
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient fluorescence of alkali metal carbazolides†
Michelle
Kaiser
,
Maximilian P.
Müller
,
Frederic
Krätschmer
,
Mark
Rutschmann
and
Alexander
Hinz
 *
*
Karlsruhe Institute of Technology (KIT), Institute of Inorganic Chemistry (AOC), Engesserstr. 15, Geb. 30.45, 76131 Karlsruhe, Germany. E-mail: alexander.hinz@kit.edu
First published on 24th March 2023
Abstract
The sterically demanding 1,8-bis(3,5-ditertbutylphenyl)-3,6-di-tert-butylcarbazole (dtbpCbz)–H is deprotonated with various metal bases to obtain a complete series of thermally robust alkali metal compounds of the type [(dtbpCbz)M] (M = Li, Na, K, Rb, Cs). These carbazolides can either be solely coordinated by carbazole ligands (1a–5a) or bear additional toluene ligands (1b–5b). In the cases of [(dtbpCbz)Li] (1a), [(dtbpCbz)K] (3a) and [(dtbpCbz)Rb] (4a), monomeric species were identified by single crystal XRD. Additionally, a carbazolide anion was incorporated into [NBu4][dtbpCbz] (6). All compounds were strongly luminescent, so PL and PLE measurements were conducted, revealing emissions in the range of 460–580 nm. The excited states possess a lifetime of up to 21 ns, so fluorescence phenomena were observed. Quantum yields at ambient temperature were measured up to 29% in the solid state and up to 100% in solution. In the series of 1a–4a, the emission maxima shift to higher energy as the alkaline earth metal cation becomes heavier, and the lifetimes of the excited state decrease. The Cs compounds show low quantum yields and lifetimes of the excited state.
Introduction
In recent years, derivatives of carbazole have been studied intensively with a focus on their optical properties with the goal of applications as materials in OLEDs and OFETs.1–3 Depending on substitution patterns, such as N-substitution, extension of aromatic frameworks, and energy level manipulation of HOMO and LUMO by substituents on carbazole, interesting properties such as redox activity and luminescence phenomena have been discovered.4,5 For instance, Hou and co-workers developed carbazole-substituted aromatic (E) and (Z) enynes, which emit almost “pure” white light with a high luminescence efficiency, so they can accordingly act as a single-emitting-component WOLED (white organic light-emitting device) (Scheme 1, A).4 Apart from being a useful building block in organic materials, carbazolides can also act as anionic ligands. A landmark example of this class of compounds is carbene-coordinated Cu(I) carbazolides [(HCbz)Cu(CAAC)] (HCbz = parent carbazolide, CAAC = cyclic alkyl amino carbene) which have long lifetimes of the excited state and near-quantitative quantum yields.6,7 Fu and Peters investigated the copper-catalyzed photochemical coupling of carbazoles with haloarenes and haloalkanes and established a radical reaction pathway.8,9 The excited lithium carbazolide [(HCbz)Li] was identified as the active reducing species in this transformation and achieved a quantum yield of 10%.10 | ||
| Scheme 1 Selected examples of the compounds with a carbazole moiety. | ||
The carbazole scaffold has also been widely used for the preparation of pincer ligands11–13 which are applied in the chemistry of main group elements, transition metals and f-block elements.14–16 Examples of various tridentate carbazolides span a broad range of donor atoms and could be tuned for complexation of various elements. Bezuidenhout and Kunz developed bis(carbene)carbazolides,11,12 while the group of Gade focused on another bis(phosphine)-carbazole-based PNP pincer ligand,14,17 and carbazole-based NNN pincer ligands with bis(imino) functionalities were employed by Sarazin and Twamley.13,18–20 However, the luminescence properties of those compounds were not routinely investigated.
Hohloch, Guldi and Munz have only recently reported on Li and Mg (B) complexes of a carbazole-based CNC pincer ligand and observed quantum yields of up to 16% in solution with the corresponding precursor compounds, the protonated ligand and the monodeprotonated ligand, being non-luminescent.21 Recently, main group metal compounds with various different ligand sets have been discovered, including Mg 2,2′-pyridylpyrrolide complexes,22 phosphine-functionalised diamido compounds of alkaline earth metals23 and dinuclear Al complexes24 that feature luminescence with high quantum yields.
Monodentate carbazolides of the alkali metals frequently aggregate in the absence of strong donors, similar to the parent carbazole.25,26 For instance, Aldridge reported that the deprotonation of phenyl-substituted 1,8-diphenyl-3,6-dimethylcarbazole with a potassium base afforded a dimeric product.27 The frequently used 1,2,6,8-tetra-tert-butylcarbazolide can form either a dimer with Li as the counterion or a monomeric compound with additional coordinated THF molecules (C).28
In previous works, our group prepared potassium carbazolide [(dtbpCbz)K] (3a) by deprotonation of the (dtbpCbz)–H ligand with benzyl potassium.29–32 It was noted that the compound is highly luminescent, but at that time, metathesis reactions were the focus of the study. In order to gain detailed insights into the luminescence behaviour of the carbazolide entity and the influence of the coordinated group 1 element, two series of compounds were prepared: (a) unsolvated complexes [(dtbpCbz)M], M = Li (1a), Na (2a), K (3a), Rb (4a), and Cs (5a) and (b) toluene solvates [(dtbpCbz)M(Tol)n], M = Li (1b), Na (2b), K (3b), Rb (4b), and Cs (5b). For comparison, tetrabutylammonium carbazolide [NBu4][dtbpCbz] (6) was included in this study as well.
Results and discussion
The route for synthesis is straightforward starting from the protic ligand dtbpCbz–H (Scheme 2). Deprotonation can be carried out with alkali metal bases such as alkyls or amides. This reaction was conducted in n-hexane, and the unsolvated compounds [(dtbpCbz)M] (1a–5a) were formed. When the deprotonation reaction was carried out in a coordinating solvent such as benzene, toluene or THF, solvated products were obtained, and the series of toluene solvates (1b–5b) was investigated in this contribution. The coordinated solvents can be removed in vacuo at elevated temperatures. | ||
| Scheme 2 General reaction scheme for the synthesis of the alkali metal and the tetrabutylammonium carbazolides. M[base] = LiMe, Li[N(SiMe3)2], Na[N(SiMe3)2], K[N(SiMe3)2], Rb[N(SiMe3)2], and Cs[N(SiMe3)2]. | ||
The “naked” carbazolide salt [NBu4][dtbpCbz] (6) was prepared by salt metathesis of [(dtbpCbz)K] with [NBu4]I in THF. The solubility shows a remarkable trend: the lithium carbazolides 1a and 1b are highly soluble in both toluene and n-hexane. The solubility of the carbazolide decreases considerably as the alkali metal in it becomes heavier, and the caesium carbazolide is moderately soluble in toluene but virtually insoluble in n-hexane. Surprisingly, the tetrabutylammonium carbazolide 6 is only sparingly soluble even in THF. All compounds appear yellow and show visible luminescence. The carbazolide compounds are thermally robust. The lithium and sodium carbazolides melt at 309 °C (1a), 286 °C (1b), 173 °C (2a) and 201 °C (2b), respectively, while the carbazolides with organic counterions decompose at 235 °C. The potassium, rubidium and caesium compounds appear to be stable up to 350 °C.
For all compounds 1a–4a, 1b–5b and 6, single crystals suitable for X-ray structure elucidation could be obtained, but 5a could not be crystallised from non-coordinating solvents due to its insolubility. For [(dtbpCbz)Li] (1a), [(dtbpCbz)K] (3a) and [(dtbpCbz)Rb] (4a), a monomeric structure was found in which the metal is coordinated with the plane of the carbazole scaffold in between the flanking arene moieties in the coordination mode this ligand was designed for. Surprisingly, the Na-carbazolide (2a) differed in its behaviour and formed a dimer in the solid state. The molecular structures of [(dtbpCbz)Li] (1a) and [(dtbpCbz)Rb] (4a) are depicted in Fig. 1 (top).
 | ||
| Fig. 1 Molecular structures of the unsolvated alkali metal carbazolides 1a, 4a, 2a and 6. | ||
The alkali metals are coordinated by the N donor as a σ-bonding ligand of the carbazole scaffold and a π-interaction with the flanking aryl ligands. The N–M bond length increases from 1.877(4) Å for 1a (Li) to 2.6122(13) Å for 3a (K) and to 2.740(2) Å for 4a (Rb), reflecting the increasing ionic radius of the alkali metal ion. This also has an impact on the dihedral angle between the carbazole plane and the flanking arenes. In the Li compound, both arenes are tilted in the same direction (119.9°, 54.4°) to reduce the size of the “pocket” formed by the ligand and establish π-interactions. In contrast, the flanking arenes in [(dtbpCbz)K] (3a, 93.9°, 96.1°) and [(dtbpCbz)Rb] (4a, 96.9°, 97.1°) are tilted in opposite directions, but they differ only slightly from 90°. This shows that K+ is a near-perfect fit for the dtbpCbz ligand. The Rb ion fits nearly as well, but the arenes are slightly more bent outwards to accommodate the ion. This is also reflected in the distance of the arene ipso-C atoms which increases from 5.510 Å in 1a to 5.652 Å in 4a.
The sodium complex [(dtbpCbz)Na] (2a) surprisingly crystallised as an unsymmetric dimer (Fig. 1, bottom). Two essentially coplanarly arranged carbazolide anions are bridged by Na cations which are bent out of the carbazole plane. There is a close N–Na contact (Na1–N1 2.338(2), Na2–N2 2.331(2) Å) and an interaction with one of the flanking arenes. Furthermore, Na1 has a contact with a six-membered ring (Na1-centroid 2.643 Å) of the other carbazole ligands, while Na2 features a similar interaction with the central five-membered heterocycle (Na1-centroid 2.640 Å) of the respective other carbazoles.
By performing the synthesis in toluene instead of n-hexane, the toluene-solvated alkali metal complexes were obtained. The number of coordinating toluene molecules per moiety increases with the size of the alkali metal cation from 1 in (1b), to 1.5 in (2b), and to 2 for K (3b), Rb (4b) and Cs (5b). For 3b, the data sets obtained by XRD are of poor quality and are only suitable for determining the number of coordinating toluene molecules. Therefore, 3b is not included in the following structural discussion.
Compared to the unsolvated alkali metal carbazolides (1a–4a), the metal ions in 1b–5b are significantly shifted out of the carbazole plane (Fig. 2), thus resulting in less steric shielding by the ligand. The Li–N contacts remain of the same length (1.877 Å), which is shorter than those in Kays’ THF-containing Li carbazolide complexes (2.028(6) Å),28 while the Rb–N are slightly elongated (2.740, 2.823 Å). The metal atoms are bent out of the carbazole plane by 24.3° (1b), 42.2° (2b), 42.6° (4b) and 39.2° (5b). Consequently, they deviate from the planes by 0.84 Å, 1.518 Å, 1.90 Å and 1.84 Å, respectively.
 | ||
| Fig. 2 Molecular structures of the toluene-solvated alkali metal carbazolides of 1b, 2b, 4b, and 5b. | ||
In 6, the carbazolyl ligand is present as a “naked” anion with the tetrabutylammonium as the counterion outside the ligand pocket. The flanking arene moieties are twisted in the same direction and show dihedral angles to the carbazole planes of 26.8° and 21.4°.
The 1H NMR spectra of the series of alkali metal carbazolides show three sets of resonances with a monotonic trend in the sequence from 1a to 5a. The singlet of the arene tertbutyl groups (1a: 1.63 ppm, 2a: 1.66 ppm, 3a: 1.69 ppm, 4a: 1.70 ppm, and 5a: 1.71 ppm) and the doublet of the C4,5H protons are shifted downfield (1a: 8.70 ppm, 2a: 8.71 ppm, 3a: 8.73 ppm, 4a: 8.80 ppm, and 5a: 8.78 ppm), while the p-CH resonance is shifted upfield (1a: 7.50 ppm, 2a: 7.46 ppm, 3a: 7.36 ppm, 4a: 7.29 ppm, 5a: 7.26 ppm) as the alkali metal ions get heavier. Due to the similarity of all the spectra, it can be concluded that the difference in coordination displayed in the solid state does not persist in the benzene solution.
The UV/vis absorption spectra of the carbazolide complexes 1–5 in the toluene solution each show a broad band in the range of 400–430 nm. According to the TD-DFT calculations, in each instance, multiple transitions contribute to this band (see ESI 5.2† for further details).
The photoluminescence emission (PL) and photoluminescence excitation (PLE) measurements of all the samples were conducted in the solid state at room temperature (Fig. 3, bottom and ESI†). The protonated ligand is a colourless solid which exhibits purple-blue photoluminescence under UV excitation, with an emission maximum at 385 nm, a quantum yield of 9% and a lifetime of the excited state of 2 ns. In contrast, all the investigated carbazolide compounds are yellow solids that feature visible green-blue photoluminescence as illustrated in Fig. 3 (top). Small differences in the behaviour of toluene-solvated and unsolvated complexes were observed and are summarised in Table 1. The PLE measurements revealed a broad and merged set of bands below 500 nm, which is consistent with the yellow colour of the compounds.
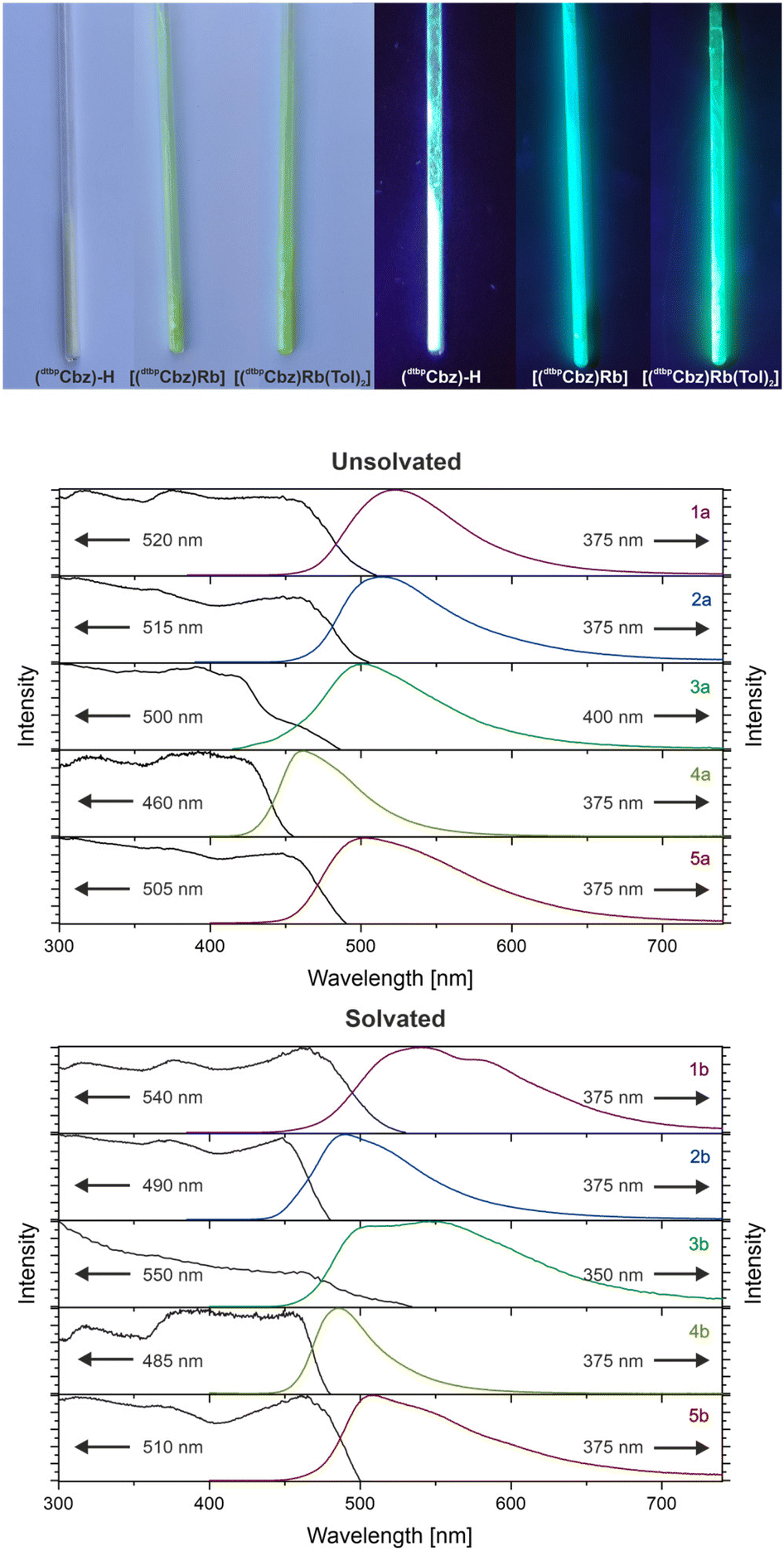 | ||
| Fig. 3 Top: comparison of solid samples of (dtbpCbz)–H and the Rb carbazolides 4a and 4b in daylight and under UV excitation. Bottom: normalised PLE and PL spectra of the solid alkali metal carbazolides at 295 K. The arrows pointing towards the right indicate the excitation wavelengths, and the arrows pointing towards the left indicate the emission maximum wavelengths. | ||
| Φ solid [%] | Φ solution [%] | λ ex [nm] | λ em,max [nm] | τ FL [ns] | |
|---|---|---|---|---|---|
| a Solvent: toluene for all except 6, where THF was used. b Measured in the solid state. | |||||
| (dtbpCbz)–H | 9 | 350 | 385 | 2 | |
| 1a | 12 | 375 | 520 | 14 | |
| 1b | 8 | 100 | 375 | 540/580 | 13 |
| 2a | 9 | 375 | 515 | 14 | |
| 2b | 12 | 87 | 375 | 490/515 | 13 |
| 3a | 18 | 400 | 500 | 15 | |
| 3b | 14 | 80 | 350 | 510/550 | 11 |
| 4a | 29 | 375 | 460 | 9 | |
| 4b | 17 | 95 | 375 | 485 | 8 |
| 5a | 3 | 375 | 505 | 3 | |
| 5b | 3 | 30 | 375 | 510 | 4 |
| 6 | 15 | 76 | 375 | 520 | 21 |
The complexes show a broad photoluminescence (PL) band upon UV excitation (Table 1) which is likely to possess an underlying fine structure as observed by Fu and Peters for the parent carbazole.10 The tetrabutylammonium salt 6 shows an emission maximum at 520 nm and a quantum yield of 15%. The energy of the maxima increases from [(dtbpCbz)Li] to [(dtbpCbz)Rb], ([(dtbpCbz)Li] (1a): 520 nm, [(dtbpCbz)Na] (2a): 515 nm, [(dtbpCbz)K] (3a): 500 nm, and [(dtbpCbz)Rb] (4a): 460 nm), and decreases for [(dtbpCbz)Cs] (5a) to 505 nm. A similar trend was observed for the solvated complexes: for [(dtbpCbz)Li(Tol)] (1b), two merged emission bands at 540 and 580 nm occur, and [(dtbpCbz)Na(Tol)1.5] (2b) features two merged bands at 490 and 515 nm, while [(dtbpCbz)K(Tol)2] (3b) has its maxima at 510 and 550 nm, [(dtbpCbz)Rb(Tol)2] (4b) at 485 nm and [(dtbpCbz)Cs(Tol)2] (5b) at 510 nm.
The quantum yields for the complexes of Li, K and Rb are higher for the unsolvated complexes, indicating that the additionally coordinated toluene molecules enable non-radiative relaxation pathways. The lighter alkali metal carbazolides (Li and Na) show quantum yields of 12% and 9%, and this number increases for the heavier homologous K and Rb up to 18% and 29%, respectively. For the solvated compounds, the quantum yields increase with the increasing atomic number as well ([(dtbpCbz)Li(Tol)] (1b): 8%, [(dtbpCbz)K(Tol)2] (3b): 14%, and [(dtbpCbz)Rb(Tol)2] (4b): 14%). Two exceptions are observed from the general trend of emission wavelength and quantum yields.
The expected outliers are both sodium compounds, as their molecular structure differs from the rest of the series in both the solvated and the unsolvated cases. The unsolvated complex is a non-centrosymmetric dimer and shows lower quantum yields as expected. The solvated complex shows higher energy emission and is the only compound that possesses a bridging toluene ligand. The unexpected exceptions are the caesium compounds which exhibit poor quantum yields and the fastest relaxation.
The Roesky group used a chiral iminophosphonamide ligand to prepare dimeric complexes of Li, Na, K, Rb and Cs and the polymeric Cs complex as a cocrystal of the dimer which exhibited both fluorescence and TADF.33 Interestingly, in this series, quantum yields of 8, 36, 21, 21, and 3% were observed at ambient temperature, where again Cs showed the lowest quantum yields and fastest relaxation. In this instance, different crystallisation processes were invoked as an explanation.
It is remarkable that the carbazolide complexes show considerably higher quantum yields for fluorescence in solution. The experiments with the metal carbazolides were conducted with toluene solutions of the samples. The identity of the luminescent species is inferred to be toluene-solvated complexes of the type [(dtbpCbz)M(Tol)n]. The measured quantum yields range from 100% for the Li compound to 87%, 80% and 95% for the Na, K and Rb complexes, respectively. The Cs compound exhibits a considerably poorer quantum yield of 30%. Due to its poor solubility, the metal-free carbazolide [NBu4][dtbpCbz] was investigated in THF, and 76% quantum yield was observed.
TD-DFT calculated absorption spectra using PBE0-D3/def2-SVP-optimized structural parameters helped to corroborate the experimental results on the non-solvated compounds (dtbpCbz)–H, 1a–5a and 6. For the free carbazole, the lowest-energy absorptions were calculated at 334, 303, and 284 nm which are in accordance with the compound being perceived as colourless.
For the carbazolide anion, the three lowest-energy absorptions are predicted at 436, 395, and 352 nm in line with a yellow compound as broad bands extend into the visible range.
The neutral carbazolide salts 1a–5a possess several calculated absorptions in the range 390–430 nm. In the experimental absorption spectra, a band with a maximum between 410 and 430 nm was found. Interestingly, especially for the heavier members of the series 3a–5a, the lowest-energy transition dominated by the HOMO → LUMO excitation features extremely low calculated oscillator strengths. In contrast, the second transition, which has HOMO → LUMO+1 as its main component, has a stronger oscillator strength by a factor of up to 83 for 5a (see ESI 5.2†). The sodium complex 2a is an outlier in this series due to its dimeric structure instead of a monomeric structure, but as the excitations largely occur from one ligand backbone to the arenes flanking the N-coordinated metal atom, essentially two independent monomeric moieties are present and no significant change in absorption bands occurs. It is interesting to note that the dimeric structure of the sodium complex 2a is unlikely to persist in solution, as in the NMR spectra of benzene only one set of tert-butyl and ortho-CH groups on the flanking arene moieties is observable.
As models for the emission spectra, carbazole (dtbpCbz)–H and the carbazolide ion 6 were considered. This simple model also nicely reproduces the predicted emissions which are calculated to be 376 and 509 nm, respectively. With the carbazolide anion as a model, the two least-energy bands are dominated by the HOMO → LUMO and HOMO → LUMO+1 transitions (Fig. 4). In both the LUMO and LUMO+1, there is a contribution from a π-bonding component between the carbazole moiety and the flanking arenes. Consequently, in the excited states, arene moieties are twisted more towards a coplanar arrangement with the carbazole scaffold.
 | ||
| Fig. 4 Selected orbitals of [dtbpCbz]H and [dtbpCbz]−, 4a and 5a. | ||
For the metal-containing carbazolides 1a–5a, the LUMO is dominated by s orbitals of the metal ion, but the LUMO+1 still features the aforementioned π-bonding contribution. The calculated emissions for the transition from the first excited state to the ground state yield considerably low energies (667–772 nm, see ESI 5.3†). This was not observed in the non-solvated complexes 1a–5a, possibly due to the negligible oscillator strength of the HOMO → LUMO transition. However, the transition from the second excited state to the ground state reasonably reproduces the observed energies (473–482 nm). Thus, in each instance, not the first, but the second excited state appears to be responsible for the observed emission. The optimised second excited states show a clear shift of the metal ion towards one of the flanking arenes but otherwise show little distortion of the ligand scaffold (see Fig. 5).
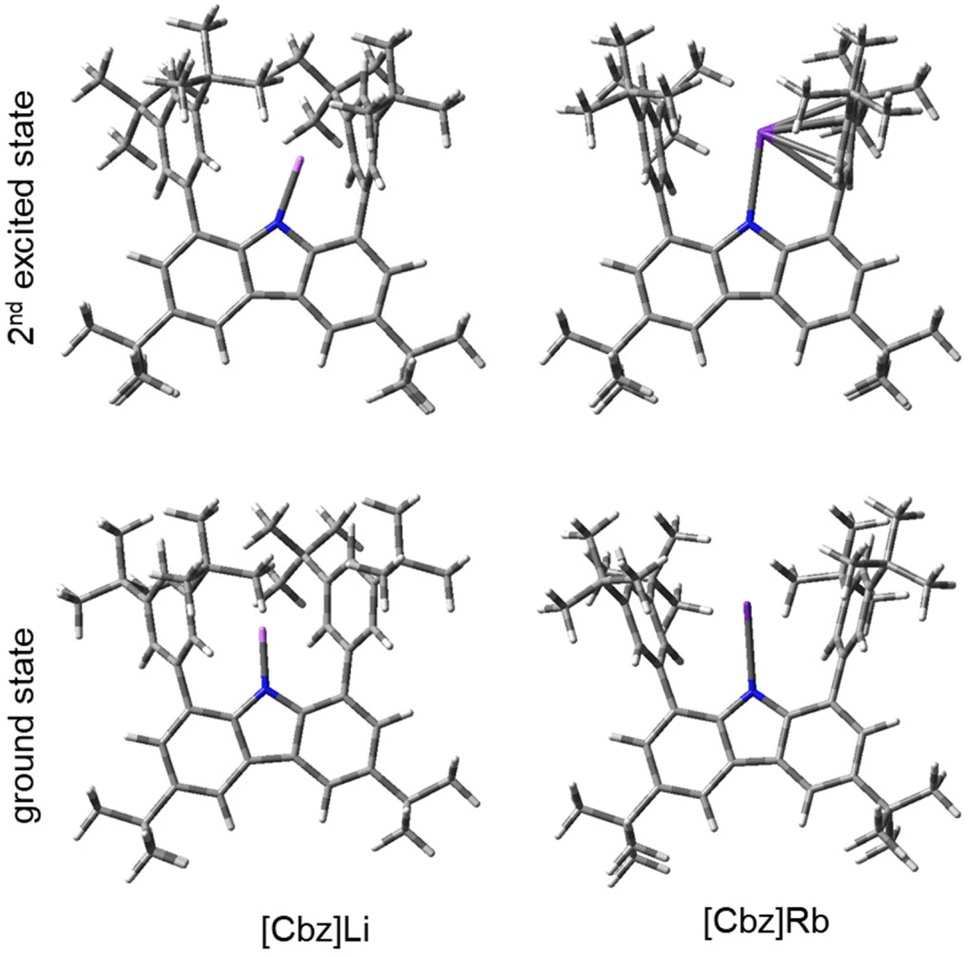 | ||
| Fig. 5 Optimised geometries of the ground state and the 2nd excited state of [Cbz]Li and [Cbz]Rb. | ||
The toluene-solvated compounds, 1b, 3b, 4b and 5b, show emissions at a lower energy level than their non-solvated counterparts (Fig. S51–S55†). The sodium complex 2b is an outlier in this respect as well, and the emission of the toluene solvate occurs at a higher energy level than that of the toluene-free compound. This is likely to be its unique coordination mode in the solid state.
Within the series 1b–5b in the emission spectra of the toluene solvates 1b–3b, two bands are discernible. An explanation for this can be provided by analysing the process using the example of 1b. The electronic excitation originates from the HOMO into the unoccupied orbitals at energies of 423, 405 and 396 nm for the first three transitions, and the geometric distortions of the molecules are marginal (Fig. S59†). The HOMO → LUMO transition has the smallest oscillator strength among them (ESI 5.2.8†). The HOMO is localised on the π system of the carbazole scaffold. LUMO and LUMO+1 are dominated by the π orbitals of the coordinated toluene, while LUMO+2 has the contributions from carbazole and toluene π orbitals (Fig. 6).
 | ||
| Fig. 6 Selected orbitals of [(dtbpCbz)Li(Tol)] (1b). | ||
The emission from the LUMO is predicted at low energy (854 nm), which was not observed experimentally. However, the emissions from LUMO+1 and LUMO+2 are calculated to be 539 and 495 nm respectively, which matches the experimental data. It also reproduces the observation of lower emission energies for the toluene solvates compared to the toluene-free compounds (calculated: 539 and 495 nm for 1bvs. 476 nm for 1a; observed: 580 and 540 nm for 1bvs. 520 nm for 1a). Toluene features the unoccupied orbitals lowest in energy, and it dominates LUMO+1 and contributes to LUMO+2. Thus, the introduction of additional toluene ligands can lead to two emission bands at a lower energy level than that in the toluene-free complexes.
Previous investigations by the Roesky group also reported a series of alkali metal compounds33 of which the caesium compound showed the fastest relaxation of the excited state. They proposed that different coordination behaviour is a cause for this phenomenon, which certainly can play a role. However, for the caesium compound 5a, another phenomenon could contribute to the relaxation of the excited state. The involvement of the d atomic orbitals of the metal atom in the LUMO+1 is a discernible discrepancy between 5a and all the other studied carbazolide compounds, visible in Fig. 4, but only with small numerical differences (see ESI 5.1†). The astonishingly short lifetime of the excited state in this case may be due to exactlythis interaction. It is interesting to note that in the work of Fu and Peters, the lithium salt of the parent carbazolide only showed aggregation-induced changes of absorption and emission behaviour, i.e. the effects would only be present at high concentrations.10 In the present series of carbazolides, efficient emission is found even at low concentrations.
Conclusions
In conclusion, the synthesis and luminescence properties of a series of carbazolide complexes are presented. Both quasi-monocoordinated complexes of the type [(dtbpCbz)M] and the corresponding toluene solvates were investigated. In both series, the Na+ complexes showed unexpected behaviour, forming a dimeric complex without toluene and an arene-bridged complex with toluene. All compounds show efficient fluorescence, with emission maxima in the range of 540–460 nm. The introduction of the toluene ligands causes the emissions to shift to lower energies. The emission energy increases in the sequence [(dtbpCbz)]− < [(dtbpCbz)Li] < [(dtbpCbz)Na] < [(dtbpCbz)K] < [(dtbpCbz)Rb]. An outlier in this trend is the caesium compound which is comparable to the potassium complex. The solid-state quantum yields increase as the metal atom becomes heavier reaching 29% for [(dtbpCbz)Rb], again with the exception of the caesium complex, which exhibits the lowest quantum yield of 3%. In toluene solution, both the lithium and the rubidium carbazolides show quantum yields that approach 1. The lifetimes of the excited states decrease with increasing atomic mass of the metal.Author contributions
M. K. and M. P. M. carried out the synthetic work and characterisation. F. K. and M. R. conducted luminescence measurements. A. H. contributed to crystallographic and computational work. The manuscript was written through the contributions of all the authors. All the authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Fonds der Chemischen Industrie through a Liebig Fellowship for A. H. and a Kekule Fellowship for M. K. and M. P. M. and by the German Research Foundation (DFG) through the Emmy Noether Programme (HI2063/1-1) is acknowledged. We thank Prof. Frank Breher, Prof. Peter Roesky and Prof. Claus Feldmann for their continuous support, and Prof. Florian Weigend for the valuable discussions. This work was carried out with the support of the Karlsruhe Nano Micro Facility (KNMF), a Helmholtz Research Infrastructure at Karlsruhe Institute of Technology (KIT), and Prof. Dieter Fenske is gratefully acknowledged for his help with XRD. We acknowledge the support by the state of Baden-Württemberg through bwHPCand DFG through grant no. INST 40/467-1FUGG (JUSTUScluster).References
- G. Sathiyan, E. K. T. Sivakumar, R. Ganesamoorthy, R. Thangamuthu and P. Sakthivel, Review of carbazole based conjugated molecules for highly efficient organic solar cell application, Tetrahedron Lett., 2016, 57, 243–252 CrossRef CAS.
- H. Jiang, J. Sun and J. Zhang, A review on synthesis of carbazole-based chromophores as organic light-emitting materials, Curr. Org. Chem., 2012, 16, 2014–2025 CrossRef CAS.
- K. R. Justin Thomas, J. T. Lin, Y.-T. Tao and C.-W. Ko, Light-emitting carbazole derivatives: Potential electroluminescent materials, J. Am. Chem. Soc., 2001, 123, 9404–9411 CrossRef PubMed.
- Y. Liu, M. Nishiura, Y. Wang and Z. Hou, π-Conjugated aromatic enynes as a single-emitting component for white electroluminescence, J. Am. Chem. Soc., 2006, 128, 5592–5593 CrossRef CAS PubMed.
- K. Karon and M. Lapkowski, Carbazole electrochemistry: A short review, J. Solid State Electrochem., 2015, 19, 2601–2610 CrossRef CAS.
- R. Hamze, J. L. Peltier, D. Sylvinson, M. Jung, J. Cardenas, R. Haiges, M. Soleilhavoup, R. Jazzar, P. I. Djurovich, G. Bertrand and M. E. Thompson, Eliminating nonradiative decay in Cu(I) emitters: > 99% quantum efficiency and microsecond lifetime, Science, 2019, 363, 601–606 CrossRef CAS PubMed.
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. Abdi Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, High-performance light-emitting diodes based on carbene-metal-amides, Science, 2017, 356, 159–163 CrossRef CAS PubMed.
- S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Photoinduced Ullmann C–N coupling: Demonstrating the viability of a radical pathway, Science, 2012, 338, 647–651 CrossRef CAS PubMed.
- A. C. Bissember, R. J. Lundgren, S. E. Creutz, J. C. Peters and G. C. Fu, Transition-metal-catalyzed alkylations of amines with alkyl halides: Photoinduced, copper–catalyzed couplings of carbazoles, Angew. Chem., Int. Ed., 2013, 52, 5129–5133 CrossRef CAS PubMed.
- J. M. Ahn, T. S. Ratani, K. I. Hannoun, G. C. Fu and J. C. Peters, Photoinduced, copper-catalyzed alkylation of amines: A mechanistic study of the cross-coupling of carbazole with alkyl bromides, J. Am. Chem. Soc., 2017, 139, 12716–12723 CrossRef CAS PubMed.
- D. I. Bezuidenhout, G. Kleinhans, G. Guisado-Barrios, D. C. Liles, G. Ung and G. Bertrand, Isolation of a potassium bis (1, 2, 3-triazol-5-ylidene) carbazolide: A stabilizing pincer ligand for reactive late transition metal complexes, Chem. Commun., 2014, 50, 2431–2433 RSC.
- R. Jordan and D. Kunz, The fascinating flexibility and coordination modes of a pentamethylene connected macrocyclic CNC pincer ligand, Molecules, 2021, 26, 1669 CrossRef CAS PubMed.
- P. M. Chapple, S. Kahlal, J. Cartron, T. Roisnel, V. Dorcet, M. Cordier, J.-Y. Saillard, J.-F. Carpentier and Y. Sarazin, Bis (imino) carbazolate: A master key for barium chemistry, Angew. Chem., Int. Ed., 2020, 59, 9120–9126 CrossRef CAS PubMed.
- J. C. Ott, H. Wadepohl, M. Enders and L. H. Gade, J. Am. Chem. Soc., 2018, 140, 17413 ( J. Am. Chem. Soc. , 2018 , 140 , 17413–17417 ) CrossRef CAS PubMed.
- G. Kleinhans, M. M. Hansmann, G. Guisado-Barrios, D. C. Liles, G. Bertrand and D. I. Bezuidenhout, Nucleophilic T-shaped (LXL) Au(I)-pincer complexes: Protonation and alkylation, J. Am. Chem. Soc., 2016, 138, 15873–15876 CrossRef CAS PubMed.
- C. Cheng, B. G. Kim, D. Guironnet, M. Brookhart, C. Guan, D. Y. Wang, K. Krogh-Jespersen and A. S. Goldman, Synthesis and characterization of carbazolide-based iridium PNP pincer complexes. Mechanistic and computational investigation of alkene hydrogenation: Evidence for an Ir(III)/Ir(V)/Ir(III) catalytic Cycle, J. Am. Chem. Soc., 2014, 136, 6672–6683 CrossRef CAS PubMed.
- J. C. Ott, H. Wadepohl and L. H. Gade, Opening up the valence shell: AT-shaped iron(I) metalloradical and its potential for atom abstraction, Angew. Chem., Int. Ed., 2020, 59, 9448–9452 CrossRef CAS PubMed.
- P. M. Chapple, M. Cordier, V. Dorcet, T. Roisnel, J.-F. Carpentier and Y. Sarazin, A versatile nitrogen ligand for alkaline-earth chemistry, Dalton Trans., 2020, 49, 11878–11889 RSC.
- P. M. Chapple, T. Roisnel, M. Cordier, J.-F. Carpentier and Y. Sarazin, Heteroleptic carbazolato-barium hydroborates and a related separated ion pair, Polyhedron, 2022, 217, 115731 CrossRef CAS.
- J. Zou, D. J. Berg, D. Stuart, R. McDonald and B. Twamley, Carbazole-bis(oxazolines) as monoanionic, tridentate chelates in lanthanide chemistry: Synthesis and structural studies of thermally robust and kinetically stable dialkyl and dichloride complexes, Organometallics, 2011, 30, 4958–4967 CrossRef CAS.
- P. Pinter, C. M. Schüßlbauer, F. A. Watt, N. Dickmann, R. Herbst-Irmer, B. Morgenstern, A. Grünwald, T. Ullrich, M. Zimmer, S. Hohloch, D. M. Guldi and D. Munz, Bright luminescent lithium and magnesium carbene complexes, Chem. Sci., 2021, 12, 7401–7410 RSC.
- R. Kumar, S. Pahar, J. Chatterjee, S. R. Dash, R. G. Gonnade, K. Vanka and S. S. Sen, Luminescent magnesium complexes with intra- and inter-ligand charge transfer, Chem. Commun., 2022, 58, 11843–11846 RSC.
- S. Bestgen, C. Schoo, B. L. Neumeier, T. J. Feuerstein, C. Zovko, R. Köppe, C. Feldmann and P. W. Roesky, Intensely photoluminescent diamidophosphines of the alkaline-earth metals, aluminum, and zinc, Angew. Chem., Int. Ed., 2018, 57, 14265–14269 CrossRef CAS PubMed.
- F. L. Portwich, Y. Carstensen, A. Dasgupta, S. Kupfer, R. Wyrwa, H. Görls, C. Eggeling, B. Dietzek, S. Gräfe, M. Wächtler and R. Kretschmer, A highly fluorescent dinuclear aluminium complex with near-unity quantum yield, Angew. Chem., Int. Ed., 2022, 61, e202117499 CrossRef CAS PubMed.
- R. Hacker, E. Kaufmann, P. von Ragué Schleyer, W. Mahdi and H. Dietrich, Die Röntgenstruktur von N-Lithiocarbazol-Dimer: Experimentelle Bestätigung der theoretischen Analyse von Strukturen des N-Lithiopyrrol-Typs, Chem. Ber., 1987, 120, 1533–1538 CrossRef CAS.
- R. Dinnebier, H. Esbak, F. Olbrich and U. Behrens, Structure determination of unsolvated potassium, rubidium, and cesium carbazolates, Organometallics, 2007, 26, 2604–2608 CrossRef CAS.
- N. D. Coombs, A. Stasch, A. Cowley, A. L. Thompson and S. Aldridge, Bulky aryl functionalized carbazolyl ligands: Amido alternatives to the 2,6-diarylphenyl ligand class?, Dalton Trans., 2008, 332–337 RSC.
- R. S. Moorhouse, G. J. Moxey, F. Ortu, T. J. Reade, W. Lewis, A. J. Blake and D. L. Kays, Structural diversity in alkali metal complexes of sterically demanding carbazol-9-yl ligands, Inorg. Chem., 2013, 52, 2678–2683 CrossRef CAS PubMed.
- A. Hinz, Pseudo-one-coordinate tetrylenium salts bearing a bulky carbazolyl substituent, Chem. – Eur. J., 2019, 25, 3267–3271 CrossRef CAS PubMed.
- M. Kaiser, J. Göttlicher, T. Vitova and A. Hinz, Towards heteroleptic dicoordinate CuII complexes, Chem. – Eur. J., 2021, 27, 7998–8002 CrossRef CAS PubMed.
- A. Hinz, A Mono-substituted silicon(II) cation: A crystalline “supersilylene”, Angew. Chem., Int. Ed., 2020, 59, 19065–19069 CrossRef CAS PubMed.
- M. Kaiser, L. Winkler and A. Hinz, Complexes of 3d metals with a bulky carbazolyl ligand, Z. Anorg. Allg. Chem., 2022, 648, e202200131 CrossRef CAS.
- T. J. Feuerstein, B. Goswami, P. Rauthe, R. Köppe, S. Lebedkin, M. M. Kappes and P. W. Roesky, Alkali metal complexes of an enantiopure iminophosphonamide ligand with bright delayed fluorescence, Chem. Sci., 2019, 10, 4742–4749 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2206682–2206689. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi00348e |
| This journal is © the Partner Organisations 2023 |