
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cobalt pincer-type complexes demonstrating unique selectivity for the hydroboration reaction of olefins under mild conditions†
Dariusz
Lewandowski
and
Grzegorz
Hreczycho
 *
*
Faculty of Chemistry, Adam Mickiewicz University in Poznań, Uniwersytetu Poznańskiego St. 8, 61-614 Poznań, Poland. E-mail: g.h@amu.edu.pl
First published on 22nd May 2023
Abstract
The hydroboration of alkenes catalyzed using an inexpensive and easily accessible Earth-abundant cobalt pincer complex based on a PN5P triazine backbone is reported. The presented protocol allows efficient anti-Markovnikov functionalization of alkenes bearing a wide range of functional groups (unsaturated amines, carbonates, ethers, vinylarenes, vinylsilanes, and natural products) under mild conditions with high yields. Moreover, the presented procedure exhibits unique selectivity distinguishing vinylsilyl groups from other alkenyl or alkyne groups, leading to an interesting class of bifunctional compounds that have potential application in the synthesis of advanced materials. Their application potential was demonstrated by several model transformations.
Introduction
Hydroboration, the addition of a boron–hydrogen bond to an unsaturated carbon–carbon or carbon–heteroatom bond, was discovered in 1956 by H. C. Brown.1 Since its discovery, it has become an important tool used in organic chemistry, as evidenced by the numerous investigations published each year focused on the synthesis and transformation of the boron group.2–7Organoboron compounds can be synthesized directly from unsaturated substrates in the presence of more reactive boron hydrides such as trihydridoboron (BH3) and dialkyl boron hydrides (HBR2) without the need for a catalyst, but the use of these reactants is associated with safety hazards and the high instability of the resulting products.8–10 As a result, in practice, less reactive dioxaborolane derivatives (pinacolborane, catecholborane) are commonly used, which usually require the assistance of a catalyst.4
In recent years, researchers have developed a variety of catalysts for hydroboration reactions, many of which use noble metals like rhodium and iridium.11,12 However, there is now a growing focus on using first-row d-block metals for catalysis.13–15 Specifically, there is significant interest in cobalt catalysts that utilize pincer ligands, which have shown promise as an alternative to traditional precious metal catalysts.16
Recently, there have been several articles highlighting the exceptional catalytic performance of cobalt complexes in olefin hydroboration reactions, particularly when used in low catalyst concentrations.17–22 These studies are headlined by unprecedented research by Huang's group. In their work, they reported a highly efficient Co-complex [(iPrPNN)CoCl2] with high activity and exhibiting excellent regio- and chemoselectivity toward anti-Markovnikov products.23 Similarly, Zheng's team demonstrated the possibility of hydroboration of vinylarenes using a cobalt(II) coordination polymer (CP) based on the pincer moiety for achieving excellent activity and observing the Markovnikov selectivity.24 This unusual hydroboration selectivity has also been reported for the bipyridyl–oxazoline cobalt catalyst [(tBuBPO)CoCl2].25 Moreover, an interesting example of the pincer cobalt complex utility is their use as catalysts in asymmetric hydroboration reactions.14,26–30 Exciting developments have emerged from Prof. Lu's group, who have achieved control over the enantioselectivity of hydroboration reactions by introducing a rigid imine group in place of a flexible amine group in the ligand structure.31 Additionally, other promising methods for synthesizing organoboron compounds include the sequential hydrosilylation–hydroboration reaction of terminal acetylenes32–34 and the use of the cobalt-catalyzed isomerization–hydroboration reaction of internal alkenes.35–37
In the case of bifunctional terminal olefins or unconjugated (1,n-) dienes, there are known reports of dihydroboration38–40 or hydroboration with the retention of the internal double bond,17,18,41,42 as well as the synthesis of gem-bis(boryl)alkanes using a chain-walking mechanism.43 As far as we know, there is a lack of examples allowing regioselective monohydroboration of terminal uncoupled dienes as well as uncoupled enynes.
Therefore, we decided to examine how pincer cobalt complexes based on the triazine backbone would perform in olefin hydroboration reactions with a strong focus on chemoselectivity. Recently, we have shown the possibility of their use in the dehydrogenative silylation reactions of silylacetylenes44 or dehydrogenative coupling of silanes with alcohols.45 Moreover, we have demonstrated the possibility of controlling the direction of the reaction of silylacetylenes with pinacolborane toward hydroboration or dehydrogenative borylation products depending on the substituents at the triazine ring.46 Now we present an efficient anti-Markovnikov selective protocol for the hydroboration of a broad range of alkenes (amines, carbonates, ethers, vinylarenes, vinylsilanes, natural products) under mild conditions using an inexpensive and Earth-abundant cobalt complex as a catalyst. Moreover, the presented procedure allows selective functionalization of alkenyl(vinyl)silanes and alkynyl(vinyl)silanes with the retention of the alkenyl or vinyl group, respectively, leading to interesting bifunctional precursors of modern materials, in which the free alkenyl group allows further modification of the obtained polymers or their crosslinking.
Results and discussion
In a two-step synthesis, a series of PNP ligands based on the triazine ring were obtained from commercially available substrates. Subsequently, they were utilized in the synthesis of the corresponding complexes from the cobalt chloride precursor (Scheme 1).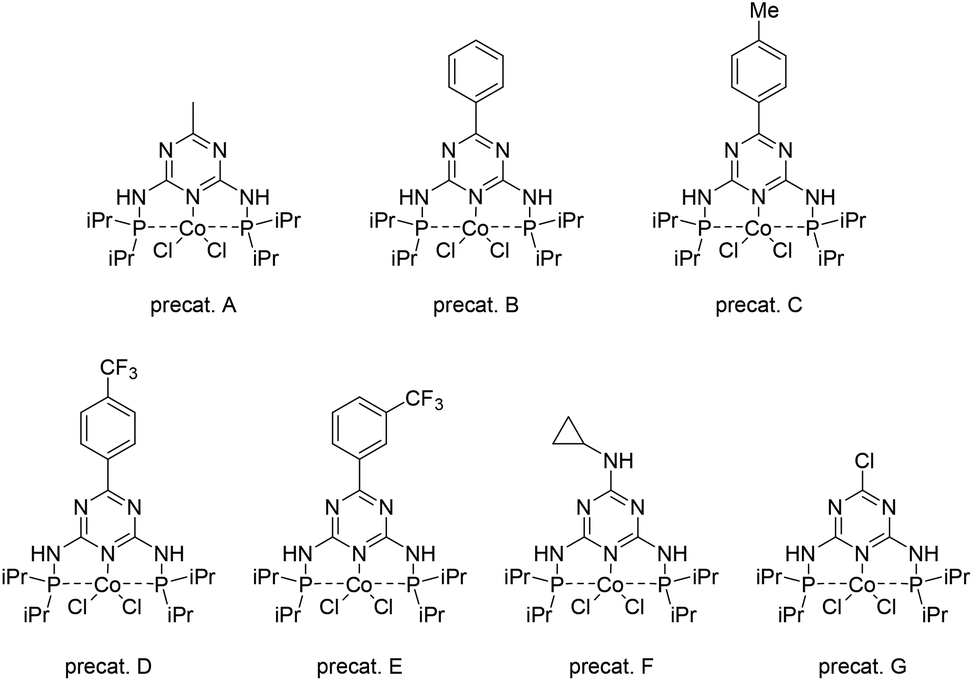 | ||
| Scheme 1 Structures of the obtained pincer cobalt complexes. | ||
First, we checked the activity of the most promising cobalt precatalyst F in a model hydroboration reaction of dimethylphenylvinylsilane with pinacolborane (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Activator | Solvent | Conversiona | Selectivityb A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B :B |
|
|
a Conversion of vinylsilane determined by GC with n-dodecane as an internal standard.
b Selectivity of [A]:[B] products determined by GC.
c Reaction performed at 40 °C.
d Reaction performed at 25 °C.
e CoCl2 instead of cat. F.
f Catalyst-free.
|
|||||
| 1 | — | Chlorobenzene | 66% | 99 | 1 |
| 2 | Cs2CO3 | Chlorobenzene | 98% | 99 | 1 |
| 3 | Cs2CO3 | Chlorobenzene | 80%c | 99 | 1 |
| 4 | Cs2CO3 | Chlorobenzene | 26%d | 84 | 16 |
| 5 | — | Chlorobenzene | Tracee | — | — |
| 6 | Cs2CO3 | Chlorobenzene | Tracee | — | — |
| 7 | — | THF | 95% | 98 | 2 |
| 8 | Cs2CO3 | THF | 99% | 90 | 10 |
| 9 | — | Toluene | 16% | 94 | 6 |
| 10 | Cs2CO3 | Toluene | 98% | 91 | 9 |
| 11 | — | Solvent-free | 97% | 99 | 1 |
| 12 | — | Solvent-free | Tracef | — | — |
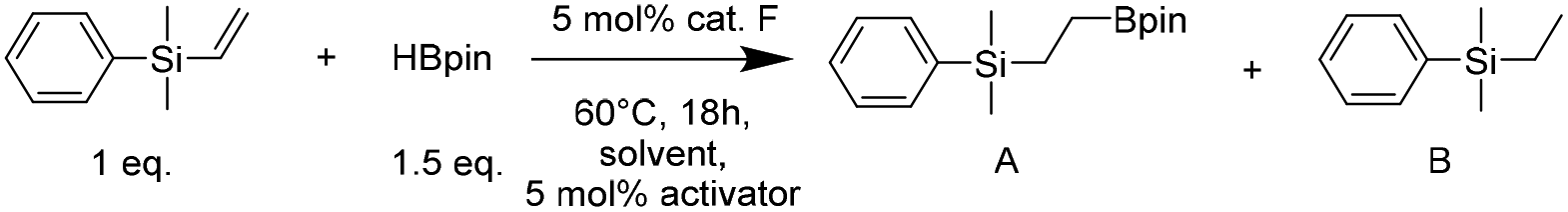
In the initial reaction carried out in chlorobenzene, using pinacolborane as the substrate and simultaneous precatalyst activator, we obtained 66% conversion of vinylsilane (entry 1). Therefore, to increase the conversion we added cesium carbonate as an additional activator, which allowed us to achieve a full conversion of the substrate (entry 2). Decreasing the reaction temperature to 40 °C (entry 3) or room temperature (entry 4) reduced the conversion rate, leading to 80% and 26% transformation of dimethylphenylvinylsilane, respectively. Experiments in other solvents (entries 7–10) gave results analogous to the model reaction in chlorobenzene. However, a reaction carried out under solvent-free conditions (entry 11) showed the possibility of running the process without the addition of an activator, giving a selectively anti-Markovnikov hydroboration product. A catalyst-free and solvent-free control experiment yielded only trace amounts of the product (entry 12). Consequently, in the next step, we decided to check the catalytic activity of other synthesized cobalt precatalysts (ESI Table S1†) under solvent-free conditions. Among them, the best results were obtained using complex F (ESI Table S2†), for which we also optimized the amount of used pinacolborane to 1.1 equivalents.
With the optimized reaction conditions in hand, we decided to investigate other vinylsilanes in the hydroboration reaction with pinacolborane (Scheme 2). Experiments with vinylsilanes having both aliphatic (1a and 1b) and aromatic (1c) substituents afforded the desired products with high conversions and yields. The developed method also allows selective functionalization of sensitive alkoxysilanes (1d–1g), belonging to a group of important polymer precursors commonly used in materials science. The proposed procedure is fully compatible with vinylsilanes having the Si–H group (1h) capable of further use of the obtained products in hydrosilylation reactions. Interestingly, in the case of 2-methylallyldimethyl(vinyl)silane, the developed catalytic system permitted an exclusive functionalization of the vinyl group (1i), indicating the lack of activity of the presented protocol towards the functionalization of 2-methylallyl derivatives. Moreover, using multi-vinyl substituted silanes we were able to obtain branched derivatives (1j–1m) which are interesting building blocks for the synthesis of dendrimers.
 | ||
| Scheme 2 Cobalt-catalyzed solvent-free hydroboration of vinylsilanes. aConversion of vinylsilane determined by GC with n-dodecane as an internal standard; the isolated yields are given in parentheses. bReaction performed at 80 °C. c5 mol% of catalyst per vinyl group. | ||
Unfortunately, attempts to hydroborate allyltrimethylsilane or allyltriisopropylsilane resulted in low conversions (<7%). Similarly, in the case of non-silylated alkenes (allylbenzene and decene), we observed only partial conversions of the substrate, suggesting a strong influence of the β-silicon effect on the performed reaction.
As a consequence, we changed our strategy and used the addition of an activator (NaHBEt3). Preliminary results for the model hydroboration reaction of triisopropylallylsilane provided the desired product with full conversion (ESI Table S4†). For this reason, we conducted an optimization process, the details of which are provided in the ESI (Tables S4–S8†). Among the tested activators, all of them allowed the successful activation of the complex. Finally, we decided to use sodium triethylboron hydride solution due to its commercial availability and ease of handling. Ultimately, optimal conditions were achieved by running the reactions at 40 °C and reducing the amount of precatalyst F used to 1 mol% and the activator to 2 mol%.
Under such developed conditions, we tested a wide range of alkenes in the hydroboration reaction (Scheme 3). As a result, we functionalized with good yields of allylsilanes (2a and 2b), vinylsilanes (2c), aliphatic alkenes (2d and 2e), haloalkenes (2f), unsaturated amines (2g and 2h), ethers (2i), carbonates (2j), or vinylarenes (2k).
 | ||
| Scheme 3 Cobalt-catalyzed hydroboration of alkenes with sodium triethylboron hydride as a precatalyst activator. aConversion of olefin determined by GC with n-dodecane as an internal standard; the isolated yields are given in parentheses. b5 mol% cat. c2.5 mol% cat. | ||
Moreover, the excellent results of the hydroboration of allylbenzene (2l) and pentafluoroallylbenzene (2m) prompted us to investigate other naturally occurring derivatives of allylbenzenes such as estragole (2n) and methyl eugenol (2o). These compounds, due to their biological activity, are often a moiety of newly designed drugs47–50 and for these reasons, the obtained products provide interesting biorelevant scaffolds for the synthesis of pharmaceuticals. Interestingly, under the developed conditions, we selectively functionalized 2-allylphenol (2p), leaving the hydroxyl group free, while there are scientific reports of catalyst-free dehydrocoupling of pinacol with nucleophiles (including phenols).51
To test the limitations of the developed method, we performed reactions with four unsaturated hexane derivatives under the optimized conditions. The reaction involving 1-hexene (2r) achieved a complete conversion of the substrate into the desired product. However, for trans-3-hexene and cis-3-hexene, no product formation was observed. This indicates that the described method enables only the functionalization of terminal olefins, which eventually allowed us to selectively functionalize mono hydroborate cis-1,4-hexadiene (2s).
Interestingly, the different activities of the presented catalytic systems allow selective functionalization of the vinyl group in alkenyl(vinyl)silane derivatives. To confirm this concept, we carried out the hydroboration of allyldimethyl(vinyl)silane under the two developed conditions (Scheme 4). When the reaction was carried out under solvent- and activator-free conditions, we selectively modified the vinyl group leaving the allyl double bond intact (3a), while the use of an activator led to a double hydroboration product (3b).
 | ||
| Scheme 4 Comparison of the selectivity of the two developed hydroboration methods. | ||
This unique selectivity provides a number of interesting opportunities that may find application in the synthesis of highly complex precursors of advanced materials. The applicability of this approach was demonstrated in two model functionalization reactions of the monohydroborated product involving an intact allylic group (Scheme 5). To the best our knowledge, this is the first example of a catalytic hydroboration that distinguishes the vinyl group of silanes from other alkenyl groups and allows selective functionalization without further isomerization or reduction of the obtained molecule.
 | ||
| Scheme 5 Functionalization of the monohydroborated product using hydrosililation and O-sililation52 reactions. | ||
Finally, we conducted tests to check the selectivity of our system in the competitive hydroboration reaction of vinylsilane in the presence of triisopropylsilylacetylene (Scheme 6). The results showed almost full conversion of acetylene (95%) while vinylsilane gave only a trace amount of the product (4%). This prompted us to modify the bifunctional (methylphenyl(vinyl)silyl)acetylene molecule, which allowed selective functionalization of the acetylene group. This is in accordance with our previous reports in which we demonstrated the possibility of the hydroboration of silylacetylenes on the described catalyst.46
 | ||
| Scheme 6 Inter- and intramolecular competition hydroboration experiment. | ||
To prove the synthetic potential of the developed method, we tested its scalability by performing two gram-scale reactions for both the solvent-free hydroboration of vinylsilanes and the reaction with an activator. In both cases, we observed quantitative conversion which resulted in excellent isolation yields (Scheme 7).
 | ||
| Scheme 7 Gram-scale hydroboration reactions. | ||
Bifunctional organometallic compounds having moieties with different reactivities are interesting building blocks in synthetic chemistry. Therefore, we decided to demonstrate the possibilities offered by the developed method by performing two one-pot functionalizations (Scheme 8). In the first synthetic pathway, we modified the hydroboration product through a hydrolysis and condensation reaction of the organosilicon moiety, while in the second, we introduced a new functional group through the transformation of the organoboron moiety. The high compatibility of the developed method with commonly used reactions allows omitting the necessity to isolate the hydroboration products perfectly fits into the tenets of modern chemistry and increases its application potential.
 | ||
| Scheme 8 One-pot functionalization of hydroboration products. | ||
To confirm the reaction mechanism, we conducted a series of studies. First, if sodium triethylboron hydride is utilized as an activator, there is a possibility of BH3 generation, which can act as a hidden catalyst.53–56 Our experiment under standard conditions using the addition of tetramethylethylenediamine as a complexing agent for potentially forming borane showed no effect of TMEDA on substrate conversion (Scheme 9a). In the case of using BH3·THF instead of NaHBEt3, we observed only partial conversion of the substrate (Scheme 9b). Therefore, we cannot fully exclude a mechanism based on the decomposition of HBpin to BH3, which then undergoes addition to the double bond with simultaneous transborylation with LCoH2Bpin, but it is certainly a minor phenomenon.
 | ||
| Scheme 9 Experiments verifying the potential formation of borane as a hidden reaction catalyst. | ||
To rule out the possibility of a heterogeneous mechanism driven by catalytically active cobalt nanoparticles, which may have been formed due to the reduction of the complex with sodium triethylboron hydride, we conducted the hydroboration reaction under the developed conditions by introducing a drop of mercury into the reaction vessel (refer to the ESI for details†). GC-MS analysis of the experiment confirmed complete substrate conversion, thereby ruling out the occurrence of a heterogeneous mechanism in the reaction.
Therefore, we conducted NMR tests in an attempt to detect the active form of the catalyst. Since our precatalysts are paramagnets, the NMR tests performed clearly indicate that there is a change in the oxidation state of cobalt during activation. In earlier studies, Chirik et al. investigated the activation of pinacolborane complexes with NaHBEt3, in which cobalt is reduced from its second oxidation state to the first oxidation state, followed by an oxidative addition by HBpin to the third oxidation state (Scheme 10a).57 However, in the case of our precatalysts, such an activation mechanism for both the reactions with pinacolborane and with the addition of NaHBEt3 leads to the same active form of the complex, thus not explaining the differences in the activity that we observed. Moreover, in the structure of the ligand from the Chirik group, the acidic NH protons were not present. Thus, for our precatalysts, the addition of NaHBEt3 is probably responsible for the deprotonation of the NH groups of the complex, in a manner analogous to that proposed in the studies of the Kempe group (Scheme 10b), in which they postulate that the anionic nature of the catalyst affects its high activity.58 In our study, we did not observe a difference in the catalytic activity depending on the activator used or its counterion (see the ESI, Table S8†), therefore we believe that the deprotonated form of the complex is fully responsible for its high catalytic activity in the olefin hydroboration reaction. The 11B NMR spectrum acquired for structure 2 (Scheme 10c) indicates the presence of several diverse boron nuclei (see the ESI†), probably originating from BEt3 adducts with the anionic form of the complex, which is in agreement with the literature reports by the Pawluć group.59 In the case of the non-anionic form, its lower catalytic activity is probably compensated by the β-silicon effect, stabilizing the alkyl complex and remarkably accelerating the reaction rate.60–62
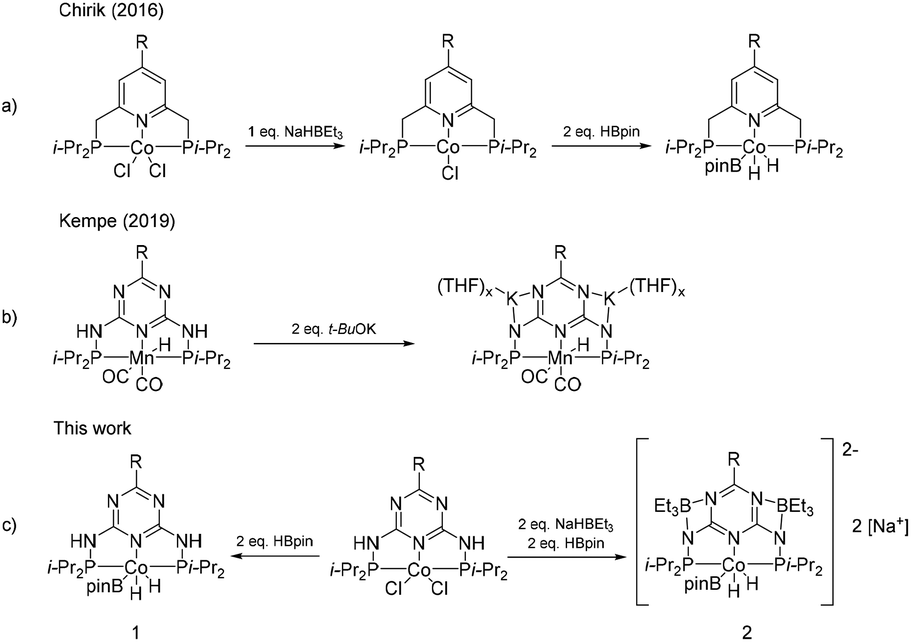 | ||
| Scheme 10 Mechanisms of the activation of pincer complexes.57,58 | ||
Based on this and our previous studies on pincer cobalt complexes, as well as literature reports,44–46,57,63–67 we postulate that in the reaction mixture, as a result of activation with pinacolborane or sodium triethylboron hydride, the two forms of the cobalt complex (Co(I) and Co(III)) coexist in equilibrium. Either of them can catalyze the process; however, based on recent reports,66 we assume that the main catalytically active form is Co(I) (LCo-H). First, the alkene undergoes 1,2-insertion into a Co–H bond, which is followed by transmetalation with the pinacolborane moiety, resulting in the regeneration of the active form (Scheme 11). In the case of the hydroboration of vinylsilanes using pinacolborane alone as an activator, we assume that the active form of the complex is LCo-H being in equilibrium with structure 1 (Scheme 9c and see the ESI†), while for activation with the addition of NaHBEt3, we are unable to conclusively determine the active form of the complex; however, we postulate analogous structures with a deprotonated ligand (Scheme 9c and see the ESI†).
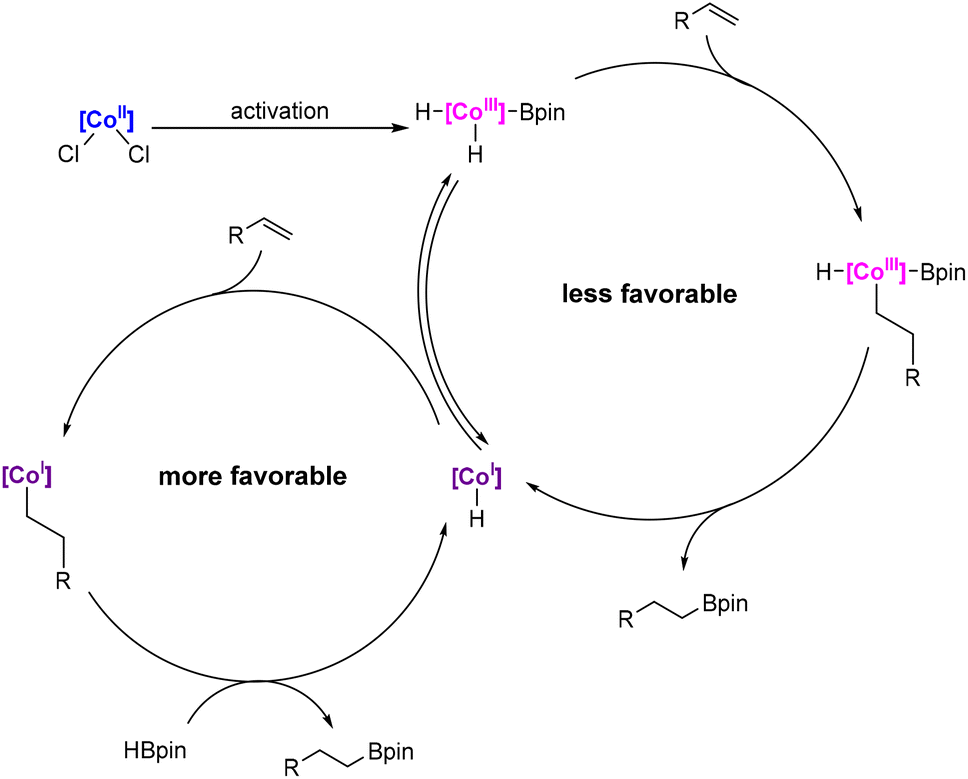 | ||
| Scheme 11 Proposed catalytic cycle. | ||
Conclusions
In conclusion, we have developed new highly efficient catalytic methods for anti-Markovnikov hydroboration of terminal alkenes using a cheap catalyst based on Earth-abundant cobalt and proceeding under mild conditions. The proposed protocols allow the functionalization of a wide range of alkene derivatives (silanes, amines, ethers, natural products) showing high chemo- and regioselectivity, with the obtained compounds being interesting synthons for the synthesis of fine chemicals, pharmaceuticals, and polymers. We also demonstrated the possibility of selective functionalization of the vinyl group in alkenyl(vinyl)silanes or of the alkynyl group in alkynyl(vinyl)silanes based on the developed solvent and activator-free method. Moreover, we proved the high scalability and compatibility of the described methodology with other procedures allowing further one-pot modification of the obtained products highlighting the high applicability character of the developed protocol.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Science Centre Grant UMO-2018/30/E/ST5/00045.References
- H. C. Brown and B. C. S. Rao, A New Technique For The Conversion Of Olefins Into Organoboranes And Related Alcohols, J. Am. Chem. Soc., 1956, 78(21), 5694–5695 CrossRef CAS
.
- A. Suzuki, Carbon-carbon bonding made easy, Chem. Commun., 2005, 4759–4763 RSC
- C. Sandford and V. K. Aggarwal, Stereospecific functionalizations and transformations of secondary and tertiary boronic esters, Chem. Commun., 2017, 53, 5481–5494 RSC
- S. J. Geier, C. M. Vogels, J. A. Melanson, S. A. Westcott and S. J. Geier, The transition metal-catalysed hydroboration reaction, Chem. Soc. Rev., 2022, 51, 8877–8922 RSC
- J. W. B. Fyfe and A. J. B. Watson, Recent Developments in Organoboron Chemistry: Old Dogs, New Tricks, Chem, 2017, 3, 31–55 CAS
- Z. He, Y. Hu, C. Xia and C. Liu, Recent advances in the borylative transformation of carbonyl and carboxyl compounds, Org. Biomol. Chem., 2019, 17, 6099–6113 RSC
- A. Suzuki, New synthetic transformations via organoboron compounds, Pure Appl. Chem., 1994, 66, 213–222 CrossRef CAS
- H. C. Brown and G. Zweifel, Hydroboration. VIII. Bis-3-methyl-2-butylborane, J. Am. Chem. Soc., 1961, 83, 1241–1246 CrossRef CAS
- G. Zweifel, N. R. Ayyangar and H. C. Brown, Hydroboration. XVII. An Examination of Several Representative Dialkylboranes as Selective Hydroborating Agents, J. Am. Chem. Soc., 1963, 85, 2072–2075 CrossRef CAS
- E. F. Knights and H. C. Brown, 9-Borabicyclo[3.3.1]nonane as a Convenient Selective Hydroborating Agent, J. Am. Chem. Soc., 1968, 90, 5281–5283 CrossRef CAS
- K. Burgess and M. J. Ohlmeyer, Transition-Metal-Promoted Hydroborations of Alkenes, Emerging Methodology for Organic Transformations, Chem. Rev., 1991, 91, 1179–1191 CrossRef CAS
- C. M. Crudden and D. Edwards, Catalytic Asymmetric Hydroboration: Recent Advances and Applications in Carbon-Carbon Bond-Forming Reactions, Eur. J. Org. Chem., 2003, 4695–4712 CrossRef CAS
- J. V. Obligacion and P. J. Chirik, Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroboration, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS PubMed
- Z. Zuo, H. Wen, G. Liu and Z. Huang, Cobalt-Catalyzed Hydroboration and Borylation of Alkenes and Alkynes, Synlett, 2018, 1421–1429 CAS
- J. Chen, J. Guo and Z. Lu, Recent Advances in Hydrometallation of Alkenes and Alkynes via the First Row Transition Metal Catalysis, Chin. J. Chem., 2018, 36, 1075–1109 CrossRef CAS
- K. Junge, V. Papa and M. Beller, Cobalt–Pincer Complexes in Catalysis, Chem. – Eur. J., 2019, 25, 122–143 CrossRef CAS PubMed
- R. Agahi, A. J. Challinor, N. B. Carter and S. P. Thomas, Earth-Abundant Metal Catalysis Enabled by Counterion Activation, Org. Lett., 2019, 21, 993–997 CrossRef CAS PubMed
- J. Pecak, S. Fleissner, L. F. Veiros, E. Pittenauer, B. Stöger and K. Kirchner, Synthesis and Catalytic Reactivity of Cobalt Pincer Nitrosyl Hydride Complexes, Organometallics, 2021, 40, 278–285 CrossRef CAS PubMed
- A. M. Poitras, L. K. Oliemuller, G. P. Hatzis and C. M. Thomas, Highly Selective Hydroboration of Terminal Alkenes Catalyzed by a Cobalt Pincer Complex Featuring a Central Reactive N-Heterocyclic Phosphido Fragment, Organometallics, 2021, 40, 1025–1031 CrossRef CAS
- A. D. Ibrahim, S. W. Entsminger and A. R. Fout, Insights into a Chemoselective Cobalt Catalyst for the Hydroboration of Alkenes and Nitriles, ACS Catal., 2017, 7, 3730–3734 CrossRef CAS
- G. Zhang, J. Wu, M. Wang, H. Zeng, J. Cheng, M. C. Neary and S. Zheng, Cobalt-Catalyzed Regioselective Hydroboration of Terminal Alkenes, Eur. J. Org. Chem., 2017, 5814–5818 CrossRef CAS
- W. N. Palmer, T. Diao, I. Pappas and P. J. Chirik, High-activity cobalt catalysts for alkene hydroboration with electronically responsive terpyridine and α-diimine ligands, ACS Catal., 2015, 5, 622–626 CrossRef CAS
- L. Zhang, Z. Zuo, X. Leng and Z. Huang, A cobalt-catalyzed alkene hydroboration with pinacolborane, Angew. Chem., Int. Ed., 2014, 53, 2696–2700 CrossRef CAS PubMed
- G. Zhang, J. Wu, S. Li, S. Cass and S. Zheng, Markovnikov-Selective Hydroboration of Vinylarenes Catalyzed by a Cobalt(II) Coordination Polymer, Org. Lett., 2018, 20, 7893–7897 CrossRef CAS PubMed
- J. Peng, J. H. Docherty, A. P. Dominey and S. P. Thomas, Cobalt-catalysed Markovnikov selective hydroboration of vinylarenes, Chem. Commun., 2017, 53, 4726–4729 RSC
- J. Guo, Z. Cheng, J. Chen, X. Chen and Z. Lu, Iron-and Cobalt-Catalyzed Asymmetric Hydrofunctionalization of Alkenes and Alkynes, Acc. Chem. Res., 2021, 54, 2701–2716 CrossRef CAS PubMed
- F. Paquin, J. Rivnay, A. Salleo, N. Stingelin and C. Silva, Multi-phase semicrystalline microstructures drive exciton dissociation in neat plastic semiconductors, J. Mater. Chem. C, 2015, 3, 10715–10722 RSC
- L. Zhang, Z. Zuo, X. Wan and Z. Huang, Cobalt-catalyzed enantioselective hydroboration of 1,1-disubstituted aryl alkenes, J. Am. Chem. Soc., 2014, 136, 15501–15504 CrossRef CAS PubMed
-
R. V. Chaudhari, Fundamentals of Homogeneous Catalysis, Elsevier Inc., 2016 Search PubMed
- X. Chen, Z. Cheng and Z. Lu, Cobalt-Catalyzed Asymmetric Markovnikov Hydroboration of Styrenes, ACS Catal., 2019, 9, 4025–4029 CrossRef CAS
- H. Zhang and Z. Lu, Dual-Stereocontrol Asymmetric Cobalt-Catalyzed Hydroboration of Sterically Hindered Styrenes, ACS Catal., 2016, 6, 6596–6600 CrossRef CAS
- Z. Cheng, J. Guo, Y. Sun, Y. Zheng, Z. Zhou and Z. Lu, Regio-controllable Cobalt-Catalyzed Sequential Hydrosilylation/Hydroboration of Arylacetylenes, Angew. Chem., Int. Ed., 2021, 60, 22454–22460 CrossRef CAS PubMed
- Z. Zuo, J. Yang and Z. Huang, Cobalt-Catalyzed Alkyne Hydrosilylation and Sequential Vinylsilane Hydroboration with Markovnikov Selectivity, Angew. Chem., Int. Ed., 2016, 55, 10839–10843 CrossRef CAS PubMed
- J. Guo and Z. Lu, Highly Chemo-, Regio-, and Stereoselective Cobalt-Catalyzed Markovnikov Hydrosilylation of Alkynes, Angew. Chem., Int. Ed., 2016, 55, 10835–10838 CrossRef CAS PubMed
- T. Ogawa, A. J. Ruddy, O. L. Sydora, M. Stradiotto and L. Turculet, Cobalt- and Iron-Catalyzed Isomerization-Hydroboration of Branched Alkenes: Terminal Hydroboration with Pinacolborane and 1,3,2-Diazaborolanes, Organometallics, 2017, 36, 417–423 CrossRef CAS
- M. L. Scheuermann, E. J. Johnson and P. J. Chirik, Alkene isomerization-hydroboration promoted by phosphine-ligated cobalt catalysts, Org. Lett., 2015, 17, 2716–2719 CrossRef CAS PubMed
- A. J. Ruddy, O. L. Sydora, B. L. Small, M. Stradiotto and L. Turculet, (N-Phosphinoamidinate)cobalt-Catalyzed Hydroboration: Alkene Isomerization Affords Terminal Selectivity, Chem. – Eur. J., 2014, 20, 13918–13922 CrossRef CAS PubMed
- A. K. Jaladi, W. K. Shin and D. K. An, Alkene hydroboration with pinacolborane catalysed by lithium diisobutyl-: Tert-butoxyaluminum hydride, RSC Adv., 2019, 9, 26483–26486 RSC
- T. Iwasaki, X. Min, A. Fukuoka, L. Zhu, R. Qiu, T. Yang, M. Ehara, A. Sudalai and N. Kambe, Ni-Catalyzed Dimerization and Hydroperfluoroarylation of 1,3-Dienes, J. Org. Chem., 2018, 83, 9267–9277 CrossRef CAS PubMed
-
A. Yusuke, K. Nagashima and F. Naoki, Crosslinking Agent, JP5680449B2, 2012
- D. Lu, C. Chen, L. Zheng, J. Ying and Z. Lu, Regio- and Stereoselective Cobalt-Catalyzed Hydroboration of Vinylcyclopropanes to Access Homoallylic Boronates, Organometallics, 2023 DOI:10.1021/acs.organomet.2c00592
- C. Chen, H. Wang, T. Li, D. Lu, J. Li, X. Zhang, X. Hong and Z. Lu, Cobalt-Catalyzed Asymmetric Sequential Hydroboration/Isomerization/Hydroboration of 2-Aryl Vinylcyclopropanes, Angew. Chem., Int. Ed., 2022, 61, e202205619 CAS
- M. Hu and S. Ge, Versatile cobalt-catalyzed regioselective chain-walking double hydroboration of 1,n-dienes to access gem-bis(boryl)alkanes, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed
- H. Stachowiak, K. Kuciński, F. Kallmeier, R. Kempe and G. Hreczycho, Cobalt-Catalyzed Dehydrogenative C−H Silylation of Alkynylsilanes, Chem. – Eur. J., 2022, 28, e202103629 CrossRef CAS PubMed
- E. Szafoni, K. Kuciński and G. Hreczycho, Green Chemistry Letters and Reviews Cobalt-catalyzed synthesis of silyl ethers via cross- dehydrogenative coupling between alcohols and hydrosilanes, Green Chem. Lett. Rev., 2022, 15, 757–764 CrossRef CAS
- D. Lewandowski, T. Cytlak, R. Kempe and G. Hreczycho, Ligand-controlled Cobalt-Catalyzed formation of Carbon–Boron bonds: Hydroboration vs C–H/B–H dehydrocoupling, J. Catal., 2022, 413, 728–734 CrossRef CAS
- F. P. Jørgensen, D. Madsen, M. Meldal, J. V. Olsen, M. Petersen, J. Granhøj and M. Bols, Synthesis of Shld Derivatives, Their Binding to the Destabilizing Domain, and Influence on Protein Accumulation in Transgenic Plants, J. Med. Chem., 2019, 62, 5191–5216 CrossRef PubMed
- L. Rivail, M. Giner, M. Gastineau, M. Berthouze, J. L. Soulier, R. Fischmeister, F. Lezoualc'h, B. Maigret, S. Sicsic and I. Berque-Bestel, New insights into human 5-HT 4 receptor binding site: Exploration of a hydrophobic pocket, Br. J. Pharmacol., 2004, 143, 361–370 CrossRef CAS PubMed
- M. V. B. Reddy, H. Y. Hung, P. C. Kuo, G. J. Huang, Y. Y. Chan, S. C. Huang, S. J. Wu, S. L. Morris-Natschke, K. H. Lee and T. S. Wu, Synthesis and biological evaluation of chalcone, dihydrochalcone, and 1,3-diarylpropane analogs as anti-inflammatory agents, Bioorg. Med. Chem. Lett., 2017, 27, 1547–1550 CrossRef PubMed
- C. Choi, J. H. Li, M. Vaal, C. Thomas, D. Limburg, Y. Q. Wu, Y. Chen, R. Soni, C. Scott, D. T. Ross, H. Guo, P. Howorth, H. Valentine, S. Liang, D. Spicer, M. Fuller, J. Steiner and G. S. Hamilton, Use of parallel-synthesis combinatorial libraries for rapid identification of potent FKBP12 inhibitors, Bioorg. Med. Chem. Lett., 2002, 12, 1421–1428 CrossRef CAS PubMed
- E. A. Romero, J. L. Peltier, R. Jazzar and G. Bertrand, Catalyst-free dehydrocoupling of amines, alcohols, and thiols with pinacol borane and 9-borabicyclononane (9-BBN), Chem. Commun., 2016, 52, 10563–10565 RSC
- K. Kuciński and G. Hreczycho, O-Metalation, of silanols and POSS silanols over Amberlyst-15 catalyst: A facile route to unsymmetrical siloxanes, borasiloxanes and germasiloxanes, Inorg. Chim. Acta, 2019, 490, 261–266 CrossRef
- A. D. Bage, T. A. Hunt and S. P. Thomas, Hidden Boron Catalysis: Nucleophile-Promoted Decomposition of HBpin, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS PubMed
- A. D. Bage, K. Nicholson, T. Langer and S. P. Thomas, The Hidden Role of Boranes and Borohydrides in Hydroboration Catalysis, ACS Catal., 2020, 10, 13479–13486 CrossRef CAS
- E. Nieto-Sepulveda, A. D. Bage, L. A. Evans, T. A. Hunt, A. G. Leach, S. P. Thomas and G. C. Lloyd-Jones, Kinetics and Mechanism of the Arase-Hoshi R2BH-Catalyzed Alkyne Hydroboration: Alkenylboronate Generation via B-H/C-B Metathesis, J. Am. Chem. Soc., 2019, 141, 18600–18611 CrossRef CAS PubMed
- K. Shirakawa, A. Arase and M. Hoshi, Preparation of (E)-1-alkenylboronic acid pinacol esters via transfer of alkenyl group from boron to boron, Synthesis, 2004, 1814–1820 CAS
- J. V. Obligacion, S. P. Semproni, I. Pappas and P. J. Chirik, Cobalt-Catalyzed C(sp2)-H Borylation: Mechanistic Insights Inspire Catalyst Design, J. Am. Chem. Soc., 2016, 138, 10645–10653 CrossRef CAS PubMed
- F. Freitag, T. Irrgang and R. Kempe, Mechanistic Studies of Hydride Transfer to Imines from a Highly Active and Chemoselective Manganate Catalyst, J. Am. Chem. Soc., 2019, 141, 11677–11685 CrossRef CAS PubMed
- Ł. Banach, D. Brykczyńska, A. Gorczyński, B. Wyrzykiewicz, M. Skrodzki and P. Pawluć, Markovnikov-selective double hydrosilylation of challenging terminal aryl alkynes under cobalt and iron catalysis, Chem. Commun., 2022, 58, 13763–13766 RSC
- D. D. Roberts and M. G. McLaughlin, Strategic Applications of the β-Silicon Effect, Adv. Synth. Catal., 2022, 364, 2307–2332 CrossRef CAS
- B. E. Haines, R. Sarpong and D. G. Musaev, Generality and Strength of Transition Metal β-Effects, J. Am. Chem. Soc., 2018, 140, 10612–10618 CrossRef CAS PubMed
- B. Chiavarino, M. E. Crestoni and S. Fornarini, Radiolytic silylation of alkenes and alkynes by gaseous R3Si+ions. Stereochemical evidence for the β-silyl effect, J. Am. Chem. Soc., 1998, 120, 1523–1527 CrossRef CAS
- J. V. Obligacion, S. P. Semproni and P. J. Chirik, Cobalt-Catalyzed C−H Borylation, J. Am. Chem. Soc., 2014, 134, 4133–4136 CrossRef PubMed
- J. V. Obligacion and P. J. Chirik, Mechanistic Studies of Cobalt-Catalyzed C(sp2)-H Borylation of Five-Membered Heteroarenes with Pinacolborane, ACS Catal., 2017, 7, 4366–4371 CrossRef CAS PubMed
- H. Li, J. V. Obligacion, P. J. Chirik and M. B. Hall, Cobalt Pincer Complexes in Catalytic C-H Borylation: The Pincer Ligand Flips Rather Than Dearomatizes, ACS Catal., 2018, 8, 10606–10618 CrossRef CAS PubMed
- B. Lee, T. P. Pabst, G. Hierlmeier and P. J. Chirik, Exploring the Effect of Pincer Rigidity on Oxidative Addition Reactions with Cobalt(I) Complexes, Organometallics, 2023, 42(8), 708–718 CrossRef CAS
- E. Szafoni, K. Kuciński and G. Hreczycho, Cobalt-Catalyzed Dehydrogenative Cross-Coupling Reaction: Selective Access to Dihydrosiloxanes, Hydrosiloxanes and Functionalized Silsesquioxanes, J. Catal., 2023, 423, 1–9 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra and characterization data. See DOI: https://doi.org/10.1039/d3qi00478c |
| This journal is © the Partner Organisations 2023 |