|---|
| Method I | 64% | 23% | 58% | 19% |
| Method II | 90% | 85% | 94% | 91% |
The procedure developed for the effective formation of G0-2D-2H was transferred to other DDSQ compounds, e.g. to obtain G1-2D-4H (0.6 equiv. Red-Al® on the reactive group). Unfortunately, the isolation yield was unsatisfactory, i.e. only 23%. In this case, a different method of isolation was developed which includes a combination of different techniques. One of the procedures was a dropwise addition of the reaction mixture to cold isopropanol (iPrOH) to decompose Red-Al® followed by solvent evaporation. The crude product was filtered on a chromatography column with DCM as eluent. Another method was based also on iPrOH used for Red-Al® decomposition and washing of resulting precipitate with DCM to extract G1-2D-4H. However, none of the presented protocols provided a positive result in the increase of G1-2D-4H yield. Finally, as the crucial aspect is decomposition of Red-Al® and the fact that DDSQ compounds are not soluble in water, the extraction was performed using toluene and 10% acetic acid water solution. Surprisingly, this procedure enabled the isolation of G1-2D-4H in 85% yield and was described as Method II, shown in Fig. 4. All DDSQ-based dendrimers were isolated using both methods to compare their impact on the isolation yield, which was presented in Table 1. As a result, Method II based on the extraction using toluene and 10% acetic acid water solution significantly affected the isolation yields of verified compounds when compared to Method I.
 | ||
| Fig. 4 Method II for isolating products from a reaction mixture from a reaction in which the reducing agent was Red-Al® for phenyl derivatives of DDSQ and T8. | ||
After optimizing the conditions for various SQ derivatives, we decided to apply the developed methodology for the functionalization of more challenging structures, namely spherosilicates bearing a significantly greater number of reactive Si–H species. The Pt-catalyzed hydrosilylation of trichlorovinylsilane with octakis(dimethyl-siloxy)silsesquioxane (octaT8-OSiH) followed by subsequent reduction of the Si–Cl intermediate in the presence of Red-Al® gave a mixture of isomers (β:α = 64:36). This phenomenon was expected and also observed for other classes of SQs. However, the opposite approach based on the application of octakis(dimethylvinylsiloxy)silsesquioxane (octaT8-OSiVi) and trichlorosilane resulted in the regioselective formation of β isomer. The same behaviour was observed when the dichloromethylsilane and octaT8-OSiVi were used, which was corroborated by 1H, 13C and 29Si NMR analyses (Fig. S35–S37†). The outcomes of these experiments clearly indicated that the application of the chlorohydrosilanes and vinyl-SQ derivatives always leads to the anti-Markovnikov addition products (β isomer), regardless of the type of SQ structure and the number of reactive groups bonded to the SQ cage. Surprisingly, this observation has not been reported so far. The isolation procedure for octa-T8 derivatives relied on the decomposition of Red-Al® in 2-propanol followed by evaporation of the volatiles and subsequent product extraction by n-hexane. This simple methodology allowed us to isolate products with satisfactory yields i.e. >80% The synthesized novel derivative (G1-8T-16H), due to the easily accessible hydrosilyl units, opens possibilities for the synthesis of functionalized dendritic SQ-derivatives bearing sixteen organic groups around the cage, as well as the formation of a higher generation through the above-described one-pot protocol.
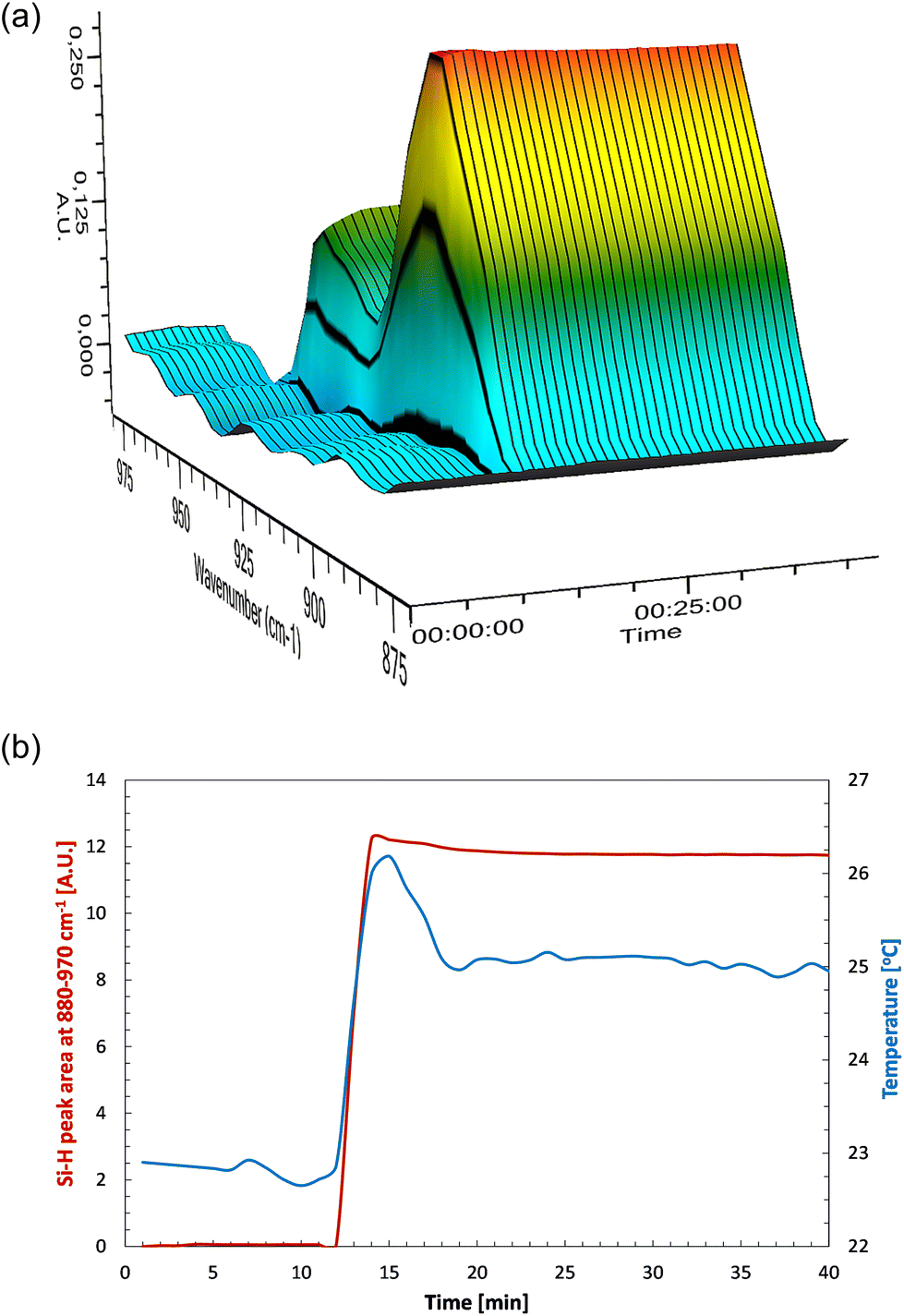
While searching for the most effective purification methodology enabling efficient product isolation, two methods were developed (Fig. 3 and 4) depending on the type of the SQ core. Method 1 involves the use of MeCN, a good precipitating solvent for silsesquioxanes. During the isolation of respective SQ derivatives, some crystal material of the following systems were obtained: G0-4D-4H and G1-2D-4H, which were amenable to XRD analysis. These two systems were found to be more susceptible to crystallization in MeCN when compared to other compounds. Perspective views of the molecules G0-4D-4H and G1-2D-4H are shown in Fig. 5a and b. The molecule G1-2D-4H is Ci-symmetrical, lying across the inversion center in the space group Pbca. As observed in similar structures, the geometry around Si atom is quite stable, as shown by the mean values and their esd's (Table 2). On the other hand, the range of Si–O–Si angles is quite extensive. In principle, the overall geometries and shapes of both open and closed structures are similar to those shown by us earlier.56,57
 | ||
| Fig. 5 Perspective view of: (a) the complex G0-4D-4H; ellipsoids are drawn at the 50% probability level; only the molecule of larger occupancy is shown. Hydrogen atoms are shown as spheres of arbitrary radii; (b) one of the symmetry-independent molecules of complex G1-2D-4H; ellipsoids are drawn at the 50% probability level; hydrogen atoms are shown as spheres of arbitrary radii. The unlabeled part is related to the labelled one by symmetry operation −x, −y, 2 − z. | ||
| G0-4D-4H | G1-2D-4H | |
|---|---|---|
| 〈Si–O〉 | 1.620(12) | 1.615(9) |
| 〈Si–C〉 | 1.858(23) | 1.845(8) |
| 〈Si–O–Si〉 | 147(7) | 155(7) |
| Max (Si–O–Si) | 162.2(2) | 162.8(2) |
| Min (Si–O–Si) | 138.5(2) | 141.7(2) |
| 〈O–Si–O〉 | 109.3(12) | 109.2(14) |
| 〈O–Si–C〉 | 109.2(18) | 109.4(14) |
| 〈C–Si–C〉 | 111(3) | 114.9(4) |
In the elaboration of the synthetic protocol for the DDSQ-based dendrimers based on Red-Al® instead of LiAlH4, some tests were performed to obtain mono-T8 systems with Ph inert groups, which exhibited some failures in the formation. It was decided to verify the methods using LiAlH4 and Red-Al® for these derivatives. In the beginning, a similar procedure to gain G0-1Ph-1H was conducted using LiAlH4 by analogy to the G0-1iBu-1H system. Unfortunately, similar lack of Si–H bond was observed on 1H NMR spectra. Analogous reduction protocol based on Red-Al® was applied and the desired products (G0-1Ph-1H and G1-1Ph-2H) were obtained with good yields. Both methods were validated to obtain G1-1Ph-2H. Interestingly, for this system, the use of LiAlH4 and Red-Al® made it possible to obtain a product with a yield of 45% and 60%, respectively, showing improvement for Red-Al®. The G1-1Ph-2H is again a good example of the fact that silsesquioxanes, due to their differences in architectures and presence of diverse inert groups should be approached individually. This concerns the synthetic protocol conditions for their formation as well as their isolation and purification.35,58
The reactivity of Si–H modified SQ-based dendrimers
Silsesquioxane dendrimers of G0 and G1 generation possessing Si–H bond can be modified to increase their generation by a sequence of hydrosilylation and reduction as well as hydrosilylation reaction with functional alkenes.49,59 To verify the reactivity of the Si–H bond of selected compounds, the G1-4D-8H was reacted with allyl glycidyl ether via hydrosilylation reaction with elaborated reaction conditions, i.e. the following stoichiometry: [G1-4D-8H]:[allyl glycidyl ether]:([Pt2(dvds)3]) = 1:9:8 × 10−4, in 24 h and at 95 °C. The FT-IR performed for the reaction mixture enabled complete confirmation of G1-4D-8H, the disappearance of bands derived from Si–H bond stretching vibrations in the range of = 2124 and 900 cm−1.49,57 This was also validated by the results in 1H NMR spectra and the lack of resonance lines attributed to the proton of Si–H moiety. Interestingly, the 1H NMR analysis revealed the increase in integration of protons derived from the –CH2–CH2– bridge present in the structure from 16 to 32. This may indicate that the Si–CH2–CH2– resonance generated in this process has a similar chemical environment to the pre-existing –CH2–CH2– bridge in the structure, leading to overlap in the resonances and requiring integration for accurate identification. This process was selective towards the exclusive formation of β-addition product. Additionally, 29Si NMR analysis displayed the shift of signal at −29.37 ppm originating from the silicon in –Si(CH3)H2 moiety to 5.48 ppm which is a confirmation of –Si(CH3)(CH2–)2 group formation. The verification of catalytic reactivity of Si–H groups was performed for the DDSQ—based dendrimers with allyl glycidyl ether. The analogous reactions were applied to T8-based dendrimer with symmetrically octasubstituted T8 core: G1-8T-16H, allyl glycidyl ether and 1-octene resulting in the formation of G1-16T-16epoxy and G1-16T-16octyl, respectively. However, in this case, the process turned out to be less selective as up to 13% α-addition products were detected. Nevertheless, the exemplary experiments confirmed that the incorporated Si–H units were susceptible to further functionalization via hydrosilylation reaction. Therefore, the scope of the functional alkenes can be easily extended thanks to a wide gamut of commercially available chemicals and known, highly tolerant catalytic systems. The epoxy-modified SQ-based dendrimers, due to the presence of oxirane rings are susceptible to further modifications, e.g. ring opening.60 It is a procedure leading to the formation of modifiers applied in epoxy resin or nanocomposites.60–62
The second generation of carbosilane dendrimers with silsesquioxane cores
The essence of dendritic systems is the possibility to create compounds with the highest generation. Consequently, the next step of our research were attempts at obtaining the G2 generation of the studied systems. Isobutyl derivative (G1-1iBu-2H) was used as a model reagent. The same hydrosilylation/reduction sequence protocol was applied with LiAlH4 as a reducing reagent to the synthesized G1 dendritic system. Hydrosilylation conditions: [G1-1iBu-2H]:[Cl2MeSiVi]:[Pt2(dvds)3] = 1:4:4 × 10−4; reduction conditions: LiAlH4 1.2 equiv. per one –Si–Cl group. The 1H NMR analysis of the reaction mixture revealed the presence of two multiplets deriving from Si–H bond in a range of 3.70–3.80 ppm (G2-1iBu-4H). These resonance lines were slightly low-field-shifted when compared to the placement of protons of Si–H in G1-1iBu-2H (3.69–3.73 ppm). Moreover, the 29Si NMR analysis of G2-1iBu-4H displayed the presence of three peaks at ca. −2.60, −3.30 and −4.30 ppm, deriving from the created –Si(CH3)–C– moiety. Additionally, there were four peaks derived from Si–H groups, i.e. at −29.19, −29.27, −29.63 and −29.66 ppm. These results may suggest that a mixture of β- and α-addition products was obtained. These are isomers of statistical substitution and three possible products may be obtained, because of different addition of each Si–H double bond of olefin. The 13C NMR supported by the DEPT-135 NMR technique enabled analysis of the reaction mixture and selection and assignment of peaks to respective β- and α-addition products, as there are three possible placement of substituents (Scheme 4, Fig. S11 and S13 in ESI†).
 | ||
| Scheme 4 Three different possibilities of substitution in the G2-1iBu-4H system. | ||
Although the product of double β-addition is the preferred substitution position in the synthesis of dendrimers during the hydrosilylation reaction, all of the products in this mixture have the G2 generation. For the β, β-product, a longer flexible, ethylene bridge reduces steric hindrance in subsequent generations. On the other hand, for the α, β- or α, α – systems, the reactive groups are available for further modification, though the steric hindrance may be an obstacle to further modification which may not proceed easily. Moreover, the same result was obtained when G1-8T-16H was used as a starting material in the synthesis of the SQ-based second dendrimer generation (G2-16T-32H) which was depicted in Scheme 5. This in turn confirms the previously emphasized conclusion describing the crucial role of the Si–H and Si–Vi location in reagents on the reaction regioselectivity. Nevertheless, the presence of α-regioisomers does not exclude their susceptibility to further functionalization through catalytic reactions.47,63,64
 | ||
| Scheme 5 Synthetic path to obtain G2-16T-32H. | ||
The GPC analysis may enable the determination of the dispersity of dendritic systems. The obtained chromatograms confirmed the monodispersity of all the obtained systems. It should be also emphasized that no increase in the value of the polydispersity index (PDI) was observed in any of the example. Its value is in the range of 1.08–1.24, which proves the high selectivity of the process without the formation of any duplex systems (see ESI†). Such relationships were observed for silsesquioxane-based dendritic systems.65–68
Conclusions
To conclude, an effective one-pot synthesis of dendrimers with a different type of silsesquioxane core, from mono-T8, octa-T8 SQ to di- and tetrafunctional DDSQ, was presented. The synthetic protocol was based on the reaction sequence, i.e. hydrosilylation followed by the reduction. To obtain the β-product, the placement of Si–H and Si–Vi reactive groups was verified in regards to respective products. Within the scope of our studies, two reducing agents were tested: LiAlH4 and Red-Al®. The systems with a mono-functional SQ core, LiAlH4 revealed to be good reducing agents, however, for dendrimers with a DDSQ and octa-T8 core, the Red-Al® was preferable. These studies are another example35 that macromolecular systems, despite similar structures, should be approached individually, which was also observed when purifying the obtained compounds. In addition, the structures of the compounds G0-4D-4H and G1-2D-4H were confirmed by XRD analysis. Hydrogen derivatives of the G0 and G1 generations are characterized by the presence of a reactive Si–H bond, which was confirmed in the hydrosilylation reaction with 1-octene and allyl glycidyl ether. Additionally, reactive G1 generation systems were developed to obtain higher generation dendrimers. Despite the non-selectivity of the hydrosilylation, reactive G2 generation dendritic systems with mixed α and β substitution were obtained. All synthesized compounds can be modified in catalytic processes, such as hydrosilylation.Author contributions
B.D. and R.J. conceived the experiment and designed the study; A.M., R.J., J.D. participated in the planning of the study; M.D performed GPC analysis; M.K. carried out the X-ray analysis and description; all authors discussed the results and contributed to the interpretation of data. B.D, A.M, and R.J. co-wrote the manuscript. All authors read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Science Centre (Poland) Project: OPUS UMO 2016/23/B/ST5/00201, Sonatina UMO-2019/32/C/ST4/00178 and NCBR POWR.03.05.00-00-Z303/17. The authors are grateful to Mr Jan Jarożek for the project of the graphical abstract.References
- M. A. Mintzer and M. W. Grinstaff, Biomedical applications of dendrimers: A tutorial, Chem. Soc. Rev., 2011, 40, 173–190 RSC.
- E. Abbasi, S. F. Aval, A. Akbarzadeh, M. Milani, H. T. Nasrabadi, S. W. Joo, Y. Hanifehpour, K. Nejati-Koshki and R. Pashaei-Asl, Dendrimers: Synthesis, applications, and properties, Nanoscale Res. Lett., 2014, 9, 1–10 CrossRef CAS PubMed.
- C. Kim and J. H. Hong, Carbosilane and carbosiloxane dendrimers, Molecules, 2009, 14, 3719–3730 CrossRef CAS PubMed.
- A. M. Caminade, Inorganic dendrimers: Recent advances for catalysis, nanomaterials, and nanomedicine, Chem. Soc. Rev., 2016, 45, 5174–5186 RSC.
- A. W. Van Der Made and P. W. N. M. Van Leeuwen, Silane dendrimers, J. Chem. Soc., Chem. Commun., 1992, 1400–1401 RSC.
- L. Zhou and J. Roovers, Novel Carbosilane Dendritic Macromolecules, Macromolecules, 1993, 26, 963–968 CrossRef CAS.
- E. A. Rebrov, G. M. Ignat'eva, A. I. Lysachkov, N. V. Demchenko and A. M. Muzafarov, Divergent synthesis of segmented carbosilane dendrimers, Polym. Sci., Ser. A, 2007, 49, 483–495 CrossRef.
- J. Losada, P. García-Armada, V. Robles, Á. M. Martínez, C. M. Casado and B. Alonso, Carbosilane based dendritic cores functionalized with interacting ferrocenyl units: Synthesis and electrocatalytical properties, New J. Chem., 2011, 35, 2187–2195 RSC.
- A. I. Ryzhkov, F. V. Drozdov, G. V. Cherkaev and A. M. Muzafarov, Synthesis of Carbosilane and Carbosilane-Siloxane Dendrons Based on Limonene, Polymers, 2022, 14, 3279 CrossRef CAS PubMed.
- D. Seyferth, D. Y. Son, A. L. Rheingold and R. L. Ostrander, Synthesis of an Organosilicon Dendrimer Containing 324 Si-H Bonds, Organometallics, 1994, 13, 2682–2690 CrossRef CAS.
- V. G. Vasil'ev, E. Y. Kramarenko, E. A. Tatarinova, S. A. Milenin, A. A. Kalinina, V. S. Papkov and A. M. Muzafarov, An unprecedented jump in the viscosity of high-generation carbosilane dendrimer melts, Polymer, 2018, 146, 1–5 CrossRef.
- S. A. Milenin, E. V. Selezneva, P. A. Tikhonov, V. G. Vasil, A. I. Buzin, N. K. Balabaev, A. O. Kurbatov, M. V. Petoukhov, E. V. Shtykova, L. A. Feigin, E. A. Tatarinova, E. Y. Kramarenko, S. N. Chvalun and A. M. Muzafarov, Hybrid Polycarbosilane-Siloxane Dendrimers : Synthesis and Properties, Polymers, 2021, 13, 606 CrossRef CAS PubMed.
- C. Kim, S. Son and B. Kim, Dendritic carbosilanes containing hydroxy groups on the periphery, J. Organomet. Chem., 1999, 588, 1–8 CrossRef CAS.
- J. W. Kriesel, S. König, M. A. Freitas, A. G. Marshall, J. A. Leary and T. D. Tilley, Synthesis of highly charged organometallic dendrimers and their characterization by electrospray mass spectrometry and single-crystal X-ray diffraction, J. Am. Chem. Soc., 1998, 120, 12207–12215 CrossRef CAS.
- M. Müllerová, S. Šabata, J. Matoušek, M. Kormunda, J. Holubová, R. Bálková, R. Petričkovič, M. Koštejn, J. Kupčík, R. Fajgar and T. Strašák, Organoclays with carbosilane dendrimers containing ammonium or phosphonium groups, New J. Chem., 2017, 42, 1187–1196 RSC.
- T. Strašák, J. Malý, D. Wróbel, M. Malý, R. Herma, J. Čermák, M. Müllerová, L. Č. Štástná and P. Cuřínová, Phosphonium carbosilane dendrimers for biomedical applications-synthesis, characterization and cytotoxicity evaluation, RSC Adv., 2017, 7, 18724–18744 RSC.
- I. Relaño-Rodríguez, M. S. Espinar-Buitrago, V. Martín-Cañadilla, R. Gómez-Ramirez, J. L. Jiménez and M. A. Muñoz-Fernández, Nanotechnology against human cytomegalovirus in vitro: polyanionic carbosilane dendrimers as antiviral agents, J. Nanobiotechnol., 2021, 19, 1–9 CrossRef PubMed.
- R. Andrés, E. De Jesús, J. L. G. Fierro and P. Terreros, Bifunctional carbosilane dendrons for the immobilization of zirconocene catalysts on silica, New J. Chem., 2011, 35, 2203–2211 RSC.
- D. Sepúlveda-Crespo, R. Gómez, F. J. De La Mata, J. L. Jiménez and M. Á. Muñoz-Fernández, Polyanionic carbosilane dendrimer-conjugated antiviral drugs as efficient microbicides: Recent trends and developments in HIV treatment/therapy, Nanomedicine, 2015, 11, 1481–1498 CrossRef PubMed.
- C. Llamazares, N. Sanz Del Olmo, J. Soliveri, F. J. de la Mata, J. L. Copa-Patiño and S. García-Gallego, Insight on the structure-to-activity of carbosilane metallodendrimers in the fight against staphylococcus aureus biofilms, Antibiotics, 2021, 10, 589 CrossRef CAS PubMed.
- N. Sanz del Olmo, M. Maroto-Diaz, S. Quintana, R. Gómez, M. Holota, M. Ionov, M. Bryszewska, M. J. Carmena, P. Ortega and F. Javier de la Mata, Heterofunctional ruthenium(II) carbosilane dendrons, a new class of dendritic molecules to fight against prostate cancer, Eur. J. Med. Chem., 2020, 207, 112695 CrossRef CAS PubMed.
- N. Rabiee, S. Ahmadvand, S. Ahmadi, Y. Fatahi, R. Dinarvand, M. Bagherzadeh, M. Rabiee, M. Tahriri, L. Tayebi and M. R. Hamblin, Carbosilane dendrimers: Drug and gene delivery applications, J. Drug Delivery Sci. Technol., 2020, 59, 101879 CrossRef CAS.
- E. Weisheim, B. Neumann, H. G. Stammler and N. W. Mitzel, Dendrimers with 1, 3, 5-Trisilacyclohexane as Core Unit, J. Inorg. Gen. Chem., 2016, 642, 329–334 CAS.
- Z. Li, J. Hu, L. Yang, X. Zhang, X. Liu, Z. Wang and Y. Li, Integrated POSS-dendrimer nanohybrid materials: Current status and future perspective, Nanoscale, 2020, 12, 11395–11415 RSC.
- M. Laird, N. Herrmann, N. Ramsahye, C. Totée, C. Carcel, M. Unno, J. R. Bartlett and M. Wong Chi Man, Large Polyhedral Oligomeric Silsesquioxane Cages: The Isolation of Functionalized POSS with an Unprecedented Si18O27 Core, Angew. Chem., Int. Ed., 2021, 60, 3022–3027 CrossRef CAS PubMed.
- B. Dudziec and B. Marciniec, Double-decker Silsesquioxanes: Current Chemistry and Applications, Curr. Org. Chem., 2017, 21, 2794–2813 CAS.
- B. Dudziec, P. Żak and B. Marciniec, Synthetic routes to silsesquioxane-based systems as photoactive materials and their precursors, Polymers, 2019, 11, 504–543 CrossRef PubMed.
- M. Wang, H. Chi, K. S. Joshy and F. Wang, Progress in the synthesis of bifunctionalized polyhedral oligomeric silsesquioxane, Polymers, 2019, 11, 2098–2118 CrossRef CAS PubMed.
- K. Matsumoto, K. Nishi, K. Ando and M. Jikei, Synthesis and properties of aromatic polyamide dendrimers with polyhedral oligomeric silsesquioxane cores, Polym. Chem., 2015, 6, 4758–4765 RSC.
- K. Naka and Y. Irie, Synthesis of single component element-block materials based on siloxane-based cage frameworks, Polym. Int., 2017, 66, 187–194 CrossRef CAS.
- X. Ma, F. Tao, Y. Zhang, T. Li, F. M. Raymo and Y. Cui, Detection of nitroaromatic explosives by a 3D hyperbranched σ-π Conjugated polymer based on a POSS scaffold, J. Mater. Chem. A, 2017, 5, 14343–14354 RSC.
- J. Jiang, Y. Wang, L. Jin, C. H. Hsu, S. Zhang, J. Mao, W. Yin, T. Li, B. Ni, Z. Su, J. Huang, C. Wesdemiotis, K. Yue, W. Zhang and S. Z. D. Cheng, Modularly Constructed Polyhedral Oligomeric Silsesquioxane-Based Giant Molecules for Unconventional Nanostructure Fabrication, ACS Appl. Nano Mater., 2020, 3, 2952–2958 CrossRef CAS.
- L. Song, S. Peng and Y. Shu, Preparation of a novel functionated POSS nano-particle bearing the perfluoro aryl ether dendron, Adv. Mater. Res., 2011, 148–149, 1212–1216 CAS.
- X. Feng, R. Zhang, Y. Li, Y. L. Hong, D. Guo, K. Lang, K. Y. Wu, M. Huang, J. Mao, C. Wesdemiotis, Y. Nishiyama, W. Zhang, T. Miyoshi, T. Li and S. Z. D. Cheng, Hierarchical Self-Organization of ABn Dendron-like Molecules into a Supramolecular Lattice Sequence, ACS Cent. Sci., 2017, 3, 860–867 CrossRef CAS PubMed.
- A. Mrzygłód, M. Kubicki and B. Dudziec, Vinyl- and chloromethyl-substituted mono-T8 and double-decker silsesquioxanes as specific cores to low generation dendritic systems, Dalton Trans., 2022, 51, 1144–1149 RSC.
- Y. Pu, L. Zhang, H. Zheng, B. He and Z. Gu, Synthesis and drug release of star-shaped poly(benzyl L-aspartate)-block- poly(ethylene glycol) copolymers with POSS cores, Macromol. Biosci., 2014, 14, 289–297 CrossRef CAS PubMed.
- L. Ropartz, R. E. Morris, G. P. Schwarz, D. F. Foster and D. J. Cole-Hamilton, Dendrimer-bound tertiary phosphines for alkene hydroformylation, Inorg. Chem. Commun., 2000, 3, 714–717 CrossRef CAS.
- I. P. Coupar, P.-A. Jaffres and R. E. Morris, Synthesis and characterisation of silanol-functionalised dendrimers, J. Chem. Soc., Dalton Trans., 1999, 309, 2183–2187 RSC.
- L. Ropartz, R. E. Morris, D. F. Foster and D. J. Cole-Hamilton, Increased selectivity in hydroformylation reactions using dendrimer based catalysts; a positive dendrimer effect, Chem. Commun., 2001, 361–362 RSC.
- L. Ropartz, D. F. Foster, R. E. Morris, A. M. Z. Slawin and D. J. Cole-Hamilton, Hydrocarbonylation reactions using alkylphosphine-containing dendrimers based on a polyhedral oligosilsesquioxane core, J. Chem. Soc., Dalton Trans., 2002, 1997–2008 RSC.
- N. R. Vautravers and D. J. Cole-Hamilton, Diphenylphosphine containing macromolecules in the methoxycarbonylation of ethene: The effect of macromolecular architecture on the selectivity of the reaction, J. Chem. Soc., Dalton Trans., 2009, 2130–2134 RSC.
- J. Losada, M. P. G. Armada, I. Cuadrado, B. Alonso, B. González, C. M. Casado and J. Zhang, Ferrocenyl and permethylferrocenyl cyclic and polyhedral siloxane polymers as mediators in amperometric biosensors, J. Organomet. Chem., 2004, 689, 2799–2807 CrossRef CAS.
- M. Herrero, B. Alonso, J. Losada, P. García-Armada and C. M. Casado, Ferrocenyl dendrimers based on octasilsesquioxane cores, Organometallics, 2012, 31, 6344–6350 CrossRef CAS.
- L. Fernández, M. Herrero, B. Alonso, C. M. Casado and M. P. G. Armada, Three-dimensional electrocatalytic surface based on an octasilsesquioxane dendrimer for sensing applications, J. Electroanal. Chem., 2019, 839, 16–24 CrossRef.
- D. Troegel and J. Stohrer, Recent advances and actual challenges in late transition metal catalyzed hydrosilylation of olefins from an industrial point of view, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS.
- T. K. Meister, K. Riener, P. Gigler, J. Stohrer, W. A. Herrmann and F. E. Kühn, Catalysis Revisited-Unraveling Principles of Catalytic Olefin Hydrosilylation, ACS Catal., 2016, 6, 1274–1284 CrossRef CAS.
- B. Marciniec, C. Pietraszuk, P. Pawluć and H. Maciejewski, Inorganometallics (Transition Metal–Metalloid Complexes) and Catalysis, Chem. Rev., 2022, 122(3), 3996–4090 CrossRef CAS PubMed.
- D. Brząkalski, M. Walczak, J. Duszczak, B. Dudziec and B. Marciniec, Chlorine-Free Catalytic Formation of Silsesquioxanes with Si-OH and Si-OR Functional Groups, Eur. J. Inorg. Chem., 2018, 4905–4910 CrossRef.
- J. Duszczak, K. Mituła, R. Januszewski, P. Żak, B. Dudziec and B. Marciniec, Highly Efficient Route for the Synthesis of a Novel Generation of Tetraorganofunctional Double-decker Type of Silsesquioxanes, ChemCatChem, 2019, 11, 1086–1091 CAS.
- M. Morán, C. M. Casado, I. Cuadrado and J. Losada, Ferrocenyl Substituted Octakis(dimethylsiloxy)octasilsesquioxanes: A New Class of Supramolecular Organometallic Compounds. Synthesis, Characterization, and Electrochemistry, Organometallics, 1993, 12, 4327–4333 CrossRef.
- J. Duszczak, A. Mrzygłód, K. Mituła, M. Dutkiewicz, R. Januszewski, M. Rzonsowska, B. Dudziec, M. Nowicki and M. Kubicki, Distinct insight into the use of difunctional double-decker silsesquioxanes as building blocks for alternating A-B type macromolecular frameworks, Inorg. Chem. Front., 2023, 10, 888–899 RSC.
- K. Mituła, M. Dutkiewicz, B. Dudziec, B. Marciniec and K. Czaja, A library of monoalkenylsilsesquioxanes as potential comonomers for synthesis of hybrid materials, J. Therm. Anal. Calorim., 2018, 132, 1545–1555 CrossRef.
- Y. Morimoto, K. Watanabe, N. Ootake, J. Inagaki, K. Yoshida and K. Ohguma, Silsesquioxane Derivative And Production Process For The Same, US 7449539B2, 2008 Search PubMed.
- K. Yoshida, Y. Morimoto, K. Watanabe and N. Ootake, Silsesquioxane Derivative And Process For Producing The Same, US 7319129B2, 2008 Search PubMed.
- R. Januszewski, M. Dutkiewicz and I. Kownacki, An efficient methodology for the synthesis of unique functional polyolefins, Mater. Des., 2021, 206, 109801 CrossRef CAS.
- K. Mituła, J. Duszczak, D. Brząkalski, B. Dudziec, M. Kubicki and B. Marciniec, Tetra-functional double-decker silsesquioxanes as anchors for reactive functional groups and potential synthons for hybrid materials, Chem. Commun., 2017, 53, 10370–10373 RSC.
- M. Walczak, R. Januszewski, M. Majchrzak, M. Kubicki, B. Dudziec and B. Marciniec, Unusual: Cis and trans architecture of dihydrofunctional double-decker shaped silsesquioxane and synthesis of its ethyl bridged π-conjugated arene derivatives, New J. Chem., 2017, 41, 3290–3296 RSC.
- M. Rzonsowska, K. Mituła, J. Duszczak, M. Kasperkowiak, R. Januszewski, A. Grześkiewicz, M. Kubicki, D. Głowacka and B. Dudziec, Unexpected and frustrating rearrangements of double-decker silsesquioxanes, Inorg. Chem. Front., 2022, 9, 379–390 RSC.
- A. Sellinger and R. M. Laine, Silsesquioxanes as Synthetic Platforms. 3. Photocurable, Liquid Epoxides as Inorganic/Organic Hybrid Precursors, Chem. Mater., 1996, 8, 1592–1593 CrossRef CAS.
- M. Dutkiewicz, H. Maciejewski and B. Marciniec, Synthesis of azido-, hydroxy- and nitro-, hydroxy-functionalized spherosilicates via oxirane ring-opening reactions, Synthesis, 2012, 44, 881–884 CrossRef CAS.
- M. Szołyga, M. Dutkiewicz, M. Nowicki, K. Sałasińska, M. Celiński and B. Marciniec, Phosphorus-containing silsesquioxane derivatives as additive or reactive components of epoxy resins, Materials, 2020, 13, 1–23 CrossRef PubMed.
- D. Czarnecka-Komorowska, T. Sterzynski and M. Dutkiewicz, Polyoxymethylene/Polyhedral Oligomeric Silsesquioxane Composites: Processing, Crystallization, Morphology and Thermo- Mechanical Behavior, Int. Polym. Process., 2016, 31, 598–606 CrossRef CAS.
- M. Zaranek and P. Pawluć, Markovnikov Hydrosilylation of Alkenes : How an Oddity Becomes the Goal, ACS Catal., 2018, 8, 8965–9876 Search PubMed.
- B. Cheng, P. Lu, H. Zhang, X. Cheng and Z. Lu, Highly Enantioselective Cobalt-Catalyzed Hydrosilylation of Alkenes, J. Am. Chem. Soc., 2017, 139, 9439–9442 CrossRef CAS PubMed.
- K. Wada, N. Watanabe, K. Yamada, T. Kondo and T. A. Mitsudo, Synthesis of novel starburst and dendritic polyhedral oligosilsesquioxanes, Chem. Commun., 2005, 95–97 RSC.
- S. Yuasa, H. Imoto and K. Naka, Synthesis and properties of hyperbranched polymers by polymerization of an AB3-type incompletely condensed cage silsesquioxane (IC-POSS) monomer, Polym. J., 2018, 50, 879–887 CrossRef CAS.
- I. M. Saez and J. W. Goodby, Supermolecular liquid crystal dendrimers based on the octasilsesquioxane core, Liq. Cryst., 1999, 26, 1101–1105 CrossRef CAS.
- Y. Irie and K. Naka, Single component transparent free-standing films based on polyhedral octasilicate-core dendrimers bearing carbazole terminal groups and their emission properties, J. Polym. Sci., Part A: Polym. Chem., 2015, 54, 628–633 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data of isolated compounds. CCDC 2184824 and 2184825. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi00500c |
| This journal is © the Partner Organisations 2023 |