
Contrasting-functionality-decked robust MOF for moisture-tolerant and variable-temperature CO2 adsorption with in-built urea group mediated mild condition cycloaddition†
Manpreet
Singh
 ab,
Partha Pratim
Mondal
a,
Sonal
Rajput
a and
Subhadip
Neogi
*ab
ab,
Partha Pratim
Mondal
a,
Sonal
Rajput
a and
Subhadip
Neogi
*ab
aInorganic Materials & Catalysis Division, CSIR-Central Salt and Marine Chemicals Research Institute (CSIR-CSMCRI), Bhavnagar, Gujarat-364002, India. E-mail: sneogi@csmcri.res.in; subhadip79@gmail.com
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
First published on 4th May 2023
Abstract
The dire need to reduce the atmospheric carbon dioxide (CO2) concentration has attracted worldwide attention to the capture of this greenhouse gas and its conversion into useful chemicals. Nevertheless, it is still difficult to achieve variable-temperature and humid-condition adsorption with mild condition fixation of CO2 in metal–organic frameworks (MOFs) due to difficulties in positioning assorted task-specific sites. We introduced open metal site (OMS), hydrogen-bond operative functionality, and free amine moiety inside the pore wall of a mixed-ligand robust Cd(II) framework. Two-fold interpenetration generated high-density acid–base functionalization promotes appreciable CO2 adsorption in the guest-free structure at elevated temperature with considerable MOF–CO2 interaction. The aqua-robust MOF exhibits minimum loss in CO2 uptake during multiple capture–release cycles under variable temperature and retains the adsorption capacities even upon exposure to 75% RH. The atomistic-level snapshots of temperature-induced inclusion of gas molecules inside this microporous vessel are rationalized from simulation studies, and portray diverse CO2-philic sites. Particularly, the four-fold increased CO2 adsorption compared to that of an un-functionalized MOF validates the prime role of pore surface engineering. Moreover, the CO2 selectivity shows a drastic improvement upon gradually increasing the temperature, attaining a CO2/N2 value of 380 at 313 K. The framework further demonstrates solvent-free CO2 conversion to cyclic carbonates in high yield with broad substrate scope and satisfactory reusability under less harsh conditions and in a rather short time. In addition to typical OMS/co-catalyst synergism, the mutual participation of antagonistic active sites in substrate interaction and activation is validated by juxtaposing the performance of a urea-free isoskeletal framework and by the relative fluorescence modification in the presence of epoxide. The results corroborate the unique organic-functionality-mediated cycloaddition mechanism, which provides important structure–function synergy in this unconventional route to non-redox CO2 fixation.
Introduction
The rising concentration of the major greenhouse gas, carbon dioxide (CO2), in the atmosphere is recognized to be the prime contributor to worldwide climate destabilization.1,2 Although the use of renewable energy sources and energy-efficient industrial processes has been proposed to mitigate this global issue, achieving total freedom from fossil-fuel-based energy seems difficult in the near future from a practical perspective.3,4 Therefore, CO2 capture from point sources is a more viable option, particularly for developing countries.5,6 So far, CO2 capture and removal has been overwhelmingly dominated by aqueous-amine-solution-based chemisorption, which presents intrinsic difficulties such as energy-intensive adsorbent regeneration, leakage of toxic amines and poor recyclability.7,8 Thus, the development of recyclable adsorbent material with high stability and selectivity is becoming the need of the hour.9–12 The adsorbents must cover certain important parameters, including (1) maintenance of selective CO2 adsorption over a wide range of temperatures, (2) stable adsorption/desorption capacity (non-hysteretic) during repeated cycles, and (3) prospect to rationalize the molecular-level adsorption mechanism.13–16 Additionally, adsorption capacity is often reduced upon moisture exposure; thus, the choice of material is very crucial, as most of commercial adsorbents show very poor performance for CO2 capture under humid conditions.17–19 Hence, tailor-made materials with molecule-specific interactions on their surface have gained immense importance and popularity.20The newly developed porous hybrid structures (metal–organic frameworks: MOFs), because of their remarkable surface area, excellent thermo-chemical stability, the simplicity of fine-tuning the pore chemistry, and the ability to affix task-specific functionalities, are the best potential candidates.21–27 Nevertheless, decreased MOF–CO2 interaction with increasing adsorption temperature severely reduces the CO2 uptake in MOFs, and only a handful of examples exist for humid-condition CO2 uptake because of MOF instability and/or pore blockage by water molecules.13,28 Thus, the fabrication of robust and tailor-made CO2-phillic frameworks is of utmost importance.29–31 The successful implementation of this strategy requires the judicious amalgamation of multiple task-specific sites in such a way that they do not hinder MOF formation and/or block the effective pore space during CO2 adsorption.32,33
On a similar note, the valorization of CO2 to chemicals is an alternative effective way to overcome major environmental problems, which has sparked extensive research on the use of captured CO2 as a C1 feedstock for value-added chemicals.29,34–36 While high-energy redox processes generally involve the production of methanol, methane, ethanol, or ethylene, non-redox reactions of CO2 lead to the formation of urea, polycarbonate, cyclic carbonates, etc.37–40 In particular, catalytic CO2 fixation to cyclic carbonates is a green and atom-economic approach, and of particular industrial importance for their wide applications in polymer synthesis, the electrolytic elements of batteries, pharmaceuticals, polar aprotic solvents, and as precursors to fine chemicals.41,42 However, homogeneous CO2 cycloaddition presents inherent product separation difficulties, whereas most heterogeneous catalyses suffer from very low conversion because of the high thermodynamic stability and kinetically inert nature of CO2.43,44 In this context, the major advantages of MOFs include: (1) open metal sites (OMSs) as Lewis acid sites, which provide electrostatic interactions with substrates, (2) heteroatom-bearing Lewis basic functionalities on organic struts, which confer favourable interactions with the reactants, and (3) optimized framework pores, which provide micro-reactor type environments to augment collisions between incoming CO2 gas and epoxides to facilitate cycloaddition reaction.40,45–52 Nevertheless, non-redox CO2 fixation in most MOFs often requires the use of hazardous solvents, harsh temperatures and/or pressures, and long reaction time. Additionally, the creation of high-density OMSs is hindered by thermo-chemical instability as well as lack of solvent-bound metal nodes, whilst pendent groups in organic struts not only block the micropores, but also lead to unwanted structures through coordination with metal ions. The above discussion clearly indicates that the rational design of an acid–base bifunctional MOF, encompassing unsaturated metal nodes as well as built-in Lewis basic sites and featuring suitably-sized cavities, is imperative for high-temperature adsorption and non-redox CO2 conversion under mild conditions.
Building on the above discussion, and our quest to develop multi-functional, robust frameworks for assorted sustainable applications,53–59 we constructed a bipillared-layer Cd(II)-organic framework (Scheme 1) from the amine-based tri-carboxylate ligand 4,4′,4′′-tricarboxytriphenylamine (H3TCA) and the urea-moiety-grafted pyridyl linker 1,3-di(pyridin-4-yl)urea (L). The guest-free MOF exhibits high CO2 adsorption at diverse temperatures and shows appreciable CO2–framework interaction with trivial loss during multiple capture–release cycles under humid conditions. Remarkably, gradually raising the temperature from 273 to 298 to 313 K leads to significant improvement in the CO2/N2 and CO2/CH4 selectivities, which was also validated by the presence of multiple CO2 attraction sites inside the optimum-sized MOF cavities based on simulation studies. This Lewis acid–base functionalized framework further demonstrates effective CO2 cycloaddition under mild, solvent-free conditions with a high yield, broad substrate scope and good reusability. The synergistic role of the contrasting functionalities is demonstrated using rationally designed control experiments that substantiate the route to CO2 fixation. Overall, this study highlights a rare demonstration of variable-temperature and selective scavenging of CO2 under humid conditions and organic functionality-assisted non-redox conversion of a major greenhouse gas to chemicals.
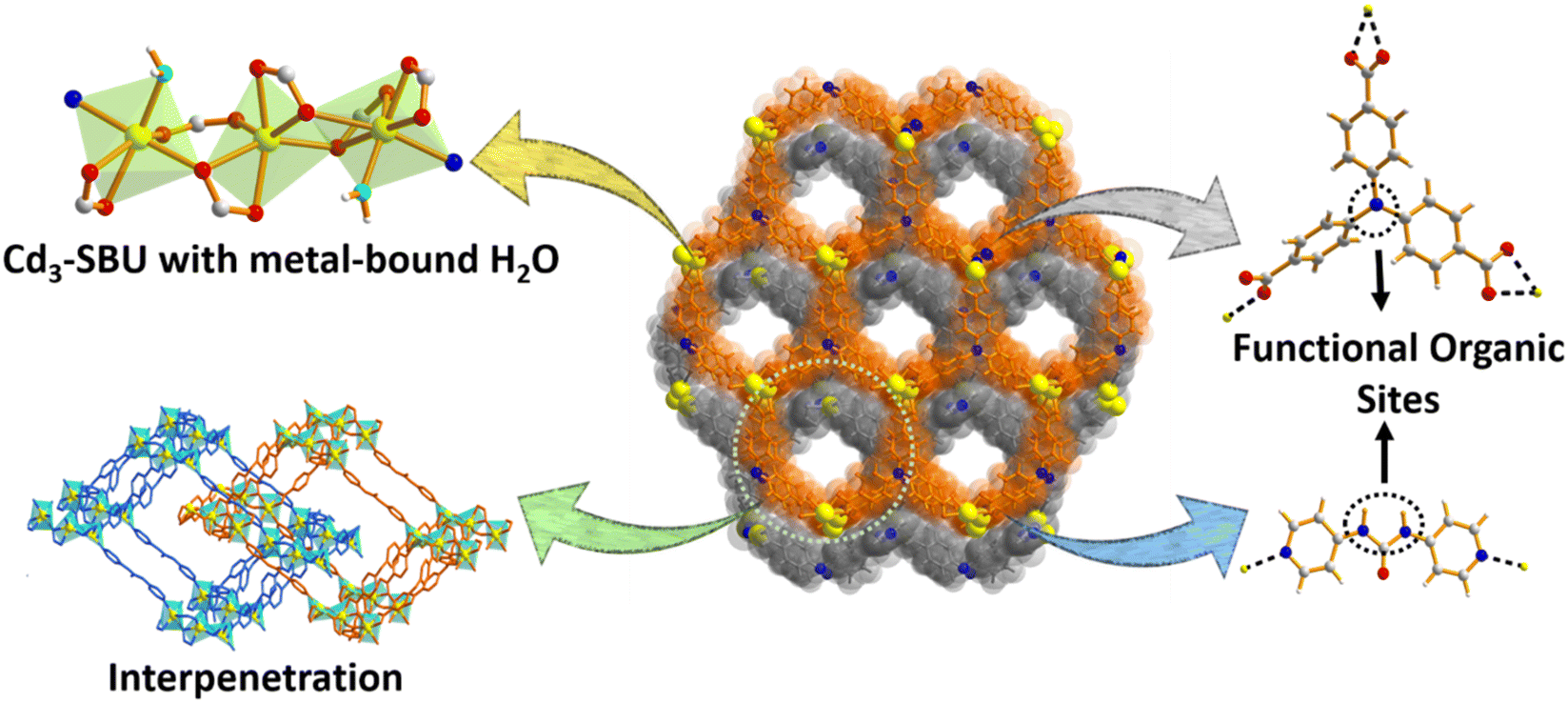 | ||
| Scheme 1 Structural and functional attributes of the MOF for CO2 capture and fixation. | ||
Results and discussion
Structural characterization and stability aspects
Single-crystal X-ray diffraction analysis revealed that the MOF exhibits the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , and the asymmetric unit (Fig. S1a, ESI†) comprises three Cd(II) ions, two deprotonated H3TCA ligands, one pyridyl linker (L) and two metal-bound molecules of the solvent water. Six carboxylate groups adjoin three Cd(II) centers to form a trinuclear [Cd3(COO)6] unit, in which two of the terminal Cd(II) centres are attached to the aqua molecules and provide the chance to create open metal sites (OMSs) upon activation (Scheme 1). The six-coordination of the middle Cd(II) ion is satisfied by oxygen atoms from four carboxylate groups, while terminal Cd(II) ions are attached to both carboxylate and pyridine ligands. The [Cd3(COO)6] units are connected through a C3-symmetric tripodal ligand and generate a two-dimensional (2D) bilayer (Fig. 1a) with hexagonal pores (15.98 × 18.77 Å2) (distance refers atom-to-atom connection). The linkers connect these bilayers to a 3D pillar-bilayer structure with rhombus channels (22.33 × 25.11 Å2) when viewed along b axis.45 Importantly, the urea group of the linker the and water-bound metal centers are projected inward the pores (Fig. 1b), providing an appropriate Lewis acid–base environment for the interaction/activation of incoming guest molecules. The large cavity dimensions lead to the two-fold interpenetration (Scheme 1) of the overall framework, resulting in a (3,8)-connected 2-nodal 3D net (Fig. 1c) with Schlafli symbol (436248)(43)2.60
, and the asymmetric unit (Fig. S1a, ESI†) comprises three Cd(II) ions, two deprotonated H3TCA ligands, one pyridyl linker (L) and two metal-bound molecules of the solvent water. Six carboxylate groups adjoin three Cd(II) centers to form a trinuclear [Cd3(COO)6] unit, in which two of the terminal Cd(II) centres are attached to the aqua molecules and provide the chance to create open metal sites (OMSs) upon activation (Scheme 1). The six-coordination of the middle Cd(II) ion is satisfied by oxygen atoms from four carboxylate groups, while terminal Cd(II) ions are attached to both carboxylate and pyridine ligands. The [Cd3(COO)6] units are connected through a C3-symmetric tripodal ligand and generate a two-dimensional (2D) bilayer (Fig. 1a) with hexagonal pores (15.98 × 18.77 Å2) (distance refers atom-to-atom connection). The linkers connect these bilayers to a 3D pillar-bilayer structure with rhombus channels (22.33 × 25.11 Å2) when viewed along b axis.45 Importantly, the urea group of the linker the and water-bound metal centers are projected inward the pores (Fig. 1b), providing an appropriate Lewis acid–base environment for the interaction/activation of incoming guest molecules. The large cavity dimensions lead to the two-fold interpenetration (Scheme 1) of the overall framework, resulting in a (3,8)-connected 2-nodal 3D net (Fig. 1c) with Schlafli symbol (436248)(43)2.60
 | ||
| Fig. 1 (a) View of the nearly hexagonal pores in a single net of the MOF. (b) Free Lewis-basic-site and metal-bound-water (green) decorated pores along the b axis. (c) Topological representation and (d) void view (along the a axis) of the two-fold entangled structure. | ||
Despite the interpenetration, cavities with dimensions of 6.67 × 10.61 Å2 still exist along the a axis (Fig. 1d), which accounts for the large 1689.8 Å3 (44%) solvent-accessible void volume per unit cell.61 It is worth mentioning that the number of contrasting task-specific functionalities (urea moieties, amine groups and OMSs) consequently doubles in the overall structure, which might be advantageous for the selective adsorption and transformation of CO2 molecules. The disordered guest molecules were determined using the combined results of thermogravimetric analysis (TGA), FT-IR, PLATON calculations and elemental analysis, which support the molecular formula [Cd3(TCA)2L(H2O)2]·2DMA·11H2O (for calculations, see ESI†). The powder X-ray diffraction (PXRD) pattern of the as-synthesized MOF perfectly matches the simulated one (Fig. 2a), attesting to high crystallinity as well as phase purity. The crystalline material was dipped in various solvents (methanol, chloroform, acetonitrile, acetone and toluene) for a day, and the PXRD patterns were found to be consistent with that of the pristine MOF in all cases (Fig. 2a), affirming its solvent stability. Noticeably, the framework is capable of preserving its structural integrity in open-air-conditions and exposure to water (Fig. 2a). Presumably, the close-packed structure with a large aromatic cloud from the organic struts and strong interactions among layers and pillars in the interpenetrated net provides rigidity to this 3D MOF for hydrolytic stability. TGA data showed a two-step weight loss (Fig. S2, ESI†), with a first weight loss of 11.93% (calc. 11.97%) around 120 °C, corresponding to the low-boiling lattice guest (water). A second weight loss (9.34%, calc. 9.77%) up to 300 °C is attributed to the loss of the high-boiling guest DMA.
 | ||
| Fig. 2 (a) Powder X-ray diffraction (PXRD) patterns of the MOF as-synthesized, activated, simulated and after being subjected to several organic solvents, water and open air. (b) Elemental mapping of the MOF crystal, showing even distribution of elements (mix, Cd, O, N, and C) throughout the surface. XPS spectra acquired at a high resolution showing (c) Cd 3d and (d) C 1s. | ||
The FT-IR spectrum revealed a peak at 1640 cm−1 (>C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) due to the presence of DMA guest molecules (Fig. S3, ESI†), whereas the solvent water molecules gave rise to a broad peak around 3500 cm−1. The C–N stretch for the tertiary amine in the framework could be found at 1210 cm−1, and the N–H bend for the urea functionality appeared at 1610 cm−1. The peaks at 1384 and 1596 cm−1 represent the antisymmetric stretching of metal-bound carboxylates. Scanning electron microscopy (SEM) with energy dispersive X-ray analysis (EDX, Fig. 2b) indicated homogenous distribution of all the constituent elements of the MOF (C, N, O, and Cd), which was also observed from the X-ray photoelectron spectroscopy (XPS) results (Fig. S5a, ESI†). The high-resolution C 1s XPS (Fig. 2d) was fitted to peaks at binding energies (BEs) of 284.5, 285.63, and 287.7 eV, corresponding to CC, C–N, and C–O bonding, respectively.62 Two major spin–orbit orientations (Fig. 2c) corresponding to Cd 3d5/2 and Cd 3d3/2 were fitted at 405.54 eV and 412.27 eV, respectively.54 The O–Cd and O–C bonding in the deconvoluted O 1s spectrum (Fig. S5b, ESI†) were found at 530.3 eV and 531.9 eV, while the high-resolution N 1s (Fig. S5c, ESI†) exhibited two peaks at 399.59 and 400.62 eV, attributed to the tertiary nitrogen of the tripodal ligand and N–H binding in the N-donor linker, respectively.55,63
O stretching) due to the presence of DMA guest molecules (Fig. S3, ESI†), whereas the solvent water molecules gave rise to a broad peak around 3500 cm−1. The C–N stretch for the tertiary amine in the framework could be found at 1210 cm−1, and the N–H bend for the urea functionality appeared at 1610 cm−1. The peaks at 1384 and 1596 cm−1 represent the antisymmetric stretching of metal-bound carboxylates. Scanning electron microscopy (SEM) with energy dispersive X-ray analysis (EDX, Fig. 2b) indicated homogenous distribution of all the constituent elements of the MOF (C, N, O, and Cd), which was also observed from the X-ray photoelectron spectroscopy (XPS) results (Fig. S5a, ESI†). The high-resolution C 1s XPS (Fig. 2d) was fitted to peaks at binding energies (BEs) of 284.5, 285.63, and 287.7 eV, corresponding to CC, C–N, and C–O bonding, respectively.62 Two major spin–orbit orientations (Fig. 2c) corresponding to Cd 3d5/2 and Cd 3d3/2 were fitted at 405.54 eV and 412.27 eV, respectively.54 The O–Cd and O–C bonding in the deconvoluted O 1s spectrum (Fig. S5b, ESI†) were found at 530.3 eV and 531.9 eV, while the high-resolution N 1s (Fig. S5c, ESI†) exhibited two peaks at 399.59 and 400.62 eV, attributed to the tertiary nitrogen of the tripodal ligand and N–H binding in the N-donor linker, respectively.55,63
Porosity measurement and gas adsorption studies
Before proceeding to the projected applications, the framework was activated by soaking the finely ground crystals in acetone (for 3 days) and subsequent heating at 120 °C under vacuum for 6 h. The PXRD pattern of the activated MOF (Fig. 2a) showed similar peaks to that of the as-synthesized framework, indicating maintenance of structural integrity and crystallinity. Additionally, peaks corresponding to DMA molecules were absent in the FT-IR spectrum (Fig. S3, ESI†), and TGA revealed no weight loss up to 300 °C (Fig. S2, ESI†), indicating complete expulsion of the solvent molecules. Further, an SEM image of the activated crystal revealed an unaltered block-shaped morphology (Fig. S4, ESI†). The nitrogen adsorption isotherm of the activated MOF (Fig. S8a, ESI†) at 77 K indicated a maximum uptake of ∼149 cm3 g−1 (P/P0 = 1), which corresponds to a Brunauer–Emmett–Teller (BET) specific surface area of 503 m2 g−1. The pore size distribution (PSD) calculation, which was based on the non-local density functional theory (NLDFT) models, gave an average pore diameter of 1.022 nm (Fig. S8a, inset, ESI†), which is attributed to the two-fold-interpenetration-induced generation of small-sized micropores. Although several high-surface-area MOFs are available in the literature, the present value is still higher than that of some well-known frameworks like TMOF-1 (Table S4, entry 9, ESI†), MAC-4-OH (Table S4, entry 27, ESI†), {[Co(BDC)(L)·2H2O]·xG}(Table S4, entry 29, ESI†), Ni-MOF-1 (Table S4, entry 31, ESI†), CSMCRI-7 (Table S4, entry 32, ESI†) and CSMCRI-13 (Table S4, entry 33, ESI†). The presence of the OMS-, urea- and amine-decorated 1D channels, together with the high thermo-chemical stability and microporous nature of the MOF, prompted us to use it for sorption studies of various small gas molecules (CO2, N2, and CH4). The gas adsorption studies on the activated framework were performed at different temperatures at absolute pressures of up to 1.0 bar. As divulged in Fig. 3a and b, the framework begins to adsorb CO2 in the low-pressure region, and the maximum uptake at 273 K and 298 K reaches 53.06 cm3 g−1 (2.37 mmol g−1, 10.42 wt%) and 28.39 cm3 g−1 (1.27 mmol g−1, 5.57 wt%), respectively. It can be noted that the adsorption capacity is comparable and/or superior to that of other reported frameworks (Fig. 4c). For instance, the CO2 adsorption capacities of [Cd(bpydc)2(DMF)2·2DMF]n (Table S4,† entry 3), [Zn(bpydc)(DMF)·DMF]n (JMS-4) (entry 3), [CoII4(μ-OH2)4(MTB)2·(H2O)4]n·13nDMF·11nH2O (entry 5), {[Zn(SDB)(3,3′-L)0.5]·xG}n (entry 23), {[Zn2(SDB)2(4,4′-L)]·xG}n (entry 23), CSMCRI-7 (entry 32) and CSMCRI-8 (entry 32) are comparable or lower to the that of the present MOF. Further, the adsorption–desorption pathways at both temperatures exhibit minimal hysteresis with no apparent saturation at pressures up to 1.0 bar, which indicate the possibility of greater adsorption capacity in the high-pressure range. The adsorption isotherms for CH4 and N2 were also recorded (Fig. 3a and b) and showed uptake capacities of 14.70 cm3 g−1 and 0.52 cm3 g−1 at 273 K, and 4.53 cm3 g−1 and 0.19 cm3 g−1 at 298 K. Furthermore, the adsorption data for this MOF were investigated at elevated temperature. As depicted in Fig. 3c, the framework still exhibits a type-I isotherm with negligible hysteresis and an uptake of 20.63 cm3 g−1 (0.92 mmol g−1, 4.05 wt%) of CO2 at 313 K. Although the adsorption capacity was reduced as a result of the temperature effect, the value is still comparable to those in the handful of reports on CO2 adsorption in crystalline frameworks at 313 K.11,13,28,64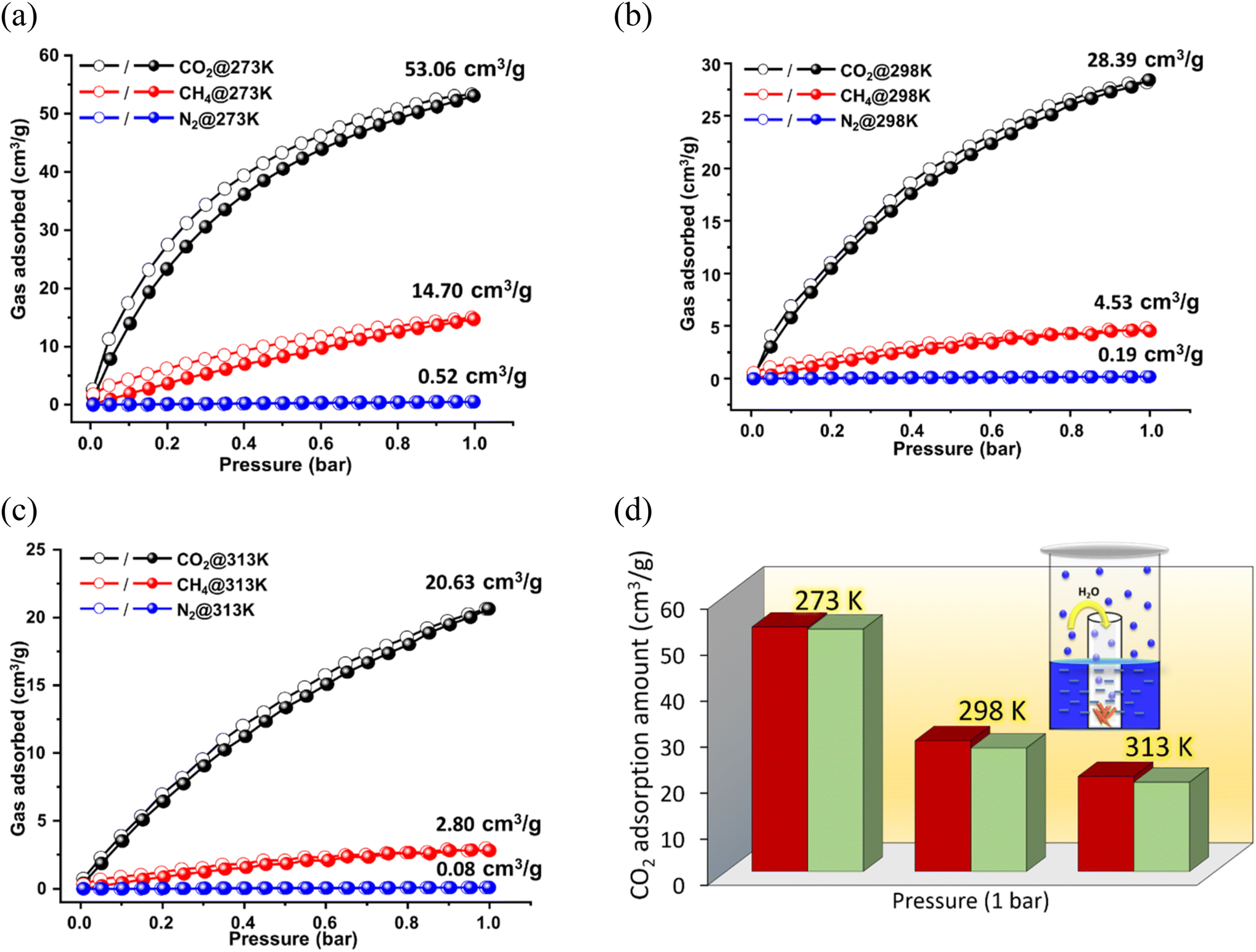 | ||
| Fig. 3 Gas adsorption isotherms for CO2, CH4, and N2 at (a) 273 K, (b) 298 K, and (c) 313 K. (d) Amount of CO2 adsorbed before (red) and after (green) exposure of the MOF to humidity at different temperatures (inset shows a schematic of water vapour exposure). | ||
 | ||
| Fig. 4 (a) Multicyclic CO2 adsorption at 273 K (black), 298 K (red), and 313 K (green). (b) Selectivity toward CO2 over other gases (CH4 and N2) at three temperatures. Comparison of (c) CO2 uptake capacity with CO2/N2 selectivity and (d) the Qst values of some benchmark MOFs as well as other MOFs. | ||
Nonetheless, further studies indicated negligible uptake of CH4 (2.80 cm3 g−1) and N2 (0.08 cm3 g−1) at this temperature. Taking advantage of the hydrolytic stability of the MOF, and in light of the importance of CO2 adsorption under humid conditions, we examined the CO2 adsorption of a water-vapor-exposed sample. For this purpose, the MOF was exposed to 75% relative humidity (RH) overnight (Fig. 3d), and thereafter degassed under mild conditions (50 °C, 2 h) to ensure that complete expulsion of the aqua molecules from the water-encapsulated structure did not ensue. The CO2 isotherms were separately recorded using these samples at 273, 298, and 313 K (Fig. S8c, ESI†), which showed a minor drop in the uptake capacities (Fig. 3d) with no alteration in the MOF integrity (Fig. S8d, ESI†). To check the reusability of this material, we tested the CO2 sorption capacity for five consecutive cycles at three different temperatures (degassing the same sample prior to each cycle). Pleasingly, all the experiments showed complete sorption recurrence with no hysteresis (Fig. 4a), indicating the great prospects of this material in retaining adsorption capacity at diverse analysis temperatures. To explore the effect of functionality on CO2 adsorption, we synthesized a TCA-based Cd(II) framework by replacing the urea-functionalized linker with 4,4′-bipyridine (bpy). The CO2 adsorption capacity for this un-functionalized framework was only 15.1 cm3 g−1 of uptake at 273 K (Fig. S8e, ESI†), which is four times less than that of the present MOF and clearly validates the essential role of the structural aspect and CO2-specific sites in this tri-functionalized framework.
Adsorption selectivity and computational insights
To further understand the potential of the MOF for gas separation, the selectivity for CO2 (S) over other gases was determined at three different temperatures using the ideal adsorbed solution theory (IAST).65 At the onset, all the adsorption isotherms were fitted using a single-site Langmuir (SSL) equation (see ESI† for details), and fitting parameters were computed.28,66 Subsequently, this SSL fitting equation provided the saturation capacities (qm) and affinity coefficients for the different gases at different temperatures. To our delight, the adsorption selectivity (Fig. 4b) for CO2/N2 was found to be 237 (273 K), 301 (298 K), and 380 (313 K), while that of CO2/CH4 was found to be 8 (273 K), 11 (298 K), and 18 (313 K). The systems tabulated in Table S4,†i.e., [NH2(CH3)2]2[Cd3(BTA)(BTC)2(H2O)]2 (entry 2), [Zn2(btm)2]·4H2O (entry 7), CSMCRI-16 (entry 10), and IITKGP-5 (entry 37) have a lesser value of selectivity towards CO2 adsorption. Despite the obvious reduction in the adsorption capacity, the selectivity values greatly increased upon elevating the temperature, which is unique in comparison to porous carbon and/or zeolitic materials. These values further substantiate that polar CO2 molecules (quadrupole moment: 13.4 × 10−40 C m2; polarizability: 26.3 × 10−25 cm3) interact strongly with the available interaction sites (OMS, urea, and amine functionalities) inside the MOF channels and are preferentially adsorbed.53,67 To confirm this verdict, we measured the isosteric heat of CO2 adsorption (Qst) using the Clausius–Clapeyron equation for the data at 273 and 298 K.68 The Qst value (Fig. S7, ESI†) at zero loading was found to be 32.2 kJ mol−1, and maintains a steady curve at higher coverage, even though the sites for maximum affinity become saturated. Comparative Qst data of recent as well as some benchmark MOFs possessing OMSs are presented in Fig. 4d and Table S4 of the ESI,† which show that the present value is ideal for physisorption-based CO2 adsorbents.69–71 However, the relatively superior uptake of CH4 in spite of its larger kinetic diameter (3.8 Å) compared to that of N2 (3.64 Å) can be explained based on the greater polarizability (26 × 10−25 cm3) of CH4 molecule compared to that of N2 (17.6 × 10−25 cm3). These influences improved interaction with the available π-electron cloud in the framework, causing it to be adsorbed more than N2.28,72,73To gain molecular-level understanding of the highly preferred uptake of CO2 over other gases in this MOF and determine the actual binding interactions of the adsorbed CO2 gas with three different CO2-philic sites (the bridgehead nitrogen in the carboxylate ligand, the free urea site of the linker, and the coordination-frustrated Cd(II) centre), computational studies employing a configurational bias Monte Carlo (CBMC) simulation were carried out. Analysis of the CO2 sorption was calculated using a framework consisting of (1 × 1 × 1) unit cells at three experimental temperatures (273 K, 298 K, 313 K) at pressures of up to 1.0 bar (Fig. 5). The patterns seen in the experiments are in exact accordance with the trends shown in the models of CO2 capture at these temperatures (Fig. 5a–c). A progressive reduction in the CO2 uptake amount in the unit cell is evidenced upon gradual increase in the temperature from 273 to 313 K.
 | ||
| Fig. 5 Snapshots of the location of CO2 molecules in the framework configurational bias derived from Monte Carlo simulation at (a) 273 K, (b) 298 K, and (c) 313 K at a pressure of 1 bar. Interaction between CO2 molecule and different functionalities, including the (d) OMS, (e) urea, and (f) amine sites of the framework with their interaction distances. | ||
It is clear that strong dipole–quadrupole and quadrupole–quadrupole interactions exist between incoming CO2 gas molecules and the functional adsorptive sites in the framework. To validate the CO2–framework interactions, we closely examined these structures. The unsaturated metal centers of the [Cd3(COO)6] units interact with the oxygen atoms of the CO2 molecule (Cd⋯O–C = 3.05 Å) (Fig. 5d). Moreover, the CO2 molecule exhibits interaction (Fig. 5e and f) with the free basic nitrogen of the TCA3− ligand (N⋯C–O = 3.6 Å) and the –NH group of urea (N–H⋯O–C = 3.01 Å) inside the channel. These binding distances are in good agreement with those of literature reports.13,74–76 Further insights regarding the adsorptive locations were obtained from the density distribution map (Fig. S9, ESI†), which demonstrates that the majority of the of CO2 molecules lie inside the available porous channels of the framework. These results are also in accordance with the number of CO2 molecules present per unit cell at 273 K (7), 298 K (4), and 313 K (3).
Solvent-free non-redox fixation of CO2
The presence of the exposed OMS as a Lewis acid site and the urea and amine moieties as Lewis basic sites, together with the appreciable CO2 adsorption, indicated that the framework might serve as a catalyst for CO2 conversion. Based on this conjecture, we employed this tri-functionalized MOF for effective CO2 cycloaddition reaction (Scheme 2) with the model substrate styrene oxide (SO). The optimization was performed under solvent-free conditions (Table 1), and provided excellent conversion of SO (99.9%) with 100% selectivity. Further experiments were conducted to ensure maximum conversion under mild conditions. However, the catalyst-free reaction did not lead to any styrene carbonate (SC) formation. Although the use of Cd(NO3)2·4H2O resulted in a conversion of nearly 17.2%, the use of the organic struts (H3TCA and L) showed no conversion (Table 1, entry 2). Subsequent screening studies were conducted to establish the need for the co-catalyst tetrabutylammonium bromide (TBAB), which is known to assist in the ring-opening step in this reaction.77 However, TBAB alone and/or mixed with 4,4′,4′′-tricarboxytriphenylamine provided little conversion. In contrast, the combination L/Bu4N+Br− resulted in better conversion due to the presence of the urea moiety as a potential interaction site (Table 1, entry 5).45,46 To our delight, the use of the activated MOF (0.25 mol%) and TBAB (0.2 mol%) afforded remarkable >99% conversion in the absence of solvent, which attests to the synergistic effect of the catalyst and co-catalyst in this reaction.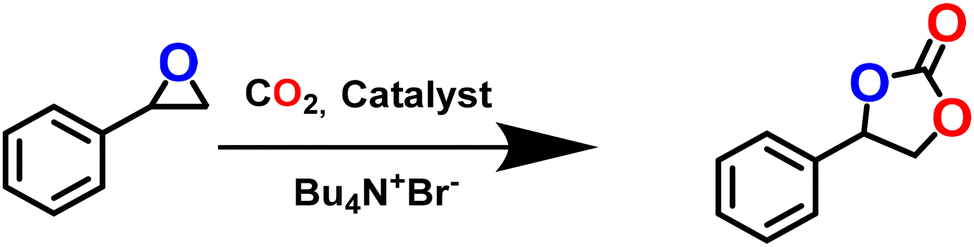 | ||
| Scheme 2 Cycloaddition reaction of CO2 and styrene oxide. | ||
| Entry | Catalyst/precursors | Temperature (°C) | Selectivity (%) | Conversion (%) | Time (h) |
|---|---|---|---|---|---|
| a Reaction conditions: epoxide: 26.46 mmol; CO2 pressure: 0.5 MPa. b CO2 pressure: 0.1 MPa. | |||||
| 1 | Blank | 70 | — | — | 10 |
| 2 | H3TCA/L | 70 | — | — | 10 |
| 3 | Cd(NO3)2·4H2O | 70 | 93 | 17.2 | 10 |
| 4 | Bu4N+Br− | 70 | 94 | 15.7 | 10 |
| 5 | L /Bu4N+Br− | 70 | 98 | 53.2 | 10 |
| 6 | H3TCA/Bu4N+Br− | 70 | 97 | 20.2 | 10 |
| 7 | Cd(NO3)2·4H2O/Bu4N+Br− | 70 | 97 | 22.5 | 10 |
| 8 | As-synthesised MOF | 70 | 97 | 23.4 | 10 |
| 9 | Activated MOF/Bu4N+I− | 70 | 100 | 70 | 10 |
| 10 | Activated MOF/Bu4N+Br− | 70 | 100 | 99.9 | 4 |
| 11 | Activated MOF/Bu4N+Br− | 50 | 100 | 99.9 | 4 |
| 12. | Activated MOF/Bu4N+Br− | RTb | 100 | 20 | 8 |
| 13. | Activated MOF/Bu4N+Br− | RTb | 100 | 38 | 24 |
| 14. | Activated MOF/KCl | 50 | 100 | 42 | 4 |
| 15. | Activated MOF/KBr | 50 | 100 | 73 | 4 |
| 16. | Activated MOF/KI | 50 | 100 | 71 | 4 |
All the parameters, including the amount of catalyst and co-catalyst, CO2 pressure, reaction temperature, and time and were subsequently optimized. Upon gradually increasing the catalyst loading (0.05 to 0.25 mol%), the conversion reached a maximum value of 99.9% (Fig. 6a), and further increase led to a slight reduction in the conversion due to alteration in the mass transfer process.78 Next, the outcome of cycloaddition with varying the co-catalyst amount (0.05 to 0.15 mol%) (Fig. 6b) was studied, which revealed a significant increase from 28 to 76%. Further increasing the amount of TBAB to 0.2 mol% led to 99.9% conversion. We also examined the performance of other co-catalysts, like TBAI (Bu4N+I−) and KX salts (X = Cl, Br, I) under the optimized conditions. The co-catalyst TBAI resulted in decreased conversion (70%) (Table 1, entry 9) in comparison to TBAB. In the case of the halide salts, we observed that the conversion of styrene oxide was lowest in the presence of KCl (42%) (Table 1, entry 14), while KBr (73%) and KI (71%) produced comparable conversion (Table 1, entries 15 and 16). These findings are in accordance with previous literature reports.39,79,80 Given that the MOF possesses optimum-sized cavities, we speculate that the larger sized I− experiences steric hindrance in entering the channels for synergic catalysis45,73 which explains the lower conversion of the product for TBAI and KI. This fact was additionally proved when bulkier epoxide substrates were considered (vide infra). Although reduction of the CO2 pressure from 1.0 MPa to 0.5 MPa did not make any difference (Fig. 6c), further lowering it to 0.3 MPa resulted in reduced conversion (65%), demonstrating the enrichment effect of CO2 in the reaction vessel towards the formation of the desired product. On the other hand, monitoring of the reaction at temperatures from 30 to 60 °C showed a gradual increase in the SC conversion (%) with temperature, and product formation remained constant beyond 50 °C (Fig. 6d). The time-dependent reaction analysis revealed maximum conversion within 4 h (Fig. S10, ESI†). Thus, the optimum cycloaddition conditions were set as: 0.5 MPa CO2 pressure, 50 °C temperature, and 4 h duration, with no formation of diols and dimers as side products.45,73,81 A comparison with literature data revealed that present non-redox CO2 fixation performance is among the best reported for MOFs (Table S5, ESI†) with milder catalytic conditions. Moreover, the CO2 conversion can reach equilibrium within a rather short time compared to other materials, indicating that this MOF can efficiently accelerate the CO2 conversion process in a less-energy-consuming and time-saving manner. This effective CO2 conversion performance may be attributed to the modification of the microenvironment in the 1D channels via the introduction of multiple task-specific sites, which include two different functional organic groups and an OMS. Particularly, the urea group and OMS should be the key factors to actively interact and activate SO, as is detailed in the mechanistic validation below.46,82,83 The catalytic performances of the various systems presented in Table S5† clearly indicate that this MOF can perform the non-redox transformation of CO2 into the corresponding cyclic carbonates under relatively milder conditions than Mg-MOF-74 (entry 1), NH2-MIL-101(Al) (entry 2), Ni@ZrOF (entry 14), CSMCRI-13 (entry 15) and DUT-52(Zr) (entry 17).
 | ||
| Fig. 6 Effects of various reaction parameters on the conversion and selectivity of styrene oxide: (a) dosage of catalyst, (b) amount of co-catalyst, (c) CO2 pressure, and (d) temperature. | ||
The catalyst activity under the optimized conditions was further extended by considering various other epoxides. As detailed in Table 2, complete maintenance of catalytic efficacy was observed for a wide range of aromatic and aliphatic substrates (Fig. S11–S15, ESI†). Nevertheless, cyclohexene oxide exhibits low conversion due to the steric crowding, which deters the SN2 attack of the Br− ion on the intermediate species (Scheme 3).84 From the results, it was further noticed that the conversion decreases with increasing alkyl chain length of the epoxides (Table 2, entry 6). This fact can be attributed to limited diffusion of larger epoxides in the narrow voids of the microporous framework, and alternately, indicates size-selectivity. To check the reusability performance of the catalyst over multiple cycles, we recovered the MOF by centrifugation, washed it with acetone and then reused the powdered material after drying in vacuum at 100 °C. The spent catalyst showed almost constant SC conversion over five consecutive cycles (Fig. 7a). The slight reduction in the conversion might stem from catalyst loss during the recovery process. The PXRD pattern of the recovered MOF shows precise agreement with that of the unused catalyst (Fig. S16, ESI†). The FT-IR (Fig. S17, ESI†) and FE-SEM analyses (Fig. S19a, ESI†) of the MOF after multiple runs show no alteration in its structural and/or morphological properties, attesting to its great potential for repeated cycloaddition. Also, elemental mapping (SEM-EDX) shows a uniform distribution of elements in the selected area of the MOF crystal after catalysis (Fig. S19b, ESI†). To check for any leaching of metal ions during CO2 fixation, the MOF was separated after reaction, and the filtrate was dried and analyzed using inductively coupled plasma mass spectrometry (ICP-MS), which showed a negligible Cd(II) ion concentration (0.003 ppm). Furthermore, the XPS spectra (Fig. S18e, ESI†) of the recovered MOF was found to be similar to that of the fresh catalyst. The N2 adsorption isotherm at 77 K did not reveal any significant differences (Fig. S20, ESI†), corroborating the maintenance of the porous structure. In a nutshell, this MOF exemplifies an efficient heterogeneous catalyst for non-redox conversion of CO2 under mild conditions.
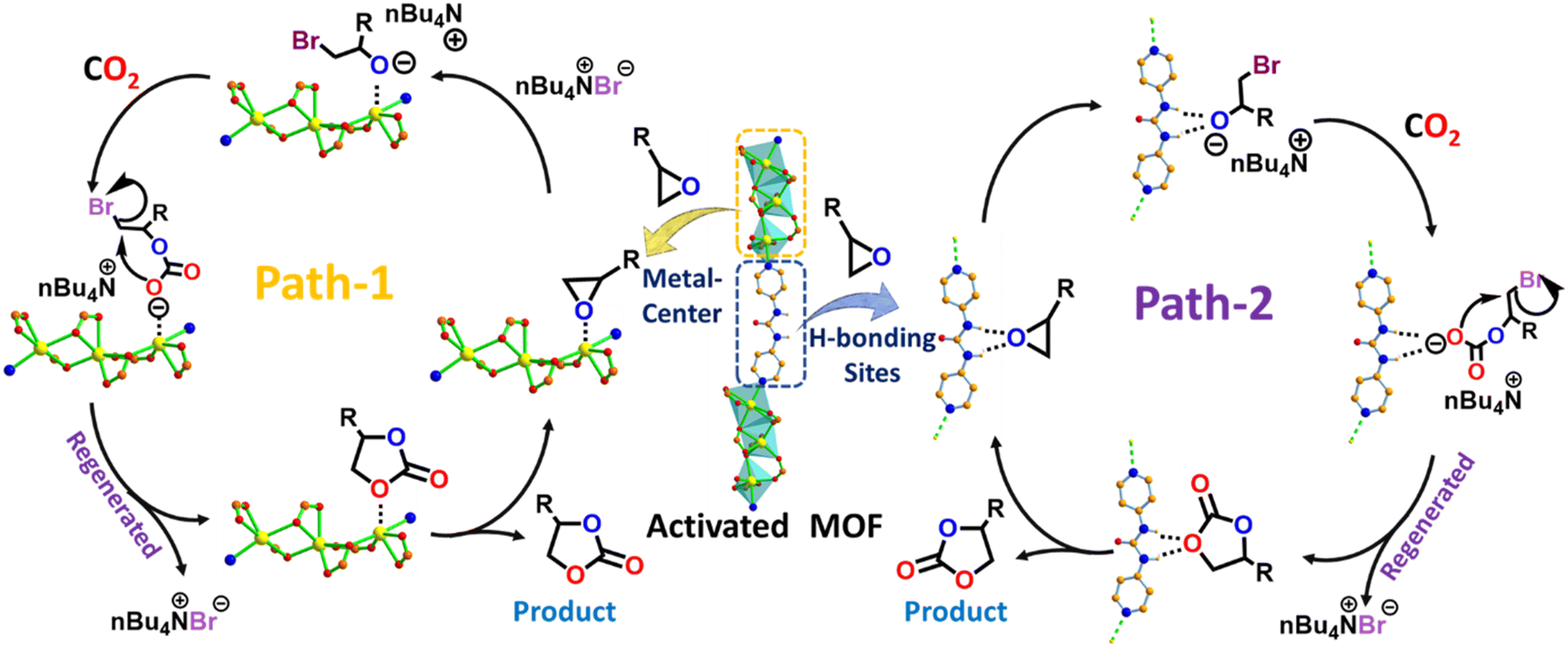 | ||
| Scheme 3 Proposed reaction pathway for CO2 cycloaddition with epoxide using the OMS and functional organic site of guest-free MOF in the presence of the co-catalyst. | ||
 | ||
| Fig. 7 (a) Multicycle catalytic performance of the activated framework towards the cycloaddition reaction. (b) Change in the luminescence intensity of the activated framework upon incremental addition of styrene oxide. | ||
| Entry | Reactant | Model structure | Product | Model structure | Yield (%) | TON |
|---|---|---|---|---|---|---|
| a Reaction conditions: epoxide: 26.46 mmol; MOF: 0.25 mol%; Bu4N+Br−: 0.2 mol%; CO2 pressure: 0.5 MPa; temp: 50 °C; time: 4 h; selectivity >99%. TON = (moles of product/moles of catalyst). | ||||||
| 1 |

|

|

|

|
99.9 | 498 |
| 2 |

|

|

|

|
99.9 | 498 |
| 3 |

|
|

|

|
94.5 | 471 |
| 4 |
|

|
|

|
97.1 | 484 |
| 5 |
|
|
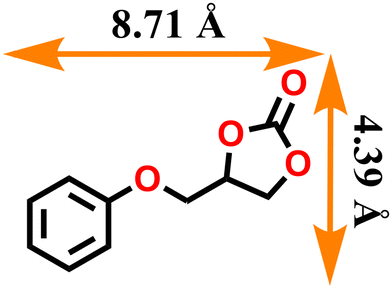
|

|
99.9 | 498 |
| 6 |

|
|
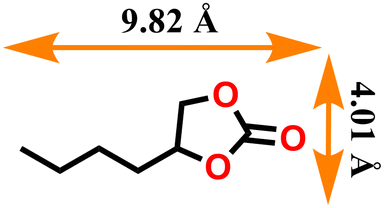
|
|
68 | 339 |
| 7 |
|

|

|

|
18 | 90 |
Mechanistic validation of CO2 cycloaddition reaction
Given that this MOF contains multiple potential substrate interaction sites, as confirmed during the CO2 adsorption studies, we conducted a series of control experiments to validate the exact catalytic pathway of the cycloaddition reaction. At the onset, the role of classical OMS was probed by comparing the activities of the as-synthesized and activated MOFs in the presence of TBAB under the optimized conditions. As indicated in Table 1 (entry 8), the guest-loaded MOF provided hardly any SO conversion, which was ascribed to the unavailability of the OMSs for substrate activation. Based on this observation, coupled with prior literature reports, we propose the OMS-mediated tentative mechanism shown as Path-1 in Scheme 3. Here, the cycloaddition reaction is initiated by the activation of the epoxide through interaction from the Lewis-acidic Cd(II) centre. Subsequently, the Br− of TBAB participates in a nucleophilic attack on the less-crowded carbon atom of the epoxide, which leads to ring opening.39,55,79,85 Finally, an intramolecular ring closure reaction produces the expected cyclic carbonate with the regeneration of the MOF catalyst, which takes part in activation of another epoxide molecule.On the other hand, urea group in the framework can also principally act as an efficient activation site for the epoxide via hydrogen-bonding interactions.86,87 Preliminarily, CO2 fixation using the linker in the presence of a co-catalyst revealed 53.2% conversion for the L/TBAB combination (Table 1, entry 5). This experiment indicates that reaction must have proceeded via using the urea site of the organic strut. In order to more explicitly rationalize the preferred interaction site between the amine and the urea functionality, a comparison of the catalytic activity of an isoskeletal MOF that lacks the urea moiety was carried out. To this end, we used one of our previous systems, which forms an identical 2-fold interpenetrated network, containing the TCA3− ligand and aqua-molecule-bound [Cd3(COO)6] secondary building unit, but without a urea moiety in the linker (Fig. S1, ESI†). This MOF exhibited the following optimized conditions for CO2 cycloaddition: 1.0 MPa CO2 pressure, 65 °C temperature, and 6 h duration, and a higher loading of the catalyst (0.35 mol%) and co-catalyst (0.36 mol%).45 This fact clearly demonstrates that urea functionalization has a positive impact in effectively catalyzing CO2 with epoxide under milder conditions in the present system. The role of the urea moiety in substrate activation was further proven by fluorescence modification of the MOF in the presence of a model epoxide. First, 2 mg of finely ground as-synthesized MOF powder was well-dispersed (in 2 mL toluene) in a cuvette. Freshly prepared SO solution (1 mM in toluene) was then incrementally added to the cuvette under stirring conditions. To our delight, the luminescence intensity of the MOF considerably decreased (Fig. 7b), which signifies strong host–guest interaction. In sharp contrast, the isoskeletal MOF without the urea unit did not show much influence on the spectral intensity (Fig. S21b, ESI†), which instead demonstrates the obvious interaction and activation of the epoxide molecule by the urea functionality in the present system through spatially controlled and pore-wall-aligned H-bonding sites in the present framework.46,88 Building on these, we propose the urea-group-triggered CO2 cycloaddition shown as Path-2 in Scheme 3. Here, the epoxide molecule is activated through formation of two-point H-bonding interactions. Subsequently, the bromide ion (Br−) of the co-catalyst attacks in SN2 fashion at the less-hindered carbon atom of the epoxide, resulting in ring opening. After coupling between CO2 and the ring-opened epoxy species, the thus-formed alkyl carbonate anion subsequently attacks the electrophilic β-carbon atom of the ring-opened epoxide, leading to ring closure via elimination of Br−.46 Although the cycloaddition reaction using OMS-based Lewis-acidic sites is well explored, the present version through H-bond-donating sites in the framework is hitherto unprecedented. To the best of our knowledge, this work represents a unique demonstration of CO2 cycloaddition, in which the mutual participation of contrasting functionalities in the MOF facilitates substrate interaction and activation, which in turn paves catalysis of the reaction under less-harsh conditions.
Conclusions
In conclusion, we successfully introduced a Lewis acid centre, H-bond operative site, and free amine moiety inside the microporous channels of a mixed-ligand Cd(II) MOF. The two-fold interpenetration of this three-dimensional (3D) pillar-bilayer framework leads to the creation of suitably sized unidirectional porous channels and doubles the number of active sites. Benefiting from these outstanding structural attributes and pore functionalization, the activated MOF shows high CO2 adsorption at 273, 298 and 313 K, with the adsorption capacity at every temperature remaining unaltered for up to five uptake–release cycles. Interestingly, the selectivity for CO2 over N2 and CH4 displays a remarkable improvement upon gradually increasing the temperature, while variable-temperature CO2 adsorption under humid conditions revealed remarkable sorption recurrences, making this MOF a potential material for practical CO2 sequestration. A configurational bias Monte Carlo simulation gave molecular-level insights into the sequential insertion of CO2 molecules inside the multi-functionalized porous channels and portrays structure–property synergy through depicting the individual roles of the CO2-philic sites. In situ grafted Lewis-basic and H-bond operative sites, together with activation-generated open metal centres, promote the performance of the MOF as an excellent heterogeneous catalyst in the solvent-free fixation of CO2 to cyclic carbonates with high yield, broad substrate scope and multicyclic reusability. In addition to the mild catalytic conditions and conventional catalyst–co-catalyst synergism, the mutual participation of the contrasting functionalities towards substrate interaction and activation was validated via the fluorescence modification of the framework in the presence of epoxide and comparison with the catalytic activity of an iso-structural MOF, which confirm the unique organic-functionality-driven mechanistic route to CO2 valorization. Given that the adsorption and chemical fixation of CO2 are highly important global agendas for environmental remediation, this one-of-a-kind tri-functional MOF satisfies important prerequisites for real-time CO2 capture and demonstrates unconventional organic-functionality-actuated CO2 fixation, which promises to unlock new avenues to resolve modern-day issues of concern.Experimental section
Detailed description of the synthesis of the linkers and framework has been provided in the ESI.†Author contributions
Manpreet Singh: conceptualization, methodology, formal analysis, data curation, writing-original draft, writing-review and editing. Sonal Rajput: methodology, formal analysis, validation, data curation, writing-original draft. Partha Pratim Mondal: formal analysis, validation, data curation, writing-review and editing. Subhadip Neogi: funding acquisition, supervision, writing-review and editing.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
M. S. acknowledges UGC, Delhi, for providing a senior research fellowship. P. P. M. acknowledges the DST-SERB (grant no. CRG/2021/002529). S. R. acknowledges the CSIR (grant no. MLP-0054). S. N. acknowledges the DST-SERB (grant no. CRG/2021/002529), DST (DST/TM/EWO/MI/CCUS/21(C1), and CSIR (grant no. MLP-0054). The analytical support from AESD & CIF (CSMCRI), and crystallographic help from Dr E. Suresh are gratefully acknowledged. We acknowledge Mr Nilanjan Seal for valuable suggestions. CSMCRI communication no. 161/2022.References
- A. S. Palakkal and R. S. Pillai, Evaluating the Performance of Cr-Soc-MOF Super-Adsorbents for CO2 Capture from Flue Gas under Humid Condition through Molecular Simulation, Sep. Purif. Technol., 2022, 295, 121298 CrossRef CAS
.
- A. S. Palakkal and R. S. Pillai, Fluorinated Metal Organic Frameworks, MFFIVE-Ni-L (M=Fe/Al, L=pyr), with Coordinatively Unsaturated Metal Site for CO2 Separation from Flue Gas in the Presence of Humidity by Computational Methods, Dalton Trans., 2021, 50, 466–471 RSC
- M. J. Burke and J. C. Stephens, Political Power and Renewable Energy Futures: A Critical Review, Energy Res. Soc. Sci., 2018, 35, 78–93 CrossRef
- X. Yao, K. E. Cordova and Y.-B. Zhang, Flexible Metal–Organic Frameworks as CO2 Adsorbents en Route to Energy-Efficient Carbon Capture, Small Struct., 2022, 3, 2100209 CrossRef CAS
- M. Román, Carbon Capture and Storage in Developing Countries: A Comparison of Brazil, South africa and India, Glob. Environ. Change, 2011, 21, 391–401 CrossRef
- T. Wilberforce, A. G. Olabi, E. T. Sayed, K. Elsaid and M. A. Abdelkareem, Progress in carbon capture technologies, Sci. Total Environ., 2021, 761, 143203 CrossRef CAS PubMed
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. He and S.-E. Park, Industrial Carbon Dioxide Capture and Utilization: State of the Art and Future Challenges, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC
- G. T. Rochelle, Amine Scrubbing for CO2 Capture, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed
- M. A. Habib, M. Nemitallah and R. Ben-Mansour, Recent Development in Oxy-Combustion Technology and its Applications to Gas Turbine Combustors and ITM Reactors, Energy Fuels, 2013, 27, 2–19 CrossRef CAS
- M. Singh, N. Borkhatariya, P. Pramanik, S. Dutta, S. K. Ghosh, P. Maiti, S. Neogi and S. Maiti, Microporous Carbon Derived from Cotton Stalk Crop-Residue Across Diverse Geographical Locations as Efficient and Regenerable CO2 Adsorbent with Selectivity, J. CO2 Util., 2022, 60, 101975 CrossRef CAS
- H. A. Patel, S. Hyun Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Unprecedented High-Temperature CO2 Selectivity in N2-Phobic Nanoporous Covalent Organic Polymers, Nat. Commun., 2013, 4, 1357 CrossRef PubMed
- A. Helal, M. Fettouhi, M. E. Arafat, M. Y. Khan and M. A. Sanhoob, Nickel based Metal-Organic Framework as Catalyst for Chemical Fixation of CO2 in Oxazolidinone Synthesis, J. CO2 Util., 2021, 50, 101603 CrossRef CAS
- P. Das and S. K. Mandal, Unprecedented High Temperature CO2 Selectivity and Effective Chemical Fixation by a Copper-Based Undulated Metal–Organic Framework, ACS Appl. Mater. Interfaces, 2020, 12, 37137–37146 CrossRef CAS PubMed
- J. Qiao, X. Liu, X. Liu, X. Liu, L. Zhang and Y. Liu, Two Urea-Functionalized pcu Metal–Organic Frameworks based on A Pillared-Layer Strategy for Gas Adsorption and Separation, Inorg. Chem. Front., 2020, 7, 3500–3508 RSC
- M. Singh and S. Neogi, Largely Entangled Diamondoid Framework with High-Density Urea and Divergent Metal Nodes for Selective Scavenging of CO2 and Molecular Dimension-Mediated Size-Exclusive H-Bond Donor Catalysis, Inorg. Chem., 2023, 62, 871–884 CrossRef CAS PubMed
- A. S. Palakkal and R. S. Pillai, Tuning the Ultra-Micropore Size of Fluorinated MOFs (M′F6-Ni-L) for CO2 Capture from Flue Gases by Advanced Computational Methods, J. Phys. Chem. C, 2020, 124, 16975–16989 CrossRef CAS
- H.-M. Wen, C. Liao, L. Li, A. Alsalme, Z. Alothman, R. Krishna, H. Wu, W. Zhou, J. Hu and B. Chen, A Metal–Organic Framework with Suitable Pore Size and Dual Functionalities for Highly Efficient Post-Combustion CO2 Capture, J. Mater. Chem. A, 2019, 7, 3128–3134 RSC
- A. Pal, S. Chand, D. G. Madden, D. Franz, L. Ritter, B. Space, T. Curtin, S. Chand Pal and M. C. Das, Immobilization of a Polar Sulfone Moiety onto the Pore Surface of a Humid-Stable MOF for Highly Efficient CO2 Separation under Dry and Wet Environments through Direct CO2−Sulfone Interactions, ACS Appl. Mater. Interfaces, 2020, 12, 41177–41184 CrossRef CAS PubMed
- K. A. Adegoke, K. O. Oyedotun, J. O. Ighalo, J. F. Amaku, C. Olisah, A. O. Adeola, K. O. Iwuozor, K. G. Akpomie and J. Conradie, Cellulose Derivatives and Cellulose-Metal-Organic Frameworks for CO2 Adsorption and Separation, J. CO2 Util., 2022, 64, 102163 CrossRef CAS
- E. González-Zamora and I. A. Ibarra, CO2 Capture under Humid Conditions in Metal–Organic Frameworks, Mater. Chem. Front., 2017, 1, 1471–1484 RSC
- A. S. Palakkal and R. S. Pillai, Discovering the Impact of Targeted Defects in SP-MOF for CO2 Capture from Flue Gas in Presence of Humidity through Computational Modelling, J. CO2 Util., 2022, 66, 102264 CrossRef CAS
- J. H. Choe, H. Kim and C. S. Hong, MOF-74 Type Variants for CO2 Capture, Mater. Chem. Front., 2021, 5, 5172–5185 RSC
- L. Feng, K.-Y. Wang, G. S. Day, M. R. Ryder and H.-C. Zhou, Destruction of Metal–Organic Frameworks: Positive And Negative Aspects of Stability and Lability, Chem. Rev., 2020, 120, 13087–13133 CrossRef CAS PubMed
- S. C. Pal, D. Mukherjee and M. C. Das, pH-stable Luminescent Metal–Organic Frameworks for the Selective Detection of Aqueous-Phase FeIII and CrVI ions, Inorg. Chem., 2022, 61, 12396–12405 CrossRef CAS PubMed
- R. Sahoo, S. Mondal, D. Mukherjee and M. C. Das, Metal–Organic Frameworks for CO2 Separation from Flue and Biogas Mixtures, Adv. Funct. Mater., 2022, 2207197 CrossRef CAS
- J.-L. K. Gbe, K. Ravi, M. Singh, S. Neogi, M. Grafouté and A. V. Biradar, Hierarchical Porous Nitrogen-doped Carbon Supported MgO as An Excellent Composite for CO2 Capture at Atmospheric Pressure and Conversion to Value-added Products, J. CO2 Util., 2022, 65, 102222 CrossRef CAS
- J. H. Choe, D. W. Kang, M. Kang, H. Kim, J. R. Park, D. W. Kim and C. S. Hong, Revealing An Unusual Temperature-Dependent CO2 Adsorption Trend and Selective CO2 Uptake Over Water Vapors in a Polyamine-Appended Metal–Organic Framework, Mater. Chem. Front., 2019, 3, 2759–2767 RSC
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Evaluating Metal–Organic Frameworks for Post-Combustion Carbon Dioxide Capture via Temperature Swing Adsorption, Energy Environ. Sci., 2011, 4, 3030–3040 RSC
- H. He, Q. Sun, W. Gao, J. A. Perman, F. Sun, G. Zhu, B. Aguila, K. Forrest, B. Space and S. Ma, A Stable Metal–Organic Framework featuring A Local Buffer Environment for Carbon Dioxide Fixation, Angew. Chem., Int. Ed., 2018, 57, 4657–4662 CrossRef CAS PubMed
- H. J. Jun, D. K. Yoo and S. H. Jhung, Metal-Organic Framework (MOF-808) Functionalized with Ethyleneamines: Selective Adsorbent to Capture CO2 Under Low Pressure, J. CO2 Util., 2022, 58, 101932 CrossRef CAS
- X. Y. Li, L. N. Ma, Y. Liu, L. Hou, Y. Y. Wang and Z. H. Zhu, Honeycomb metal-organic framework with lewis acidic and basic bifunctional sites: selective adsorption and CO2 catalytic fixation, ACS Appl. Mater. Interfaces, 2018, 10, 10965–10973 CrossRef CAS PubMed
- X.-L. Yang, Y.-T. Yan, W.-J. Wang, Z.-Z. Hao, W.-Y. Zhang, W. Huang and Y.-Y. Wang, A 2-Fold Interpenetrated Nitrogen-Rich Metal–Organic Framework: Dye Adsorption and CO2 Capture and Conversion, Inorg. Chem., 2021, 60, 3156–3164 CrossRef CAS PubMed
- Y.-Z. Li, G.-D. Wang, H.-Y. Yang, L. Hou, Y.-Y. Wang and Z. Zhu, Novel cage-like MOF for gas separation, CO2 conversion and selective adsorption of an organic dye, Inorg. Chem. Front., 2020, 7, 746–755 RSC
- Y. Yuan, J. Li, X. Sun, G. Li, Y. Liu, G. Verma and S. Ma, Indium–Organic Frameworks Based on Dual Secondary Building Units Featuring Halogen-Decorated Channels for Highly Effective CO2 Fixation, Chem. Mater., 2019, 31, 1084–1091 CrossRef CAS
- G. Singh and C. M. Nagaraja, Highly efficient metal/solvent-free chemical fixation of CO2 at atmospheric pressure conditions using functionalized porous covalent organic frameworks, J. CO2 Util., 2021, 53, 101716 CrossRef CAS
- L. Shang, X.-L. Chen, L. Liu, M. Cai, R.-K. Yan, H.-L. Cui, H. Yang and J.-J. Wang, Catalytic Performance of MOFs Containing Trinuclear Lanthanides Clusters in the Cycladdition Reaction Of CO2 And Epoxide, J. CO2 Util., 2022, 65, 102235 CrossRef CAS
- S. Subramanian, J. Oppenheim, D. Kim, T. S. Nguyen, W. M. H. Silo, B. Kim, W. A. Goddard and C. T. Yavuz, Catalytic Non-Redox Carbon Dioxide Fixation in Cyclic Carbonates, Chem, 2019, 5, 3232–3242 CAS
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Direct Conversion of CO2 into Liquid Fuels with High Selectivity over a Bifunctional Catalyst, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS PubMed
- B. Parmar, P. Patel, R. S. Pillai, R. K. Tak, R. I. Kureshy, N. U. Khan and E. Suresh, Cycloaddition of CO2 with an Epoxide-bearing Oxindole Scaffold by a Metal-Organic Framework-Based Heterogeneous Catalyst under Ambient Conditions, Inorg. Chem., 2019, 58, 10084–10096 CrossRef CAS PubMed
- U. Patel, P. Patel, B. Parmar, A. Dadhania and E. Suresh, Synergy of Dual Functional Sites for Conversion of CO2 in a Cycloaddition Reaction under Solvent-Free Conditions by a Zn(II)-based Coordination Network with a Ladder Motif, Cryst. Growth Des., 2021, 21, 1833–1842 CrossRef CAS
- J. Liang, Y. B. Huang and R. Cao, Metal-Organic Frameworks and Porous Organic Polymers for Sustainable Fixation of Carbon Dioxide into Cyclic Carbonates, Coord. Chem. Rev., 2019, 378, 32–65 CrossRef CAS
- A. K. Gupta, N. Guha, S. Krishnan, P. Mathur and D. K. Rai, A Three-Dimensional Cu(II)-MOF with Lewis Acid−Base Dual Functional Sites for Chemical Fixation of CO2 via Cyclic Carbonate Synthesis, J. CO2 Util., 2020, 39, 101173 CrossRef CAS
- D. Kim, Y. Moon, D. Ji, H. Kim and D. Cho, Metal-Containing Ionic Liquids As Synergistic Catalysts for the Cycloaddition Of CO2: A Density Functional Theory and Response Surface Methodology Corroborated Study, ACS Sustainable Chem. Eng., 2016, 4, 4591–4600 CrossRef CAS
- A. Decortes, M. Martínez Belmonte, J. Benet-Buchholz and A. W. Kleij, Efficient Carbonate Synthesis under Mild Conditions through Cycloaddition of Carbon Dioxide to Oxiranes using a Zn(salphen) Catalyst, Chem. Commun., 2010, 46, 4580–4582 RSC
- N. Seal, M. Singh, S. Das, R. Goswami, B. Pathak and S. Neogi, Dual-Functionalization Actuated Trimodal Attribute in an Ultra-Robust MOF: Exceptionally Selective Capture and Effectual Fixation Of CO2 with Fast-Responsive, Nanomolar Detection of Assorted Organo-Contaminants in Water, Mater. Chem. Front., 2021, 5, 979–994 RSC
- L.-Q. Wei and B.-H. Ye, Efficient Conversion of CO2 via Grafting Urea Group into a [Cu2(COO)4]-based Metal–Organic Framework with Hierarchical Porosity, Inorg. Chem., 2019, 58, 4385–4393 CrossRef CAS PubMed
- R. Abazari, S. Sanati, A. Morsali, A. M. Kirillov, A. M. Z. Slawin and C. L. Carpenter-Warren, Simultaneous Presence of Open Metal Sites and Amine Groups on A 3d Dy(III)-Metal–Organic Framework Catalyst for Mild and Solvent-Free Conversion Of CO2 fo Cyclic Carbonates, Inorg. Chem., 2021, 60, 2056–2067 CrossRef CAS PubMed
- D. H. Le, R. P. Loughan, A. Gładysiak, N. Rampal, I. A. Brooks, A.-H. A. Park, D. Fairen-Jimenez and K. C. Stylianou, Lanthanide Metal–Organic Frameworks for the Fixation of CO2 under Aqueous-Rich and Mixed-Gas Conditions, J. Mater. Chem. A, 2022, 10, 1442–1450 RSC
- L. Gao, Y. Zhou, Z. Li, J. He, Y. Qu, X. Zou, B. Liu, C. Ma, J. Sun and K. Guo, Nicotinamide Onium Halide Bidentate Hybrid H–bond Donor Organocatalyst for CO2 Fixation, J. CO2 Util., 2022, 65, 102196 CrossRef CAS
- M. Saghian, S. Dehghanpour and M. Sharbatdaran, Amine-Functionalized Frameworks as Highly Actives Catalysts for Chemical Fixation of CO2 under Solvent and Co-catalyst free Conditions, J. CO2 Util., 2020, 41, 101253 CrossRef CAS
- M. Jia, J. Li, J. Gu, L. Zhang and Y. Liu, Inquiry for the Multifunctional Design of Metal–Organic Frameworks: In Situ Equipping Additional Open Metal Sites (OMSs) inducing High CO2 Capture/Conversion Abilities, Mater. Chem. Front., 2021, 5, 1398–1404 RSC
- X. Zhang, X. Wang, W. Fan, Y. Wang, X. Wang, K. Zhang and D. Sun, A Multifunctional Zr-MOF for the Rapid Removal of Cr2O72−, Efficient Gas Adsorption/Separation, and Catalytic Performance, Mater. Chem. Front., 2020, 4, 1150–1157 RSC
- M. Singh and S. Neogi, Selective and Multicyclic CO2 Adsorption with Visible Light-Driven Photodegradation of Organic Dyes in a Robust Metal–Organic Framework Embracing Heteroatom-Affixed Pores, Inorg. Chem., 2022, 61, 10731–10742 CrossRef CAS PubMed
- M. Singh and S. Neogi, Urea-Engineering Mediated Hydrogen-Bond Donating Friedel–Crafts Alkylation of Indoles and Nitroalkenes in a Dual-Functionalized Microporous Metal–Organic Framework with High Recyclability and Pore-Fitting-Induced Size-Selectivity, Inorg. Chem. Front., 2022, 9, 1897–1911 RSC
- N. Seal, K. Karthick, M. Singh, S. Kundu and S. Neogi, Mixed-Ligand-Devised Anionic MOF with Divergent Open Co(II)-Nodes as Chemo-Resistant, Bi-Functional Material for Electrochemical Water Oxidation and Mild-Condition Tandem CO2 Fixation, Chem. Eng. J., 2022, 429, 132301 CrossRef CAS
- M. Singh, G. Kumar and S. Neogi, Devising Mixed-Ligand based robust Cd(II)-Framework from Bi-Functional Ligand for Fast Responsive Luminescent Detection of Fe3+ And Cr(VI) Oxo-Anions in Water with High Selectivity and Recyclability, Front. Chem., 2021, 9, 651866 CrossRef CAS PubMed
- N. Seal and S. Neogi, Lewis Acid-Base Integrated Robust Metal-Organic Framework and Reconfigurable Composite for Solvent-Free Biginelli Condensation and Tandem Catalysis with Size Selectivity, Mater. Today Chem., 2022, 26, 101064 CrossRef CAS
- R. Goswami, B. D. Bankar, S. Rajput, N. Seal, R. S. Pillai, A. V. Biradar and S. Neogi, In situ fabricated MOF–Cellulose Composite as An Advanced ROS Deactivator-Convertor: Fluoroswitchable Bi-phasic tweezers for Free Chlorine Detoxification and Size-Exclusive Catalytic Insertion of Aqueous H2O2, J. Mater. Chem. A, 2022, 10, 4316–4332 RSC
- R. Goswami, K. Karthick, S. Das, S. Rajput, N. Seal, B. Pathak, S. Kundu and S. Neogi, Brønsted Acid-Functionalized Ionic Co(II) Framework: A Tailored Vessel for Electrocatalytic Oxygen Evolution and Size-Exclusive Optical Speciation of Biothiols, ACS Appl. Mater. Interfaces, 2022, 14, 29773–29787 CrossRef CAS PubMed
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Applied Topological Analysis of Crystal Structures with the Program Package TOPOSPRO, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS
- A. Spek, Single-Crystal Structure Validation with the Program Platon, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS
- C.-A. Tao, J. Wang, S. Qin, Y. Lv, Y. Long, H. Zhu and Z. Jiang, Fabrication of pH-Sensitive Graphene Oxide–drug Supramolecular Hydrogels as Controlled Release Systems, J. Mater. Chem., 2012, 22, 24856–24861 RSC
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Cobalt-Modified Covalent Organic Framework as a Robust Water Oxidation Electrocatalyst, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS
- F. Chen, J. Wang, L. Guo, X. Huang, Z. Zhang, Q. Yang, Y. Yang, Q. Ren and Z. Bao, Carbon Dioxide Capture in Gallate-based Metal-Organic Frameworks, Sep. Purif. Technol., 2022, 292, 121031 CrossRef CAS
- A. L. Myers and J. M. Prausnitz, Thermodynamics of mixed-gas adsorption, AIChE J., 1965, 11, 121 CrossRef CAS
- H. R. Mahdipoor, R. Halladj, E. Ganji Babakhani, S. Amjad-Iranagh and J. Sadeghzadeh Ahari, Synthesis, Characterization, and CO2 Adsorption Properties of Metal Organic Framework Fe-BDC, RSC Adv., 2021, 11, 5192–5203 RSC
- J. Wang, Y. Zhang, Y. Su, X. Liu, P. Zhang, R.-B. Lin, S. Chen, Q. Deng, Z. Zeng, S. Deng and B. Chen, Fine Pore Engineering in a Series of Isoreticular Metal-Organic Frameworks for Efficient C2H2/CO2 Separation, Nat. Commun., 2022, 13, 200 CrossRef CAS PubMed
- A. Nuhnen and C. Janiak, A Practical Guide to Calculate the Isosteric Heat/Enthalpy of Adsorption via Adsorption Isotherms in Metal–Organic Frameworks, MOFs, Dalton Trans., 2020, 10295–10307, 10.1039/D0DT01784A
- Z. Li, P. Liu, C. Ou and X. Dong, Porous Metal–Organic Frameworks for Carbon Dioxide Adsorption and Separation at Low Pressure, ACS Sustainable Chem. Eng., 2020, 8, 15378–15404 CrossRef CAS
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Tuning the Adsorption Properties of UiO-66 via Ligand Functionalization, Langmuir, 2012, 28, 15606–15613 CrossRef CAS PubMed
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Carbon Dioxide Capture in Metal–Organic Frameworks, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed
- X. Wu, B. Yuan, Z. Bao and S. Deng, Adsorption of Carbon Dioxide, Methane and Nitrogen on an Ultramicroporous Copper Metal–Organic Framework, J. Colloid Interface Sci., 2014, 430, 78–84 CrossRef CAS PubMed
- M. Singh, P. Solanki, P. Patel, A. Mondal and S. Neogi, Highly Active Ultrasmall Ni Nanoparticle Embedded inside a Robust Metal–Organic Framework: Remarkably Improved Adsorption, Selectivity, and Solvent-Free Efficient Fixation of CO2, Inorg. Chem., 2019, 58, 8100–8110 CrossRef CAS PubMed
- H.-H. Wang, L. Hou, Y.-Z. Li, C.-Y. Jiang, Y.-Y. Wang and Z. Zhu, Porous MOF with Highly Efficient Selectivity and Chemical Conversion for CO2, ACS Appl. Mater. Interfaces, 2017, 9, 17969–17976 CrossRef CAS PubMed
- J. Dai, D. Xie, Y. Liu, Z. Zhang, Y. Yang, Q. Yang, Q. Ren and Z. Bao, Supramolecular Metal–Organic Framework for CO2/CH4 and CO2/N2 Separation, Ind. Eng. Chem. Res., 2020, 59, 7866–7874 CrossRef CAS
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Microporous Metal-Organic Framework with Potential for Carbon Dioxide Capture at Ambient Conditions, Nat. Commun., 2012, 3, 954 CrossRef PubMed
- J. F. Kurisingal, Y. Rachuri, A. S. Palakkal, R. S. Pillai, Y. Gu, Y. Choe and D.-W. Park, Water-Tolerant DUT-Series Metal–Organic Frameworks: A Theoretical–Experimental Study for the Chemical Fixation of CO2 and Catalytic Transfer Hydrogenation of Ethyl Levulinate to γ-Valerolactone, ACS Appl. Mater. Interfaces, 2019, 11, 41458–41471 CrossRef CAS PubMed
- A. C. Kathalikkattil, J. Tharun, R. Roshan, H.-G. Soek and D.-W. Park, Efficient Route for Oxazolidinone Synthesis using Heterogeneous Biopolymer Catalysts from Unactivated Alkyl Aziridine and CO2 under Mild Conditions, Appl. Catal., A, 2012, 447–448, 107–114 CrossRef CAS
- P. Patel, B. Parmar, R. S. Pillai, A. Ansari, N.-U. H. Khan and E. Suresh, CO2 Fixation by Cycloaddition of Mono/Disubstituted Epoxides using Acyl Amide Decorated Co(II) MOF as A Synergistic Heterogeneous Catalyst, Appl. Catal., A, 2020, 590, 117375 CrossRef CAS
- F. Jutz, J.-D. Grunwaldt and A. Baiker, Mn(III)(salen)-Catalyzed Synthesis of Cyclic Organic Carbonates from Propylene and Styrene Oxide in “Supercritical” CO2, J. Mol. Catal. A: Chem., 2008, 279, 94–103 CrossRef CAS
- N. Seal and S. Neogi, Intrinsic-Unsaturation-Enriched Biporous and Chemorobust Cu(II) Framework for Efficient Catalytic CO2 Fixation and Pore-Fitting Actuated Size-Exclusive Hantzsch Condensation with Mechanistic Validation, ACS Appl. Mater. Interfaces, 2021, 13, 55123–55135 CrossRef CAS PubMed
- S. Arayachukiat, C. Kongtes, A. Barthel, S. V. C. Vummaleti, A. Poater, S. Wannakao, L. Cavallo and V. D'Elia, Ascorbic Acid as a Bifunctional Hydrogen Bond Donor for the Synthesis of Cyclic Carbonates from CO2 under Ambient Conditions, ACS Sustainable Chem. Eng., 2017, 5, 6392–6397 CrossRef CAS
- S. Sopeña, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Pushing the Limits with Squaramide-Based Organocatalysts in Cyclic Carbonate Synthesis, ACS Catal., 2017, 7, 3532–3539 CrossRef
- Z. Xue, J. Jiang, M.-G. Ma, M.-F. Li and T. Mu, Gadolinium-based Metal–Organic Framework as an Efficient and Heterogeneous Catalyst to Activate Epoxides for Cycloaddition of CO2 and Alcoholysis, ACS Sustainable Chem. Eng., 2017, 5, 2623–2631 CrossRef CAS
- R. Babu, S.-H. Kim, J. F. Kurisingal, H.-J. Kim, G.-G. Choi and D.-W. Park, A Room Temperature Synthesizable Zeolitic Imidazolium Framework Catalyst for the Solvent-Free Synthesis of Cyclic Carbonates, J. CO2 Util., 2018, 25, 6–13 CrossRef CAS
- F. Afshariazar, A. Morsali and P. Retailleau, Investigation of the Influence of Functionalization Strategy on Urea 2D MOF Catalytic Performance, Inorg. Chem., 2023, 62, 3498–3505 CrossRef CAS PubMed
- P. Patel, U. Patel, B. Parmar, A. Dadhania and E. Suresh, Regioselective Ring-Opening of Spiro-Epoxyoxindoles by a Dual-Ligand Zinc-Based Metal–Organic Framework as an Efficient Heterogeneous Catalyst, ACS Appl. Nano Mater., 2022, 5, 3712–3721 CrossRef CAS
- Y. Wang and J. Duan, Urea and Thiourea-Functionalized, Pyridinium-based Ionic Polymers Convert CO2 to Cyclic Carbonate under Mild Conditions, ACS Appl. Polym. Mater., 2022, 4, 5851–5860 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Materials and physical measurements; experimental details; single-crystal X-ray crystallography; asymmetric unit; TGA curves; FT-IR spectra; nitrogen adsorption–desorption isotherms; fitting parameters; calculations; heat of adsorption, PXRD patterns; FE-SEM images; NMR spectra; crystal data; XPS data and refinement parameters; comparative tables. CCDC 2256894. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi00428g |
| This journal is © the Partner Organisations 2023 |