
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diruthenium complexes as pH-responsive delivery systems: a quantitative assessment†
Isabel
Coloma
a,
Miguel
Cortijo
 *a,
María José
Mancheño
b,
María Eugenia
León-González
c,
Crisanto
Gutierrez
d,
Bénédicte
Desvoyes
*d and
Santiago
Herrero
*a
*a,
María José
Mancheño
b,
María Eugenia
León-González
c,
Crisanto
Gutierrez
d,
Bénédicte
Desvoyes
*d and
Santiago
Herrero
*a
aInorganic Chemistry Department, Faculty of Chemistry, Universidad Complutense de Madrid, E-28040 Madrid, Spain. E-mail: miguelcortijomontes@ucm.es; sherrero@ucm.es
bOrganic Chemistry Department, Faculty of Chemistry, Universidad Complutense de Madrid, E-28040 Madrid, Spain
cAnalytical Chemistry Department, Faculty of Chemistry, Universidad Complutense de Madrid, E-28040 Madrid, Spain
dCentro de Biología Molecular Severo Ochoa, CSIC-UAM, E-28049 Madrid, Spain. E-mail: bdesvoyes@cbm.csic.es
First published on 18th May 2023
Abstract
The controlled release of biologically active species from diruthenium compounds is crucial for the development of selective drug delivery systems based on such complexes, which in addition display antineoplastic properties by themselves. In the present work, we analyse in detail the kinetics of the pH-triggered release of the auxin-related hormones 2,4-D (2,4-dichlorophenoxyacetate) and NAA (1-naphthaleneacetate) from the metal–metal bonded tris(formamidinato) Ru25+ complexes [Ru2Cl(μ-DPhF)3(μ-2,4-D)] (Ru2,4-D), [Ru2Cl(μ-DPhF)3(μ-NAA)] (RuNAA), [Ru2Cl(μ-DAniF)3(μ-2,4-D)] (Ru′2,4-D) and [Ru2Cl(μ-DAniF)3(μ-NAA)] (Ru′NAA) (DPhF = N,N′-diphenylformamidinate, DAniF = N,N′-bis(p-methoxy)phenylformamidinate). Moreover, the synthesis and complete characterisation of [Ru2Cl(μ-DAniF)3(μ-IAA)] (Ru′IAA, IAA = indole-3-acetate), Ru′2,4-D and Ru′NAA, including the crystal structure of the two latter ones, is reported. The release of auxins is studied through a fluorimetric quantitative assay, which allows determining the influence of different formamidinate ancillary ligands and the nature of the outgoing auxin ligand in the release process. Chemometrics is employed to evaluate the statistical significance of the variables. The release of auxins is slower at physiological pH and occurs faster at slightly acidic pH values. Compounds containing DPhF ancillary ligands and NAA outgoing ligand present a slower dissociation of the auxin, which is not complete in the first 24 h. The release rate is also correlated with the bond distance O1(auxin)–Ru1(hexacoordinated). A mechanism of the substitution reaction is tentatively proposed based on these findings. Overall, these results pave the way towards new systems for the controlled delivery of antineoplastic drugs under mild-acidic conditions like those surrounding solid tumours.
Introduction
Cisplatin1 and other platinum-based drugs, such as oxaliplatin2 and carboplatin3 have been widely used in clinical treatment of different human cancers: e.g., ovarian, head, neck, colorectal, lung or bladder. They are efficient and economically affordable, but suffer from two major limitations: a poor selectivity, which leads to dose-limiting side effects and drug resistance development.4–6 Consequently, significant efforts have been made to prepare more efficient drugs, with fewer side effects using metals different than platinum. In this regard, the search of new ruthenium-based metallodrugs has been very fruitful and has led to the discovery of the therapeutic or biomedical properties of several species.7,8The imidazolium salt of trans-tetrachlorido(dimethylsulfoxide)imidazoleruthenate(III) (NAMI-A) was the first ruthenium derivative to enter in clinical tests, but its use was abandoned due to significant side effects in phase II trials.9–11 Later on, the indazolium salt of the trans-[tetrachloridobis(1H-indazole)ruthenate(III)] complex (KP1019) entered in clinical phase I trials and did not show severe side effects.12,13 However, its poor solubility in aqueous media hampered further clinical tests. Its analogous sodium salt, (N)KP1339, with improved solubility, underwent clinical phase I trials showing minor adverse effects, but only moderate anti-tumour activity.14–16 Current interests have been expanded to Ru(II) polypyridyl species such as TLD1433,7,17,18 which recently entered in clinical phase II trials, and Ru(II)-arene derivatives such as RM175 and RAPTA-C19 or Ru1080.20
Another promising family of ruthenium compounds for medicinal purposes is related to [Ru2Cl(O2CMe)4],21 characterised by its paddlewheel structure, a 2.5 metal–metal bond order and an intermediate oxidation state for the ruthenium centres of 2.5.22 The first [Ru2Cl(carboxylate)4] compounds that showed antineoplastic activity were published at the end of the last century.23 Increasing water-solubility of the compounds improved their cytotoxic properties.24 An important step forward was the incorporation of biologically active carboxylate ligands to the diruthenium core to search synergy between the bimetal centre and the ligands against cancer cells and avoid drug resistance.25–29
Nevertheless, the lack of specificity is one of the most important drawbacks of cancer chemotherapy.30 To overcome the poor selectivity, employing prodrugs that can be activated or drugs that can be delivered in a specific region have become promising strategies.31 For example, solid tumour environment (the most common form of cancer) is usually characterised by hypoxia32 and a slight acidity,33–35 properties that can be used to activate the prodrugs or to deliver the drugs. Carboxylatodiruthenium(II,III) compounds are also interesting under this point of view since they have redox potentials physiologically accessible.22,36,37 They can be reduced under hypoxia conditions, and also probably in the presence of high levels of glutathione,38 in the tumour environment to produce Ru24+ species21 that are much more reactive. In order to gain insight into the parameters that could influence the pH-dependent release of biomolecules, as a proof of concept, we recently designed a new family of tris(formamidinato) Ru25+ complexes carrying carboxylate species such as auxins phytohormones. We demonstrated that auxins were readily liberated by lowering the pH keeping their biological activity in plants.39 We used, the auxins indole-3-acetate (IAA), 2,4-dichlorophenoxyacetate (2,4-D), and 1-naphthaleneacetate (NAA), to prepare [Ru2Cl(μ-DPhF)3(μ-IAA)] (RuIAA), [Ru2Cl(μ-DPhF)3(μ-2,4-D)] (Ru2,4-D) and [Ru2Cl(μ-DPhF)3(μ-NAA)] (RuNAA) (DPhF = N,N′-diphenylformamidinate). The presence of three formamidinate ligands confers great stability to the diruthenium core,40,41 which is a great advantage over the tetracarboxylato complexes usually employed. This stability39 avoids the decomposition of the molecule prior to the auxin release, which is time-dependent and only occurs rapidly under slightly acidic pH conditions (pH = 6.5).
As stated before, this type of on-demand delivery systems, able to recognize and respond to small pH changes, are of special interest not only in the context of cancer chemotherapy, but also in bacterial infection, inflammation or even in neurological diseases or ischemic strokes.34,42 To gain insight of the governing factors that can influence the delivery of the bioactive species covalently linked to the metal and our ability to electronically tune the diruthenium scaffold, we were prompted to assess quantitatively the amount of various auxin phytohormones (Aux) released from different tris(formamidinato)diruthenium complexes in biological assays. Therefore, we study the impact of the donor capacity of the formamidinate ancillary ligands by comparing the auxin release properties of [Ru2Cl(μ-DAniF)3(μ-Aux)] (Ru′Aux; DAniF = N,N′-bis(p-methoxy)phenylformamidinate) with those of the previously characterised complexes (RuAux)39 bearing less donor DPhF ligands. Thus, a biological set up was implemented to quantitatively assess the auxin activity of the RuAux and Ru′Aux complexes and, therefore, compare the release from both families of compounds. Chemometric tools were used to perform a multiparametric statistical analysis and obtain relevant information from the experimental data.
Experimental
Materials and physical measurements
[Ru2Cl(μ-O2CMe)4],43N,N′-bis(p-methoxy)phenylformamidine (HDAniF),44Ru2,4-D and RuNAA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39 were prepared according to published procedures. [Ru2Cl(μ-DAniF)3(μ-O2CMe)] was also prepared following a reported procedure,45 slightly modified to avoid the use of benzene in the purification steps, employing acetone and toluene instead. The rest of reactants and solvents were obtained from commercial sources and used as received. Elemental analyses were carried out by de Microanalytical Service of the Complutense University of Madrid. FT-IR spectra were collected employing a PerkinElmer Spectrum 100 instrument including a universal ATR accessory. Electronic spectra of ∼10−4 M dichloromethane or DMSO/buffer solutions of the compounds were recorded using a Cary 5G spectrometer. Electrospray ionization (ESI) mass spectra were collected by the Mass Spectrometry Service of the Complutense University of Madrid using an ion trap-Bruker Esquire-LC spectrometer. Variable-temperature magnetization measurements were performed on a Quantum Design MPMXL SQUID magnetometer, operating under a 0.5 T magnetic field, using 17.63, 15.45 and 23.23 mg of Ru′IAA, Ru′2,4-D and Ru′NAA. All data were corrected for the diamagnetic contribution of both sample holder and compound.
39 were prepared according to published procedures. [Ru2Cl(μ-DAniF)3(μ-O2CMe)] was also prepared following a reported procedure,45 slightly modified to avoid the use of benzene in the purification steps, employing acetone and toluene instead. The rest of reactants and solvents were obtained from commercial sources and used as received. Elemental analyses were carried out by de Microanalytical Service of the Complutense University of Madrid. FT-IR spectra were collected employing a PerkinElmer Spectrum 100 instrument including a universal ATR accessory. Electronic spectra of ∼10−4 M dichloromethane or DMSO/buffer solutions of the compounds were recorded using a Cary 5G spectrometer. Electrospray ionization (ESI) mass spectra were collected by the Mass Spectrometry Service of the Complutense University of Madrid using an ion trap-Bruker Esquire-LC spectrometer. Variable-temperature magnetization measurements were performed on a Quantum Design MPMXL SQUID magnetometer, operating under a 0.5 T magnetic field, using 17.63, 15.45 and 23.23 mg of Ru′IAA, Ru′2,4-D and Ru′NAA. All data were corrected for the diamagnetic contribution of both sample holder and compound.
X-ray diffraction data collection and structure refinement
Suitable single crystals of Ru′2,4-D and Ru′NAA were measured using a Bruker Smart-CCD (Mo Kα radiation, λ = 0.71073 Å) and a D8 Venture (Cu Kα radiation, λ = 1.54178 Å) diffractometer, respectively. The Ru′2,4-D crystal was kept at 296 K during the data collection, while the Ru′NAA crystal was kept at 100 K. The structures were solved using Olex2 software,46 with the SHELXT47 structure solution program using intrinsic phasing and refined with the SHELXL48 refinement package using least squares minimization. All non-hydrogen atoms were anisotropically refined. The hydrogen atoms were included with fixed isotropic contributions at their calculated positions determined by molecular geometry. CCDC 2235327 and 2235329 contain the crystallographic data for Ru′2,4-D and Ru′NAA.† More detailed information can be found in the synthesis and characterisation section and in Tables S1–S4 and Fig. S1–S3 in the ESI.†Synthesis of starting materials
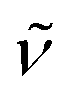 (cm−1): 3005w, 2957w, 2835w, 1607m, 1578w, 1540s, 1498vs, 1438s, 1313m, 1292s, 1242vs, 1212vs, 1167s, 1105m, 1026s, 940m, 823s, 790m, 766m, 731m, 692s. UV/Vis/NIR (CH2Cl2): λmax/nm (ε/M−1 cm−1) ∼380 sh (6100), 511 (4000), 594 (4300). MS (ESI+): m/z 1028.1, [M − Cl]+ (100).
(cm−1): 3005w, 2957w, 2835w, 1607m, 1578w, 1540s, 1498vs, 1438s, 1313m, 1292s, 1242vs, 1212vs, 1167s, 1105m, 1026s, 940m, 823s, 790m, 766m, 731m, 692s. UV/Vis/NIR (CH2Cl2): λmax/nm (ε/M−1 cm−1) ∼380 sh (6100), 511 (4000), 594 (4300). MS (ESI+): m/z 1028.1, [M − Cl]+ (100).
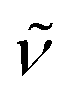 (cm−1): 3042w, 2944w, 2833w, 1602m, 1539w, 1498vs, 1462m, 1440m, 1294m, 1242vs, 1210vs, 1170s, 1108m, 1025s, 935m, 824s, 790m, 768m, 728m. UV/Vis/NIR (CH2Cl2): λmax/nm (ε/M−1 cm−1) ∼534 sh (4800), 635 (5200), 837 (4700).
(cm−1): 3042w, 2944w, 2833w, 1602m, 1539w, 1498vs, 1462m, 1440m, 1294m, 1242vs, 1210vs, 1170s, 1108m, 1025s, 935m, 824s, 790m, 768m, 728m. UV/Vis/NIR (CH2Cl2): λmax/nm (ε/M−1 cm−1) ∼534 sh (4800), 635 (5200), 837 (4700).
Synthesis of final products
 (cm−1): 3276w, 3039w, 2997w, 2957w, 2905w, 2833w, 1606m, 1540m, 1497vs, 1460s, 1439s, 1412s, 1313m, 1291s, 1239vs, 1169vs, 1107s, 1027vs, 938m, 825vs, 790s, 765s, 745s, 724s. UV/Vis/NIR (CH2Cl2): λmax/nm (ε/M−1 cm−1) ∼385 sh (6000), 498 (4100), 594 (5000). MS (ESI+): m/z 1143.2, [M − Cl]+ (100).
(cm−1): 3276w, 3039w, 2997w, 2957w, 2905w, 2833w, 1606m, 1540m, 1497vs, 1460s, 1439s, 1412s, 1313m, 1291s, 1239vs, 1169vs, 1107s, 1027vs, 938m, 825vs, 790s, 765s, 745s, 724s. UV/Vis/NIR (CH2Cl2): λmax/nm (ε/M−1 cm−1) ∼385 sh (6000), 498 (4100), 594 (5000). MS (ESI+): m/z 1143.2, [M − Cl]+ (100).
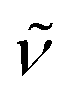 (cm−1): 3039w, 3009w, 2959w, 2896w, 2832w, 1606m, 1551s, 1498vs, 1455s, 1438s, 1421s, 1394w, 1340m, 1319m, 1291s, 1241vs, 1210vs, 1172vs, 1107s, 1067m, 1027vs, 961m, 937m, 882w, 824vs, 807s, 790s, 762s, 716m, 727m, 697m. UV/Vis/NIR (CH2Cl2): λmax/nm (ε/M−1 cm−1) ∼385 sh (5300), 478 (4400), 603 (4800). MS (ESI+): m/z 1188.1, [M − Cl]+ (100) Crystals of Ru′2,4-D suitable for single-crystal X-ray diffraction analysis were obtained by slow diffusion of hexane into a dichloromethane solution of the compound.
(cm−1): 3039w, 3009w, 2959w, 2896w, 2832w, 1606m, 1551s, 1498vs, 1455s, 1438s, 1421s, 1394w, 1340m, 1319m, 1291s, 1241vs, 1210vs, 1172vs, 1107s, 1067m, 1027vs, 961m, 937m, 882w, 824vs, 807s, 790s, 762s, 716m, 727m, 697m. UV/Vis/NIR (CH2Cl2): λmax/nm (ε/M−1 cm−1) ∼385 sh (5300), 478 (4400), 603 (4800). MS (ESI+): m/z 1188.1, [M − Cl]+ (100) Crystals of Ru′2,4-D suitable for single-crystal X-ray diffraction analysis were obtained by slow diffusion of hexane into a dichloromethane solution of the compound.
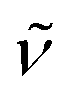 (cm−1): 3036w, 2954w, 2833w, 1606m, 1541m, 1499vs, 1407m, 1320m, 1292m, 1242vs, 1212vs, 1170s, 1107m, 1030s, 939m, 826s, 790s, 767s, 715m. UV/Vis/NIR (CH2Cl2): λmax/nm (ε/M−1 cm−1) ∼360 sh (11000), 500 (6100), 590 (6700). MS (ESI+): m/z 1154.2, [M − Cl]+ (100). Crystals of Ru′NAA suitable for single-crystal X-ray diffraction determination were obtained by slow diffusion of hexane into a THF solution of the compound.
(cm−1): 3036w, 2954w, 2833w, 1606m, 1541m, 1499vs, 1407m, 1320m, 1292m, 1242vs, 1212vs, 1170s, 1107m, 1030s, 939m, 826s, 790s, 767s, 715m. UV/Vis/NIR (CH2Cl2): λmax/nm (ε/M−1 cm−1) ∼360 sh (11000), 500 (6100), 590 (6700). MS (ESI+): m/z 1154.2, [M − Cl]+ (100). Crystals of Ru′NAA suitable for single-crystal X-ray diffraction determination were obtained by slow diffusion of hexane into a THF solution of the compound.
Biological assays
Arabidopsis thaliana seeds carrying pDR5::GUS construct49 were sown as described earlier on plates containing 1/2 Murashige and Skoog salts supplemented with 0.5 g L−1 MES (2-(N-morpholino)ethanesulfonic acid) pH 5.7, 1% sucrose, and 1% plant agar (Duchefa).39 Plants were grown during 5 days at 21 °C under long day conditions (16 h light/8 h dark) and 60% moisture. Fresh solutions of dichlorophenoxyacetic acid (H2,4-D), potassium 1-naphthaleneacetate (KNAA), and each diruthenium compound (Ru2,4-D, RuNAA, Ru′2,4-D, and Ru′NAA) were prepared at 5 mM in DMSO for 3 replicates. Plants were transferred to six-well plates containing each 5 μM of KNAA, RuNAA, Ru′NAA, H2,4-D, Ru2,4-D or Ru′2,4-D in a 10 mM MOPS (3-(N-morpholino)propanesulfonic acid) KOH buffer at pH 6.5, 7.0, and 7.5. Ru′IAA was not used due to the lack of long-term stability in solution. Plants were incubated during 3, 6, and 24 h at room temperature. At each time point, plants were harvested, frozen in liquid nitrogen and kept at −80 °C. Frozen plants were grinded with glass beads using a Silamat device and protein extract was made by adding to each sample 200 μL of extraction buffer: 50 mM NaH2PO4/Na2HPO4 pH 7, 10 mM ethylendiaminetetraacetic acid (EDTA), 0.1% sodium dodecyl sulphate (SDS), 0.1% Triton-X100, 10 mM β-mercaptoethanol, 1 mM phenylmethanesulfonyl fluoride (PMSF) and protease inhibitors cocktail (Sigma, P9599). Extracts were left on ice during 30 min and centrifuged during 10 min at 21000g. Supernatant was used to perform the quantitative β-glucuronidase (GUS) enzyme activity assay as follows: in a 96 well black microplate, 5 μL of each plant extract was incubated during 1 h at 37 °C with 95 μL of 0.4 mM 4-methylumbelliferyl-β-D-glucuronide (4-MUG) dissolved in extraction buffer without detergents, proteases inhibitors and β-mercaptoethanol. Two technical duplicates were performed for each plant extract of three biological replicates. The reaction was stopped by adding 200 μL of a 0.2 M solution of Na2CO3 and fluorescence was measured in a microplate reader (ClarioStarPlus, BMGLabtec) with excitation set at 355 nm and emission at 450 nm. A standard curve with 4-methylumbelliferone (4-MU) from 0 to 2000 nM was added to each microplate to calculate the enzymatic activity. Gain was adjusted to the well with the highest fluorescence intensity. Protein quantification was performed with Bradford colorimetric assay (BioRad).50 Enzymatic activity was expressed in 4-MU nmol min−1 μg−1 protein.
Chemometrics
Chemometric tools were used to obtain statistically relevant information from the experimental data. In particular, the multivariate chemometric tools employed were the analysis of multifactorial variance (ANOVA) and the analysis of response surfaces.51 Multivariate ANOVA refers to the statistical analysis that allows to determine whether more than one factor or independent variable (e.g., time and pH) has a significant impact on a quantitative dependent variable (i.e., auxin activity). A correlation model was built to evaluate how the dependent quantitative variable, i.e. auxin activity differs in terms of the independent variables i.e. pH and time.Results and discussion
Synthesis and characterisation
The syntheses of Ru2,4-D,39RuNAA,39Ru′IAA, Ru′2,4-D and Ru′NAA are accomplished as shown in Scheme 1. The starting material in the synthesis of Ru2,4-D and RuNAA is the paddlewheel [Ru2Cl(μ-DPhF)3(μ-O2CMe)]52 complex, while the starting material in the synthesis of Ru′IAA, Ru′2,4-D and Ru′NAA is the paddlewheel [Ru2Cl(μ-DAniF)3(μ-O2CMe)] complex. The reactions of these two starting materials with an excess of hydrochloric acid, in methanol at room temperature, yield [Ru2Cl2(μ-DPhF)3] or [Ru2Cl2(μ-DAniF)3]. Similar procedures have been employed to prepare [Ru2X2(μ-DPhF)3] (X = Cl, Br, I) compounds showing an open-paddlewheel structure.53 The reactions at room temperature, in an acetone/water mixture, of NaIAA, H2,4-D or KNAA with either [Ru2Cl2(μ-DPhF)3] or [Ru2Cl2(μ-DAniF)3] gave rise to the final products. Although a colour change is immediately observed upon reagents mixing in the last step, the reactions were left to stir for 3 days in order to increase the yields. This synthetic approach avoids the use of refluxing solvents54,55 or microwave radiation56 to replace the acetate ligand by another O,O′-donor bridging ligand in [Ru2Cl(μ-DPhF)3(μ-O2CMe)] and [Ru2Cl(μ-DAniF)3(μ-O2CMe)], and are imperative when working with temperature-sensitive bioactive ligands.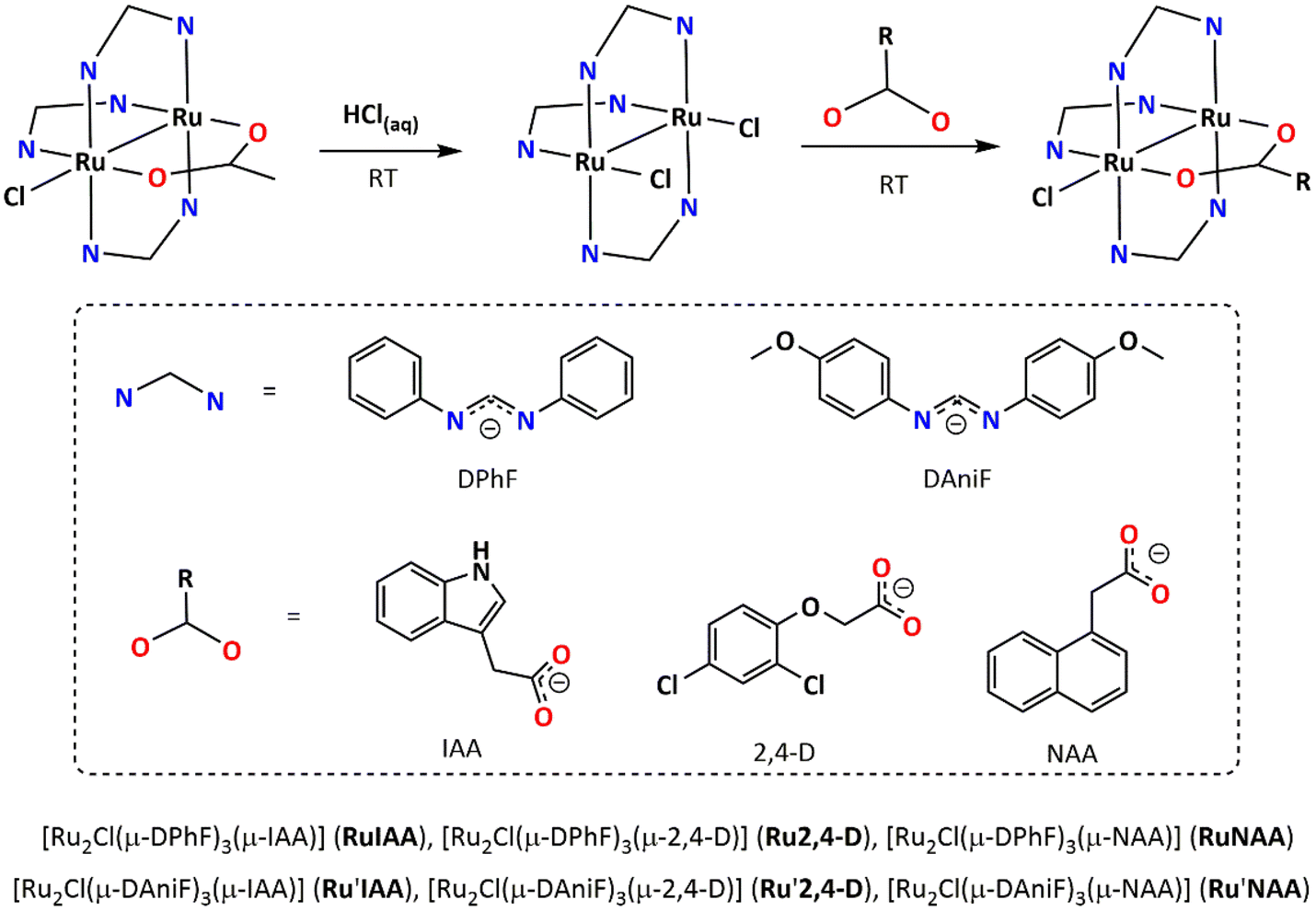 | ||
| Scheme 1 Scheme of synthesis followed to prepare RuAux and Ru′Aux compounds. | ||
The characterisation data obtained for Ru2,4-D and RuNAA were consistent with those reported elsewhere.39Ru′IAA, Ru′2,4-D and Ru′NAA were characterised by elemental analysis, mass spectrometry, IR and UV-vis spectroscopies and magnetization measurements at variable temperature. Moreover, single crystal X-ray diffraction allowed determining the crystal structure of Ru′2,4-D and Ru′NAA.
The experimental elemental analyses are in accordance with the expected composition in all cases. The ESI+ mass spectra of Ru′IAA, Ru′2,4-D and Ru′NAA (Fig. S4†) show the [M − Cl]+ fragments with isotope pattern distributions that agree well with the calculated ones. The molecular peak is not observed or is practically imperceptible in all complexes.
Despite the structural resemblance between [Ru2Cl(μ-DAniF)3(μ-O2CMe)] and [Ru2Cl2(μ-DAniF)3], their IR spectra can be readily distinguished based on the presence or absence of the symmetric (∼1440 cm−1) and antisymmetric (∼1540 cm−1) O−C−O stretching bands. The symmetric O−C−O stretching band observed in the IR spectrum of [Ru2Cl(μ-DAniF)3(μ-O2CMe)] is not present in that of the open-paddlewheel [Ru2Cl2(μ-DAniF)3] complex, which is formed by release of an acetate ligand in presence of HCl. However, the antisymmetric O−C−O stretching band seems to not disappear in the spectrum of [Ru2Cl2(μ-DAniF)3] and only diminishes its intensity. This is explained by an overlap in this region of the antisymmetric O−C−O and the N−C−N stretching bands of the formamidinate ligands.
The IR spectra of Ru′IAA, Ru′2,4-D and Ru′NAA are very similar to that of [Ru2Cl(μ-DAniF)3(μ-O2CMe)]. As expected, they show a symmetric O−C−O stretching band, in the 1407–1421 cm−1 range, and an antisymmetric O−C−O stretching band overlapped with the N−C−N stretching band, in the 1540–1551 cm−1 range. It must be noted that the separation between the symmetric and antisymmetric O−C−O stretching band is very similar in all the complexes, which suggests the same bridging coordination mode for the carboxylate ligands.57 The IR spectra of [Ru2Cl(μ-DAniF)3(μ-O2CMe)], [Ru2Cl2(μ-DAniF)3], Ru′IAA, Ru′2,4-D and Ru′NAA are shown in the ESI (Fig. S5†) together with an assignment of the most relevant bands found for all the compounds (Table S5†).
Dichloromethane solutions (∼10−4 M) of Ru′IAA, Ru′2,4-D and Ru′NAA exhibit the same electronic spectra profile than their precursor [Ru2Cl(μ-DAniF)3(μ-O2CMe)] (Fig. S6†): two maxima, ascribed to π(RuO/N,Ru2) → π*(Ru2) (478–511 nm) and π*(Ru2) → σ*(RuO/N) (590–603 nm) transitions, and two shoulders. The first one is ascribed to a π(Cl) → π*(Ru2) LMCT transition (360–385 nm) and the second one, which can only be guessed from a slight change of slope at ca. 690 nm, to a δ(Ru2) → π*(Ru2) transition. Remarkably, the electronic spectra profile of these compounds differs with that of [Ru2Cl(μ-DPhF)3(μ-O2CMe)], RuIAA, Ru2,4-D and RuNAA,39 which display one maximum and three shoulders (Fig. S6 and Table S6†), showing that the electronic structure of this type of compounds is highly dependent on the donor character of the formamidinate ligands. Both profiles are typical of high spin diruthenium complexes, although the first one is usually observed when a relatively high electron density is surrounding the Ru atoms.54,55,58–60
The stability of RuNAA and Ru′NAA in DMSO/water solution was studied via UV-vis spectroscopy using an HEPES KOH (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer at pH 6.5. As expected,41 no significant changes in the electronic profiles were observed over 24 hours (Fig. S7†). The most likely explanation to this observation is that in presence of an excess of the buffer, diruthenium species would certainly form [Ru2Cl(formamidinate)3(4-(2-hydroxyethyl)-1-piperazineethanesulfonate)] species with the same [Ru2Cl(N,N′)3(O,O′)] coordination environment around the metal and very similar electronic spectra profile. Nevertheless, the release of carboxylate ligands could be confirmed by mass spectrometry. A 120% increase of the auxin ligand peak intensity is observed when the solution is slightly acidified to pH 6.5 (Fig. S8†).
Variable temperature magnetization measurements (Fig. S9–S11†) confirmed the presence of three unpaired electrons in Ru′IAA, Ru′2,4-D and Ru′NAA. Their χM·T values at room temperature (Table 1) are close or slightly larger than 1.87 cm3 K mol−1, which is the spin contribution expected for a quartet state that arises from a σ2δ2π4(δ*π*)3 electronic configuration. Lowering the temperature results in a decrease of the χM·T product, which is especially pronounced below 100 K and mainly ascribed to a strong zero-field splitting.61 Thus, the magnetic data were well fitted considering a quadruplet state undergoing an axial zero-field splitting (D) (see eqn (S1)–(S4) in ESI†).
Additionally, the data were fitted introducing an intermolecular magnetic exchange term (zJ) in the model as a perturbation on the molecular field (eqn (S4)†). However, it only led to a better-quality fit and unneglectable values of zJ in the case of Ru′2,4-D. A zJ value of −0.7 cm−1 was obtained in this case, indicating the existence of intermolecular antiferromagnetic interactions. Table 1 shows the parameters obtained from the best fit of the data. These parameters are similar to those obtained previously for RuIAA, Ru2,4-D and RuNAA.39
Single crystals of Ru′2,4-D and Ru′NAA suitable for X-ray diffraction were obtained by slow diffusion of hexane in a solution of the corresponding complex in CH2Cl2 and THF, respectively. The crystal structure determinations confirmed that the two compounds exhibit a paddlewheel structure formed by diruthenium units bridged by three formamidinate ligands (DAniF) and one carboxylate (2,4-D or NAA) at the equatorial positions with a chloride ligand coordinated at one axial position. Fig. 1 shows a paddlewheel unit of Ru′NAA, which has been chosen as a representative example. A view of the asymmetric unit of Ru′2,4-D can be found in the ESI (Fig. S1†). Main bond distances and angles are shown in Tables S2 and S4.† Particularly, the Ru–Ru distance is 2.3288(8) Å for Ru′2,4-D and 2.3155(6) Å for Ru′NAA. These distances indicate that the Ru–Ru bond order is 2.5, which is the case expected for a σ2δ2π4(δ*π*)3 electronic configuration.61
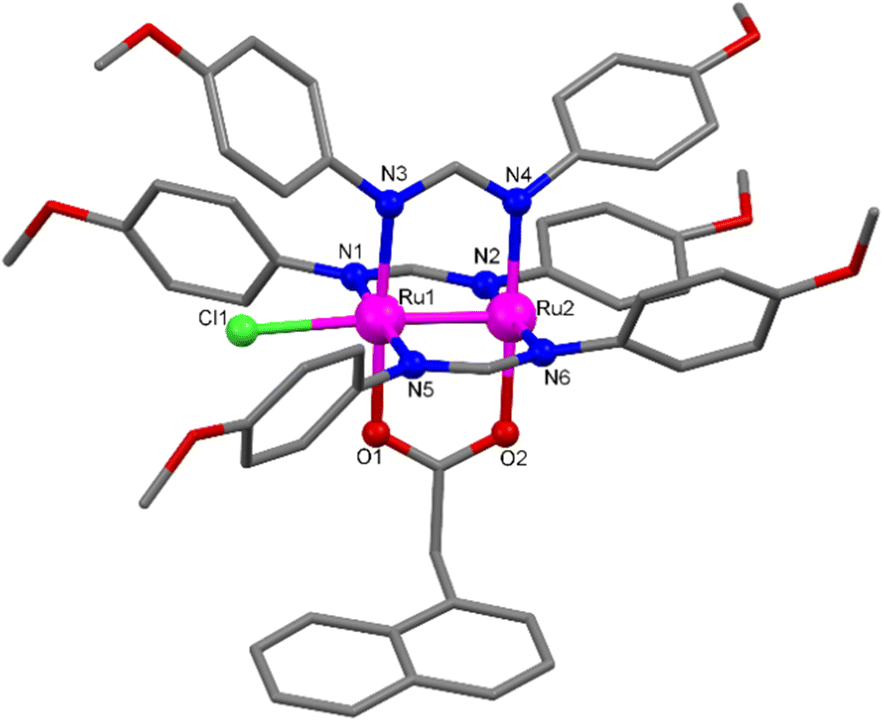 | ||
| Fig. 1 View of the asymmetric unit of Ru′NAA with selected atoms labelled. Ru atoms are shown in pink, N atoms in blue, O atoms in red and C atoms in grey. H atoms are omitted for clarity. | ||
Two C–H⋯π intramolecular T-interactions can be found in the asymmetric unit of Ru′2,4-D (Fig. S2†). The first one involves one hydrogen of the ring bonded to N2 and the aromatic π cloud of the ring bonded to N4 (2.757 Å). The second one involves one hydrogen of the ring bonded to N4 and the aromatic π cloud of the ring attached to N6 (2.869 Å). Regarding intermolecular interactions, only weak C–H⋯O interactions (C⋯O distance of 3.169 Å) in the crystal structure of Ru′2,4-D (Fig. S2†) and weak C–H⋯O (C⋯O distance of 3.210 Å) and C–H⋯Cl (C⋯Cl distance of 3.415 Å) interactions in the structure of Ru′NAA are observed (Fig. S3†).
Biological assays
A biological assay was set up to quantitatively measure and compare the activity of two auxin ligands (NAA and 2,4-D) dissociated from the two characterised families of diruthenium compounds (RuAux and Ru′Aux). For this purpose, in vivo release assays were carried out in Arabidopsis transgenic plants that carry a highly sensitive reporter of auxin activity, which in presence of the hormone drives the expression of the enzyme β-glucuronidase (GUS). The GUS activity was measured using the substrate 4-methylumbelliferyl-β-D-glucuronide (4-MUG) that liberates a fluorescent product (4-methylumbelliferone, 4-MU) that can be quantitatively detected by fluorimetry.Plants were treated with the RuAux and Ru′Aux complexes and with their corresponding free hormone in buffered solution at different pH (6.5, 7.0 and 7.5). The activity was measured after 3, 6 and 24 h.
The multifactorial ANOVA analysis (Table S7 and Fig. S12†) shows significant statistical differences (p-value less than 0.05) in the effect of pH and time over the auxin activity. The multifactorial analysis also shows the interrelationship between pH and the time conditions used in the activity measurements (Fig. S13†). The analysis of variance indicates that there are significant differences between the independent variables evaluated. In order to evaluate at what pH or time levels these differences occur, a multiple range statistical test (least significant difference) has been carried out. Although all the variables studied are statistically significant, the effect on the instrumental response (auxin activity) is different depending on the compound, time and pH values considered. Then, it was decided to make a joint study of the two variables on the activity. Since many variables can affect the response of the system under study, the selection of those with the greatest effects was, therefore, a major challenge. Once these variables were determined, it was established a correlation model that allows evaluating how the dependent quantitative variables can differ depending on the independent variable studied, in this case time and pH. Its representation on a response surface (two-dimensional representation) allows an overall evaluation of the impact of the independent variables on the measured instrumental response.62,63 The regression model obtained can be expressed with an equation of the type:
 | (1) |
Response surfaces are shown in Fig. 2. Fitted parameters values, goodness of fit (R2) and standard error of estimation (SEE) obtained from normalized second order polynomial model described according to eqn (1) for each evaluated compound are listed in Table 2.
 | ||
| Fig. 2 Normalized response surfaces of auxin activity (4-MU nmol min−1 μg−1 protein) for pH and time (h): (a) 2,4 D; (b) Ru2,4-D; (c) Ru′2,4-D; (d) NAA; (e) RuNAA; (f) Ru′NAA. The effect of time and pH on auxin activity is studied by response surface analysis once normalized variables between −1 and 1. | ||
| Coefficients | R 2 | SEE | ||||||
|---|---|---|---|---|---|---|---|---|
| b 0 | b 1 | b 2 | b 12 | b 11 | b 22 | |||
| The p-values are given in parentheses (in bold p < 0.05 indicates that the effects are significantly different from zero at 95% confidence level). | ||||||||
| 2,4-D | 0.0783 | −0.9108 (0.0000) | 0.1680 (0.0000) | −0.0289 (0.0143) | 0.0451 (0.1165) | 0.1467 (0.0003) | 0.9124 | 0.067 |
| Ru2,4-D | 0.0790 | −0.6871 (0.0000) | 0.1422 (0.0000) | −0.0778 (0.0000) | 0.0033 (0.8159) | 0.0754 (0.0003) | 0.9630 | 0.035 |
| Ru′2,4-D | 0.1301 | −0.0643 (0.0001) | 0.1846 (0.0000) | −0.0863 (0.0000) | −0.0018 (0.9351) | 0.0736 (0.0117) | 0.9424 | 0.0529 |
| NAA | 0.1409 | −0.0760 (0.0000) | 0.2091 (0.0000) | −0.0747 (0.0000) | 0.0120 (0.5275) | 0.0883 (0.0010) | 0.9651 | 0.0456 |
| RuNAA | 0.0660 | −0.0740 (0.0000) | 0.1221 (0.0000) | −0.0908 (0.0000) | 0.0218 (0.0267) | 0.0687 (0.0000) | 0.9820 | 0.0223 |
| Ru′NAA | 0.1173 | −0.0443 (0.0104) | 0.1346 (0.0000) | −0.0879 (0.0001) | 0.0551 (0.0629) | 0.0121 (0.7192) | 0.8595 | 0.065 |
As can be seen in Fig. 2 all graphs show curved surfaces corresponding to variations in instrumental response over time. In other words, the auxin activity does not have a linear dependence relationship neither the pH nor the time. On the one hand, the b1 coefficient is more negative in the compounds that contain DPhF as ancillary ligands, Ru2,4-D and RuNAA (Table 2). At the same pH the auxin activity is higher for the compounds with DAniF. On the other hand, although the b2 coefficient is lower for free 2,4-D auxin than for free NAA, diruthenium compounds containing NAA have the lowest b2 coefficients (Table 2), which indicates that 2,4-D is better outgoing ligand than NAA.
The quadratic coefficients of time are statistically significant at all pH values evaluated except for Ru′NAA. While the variation of the response with the pH to the different times evaluated is linear to any of the evaluated times (the quadratic coefficient is not statistically significant) except for RuNAA. Besides, the interaction between pH and time is significant (see b12, Table 2), except for 2,4-D, and, in all cases, b12 has a negative value as the b1 coefficient corresponding to the pH, which indicates that at high pH values and long times the contribution to the response will be lower. As it can be observed at 24 h (factor2 = +1) the response is the lowest at pH 7.5 (factor1 = +1) and the highest at pH 6.5 (factor1 = −1). As it can also be seen in Fig. 2, the lowest auxin activity at 24 h is obtained for RuNAA.
The registered activity is governed by the free/liberated auxin. As mentioned above, the release of the auxin depends on the pH and time, that is not linear, and also depends on the compound analysed. The comparison at all times and all pH of the auxin activity of diruthenium compounds reveals that the greatest activity is found for the Ru′2,4-D, that contains DAniF as ancillary ligands (Fig. 3).
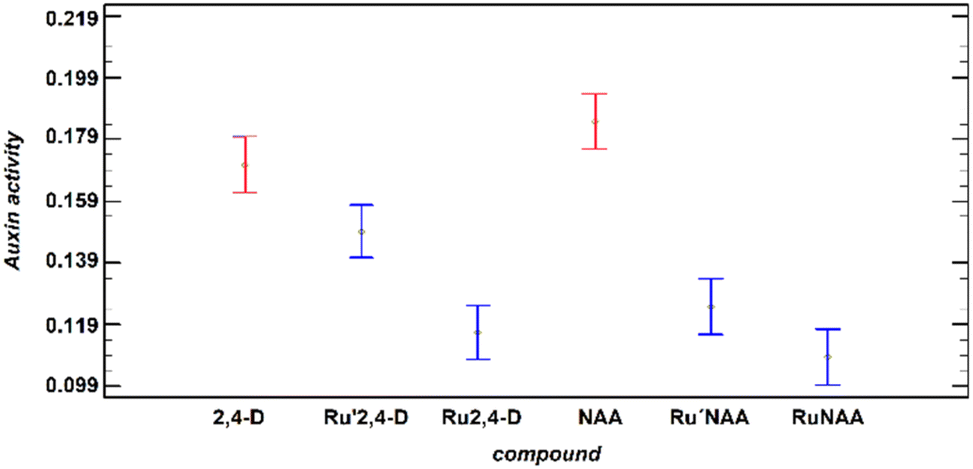 | ||
| Fig. 3 Auxin activity (4-MU nmol min−1 μg−1 protein) at all times and all pH observed for the free auxins and Ru′Aux and RuAux compounds. The interval has been estimated using the LSD statistic at 95.0% probability. | ||
The change of the formamidinate to the less donor ligand DPhF (Ru2,4-D) produces a significant deceleration of the 2,4-D release. This result means that the “Ru2Cl(DPhF)3” moiety can be used as a pH-responsive delivery system for RCOO− substances coordinated as a bridge to the diruthenium species through the carboxylate group.
The change of the outgoing ligand from Ru′2,4-D to Ru′NAA also implies a significant slowdown. In fact, there is a relationship between the auxin activity, that is directly related to the rate of the release, and the Ru1–O1carboxylate distance (Ru1 is the hexacoordinated metal centre) in diruthenium derivatives (Table 3). The distance is longer, and faster the release, for 2,4-D derivatives which could mean that this bond is broken in the rate determining step (see Scheme 2) of the substitution of the auxin by preferably another bridging ligand. Interestingly, the change of the formamidinate does not provide a significant variation of the Ru1–O1 distance.
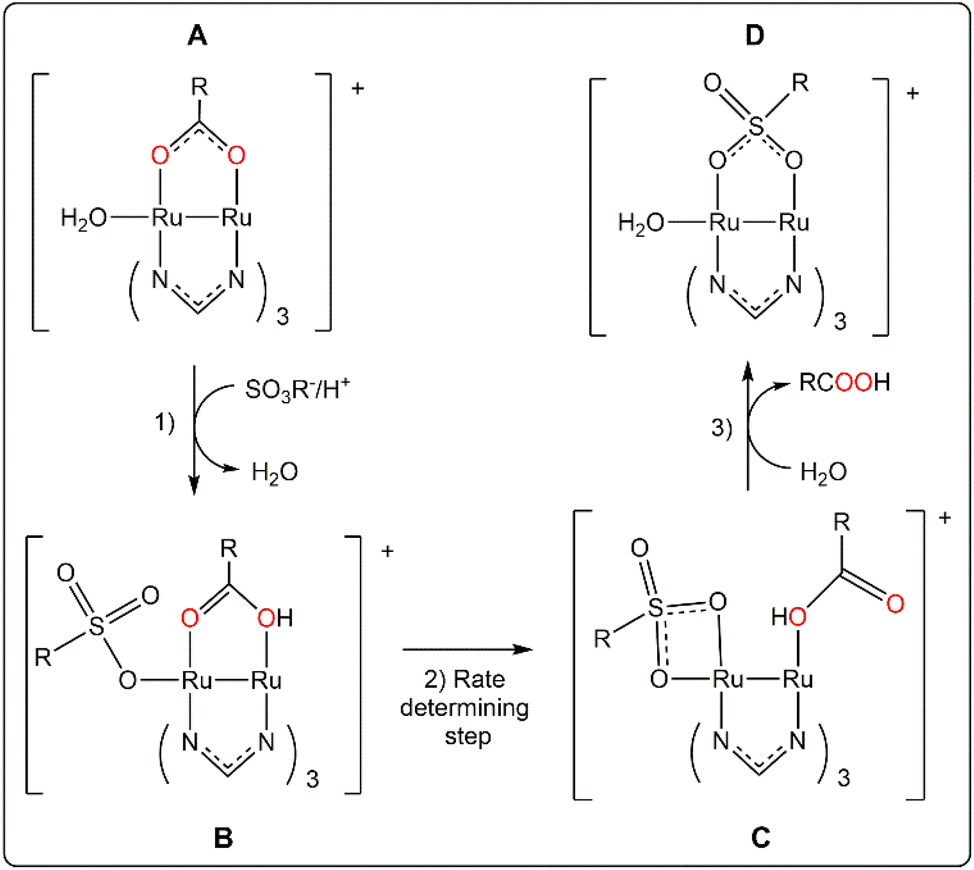 | ||
| Scheme 2 Scheme of the proposed mechanism for the acid-catalyzed substitution reaction of the carboxylate in RuAux and Ru′Aux compounds. | ||
| Compound | Ru2,4-D | RuNAA | Ru′2,4-D | Ru′NAA |
|---|---|---|---|---|
| Ru1–O1 (Å) | 2.115(3) | 2.082(3) | 2.113(5) | 2.079(4) |
At all pH, the activity found for the diruthenium compounds is significantly lower than those for the free auxins. However, at 6 h the variation of the activity with respect to the pH is similar for the diruthenium compounds and the corresponding free auxins (Fig. 4, up).
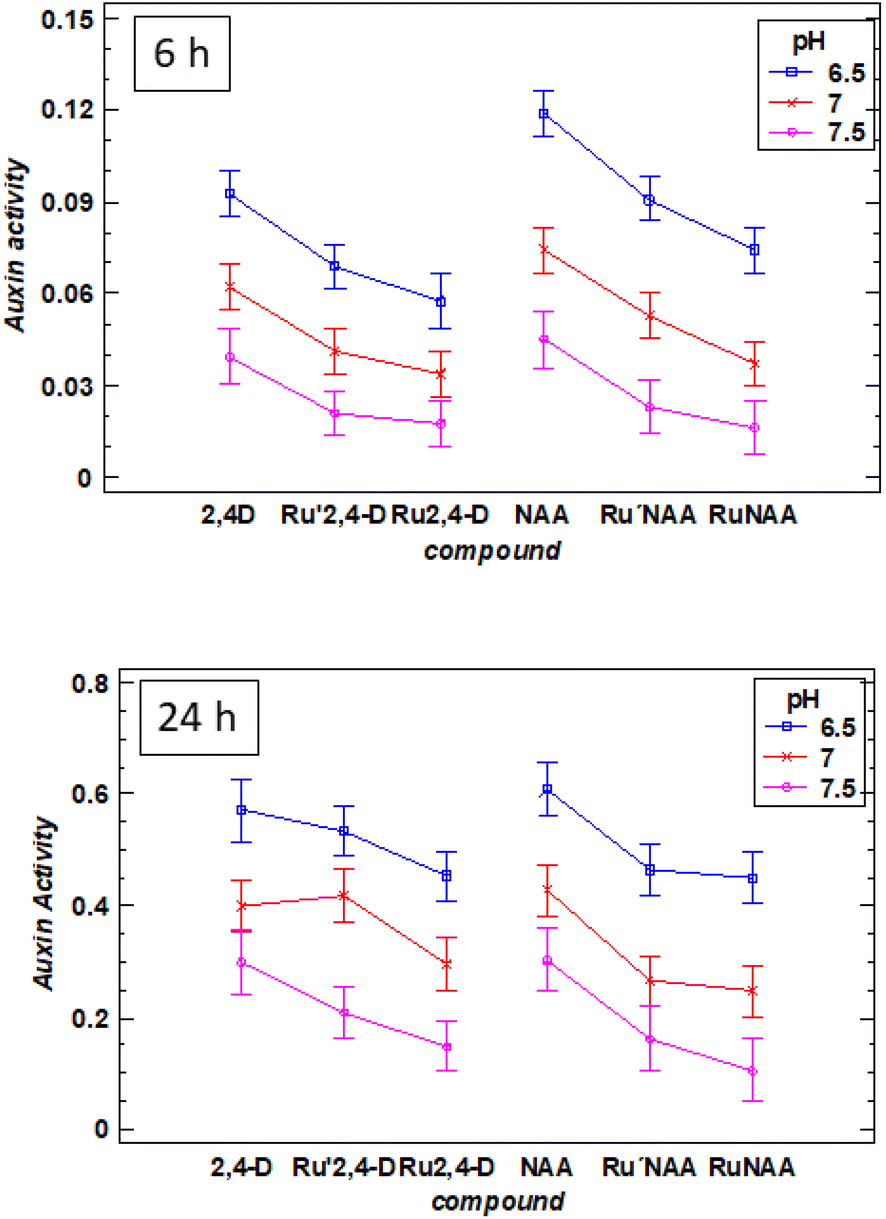 | ||
| Fig. 4 Variation of auxin activity (4-MU nmol min−1 μg−1 protein) as a function of pH (6.5, 7.0, 7.5) of 2,4-D, Ru′2,4-D, Ru2,4-D, NAA, Ru′NAA and RuNAA at 6 h (up) and 24 h (down). The interval has been estimated using the LSD statistic at 95.0% probability. | ||
At 24 h two extreme behaviors can be observed: Ru′2,4-D and RuNAA. Ru′2,4-D presents a similar auxin activity than the free 2,4-D, which means that most of the auxin has been released in the first 24 h (Fig. 4, down). However, Ru2,4-D, Ru′NAA and RuNAA have a significant difference in the auxin activity with respect to the free auxin. Moreover, the release of NAA from RuNAA is clearly dependent on pH, being especially slow at 7.5.
These data give additional information about a possible release mechanism that is hypothesized in Scheme 2.
Starting species A are proposed considering that their concentration in aqueous solution should be significant despite that chloride is better ligand than water for the ruthenium centres.64,65 The coordination of ligands at both axial positions is possible,54 but less likely with three formamidinate ligands.40
Species B are formed by substitution of the water axial ligand by the incoming ligand that in our experiment could be 3-(N-morpholino)propanesulfonate (two thousand times more concentrated than the diruthenium species). Even weakly binding anions can be bonded at certain degree to diruthenium paddlewheel species at their axial positions in aqueous solution.66,67 Previous reported calculations indicate that substitution of a water molecule at the axial position by an amino acid in tetracetatodiruthenium(II,III) compounds is kinetically favourable in acidic media.68 The coordination of the incoming ligand at the axial position can initiate the decomplexation of the bridging carboxylate by destabilization of the paddlewheel structure.68,69 This effect can be associated to the elongation of the Ru1–O1 bond distance above mentioned. The key formation of species B can explain why Ru25+ carboxylate compounds with paddlewheel structure are not reactive when the accessibility to the axial position is blocked by steric hindrance.29,70 The protonation of the bridging carboxylate of the species B facilitates the Ru1–O1 bond breaking off and explains the pH dependence of the release that is not correlated with the pKa of the auxin conjugate acid.
Already in 1988, Everhart and Earley suggested on the basis of kinetics studies that the substitution of a carboxylate in paddlewheel diruthenium(II,III) compounds could involve an associative activation by coordination of the incoming ligand. And, despite it was not known at that time any open-paddlewheel structure, they proposed an open-paddlewheel species as an intermediate compound.71 Open-paddlewheel structures are highly reactive in solution and tend to use any species that can form a bridge including a tetrafluoroborate53 or even promote the combination of species in the reaction media to act as the fourth bidentate ligand of the paddlewheel structure.72 Otherwise, they are quite unstable and decompose readily unless there is a strong donor solvent or ligand that stabilize them by coordination to the axial position.53,73
Species C would be the intermediate compound. They are proposed considering that the chelate coordination of the entering ligand is similar to the nitrate ligands in [Ru2(NO3)2(DPhF)3].53 The stability of these species would increase with the donor capacity of the formamidinate ligands. A small decrease of the activation barrier by DAniF ligands should increase the release rate as it is observed. Finally, the recovery of the quite more stable paddlewheel structure, species D, would be the final step.
Conclusions
The quantitative study presented herein, strongly supports the applicability of diruthenium complexes as pH-responsive drug delivery systems. It is confirmed that the release of the carboxylate ligands is affected by the electronic nature of the formamidinate ligands. In particular, those compounds with DPhF ancillary ligands (less donor) fit better for the design of diruthenium drug carriers. The nature of the carboxylate outgoing group also influences its pH-dependence release. The Ru1–O1 bond distance can be used as a guide to expect a faster or slower release. In any case, it should be considered that the presence of appreciable chloride concentration will slow down the release of the drug.64,67 The data obtained in this study together with the reported findings in the literature have made possible to hypothesize a mechanism of the substitution reaction of the carboxylate by other bridging ligand in Ru25+ paddlewheel tris(formamidinato) complexes.We are examining the efficiency of our synthetic method to prepare similar complexes bearing anti-tumour prodrugs coordinated to the diruthenium core through a carboxylate group. Work in this direction is currently underway.
Author contributions
S. H., B. D. and M. C. designed the work, I. C. carried out the synthesis and general characterisation of the compounds, B. D. and I. C. carried out the biological assays, M. C. and I. C. were responsible for the crystal structure determinations and the analysis of the magnetic properties, M. E. L.-G. and M. J. M. were responsible of the chemometric analysis, S. H., B. D. and M. C. directed the project with the support of C. G., M. C., I. C., S. H. and B. D. wrote the manuscript with the input of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Comunidad de Madrid (B2017/BMD-3770-CM) to S. H. and the Spanish Ministry of Science and Innovation (RTI2018-094793-B-I00) to C. G. and by institutional grants from Fundación Ramón Areces and Banco de Santander to the Centro de Biología Molecular Severo Ochoa. I. C. acknowledges a predoctoral grant (CT82/20-CT83/20) from Complutense University of Madrid and Banco de Santander.Notes and references
- P. Szturz, V. Cristina, R. G. Herrera Gómez, J. Bourhis, C. Simon and J. B. Vermorken, Cisplatin Eligibility Issues and Alternative Regimens in Locoregionally Advanced Head and Neck Cancer: Recommendations for Clinical Practice, Front. Oncol., 2019, 9, 464 CrossRef PubMed.
- A. Stein and D. Arnold, Oxaliplatin: a review of approved uses, Expert Opin. Pharmacother., 2012, 13, 125–137 CrossRef CAS PubMed.
- H. Calvert, The clinical development of carboplatin – A personal perspective, Inorg. Chim. Acta, 2019, 498, 118987 CrossRef CAS.
- A. C. L. Mortensen, T. Mohajershojai, M. Hariri, M. Pettersson and D. Spiegelberg, Overcoming Limitations of Cisplatin Therapy by Additional Treatment With the HSP90 Inhibitor Onalespib, Front. Oncol., 2020, 10, 532285 CrossRef PubMed.
- R. Oun, Y. E. Moussa and N. J. Wheate, The side effects of platinum-based chemotherapy drugs: a review for chemists, Dalton Trans., 2018, 47, 6645–6653 RSC.
- B. H. Ali, Amelioration of Oxaliplatin Neurotoxicity by Drugs in Humans and Experimental Animals: A MiniReview of Recent Literature, Basic Clin. Pharmacol. Toxicol., 2010, 106, 272–279 CAS.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, The development of anticancer ruthenium(II) complexes: from single molecule compounds to nanomaterials, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- S. Pete, N. Roy and P. Paira, A review on homo multinuclear anticancer Metallotherapuetics, Inorg. Chim. Acta, 2021, 517, 120184 CrossRef CAS.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, A Phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent, Clin. Cancer Res., 2004, 10, 3717 CrossRef CAS PubMed.
- L. Gu, X. Li, Q. Ran, C. Kang, C. Lee and J. Shen, Antimetastatic activity of novel ruthenium(III) pyridine complexes, Cancer Med., 2016, 5, 2850–2860 CrossRef CAS PubMed.
- K. Lin, Z.-Z. Zhao, H.-B. Bo, X.-J. Hao and J.-Q. Wang, Applications of Ruthenium Complex in Tumor Diagnosis and Therapy, Front. Pharmacol., 2018, 9, 1323 CrossRef CAS PubMed.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, From bench to bedside – preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A), J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, KP1019, A New Redox-Active Anticancer Agent – Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients, Chem. Biodivers., 2008, 5, 2140–2155 CrossRef CAS PubMed.
- A. K. Bytzek, G. Koellensperger, B. K. Keppler and C. G. Hartinger, Biodistribution of the novel anticancer drug sodium trans-[tetrachloridobis(1H-indazole)ruthenate(III)] KP-1339/IT139 in nude BALB/c mice and implications on its mode of action, J. Inorg. Biochem., 2016, 160, 250–255 CrossRef CAS PubMed.
- E. Alessio and L. Messori, NAMI-A and KP1019/1339, Two Iconic Ruthenium Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry, Molecules, 2019, 24, 1995 CrossRef CAS PubMed.
- H. A. Burris, S. Bakewell, J. C. Bendell, J. Infante, S. F. Jones, D. R. Spigel, G. J. Weiss, R. K. Ramanathan, A. Ogden and D. Von Hoff, Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: a first-in-human, open-label, dose-escalation phase I study with expansion cohort, ESMO Open, 2016, 1, e000154 CrossRef PubMed.
- D. A. Smithen, H. Yin, M. H. R. Beh, M. Hetu, T. S. Cameron, S. A. McFarland and A. Thompson, Synthesis and Photobiological Activity of Ru(II) Dyads Derived from Pyrrole-2-carboxylate Thionoesters, Inorg. Chem., 2017, 56, 4121–4132 CrossRef CAS PubMed.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- C. Sonkar, S. Sarkar and S. Mukhopadhyay, Ruthenium(II)–arene complexes as anti-metastatic agents, and related techniques, RSC Med. Chem., 2022, 13, 22–38 RSC.
- Y. Xu, C. Li, S. Lu, Z. Wang, S. Liu, X. Yu, X. Li and Y. Sun, Construction of emissive ruthenium(II) metallacycle over 1000 nm wavelength for in vivo biomedical applications, Nat. Commun., 2022, 13, 2009 CrossRef CAS PubMed.
- D. de Oliveira Silva, Frontiers in Anti-Cancer Drug Discovery, Bentham Science, 2014, vol. 4, pp. 88–156 Search PubMed.
- M. A. S. Aquino, Recent developments in the synthesis and properties of diruthenium tetracarboxylates, Coord. Chem. Rev., 2004, 248, 1025–1045 CrossRef CAS.
- B. K. Keppler, M. R. Berger and M. E. Heim, New tumor-inhibiting metal complexes, Cancer Treat. Rev., 1990, 17, 261–277 CrossRef CAS PubMed.
- C. E. J. Van Rensburg, E. Kreft, J. C. Swarts, S. R. Dalrymple, D. M. MacDonald, M. W. Cooke and M. A. S. Aquino, Cytotoxicity of a series of water-soluble mixed valent diruthenium tetracarboxylates., Anticancer Res., 2002, 22, 889–892 CAS.
- G. Ribeiro, M. Benadiba, A. Colquhoun and D. de Oliveira Silva, Diruthenium(II,III) complexes of ibuprofen, aspirin, naproxen and indomethacin non-steroidal anti-inflammatory drugs: Synthesis, characterization and their effects on tumor-cell proliferation, Polyhedron, 2008, 27, 1131–1137 CrossRef CAS.
- M. Benadiba, I. d. M. Costa, R. L. S. R. Santos, F. O. Serachi, D. de Oliveira Silva and A. Colquhoun, Growth inhibitory effects of the Diruthenium-Ibuprofen compound, [Ru2Cl(Ibp)4], in human glioma cells in vitro and in the rat C6 orthotopic glioma in vivo, J. Biol. Inorg. Chem., 2014, 19, 1025–1035 CrossRef CAS PubMed.
- S. R. Alves Rico, A. Z. Abbasi, G. Ribeiro, T. Ahmed, X. Y. Wu and D. de Oliveira Silva, Diruthenium(II,III) metallodrugs of ibuprofen and naproxen encapsulated in intravenously injectable polymer–lipid nanoparticles exhibit enhanced activity against breast and prostate cancer cells, Nanoscale, 2017, 9, 10701–10714 RSC.
- S. R. Alves, A. Colquhoun, X. Y. Wu and D. de Oliveira Silva, Synthesis of terpolymer-lipid encapsulated diruthenium(II,III)-anti-inflammatory metallodrug nanoparticles to enhance activity against glioblastoma cancer cells, J. Inorg. Biochem., 2020, 205, 110984 CrossRef CAS PubMed.
- E. Barresi, I. Tolbatov, T. Marzo, E. Zappelli, A. Marrone, N. Re, A. Pratesi, C. Martini, S. Taliani, F. Da Settimo and D. La Mendola, Two mixed valence diruthenium(II,III) isomeric complexes show different anticancer properties, Dalton Trans., 2021, 50, 9643–9647 RSC.
- V. Schirrmacher, From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review), Int. J. Oncol., 2019, 54, 407–419 CrossRef CAS PubMed.
- S. Y. Lee, C. Y. Kim and T.-G. Nam, Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives, Drug Des., Dev. Ther., 2020, 14, 5375–5392 CrossRef CAS PubMed.
- A. Emami Nejad, S. Najafgholian, A. Rostami, A. Sistani, S. Shojaeifar, M. Esparvarinha, R. Nedaeinia, S. Haghjooy Javanmard, M. Taherian, M. Ahmadlou, R. Salehi, B. Sadeghi and M. Manian, The role of hypoxia in the tumor microenvironment and development of cancer stem cell: a novel approach to developing treatment, Cancer Cell Int., 2021, 21, 62 CrossRef CAS PubMed.
- G. Hao, Z. P. Xu and L. Li, Manipulating extracellular tumour pH: an effective target for cancer therapy, RSC Adv., 2018, 8, 22182–22192 RSC.
- H. Ding, P. Tan, S. Fu, X. Tian, H. Zhang, X. Ma, Z. Gu and K. Luo, Preparation and application of pH-responsive drug delivery systems, J. Controlled Release, 2022, 348, 206–238 CrossRef CAS PubMed.
- S. Abdella, F. Abid, S. H. Youssef, S. Kim, F. Afinjuomo, C. Malinga, Y. Song and S. Garg, pH and its applications in targeted drug delivery, Drug Discovery, 2023, 28, 103414 CAS.
- M. A. S. Aquino, Diruthenium and diosmium tetracarboxylates: synthesis, physical properties and applications, Coord. Chem. Rev., 1998, 170, 141–202 CrossRef CAS.
- A. Terán, M. Cortijo, Á. Gutiérrez, A. E. Sánchez-Peláez, S. Herrero and R. Jiménez-Aparicio, Ultrasound-assisted synthesis of water-soluble monosubstituted diruthenium compounds, Ultrason. Sonochem., 2021, 80, 105828 CrossRef PubMed.
- M. K. Viswanadh, N. Agrawal, S. Azad, A. Jha, S. Poddar, S. K. Mahto and M. S. Muthu, Novel redox-sensitive thiolated TPGS based nanoparticles for EGFR targeted lung cancer therapy, Int. J. Pharm., 2021, 602, 120652 CrossRef CAS PubMed.
- I. Coloma, M. Cortijo, I. Fernández-Sánchez, J. Perles, J. L. Priego, C. Gutiérrez, R. Jiménez-Aparicio, B. Desvoyes and S. Herrero, pH- and Time-Dependent Release of Phytohormones from Diruthenium Complexes, Inorg. Chem., 2020, 59, 7779–7788 CrossRef CAS PubMed.
- A. Inchausti, A. Terán, A. Manchado-Parra, A. de Marcos-Galán, J. Perles, M. Cortijo, R. González-Prieto, S. Herrero and R. Jiménez-Aparicio, New insights into progressive ligand replacement from [Ru2Cl(O2CCH3)4]: synthetic strategies and variation in redox potentials and paramagnetic shifts, Dalton Trans., 2022, 51, 9708–9719 RSC.
- A. Terán, G. Ferraro, A. E. Sánchez Peláez, S. Herrero and A. Merlino, Effect of equatorial ligand substitution on the reactivity with proteins of paddlewheel diruthenium complexes: structural studies, Inorg. Chem., 2023, 62, 670–674 CrossRef PubMed.
- H. Ding, in Applied Biopharmaceutics & Pharmacokinetics, ed. L. Shargel and A. B. C. Yu, McGraw-Hill Education, New York, NY, 7th edn, 2016 Search PubMed.
- R. W. Mitchell, A. Spencer and G. Wilkinson, Carboxylato-triphenylphosphine complexes of ruthenium, cationic triphenylphosphine complexes derived from them, and their behaviour as homogeneous hydrogenation catalysts for alkenes, J. Chem. Soc., Dalton Trans., 1973, 846–854 RSC.
- R. M. Roberts, The reaction of diarylformamidines with ethyl malonate, J. Org. Chem., 1949, 14, 277–284 CrossRef CAS PubMed.
- P. A. Angaridis, F. A. Cotton, C. A. Murillo, A. S. Filato and M. A. Petrukhina, Inorganic Syntheses, John Wiley & Sons, Ltd, 2014, vol. 36, pp. 114–121 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- T. Ulmasov, J. Murfett, G. Hagen and T. J. Guilfoyle, Aux/IAA Proteins Repress Expression of Reporter Genes Containing Natural and Highly Active Synthetic Auxin Response Elements, Plant Cell, 1997, 9, 1963–1971 CAS.
- M. M. Bradford, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- C. Mongay Fernández, Quimiometría, Universitat De València, 2014 Search PubMed.
- M. C. Barral, S. Herrero, R. Jiménez-Aparicio, M. R. Torres and F. A. Urbanos, Preparation, characterization, and crystal structure of [Ru2Cl(μ-O2CMe)(μ-PhNCHNPh)3]: a way to tris(formamidinato)diruthenium(II,III) complexes, Inorg. Chem. Commun., 2004, 7, 42–46 CrossRef CAS.
- M. C. Barral, T. Gallo, S. Herrero, R. Jiménez-Aparicio, M. R. Torres and F. A. Urbanos, The First Open-Paddlewheel Structures in Diruthenium Chemistry: Examples of Intermediate Magnetic Behaviour between Low and High Spin in Ru25+ Species, Chem. – Eur. J., 2007, 13, 10088–10095 CrossRef CAS PubMed.
- M. C. Barral, T. Gallo, S. Herrero, R. Jiménez-Aparicio, M. R. Torres and F. A. Urbanos, Equatorially Connected Diruthenium(II,III) Units toward Paramagnetic Supramolecular Structures with Singular Magnetic Properties, Inorg. Chem., 2006, 45, 3639–3647 CrossRef CAS PubMed.
- E. Añez, S. Herrero, R. Jiménez-Aparicio, J. L. Priego, M. R. Torres and F. A. Urbanos, Controlling the electronic density of the [Ru2(DPhF)3(O2CR)]+ core to obtain one-dimensional compounds, Polyhedron, 2010, 29, 232–237 CrossRef.
- S. Herrero, R. Jiménez-Aparicio, J. Perles, J. L. Priego, S. Saguar and F. A. Urbanos, Microwave methods for the synthesis of paddlewheel diruthenium compounds with N,N-donor ligands, Green Chem., 2011, 13, 1885–1890 RSC.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Part B, John Wiley & Sons, Ltd, Hoboken, New Jersey, 2009, pp. 1–273 Search PubMed.
- C. Lin, T. Ren, E. J. Valente, J. D. Zubkowski and E. T. Smith, Continuous Spectroscopic and Redox Tuning of Dinuclear Compounds: Chlorotetrakis(μ-N,N′-diarylformamidinato)diruthenium(II,III), Chem. Lett., 1997, 26, 753–754 CrossRef.
- W.-Z. Chen and T. Ren, Selective Ligand Modification on the Periphery of Diruthenium Compounds: Toward New Metal-Alkynyl Scaffolds, Organometallics, 2005, 24, 2660–2669 CrossRef CAS.
- J. L. Bear, B. Han, S. Huang and K. M. Kadish, Effect of Axial Ligands on the Oxidation State, Structure, and Electronic Configuration of Diruthenium Complexes. Synthesis and Characterization of Ru2(dpf)4Cl, Ru2(dpf)4(C⋮CC6H5), Ru2(dpf)4(C⋮CC6H5)2, and Ru2(dpf)4(CN)2 (dpf = N,N′-Diphenylformamidinate), Inorg. Chem., 1996, 35, 3012–3021 CrossRef CAS.
- M. Cortijo, R. González-Prieto, S. Herrero, J. L. Priego and R. Jiménez-Aparicio, The use of amidinate ligands in paddlewheel diruthenium chemistry, Coord. Chem. Rev., 2019, 400, 213040 CrossRef CAS.
- G. Moreno-Martin, J. Sanz-Landaluze, M. E. León-Gonzalez and Y. Madrid, In-vivo solid phase microextraction for quantitative analysis of volatile organoselenium compounds in plants, Anal. Chim. Acta, 2019, 1081, 72–80 CrossRef CAS PubMed.
- E. Gómez-Mejía, L. H. Mikkelsen, N. Rosales-Conrado, M. E. León-González and Y. Madrid, A combined approach based on matrix solid-phase dispersion extraction assisted by titanium dioxide nanoparticles and liquid chromatography to determine polyphenols from grape residues, J. Chromatogr. A, 2021, 1644, 462128 CrossRef PubMed.
- R. L. S. R. Santos, R. van Eldik and D. de Oliveira Silva, Thermodynamics of Axial Substitution and Kinetics of Reactions with Amino Acids for the Paddlewheel Complex Tetrakis(acetato)chloridodiruthenium(II,III), Inorg. Chem., 2012, 51, 6615–6625 CrossRef CAS PubMed.
- M. C. Barral, R. González-Prieto, S. Herrero, R. Jiménez-Aparicio, J. L. Priego, M. R. Torres and F. A. Urbanos, Anionic dihalotetraacetatodiruthenium(II,III) compounds, Polyhedron, 2005, 24, 239–247 CrossRef CAS.
- K. Dunlop, R. Wang, T. Stanley Cameron and M. A. S. Aquino, Coordination of weakly binding anions to [Ru2(μ-O2CCH3)4]+ in aqueous solution, J. Mol. Struct., 2014, 1058, 122–129 CrossRef CAS.
- R. L. S. R. Santos, R. van Eldik and D. de Oliveira Silva, Kinetic and mechanistic studies on reactions of diruthenium(II,III) with biologically relevant reducing agents, Dalton Trans., 2013, 42, 16796–16805 RSC.
- I. Tolbatov and A. Marrone, Kinetics of Reactions of Dirhodium and Diruthenium Paddlewheel Tetraacetate Complexes with Nucleophilic Protein Sites: Computational Insights, Inorg. Chem., 2022, 61, 16421–16429 CrossRef CAS PubMed.
- I. Tolbatov and A. Marrone, Reaction of dirhodium and diruthenium paddlewheel tetraacetate complexes with nucleophilic protein sites: A computational study, Inorg. Chim. Acta, 2022, 530, 120684 CrossRef CAS.
- E. Barresi, I. Tolbatov, A. Pratesi, V. Notarstefano, E. Baglini, S. Daniele, S. Taliani, N. Re, E. Giorgini, C. Martini, F. Da Settimo, T. Marzo and D. La Mendola, A mixed-valence diruthenium(II,III) complex endowed with high stability: from experimental evidence to theoretical interpretation, Dalton Trans., 2020, 49, 14520–14527 RSC.
- M. Everhart and J. E. Earley, Kinetics of replacement of bridging ligands in a diruthenium(II,III) cation, Polyhedron, 1988, 7, 1393–1396 CrossRef CAS.
- M. C. Barral, S. Herrero, R. Jiménez-Aparicio, J. L. Priego, M. R. Torres and F. A. Urbanos, Activation of isocyanate ligands in Ru25+ complexes, J. Mol. Struct., 2008, 890, 221–226 CrossRef CAS.
- G. Lozano, R. Jimenez-Aparicio, S. Herrero and E. Martinez-Salas, Fingerprinting the junctions of RNA structure by an open-paddlewheel diruthenium compound, RNA, 2016, 22, 330–338 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2235327 (Ru′2,4-D) and 2235329 (Ru′NAA). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi00399j |
| This journal is © the Partner Organisations 2023 |