
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the potential as molecular quantum-dot cellular automata of a mixed-valence Ru2 complex deposited on a Au(111) surface
Nicolás Montenegro-Pohlhammer a,
Carlos M. Palominob and
Carmen J. Calzado*b
a,
Carlos M. Palominob and
Carmen J. Calzado*b
aLaboratory of Theoretical Chemistry, Faculty of Chemistry and Biology, University of Santiago de Chile (USACH), 9170022, Santiago, Chile
bDepartamento de Química Física, Universidad de Sevilla, c/ Prof. García González, s/n 41012, Sevilla, Spain. E-mail: calzado@us.es
First published on 21st March 2023
Abstract
A key requirement for the implementation of molecular quantum dot cellular automata (mQCA) is the ordered attachment of molecules on surfaces. In this work we explore by means of state-of-the art quantum chemistry calculations different aspects of the deposition of a mixed-valence Ru2+ complex on Au(111), underlined as a promising component in mQCA. Our results provide information about the geometry adopted by the Ru2+ molecule and the counterion PF6− on the metal surface and the electronic structure of the doublet ground state, with the excess charge localized on one side of the molecule, not only on the Ru centre, but also involving a part of the 12-carbon bridging chain, which could explain the pair of bright/dim features of STM images. Our calculations also test the functionality as the mQCA of the Ru2+ molecule, evaluating the response curve under an applied electric field as perturbation, and the electronic distribution of two neighbouring Ru2+ molecules forming a four-dot cell, with a diagonal organization of the two excess charges. To the best of our knowledge, this is the first study devoted to the analysis of the electronic structure and mQCA functionality of deposited mixed-valence molecules.
1. Introduction
The concept of quantum-dot cellular automata (QCA) was introduced by von Neumann and Ulam in the 1940s1 and experimentally demonstrated by Lent and coworkers in 1993,2 as a possible alternative to current electronics.3 A QCA cell consists of either four quantum dots positioned at the corners of a square or two dots (half-cell) arranged side by side or head to tail. A quantum dot is a region in the cell where charge can localize. In the case of the four-dot QCA, there are two extra electrons (or holes) which can move among the dots. These two charges are favourable for adopting a diagonal arrangement due to Coulomb repulsion, resulting in two degenerate states that can be used to encode logic “0” and “1” states (Fig. 1). In the two-dot QCA, an extra electron (hole) can be localized in one of the two sites. The central idea is that the Coulomb interaction between neighbouring cells is sufficient to transmit the binary signal with no current flow and low power dissipation. Then, QCA devices are expected to achieve a relevant saving of energy and a significant increase in processing speed.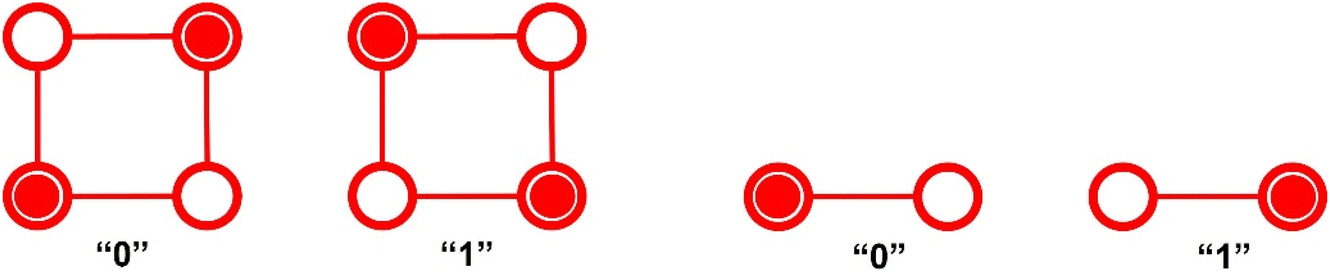 | ||
| Fig. 1 Schematic four-dot QCA (left) and two-dot half-QCA (right). Coulomb repulsion keeps the electrons (in red) at diametrical sites. Binary information “0” or “1” is encoded in the charge distribution. | ||
QCA has been experimentally demonstrated using metal islands of about 60 nm as dots but only at very low temperature (100 mK).4,5 To improve the operation temperature and reduce the size of QCA devices, it has been suggested there is a possibility of replacing the dots by a single molecule with two or more redox active centres, known as molecular QCA (mQCA).6–9 The candidate molecule needs to have bistable charge configurations that can be switched by Coulomb interactions. Several candidate systems have been proposed, including organic and organometallic mixed-valence (MV) complexes, zwitterionic boron–allyl complexes, metal cluster carboxylates, and double-cage fluorinated fullerene anions.10–23 Together with the experimental characterization, much effort has been devoted to the development of theoretical models24–29 aimed to establish the parameters controlling their functionality, such as the intracell Coulomb interaction, the electron transfer integral and the vibronic coupling.
Mixed-valence molecules are by far the most studied. Let us consider a complex with two redox sites, with one excess electron that can move between them. When this electron is localized on one of the redox sites, e.g., the left side, the molecule represents a “1” bit. The electron transfer to the right site changes the ground state configuration, and the molecule represents now a “0” bit. A moderate coupling between the redox sites is required to obtain a rapid exchange of the charge between them. This requirement can be fulfilled by a class II MV complex in Robin–Day classification. Additionally, the operation as mQCA requires the molecules to be attached to a substrate in ordered arrays, which in some cases implies functionalization with specific anchor ligands.13,18–20,30
Recently, Guo and Kandel31 investigated by means of scanning tunneling microscopy (STM) the deposition of the dinuclear complex trans-[Cl(dppe)2Ru(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)6Ru(dppe)2Cl] Ru2 on a Au(111) surface (dppe = diphenylphosphinoethane) and its potential as a two-charge-container QCA cell. The dinuclear complex consists of two Ru-centred phenylphosphine groups connected by a linear 12-carbon chain (Fig. 2). The STM images indicated that the Ru2 molecules formed monolayers of ordered stripes on Au(111) at low temperature, where the molecules lined up side-by-side into rows. The mixed valence Ru2+ complex was obtained by mixing Ru2 once deposited on the gold surface with ferrocenium hexafluorophosphate, [Fe(η5-C5H5)2][PF6]. The oxidation to Ru2+ introduced an additional positive charge on the complex that can tunnel between the two metal-centred groups. The oxidized Ru2+ molecules maintained the organization in stripes, but the two dots appeared to have different contrasts in STM images. This has been related to the localization of the extra charge in one of the two metal-centred redox sites. The STM images showed also additional small features assigned to the PF6− anions, which compensated the extra charge of Ru2+.
C)6Ru(dppe)2Cl] Ru2 on a Au(111) surface (dppe = diphenylphosphinoethane) and its potential as a two-charge-container QCA cell. The dinuclear complex consists of two Ru-centred phenylphosphine groups connected by a linear 12-carbon chain (Fig. 2). The STM images indicated that the Ru2 molecules formed monolayers of ordered stripes on Au(111) at low temperature, where the molecules lined up side-by-side into rows. The mixed valence Ru2+ complex was obtained by mixing Ru2 once deposited on the gold surface with ferrocenium hexafluorophosphate, [Fe(η5-C5H5)2][PF6]. The oxidation to Ru2+ introduced an additional positive charge on the complex that can tunnel between the two metal-centred groups. The oxidized Ru2+ molecules maintained the organization in stripes, but the two dots appeared to have different contrasts in STM images. This has been related to the localization of the extra charge in one of the two metal-centred redox sites. The STM images showed also additional small features assigned to the PF6− anions, which compensated the extra charge of Ru2+.
 | ||
| Fig. 2 Representation of the Ru2 molecule: dark green, grey, white, orange and light green correspond to Ru, C, H, P and Cl, respectively. | ||
It is the aim of this work to explore by means of state-of-the-art quantum chemistry calculations the deposition of the Ru2+ complex on Au(111) and those features that highlight its potential as mQCA. We have optimized the molecule on the gold surface in the presence of the counterion by means of periodic density functional calculations. Once optimized, we analyzed the low-lying states of the oxidized Ru2+ molecule by wave-function based approaches and found that the active redox sites do involve not only the metal centres but also part of the 12-carbon wire. The PF6− anion on the surface is placed close to one of the metal centres, compensating the extra charge of the oxidized Ru2+ cation, in line with the experimental STM images.
Finally the functional suitability of Ru2+ as mQCA cells was investigated by simulating the impact of an external electric field on the charge distribution of the molecule with the geometry adopted on the Au(111) surface, and by simulating the electronic behavior of two paired molecules, which showed the expected antipodal charge configuration.
These results confirm the stability of the Ru2+ molecule deposited on the metal substrate, the charge localization in the effective dots, the antiparallel alignment of the charge on neighboring molecules on the surface and the capability of switching upon application of an electric field.
2. Description of the models and computational details
Since there are no available X-ray data for the Ru2 system – in fact difficulties in obtaining crystals suitable for structure determination have been mentioned in the literature32–34 – we have used the X-ray crystallographic data of a similar dinuclear Ru(II) complex33trans-[(L)(dppe)2Ru(CC)6Ru(dppe)2(L)][PF6]2, with L = NCCH2CH2NH2 to build the computational models. The terminal NCCH2CH2NH2 ligands were replaced by chlorine atoms,33 with a Ru–Cl distance of 2.4 Å, as in many other Ru–Cl bonds.35 These bond distances were optimized in the subsequent calculations.
We have optimized first the isolated molecule in the mixed-valence states, using a modified B3LYP exchange–correlation functional, EXC = (1 − a)(ELSDAX + ΔEB88X) + aEHFX + ELYPC, with 35% of exact exchange (a = 0.35) and LYP correlation.36 This functional has been suggested as a practical protocol to deal with mixed-valence systems in a set of benchmark studies.37,38 Additionally, the molecule has been optimized with the rPBE functional to compare with the results obtained from periodic calculations, where the modified B3LYP functional is not available. The ORCA code39 has been employed to run the optimizations, with the basis sets of quality def2SVP for all atoms.
Once optimized, the molecule was deposited on the Au(111) surface, represented by a three-layer slab, with a total of 336 Au atoms. This model of the surface based on a three-layer slab has been extensively used in studies dealing with the deposition of molecules on metal surfaces.40–45 Both the molecule and the upper layer of the gold slab were optimized with the rPBE functional and basis set of DZP quality for all atoms, except Au atoms represented by SZP basis, using periodic conditions with the SIESTA code.46 The mesh cut-off energy was set to 300 Rydberg for both the real and reciprocal space grids in all calculations, with a self-consistency tolerance for the convergence of the energy and the density matrix of 1.0 × 10−4 eV.
Next, we performed CASSCF calculations on the optimized geometry of the isolated molecule and the molecule deposited on the metal surface to extract information on the nature and energy of its low-lying states and the extension of the electronic localization of the redox sites. The basis sets of quality def2SVP were employed for all the atoms. State-average CASSCF(7/4) calculations for the four lowest doublet roots were carried out with the ORCA code.39 The active space contains seven electrons, distributed on the Ru 4dxz and 4dxy orbitals combined with the πz and πy orbitals of the carbon bridging chain.
Finally, we analyse the functionality as the QCA of the deposited Ru2+ molecule. First, we simulated the response curve of the complex under an external electric field, with the aim of testing the functional suitability of Ru2+ as QCA cells. State-average CASSCF(7/4) calculations of the two-lowest quasi-degenerated doublet states are performed in the presence of an external electric field oriented along the long axis of the molecule (field in the x axis, almost parallel to the intramolecular Ru–Ru axis) with increasing strength. The spin density on each effective redox site is evaluated from Mulliken spin population analysis. The uniform electric field is generated by the TITAN code,47 as a set of point charges distributed in two circular plates separated by 90 Å on opposite sides of the molecule. Each plate contains 33 circular rings, separated by 2.8 Å, where the point charges are homogeneously distributed.
Additionally, we calculated the electronic structure of the cell composed of two Ru2+ molecules, parallel to each other, with the Ru centres at the vertices of a rhombus, following the packing observed in the STM images of the Ru2+[PF6−] monolayer on Au(111).31 In this case, state average CASSCF calculations of the four lowest triplet roots are performed, with active spaces containing 14 electrons in 8 active orbitals.
3. Results and discussion
3.1. Isolated Ru2+ molecules
The optimization of the isolated Ru2+ molecule with the modified B3LYP functional provides a quite symmetric structure, with only small differences between both Ru centres. A similar structure results from the optimization with the rPBE functional (Fig. 3). The 12-carbon wire presents a linear configuration with alternating short/long bonds. The C–C distances are ca. 1.283 Å/1.324 Å, which could be compared with the distances in the homovalent molecule, 1.270 Å/1.342 Å, obtained at the same level of calculation. Hence, the oxidation reduces the bond length alternation on the π-conjugated linker. This trend has been experimentally observed on one-electron oxidized dinuclear metal M complexes with all-carbon –(CC)2– as a linker, with M = Fe, Mo, Re and Ru as the metal centre.48 The terminal carbon atoms of the bridge occupy the axial positions of the Ru coordination spheres, with Ru–C distances of 1.918 Å and 1.925 Å for Ru(1) and Ru(2), respectively (1.891 Å and 1.898 Å for the rPBE functional). The carbon wire is symmetrically aligned with the x axis, with its π orbitals placed on the xz (πz) and xy (πy) planes.
 | ||
| Fig. 3 (left) C–C and C–Ru bond distances in the Ru2+ molecule from rPBE calculations on the optimized geometries for the isolated (top) and deposited molecule on Au(111) in the presence of the PF6− counterion (bottom). (right) Singly occupied orbitals of the lowest doublet state from state average CASSCF(7/4) calculations. | ||
At this level of calculation, the singly occupied molecular orbital (SOMO) is completely delocalized, resulting from the antibonding combination of the Ru 4dxy orbitals with the highest occupied πy orbital of the carbon chain (Fig. 4, top). The SOMO is close in energy to a doubly occupied orbital corresponding to an equivalent combination involving the Ru 4dxz orbitals and the occupied πz orbital of the carbon chain (HOMO−1 in Fig. 4). The nature of the SOMO indicates that the unpaired electron is highly delocalized between both metal centres with a significant participation of the carbon chain. The partial oxidation of the bridge is in line with the decrease of the bond length alternation observed for the optimized geometry of the Ru2+ molecule. This behaviour has been previously found in mixed-valence diruthenium compounds with polyynediyl linkers, where the radical centre tends to localize on the π-conjugated bridge.48,49 In fact, a three state model has been suggested to analyse the behaviour of these MV complexes,50 an extension to the general Marcus–Hush two-state model, involving additionally a bridge-localized state.
 | ||
| Fig. 4 Front and side views of the (left) SOMO and (right) HOMO−1 for the isolated Ru2+ molecule at the B3LYP* level, similar for the rPBE description. | ||
A similar description is obtained at the CASSCF level, and the lowest doublet is represented by a single configuration, where the singly occupied orbital presents the same shape as the SOMO from the B3LYP* (or rPBE) calculation (Fig. 3), i.e., the unpaired electron occupies a molecular orbital resulting from the antibonding combination of the HOMO of the polyynediyl-bridge and the 4dxy Ru orbitals. The first-excited doublet state is separated by 399 cm−1 to the ground state. The wavefunction of this excited state is dominated by a configuration where the singly occupied orbital involves the Ru 4dxz orbitals and the occupied πz orbitals, in consonance with the nature of the HOMO−1 of the DFT description.
Therefore, independent of the approach, the isolated Ru2+ molecule presents a marked charge delocalization and could then be classified as a class III Robin–Day mixed-valence compound. Hence, using concepts of the Marcus–Hush electron transfer theory51–56 for the isolated Ru2+ molecule, the resonance energy (2|J|, with J being the electronic coupling between the diabatic states) is larger than the reorganization energy (λ), 2|J|/λ > 1, and the ground state potential-energy surface shows a single minimum.
3.2. Deposited Ru2+ molecules on Au(111)
The complex is now deposited on the Au(111) surface and optimized in the presence of the PF6− anion with periodic rPBE calculations. Different conformations have been tested for both the molecule and the counterion, and the most stable one is shown in Fig. 5. The z axis corresponds to the normal to the surface. The molecule lies flat, with its long axis (x) parallel to the surface. The molecule on the surface is arranged in such a way that the intramolecular metal–metal axis forms an angle of ∼8° with respect to the [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] direction.
0] direction.
 | ||
| Fig. 5 Top and side views of the Ru2+ molecule and PF6− ions deposited on the Au(111) surface. Au, Ru, C, F, Cl, P and H are represented by yellow, green, grey, cyan, light green, blue and white balls, respectively. | ||
The molecule presents distortions both in the carbon chain, now slightly bent toward the surface although with similar C–C distances to those in the isolated molecule (Fig. 3), and the phenyl groups close to the metal surface, which adopt a quasi-planar orientation with respect to the surface, favoring the contacts with the Au atoms (with C⋯Au distances of about 2.6 Å). The PF6− group is close to one of the two Ru centres, slightly shifted with respect to the carbon chain, in line with the description resulting from the interpretation of the STM images.31 The Ru–C distances are 1.877 Å and 1.914 Å, and the shortest one corresponds to the metal Ru(1) close to the PF6− group. These Ru–C bond distances can be compared with those in the isolated molecule (1.891 Å and 1.898 Å at the same level of calculation). Hence the deposition differentiates the two metal centres, which display dissimilar metal–C bond distances, in contrast to those of the isolated molecule. Both the orientation with respect to the surface and the presence of the counterion next to one end group of Ru2+ are in agreement with the description based on the STM images of the Ru2+[PF6−] monolayer on Au(111).31
With the optimized geometry of the deposited molecule, we performed a new set of state-averaged CASSCF(7/4) calculations of the four lowest doublet roots. The ground state is described by a single configuration, the singly occupied orbital being localized in a part of the molecule, close to the PF6− ion, distributed on Ru(1) and the closest six carbon atoms of the C12 wire. The participation of the carbon atoms decreases as the distance to Ru(1) increases (Fig. 3). The first excited doublet state, as in the case of the isolated molecule, is close in energy (474 cm−1 above the ground state), showing a similar localization of the unpaired electron on a side of the molecule. In the second and third excited doublet states, the hole is localized on the opposite side of the molecule, around Ru(2), and the energy difference with the ground state gives an estimate of the reorganization energy of the deposited molecule in the presence of the counterion (λ ∼ 1.97 eV). It is important to note that this value can be affected by different factors such as the quality of the basis sets, nature and size of the active space and number of averaged roots in the CASSCF approach. It should be considered just as a rough estimate of λ. We can then describe Ru2+ once deposited on the gold surface as a class II mixed-valence compound, where the mobile hole localizes in an effective redox site, involving Ru(1) and the closest six carbon atoms of the 12-carbon chain. Hence, the Ru(2) end side group has a formal charge of +2e while the Ru(1) effective site has a charge of +3e, carrying the hole (or the unpaired electron). The PF6− counterion is placed next to Ru(1) optimizing the electrostatic interactions. Then, both the deposition and the presence of the counterion drive the localization of the charge in this system. In other words, the deposition and the presence of the counterion enhance the reorganization energy, being finally larger than the resonance energy (2|J|/λ < 1), giving rise to a double-welled potential-energy surface and the localization of the charge.51–56 As previously suggested,33,57 a mixed-valence compound classified as class III in solution or the gas-phase (here, isolated) should not be excluded as a potential candidate of mQCA, since the perturbation introduced by the interaction with the substrate, the counterion or the applied voltage could induce the charge localization, as is the case for the Ru2+ complex on Au(111). Note that the localization of the extra charge on the redox sites is just one of the requirements. In fact, the key factor for mQCA operation is that the polarization of the molecule can be switched by the action of the field induced by the neighbouring polarized driver cell.
3.3. Functionality as mQCA
Finally, we analyse the potential of this system for mQCA operation. A key requirement is that the molecular charge distribution can be switched by the coulombic perturbation produced by a neighbouring molecule switching from one state to another. The Coulomb field produced by the neighbouring molecules can be considered as the input to the molecular device, while the resulting charge distribution can be considered as its output. We model the input as an external electric field, parallel to the x axis, hence to the main axis of the carbon chain (slightly tilted when the molecule is deposited on the Au(111) surface), and we control the electronic response of the molecule to this perturbation. In fact, the electric-field driven electron-transfer in mixed valence molecules has been experimentally demonstrated18,20,33,58,59 and theoretically simulated in different MV systems.11,21,28,60–63 Other aspects, out of the scope of this work, have to be considered for practical applications of mQCA cells in energy gain and power dissipation, state latching, tunnelling effects, clocked QCA, etc.57,64,65Fig. 6 shows how the spin density on the effective redox sites changes with the applied electric field, i.e., the response curve to a coulombic perturbation. We consider that the effective redox site contains the Ru ion plus the six closest carbon atoms of the polyynediyl chain, the spin density being greater on the chain part than in the Ru atom, as previously reported for parent dinuclear Ru mixed valence complexes bridged by polyynediyl linkers.49 Note that the response curve is asymmetric, due to the presence of the counterion. In zero electric field, the unpaired electron (the hole) is localized on the effective redox site around Ru(1), placed in the positive part of the x axis in our internal coordinate system. In a simplified picture, this end side of the molecule can be considered as the positive pole (carrying the hole), while the opposite side around Ru(2) is the negative pole (with one excess electron). In the presence of a positive electric field along the x axis, the electronic distribution of the molecule is a bit more concentrated around the Ru(1) site, and the charge distribution is in fact stabilized by the applied field (Fig. 6). However, a negative electric field pushes the hole (the positive charge) to the opposite end of the molecule, changing the electronic distribution of the molecule. For an applied field larger than 0.002 a.u. (0.103 V Å−1), we observe the electron transfer from the Ru(1) to Ru(2) effective site. The molecule switches the ground state under the effect of the external electric field (Fig. 6).
 | ||
| Fig. 6 Response curve of the Ru2+ molecule under an applied electric field (in atomic units, 1 a.u. = 51.4 V Å−1) along the long axis of the molecule. Blue (red) area corresponds to positive (negative) electric fields. The insets show the shape of the singly occupied orbital of the dominant configuration of the ground state CASSCF(7/4) wavefunction. | ||
A further step should be the nuclear relaxation of the molecule, promoted by this new electronic distribution. The nuclear relaxation will act as a kind of self-trapping mechanism, enhancing the localization of the unpaired electron around the Ru(2) centre.
Additionally, we analyzed the electronic structure of the ground triplet state of a cell composed of two Ru2+ molecules, as a model of a four-dot two-electron QCA cell. They are disposed in a similar orientation to that observed in the STM images of the deposited Ru2 on Au(111). The intermolecular Ru–Ru distance is fixed to 14.949 Å, close to the intramolecular Ru–Ru distance of 17.753 Å (Fig. 7). The geometry employed is not optimized and similar conformations can be envisaged; thus these calculations should be considered just as a proof of concept. The CASSCF calculation of the lowest triplet states gives four roots close in energy, representing the four possible distributions of the unpaired electrons on the symmetric and antisymmetric combination of the 4dxz and 4dxy orbitals of Ru(1), with a relevant participation of the πz and πx orbitals, respectively, of the closest carbon atoms of the bridge chain, as observed for the single molecule. Fig. 7 presents one of the singly occupied orbitals of the dominant configuration of the ground triplet. It consists of the antisymmetric combination of the SOMO of the Ru2+ molecule. The other singly occupied orbital of the triplet wavefunction is the corresponding symmetric combination. The molecules in the cell exhibit antipodal charge densities, compliant with the molecular QCA requirements.
 | ||
| Fig. 7 A cell composed of two Ru2+ molecules, in disposition like that observed in STM images. Singly occupied orbital of the ground triplet state, mainly localized on Ru(1) sites and the closest carbon atoms of the bridge chain. | ||
4. Conclusions
The STM images of the in situ oxidized Ru2 molecule deposited on Au(111) show two features of different contrasts (bright and dim), associated with the asymmetric charge distribution between the two end groups of the Ru2+ molecule.31 Moreover, the Ru2+ complexes are organized on the surface forming rows by alternating polarization of neighbouring molecules similar to a QCA binary wire. We have explored by combining different state-of-the-art quantum chemistry approaches the deposition of the mixed-valence Ru2+ molecule on Au(111). Our calculations confirm the presence of two different end groups on the molecule, one of them close to the PF6− counterion, which corresponds to the Ru centre carrying the excess hole, i.e., the extra positive charge of the molecule. The interaction with the substrate deforms the molecule and differentiates the two Ru centres. The analysis of the wavefunction of the doublet ground state corroborates the charge localization, and the unpaired electron is localized in the end group of the molecule close to the counterion. The redox site involves not only the Ru atom, but also the closest carbon atoms of the polyynediyl chain. The bright/dim features of the STM images could then be associated with these effective redox sites.We have demonstrated that the polarization of the molecule can be switched by the action of the field induced by the neighbouring polarized driver cell. In fact, an electric field stronger than 0.002 a.u. (0.103 V Å−1) is sufficient to promote the switching. Additionally, our results evidenced that a cell formed by two Ru2+ molecules present a charge distribution with two holes in the diagonal positions, in agreement with the alternating polarization observed along the molecular rows in the STM images.
In summary, this theoretical study corroborates the interpretation of the STM images proposed by Guo and Kandel,31 and presents evidence of the role of the substrate in the localization of the charge and the potential of this system as a candidate for mQCA functionality.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support through grant PGC2018-101689-B-I00 funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”. C. J. C. thanks Dr Nadia Ben Amor for valuable discussions on the preliminary calculations of this system. The technical support of the Supercomputing Team of the Centro Informático Científico de Andalucía (CICA) and the access to the computational facilities of the “Centro de Servicios de Informática y Redes de Comunicaciones” (CSIRC, Universidad de Granada, Spain) and NLHPC(ECP-02) are also acknowledged.References
- J. V. Neumann and A. W. Burks, Theory of Self-Reproducing Automata, University of Illinois Press, 1966 Search PubMed.
- C. S. Lent, P. D. Tougaw, W. Porod and G. H. Bernstein, Quantum cellular automata, Nanotechnology, 1993, 4, 49–57 CrossRef.
- P. D. Tougaw and C. S. Lent, Logical devices implemented using quantum cellular automata, J. Appl. Phys., 1994, 75, 1818–1825 CrossRef.
- A. O. Orlov, I. Amlani, G. H. Bernstein, C. S. Lent and G. L. Snider, Realization of a Functional Cell for Quantum-Dot Cellular Automata, Science, 1997, 277, 928–930 CrossRef CAS.
- I. I. Amlani, A. O. Orlov, G. Toth, G. H. Bernstein, C. S. Lent and G. L. Snider, Digital logic gate using quantum-Dot cellular automata, Science, 1999, 284, 289–291 CrossRef CAS PubMed.
- C. S. Lent, Bypassing the Transistor Paradigm, Science, 2000, 288, 1597–1599 CrossRef CAS.
- N. Hush, Cool computing, Nat. Mater., 2003, 2, 134–135 CrossRef CAS PubMed.
- C. S. Lent, B. Isaksen and M. Lieberman, Molecular Quantum-Dot Cellular Automata, J. Am. Chem. Soc., 2003, 125, 1056–1063 CrossRef CAS PubMed.
- K. Hennessy and C. S. Lent, Clocking of molecular quantum-dot cellular automata, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct. – Process., Meas., Phenom., 2001, 19, 1752–1755 CrossRef CAS.
- X. Wang, L. Yu, V. S. S. Inakollu, X. Pan, J. Ma and H. Yu, Molecular Quantum Dot Cellular Automata Based on Diboryl Monoradical Anions, J. Phys. Chem. C, 2018, 122, 2454–2460 CrossRef CAS.
- T. Groizard, S. Kahlal and J.-F. Halet, Zwitterionic Mixed-Valence Species for the Design of Neutral Clocked Molecular Quantum-Dot Cellular Automata, Inorg. Chem., 2020, 59, 15772–15779 CrossRef CAS PubMed.
- J. Ferrando-Soria, J. Vallejo, M. Castellano, J. Martínez-Lillo, E. Pardo, J. Cano, I. Castro, F. Lloret, R. Ruiz-García and M. Julve, Molecular magnetism, quo vadis? A historical perspective from a coordination chemist viewpoint, Coord. Chem. Rev., 2017, 339, 17–103 CrossRef CAS.
- J. A. Christie, R. P. Forrest, S. A. Corcelli, N. A. Wasio, R. C. Quardokus, R. Brown, S. A. Kandel, Y. Lu, C. S. Lent and K. W. Henderson, Synthesis of a Neutral Mixed-Valence Diferrocenyl Carborane for Molecular Quantum-Dot Cellular Automata Applications, Angew. Chem., Int. Ed., 2015, 54, 15448–15451 CrossRef CAS PubMed.
- Y. Lu and C. Lent, Self-doping of molecular quantum-dot cellular automata: mixed valence zwitterions, Phys. Chem. Chem. Phys., 2011, 13, 14928–14936 RSC.
- B. Schneider, S. Demeshko, S. Neudeck, S. Dechert and F. Meyer, Mixed-Spin [2 × 2] Fe4 Grid Complex Optimized for Quantum Cellular Automata, Inorg. Chem., 2013, 52, 13230–13237 CrossRef CAS PubMed.
- J. Jiao, G. J. Long, F. Grandjean, A. M. Beatty and T. P. Fehlner, Building Blocks for the Molecular Expression of Quantum Cellular Automata. Isolation and Characterization of a Covalently Bonded Square Array of Two Ferrocenium and Two Ferrocene Complexes, J. Am. Chem. Soc., 2003, 125, 7522–7523 CrossRef CAS PubMed.
- J. Jiao, G. J. Long, L. Rebbouh, F. Grandjean, A. M. Beatty and T. P. Fehlner, Properties of a Mixed-Valence (FeII)2(FeIII)2 Square Cell for Utilization in the Quantum Cellular Automata Paradigm for Molecular Electronics, J. Am. Chem. Soc., 2005, 127, 17819–17831 CrossRef CAS PubMed.
- Z. Li, A. M. Beatty and T. P. Fehlner, Molecular QCA Cells. 1. Structure and Functionalization of an Unsymmetrical Dinuclear Mixed-Valence Complex for Surface Binding, Inorg. Chem., 2003, 42, 5707–5714 CrossRef CAS PubMed.
- Z. Li and T. P. Fehlner, Molecular QCA Cells. 2. Characterization of an Unsymmetrical Dinuclear Mixed-Valence Complex Bound to a Au Surface by an Organic Linker, Inorg. Chem., 2003, 42, 5715–5721 CrossRef CAS PubMed.
- H. Qi, S. Sharma, Z. Li, G. L. Snider, A. O. Orlov, C. S. Lent and T. P. Fehlner, Molecular Quantum Cellular Automata Cells. Electric Field Driven Switching of a Silicon Surface Bound Array of Vertically Oriented Two-Dot Molecular Quantum Cellular Automata, J. Am. Chem. Soc., 2003, 125, 15250–15259 CrossRef CAS PubMed.
- V. Arima, M. Iurlo, L. Zoli, S. Kumar, M. Piacenza, F. Della Sala, F. Matino, G. Maruccio, R. Rinaldi and F. Paolucci, et al., Toward quantum-dot cellular automata units: thiolated-carbazole linked bisferrocenes, Nanoscale, 2012, 4, 813–823 RSC.
- A. Palii, S. Aldoshin and B. Tsukerblat, Mixed-valence clusters: Prospects for single-molecule magnetoelectrics, Coord. Chem. Rev., 2021, 426, 213555 CrossRef CAS.
- T. Groizard, S. Kahlal and J.-F. Halet, Theoretical studies of mixed-valence organometallic species for potential utilization as quantum cellular automata, J. Organomet. Chem., 2017, 844, 35–42 CrossRef CAS.
- B. Tsukerblat, A. Palii, S. Zilberg, D. Korchagin, S. Aldoshin and J. M. Clemente-Juan, Vibronic recovering of functionality of quantum cellular automata based on bi-dimeric square cells with violated condition of strong Coulomb repulsion, J. Chem. Phys., 2022, 157, 074308 CrossRef CAS PubMed.
- A. Palii, A. Rybakov, S. Aldoshin and B. Tsukerblat, Semiclassical versus quantum-mechanical vibronic approach in the analysis of the functional characteristics of molecular quantum cellular automata, Phys. Chem. Chem. Phys., 2019, 21, 16751–16761 RSC.
- B. Tsukerblat, A. Palii, J. M. Clemente-Juan and E. Coronado, Mixed-valence molecular four-dot unit for quantum cellular automata: Vibronic self-trapping and cell-cell response, J. Chem. Phys., 2015, 143, 134307 CrossRef PubMed.
- A. Palii, S. Aldoshin and B. Tsukerblat, Towards the design of molecular cells for quantum cellular automata: critical reconsideration of the parameter regime for achieving functionality, Dalton Trans., 2022, 51, 286–302 RSC.
- E. P. Blair, S. A. Corcelli and C. S. Lent, Electric-field-driven electron-transfer in mixed-valence molecules, J. Chem. Phys., 2016, 145, 014307 CrossRef PubMed.
- Y. Lu and C. S. Lent, A metric for characterizing the bistability of molecular quantum-dot cellular automata, Nanotechnology, 2008, 19, 155703 CrossRef PubMed.
- N. A. Wasio, R. C. Quardokus, R. P. Forrest, S. A. Corcelli, Y. Lu, C. S. Lent, F. Justaud, C. Lapinte and S. A. Kandel, STM Imaging of Three-Metal-Center Molecules: Comparison of Experiment and Theory For Two Mixed-Valence Oxidation States, J. Phys. Chem. C, 2012, 116, 25486–25492 CrossRef CAS.
- S. Guo and S. A. Kandel, Scanning Tunneling Microscopy of Mixed Valence Dinuclear Organometallic Cations and Counterions on Au(111), J. Phys. Chem. Lett., 2010, 1, 420–424 CrossRef CAS.
- S. Rigaut, J. Perruchon, L. Le Pichon, D. Touchard and P. H. Dixneuf, J. Organomet. Chem., 2003, 670, 37–44 CrossRef CAS.
- H. Qi, A. Gupta, B. C. Noll, G. L. Snider, Y. Lu, C. Lent and T. P. Fehlner, Dependence of Field Switched Ordered Arrays of Dinuclear Mixed-Valence Complexes on the Distance between the Redox Centers and the Size of the Counterions, J. Am. Chem. Soc., 2005, 127, 15218–15227 CrossRef CAS PubMed.
- B. Chaudret, G. Commenges and R. Poilblanc, Bis(diphenylphosphino)methane complexes of ruthenium(0) and ruthenium(II), J. Chem. Soc., Dalton Trans., 1984, 1635–1639 RSC.
- A. A. Adeniyi and P. A. Ajibade, Exploring the Ruthenium-Ligands Bond and Their Relative Properties at Different Computational Methods, J. Chem., 2016, 2016, 3672062 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- M. Renz, K. Theilacker, C. Lambert and M. Kaupp, A reliable quantum-chemical protocol for the characterization of organic mixed-valence compounds, J. Am. Chem. Soc., 2009, 131, 16292–16302 CrossRef CAS PubMed.
- M. Kaupp, M. Renz, M. Parthey, M. Stolte, F. Würthner and C. Lambert, Computational and spectroscopic studies of organic mixed-valence compounds: where is the charge?, Phys. Chem. Chem. Phys., 2011, 13, 16973–16986 RSC.
- F. Neese, The ORCA program system, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- C. Fourmental, S. Mondal, R. Banerjee, A. Bellec, Y. Garreau, A. Coati, C. Chacon, Y. Girard, J. Lagoute and S. Rousset, et al., Importance of Epitaxial Strain at a Spin-Crossover Molecule–Metal Interface, J. Phys. Chem. Lett., 2019, 10, 4103–4109 CrossRef CAS PubMed.
- S. Gueddida and M. Alouani, Spin crossover in a single Fe(phen)2(NCS)2 molecule adsorbed onto metallic substrates: An ab initio calculation, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 144413 CrossRef.
- S. Gueddida, M. Gruber, T. Miyamachi, E. Beaurepaire, W. Wulfhekel and M. Alouani, Exchange Coupling of Spin-Crossover Molecules to Ferromagnetic Co Islands, J. Phys. Chem. Lett., 2016, 7, 900–904 CrossRef CAS PubMed.
- Y. Zhang, Surface effects on temperature-driven spin crossover in Fe(phen)2(NCS)2, J. Chem. Phys., 2020, 153, 134704 CrossRef CAS PubMed.
- Y. Zhang, Fe(phen)2(NCS)2 on Al(100): influence of AlN layer on spin crossover barrier, Phys. Chem. Chem. Phys., 2021, 23, 23758–23767 RSC.
- S. Javaid, S. Lebègue, B. Detlefs, F. Ibrahim, F. Djeghloul, M. Bowen, S. Boukari, T. Miyamachi, J. Arabski and D. Spor, et al., Chemisorption of manganese phthalocyanine on Cu(001) surface promoted by van der Waals interactions, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 155418 CrossRef.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, The SIESTA method forab initioorder-Nmaterials simulation, J. Phys.: Condens. Matter, 2002, 14, 2745–2779 CrossRef CAS.
- T. Stuyver, J. Huang, D. Mallick, D. Danovich and S. Shaik, TITAN: A Code for Modeling and Generating Electric Fields-Features and Applications to Enzymatic Reactivity, J. Comput. Chem., 2020, 41, 74–82 CrossRef CAS PubMed.
- M. I. Bruce, B. G. Ellis, P. J. Low, B. W. Skelton and A. H. White, Syntheses, structures, and spectro-electrochemistry of {Cp*(PP)Ru}CCCC{Ru(PP)Cp*} (PP = dppm, dppe) and their mono- and dications, Organometallics, 2003, 22, 3184–3198 CrossRef CAS.
- M. I. Bruce, K. Costuas, T. Davin, B. G. Ellis, J.-F. Halet, C. Lapinte, P. J. Low, M. E. Smith, B. W. Skelton and L. Toupet, et al., Iron versus Ruthenium: Dramatic Changes in Electronic Structure Result from Replacement of One Fe by Ru in [{Cp*(dppe)Fe}-CC-CC-{Fe(dppe)Cp*}]n+ (n = 0, 1, 2), Organometallics, 2005, 24, 3864–3881 CrossRef CAS.
- Y. Tanaka and M. Akita, Organometallic radicals of iron and ruthenium: Similarities and dissimilarities of radical reactivity and charge delocalization, Coord. Chem. Rev., 2019, 388, 334–342 CrossRef CAS.
- N. S. Hush, Adiabatic Rate Processes at Electrodes. I. Energy–Charge Relationships, J. Chem. Phys., 1958, 28, 962–972 CrossRef CAS.
- N. S. Hush, Intervalence-Transfer Absorption. Part 2. Theoretical Considerations and Spectroscopic Data, Prog. Inorg. Chem., 1967, 391–444 CAS.
- N. S. Hush, Adiabatic theory of outer sphere electron-transfer reactions in solution, Trans. Faraday Soc., 1961, 57, 557–580 RSC.
- N. S. Hush, Homogeneous and heterogeneous optical and thermal electron transfer, Electrochim. Acta, 1968, 13, 1005–1023 CrossRef CAS.
- R. A. Marcus, On the Theory of Oxidation–Reduction Reactions Involving Electron Transfer. I, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- R. A. Marcus, Theory of Electron-Transfer Reaction Rates of Solvated Electrons, J. Chem. Phys., 1965, 43, 3477–3489 CrossRef CAS.
- E. Rahimi and J. R. Reimers, Molecular quantum cellular automata cell design trade-offs: latching vs. power dissipation, Phys. Chem. Chem. Phys., 2018, 20, 17881–17888 RSC.
- Y. Lu, R. Quardokus, C. S. Lent, F. Justaud, C. Lapinte and S. A. Kandel, Charge Localization in Isolated Mixed-Valence Complexes: An STM and Theoretical Study, J. Am. Chem. Soc., 2010, 132, 13519–13524 CrossRef CAS PubMed.
- A. Aviram and P. Roland, The Effect of Electric Fields on Double-Well-Potential Molecules, Ann. N. Y. Acad. Sci., 1998, 852, 339–348 CrossRef CAS.
- N. Liza, Y. Lu and E. P. Blair, Designing boron-cluster-centered zwitterionic Y-shaped clocked QCA molecules, Nanotechnology, 2022, 33, 465201 CrossRef PubMed.
- N. Liza, D. Murphey, P. Cong, D. W. Beggs, Y. Lu and E. P. Blair, Asymmetric, mixed-valence molecules for spectroscopic readout of quantum-dot cellular automata, Nanotechnology, 2022, 33, 115201 CrossRef CAS PubMed.
- N. S. Hush, A. T. Wong, G. B. Bacskay and J. R. Reimers, Electron and energy transfer through bridged systems. 6. Molecular switches: the critical field in electric field activated bistable molecules, J. Am. Chem. Soc., 1990, 112, 4192–4197 CrossRef CAS.
- A. Farazdel, M. Dupuis, E. Clementi and A. Aviram, Electric-field induced intramolecular electron transfer in spiro .pi.-electron systems and their suitability as molecular electronic devices. A theoretical study, J. Am. Chem. Soc., 1990, 112, 4206–4214 CrossRef CAS.
- J. Timler and C. S. Lent, Power gain and dissipation in quantum-dot cellular automata, J. Appl. Phys., 2002, 91, 823–831 CrossRef CAS.
- P. Cong and E. P. Blair, Robust Electric-Field Input Circuits for Clocked Molecular Quantum-Dot Cellular Automata, IEEE Trans. Nanotechnol., 2022, 21, 424–433 CAS.
| This journal is © the Partner Organisations 2023 |