
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnti-myeloma pro-apoptotic Pt(II) diiodido complexes†
Lukáš
Masaryk‡
 a,
Denisa
Weiser Drozdková‡
bc,
Karolina
Słoczyńska
d,
Ján
Moncol’
e,
David
Milde
f,
Radka
Křikavová
a,
Justyna
Popiół
d,
Elżbieta
Pękala
d,
Katarína
Ondrušková
b,
Ivan
Nemec
*ah,
Kateřina
Smešný Trtková
*bcg and
Pavel
Štarha
*a
a,
Denisa
Weiser Drozdková‡
bc,
Karolina
Słoczyńska
d,
Ján
Moncol’
e,
David
Milde
f,
Radka
Křikavová
a,
Justyna
Popiół
d,
Elżbieta
Pękala
d,
Katarína
Ondrušková
b,
Ivan
Nemec
*ah,
Kateřina
Smešný Trtková
*bcg and
Pavel
Štarha
*a
aDepartment of Inorganic Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 12, 77146 Olomouc, Czech Republic. E-mail: pavel.starha@upol.cz; Tel: +420 585 634 348
bDepartment of Clinical and Molecular Pathology, Faculty of Medicine and Dentistry, Palacký University Olomouc, Hněvotínská 3, 77515 Olomouc, Czech Republic. E-mail: katerina.smesny@upol.cz; Tel: +420 585 632 455
cInstitute of Molecular and Clinical Pathology and Medical Genetics, Faculty of Medicine, University of Ostrava, Syllabova 19, 703 00 Ostrava, Vítkovice, Czech Republic
dDepartment of Pharmaceutical Biochemistry, Faculty of Pharmacy, Jagiellonian University Medical College, Medyczna 9, 30-688 Kraków, Poland
eDepartment of Inorganic Chemistry, Faculty of Chemical and Food Technology, Slovak University of Technology in Bratislava, 81237 Bratislava, Slovakia
fDepartment of Analytical Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 12, 77146 Olomouc, Czech Republic
gInstitute of Molecular and Translational Medicine, Faculty of Medicine and Dentistry, Palacký University Olomouc, Hněvotínská 5, 77515 Olomouc, Czech Republic
hCentral European Institute of Technology, Brno University of Technology, Purkyňova 123, 61200 Brno, Czech Republic. E-mail: ivan.nemec@upol.cz; Tel: +420 541 149 269
First published on 3rd May 2023
Abstract
Platinum-based agents unwaveringly hold their prominent position in cancer treatment. Current research emphasizes finding novel complexes for hard-to-treat cancers with minimum side effects, capable of overcoming resistance. This work presents easy-to-prepare diiodidoplatinum(II) complexes cis-[PtI2(Ln)2] (1–7) with imidazole derivatives (Ln), which were considerably effective against multiple myeloma cell lines U266B1 and KMS12-PE. The leading compound 6 (at 3 μM concentration) extraordinarily reduced viability of U266B1 and KMS12-PE myeloma cells to 3.0% and 1.1%, respectively, and exceeded the conventional platinum-based anticancer drug cisplatin (93.1% and 88.3%, respectively) that is used clinically for the combination therapy of multiple myeloma. Complex 6 was significantly more effective in inducing apoptosis in KMS12-PE cells without interleukin-6 (IL-6) expression than in U266B1 cells with IL-6 expression. Complex 6 also induced apoptosis in co-culture of KMS12-PE with non-cancerous stromal fibroblasts (HS-5), and displayed markedly lower activity in the HS-5 stromal fibroblast cells than in myeloma cells, pointing out its pharmocologically prospective selectivity towards the cancer cells over the normal ones. No caspase 3/7 activity was detected in apoptotic KMS12-PE cells treated by complex 6 indicating a different mechanism of apoptosis action from cisplatin. This work demonstrates that simple non-classical platinum(II) complexes represent a new perspective for a monotherapy of hard-to-treat multiple myeloma.
Introduction
Cancer remains a leading cause of death worldwide.1 Despite massive attention, unceasing research and undisputable advancement over the years, some types of cancer are, unfortunately, still incurable. Multiple myeloma (MM), also referred to as plasma cell myeloma, is a unique hematological disorder characterized by proliferation of plasma cells in the bone marrow (BM), and is strongly tied to monoclonal protein production and overexpression of cytokines, such as interleukin-6 (IL-6), responsible for the destruction of bone tissue.2–4 Like their normal counterparts, myeloma cells mainly proliferate and survive within the BM by physiological and functional interactions with bone marrow stromal cells (BMSCs) and the surrounding BM microenvironment. The latter plays a central role in the pathogenesis of MM and these interactions have been shown to be important in myeloma cell survival and progression.5–7Moreover, it has been established that the lower response of myeloma cells to conventional treatment, such as glucocorticoids or cytotoxic chemotherapeutics, is connected with the presence of BMSCs. Beneficially, some of the newly approved anti-myeloma drugs are able to circumvent the protective effects of the BM microenvironment.3 This fact demonstrates that in recent years, much progress has been made in the treatment of symptomatic MM, particularly with the development of proteasome inhibitors (e.g. bortezomib, carfilzomib and ixazomib), immunomodulatory drugs (IMiDs; e.g. thalidomide, lenalidomide and pomalidomide), and monoclonal antibodies (e.g. daratumumab, elotuzumab or isatuximab) with pleiotropic anti-myeloma properties.8 Additionally, treatment regimens (e.g. the proteasome inhibitor-based regimen) nowadays usually comprise targeted drug therapy that combines varied classes of drugs to fight the growth of MM.9
Despite these possibilities, MM still remains incurable, and most patients develop resistance to proteasome inhibitors and IMiDs and almost all patients with MM eventually relapse.10 Intensive chemotherapy modes were originally developed to improve response to treatment and overcome chemotherapeutic resistance in patients with relapsed or refractory MM. However, these intensive salvage regimens, such as DCEP which uses dexamethasone with continuous infusions of cyclophosphamide, etoposide, and cisplatin, are associated with significant toxicity.11
Conventional platinum-based drugs (e.g. cisplatin) are not effective for monotherapy of MM but are advantageous in advanced combination treatment (see above). Their irreplaceable role even in the therapy of hard-to-treat cancers is the reason why these metallotherapeutics still represent prominent antineoplastic agents worldwide,12 despite the fact that their application is limited by adverse side effects (e.g. nephro- and hepatotoxicity) and a lack of efficacy against many types of tumours.13 These limitations still motivate the search for new, more effective and safer (metallo)drugs with different mechanism of action (MoA) based not only on platinum but also other transition metals.14 Recent success of such research has brought imifoplatin, i.e. Pt(II) pyrophosphato complex PT-112,15 which is currently evaluated in clinical trials on oncological patients.16 Imifoplatin proves that variation of carrier/leaving ligands in conventional platinum-based drugs can still be a viable design strategy, as this complex practically represents a derivative of oxaliplatin with oxalate substituted with pyrophosphate.
Analogical strategy, this time replacing chlorides by iodides in cisplatin analogues, has also resulted in complexes with high pharmacological potential. Indeed, in the last decade, various cis-diiodidoplatinum(II) complexes have been shown to exhibit higher cytotoxicity connected with different mechanistic effects as compared with chlorido (including cisplatin) and carboxylato analogues.17 The MoA of Pt(II) diiodido complexes likely involves interactions with biomolecules other than DNA, and the formation of protein/peptide adducts has to be considered as well.17–20 In connection to this, we hypothesized that different MoA of diiodidoplatinum(II) complexes from cisplatin could be advantageous in terms of potency against hard-to-treat tumours, such as MM, where cisplatin shows only limited activity. Thus, in this work, the substituted-imidazole-based diiodidoplatinum(II) complexes (1–7; Fig. 1) were studied for their in vitro antiproliferative activity against human multiple myeloma cell lines.
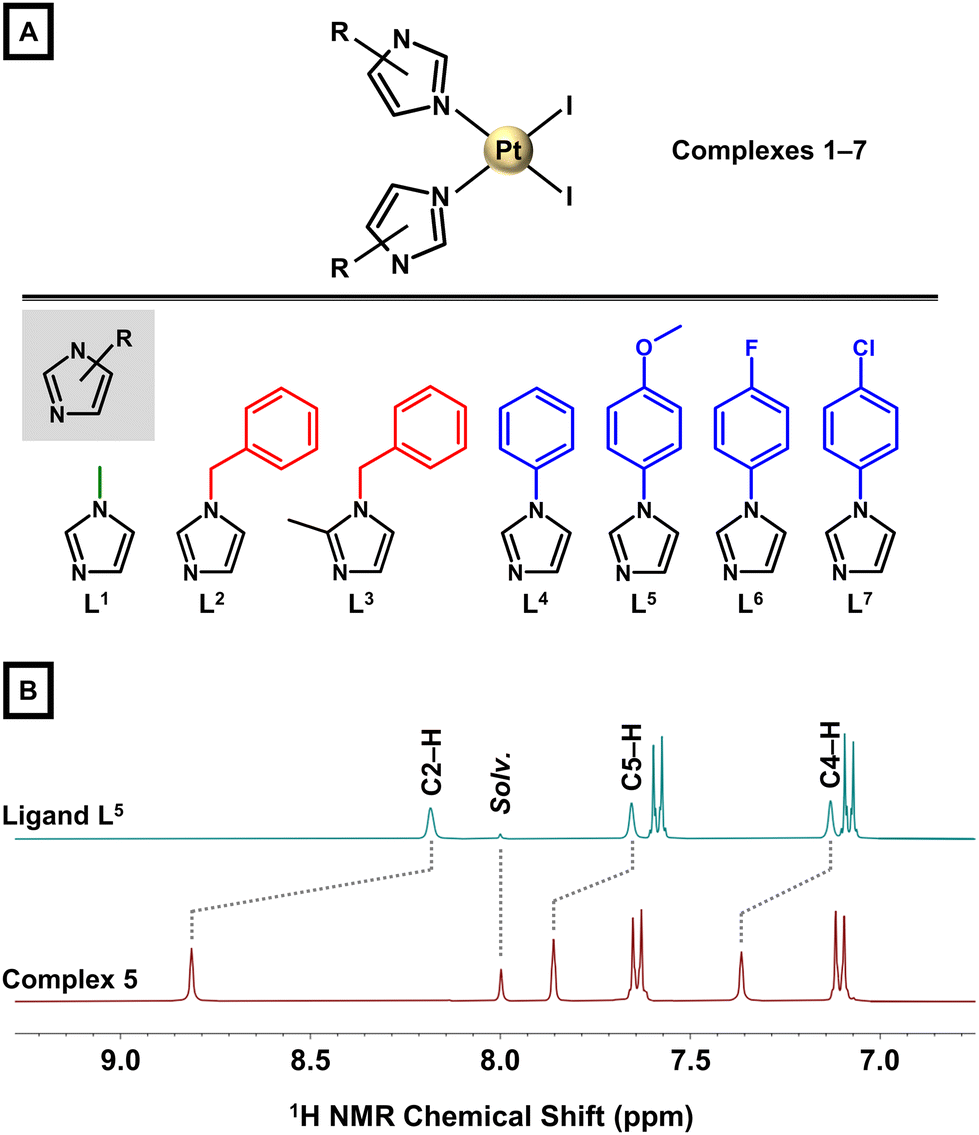 | ||
| Fig. 1 (A) The general structural formula of complexes cis-[PtI2(Ln)2] (1–7; top) and structural formulas of the used imidazole derivatives L1–L7 (bottom). (B) Part of 1H NMR spectra (DMF-d7 solutions) of the representative ligand L5 (top) and complex 5 (bottom). | ||
Results and discussion
Design and rationale
The platinum(II) anticancer drugs cisplatin, carboplatin and oxaliplatin represent leading compounds in cancer treatment used in ca. 50% of chemotherapies worldwide either in monotherapy or in combination with radiotherapy or different classes of drugs. The unparalleled efficacy of the Pt-based agents in clinic motivates unceasing search for novel metallotherapeutics with an improved pharmacological profile. Current research prominently focuses on new complexes, which could broaden the spectrum of treatable tumours, minimize side effects and circumvent the problem of ubiquitous induction of resistance to platinum drugs.14The majority of design strategies for Pt-based agents involves variation of the original structure of the conventional Pt-based drugs. Multiple combinations of leaving and carrier ligands in Pt complexes have been investigated.14,17 This work reports on seven diiodidoplatinum(II) complexes of the general formula cis-[PtI2(Ln)2] (1–7) with various imidazole derivatives (Ln) used as monodentate N-donor ligands. A family of imidazole derivatives represents both natural and synthetic compounds, including anticancer imidazole-based drugs.21,22 Cytotoxic efficacy was previously described for some Pt(II) chlorido complexes bearing various imidazole derivatives.23–25
It should be pointed out that the goals of overcoming the drawbacks of the classical platinum chemotherapy and finding agents for hard-to-treat cancers can most likely be achieved with compounds acting through diverse MoA. It is known in the literature that diiodido Pt(II) complexes of the general formula cis-[PtI2L2] containing varied N-donor heterocyclic ligands (L) exhibited higher cytotoxicity connected with different mechanistic effects as compared with their chlorido and carboxylato analogues.17 For example, complex cis-[Pt(3Braza)2I2] was more potent in MCF-7 breast carcinoma cell line than its dichlorido and oxalato analogues; 3Braza = 3-bromo-7-azaindole.26–28 Thus, we hypothesized for this work that the known cytotoxicity of dichlorido complexes with various imidazole derivatives23–25 could be improved for the new substituted-imidazole-based diiodidoplatinum(II) complexes, which could be, in contrast to cisplatin, potent against hard-to-treat human tumours (i.e. multiple myeloma in this work) as a consequence of their different MoA. Notably, anticancer Pt(II) diiodido complexes with imidazole-based ligands are scarce in the literature.29–31
Synthesis and basic characterizations
A series of seven cis-diiodidoplatinum(II) complexes, cis-[PtI2(Ln)2] (1–7), involving various N3-coordinated imidazole derivatives (L1–L7; Fig. 1A) was prepared following the well-established Dhara's method32 with [PtI4]2− as a key intermediate prepared in situ from K2[PtCl4]; L1 = 1-methyl-1H-imidazole for 1, L2 = 1-benzyl-1H-imidazole for 2, L3 = 1-benzyl-2-methyl-1H-imidazole for 3, L4 = 1-phenyl-1H-imidazole for 4, L5 = 1-(4-methoxyphenyl)-1H-imidazole for 5, L6 = 1-(4-fluorophenyl)-1H-imidazole for 6, and L7 = 1-(4-chlorophenyl)-1H-imidazole for 7. Complexes 1–7 were only negligibly soluble in H2O, methanol (MeOH), ethanol or acetone, but they were well soluble in N,N-dimethylformamide (DMF) and dimethyl sulfoxide (DMSO). The purity and composition of 1–7 was studied by elemental analysis, NMR, mass spectrometry and FTIR (see ESI, Experimental section†).The ESI+ mass spectra of 1–7 contained the peaks of the {[PtI2(Ln)2] + X}+ species with adequate m/z values and isotopic distribution; X = H+, Na+, K+ or NH4+ (see ESI, Experimental section†). All hydrogen and carbon atoms were identified in the 1H and 13C NMR spectra (ESI, Fig. S1–S7†). The largest 1H NMR coordination shifts (Δδ = 0.53–0.90 ppm) were observed for the C2–H resonances (Fig. 1B), while the other signals of the imidazole ring showed a somewhat lower change of δ (0.22–0.57 ppm). The cis-configuration was confirmed in solution, as 195Pt NMR spectra of 1–7 showed a single resonance in the region characteristic for cis-diiodidoplatinum(II) complexes (ca. −3150 ppm; ESI, Fig. S1–S7†).18,33,34 Additionally, the representative complexes 6 (97.8% at tR = 16.43 min) and 7 (96.3% at tR = 18.00 min) were analysed by reversed–phase high–performance liquid chromatography (RP–HPLC) coupled to mass spectrometry (electrospray ionization, ESI) (Fig. S8†).
Single crystals of sufficient quality were prepared for 3, 4, 5 and 7. X-ray diffraction analysis revealed that all these compounds contained cis-[PtI2(Ln)2] molecules in their crystal structure (Fig. 2 and ESI, Fig. S9 and Table S1†). The Ln ligands coordinate Pt(II) atoms by the N3 atom. The Pt atoms are tetracoordinate with the coordination polyhedron adopting the shape close to square planar. This can be documented by the trans-N–Pt–I angles adopting values close to 180° (174–179°). Also, the values of the cis-angles 86–93° were as expected for the square-planar geometry. The Pt–I bonds adopt the lengths of 2.58–2.60 Å, which are much longer than those observed for the Pt–N bonds (2.01–2.04 Å). All the metal–ligand bond lengths and angles are summarized in the caption of Fig. 2.
 | ||
| Fig. 2 An Ortep drawing (50% probability level) of the complex molecules in compounds 3 and 4 (see ESI, Fig. S9† for 5 and 7). Colour code: C (grey), H (white), I (purple), N (blue) and Pt (light grey). Selected bond lengths (in Å): 3, d(Pt1–N1) = 2.037(2), d(Pt1–N3) = 2.023(2), d(Pt1–I1) = 2.5880(1), d(Pt1–I2) = 2.5842(2); 4, d(Pt1–N1) = 2.043(2), d(Pt1–N3) = 2.034(2), d(Pt1–I1) = 2.5946(2), d(Pt1–I2) = 2.5838(2); 5, d(Pt1–N1) = 2.030(2), d(Pt1–N3) = 2.042(2), d(Pt1–I1) = 2.5794(2), d(Pt1–I2) = 2.5962(2), d(Pt2–N5) = 2.041(2), d(Pt2–N7) = 2.041(2), d(Pt2–I3) = 2.6014(2), d(Pt2–I4) = 2.5925(2); 7, d(Pt1–N1) = 2.041(3), d(Pt1–N3) = 2.039(3), d(Pt1–I1) = 2.5823(2), d(Pt1–I2) = 2.5871(2), d(Pt2–N5) = 2.030(3), d(Pt2–N7) = 2.035(3), d(Pt2–I3) = 2.5957(3), d(Pt2–I4) = 2.5976(2). | ||
Besides rare reports on biological investigations of cis-Pt(II)-diiodido complexes with imidazole-based N-donor ligands, also structural information on this type of compounds is rather scarce. The search for this structural motif in the Cambridge Structural Database (version 5.4.1 November 2019)35 gives back only two deposited crystal structures of cis-Pt(II)-diiodido complexes, both with bidentate bis-imidazole ligands.31,36 In other words, complexes 3, 4, 5 and 7 represent the first crystallographically characterized complexes of the cis-[PtI2L2] type with two monodentate imidazoles.
Stability in solution
It has been established that MoA of square planar Pt(II) complexes, in particular dichlorido ones, involves activation via intracellular hydrolysis. MoA of diiodido complexes, with their significantly slower aquation, has been shown to differ even in this initial aspect of pharmacokinetics.17 Therefore, 1–7 were also studied for their behaviour in a pseudo-physiological environment, mimicking the conditions of the biological studies.
1H NMR experiments proved that complexes 1–7 did not undergo any chemical changes when dissolved in DMF-d7 for 24 h at room temperature (r.t.), since no new signals appeared in their spectra. In the presence of water (i.e., in 50% DMF-d7/50% PBS in D2O; Fig. 3 and ESI, Fig. S10–S16†), complexes 1–3, 5 and 6 were stable as well, as their 1H NMR spectra did not change within the whole-time interval. In contrast, 1H NMR spectra of 4 and 7 showed new signals at t = 24 h, assignable to free ligands L4 and L7. Specifically, the ratio of the parent complexes and released ligands was ca. 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 after 24 h for both complexes. Although 4 and 7 released their Ln ligands (no 1H NMR signals indicating the presence of Pt-containing species with only one Ln ligand were detected), the extent of their decomposition was acceptable with respect to the intended biological analyses.
:1 after 24 h for both complexes. Although 4 and 7 released their Ln ligands (no 1H NMR signals indicating the presence of Pt-containing species with only one Ln ligand were detected), the extent of their decomposition was acceptable with respect to the intended biological analyses.
 | ||
| Fig. 3 1H NMR spectra of representative complexes 6 and 7 in 50% DMF-d7/50% PBS in D2O (pH 7.4) without (labelled PBS) or with (labelled GSH) 2 molar equiv. of reduced glutathione (GSH), as observed at different time points (t = 0 or 24 h). 1H NMR spectra of free ligands L6 and L7 in 50% DMF-d7/50% PBS in D2O (pH 7.4) are shown for comparative purposes. Background colour code: blue = complex; red = free ligand. | ||
Interaction with reduced glutathione
Reactivity of the platinum central atom towards ligand exchange with varied biomolecules greatly influences the fate of these metallodrugs in the physiological environment. Pt(II) binds strongly to sulphur-containing molecules, such as GSH, which affects the metabolism of Pt-based drugs and can negatively influence their therapeutic efficacy.37 For example, GSH plays a role in Pt(II) detoxification, especially in resistant cancers. Accordingly, these reactions can result in off-target side-effects. On the other hand, some anticancer Pt complexes (e.g., picoplatin) are known to be inert towards GSH, allowing them to circumvent resistance of tumours effectively and act readily towards cancers known to have low sensitivity towards e.g. cisplatin and analogues.26,38 That is why the ability of 1–7 to interact with GSH was investigated.In the presence of an excess of intracellular thiol GSH (2 molar equiv.; 50% DMF-d7/50% PBS in D2O, pH 7.4), 1–3 were stable for 24 h (ESI, Fig. S10–S16†). In contrast, new signals appeared in 1H spectra of 4–7 involving phenyl-substituted imidazoles (t = 24 h; Fig. 3). These resonances can be unambiguously assigned to the released ligands L4–L7 (measured for comparative purposes under the same experimental conditions). The extent of the Ln release from phenyl-substituted imidazole-based complexes was more or less the same for 4 and 7 in the presence of GSH as compared with the results obtained for the mixture of 50% DMF-d7/50% PBS in D2O (i.e. without GSH). All the studied complexes were inert towards the covalent attack of GSH, because no signals assignable to the Pt–SG adducts were detected in the 1H NMR spectra.
Lipophilicity
Generally, antiproliferative activity and the ability of drugs to enter cells often correlate with their lipophilicity (hydrophobicity).39 For cisplatin and its iodido analogues, general pharmacokinetics is different not only due to slower hydrolysis rate, but also because of different lipophilicity.19 For 1–7, logP values were determined by the investigation of octanol/water distribution, and equal −0.69 ± 0.09 (for 1), −0.28 ± 0.11 (for 2), −0.40 ± 0.18 (for 3), −0.02 ± 0.05 (for 4), −0.07 ± 0.19 (for 5), −0.21 ± 0.15 (for 6) and −0.13 ± 0.12 (for 7). The benzyl- and phenyl-based complexes 2–7 had higher lipophilicity than 1 containing methyl-substituted imidazole, and among them, it was phenyl-based complexes 4–7, which showed higher lipophilicity than benzyl-analogues 2–3. The obtained logP values of 1–7 are similar with highly anticancer active Pt(II) diiodido complexes with various azaindoles,26,33 and indicated higher lipophilicity of the studied complexes as compared with cisplatin.40
Antiproliferative activity in primary cultures of myeloma cells
As it is mentioned above, we hypothesized that the different antiproliferative activity profile and MoA of previously reported diiodidoplatinum(II) complexes from conventional cisplatin17 could mean a possibly high biological effect of new complexes (1–7) in myeloma cells with low sensitivity towards conventional Pt-based drugs. It has to be mentioned that, despite the low sensitivity of myeloma cells towards cisplatin, this Pt drug is used clinically for the treatment of multiple myeloma within various combination therapy regimes.11,41 This allowed us to utilize cisplatin as a reference drug of our biological investigations. Complexes 1–7 were tested for their in vitro antiproliferative activity against two human patient-derived multiple myeloma cell lines – U266B1 and KMS12-PE; PE = pleural effusion due to plasma cell infiltration. Dose-dependent cell viabilities are presented in Fig. 4 and ESI, Table S2†. U266B1 and KMS12-PE were selected in order to enable comparison of the effect of 1–7 on myeloma cells with different IL-6 expression, as IL-6 helps to create an ideal microenvironment for oncogenesis and metastasis, and play a significant role in the pathophysiology of MM.42–44 | ||
| Fig. 4 Dose dependent cell viabilities of U266B1 (top) and KMS12-PE (bottom) myeloma cell lines after the treatment with complexes 1–7 and the reference drug cisplatin (CDDP); MTT assay, 24 h exposure time. | ||
Complexes 1–7 considerably reduced the MM cell viability at the applied 1–3 μM concentrations (Fig. 4 and ESI, Table S2†). More lipophilic benzyl- and phenyl-substituted complexes 2–7 were more antiproliferative active than 1. Complexes 5–7, which contain variously substituted 1-phenyl-1H-imidazoles (L5–L7) were more anti-myeloma active than 4 with unsubstituted 1-phenyl-1H-imidazole (L4). The best-performing complexes 6 and 7 were significantly more potent against both the used myeloma cells than the reference drug cisplatin (ESI, Fig. S17†), known to be involved in various clinical treatment protocols for MM.11,41 Specifically, the 2 μM and 3 μM concentrations of 6 and 7 caused a decrease in the proportion of viable cells of both cell types below 10% (e.g. to 3.0% and 1.1% in U266B1 and KMS12-PE cells, respectively, for 3 μM 6), which was significantly higher as compared with cisplatin (93.1% and 88.3% cell viability, respectively; at 3 μM concentration). Importantly, 7 significantly reduced the myeloma cell viability significantly even at 1 μM concentration to the level of 11.5% (U266B1) and 2.1% (KMS12-PE), implying its extraordinarily high anti-myeloma activity.
The studied diiodidoplatinum(II) complexes, especially the best-performing compounds 6 and 7, showed a high anti-myeloma effect. Lower myeloma cell viability inhibition was previously reported for the other Pt-based drugs oxaliplatin and carboplatin,45 Pt(II) dichlorido complexes with steroidal esters of L-methionine and L-histidine,46 or complexes derived from different d-block elements.47–51 This unprecedented anti-myeloma activity of the studied Pt diiodido complexes is particularly important for their future research, because it offers a possibility of myeloma monotherapy instead of the conventional combination therapy regimens.
Although 6 was not the most antiproliferative active compound (7 showed slightly higher anti-myeloma potency), we decided to use 6 for additional studies of MoA (apoptosis, cell cycle modification), especially because its stability was higher than determined for 7 under the used aqueous conditions.
Antiproliferative activity – comparative experiments
High anti-myeloma activity of 6, which significantly exceeded Pt-based drug cisplatin (Fig. 4 and ESI, Table S2†), was a strong incentive to continue with the investigation of its pharmacological potency, this time in non-cancerous stromal fibroblasts (HS-5) and in several solid tumour cell lines.Myeloma cells proliferate and survive in the BM through physiological and functional interactions with bone marrow stromal cells and surrounding microenvironment.5–7 BM stroma consists of different types of cells including fibroblast stromal cells.52,53 For this reason, non-cancerous stromal fibroblast cell line (HS-5), representing naturally occurring healthy cells of bone marrow physiologically connected with myeloma cells, was used to determine the in vitro biological effect of the leading compound 6 towards non-cancerous cells and its selectivity. Intriguingly, 6 reduced the HS-5 cell viability to 82.5 ± 4.4, 63.0 ± 10.4 and 51.0 ± 3.2% when applied at 1, 2 and 3 μM concentrations, respectively (Fig. 5A and ESI, Fig. S18†). Thus, only a negligible effect of 6 on the cell viability was observed in HS-5 normal fibroblasts. This is particularly important because this observation is highly suggestive of a selective effect of 6 against myeloma cells over healthy stromal fibroblasts. This difference is not connected with the amount of the complex accumulated by the treated cells, because the cellular accumulation of the used complex 6 by HS-5 cells (795 ± 260 ng Pt/106 cells) was higher as compared with both MM cell lines (232 ± 21 ng Pt/106 cells for U266B1, and 171 ± 66 ng Pt/106 cells for KMS12-PE); analysed by inductively coupled plasma-mass spectrometry (ICP-MS) at 1 μM concentration.
 | ||
| Fig. 5 (A) A comparison of in vitro antiproliferative activity results (given as cell viability percentage) obtained at 3 μM concentration of the representative complex 6 in human HS-5 stromal fibroblasts (non-treated HS-5 cells are given for comparative purposes) and myeloma cell lines U266B1 and KMS12-PE. (B) A comparison of in vitro antiproliferative activity results (given as cell viability percentage) obtained at 1 μM concentration of the representative complex 6 (and cisplatin for comparative purposes) in human solid cancer cell lines (DU-145 prostate cancer, HepG2 liver hepatocellular cancer, MCF-7 breast cancer) and myeloma cell lines U266B1 and KMS12-PE. | ||
Further for comparative purposes, in vitro antiproliferative activity was also tested in several solid tumour cell lines, specifically, in DU-145 prostate cancer, HepG2 hepatocellular cancer and MCF-7 breast cancer (1 μM concentration, MTT assay). At 1 μM concentration level (i.e. equimolar with the lowest tested concentration in the myeloma cells), complex 6 was even less effective in the used solid tumour cell lines (73–97% cell viability), which was not the case of the reference drug cisplatin (Fig. 5B and ESI, Table S3†). Thus, complex 6 was more antiproliferative active in the myeloma cell lines than in the used solid cancer cell lines at 1 μM concentration.
Altogether, these results implied high selectivity of 6 towards MM cells over normal HS-5 stromal fibroblasts, and its higher antiproliferative activity in hard-to-treat MM cells than determined in the solid tumour cells (i.e. different anticancer activity profile).
Apoptosis in primary culture of myeloma cells
The effect of the representative highly antiproliferative active complex 6 on apoptosis induction in myeloma cell lines U266B1 and KMS12-PE differing in the IL-6 expression was investigated by determination of caspase 3 and 7 activities (Fig. 6 and ESI, Fig. S19, S20, Table S4†) and by Annexin-V/7-AAD staining (ESI, Fig. S21 and Table S5†). Regarding the principle of Annexin-V/7-AAD staining, while Annexin-V is sensitive to detection of apoptosis, 7-AAD targets necrotic or late apoptotic cells. | ||
| Fig. 6 Apoptosis studies by determination of caspase 3/7 activities to semi-quantify viable, early apoptotic (EA) and late apoptotic/necrotic (LA/N) U266B1 and KMS12-PE myeloma cells (top) or their co-cultures with HS-5 stromal fibroblasts (bottom), as detected after 24 h treatment with 1–3 μM concentrations of complex 6, given with the dot-plots obtained at 1 μM concentration (insets). Colours: white – control, light grey – 1 μM, grey – 2 μM and dark grey – 3 μM. | ||
Regarding the determination of caspase 3/7 activities, the number of viable U266B1 cells (with IL-6 expression) was significantly decreased after treatment with 2 μM (p < 0.001) and 3 μM (p < 0.0001) concentrations of 6, as compared to untreated cells (Fig. 6 and ESI, Fig. S19, Table S4†). The distribution of early apoptotic (EA), late apoptotic (LA) and necrotic (N) populations of the U266B1 cells indicates a dose-dependent effect of the treatment with 6 on the induction of apoptosis (Fig. 6). Yet, the concentration of 1 μM was recognizably insufficient to trigger apoptosis in U266B1 cells.
Similarly, the number of viable KMS12-PE cells determined by the same method significantly decreased (p < 0.05 and p < 0.001, respectively) after treatment with 2 μM and 3 μM concentrations of 6 (Fig. 6 and ESI, Fig. S20, Table S4†), corresponding to the highest proportion of apoptotic cells in the EA phase (Fig. 6). More KMS12-PE cells detected in the LA and N populations corresponds to the reduced number of viable KMS12-PE cells under these experimental conditions (Fig. 4). In contrast to U266B1 cells, the 1 μM concentration of 6 appears to be effective in inducing KMS12-PE cell apoptosis (Fig. 6). In connection with the antiproliferative activity studies (vide supra), the results of the studies of apoptosis induction unambiguously proved that KMS12-PE cells without IL-6 expression are much more susceptible to the treatment by 6 than U266B1 cells with IL-6 expression, because 6 was significantly more effective in reducing cell viability and inducing apoptosis in KMS12-PE cells. Similar results were obtained by Annexin-V/7-AAD staining (ESI, Fig. S21 and Table S5†).
Notably, no caspase activity was detected in the compound 6-treated myeloma cells, which indicated that the ongoing cell death was independent of caspases. This is consistent with previous findings that in myeloma cells, the administration of drugs containing metal ions causes mitochondrial permeabilization by increasing ROS and Ca2+ levels, leading to the induction of caspase-independent apoptosis.54 This process is accompanied by the transfer of cytochrome c, apoptosis inducing factor (AIF) and endonuclease G (EndoG), from the mitochondria to the cytosol.55 AIF then binds directly to DNA, causing chromatin condensation and DNA fragmentation, which is consistent with our investigation and high sub-G1 cell cycle phase population of compound 6-treated myeloma cells (see below).
Also of importance, the caspase-independent apoptosis induced by compound 6 seems to be different from oxaliplatin45 or non-platinum49,50 complexes, which induced caspase-3-dependent apoptosis in myeloma cells. In total, our results are indicative of a different MoA of 6 in myeloma cells as compared with conventional platinum-based anticancer drugs and other types of anti-myeloma active compounds.
Apoptosis in co-culture of myeloma cells and stromal fibroblasts
With respect to the above-discussed BM composition,5–7,52,53 both myeloma cell lines U266B1 and KMS12-PE were in this study also co-cultured with non-cancerous stromal fibroblasts (HS-5), to determine the in vitro biological effect of the leading compound 6 in a mimicked BM environment. The ratio of myeloma cells (KMS12-PE or U266B1) and HS-5 fibroblasts was 20:1.
The number of viable cells in the co-culture of U266B1 myeloma cells and HS-5 stromal fibroblasts was significantly lower after treatment with 2 and 3 μM concentrations of 6 (p < 0.001 by both caspases 3/7 activities and Annexin-V staining) by comparison with the negative control (Fig. 6 and ESI, Fig. S22, Table S5†). 1 μM concentration of 6 had only a modest effect on apoptosis of co-cultured U266B1 cells.
The number of viable KMS12-PE myeloma cells co-cultured with HS-5 stromal fibroblasts was significantly reduced after application of 2 μM and 3 μM of 6, compared to unaffected viable cells in solvent (p < 0.01 for caspases 3/7 activities, p < 0.0001 for Annexin-V staining) (Fig. 6 and ESI, Fig. S23, Table S5†). Subsequent distribution of apoptotic co-cultured cells in the EA and LA/N populations was increased after treatment with 2 μM and 3 μM of 6 concentrations compared to 1 μM. Notably, the 1 μM concentration of 6 appears to be more effective in inducing apoptosis in co-cultivated KMS12-PE cells than in co-cultivated U266B1 cells, both with HS-5 fibroblasts (Fig. 6).
In total for apoptosis investigations, similar results were observed in myeloma cells alone and their co-cultures with healthy stromal fibroblasts (Fig. 6 and ESI, Tables S4, S5†). This observation is suggestive for low level of deactivation of 6 in the mimicked BM environment consisting of target myeloma cells co-cultured with non-cancerous stromal fibroblasts.
Cell cycle analysis in primary culture of myeloma cells
As it is discussed above, the viability of U266B1 cells significantly decreased only after treatment with 2 μM and 3 μM concentration of complex 6 (Fig. 4 and ESI, Table S2†) and the data also clearly indicated a dose-dependent effect of 6 on the induction of apoptosis (Fig. 6 and ESI, Tables S4, S5†). Consistently, influencing the U266B1 cell cycle by 2 μM and 3 μM concentrations of 6 leads to a significant increase (p < 0.01 and p < 0.001, respectively) in the number of cells in the sub-G1 phase of the cell cycle (Fig. 7A and ESI, Fig. S24, Table S6†). Moreover, the investigations showed that the concentration of 1 μM, proved to be insufficiently effective in inducing apoptosis (see above), causes cell cycle arrest in the G1 phase.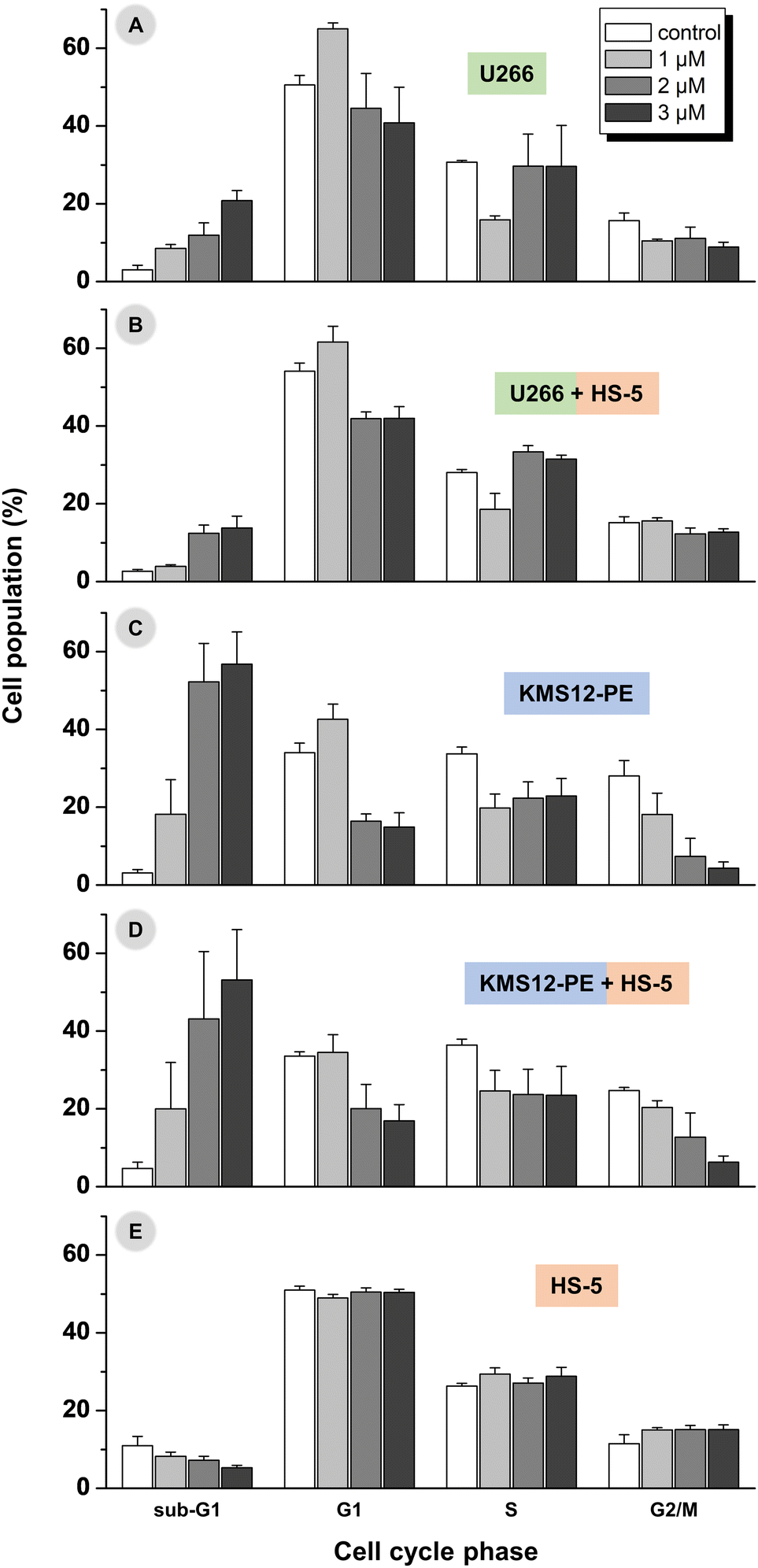 | ||
| Fig. 7 The cell cycle analysis in myeloma cell lines U266B1 (A) and KMS12-PE (C), and their co-cultures with HS-5 stromal fibroblasts (U266B1 + HS-5 in B, and KMS12-PE + HS-5 in D) after 24 h treatment with 1–3 μM concentrations of 6. The results obtained for non-cancerous stromal fibroblasts HS-5 are given for comparative purposes (E). Control = medium with DMF (the same concentration as for experiments with 6). The ratio of myeloma cells (KMS12-PE or U266B1) and HS-5 fibroblasts was 20:1. | ||
Regarding more sensitive KMS12-PE myeloma cells, concentration of 2 μM and 3 μM caused massive cell death and it is difficult to distinguish between different phases of the cell cycle (Fig. 7B and ESI, Fig. S25, Table S6†). These concentrations of 6 appear to be toxic to the KMS12-PE cells. 1 μM concentration was also effective in inducing the cell cycle alternation of the KMS12-PE cells connected with the detection of ca. 20% cell population in the sub-G1 cell cycle phase. The observed higher susceptibility of the IL-6 non-expressing KMS12-PE cells to apoptosis and cell cycle modifications, than determined for the IL-6 expressing U266B1 cells, may be explained by the expression of IL-6, which could promote survival and further proliferation in myeloma cells.4,42–44
Cell cycle analysis in co-culture of myeloma cells and stromal fibroblasts
Regarding the cell cycle modification experiments performed in the co-cultivated myeloma and HS-5 cells (Fig. 7B, D and ESI, Fig. S26, S27, Table S6†), the results were comparable with those detected in the myeloma cells alone (i.e. without stromal fibroblasts; Fig. 7A, C), which proved only a negligible effect of the presence of normal stromal fibroblasts on the biological effect of 6 against the used multiple myeloma cells.Specifically, in the co-culture of U266B1 and HS-5 cells, 6 induced significantly higher sub-G1 cell cycle phase population at 2 μM (p = 0.003) and 3 μM (p = 0.006) concentrations than observed for non-treated co-cultured cells (Fig. 7B and ESI, Fig. S26, Table S6†). The 1 μM concentration of 6 was not effective enough to induce apoptosis in the U266B1/HS-5 cell co-culture. In contrast to the results obtained for the co-culture of the U266B1 and HS-5 cells, the 1 μM concentration of 6 causes a significant increase in the number of co-cultivated KMS12-PE cells in the sub-G1 phase (p < 0.01; Fig. 7D and ESI, Fig. S27, Table S6†).
For comparative purposes, the cell cycle modification was also studied in HS-5 cells without myeloma cells (Fig. 7E and ESI, Fig. S28†), and only negligible changes were detected between the untreated cells in comparison with the cells treated by 1–3 μM concentration of 6 (ESI, Fig. S29†). This is particularly important, because it underlined the conclusion that the observed biological effects (i.e. induction of apoptosis, cell cycle alternation) connected with MoA of highly active anti-myeloma compound 6 can be unambiguously assigned to the used myeloma cells.
DNA as a target molecule?
DNA is a pharmacological target molecule for Pt-based anticancer drugs56 and also for some diiodidoplatinum(II) complexes.17,19,57–59 On the other hand, some Pt iodido complexes did not platinate DNA to a pharmacologically significant level, usually markedly less than cisplatin.20,26 This led us to investigate DNA platination in the model experiment with guanosine monophosphate (GMP) and in the cell-free medium with salmon sperm DNA.At first, the representative complexes 6 and 7 (and cisplatin for comparative purposes) were studied by 1H NMR for their possible interaction with the model nucleobase GMP, to investigate the affinity of these complexes towards DNA. For the mixtures of the complexes (6, 7) with GMP, no new 1H NMR C8–H resonance of GMP was observed up to 24 h of standing in the dark at r.t. (ESI, Fig. S30†), implying that the studied complexes do not interact covalently with the used model nucleobase. These results also suggested that DNA is most likely not a target for the studied Pt(II) diiodido complexes. The control experiment with DNA-binding anticancer drug cisplatin mixed with GMP resulted in the new resonance of the cisplatin–GMP adduct at 8.91 ppm (ESI, Fig. S30†).26,60
Additionally to the experiments with the DNA model molecule GMP, the same complexes (6, 7, cisplatin) were incubated with double-stranded DNA in 10 mM NaClO4 (24 h, 37 °C). The results proved that the new diiodidoplatinum(II) complexes 6 and 7 interacted with the used DNA molecule to a markedly lower extent than cisplatin. Specifically, only ca. 2% of the Pt(II) diiodido complexes (2.1% for 6 and 1.7% for 7) bound to DNA, while cisplatin platinated the used DNA almost quantitatively (90.7%) under the used cell-free experimental conditions.
Overall, the results of the performed studies of possible DNA interactions with GMP and salmon sperm DNA excluded DNA as the primary intracellular target for the studied complexes 6 and 7, clearly distinguishing their MoA from cisplatin. We hypothesize that the studied anti-myeloma diiodido complexes target a different molecule(s) than DNA and trigger unique responses different from conventional platinum-based drugs. In the field of Pt iodido complexes, such unusual MoA has been reported for compounds interacting with mitotic kinesin Eg520 or for complexes inducing high levels of ROS and interfering with redox homeostasis of the treated cancer cells.61
Experimental
The details of the experimental section regarding materials, synthesis of complexes 1–7, general methods, crystallography (CCDC 2211813 (3), 2211814 (4), 2211815 (5) and 2211816 (7)), studies of solvolysis, lipophilicity (logP) and interactions with biomolecules, cell cultures and antiproliferative activity testing (MTT assay) are given in ESI.†
Conclusions
Multiple myeloma unfortunately remains, despite undeniable progress in treatment in recent years, very much disabling and basically incurable disease with most patients developing resistance and relapsing. Intensive chemotherapy combination treatment still includes cisplatin thanks to its strong effect, notwithstanding its known adverse side effects.11 For these reasons, development of anticancer complexes acting through a different mechanism of action represents an attractive field of research drawing considerable attention. Recently, diiodido analogues of cisplatin derivatives have been shown to affect cancer cells in a different fashion, and might thus represent a promising group of agents able to eliminate the drawbacks of platinum chemotherapy.In this work, a series of seven platinum(II) complexes of the general formula cis-[PtI2(Ln)2] (1–7) with various imidazole derivatives (L1 = 1-methyl-1H-imidazole for 1, L2 = 1-benzyl-1H-imidazole for 2, L3 = 1-benzyl-2-methyl-1H-imidazole for 3, L4 = 1-phenyl-1H-imidazole for 4, L5 = 1-(4-methoxyphenyl)-1H-imidazole for 5, L6 = 1-(4-fluorophenyl)-1H-imidazole for 6, and L7 = 1-(4-chlorophenyl)-1H-imidazole for 7) was prepared and thoroughly characterized, including a single-crystal X-ray analysis of 3, 4, 5 and 7. Desirable characteristics related to mechanism of action different from cisplatin were confirmed by 1H NMR spectroscopy, particularly, the complexes do not form covalent adducts either with reduced glutathione or with guanosine monophosphate. Additionally, only a negligible level of DNA-platination was confirmed after incubation of the presented complexes with salmon sperm DNA in a cell-free medium, thus supplying further evidence for contrasting mechanistic behaviour with respect to cisplatin.
Antiproliferative activity of the complexes was evaluated in two multiple myeloma cell lines (U266B1 and KMS12-PE; PE = pleural effusion due to plasma cell infiltration). The anti-myeloma activity of 1–7 was significantly higher than for the reference platinum-based anticancer drug cisplatin. For example, the cell viability of U266B1 and KMS12-PE cells was extraordinarily reduced to 3.0% and 1.1%, respectively, for the leading compound 6, while remained only slightly reduced (93.1% and 88.3%, respectively) for the cisplatin-treated myeloma cells (both compounds applied at 3 μM concentration). Interestingly, complex 6 appeared to induce cell apoptosis significantly more efficiently in KMS12-PE cells without interleukin-6 (IL-6) expression than in U266B1 cells with IL-6 expression. Complex 6 also induced apoptosis in the co-cultures of myeloma cells with non-cancerous stromal fibroblasts. The observed cancer cell death was clearly not dependent on caspases, since no caspase 3/7 activity was detected in the apoptotic myeloma cells treated by 6. This investigation represents a new perspective and challenge for metal-based anti-myeloma therapy, including a monotherapy of this hard-to-treat type of cancer.
Author contributions
Conceptualization: L. M., D. W. D., I. N., K. S. T., P. Š.; methodology: L. M., D. W. D., K. S., J. M., D. M., J. P., I. N., K. S. T., P. Š.; software: —; validation: D. W. D., R. K., I. N., K. S. T.; formal analysis: L. M., D. W. D., K. S., J. M., D. M., R. K., J. P., I. N., K. S. T., P. Š.; investigation: L. M., D. W. D., K. S., J. M., D. M., J. P., K. O.; resources: K. S., J. M., D. M., E. P., I. N., K. S. T., P. Š.; data curation: L. M., D. W. D., K. S., J. M., D. M., I. N., K. S. T., P. Š.; writing – original draft: L. M., D. W. D., K. S., J. M., D. M., I. N., K. S. T., P. Š.; writing – review & editing: L. M., D. W. D., J. M., D. M., R. K., I. N., K. S. T., PŠ; visualization: L. M., D. W. D., I. N., K. S. T., P. Š.; supervision: J. M., D. M., E. P., I. N., K. S. T., P. Š.; project administration: K. S., I. N., K. S. T., P. Š.; funding acquisition: J. M., E. P., I. N., K. S. T., P. Š. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare the following competing financial interest(s): L. M., D. W. D., K. O., I. N., K. S. T. and P. Š. have filed a patent application in the Czech Republic (PV 2021-444) and international patent application (PCT/CZ2022/050095). The other authors declare no competing financial interest.Acknowledgements
This work was supported by Palacký University Olomouc (projects PrF_2021_009, PrF_2022_006 and LF_2022_005), the European Regional Development Fund – Project ENOCH (No. CZ.02.1.01/0.0/0.0/16_019/0000868), the Ministry of Education, Youth and Sports of the Czech Republic under the project CEITEC 2020 (LQ1601), and Jagiellonian University Medical College (N42/DBS/000101). This work was supported by the Slovak Research and Development Agency (APVV-19-0087) and by the Scientific Grant Agency of the Slovak Republic (Project 1/0686/23).References
- J. Ferlay, M. Ervik, F. Lam, M. Colombet, L. Mery, M. Piñeros, A. Znaor, I. Soerjomataram and F. Bray, Global Cancer Observatory: Cancer Today, International Agency for Research on Cancer, Lyon, 2020, https://gco.iarc.fr/today, accessed 09/2022 Search PubMed.
- A. Barchnicka, M. Olejniczak-Nowakowska, K. Krupa-Kotara and S. Grosickiet, The Importance of Antiangiogenic Effect in Multiple Myeloma Treatment, Adv. Clin. Exp. Med., 2018, 27, 291–297 CrossRef PubMed.
- C. S. Mitsiades, D. W. McMillin, S. Klippel, T. Hideshima, D. Chauhan, P. G. Richardson, N. C. Munshi and K. C. Anderson, The Role of the Bone Marrow Microenvironment in the Pathophysiology of Myeloma and its Significance in the Development of More Effective Therapies, Hematol./Oncol. Clin. North Am., 2007, 21, 1007–1034 CrossRef PubMed.
- T. Matthes, B. Manfroi and B. Huard, Revisiting IL-6 Antagonism in Multiple Myeloma, Crit. Rev. Oncol. Hematol., 2016, 105, 1–4 CrossRef PubMed.
- G. Bianchi and N. C. Munshi, Pathogenesis beyond the Cancer Clone(s) in Multiple Myeloma, Blood, 2015, 125, 3049–3058 CrossRef CAS PubMed.
- M. Abe, Targeting the Interplay between Myeloma Cells and the Bone Marrow Microenvironment in Myeloma, Internet J. Hematol., 2011, 94, 334–343 CrossRef PubMed.
- M. B. Meads, L. A. Hazlehurst and W. S. Daltonet, The Bone Marrow Microenvironment as a Tumor Sanctuary and Contributor to Drug Resistence, Clin. Cancer Res., 2008, 14, 2519–2526 CrossRef CAS PubMed.
- A. H.-H. Wong, E. M. Shin, V. Tergaonkar and W.-J. Chng, Targeting NF-κB Signaling for Multiple Myeloma, Cancers, 2020, 12, 2203 CrossRef CAS PubMed.
- L. Rosiñol, M. Beksac, E. Zamagni, N. W. C. J. Van de Donk, K. C. Anderson, A. Badros, J. Caers, M. Cavo, M.-A. Dimopoulos, A. Dispenzieri, H. Einsele, M. Engelhardt, C. F. de Larrea, G. Gahrton, F. Gay, R. Hájek, V. Hungria, A. Jurczyszyn, N. Kröger, R. A. Kyle, F. L. da Costa, X. Leleu, S. Lentzsch, M. V. Mateos, G. Merlini, M. Mohty, P. Moreau, L. Rasche, D. Reece, O. Sezer, P. Sonneveld, S. Z. Usmani, K. Vanderkerken, D. H. Vesole, A. Waage, S. Zweegman, P. G. Richardson and J. Bladé, Expert Review on Soft-tissue Plasmacytomas in Multiple Myeloma: Definition, Disease Assessment and Treatment Considerations, Br. J. Haematol., 2021, 194, 496–507 CrossRef PubMed.
- S. V. Rajkumar and S. Kumar, Multiple Myeloma Current Treatment Algorithms, Blood Cancer J., 2020, 10, 94 CrossRef PubMed.
- P. T. Griffin, V. Q. Ho, W. Fulp, T. Nishihori, K. H. Shain, M. Alsina and R. C. Baz, A Comparison of Salvage Infusional Chemotherapy Regimens for Recurrent/Refractory Multiple Myeloma, Cancer, 2015, 121, 3622–3630 CrossRef CAS PubMed.
- L. Kelland, The Resurgence of Platinum-Based Cancer Chemotherapy, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- R. Oun, Y. E. Moussa and N. J. Wheate, The Side Effects of Platinum-based Chemotherapy Drugs: A Review for Chemists, Dalton Trans., 2018, 47, 6645–6653 RSC.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W.-Y. Zhang and Z. Zhang, Metallodrugs are Unique: Opportunities and Challenges of Discovery and Development, Chem. Sci., 2020, 11, 12888–12917 RSC.
- R. N. Bose, L. Maurmann, R. J. Mishur, L. Yasui, S. Gupta, W. S. Grayburn, H. Hofstetter and T. Salley, Non-DNA-binding Platinum Anticancer Agents: Cytotoxic Activities of Platinum-phosphato Complexes Towards Human Ovarian Cancer Cells, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18314–18319 CrossRef CAS PubMed.
- D. D. Karp, D. R. Camidge, J. R. Infante, T. D. Ames, M. R. Price, J. Jimeno and A. H. Bryce, Phase I Study of PT-112, a Novel Pyrophosphateplatinum Immunogenic Cell Death Inducer, in Advanced Solid Tumours, EClinicalMedicine, 2022, 49, 101430 CrossRef PubMed.
- P. Štarha, J. Vančo and Z. Trávníček, Platinum Iodido Complexes: A Comprehensive Overview of Anticancer Activity and Mechanisms of Action, Coord. Chem. Rev., 2019, 380, 103–135 CrossRef.
- L. Messori, L. Cubo, C. Gabbiani, A. álvarez-Valdés, E. Michelucci, G. Pieraccini, C. Ríos-Luci, L. G. León, J. M. Padrón, C. Navarro-Ranninger, A. Casini and A. G. Quiroga, Reactivity and Biological Properties of a Series of Cytotoxic PtI2(amine)2 Complexes, Either cis or trans Configured, Inorg. Chem., 2012, 51, 1717–1726 CrossRef CAS PubMed.
- T. Marzo, S. Pillozzi, O. Hrabina, J. Kasparkova, V. Brabec, A. Arcangeli, G. Bartoli, M. Severi, A. Lunghi, F. Totti, C. Gabbiani, A. G. Quiroga and L. Messori, cis-PtI2(NH3)2: A Reappraisal, Dalton Trans., 2015, 44, 14896–14905 RSC.
- J. Kasparkova, H. Kostrhunova, V. Novohradsky, A. A. Logvinov, V. V. Temnov, N. E. Borisova, T. A. Podrugina, L. Markova, P. Štarha, A. A. Nazarov and V. Brabec, Novel cis-Pt(II) Complexes with Alkylpyrazole Ligands: Synthesis, Characterization, and Unusual Mode of Anticancer Action, Bioinorg. Chem. Appl., 2022, 2022, 1717200 Search PubMed.
- I. Ali, M. N. Lone and H. Y. Aboul-Enein, Imidazoles as Potential Anticancer Agents, MedChemComm, 2017, 8, 1742–1773 RSC.
- P. Sharma, C. LaRosa, J. Antwi, R. Govindarajan and K. A. Werbovetz, Imidazoles as Potential Anticancer Agents: An Update on Recent Studies, Molecules, 2021, 26, 4213 CrossRef CAS PubMed.
- N. Ferri, G. Facchetti, S. Pellegrino, C. Ricci, G. Curigliano, E. Pini and I. Rimoldi, Promising Antiproliferative Platinum(II) Complexes Based on Imidazole Moiety: Synthesis, Evaluation in HCT-116 Cancer Cell Line and Interaction with Ctr-1 Met-rich Domain, Bioorg. Med. Chem., 2015, 23, 2538–2547 CrossRef CAS PubMed.
- M. Gozelle, A. K. Süloğlu, G. Selmanoğlu, N. Ramazanoğlu, L. Açık and F. Gümüş, Studies on the Synthesis, Characterization, Cytotoxic Activities and Plasmid DNA Binding of Platinum(II) Complexes Having 2-Subsituted Benzimidazole Ligands, Polyhedron, 2019, 161, 298–308 CrossRef CAS.
- G. Facchetti, N. Ferri, M. G. Lupo, L. Giorgio and I. Rimoldi, Monofunctional PtII Complexes Based on 8-Aminoquinoline: Synthesis and Pharmacological Characterization, Eur. J. Inorg. Chem., 2019, 3389–3395 CrossRef CAS.
- P. Štarha, J. Vančo, Z. Trávníček, J. Hošek, J. Klusáková and Z. Dvořák, Platinum(II) Iodido Complexes of 7-Azaindoles with Significant Antiproliferative Effects: An Old Story Revisited with Unexpected Outcomes, PLoS One, 2016, 11, e0165062 CrossRef PubMed.
- P. Štarha, Z. Dvořák and Z. Trávníček, Highly and Broad-spectrum in Vitro Antitumor Active cis-Dichloridoplatinum(II) Complexes with 7-Azaindoles, PLoS One, 2015, 10, e0136338 CrossRef PubMed.
- P. Štarha, Z. Trávníček, I. Popa and Z. Dvořák, Synthesis, Characterization and in Vitro Antitumor Activity of Platinum(II) Oxalato Complexes Involving 7-Azaindole Derivatives as Coligands, Molecules, 2014, 19, 10832–10844 CrossRef PubMed.
- S. Utku, F. Gumus, S. Tezcan, M. S. Serin and A. Ozkul, Synthesis, Characterization, Cytotoxicity, and DNA Binding of Some New Platinum(II) and Platinum(IV) Complexes with Benzimidazole Ligands, J. Enzyme Inhib. Med. Chem., 2010, 25, 502–508 CrossRef CAS PubMed.
- S. Utku, F. Gumus, T. Karaoglu and A. Ozkul, Cytotoxic Activity of Platinum(II) and Platinum(IV) Complexes Bearing 5(6)-Non/chlorosubstituted-2-hydroxymethyl Benzimidazole Ligands against HEp-2 Cell Line, J. Fac. Pharm. Ankara Univ., 2007, 36, 21–30 CAS.
- M. Ravera, E. Gabano, M. Sardi, G. Ermondi, G. Caron, M. J. McGlinchey, H. Müller-Bunz, E. Monti, M. B. Gariboldi and D. Osella, Synthesis, Characterization, Structure, Molecular Modeling Studies and Biological Activity of Sterically Crowded Pt(II) Complexes Containing Bis(imidazole) Ligands, J. Inorg. Biochem., 2011, 105, 400–409 CrossRef CAS PubMed.
- S. C. Dhara, A Rapid Method for the Synthesis of cis-[Pt(NH3)2Cl2], Indian J. Chem., 1970, 8, 193–194 Search PubMed.
- P. Štarha, Z. Trávníček, J. Vančo and Z. Dvořák, In Vitro Anticancer Active cis-Pt(II)-diiodido Complexes Containing 4-Azaindoles, J. Biol. Inorg. Chem., 2019, 24, 257–269 CrossRef PubMed.
- B. M. Still, P. A. Kumar, J. R. Aldrich-Wright and W. S. Price, 195Pt NMR - Theory and Application, Chem. Soc. Rev., 2007, 36, 665–686 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- J. S. Casas, A. Castineiras, Y. Parajo, J. Sordo and J. M. Varela, (1,1′-Dimethyl-2,2′-biimidazole-N3,N3′)diiodoplatinum(II), Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 1777–1779 CrossRef.
- J. Reedijk, Why Does Cisplatin Reach Guanine-N7 with Competing S-Donor Ligands Available in the Cell?, Chem. Rev., 1999, 99, 2499–2510 CrossRef CAS PubMed.
- J. Holford, S. Y. Sharp, B. A. Murrer, M. Abrams and L. R. Kelland, In Vitro Circumvention of Cisplatin Resistance by the Novel Sterically Hindered Platinum Complex AMD473, Br. J. Cancer, 1998, 77, 366–373 CrossRef CAS PubMed.
- R. Mannhold, G. I. Poda, C. Ostermann and I. V. Tetko, Calculation of Molecular Lipophilicity: State-of-the-art and Comparison of log P Methods on More than 96,000 Compounds, J. Pharm. Sci., 2009, 98, 861–893 CrossRef CAS PubMed.
- J. J. Wilson and S. J. Lippard, In Vitro Anticancer Activity of cis-Diammineplatinum(II) Complexes with β-Diketonate Leaving Group Ligands, J. Med. Chem., 2012, 55, 5326–5336 CrossRef CAS PubMed.
- A. S. Gerrie, J. R. Mikhael, L. Cheng, H. Jiang, V. Kukreti, T. Panzarella, D. Reece, K. A. Stewart, Y. Trieu, S. Trudel and C. I. Chen, D(T)PACE as Salvage Therapy for Aggressive or Refractorymultiple Myeloma, Br. J. Haematol., 2013, 161, 802–810 CrossRef CAS PubMed.
- D. Harmer, C. Falank and M. R. Reagan, Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss, and Multiple Myeloma, Front. Endocrinol., 2019, 9, 788 CrossRef PubMed.
- S. B. Ingersoll, S. Ahmad, N. D. Thoni, F. H. Ahmed, K. A. Monahan and J. R. Edwards, Targeting the IL-6 Pathway in Multiple Myeloma and its Implications in Cancer-associated Gene Hypermethylation, Med. Chem., 2011, 7, 473–479 CrossRef CAS.
- L. Shirato, T. Otsuki, O. Yamada, M. Namba, H. Nakajima, Y. Nozawa, A. Ueki and Y. Yawata, Down Regulation of Protein Kinase C During Growth Enhancement Induced by Interleukin-6 on a Human Myeloma Cell Line, KMS-111996, Cancer Lett., 1996, 107, 131–136 CrossRef CAS PubMed.
- P. Tassone, P. Tagliaferri, E. Galea, C. Palmieri, P. Bonelli, M. L. Martelli, F. Tuccillo, M. C. Turco and S. Venuta, Oxaliplatin (L-OHP) Treatment of Human Myeloma Cells Induces in Vitro Growth Inhibition and Apoptotic Cell Death, Eur. J. Cancer, 2002, 38, 1141–1147 CrossRef CAS PubMed.
- M. Kvasnica, M. Budesinsky, J. Swaczynova, V. Pouzar and L. Kohout, Platinum(II) Complexes with Steroidal Esters of L-Methionine and L-Histidine: Synthesis, Characterization and Cytotoxic Activity, Bioorg. Med. Chem., 2008, 16, 3704–3713 CrossRef CAS PubMed.
- M. Krstić, S. P. Sovilj, S. Grgurić-Šipka, I. R. Evans, S. Borozan and J. F. Santibanez, Synthesis, Structural and Spectroscopic Characterization, in Vitro, Cytotoxicity and in Vivo Activity as Free Radical Scavengers of Chlorido(p-cymene) Complexes of Ruthenium(II) Containing N-Alkylphenothiazines, Eur. J. Med. Chem., 2011, 46, 4168–4177 CrossRef PubMed.
- R. Pettinari, F. Marchetti, A. Petrini, C. Pettinari, G. Lupidi, B. Fernández, A. R. Diéguez, G. Santoni and M. Nabissi, Ruthenium(II)-arene Complexes with Dibenzoylmethane Induce Apoptotic Cell Death in Multiple Myeloma Cell Lines, Inorg. Chim. Acta, 2017, 454, 139–148 CrossRef CAS.
- A. Wołoszyn, C. Pettinari, R. Pettinari, G. V. B. Patzmay, A. Kwiecień, G. Lupidi, M. Nabissi, G. Santoni and P. Smoleński, Ru(II)-(PTA) and -mPTA Complexes with N2-donor Ligands Bipyridyl and Phenanthroline and Their Antiproliferative Activities on Human Multiple Myeloma Cell Lines, Dalton Trans., 2017, 46, 10073–10081 RSC.
- Y. Xu, Q. Zhou, X. Feng, Y. Dai, Y. Jiang, W. Jiang, X. Liu, X. Xing, Y. Wang, Y. Ni and C. Zheng, Disulfiram/copper Markedly Induced Myeloma Cell Apoptosis through Activation of JNK and Intrinsic and Extrinsic Apoptosis Pathways, Biomed. Pharmacother., 2020, 126, 110048 CrossRef CAS PubMed.
- A. Nakaya, M. Sagawa, A. Muto, H. Uchida, Y. Ikeda and M. Kizaki, The Gold Compound Auranofin Induces Apoptosis of Human Multiple Myeloma Cells through Both Down-regulation of STAT3 and Inhibition of NF-κB Activity, Leuk. Res., 2011, 35, 243–249 CrossRef CAS PubMed.
- M. Bar-Natan, D. Stroopinsky, K. Luptakova, M. D. Coll, A. Apel, H. Rajabi, A. R. Pyzer, K. Palmer, M. R. Reagan, M. R. Nahas, R. Karp Leaf, S. Jain, J. Arnason, I. M. Ghobrial, K. C. Anderson, D. Kufe, J. Rosenblatt and D. Avigan, Bone Marrow Stroma Protects Myeloma Cells from Cytotoxic Damage via Induction of the Oncoprotein MUC1, Br. J. Haematol., 2017, 176, 929–938 CrossRef CAS PubMed.
- P. de la Puente, N. Quan, R. S. Hoo, B. Muz, R. C. Gilson, M. Luderer, J. King, S. Achilefu, N. N. Salama, R. Vij and A. K. Azab, Newly Established Myeloma-derived Stromal Cell Line MSP-1 Supports Multiple Myeloma Proliferation, Migration, and Adhesion and Induces Drug Resistance More Than Normal-derived Stroma, Haematologica, 2016, 101, 307–311 CrossRef PubMed.
- V. Innao, V. Rizzo, A. G. Allegra, C. Musolino and A. Allegra, Promising Anti-mitochondrial Agents for Overcoming Acquired Drug Resistance in Multiple Myeloma, Cells, 2021, 10, 439 CrossRef CAS PubMed.
- J. Lou, Y. Zhou, Z. Feng, M. Ma, Y. Yao, Y. Wang, Y. Deng and Y. Wu, Caspase-independent Regulated Necrosis Pathways as Potential Targets in Cancer Management, Front. Oncol., 2021, 10, 616952 CrossRef PubMed.
- V. Brabec, O. Hrabina and J. Kasparkova, Cytotoxic Platinum Coordination Compounds. DNA binding agents, Coord. Chem. Rev., 2017, 351, 2–31 CrossRef CAS.
- F. Navas, S. Perfahl, C. Garino, L. Salassa, O. Novakova, C. Navarro-Ranninger, P. J. Bednarski, J. Malina and A. G. Quiroga, Reactivity and Biological Properties of a Series of Cytotoxic PtI2(amine)2 Complexes, Either cis or trans Configured, J. Inorg. Biochem., 2015, 153, 211–218 CrossRef CAS PubMed.
- M. Chtchigrovsky, L. Eloy, H. Jullien, L. Saker, E. Ségal-Bendirdjian, J. Poupon, S. Bombard, T. Cresteil, P. Retailleau and A. Marinetti, Antitumor trans-N-Heterocyclic Carbene–Amine–Pt(II) Complexes: Synthesis of Dinuclear Species and Exploratory Investigations of DNA Binding and Cytotoxicity Mechanisms, J. Med. Chem., 2013, 56, 2074–2086 CrossRef CAS PubMed.
- A. Savić, L. Filipović, S. Aranđelović, B. Dojčinović, S. Radulović, T. J. Sabo and S. Grgurić-Šipka, Synthesis, Characterization and Cytotoxic Activity of Novel Platinum(II) Iodido Complexes, Eur. J. Med. Chem., 2014, 82, 372–384 CrossRef PubMed.
- K. J. Barnham, M. I. Djuran, P. del Socorro Murdoch, J. D. Ranford and P. J. Sadler, L-Methionine Increases the Rate of Reaction of 5′-Guanosine Monophosphate with the Anticancer Drug Cisplatin: Mixed-ligand Adducts and Reversible Methionine Binding, J. Chem. Soc., Dalton Trans., 1995, 3721–3726 RSC.
- S. Misirlić Denčić, J. Poljarević, A. M. Isakovic, I. Marković, T. J. Sabo and S. Grgurić-Šipka, Antileukemic Action of Novel Diamine Pt(II) Halogenido Complexes: Comparison of the Representative Novel Pt(II) with Corresponding Pt(IV) Complex, Chem. Biol. Drug Des., 2017, 90, 262–271 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, 1H, 13C and 195Pt NMR spectra, crystal data and structure refinement, stability studies, viability data (multiple myeloma (MM), stromal fibroblasts, solid cancer cells); flow cytometry data (apoptosis studies and cell cycle). CCDC 2211813 (3), 2211814 (4), 2211815 (5) and 2211816 (7). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi00327b |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2023 |