
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two novel Pd thiosemicarbazone complexes as efficient and selective antitumoral drugs†
Tania
Hidalgo
 a,
David
Fabra
b,
Raul
Allende‡
b,
Ana I.
Matesanz
b,
Patricia
Horcajada
*a,
Tarita
Biver
*c and
Adoracion G.
Quiroga
*b
a,
David
Fabra
b,
Raul
Allende‡
b,
Ana I.
Matesanz
b,
Patricia
Horcajada
*a,
Tarita
Biver
*c and
Adoracion G.
Quiroga
*b
aAdvanced Porous Materials Unit (APMU), IMDEA Energy Institute, Av. Ramon de la Sagra 3, 28935 Móstoles-Madrid, Spain. E-mail: patricia.horcajada@imdea.org
bDepartment of Inorganic Chemistry Universidad Autonoma de Madrid, 28049 Madrid, Spain. E-mail: adoracion.gomez@uam.es
cUniv Pisa, Dept Chem & Ind Chem DCCI, Via Moruzzi 13, I-56124 Pisa, Italy. E-mail: tarita.biver@unipi.it
First published on 29th December 2022
Abstract
Cancer is a leading cause of death worldwide, accounting for nearly 10 million deaths in 2020. Palladium complexes can be used as anticancer and pharmacological agents as a promising alternative to overcome the disadvantages of platinum drugs, controlling the speciation in solution and limiting toxicity. In this study, two novel complexes were developed and their speciation in solution was deeply investigated, demonstrating their stability in solution together with their behavior versus nucleic acid models and serum proteins via thermodynamic studies. Furthermore, both complexes were demonstrated to be efficient and more specific than the benchmark cisplatin drug because of their lower toxicity to healthy cells.
1. Introduction
Cancer is likely to remain one of the biggest global health challenges in the 21st century, becoming the first leading cause of death for people aged 45–64 years. In 2020, 2.7 million people in the European Union (EU) were diagnosed with cancer, ∼1.3 million people lost their lives, and an increase of 24% is expected by 2035 with an economic impact of €100 billion per year. Although the EU has been tackling cancer for decades, the need for a renewed commitment to cancer prevention, treatment and care is one of the main missions and actions for the European community (e.g., Europe's Beating Cancer Plan),1,2 where repurposing medicines to fight cancer is part of the plan, underpinning data sharing and collaborations.Despite the therapeutic discoveries made so far, the side effects (e.g., nausea; oto-, nephro-, hepato-toxicities) and treatment resistance to classical chemotherapeutic agents continue to be major drawbacks in cancer therapies, leading to higher inefficacies. The use of metals in cancer therapy has attracted a great deal of attention in recent years, such as in the case of cisplatin: despite its side effects, it is still one of the most successful anticancer compounds used clinically for diverse human cancers (ovarian, bladder, gastrointestinal, etc.).3 Its mechanism of action involves the aquation of its chloride ligands, as Pt(II) complexes’ geometry is labile to ligand exchange. Even though the monoaquo complex is the major species (the fast aquation), hydroxo species formation and, in general, speciation inside the cell contribute greatly to generating serious side effects. Therefore, novel approaches to pharmaceutic design are needed.
A recent tendency has focused on platinum group elements (Ru, Rh, Pd, etc.)4–7 and, more precisely, palladium complexes since they display moderate antitumoral activity associated with reduced toxicological effects. In this case, their design is based on the similarity of the metal centre to the Pt(II) analogue (in terms of the electronic structure and coordination chemistry) with a remarkably higher lability. In addition, their cytotoxic activity is more favourable due to the enhanced stability provided by the strong ligand–metal coordination [e.g., polydentate amines, carbamates, thiosemicarbazones (TSCN)], creating a chelating effect.8 This effect drives their reactivity toward activation reactions and binding affinities, promoting their biomedical applications,9–12 and, more importantly, might help to overcome the problem of their possible speciation. Bearing in mind all these promising features, the development of palladium complexes for clinical applications has lately been reinforced with many studies,13 revealing their high potential therapeutic performance, especially against triple negative breast cancer (TNBC), chemotherapy-resistant cancers or photodynamic therapy resistant prostate cancers.14 In this sense, some of us have actively participated by designing pharmacologically active TSCN to achieve a variety of efficient platinum and palladium mononuclear and polynuclear complexes.15 A similar anticancer activity of both metals using these polydentate TSCN ligands was observed, demonstrating higher specificity in some of the palladium prototypes.16 By changing the complex structure with diverse substituents in the TSCN structure, the cytotoxic profile could be modulated.
The mechanism of action of these promising metallodrugs implies the dissolution of the active species, remaining in contact with the components of the physiological media. Thus, the investigation of the compounds’ active triggers (“activation process”), their stability (“aggregation, disruption effect or just integrity”) and their pharmacological profile will provide meaningful data about specific interactions with the biological environment, such as their chemical equilibria and thermodynamics, commonly used to improve commercial product performance. In this sense, the thermodynamic process in solution has been deeply investigated, starting from the simplest aqueous media to more complex ones such as phosphate-buffered saline (PBS) and Tris-HCl/NaCl buffer or coordinating solvents such as DMSO (usually employed for in vitro assays). Although in most cases the solvent acts just as a simple spectator, in certain cases solvent coordination could trigger the inactivation of the metallodrug (e.g., cisplatin).17 It can also act as a transport solvent18 or even impact their reactivity in aqueous media.19
Here, we present the synthesis and characterization of two novel palladium complexes derived from thiosemicarbazone ligands, modulating their reactivity in one of the prototypes by a particular DMSO coordination. Moreover, their complex stability in physiological media and therapeutic performance have been evaluated, revealing not only effective cytotoxicity against tumor cells but also, and more importantly, much lower toxicity to healthy cell lines. The study also embraces the analysis of the thermodynamic features of binding with nucleic acids and serum protein models.
2. Results and discussion
2.1. Synthesis and characterization of the palladium complexes
The synthesis of the palladium(II) complexes is depicted in Chart 1. The thiosemicarbazone ligand was not commercially available and was prepared by a condensation reaction following general procedures optimized in our laboratory (see the Experimental section for details).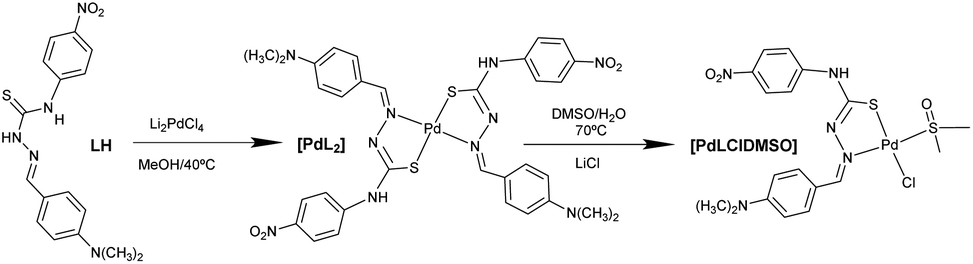 | ||
| Chart 1 Synthesis pathway of the synthesized Pd(II) complexes. | ||
Elemental analysis indicated a general formula of [Pd(L)2] and the mass spectra corroborated the formula with the molecular ion peak at 790.2 m/z [Pd(L)2]+ (see the Experimental section), whose isotopic distribution agrees with the proposed structure. Complex [Pd(L)2] was also characterized in solution by 1H- and 13C-NMR. The 1H-NMR spectra showed a similar pattern as the ligand with the expected changes in the chemical shifts induced by the coordination of the metal. To assign all the signals accurately, the two-dimensional (2D) [1H, 13C] HMQC and HMBC NMR spectra were recorded. The aromatic part of the HMBC spectra is shown in Fig. S1.†
The most significant proton shift changes were observed at H2 and H8 from the phenyl groups (see the collected proton shift assignation in the Experimental section) and in the imine proton H6 as a direct consequence of the palladium coordination. This coordination also affects the corresponding C6 chemical shift at the 13C-NMR spectra. In addition, the N–H group disappears, supporting ligand coordination in a thiolic form.
In the IR spectrum of complex [PdLCl(DMSO)], the most indicative bands are the ν(S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) vibrations corresponding to the coordinated DMSO, and the ν(Pd–Cl) band, whose values are comparable with other similar complexes found in the literature.20
O) vibrations corresponding to the coordinated DMSO, and the ν(Pd–Cl) band, whose values are comparable with other similar complexes found in the literature.20
The molecular structure of [PdLCl(DMSO)] was confirmed by single crystal X-ray diffraction (SC-XRD, Fig. 1A). The metal ion shows a square planar symmetry formed by the sulphur–nitrogen donor atoms from the mono-deprotonated TSCN, and the sulphur atom from DMSO and a chlorine atom occupy the other two sites. The central palladium ion shows bond angles close to 90°, possibly caused by the lower steric hindrance produced by the chloride and DMSO ligands.
 | ||
| Fig. 1 (A) Molecular structure of [PdLCl(DMSO)], palladium in dark blue, sulphur in yellow, carbon in grey, oxygen in red, nitrogen in blue, hydrogen in light grey, chlorine in green. (B) Hydrogen bonding network within the crystal unit, contacts in cyan lines. | ||
The found bond distances are comparable to other similar reported Pd(II)-TSCN complexes: Pd–N 2.076 Å (1.960–2.062), Pd–STSCN 2.234 Å, Pd–SDMSO 2.249 Å (2.23–2.26) and Pd–Cl 2.349 Å (2.30). Although the Pd–N distance is slightly larger than the published range, it is shorter than the sum of the covalent radii.20–22 The TSCN distances of the C–N and N–N bonds are between single and double bonding, in agreement with the tautomerization of TSCN to its thiolate form (acting as the monoanion), which results in the coordination to Pd(II).23 In addition, the crystal structure shows an infinite network stabilized by the formation of hydrogen bonds via the N4–H and the O1 of adjacent complexes (achieving dimensions ∼10.7 Å from the most peripheral moieties). This hydrogen bond is particularly strong since the three atoms (N4–H⋯O1) are aligned (180°; Fig. 1B). A table with selected distances and angles has been included in the ESI (Table S1†).
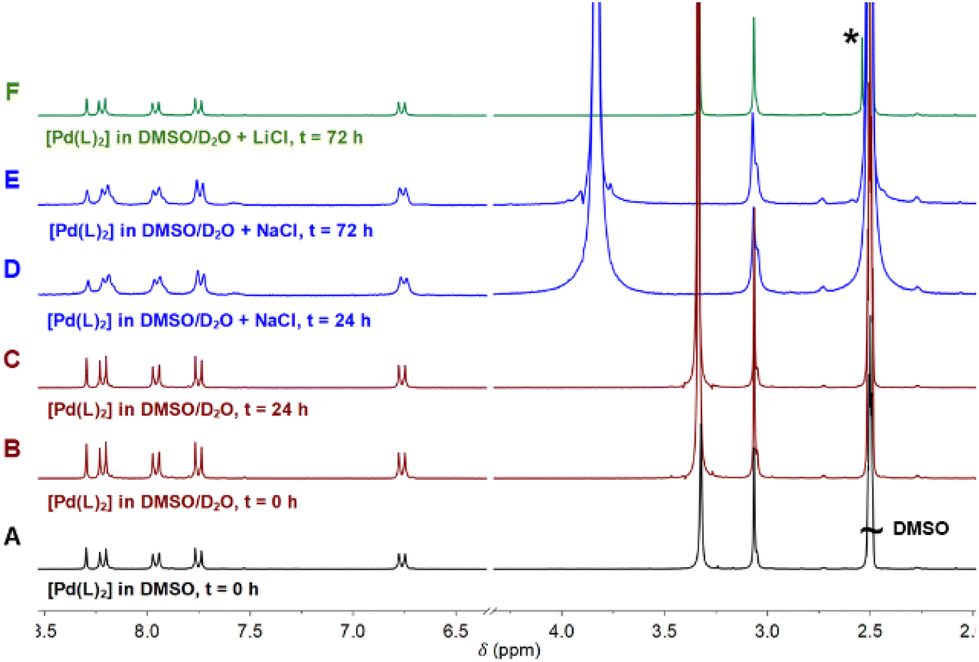 | ||
| Fig. 2 1H-NMR spectra of complex [Pd(L)2] (A) in DMSO-d6, (B) DMSO-d6 and D2O t = 0, (C) DMSO-d6 and D2O t = 24 h, (D) DMSO-d6 and D2O/H2O saturated with NaCl at t = 24 h, (E) DMSO-d6 and D2O/H2O with saturated NaCl at t = 72 h, or (F) DMSO-d6 and D2O with saturated LiCl at t = 72 h. | ||
The addition of D2O/H2O to the sample produced a very small interchange with the acidic protons (Fig. 2B and C). The sample was also stable using D2O saturated with NaCl for 3 days, with no signals of coordinated DMSO coming up in the NMR spectra (Fig. 2D and E). However, DMSO coordination emerged using LiCl after 3 days, with a small new signal arising at 2.55 ppm (Fig. 2F). The process is extremely slow: only after 3 days and increasing the temperature at 70 °C the DMSO coordination complex becomes the major product. The signal corresponding to the coordinated DMSO appears slightly deshielded compared to the free solvent (Fig. 2F, asterisk), which is a clear indication of solvent coordination.
This palladium complex reactivity toward DMSO is consistent with some platinum complexes reported in the literature, where the addition of water triggered the coordination.19
However, to the best of our knowledge, this is the first time that such behaviour is reported for palladium complexes, though such a coordination is not as quick as that observed for platinum and needs higher temperatures, longer times, and excess stabilizing counterions. Moreover, we have here accomplished the required conditions to isolate both forms and compared them. The conversion at the scaled reaction only took place in the presence of LiCl and not with NaCl. At the scaled amounts used in the reaction (see the Experimental section), the quantity of DMSO is higher and LiCl becomes a better source of chloride anions because of its higher covalency and solubility in organic solvents. On the other hand, common salts, such as NaCl (buffer present in physiological solution), are more effective in diluted solutions such as those used in UV and biological experiments.
2.2. Stability of [Pd(L)2] and [PdLCl(DMSO)] complexes in buffered solutions
Once the reactivity of the complexes was determined under the synthetic conditions, we further investigated their stability under more complex media closer to physiological ones, such as aqueous solutions and different complex buffers. In this sense, we first monitored the stability of Pd complexes in water (containing 3 or 5% DMSO) from fresh up to 24 h contact times by UV-Vis spectroscopy (Fig. S4A†), highlighting the lack of significant changes during the first 24 h for [PdLCl(DMSO)]. This is also in agreement with the hydrodynamic particle size, determined by dynamic light scattering (DLS): an average size of 15 ± 4 and 16 ± 3 nm with a positive surface charge of +27 ± 6 and +42 ± 8 mV were maintained up to 24 h for [Pd(L)2] and [PdLCl(DMSO)], respectively. However, after a 24 h contact time, [Pd(L)2] showed a gradual concentration decrease. This effect could be explained by the formation of aggregates (reported previously in TSCN complexes),24 or by the complex disruption due to the impact of other physiological parameters (e.g., pH, secondary reactions), which could lead to the formation of less soluble complexes. Here, the decrease in the particle size has been associated with the absence of aggregation after 24 h in water, supporting the complex disruption hypothesis.We also investigated the stability of compounds by adding NaCl (4μM) which is a known electrolyte existing in biological media. The spectral profile (Fig. S4B†) did not show significant changes in comparison to the Tris-HCl aqueous solution (Fig. S4A†).
To further investigate the stability of [Pd(L)2] and [PdLCl(DMSO)], their UV-Vis profiles were monitored by varying the concentration at 37 °C in Tris-HCl solutions as a common buffer used to study potential DNA interactions (Fig. S5A and B†). The findings showed that the Lambert–Beer law is obeyed in the 0–60 μM concentration range (Fig. S5C†). This behaviour suggests the absence of complex–complex interactions, also confirmed by the consistency of absorbance ratios at two different wavelengths: 299/371 nm and 300/371 nm for [Pd(L)2] and [PdLCl(DMSO)], respectively (Fig. S5D†). The molar extinction coefficient (ε) values calculated from this experiment are (at the wavelengths of maximum, 371 nm in both cases) 1.11 × 104 M−1 cm−1 for [Pd(L)2] and 1.05 × 104 M−1 cm−1 for [PdLCl(DMSO)].
For a better understanding of the target complexes and to mimic specific biological scenarios (e.g. serum, intravenous administration), the stability of both complexes was studied not only in simple aqueous solution but also in more complex conditions, including phosphate-buffered saline (PBS) and Tris-HCl/NaCl buffers under different temperatures (25–95 °C). The thermodynamics of the complex species in solution was checked by monitoring their UV-Vis spectra (Fig. S6†). The absorption profile of [Pd(L)2] complex showed a steeper concentration decrease in comparison with the [PdLCl(DMSO)] profile, being higher in Tris-HCl/NaCl compared to PBS. This change could be related to a potential degradation in the presence of Tris-HCl since NaCl concentration is lower (150 vs. 100 mM for PBS/Tris-HCl, respectively). As discussed before, no conversions were observed in the presence of NaCl at the concentrations used in the NMR stability experiment (see section 2.1.3.) because of the higher concentration of DMSO. This conversion was only detected with LiCl which is a better chloride donor in the mixture of solvents used in the reactivity study. Moreover, both complexes displayed suitable thermal stability up to 37 °C.
2.3. Cytotoxic activity
One of the requirements of effective chemotherapeutics is the large in vitro and/or in vivo growth-inhibitory capability associated with fewer side effects. Before investigating their therapeutic performance through an MTT toxicological assay (see the Experimental section and Fig. 3, where they had been compared with each precursor effect and a recognized reference drug, cisplatin), their stability in cell culture media was also evaluated. A nanoscale particle size was observed in both complexes, displaying a slight increase after 72 h (6 ± 2 nm at t = 0 in both cases to 55 ± 22 and 38 ± 12 nm at t = 72 h for [Pd(L)2] and [PdLCl(DMSO)], respectively). This average size is in agreement with the nanometric values obtained in a simpler medium such as an aqueous solution. However, the presence of biological components in the cell culture media (proteins, salts, electrolytes, serum, etc.) favors their attachment on the external surface of the complexes, resulting in a larger size and the inversion of their ζ-potential (from cationic values, +27 ± 6 and +42 ± 8 mV to a slightly negative surface, −9 ± 1 and −8 ± 1 mV for [Pd(L)2] and [PdLCl(DMSO)], respectively). Once the complex stability was confirmed, the cytotoxicity was evaluated. For this purpose, two different human cancer cells were selected: (i) PC-3 cells (“prostate cancer”), the most diagnosed cancer in males and the 2nd highest cause of male cancer-related deaths25 and (ii) colon adenocarcinoma Caco-2 cells (“colorectal cancer”), the 3rd most frequent type of cancer and well-characterized intestinal barrier model, extensively used over the past few years for preclinical studies of diverse therapeutic agents.26,27 Moreover, a promyelocytic HL-60 cell line was also chosen to evaluate the selectivity against healthy cells. In addition, this immunological cell line, able to proliferate/differentiate to myelomonocytic lineage (granulocytes, monocytes, macrophage and eosinophils),28–30 is involved in relevant biological processes, including the cellular redox homeostasis (ROS) and immune system activation.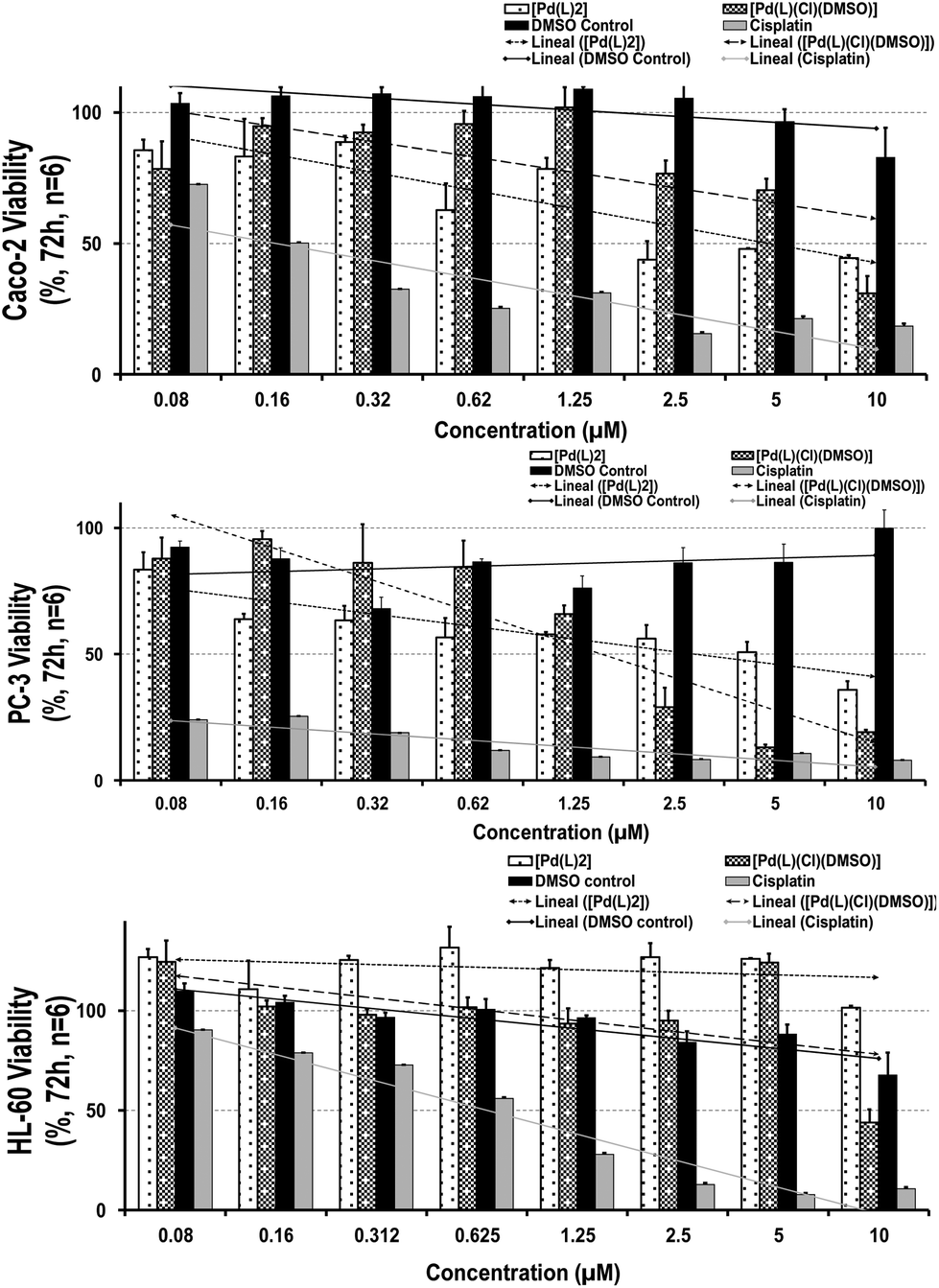 | ||
| Fig. 3 Cell viability and tendency line of (A) Caco-2, (B) PC-3 and (C) HL-60 cell lines after 72 h incubation with [Pd(L)2] (white dots), [PdLCl(DMSO)] (grey dots) complexes, the DMSO control (black) and cisplatin (grey). Note that the shown data corresponds to the average of triplicate for each concentration, obtained in two independent experiments (a total of n = 6). The vertical error bars drawn in the diagram indicate the range of fluctuations from which the standard deviations were calculated. Note the absence of cytotoxicity of the K2[PdCl4] precursor, highlighting the role of the whole Pd-complex. | ||
Remarkably, after 72 h-contact, both palladium complexes were able to decrease the cell-growth capability in tumor cells (being slightly higher for [Pd(L)2] in comparison with the DMSO control), exhibiting a stronger cytotoxic profile on prostatic than the gastrointestinal cell line. The IC50 values (50% maximum inhibitory concentration) reached a dose of 2.50 and 1.25 μM for Caco-2 and PC-3 cells, respectively, which suggests elevated responsiveness by prostatic cells (Fig. 3; Table S2†). In contrast, both complexes did not show particular toxicity to the myelomonocytic HL-60 cells (IC50 values ≥10 μM), displaying similar selectivity index values (∼0.09–0.026) than our reference compound, cisplatin. When comparing these outcomes, a higher toxicity profile was observed than that obtained by cisplatin, not only against tumor but also against healthy immune cells (even at the lowest concentration: IC50 values ∼0.16 μM in Caco-2, <0.08 μM in PC-3 and 0.62 μM in HL-60 for cisplatin), showing severe and invasive actions. Thus, their moderate antitumoral effect and similar selectivity index against cancer lines together with a gentler healthy performance of both palladium complexes make them promising chemotherapeutic agents.
To better understand the specific growth-inhibitory capability of the palladium complexes against tumor cells, their impact on the cell cycle of each cell line was investigated by flow cytometry. The general trend is the activation of cell checkpoints, arresting the cycle progression and attempting to repair the cellular damage. This process generally contributes to the growth inhibition effect (no cell death needed), resulting in the higher presence of mitotic catastrophes before entering into apoptosis (typically premature stage in S phase-arrested cells or after bypassing the G2 arrest).31–33 After 72 h of contact time, the majority of the cell population was arrested in an S-phase (∼88%, see Table S3†), with no substantial difference. This strong accumulation in the S-phase could be associated with the increase of mitotic processes, and consequently, to greater entrance into apoptosis. However, more specific molecular or gene expression analyses (e.g. caspase 3/7 enzyme assay, Cyclin A-Cdk2 activity, etc.) need to be performed to better understand the processes involved in their mechanism.
Overall, both palladium complexes are promising chemotherapeutic agents because of their antitumoral effect, similar selectivity index against cancer lines and their gentler healthy performance.
2.4. Cellular uptake
Some palladium derivatives are able to intercalate the DNA strands, blocking cellular replication. However, there are also other potential molecular mechanisms (e.g., induction of apoptosis, necrosis) that can trigger DNA damage and, consequently, tumor cell death. One of the main factors involved in successful chemotherapy is the cellular internalization of these compounds to provide the safest and most effective therapeutic dose.34 In this sense, cellular uptake was investigated in the selected tumor cell lines in order to evaluate not only the cell internalization (cytoplasmic content) but also the specific nuclear location of both complexes. For these purposes, 5 μM complexes and their respective precursors were incubated with cells for 72 h, isolating both the cytoplasmic and nuclear fractions for metal quantification by ICP-MS (more details in the Experimental section). Remarkably, all treatments were able to cross the cell membrane, reaching the cytoplasm and nuclear compartment in both cell lines, being slightly boosted for the tumor gastrointestinal cells (Caco-2; Table 1 and Fig. S7A†). Regarding the performance of complexes, [Pd(L)2] showed a larger cellular amount, mainly in the nucleus (1/0.2% vs. 12/3% for the Caco-2 and PC-3 cytoplasm/nucleus, respectively). Furthermore, the cellular uptake and nucleus targeting of the [PdLCl(DMSO)] complex were higher than those of [Pd(L)2], regardless of the cell line (0.7/0.3% vs. 0.2/1% for the Caco-2 and PC-3 cytoplasm/nucleus, respectively). Compared with the cisplatin reference, lower levels of both complexes were detected in the cytoplasm and nucleus, reaching approximately 3- and 39-times lower concentrations than cisplatin in Caco-2 and PC-3, respectively. This agrees with the previously observed reduced cytotoxic profile of the Pd-complexes when compared with cisplatin.| Metal content (μg L−1) - | ||||
|---|---|---|---|---|
| Cell uptakea (%) | ||||
| Complex | Caco-2 | PC-3 | ||
| N | C | N | C | |
| a Estimated according to the molecular weight and metal abundance in each compound. | ||||
| [Pd(L)2] | 78.60 | 7.78 | 19.11 | 1.64 |
| (12%) | (1.2%) | (2.8%) | (0.2%) | |
| [PdLCl(DMSO)] | 1.67 | 6.89 | 9.70 | 2.78 |
| (0.2%) | (0.7%) | (1.0%) | (0.3%) | |
| Cisplatin | <1.103 | >6.103 | >1.103 | <1.102 |
| (24.1%) | (>99%) | (38.4%) | (2.4%) | |
| Control - | 2.78 | 2.78 | ||
| (0.3%) | (0.3%) | |||
| K2[PdCl4] | 2.78 | 0.41 | 2.78 | |
| (0.3%) | (0.0%) | (0.3%) | ||
2.5. Interaction with biological targets
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K range.
K range.
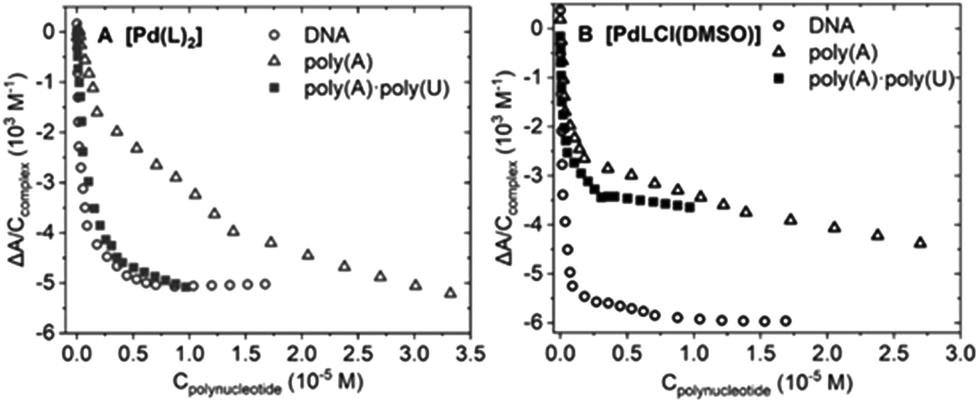 | ||
| Fig. 4 Binding isotherms of the spectrophotometric titrations of complexes (A) [Pd(L)2] and (B) [PdLCl(DMSO)] constructed with the absorbance data at 371 nm. Ccomplex = 1.06 × 10−5 M, Tris-HCl 5 mM, NaCl 100 mM, pH 7.4, 25.0 °C. | ||
K ± standard deviation) obtained from the spectrophotometric (for BSA spectrofluorometric) titrations for the interaction between [Pd(L)2] and [PdLCl(DMSO)] and the quoted biomolecules. Tris-HCl 5 mM, NaCl 100 mM, pH 7.4, 25.0 °C for all the assays
| Complex | LogK |
|||
|---|---|---|---|---|
| DNA | PolyAU | PolyA | BSA | |
| [Pd(L)2] | 6.0 ± 0.2 | 5.5 ± 0.3 | 5.4 ± 0.1 | 7.2 ± 0.1 |
| [PdLCl(DMSO)] | 6.1 ± 0.2 | 5.4 ± 0.3 | 5.1 ± 0.3 | 6.5 ± 0.1 |
:1 model (metal complex:base pair) is sufficient to describe the experimental behavior. The different binding constants are listed in Table 2.
Note that titrations with the triple-stranded RNA model poly(rU)*poly(rA)·poly(rU) were also carried out. RNA triplexes deserve high attention for many different reasons,35 ranging from telomerase function36 to viruses’ self-protection mechanisms37 and riboswitches regulating gene expression.38 However, for both palladium complexes, no significant spectral change was recorded, which indicates the lack of interaction (Fig. S8G and H†). Nevertheless, this behavior may entail that both complexes interact with double-stranded polynucleotides via groove binding. In fact, in poly(rU)·poly(rA)·poly(rU), one of the grooves is occupied by the third strand, hampering the interaction between the complex and the nucleic acid.39
To confirm the groove binding mode, ethidium bromide (EtBr) displacement tests were undertaken. EtBr is a well-known DNA intercalator reported in many procedures.40–42 Given the very different fluorescence emission of EtBr either as intercalated species or as a free probe, exchange tests are a recognized diagnostic tool to determine the polynucleotide affinity and penetration degree of non-fluorescent DNA-binding molecules.43 In the fluorescence exchange titrations with EtBr, known amounts of each complex were directly added to EtBr-saturated CT-DNA and dsRNA, a potential fluorescence reduction at the characteristic excitation (ex)/emission (em) wavelengths of the DNA-EtBr or RNA-EtBr adduct would imply the EtBr displacement from the double helix, and therefore, the intercalation of the complexes into the DNA and/or the dsRNA strand. However, in both cases the fluorescence remains constant regardless of the complex addition (Fig. S9†), ruling out intercalation in favor of external/groove binding.
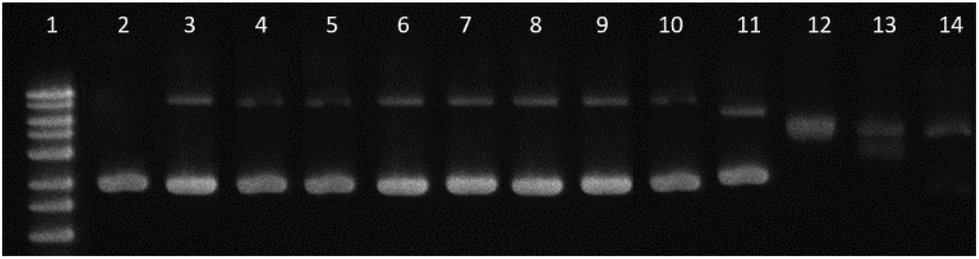 | ||
| Fig. 5 Gel electrophoresis. Lane 1: 1 kb DNA ladder; lane 2: pBR322 control; lanes 3–6: [Pd(L)2] at ri: 0.01 to 0.2; lanes 7–10: [PdLCl(DMSO)] at ri: 0.01 to 0.2; lanes 11–14: cisplatin at ri: 0.01 to 0.2. CDNA = 0.0625 μg μL−1. | ||
Furthermore, to support the minor unwinding produced by the complexes, we have used an additional direct hydrodynamic technique (viscosity).
By contrast, groove binders such as Hoechst DNA 33258 fluorescent dye do not alter the length of the DNA significantly, causing minimal changes to the viscosity of the solution. In the case of a covalent binder such as cisplatin, the relative viscosity of the solution decreases.43
Fig. 6 shows the variation in the viscosity of CT-DNA solution upon the addition of the complexes [PdL2] and [Pd(L)Cl(DMSO)]. Cisplatin and ethidium bromide (EtBr) are also added to the experiment.
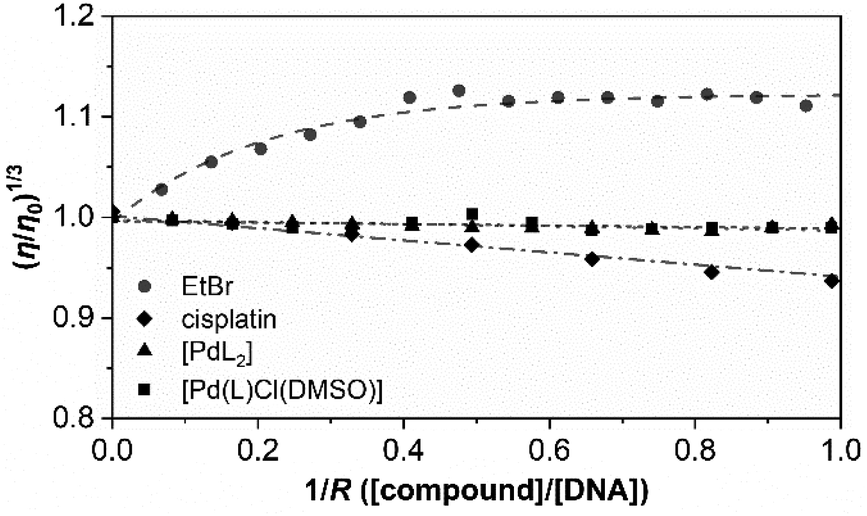 | ||
| Fig. 6 Effect of increasing amounts of complexes [PdL2] (triangles) and [Pd(L)Cl(DMSO)] (squares) on the viscosity of CT-DNA. Cisplatin and ethidium bromide (EtBr) are included as a reference (rhombi and circles, respectively). The data were collected for [DNA] = 5.69 × 10−5 M and different 1/R ratios (R = [DNA]/[compound], from 0.0 to 1.0). Tris-HCl 5 mM, NaCl 100 mM, pH 7.35, 25.0 ± 0.1 °C. | ||
The results obtained for cisplatin and EtBr are in agreement with the data reported in the literature for a covalent binder and an intercalator, respectively. The relative viscosity of CT-DNA remains certainly unchanged upon the addition of both palladium compounds. There are not very significant variations in the viscosity, and this is more in agreement with the viscosity profile of typical DNA groove binders instead of intercalators or covalent binders.
Both complexes show a very similar profile in which electrostatic interplay is discarded as both complexes are neutral. Thus, considering all the above-mentioned results, groove binding is confirmed to be the most plausible mechanism of interaction.49
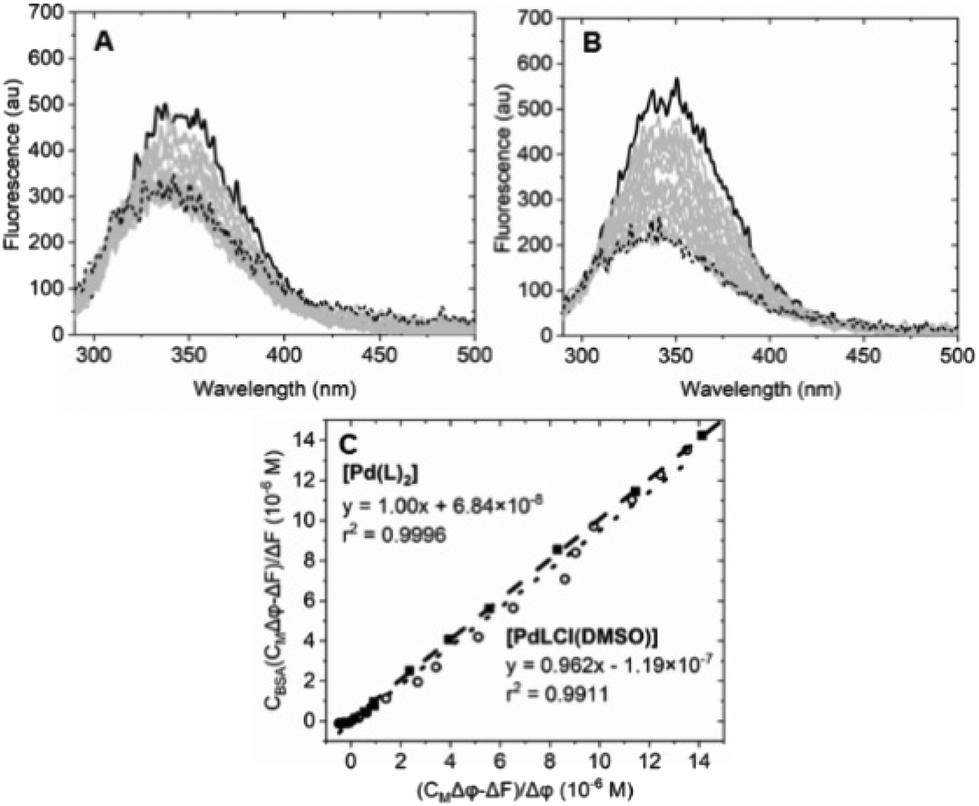 | ||
| Fig. 7 Spectrofluorimetric titration of (A) [Pd(L)2]/BSA (Ccomplex from 0 (solid) to 2.75 × 10−5 M (dotted), CBSA = 6.22 × 10−7 M); (B) spectrofluorimetric titration of [PdLCl(DMSO)]/BSA (Ccomplex from 0 (solid) to 1.41 × 10−5 M (dotted), CBSA = 6.22 × 10−7 M). (C) Plot of eqn (1) at λ = 345 nm for the [Pd(L)2]/BSA (full squares) and the [PdLCl(DMSO)]/BSA (open circles) titrations. λexc = 280 nm, λem = 345 nm. Tris-HCl 5 mM, NaCl 100 mM, pH 7.4, 25 °C. | ||
Equation applied to the titration data:
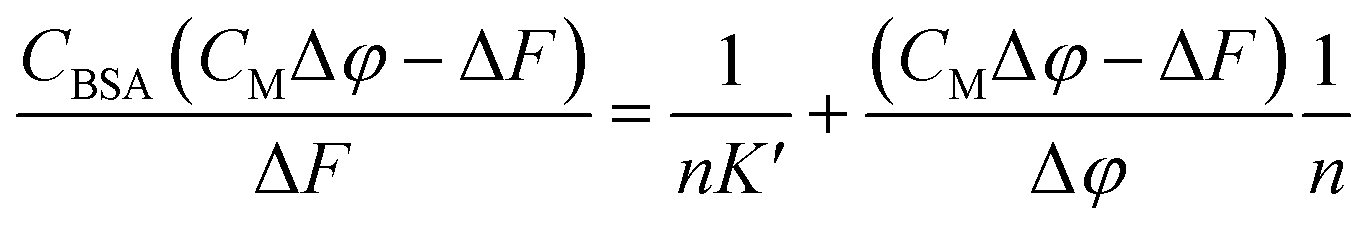 | (1) |
The binding constant could not be evaluated by eqn (1) due to an intercept too close to zero (K′ > 106): this finding confirms the very high affinity for the biosubstrate and the formation of a metal complex/BSA adduct (the quenching effect is not due to collisional quenching only).50 The same analysis indicates that a binding stoichiometry of n = 1.1 ± 0.1 holds for both systems. The confirmation of 1:1 binding allows the evaluation of K′ using the HypSpec2014® software, which considers all the wavelengths of the fluorescence titration spectra.
The binding constants for [Pd(L)2]/BSA and [PdLCl(DMSO)]/BSA interaction are listed in Table 2. The calculated constants are in line with the window for exogenous drug transportation: the binding is strong enough to actually form an adduct but, at the same time, weak enough so that the drug can be released once it gets to the target.51,52
3. Conclusion
We report on the synthesis of two novel palladium–thiosemicarbazone complexes and their speciation in solution, demonstrating their stability in buffers and clarifying the role of coordinating solvents such as DMSO. Both species strongly interact with DNA/RNA models, where the absence of interaction with the triple-stranded RNA indicates a groove binding mode. Moreover, the binding constant with serum albumin is within the transportation window. We obtained interesting results as both drugs are not only efficient in selected tumor cells from the most common human cancer disease, but more specific than cisplatin versus non-cancer cells.4. Experimental
4.1. Materials
The chemicals were purchased from Johnson and Matthey, Sigma-Aldrich and VWR. pBR322 was purchased from Fisher Scientific and Calf thymus DNA (CT-DNA), as lyophilized sodium salt, from Sigma-Aldrich. The stock solutions were prepared by dissolving known amounts in ultrapure water and were standardized spectrophotometrically (ε = 13200 M−1 cm−1 at 260 nm). Polyriboadenylic (poly(rA)), polyriboadenylic·polyribouridylic (poly(rA)·poly(rU)) and polyuridylic (poly(rU)) acids were purchased from Sigma as their potassium salts. The stock solutions (in water) were standardized spectrophotometrically (ε = 14900 M−1 cm−1 at 260 nm for poly(rA)·poly(rU) and ε = 8900 M−1 cm−1 at 260 nm for poly(rA)·poly(rU)). Poly(rU)·poly(rA)·poly(rU) was obtained at pH 7.0 by the quantitative reaction between equimolar amounts of poly(rU) and poly(rA)·poly(rU). Ethidium bromide was purchased from Sigma. Its stock solution was prepared by dissolving suitable amounts of the solid in water and was standardized spectrophotometrically (ε = 5700 M−1 cm−1 at 480 nm).
4.2. Methods
NMR spectra were recorded, at room temperature, using a two-channel 300 MHz Bruker Avance III-HD Nanobay spectrometer equipped with a 5 mm BBO 1H/X probe and Z gradients, located at SIdI. SIdI stands for Interdepartmental Investigation Service. DMSO-d6 was used as the solvent (containing 0.05 vol% tetramethylsilane (TMS) as a reference). Chemical shift values are given in ppm relative to the residual TMS signals. The following abbreviations were used: s (singlet), d (doublet) and m (multiplet). Elemental analyses were performed on a LECO CHNS-932 elemental analyzer located at SIdI. Analyses of the mass spectra were performed using FAB+ (fast atom bombardment) in a Waters VG AutoSpec mass spectrometry unit using MALDI (matrix-assisted laser desorption/ionization) with a Bruker Ultraflex III (MALDI-TOF/TOF) mass spectrometry unit, both located at SIdI. Infrared (IR) spectra were recorded using a PerkinElmer Model 283 spectrometer with an attenuated total reflectance (ATR) MIRacle Single Reflection Horizontal accessory and equipped with CsI optical windows for spectra between 600 and 200 cm−1 in Nujol mull preparations. Absorbance spectra were recorded using a Shimadzu UV-2450 double-beam UV-Vis spectrophotometer (Kyoto, Japan), whereas fluorescence spectra were recorded with an LS55 PerkinElmer spectrofluorometer (Waltham, MA, USA). Both instruments were equipped with temperature control within ±0.1 °C. For particle size and ζ-potential determinations, each complex was diluted from the DMSO stock (5 mM) with the desired media (aqueous solution, cell culture media – see Table 3: supplemented DMEM (Dulbecco's modified Eagle's medium) & RPMI (Roswell Park Memorial Institute Medium), being analyzed with a Malvern Nano-ZS, Zetasizer Nano series. Once the suspensions were prepared, an ultra-sonication step followed (ultrasound tip at 10% amplitude for 1 min), monitoring the particle size evolution from t = 0 to 24 h.| Components | Components | ||||
|---|---|---|---|---|---|
| Amino acids | DMEM | RPMI | Vitamins | DMEM | RPMI |
| Glycine | 0.40 | 0.01 | Choline chloride | 0.004 | 0.003 |
| L-Alanyl-glutamine | — | — | D-Calcium pantothenate | 0.004 | 0.00025 |
| L-Arginine hydrochloride | 0.08 | — | Folic acid | 0.004 | 0.001 |
| L-Cystine | 0.06 | 0.06 | Niacinamide | 0.004 | 0.001 |
| L-Histidine hydrochloride | 0.04 | 0.015 | Pyridoxal hydrochloride | 0.004 | 0.001 |
| L-Isoleucine | 0.10 | 0.05 | Riboflavin | 0.0004 | 0.0002 |
| L-Leucine | 0.10 | — | Thiamine hydrochloride | 0.004 | 0.001 |
| L-Lysine hydrochloride | 0.15 | — | i-Inositol | 0.007 | — |
| L-Methionine | 0.03 | 0.015 | Inorganic salts | ||
| L-Phenylalanine | 0.07 | 0.015 | Calcium chloride | 0.30 | 0.10 |
| L-Serine | 0.04 | 0.03 | Ferric nitrate | 0.0001 | — |
| L-Threonine | 0.90 | 0.02 | Magnesium sulfate | 0.10 | 0.05 |
| L-Tryptophan | 0.02 | 0.005 | Potassium chloride | 0.40 | 0.40 |
| L-Tyrosine disodium salt dihydrate | 0.12 | 0.03 | Sodium bicarbonate | 3.7 | 2.00 |
| L-Valine | 0.09 | 0.02 | Sodium chloride | 6.40 | 6.00 |
| Amino acids | DMEM | RPMI | Sodium phosphate monobasic | 0.11 | 0.80 |
| Glycine | 0.40 | 0.01 | |||
| Others | DMEM | RPMI | Others | DMEM | RPMI |
| D-Glucose | 25.00 | 4.50 | Penicillin–streptomycin | 1.00 | |
| Phenol red | 0.04 | 0.01 | MEM non-essential amino–acid solution | 1.00 | |
| Fetal bovine serum | 10.00 | ||||
4.3. Synthesis of the compounds
:EtOH 99:1 as eluents. The final product was eluted last and concentrated to dryness, isolating 712.6 mg of a yellow solid. Yield: 60%. FAB+-MS (m/z): [M + CH3CN]+: 253. 1H-RMN (300 MHz, DMSO-d6), δ (ppm): 8.07 (d, 2H, H6); 8.21 (d, 2H, H7); 10.26 (s, 1H, N–H2); 10.76 (s, 1H, N–H4). 13C-RMN (DMSO-d6), δ (ppm): 123.45, 123.70, 143.06, 145.45, 175.74. IR (cm−1): νa (NH2): 3336; νs (NH2): 3241; ν (NH): 3211; δ (NH2): 1637; νa (NO2): 1506; νs (NO2): 1334; ν (CS): 848.
HL: N1-(4-(dimethylamino)benzylidene)-N4-(4-nitrophenyl)thiosemicarbazone: an ethanolic solution (1.5 mL) of p-dimethylaminobenzaldehyde (11.3 mg, 0.076 mmol) was added dropwise to a solution of p-nitrophenylthiosemicarbazide (16.1 mg, 0.076 mmol) in AcOH 6% (2 mL) at 50 °C. The mixture was stirred and heated to the reflux temperature for 5 hours. An orange solid was isolated after cooling by filtration and washed several times with very cold portions (5 mL) of ethanol and cold methanol. Finally, the solid was recrystallized into CH2Cl2, filtered and vacuum dried. Yield: 66%. Orange solid. Elemental analysis (%) for C16H17N5O2S·H2O: calculated: C, 53.17; H, 5.30; N, 19.38. Experimental: C, 52.81; H, 4.91; N, 19.27. FAB+-MS (m/z): [M]+: 343.2; [M + H]+: 344.2. 1H-RMN (300 MHz, DMSO-d6), δ (ppm): 2.98 (s, 6H, H11); 6.74 (d, 2H, H9); 7.70 (d, 2H, H8); 8.10 (d, 3H, H2 and H6); 8.23 (d, 2H, H3); 10.29 (s, 1H, N–HA); 11.96 (s, 1H, N–HB). 13C-RMN (DMSO-d6), δ (ppm): 39.52 (C11), 111.59 (C9), 120.70 (C7), 123.59 (C2), 123.64 (C3), 129.31 (C8), 143.13 (C1), 145.25 (C6), 145.62 (C4), 151.75 (C10), 174.03 (C5). IR (cm−1): ν (NH): 3254, 3118; ν (CN): 1611; νa (NO2): 1501; νs (NO2): 1330; ν (CS): 843.
[Pd(L)2]. Dark burgundy colour. Yield: 54%. Elemental analysis (wt%) for C32H32N10O4S2Pd·3H2O: calculated: C, 48.58; H, 4.08; N, 17.70. Experimental: C, 48.13; H, 3.88; N, 17.34. MALDI-MS (m/z): [M]+: 790.2. 1H-RMN (300 MHz, DMSO-d6), δ (ppm): 3.07 (s, 6H, H11); 6.76 (d, 2H, H9); 7.75 (d, 2H, H2); 7.96 (d, 2H, H8); 8.22 (d, 2H, H3/3′); 8.30 (s, 1H, H6); 10.30 (s, 1H, N–HA). 13C-RMN (DMSO-d6), δ (ppm): 39.52 (C11), 111.22 (C9′), 117.18 (C7), 118.10 (C2/2′), 125.03 (C3), 134.75 (C8), 141.04 (C1), 146.70 (C4), 152.77 (C10), 157.89 (C6), 165.34 (C5). IR (cm−1): ν (NH): 3388; ν (CN): 1599; νa (NO2): 1502; νs (NO2): 1324; ν (C–S): 804; ν (Pd–N): 362; ν (Pd–S): 353.
[PdLCl(DMSO)]. Brown solid. Yield: 88%. Elemental analysis (wt%) for C18H22N5O3S2ClPd: calculated: C, 38.44; H, 3.94; N, 12.45. Experimental: C, 37.87; H, 3.89; N, 12.79. 1H-RMN (300 MHz, DMSO-d6), δ (ppm): 2.54 (s, H12/12′); 3.07 (s, 6H, H11); 6.76 (d, 2H, H9); 7.75 (d, 2H, H2); 7.96 (d, 2H, H8); 8.22 (d, 2H, H3); 8.30 (s, 1H, H6); 10.30 (s, 1H, N–HA). 13C-RMN (DMSO-d6), δ (ppm): 39.50 (C11/11′), 40.42 (C12/12′), 111.20 (C9), 117.17 (C7), 118.09 (C2), 124.99 (C3), 134.72 (C8), 141.03 (C1), 146.68 (C4), 152.75 (C10), 157.87 (C6), 165.34 (C5). IR (cm−1): ν (NH): 3222; ν (CN): 1; νa (NO2): 1503; νs (NO2): 1329; ν (SO): 1113; ν (C–S): 811; ν (Pd–N): 362; ν (Pd–S): 353; ν (Pd–Cl): 295.
4.4. Crystallography
Data were collected on a Bruker Kappa Apex II diffractometer. A summary of the crystal data, experimental details and refinement results is listed in Table S3.† The software package SHELXTL was used for the space group determination, structure solution, and refinement.54 The structure was solved by direct methods, completed with difference Fourier syntheses, and refined with anisotropic displacement parameters. All details can be found in CCDC 2119429† which contains the supplementary crystallographic data for this paper.4.5. Sample preparation in buffer solution of [Pd(L)2] and [PdLCl(DMSO)] complexes
The complexes were initially dissolved in DMSO (5 mM). For all experiments, the desired concentration of complexes was achieved by dilution of the DMSO stock solution with Tris-HCl aqueous buffer to reach concentration of the complex from 10−6–10−4 M. All the solutions and buffers were adjusted to pH 7.2 and 37.0 °C temperature. The studies for [Pd(L)2] and [PdLCl(DMSO)] were performed with 3 and 5% DMSO (v/v) in the final solution.The temperature stability of the compounds was evaluated by preparing 35 μM samples of each compound in Tris-HCl/NaCl or PBS buffer and recording the absorbance spectrum as the temperature was increased. All the samples had a maximum of 3% (v/v) DMSO in the final solution.
4.6. Spectrophotometric titrations
The spectrophotometric titrations were performed by adding increasing amounts of the corresponding titrant stock solution to the cuvette containing the titrand and recording the absorption or fluorescence spectrum after each addition. Small volumes were precisely measured using a Hamilton glass syringe assembled on a Mitutoyo micrometric screw so that each complete turn of the screw added 8.2 μL (1/50 of a turn was the minimum addition possible). All the spectra were corrected for dilution. Additionally, the fluorescence spectra were corrected for inner-filter effects using eqn (2)| Fcorr = Fobs × 10(Aex+Aem)/2 | (2) |
In the EtBr exchange experiments, CT-DNA or dsRNA was first saturated with EtBr, producing the characteristic emission signal of the intercalated probe; then, the molecule to be tested was added to the mixture.
In all the titrations, the DMSO content was kept negligible: the samples had a maximum of 3% DMSO (v/v) in the final solution.
4.7. Electrophoresis assay
Complexes [Pd(L)2] and [PdLCl(DMSO)] were incubated at 37 °C with 0.0625 μg μL−1 pBR322 plasmid DNA (only SC form), at different concentrations expressed as ri = complex:DNA (base pair) ratio. The ri used is from 0.01 to 0.2, in a total volume of 20 μL. After an incubation period of 24 h, the mobility of the complex-treated pBR322 samples was analyzed by gel electrophoresis at 70 V in Tris/acetate/EDTA buffer. A control of pBR322 was also incubated, and one load of 1 kb ladder was loaded in lane 1 of the gel. The gel was stained with ethidium bromide aqueous solution and DNA bands were visualized with a UV-transilluminator UVITEC Cambridge UVIDOC HD2 instrument. The studies were performed with 1% DMSO (v/v) in the final solution.
4.8. Viscosity
The viscosity experiments were carried out in an automated AND viscometer model SV-1A, at a constant temperature of 25.0 ± 0.1 °C using a water bath with thermostat control. All viscosity assays were carried out using a fixed DNA concentration of 5.69 × 10−5 M, with increasing [compound]/[CT-DNA] molar ratios (1/R from 0.0 to 1.0). The temperature was maintained constant at 25.0 ± 0.1 °C using a thermostatic water bath with a water jacket accessory. In all experiments, the desired concentration of each compound was achieved by dilution with Tris-HCl 5 mM/NaCl 100 mM buffer. The samples of the Pd complexes were additionally diluted with DMSO to avoid precipitation, reaching a final DMSO concentration of 3% (v/v). The effect of the addition of the compounds is represented by plotting the CT-DNA relative viscosity, (η/η0)1/3 against 1/R, where η0 and η are the relative viscosities of CT-DNA in the absence and presence of the compounds, respectively, and R is the [CT-DNA]/[compound] molar ratio.4.9. Cell culture and cellular viability (MTT assay)
Human colon (Caco-2, ATCC HTB-37™), prostate (PC-3, ATCC CRL-1435™) carcinoma cell lines and the promyelocytic cell line HL-60 (ATCC®CCL-240TM) were maintained in the DMEM and RPMI medium (last two), respectively, supplemented with glutamax-1 with 10% of heated-inactivated FBS, 1% penicillin/streptomycin, 1% L-glutamine and 1% non-essential amino acids. Both cell lines were routinely grown at 37 °C in a humidified 5% CO2 atmosphere. Media was changed twice per week and cells were passaged at 80% confluence (cell density at 8 × 104, ∼1 × 105 cells per cm2), being harvested by trypsinization (1% trypsin–EDTA solution).The cytotoxic activity of [Pd(L)2] and [PdLCl(DMSO)] complexes, as well as their precursors (86.55 & 60.88% of LH, 13.45 & 18.92% of K2[PdCl4], respectively), were analyzed by the colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. The adherent colon and prostate cell lines (Caco-2 & PC-3 cells) and the suspension HL-60 cells were seeded 72 h prior to the assay in 96-well plates at a density of 1 × 105 cells per well in supplemented culture media. The complex suspensions were prepared as a dilution series with cell culture media described as follows: 30 μL of each complex (from a stock prepared at 5% DMSO aqueous solution (v/v)), were added to a final volume of 300 μL per well, yielding different concentrations ranging from 10 to 0.08 μM (in triplicate). Moreover, a set of diverse control wells was left on each plate: (i) medium without cells, (ii) cells with a cytotoxic agent (Triton), (iii) untreated cells and (iv) cells containing medium with the same concentration of DMSO (DMSO control). Subsequently, all these treatments were added to the cells for 72 h, while being maintained at 37 °C under a 5% CO2 atmosphere.
Cytotoxicity was determined by adding the MTT reactant (0.5 mg mL−1 in PBS, incubated at 37 °C for 2 h) followed by washing with 100 μL of PBS, and ending with the addition of 100 μL of DMSO to each well. Absorbance was determined at λ = 539 nm under stirring. The percentage of cell viability was calculated by the absorbance measurements of control growth and test growth in the presence of the formulations at various concentration levels.
4.10. Cellular uptake
The cells were seeded in 24-well tissue culture plates at a density of 2 × 105 and 4 × 105 cells per well for PC-3 and Caco-2, respectively, and allowed to attach overnight at 37 °C. The adherent cells were incubated with each complex (5 μM) for 72 h at 37 °C. Then, excess complex-solution and dead cells were removed, detaching the adherent cells with trypsin. Cells in the suspension were centrifuged, and the pellets were resuspended in PBS, which was counted and centrifuged (7000 rpm, 5 min, 4 °C). In order to disrupt the cellular membrane, the pellets were suspended in lysis buffer (10 mM Tris, 1.5 mM MgCl2, 140 mM NaCl, pH 8.0–8.3 with 0.02% of Nonidet P40). After 15 min of ice incubation, the suspension was centrifuged at 7000 rpm for 10 min: the nuclear fraction (“pellet”) was separated from the cytoplasmic fraction (“supernatant”). The Pd content for complexes [Pd(L)2], [PdLCl(DMSO)] and its respective controls (K2[PdCl4], DMSO control and Pt amount for cisplatin,) in both fractions were measured by ICP-MS. The measurements were made after digestion with ultrapure HNO3 (65%), H2O2 and HCl, followed by evaporation and redissolution of the residue in ultrapure water to obtain a 2.0% (v/v) nitric acid solution, using an ICP-MS NexION 300 PerkinElmer instrument and 187Rhenium as an internal standard.4.11. Cell Cycle analysis
Caco-2 and PC-3 cells were treated with [Pd(L)2] and Pd(L)(Cl)(DMSO) (2.5 μM) for 72 h in 96-well plates (at a density of 10 × 104 cells per well). Negative cells (C−: exposed to any treatment) and cisplatin (as reference compound) were included as a control. Cells were trypsinized, harvested and fixed with EtOH (70%), alternating with PBS washing steps. Then, the cells were stained with PI/RNase buffer for 15 min at RT under dark conditions. Finally, the cell cycle distribution was analyzed using flow cytometry (Beckman Coulter: CytoFlex S; λEx: 488; λEm: 585/42 filter).32,33Author contributions
AGQ, TB and PH conceived and designed the research: conceptualization, data curation, methodology, resources, supervision, writing – review & editing. AIM did supervision, methodology and writing & editing. TH, DF and RA conducted the experiments and DF, TH also collaborated in the writing – review & editing.Conflicts of interest
No conflicts of interest.Acknowledgements
This contribution is part of the work of CA18202 of the NECTAR Network for Equilibria and Chemical Thermodynamics Advanced Research, supported by the COST (European Cooperation in Science and Technology). AGQ, AIM and DF acknowledge the Spanish MINECO grant PID2019-106220RB100 and MetDrugs Network RED2018-102471T for research discussion. T. B. also thanks CIRCMSB (Consorzio Inter-Universitario di Ricerca in Chimica dei Metalli nei Sistemi Biologici) and CISUP (Centro per la Integrazione della Strumentazione dell'Università di Pisa). T. H. and P. H. acknowledge the “Comunidad de Madrid” and European Regional Development Fund-FEDER 2014-2020-OE REACT-UE 1 for financial support to the VIRMOF-CM project associated with R&D projects in response to COVID-19, the Multifunctional Metallodrugs in Diagnosis and Therapy Network (MICIU, RED2018-102471-T) as well as T.H. thanks to the European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie grant agreement No 897678 (NeuroMOF). In addition, P. H. acknowledges the Spanish Ramon y Cajal Programme (grant agreement no. 2014 to 16823).References
- ECIS - European Cancer Information System, https://ecis.jrc.ec.europa.eu/.
- Cancer Tomorrow - International Agency for Research on Cancer, World Health Organisation https://gco.iarc.fr/tomorrow/en/.
- Drugs.com, https://www.drugs.com/pro/cisplatin.html#.
- Z. Liu and P. J. Sadler, Organoiridium Complexes: Anticancer Agents and Catalysts, Acc. Chem. Res., 2014, 47, 1174–1185 CrossRef CAS PubMed.
- G. Gupta, P. Kumari, J. Y. Ryu, J. Lee, S. M. Mobin and C. Y. Lee, Mitochondrial Localization of Highly Fluorescent and Photostable BODIPY-Based Ruthenium(II), Rhodium(III), and Iridium(III) Metal Complexes, Inorg. Chem., 2019, 58, 8587–8595 CrossRef CAS PubMed.
- G. Gupta, S. Cherukommu, G. Srinivas, S. W. Lee, S. H. Mun, J. Jung, N. Nagesh and C. Y. Lee, BODIPY-based Ru(II) and Ir(III) organometallic complexes of avobenzone, a sunscreen material: Potent anticancer agents, J. Inorg. Biochem., 2018, 189, 17–29 CrossRef CAS.
- R. P. Paitandi, V. Sharma, V. D. Singh, B. K. Dwivedi, S. M. Mobin and D. S. Pandey, Pyrazole appended quinoline-BODIPY based arene ruthenium complexes: their anticancer activity and potential applications in cellular imaging, Dalton Trans., 2018, 47, 17500–17514 RSC.
- A. Sengupta, A. Seitz and K. M. Merz, Simulating the Chelate Effect, J. Am. Chem. Soc., 2018, 140, 15166–15169 CrossRef CAS.
- Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, N-Heterocyclic Carbene Complexes in C–H Activation Reactions, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed.
- L. Wang, R. Liu, F. Li, Y. Meng and H. Lu, Unveiling the novel characteristics of IGPD polymer and inhibitors binding affinities using 12–6–4 LJ-type nonbonded Mn2+ model, J. Mol. Liq., 2021, 322, 114992 CrossRef CAS.
- D. Sarko, M. Eisenhut, U. Haberkorn and W. Mier, Bifunctional chelators in the design and application of radiopharmaceuticals for oncological diseases, Curr. Med. Chem., 2012, 19, 2667–2688 CrossRef CAS.
- T. Lazarević, A. Rilak and Ž D. Bugarčić, Platinum, palladium, gold and ruthenium complexes as anticancer agents: Current clinical uses, cytotoxicity studies and future perspectives, Eur. J. Med. Chem., 2017, 142, 8–31 CrossRef.
- M. Vojtek, M. P. M. Marques, I. M. P. L. V. O. Ferreira, H. Mota-Filipe and C. Diniz, Anticancer activity of palladium-based complexes against triple-negative breast cancer, Drug Discovery, 2019, 24, 1044–1058 CAS.
- European Medicines Agency, https://www.ema.europa.eu/en/medicines/human/EPAR/tookad.
- A. G. Quiroga and C. Navarro Ranninger, Contribution to the SAR field of metallated and coordination complexes: Studies of the palladium and platinum derivatives with selected thiosemicarbazones as antitumoral drugs, Coord. Chem. Rev., 2004, 248, 119–133 CrossRef CAS.
- A. I. Matesanz, E. Jimenez-Faraco, M. C. Ruiz, L. M. Balsa, C. Navarro-Ranninger, I. E. León and A. G. Quiroga, Mononuclear Pd(ii) and Pt(ii) complexes with an α-N-heterocyclic thiosemicarbazone: cytotoxicity, solution behaviour and interaction versus proven models from biological media, Inorg. Chem. Front., 2018, 5, 73–83 RSC.
- M. D. Hall, K. A. Telma, K.-E. Chang, T. D. Lee, J. P. Madigan, J. R. Lloyd, I. S. Goldlust, J. D. Hoeschele and M. M. Gottesman, Say No to DMSO: Dimethylsulfoxide Inactivates Cisplatin, Carboplatin, and Other Platinum Complexes, Cancer Res., 2014, 74, 3913–3922 CrossRef CAS PubMed.
- M. Patra, T. Joshi, V. Pierroz, K. Ingram, M. Kaiser, S. Ferrari, B. Spingler, J. Keiser and G. Gasser, DMSO-Mediated Ligand Dissociation: Renaissance for Biological Activity of N-Heterocyclic-[Ru(η6-arene)Cl2] Drug Candidates, Chem. – Eur. J., 2013, 19, 14768–14772 CrossRef CAS PubMed.
- H. P. Varbanov, D. Ortiz, D. Höfer, L. Menin, M. Galanski, B. K. Keppler and P. J. Dyson, Oxaliplatin reacts with DMSO only in the presence of water, Dalton Trans., 2017, 46, 8929–8932 RSC.
- A. P. Rebolledo, M. Vieites, D. Gambino, O. E. Piro, E. E. Castellano, C. L. Zani, E. M. Souza-Fagundes, L. R. Teixeira, A. A. Batista and H. Beraldo, Palladium(II) complexes of 2-benzoylpyridine-derived thiosemicarbazones: spectral characterization, structural studies and cytotoxic activity, J. Inorg. Biochem., 2005, 99, 698–706 CrossRef CAS PubMed.
- A. I. Matesanz, J. Perles and P. Souza, New palladium and platinum complexes with bioactive 3,5-diacetyl-1,2,4-triazol bis(4-cyclohexyl thiosemicarbazone) ligand: chemistry, antiproliferative activity and preliminary toxicity studies, Dalton Trans., 2012, 41, 12538–12547 RSC.
- L. Fuentes, A. G. Quiroga, J. A. Organero and A. I. Matesanz, Exploring DNA binding ability of two novel α-N-heterocyclic thiosemicarbazone palladium(II) complexes, J. Inorg. Biochem., 2020, 203, 110875 CrossRef CAS PubMed.
- F. H. Allen, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, in International Tables for Crystallography, 2006, pp. 790–811. DOI:10.1107/97809553602060000621.
- R. J. Glisoni, D. A. Chiappetta, L. M. Finkielsztein, A. G. Moglioni and A. Sosnik, Self-aggregation behaviour of novel thiosemicarbazone drug candidates with potential antiviral activity, New J. Chem., 2010, 34, 2047–2058 RSC.
- E. Ulukaya, F. M. Frame, B. Cevatemre, D. Pellacani, H. Walker, V. M. Mann, M. S. Simms, M. J. Stower, V. T. Yilmaz and N. J. Maitland, Differential Cytotoxic Activity of a Novel Palladium-Based Compound on Prostate Cell Lines, Primary Prostate Epithelial Cells and Prostate Stem Cells, PLoS One, 2013, 8, e64278 CrossRef CAS.
- J. Morini, G. Babini, S. Barbieri, G. Baiocco and A. Ottolenghi, The Interplay between Radioresistant Caco-2 Cells and the Immune System Increases Epithelial Layer Permeability and Alters Signaling Protein Spectrum, Front. Immunol., 2017, 8, 1–12 CrossRef.
- M. Frik, J. Fernández-Gallardo, O. Gonzalo, V. Mangas-Sanjuan, M. González-Alvarez, A. Serrano del Valle, C. Hu, I. González-Alvarez, M. Bermejo, I. Marzo and M. Contel, Cyclometalated Iminophosphorane Gold(III) and Platinum(II) Complexes. A Highly Permeable Cationic Platinum(II) Compound with Promising Anticancer Properties, J. Med. Chem., 2015, 58, 5825–5841 CrossRef CAS.
- T. Hidalgo, R. Simón-Vázquez, A. González-Fernández and P. Horcajada, Cracking the immune fingerprint of metal–organic frameworks, Chem. Sci., 2022, 13, 934–944 RSC.
- S. J. Collins, The HL-60 Promyelocytic Leukemia Cell Line: Proliferation, Differentiation, and Cellular Oncogene Expression, Blood, 1987, 70, 1233–1244 CrossRef CAS.
- Y. Guo, F. Gao, Q. Wang, K. Wang, S. Pan, Z. Pan, S. Xu, L. Li and D. Zhao, Differentiation of HL-60 cells in serum-free hematopoietic cell media enhances the production of neutrophil extracellular traps, Exp. Ther. Med., 2021, 21, 353 CrossRef CAS PubMed.
- A. Eastman, Improving anticancer drug development begins with cell culture: misinformation perpetrated by the misuse of cytotoxicity assays, Oncotarget, 2016, 8(5), 8854–8866 CrossRef.
- N. C. de Carvalho, S. P. Neves, R. B. Dias, L. d. F. Valverde, C. B. S. Sales, C. A. G. Rocha, M. B. P. Soares, E. R. dos Santos, R. M. M. Oliveira, R. M. Carlos, P. C. L. Nogueira and D. P. Bezerra, A novel ruthenium complex with xanthoxylin induces S-phase arrest and causes ERK1/2-mediated apoptosis in HepG2 cells through a p53-independent pathway, Cell Death Dis., 2018, 9, 79 CrossRef.
- N. Kuwajerwala, E. Cifuentes, S. Gautam, M. Menon, E. R. Barrack and G. P. V. Reddy, Resveratrol Induces Prostate Cancer Cell Entry into S Phase and Inhibits DNA Synthesis, Cancer Res., 2002, 62, 2488–2492 CAS.
- R. Czarnomysy, D. Radomska, O. K. Szewczyk, P. Roszczenko and K. Bielawski, Platinum and Palladium Complexes as Promising Sources for Antitumor Treatments, Int. J. Mol. Sci., 2021, 22, 8271 CrossRef CAS.
- A. Ruszkowska, M. Ruszkowski, J. P. Hulewicz, Z. Dauter and J. A. Brown, Molecular structure of a U·A-U-rich RNA triple helix with 11 consecutive base triples, Nucleic Acids Res., 2020, 48, 3304–3314 CrossRef CAS.
- C. A. Theimer, C. A. Blois and J. Feigon, Structure of the Human Telomerase RNA Pseudoknot Reveals Conserved Tertiary Interactions Essential for Function, Mol. Cell, 2005, 17, 671–682 CrossRef CAS.
- M. Mitton-Fry Rachel, J. DeGregorio Suzanne, J. Wang, A. Steitz Thomas and A. Steitz Joan, Poly(A) Tail Recognition by a Viral RNA Element Through Assembly of a Triple Helix, Science, 2010, 330, 1244–1247 CrossRef CAS.
- L. Huang and D. M. J. Lilley, Structure and ligand binding of the SAM-V riboswitch, Nucleic Acids Res., 2018, 46, 6869–6879 CrossRef CAS.
- F. Binacchi, F. Guarra, D. Cirri, T. Marzo, A. Pratesi, L. Messori, C. Gabbiani and T. Biver, On the Different Mode of Action of Au(I)/Ag(I)-NHC Bis-Anthracenyl Complexes Towards Selected Target Biomolecules, Molecules, 2020, 25, 5446 CrossRef CAS.
- M. N. Patel, P. A. Dosi and B. S. Bhatt, Square planar palladium(II) complexes of bipyridines: synthesis, characterization, and biological studies, J. Coord. Chem., 2012, 65, 3833–3844 CrossRef CAS.
- F. Macii, R. Detti, F. R. Bloise, S. Giannarelli and T. Biver, Spectroscopic Analysis of the Binding of Paraquat and Diquat Herbicides to Biosubstrates, Int. J. Environ. Res. Public Health, 2021, 18, 2412 CrossRef CAS PubMed.
- N. Korkmaz, A. Aydın, A. Karadağ, Y. Yanar, Y. Maaşoğlu, E. Şahin and Ş. Tekin, New bimetallic dicyanidoargentate(I)-based coordination compounds: Synthesis, characterization, biological activities and DNA-BSA binding affinities, Spectrochim. Acta, Part A, 2017, 173, 1007–1022 CrossRef CAS PubMed.
- A. Kellett, Z. Molphy, C. Slator, V. McKee and N. P. Farrell, Molecular methods for assessment of non-covalent metallodrug–DNA interactions, Chem. Soc. Rev., 2019, 48, 971–988 RSC.
- A. G. Quiroga, J. M. Pérez, I. López-Solera, J. R. Masaguer, A. Luque, P. Román, A. Edwards, C. Alonso and C. Navarro-Ranninger, Novel Tetranuclear Orthometalated Complexes of Pd(II) and Pt(II) Derived from p-Isopropylbenzaldehyde Thiosemicarbazone with Cytotoxic Activity in cis-DDP Resistant Tumor Cell Lines. Interaction of These Complexes with DNA, J. Med. Chem., 1998, 41, 1399–1408 CrossRef CAS PubMed.
- S. Cabrera, F. Navas, A. I. Matesanz, M. Maroto, T. Riedel, P. J. Dyson and A. G. Quiroga, Versatile Route to trans-Platinum(II) Complexes via Manipulation of a Coordinated 3-(Pyridin-3-yl)propanoic Acid Ligand, Inorg. Chem., 2019, 58, 7200–7208 CrossRef CAS.
- E. R. Jamieson and S. J. Lippard, Structure, Recognition, and Processing of Cisplatin–DNA Adducts, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed.
- G. L. Cohen, W. R. Bauer, J. K. Barton and S. J. Lippard, Binding of cis- and trans-dichlorodiammineplatinum(II) to DNA: evidence for unwinding and shortening of the double helix, Science, 1979, 203, 1014–1016 CrossRef CAS.
- Y. Zhang, J. Pan, G. Zhang and X. Zhou, Intercalation of herbicide propyzamide into DNA using acridine orange as a fluorescence probe, Sens. Actuators, B, 2015, 206, 630–639 CrossRef CAS.
- A. I. Matesanz, P. Albacete, J. Perles and P. Souza, A structural and biological study on the new 3,5-diacetyl-1,2,4-triazol bis(p-chlorophenylthiosemicarbazone) ligand and its bimetallic complexes, Inorg. Chem. Front., 2015, 2, 75–84 RSC.
- F. Macii and T. Biver, Spectrofluorimetric analysis of the binding of a target molecule to serum albumin: tricky aspects and tips, J. Inorg. Biochem., 2021, 216, 111305 CrossRef CAS PubMed.
- T. Tŏpala, A. Pascual-Álvarez, M. Moldes-Tolosa, A. Bodoki, A. Castiñeiras, J. Torres, C. Del Pozo, J. Borrás and G. Alzuet-Piña, New sulfonamide complexes with essential metal ions [Cu(II), Co(II), Ni(II) and Zn(II)]. Effect of the geometry and the metal ion on DNA binding and nuclease activity. BSA protein interaction, J. Inorg. Biochem., 2020, 202, 110823 CrossRef.
- B. Elsadek and F. Kratz, Impact of albumin on drug delivery—New applications on the horizon, J. Controlled Release, 2012, 157, 4–28 CrossRef CAS.
- R. A. Gossage, H. A. Jenkins, N. D. Jones, R. C. Jones and B. F. Yates, Pre-catalyst resting states: a kinetic, thermodynamic and quantum mechanical analyses of [PdCl2(2-oxazoline)2] complexes, Dalton Trans., 2008, 3115–3122, 10.1039/B801951G.
- SHELXTL Version 2014/7, https://shelx.uni-ac.gwdg.de/SHELX/index.php.
- M. van de Weert and L. Stella, Fluorescence quenching and ligand binding: A critical discussion of a popular methodology, J. Mol. Struct., 2011, 998, 144–150 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2119429. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2qi02424a |
| ‡ Actual address: Environment and Agronomy Department, Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA, CSIC). |
| This journal is © the Partner Organisations 2023 |