
Orange/cyan emissive sensors of Sb3+ for probing water via reversible phase transformation in rare-earth-based perovskite crystals†
Yexin
Huang
a,
Yuexiao
Pan
 *a,
Chengdong
Peng
a,
Yihong
Ding
a,
Hongzhou
Lian
b,
Liyi
Li
*c and
Jun
Lin
*b
*a,
Chengdong
Peng
a,
Yihong
Ding
a,
Hongzhou
Lian
b,
Liyi
Li
*c and
Jun
Lin
*b
aKey Laboratory of Carbon Materials of Zhejiang Province, College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, P. R. China. E-mail: yxpan@wzu.edu.cn
bState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: jlin@ciac.ac.cn
cInnovative Drug and Imaging Agent R&D Center, Research Institute of Tsinghua, Pearl River Delta, Guangzhou, P. R. China. E-mail: lily@tsinghua-gd.org
First published on 6th December 2022
Abstract
A facile strategy for water probing with visible perception is particularly important for water transport, ecosystem sustainability, and the ocean industry. Here, we have developed a simple technology to probe water content by using the response of the orange/cyan luminescence color of Sb3+ emitted from the two novel rare-earth-based perovskite crystals Rb2ScCl5·H2O and Rb3ScCl6. The reversible transformation between Rb2ScCl5·H2O and Rb3ScCl6 accompanied by the significant visible perception of the change of the luminescence color can be realized through the dehydration/hydration process in air. In addition to the favorable sensitivity to water, an in-depth study shows that the highly efficient photoluminescence (PL) of Sb3+ in both Rb2ScCl5·H2O and Rb3ScCl6 host matrixes arises from the self-trapped excitons (STEs) rather than the 5s2–5s15p1 transition of Sb3+ according to the density functional theory (DFT) calculation. It can be anticipated that this study will provide new insights into the luminescence mechanism of Sb3+ in perovskite crystals and a potential application in a sensitive sensor for probing water content.
1 Introduction
A conventional moisture indicator for the validity of a silica gel dryer often relies on the hydration/dehydration process of CoCl2/[CoCl(H2O)5]Cl·H2O accompanied by a color change (blue/pink). The reversible switch or transformation in the PL color of luminescence materials responding to water would be an alternative visual sensor for probing water content in the manufacture and transport of chemical industrial products and anti-counterfeiting. The reversible transition between luminescent CsPbBr3 and non-luminescent CsPb2Br5 has been achieved upon the exposure/removal of water by loading CsPbBr3 NCs into a porous matrix.1 The emissive light of Sb3+ can be sensitively switched between red/yellow in zero-dimensional (0D) halide single crystals (PPZ)2SbCl7·5H2O (PPZ = 1-phenylpiperazine) and Cs2InBr5·H2O:Sb3+ ascribed to the insertion/removal of the water molecules into/from the matrixes.2–4 Environmentally friendly alternatives free of Pb and organics, and possessing high PLQYs and detection sensitivity, are still in demand in the growing field for application in sensors and anti-counterfeiting.Recently, emerging luminescence materials based on Sb3+ doped all-inorganic halide perovskite crystals have become outstanding candidates for promising application in lighting and displays due to their advantages of excellent optical properties including high photoluminescence efficiency and a broad emitting band covering from the visible to near IR region.5–23 The introduction of Sb3+ dopants has led to a significant increase in the intrinsic PL efficiency of the all-inorganic halide perovskite crystals.7,8 It has been well documented that the Sb3+ ion possesses the ns2 electronic configuration and its excited state is separated into the singlet state 1P1 and triplet state (3Pn, n = 1, 2, 3). The excitation transitions from the ground 1S0 to singlet state 1P1 are allowed and one of the triplet states 3P1 is partially allowed, whereas the transitions from 1S0 to 3P2 and 3P0 are forbidden.18–23 However, it is usually hard to make a decision on whether the PL induced by Sb3+ doping is attributed to the 5s2–5s15p1 transition of Sb3+ or the STEs of the halide perovskite matrix with new trapping states produced by Sb3+ because they both are similar in PL spectra, such as broadband emission with a large Stokes shift. It has been reported that the experimental and computational studies reveal that the origin of red PL in Cs2InBr5·H2O:Sb3+ is attributed to STEs induced by a structural deformation, whereas the green emission of Cs3InCl6:Sb3+ NCs is ascribed to the inter-configurational 3P1 → 1S0 transition of Sb3+.4,13 Even though substantial efforts have been devoted to improving the PL efficiency of Sb3+-doped perovskites and the PLQYs are up to 95%, the demand for an in-depth understanding of the luminescence mechanism is booming. Therefore, further evidence should be provided to confirm the underneath mechanism because both the STEs and 3P1 → 1S0 transition of Sb3+ are actually correlated with the strong Jahn–Teller effect resulting from the introduction of exotic ions. The intense broadband cyan-green and orange-yellow photoluminescence originating from [SbCl6]3− in (Rb/Cs)3InCl6:Sb3+ and [SbCl5(H2O)]2− in Cs2InCl5·H2O:Sb3+ has been identified, respectively.5,6 The corresponding emission colors of Cs2InX5·H2O:Sb3+ (X = Cl, Br, I) can be expanded into the orange-red region through the compositional substitution of various halides.4,12 A reversible cyan/yellow emission switch is realized with Sb3+ in the transformation of the crystals of A3InX6 and A2InX5·H2O (A = Cs, Rb; X = Cl, Br) due to the dehydration/hydration process.5,13,15 Therefore, Sb3+ ions in perovskite crystals exhibit distinctly different PL, which can be switched by the addition/removal of H2O into/from the ligands of the Sb3+ octahedron.
Herein, two novel Pb-free perovskite crystals Rb2ScCl5·H2O and Rb3ScCl6 with rare-earth Sc3+ as central ions are identified for the first time, which have similar structures to those of the established Cs2ScCl5·H2O and Cs3ScCl6 crystals, respectively.24 The combination of the systematic experimental and computational results validates that the PL spectra of Sb3+ in Rb2ScCl5·H2O and Rb3ScCl6 with PLQYs up to 99% probably originate from STEs rather than the 5s2–5s15p1 transition of Sb3+ because the 5p states of Sb3+ are far higher than the bottom of the conduction band. The sensitive switch of the orange/cyan luminescence color of Sb3+ responding to water is observed, which is ascribed to the reversible transformation between the matrixes of Rb2ScCl5·H2O and Rb3ScCl6. The results of this work could offer an in-depth understanding of the luminescence mechanism of Sb3+ in perovskite crystals and provide new possibilities in water sensors, where both high PLQYs and sensitive water response are strongly desired.
2 Experimental
2.1 Raw materials
For the experiments, the raw material RbCl (99.9%) was purchased from Meryer (Shanghai, China). Sc2O3 (99.9%), and Sb2O3 (99.9%), hydrochloric acid HCl (36.5 wt%), and ethyl alcohol were purchased from Aladdin (Shanghai, China). All the starting materials for synthesis were purchased commercially and used as received without further purification.2.2 Synthesis of Sb3+ doped Rb2ScCl5·H2O single crystals via hydrothermal strategy
For the undoped Rb2ScCl5·H2O single crystals, 2 mmol of RbCl, 0.5 mmol of Sc2O3, and 3 mL of hydrochloric acid were mixed in a 15 mL Teflon liner. The obtained solution was heated in a stainless steel Parr autoclave at 150 °C for 12 h, and then naturally cooled to room temperature. The supernatant solution was removed by tilting the Teflon liner and the precipitated crystals were washed three times with ethyl alcohol and dried at 60 °C for 12 h. The similar experimental procedures were carried out to obtain the Sb3+ doped Rb2ScCl5·H2O single crystals by replacing x mmol of Sc2O3 with Sb2O3 (x = 0.0025, 0.005, 0.01, 0.015, 0.025, 0.04, 0.05, 0.075, 0.1) by keeping the stoichiometric ratios.2.3 A reversible transformation process between Sb3+ doped Rb2ScCl5·H2O and Rb3ScCl6 crystals induced by water
In a typical process, 50 mg of the as-prepared Rb2ScCl5·H2O:Sb3+ single crystals with orange emission were ground into powder and then heated at different temperatures (100, 150, 200, 220, 230, 240, and 250 °C) for 2 h in an oven. The sintered products with cyan emission were characterized by PL photographs, spectra, and XRD. The dominant component in the final products sintered at 250 °C was confirmed to be Rb3ScCl6:Sb3+. For the returning transformation procedure, 3 mg of Rb3ScCl6:Sb3+ powder was separately put in air with/without a glass lid. After 60 min, the Rb3ScCl6:Sb3+ powder exposed to air changed to be Rb2ScCl5·H2O:Sb3+ completely while that with an insulating lid maintained the insensible change. The dependence of time required for the transformation procedure on the relative humidity and the contract area was investigated.2.4 Fabrication and measurement of a WLED with Rb2ScCl5·H2O:Sb3+
A typical WLED was fabricated using a 360 nm chip, a blue phosphor BaMgAl10O17:Eu2+ (BAM:Eu), a green phosphor BaSrSiO4:Eu2+ (BSSO:Eu), and the as-prepared orange phosphor Rb2ScCl5·H2O:Sb3+. The three phosphors were thoroughly mixed with epoxy resin and coated on the surface of the LED chips. The devices were solidified at 130 °C for 1 h to produce a solid-state WLED.3 Results and discussion
Single-crystal X-ray diffraction (SCXRD) has been used to determine that the novel single crystal Rb2ScCl5·H2O adopts the orthorhombic space group Pnma with unit cell parameters of a = 14.003 Å, b = 10.045 Å, c = 7.246 Å, α = β = γ = 90° (Fig. 1a and Table S1†). The detailed crystal structure parameters of Rb2ScCl5·H2O obtained from the single crystal analysis have been compared to those of Cs2ScCl5·H2O (Table S2†). In Rb2ScCl5·H2O crystals, each rare earth Sc3+ ion in a deformed octahedron is coordinated by five Cl− ions and one H2O. The bond lengths of Sc–Cl in Rb2ScCl5·H2O range from 2.4510(14) to 2.4873(15) Å, which are longer than those from 2.4589(9) to 2.4845(5) Å in Cs2ScCl5·H2O. Besides, the angle of Cl–Sc–O is 86.591(3)° in Rb2ScCl5·H2O, which is larger than that of 85.498(17)° in Cs2ScCl5·H2O.24 The results mean the lattice of Rb2ScCl5·H2O is more easily deformed by doping Sb3+ ions and is conducive to producing STEs.25 | ||
| Fig. 1 (a and b) XRD patterns of Rb2ScCl5·H2O:x%Sb3+ (x = 0, 0.5, 1, 2, 3, 5, 8, 10, 15, and 20) compared to the corresponding simulated result of Rb2ScCl5·H2O. (c) EDS pattern, (d) SAED, (e) HRTEM, and (f–k) mapping images of the elements in Rb2ScCl5·H2O:Sb3+. | ||
In order to produce PL in Rb2ScCl5·H2O, parts of Sc3+ ions are substituted by equivalent Sb3+ ions. The powder X-ray diffraction (PXRD) measurements are carried out to analyze the phase purity after doping Sb3+ in different amounts (Fig. 1a). The results show that all the diffraction peaks of the crystals with the doping concentration of Sb3+ in 0.5–15 mol% of Sc3+ are consistent with simulated orthorhombic Rb2ScCl5·H2O, indicating the successful introduction of Sb3+ ions into the matrix of Rb2ScCl5·H2O. The sharp diffraction peaks are indicative of the high crystallinity of these crystals doped with various concentrations of Sb3+. Until the doping ratio of Sb3+ increases to 20 mol%, an extra diffraction peak can be observed at 27°, which is ascribed to the impurity of the raw material RbCl. With the Sb3+ concentration increasing, the XRD diffraction peaks shift to the lower angles (Fig. 1b), demonstrating the expanding of the crystal lattice because the ionic radius of Sb3+ (0.92 Å) is larger than that of Sc3+ (0.81 Å). Moreover, the EDS pattern of Rb2ScCl5·H2O:Sb3+ with 2 mol% Sb3+ dopants confirms the presence of the constituent elements (Rb, Sc, Cl, O, and Sb) without any impurities (Fig. 1c). The selected area electron diffraction (SAED) pattern exhibits two distinct hexagonal diffraction patterns, which correspond to the interplanar distances of the (3 0 1) and (2 5 0) planes of Rb2ScCl5·H2O (Fig. 1d). The HRTEM image shows clear lattice spacing of 0.238 and 0.163 nm, which are in good agreement with the (1 0 3) and (2 6 0) planes of the orthorhombic Rb2ScCl5·H2O crystal, respectively (Fig. 1e). The elemental mapping images are used to observe the distribution uniformity of each composing element in Rb2ScCl5·H2O:Sb3+ (Fig. 1f–k). It is obvious that the Rb, Sc, Cl, O, and Sb elements are evenly distributed in the whole crystals. XPS measurements were performed to further prove the composition and analyze the chemical states of the elements in the Rb2ScCl5·H2O:Sb3+ crystal (Fig. S1†). The binding energies at 110.98, 109.55, 407.58, 403.05, 199.70, 198.28, 532.18, and 540.31 eV match well with the corresponding spin orbitals of Rb 3d3/2, Rb 3d5/2, Sc 2p1/2, Sc 2p3/2, Cl 2p1/2, Cl 2p3/2, O 1s and Sb 3d, respectively.26–28
The PL excitation and emission spectra are illustrated to explore the optical properties of Rb2ScCl5·H2O:x%Sb3+ (x = 0, 0.5, 1, 2, 3, 5, 8, 10, 15, and 20) crystals (Fig. 2a and b). Pure Rb2ScCl5·H2O is non-emissive under ultraviolet (UV) light. For the Rb2ScCl5·H2O:xSb3+ crystals, a single excitation band at 340 nm and a strong broad orange PL emission band at 600 nm with a full width at half maximum (FWHM) of 141 nm have been observed, respectively. As the content of Sb3+ increases from 0.5 to 2 mol% of Sb3+, the PL intensity reaches a maximum. The concentration quenching effect occurs after the concentration of Sb3+ increases further, which leads to the decrease of the PL intensity. Furthermore, similar PLE spectra manifest that the excited states and dynamics are independent of the content of Sb3+. The dependence of the emission wavelength and intensity of Rb2ScCl5·H2O:xSb3+ on the concentration of Sb3+ has been explored (Fig. 2c). A slightly blue shift in the emission spectra of Rb2ScCl5·H2O:x%Sb3+ can be seen with the increasing concentration of Sb3+. It has been considered that the increase of the Sb3+ concentration extends the distance between Sb3+ and Cl− and decreases the magnitude of the crystal field, which brings about a smaller crystal field splitting and a blue shift of the Sb3+ emission spectra.29,30 The full PL excitation spectrum of Rb2ScCl5·H2O:Sb3+ (200–500 nm) monitored at 600 nm shows that there is no additional excitation peak lower than 300 nm (Fig. S2†), which corresponds to the 5p states of Sb3+ according to the DFT calculation. It is well documented that for Sb3+ with ns2 excited electronic structure, the ground state is 1S0 and the excited state can be split into four energy states 1P1, 3P0, 3P1, and 3P2. The excitation and emission of Sb3+ are usually attributed to the allowed transition between the ground state 1S0 and the excited state 3P1.31,32 However, the s–p transition of Sb3+ and STEs are exactly similar with a broad emission band and a large Stokes shift; thus, more evidence should be provided to elucidate the mechanism responsible for the luminescence of Sb3+ in Rb2ScCl5·H2O and Rb3ScCl6.
 | ||
| Fig. 2 (a and b) PL excitation (λem = 600 nm) and emission spectra (λex = 340 nm) of Rb2ScCl5·H2O:x%Sb3+ with different x values. (c) Concentration-dependent emission wavelength and intensity of Rb2ScCl5·H2O:xSb3+ (inset: PL decay curves of Rb2ScCl5·H2O:2%Sb3+ and Rb2ScCl5·H2O:20%Sb3+). (d) Linear fitting of the variation of PL intensity with excitation power density and (e) contour map of the temperature-dependent PL emission spectra of Rb2ScCl5·H2O:Sb3+. (f) Emission wavelength and intensity of Rb2ScCl5·H2O:Sb3+ as a function of measurement temperature in the range of 313–473 K. | ||
The PL lifetimes of Rb2ScCl5·H2O:Sb3+ with different Sb3+ concentrations have been measured (Fig. S3†) and all of them can be well-fitted with a singly exponential function. The PL lifetimes decrease monotonously from 1.06 to 0.34 ns with the Sb3+ concentrations increasing from 0.5 to 20 mol% (Table S3†), which is due to the increasing energy-transfer probability among the Sb3+ ions. The lifetimes of the Rb2ScCl5·H2O crystals doped with 2 and 20 mol% Sb3+ fitted from the PL decay curves monitored at 600 nm are 0.48 and 0.34 ns, respectively (Inset of Fig. 2c). Such a short decay lifetime is more likely originated from the STE emission owing to the strong Jahn–Teller distortion-induced electron–hole recombination.33
To rule out that the emission originated from permanent defects, we have investigated the function of the excitation power on the emission intensity at room temperature. The PL intensity of the emerged emission at 600 nm exhibits a linear dependence on the excitation power density up to 1000 mW cm−2, suggesting that the emission is due to the intrinsic properties of Rb2ScCl5·H2O:Sb3+ rather than permanent defects (Fig. 2d).34,35 The effect of the high temperature on luminescence can be shown by the contour map and the point-line map (Fig. 2e and f). It can be seen that the PL intensities decrease as the measurement temperature increases from 313 to 473 K, which reflects the intense non-radiative recombination process in the high-temperature region. With the increase in the temperature, the emission band appears to slightly blue-shift from 600 to 585 nm, and this may be attributed to the enhanced electron–phonon coupling at the higher temperature.8,13,19,24 After a heating–cooling cycle, the PL wavelength returns almost to the initial state and the intensity can reach 80% of the initial strength, indicating the excellent thermal stability of the Rb2ScCl5·H2O:Sb3+ crystals (Fig. S4†). Moreover, the blue-shift has been found in the normalized excitation spectrum and the emission shrinks narrower at 77 K compared to that measured at 298 K (Fig. S5†), which is attributed to the reduction of thermally populated vibrational states at a low temperature.36,37
Coordinative H2O as a part of the Sc3+ octahedron is expected to have an important influence on the PL properties of the Rb2ScCl5·H2O:Sb3+ crystals. To investigate the effect of coordinative H2O, a series of Rb2ScCl5·H2O:Sb3+ crystals have been sintered at different temperatures for 2 h in air. The digital photographs of the Rb2ScCl5·H2O:Sb3+ crystals after heating treatment taken under UV light exhibit the obvious luminescence color changing from orange to white, and finally to cyan when the sintering temperature varies from 100 to 250 °C (Fig. 3a). The sintered crystals are insulated in transparent polyvinyl chloride bags to avoid the re-absorption of water from the atmosphere and characterized by the PL spectra and XRD promptly. The heating-treated Rb2ScCl5·H2O:Sb3+ crystals show a sustained blue shift in the emission peak under 340 nm excitation with the treatment temperature increasing from 100 to 250 °C (Fig. 3b). The samples treated at 220 and 230 °C emit white light compensated by both orange and cyan emissions. The Commission International de L'Elcairage (CIE) coordinates of the samples are experienced from orange to cyan passing the white light region (Fig. S6†). The emission spectrum of the sample sintered at 230 °C can be separated into two bands centered at 590 and 605 nm from the Gaussian fitting PL spectrum (Fig. 3c). Additionally, only the cyan PL is observed in the sample treated at 250 °C.
 | ||
| Fig. 3 (a) Digital photographs of Rb2ScCl5·H2O:Sb3+ after treatment at different temperatures taken under 365 nm UV light. (b) Normalized PL emission spectra (λex = 340 nm) of Rb2ScCl5·H2O:Sb3+ after treatment at different temperatures. (c) Gaussian fitting PL spectra of Rb2ScCl5·H2O:Sb3+ excited by 340 nm after treatment at 230 °C. | ||
To elucidate the fundamental reasons responsible for the changing of PL color, the phases of Rb2ScCl5·H2O:Sb3+ crystals sintered at different temperatures have been characterized by XRD (Fig. 4a). The XRD patterns of the samples treated at a temperature lower than 200 °C can be well indexed to the pure phase of Rb2ScCl5·H2O which is almost identical to that of Rb2InCl5·H2O in previous work because the ionic radii of In3+ and Sc3+ are the same as 0.81 Å.38 The Rb2ScCl5·H2O phase is dominantly detected in the samples sintered at a temperature lower than 220 °C. An additional phase is present in those treated at a temperature higher than 230 °C. The dominant phase in the sample after heating Rb2ScCl5·H2O:Sb3+ at 250 °C is supposed to be the Rb3ScCl6 phase which is similar to Rb3InCl6 (PDF#26-0936). The minor impurity denoted by asterisks is temporarily ascribed to the RbScCl4 phase which is similar to RbInCl4 (PDF#26-0935). Accordingly, the dehydration process at a high temperature is proposed as below:
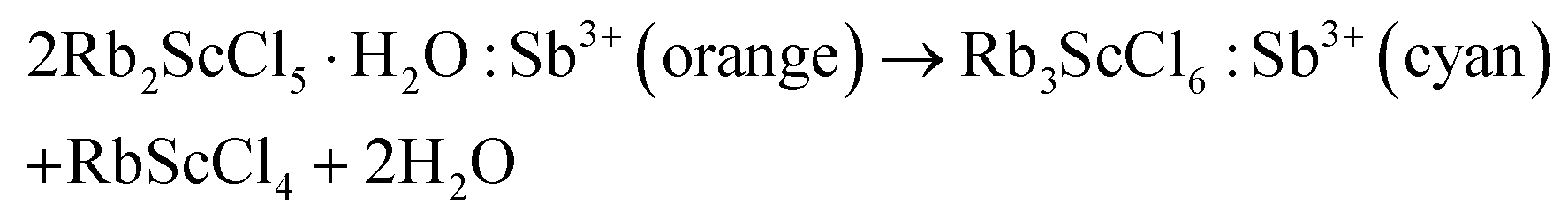
 | ||
| Fig. 4 (a) XRD patterns of Rb2ScCl5·H2O:Sb3+ treated at different temperatures. The asterisks denote the impurity of RbScCl4. Structure projection of (b) Rb3ScCl6 and (c) Rb2ScCl5·H2O crystals plotted using the software Diamond 3.1. | ||
The regular octahedron [ScCl6]3− in the Rb3ScCl6 crystals is composed of central ions Sc3+ coordinated with six same Cl− ions (Fig. 4b). When the Rb3ScCl6:Sb3+ crystals are exposed to moisture, coordinative H2O replaces in one of the Cl− ions and it is transformed into a distorted [ScCl5(H2O)]2− octahedron. Consequently, cyan Rb3ScCl6:Sb3+ is transformed into orange Rb2ScCl5·H2O:Sb3+ through the dehydration process at room temperature. In the structure of Rb2ScCl5·H2O, the individual [ScCl5(H2O)]2− octahedra are composed of central ions Sc3+ coordinated with five Cl− ions and one O2− from the H2O molecule, which breaks the symmetry and subsequently produces a Jahn–Teller distortion (Fig. 4c). It is reasonably speculated that the cyan PL is emitted from Rb3ScCl6:Sb3+ based on the two facts: (i) the crystal Rb3InCl6:Sb3+ emits cyan PL under UV light, and the structure and XRD pattern of Rb3ScCl6 are fairly similar to those of Rb3InCl6; (ii) no PL is observed in Sb3+ doped RbInCl4, and the structures of RbScCl4 and RbInCl4 are identical according to their XRD features.38 Thus, it is reasonably supposed that the bright cyan PL originated from the Rb3ScCl6:Sb3+ crystals. The TG–DTA curve of Rb2ScCl5·H2O:Sb3+ further demonstrates this transformation process (Fig. S7†). Nearly 7.8% weight loss has been observed up to 250 °C in the TG curve, which corresponds to the loss of coordinative H2O in Rb2ScCl5·H2O:Sb3+. The weight remains almost constant up to 700 °C, indicating the superior thermal stability of Rb3InCl6:Sb3+.
The PLE spectrum of the Rb3ScCl6:Sb3+ crystals displays a band at 340 nm with a small shoulder at ∼325 nm that has similar features to that of the Sb3+ doped perovskites (Fig. S8a†).39,40 The cyan emission spectrum is centered at 500 nm with an FWHM of 100 nm and its decay curve can be fitted to a short-lived PL lifetime of 0.28 ns (98%) and a long-lived PL lifetime of 0.99 ns (2%) by a biexponential function, respectively (Fig. S8b†). Nevertheless, both lifetimes are in the nanosecond scope, which further infers that the recombination routes of Sb3+ in the two perovskites are ascribed to the STEs.33
A comparison of the optical properties of Sb3+ in perovskite crystals formed as A2BX5·H2O (A = Cs, Rb; B = In, Sc; and X = Cl, Br) and A3BX6 (A = Cs, Rb; B = In, Sc; X = Cl, Br) will give insights into optimizing the PL performance of Sb3+ (Table S4†). The undoped crystals A2InX5·H2O (A = Rb, Cs; X = Cl, Br) exhibit weak PL with PLQYs lower than 33% and no PL has been observed in the primitive A3InX6 (A = Rb, Cs; X = Cl, Br) crystals. Upon doping Sb3+, the Cs2InCl5·H2O:Sb3+ crystals exhibit orange PL with wavelengths located from 560 to 610 nm and PLQYs up to 95%. The PL of Cs2InBr5·H2O:Sb3+ shifts to the deep red region (630–692 nm) compared to that of Cs2InCl5·H2O:Sb3+ due to the expansion of crystal lattices resulted from the substitution of Br− for Cl−. A similar redshift of PL from 745 to 823 nm has also been observed from the Sb3+ doped Cs2ZnX4 matrix with X varying from Cl− to Br−.41 Besides halogens, the alkali cations in A-sites and the centers of the octahedron in B-sites have also significant influences on the PL spectra and PLQYs of the STEs from Sb3+. The A3InX6:Sb3+ (A = Rb, Cs; X = Cl, Br) crystals show cyan PL with wavelengths located from 497 to 522 nm, and the maximum PLQY of 95% is obtained in Rb3InCl6:Sb3+. Thus, it is concluded that the PL of Sb3+ can be switched by the transformation between A2InX5·H2O and A3InX6 with the removal/addition of H2O ligands.
The returning process from cyan to orange PL due to the transformation from Rb3ScCl6:Sb3+ to Rb2ScCl5·H2O:Sb3+ can be realized by the hydration process under an ambient atmosphere. A series of digital photographs with a gradual change in the PL color are illustrated to depict the hydration process of Rb3ScCl6:Sb3+ stored under an ambient atmosphere and a closed atmosphere (Fig. 5a). After 60 min, the Rb3ScCl6:Sb3+ crystals exposed to air turn back to orange while those insulated from the air by a transparent glass lid maintain cyan, which definitely demonstrates the hydration process by absorbing H2O from the air. Therefore, the above results indicate that the octahedral ligands (H2O or Cl−) have a great influence on the position of the STE state and the recombination time of STEs.
 | ||
| Fig. 5 (a) Digital photographs of the cyan sample (top) exposed in and (bottom) isolated from air under an ambient atmosphere for 60 min. (b) PL emission spectra (λex = 340 nm) of Rb3ScCl6:Sb3+ changing with the aging time. (c) The times consumed in the PL color of Sb3+ based on the transformation from Rb3ScCl6 to Rb2ScCl5·H2O dependent on the contact areas (top) and relative humidity (bottom). (d) Waterfall plot of driven current-dependent EL spectra. | ||
The gradual change in the PL emission color of Sb3+ in Sc-based perovskites has been observed from 600 to 500 nm after dehydration because of the gradual distortion of octahedra (Fig. 5b). Their Commission International de L'Elcairage (CIE) coordinates shift from cyan back to orange on account of the phase transformation from Rb3ScCl6 to Rb2ScCl5·H2O (Fig. S9†). The rates of phase transformation are strongly dependent on the humidity of the environment and the contact areas of the samples exposed to the atmosphere. The times required for transformation are 1 h and 3 s by placing Rb3ScCl6:Sb3+ under an ambient atmosphere and in concentrated hydrochloric acid, respectively. The resulted products are confirmed further by XRD patterns, which indicate that the dominant phase Rb2ScCl5·H2O is accompanied by the minor impurity of RbCl (Fig. S10†). Thus, the probable reaction equation for the reversible transformation is shown below:
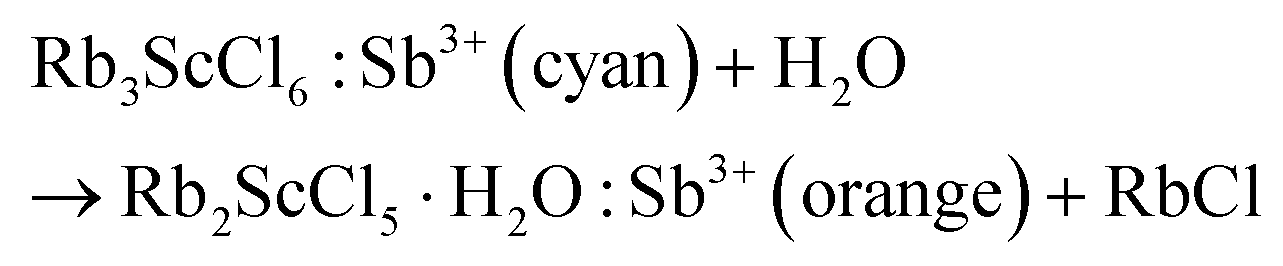
To further discuss the rate of the transformation, the times taken in the transformation from Rb3ScCl6:Sb3+ to Rb2ScCl5·H2O:Sb3+ under different contact areas and relative humidity are illustrated (Fig. 5c). As the relative humidity increases from 40% to 80%, the phase transition time decreases gradually from 102 to 29 min. Meanwhile, it shows an increasing time from 34 to 88 min with a decreasing contact area. Therefore, the larger the relative humidity and the bigger the contact area, the shorter the phase transition time required for the transformation from Rb3ScCl6:Sb3+ to Rb2ScCl5·H2O:Sb3+. Briefly, this merit enables the two novel Sb3+ doped Sc-based perovskites to act as potential candidates for a sensitive luminescent sensor for probing humidity.39
A typical WLED is fabricated with the orange phosphor Rb2ScCl5·H2O:Sb3+ combined with the green phosphor BaSrSiO4:Eu2+ (BSSO:Eu) and the blue phosphor BaMgAl10O17:Eu2+ (BAM:Eu) on a 360 nm InGaN chip. As shown in Fig. S11a,† the WLED device exhibits bright electroluminescence (EL) at an operating current of 40 mA, with a color coordinate of (0.3655, 0.3891) and a white-light correlated color temperature (CCT) of 4451 K (Fig. S11b†). Moreover, to confirm the excellent color stability, the EL spectra of the WLED driven under different operating currents are recorded as shown in Fig. 5d, and it is observed that they show similar shapes and positions except for the intensities.
To understand the Sb3+-induced optical properties and PL mechanism, the charge densities, band structure, and partial densities of states (DOS) of the primitive and Sb3+ doped Rb3ScCl6 and Rb2ScCl5·H2O crystals are analyzed by density functional theory (DFT) calculations. The band gaps of the primitive Rb3ScCl6 and Rb2ScCl5·H2O crystals are calculated to be 3.95 and 3.89 eV (Fig. S12 and S13†), respectively. After doping with Sb3+, the band gaps of the Sb3+-doped Rb3ScCl6 and Rb2ScCl5·H2O crystals are reduced to 3.18 and 3.11 eV, (Fig. 6), respectively. The results indicate that the introduction of exotic Sb3+ ions into a low-dimensional crystal lattice of Rb3ScCl6 and Rb2ScCl5·H2O would significantly decrease the bandgaps by the formation of Sb-s and Cl-p/O-p orbitals between the valence band (VB) and the conduction band (CB).8,42 Additionally, the electronic states of the valence band maximum (VBM) of the Sb3+-doped Rb3ScCl6 crystals are comprised of the Sb-s and Cl-p orbitals and the VBM of Sb3+-doped Rb2ScCl5·H2O comes from the Sb-s, Cl-p, and O-p orbitals from H2O. No orbital hybridization between Sb 5s (or 5p) orbitals and Cl 3p (or O 2p) orbitals has been observed in the calculated results from DOS, which is quite different from that in Cs2ZrCl6:Sb3+ perovskite crystals.43
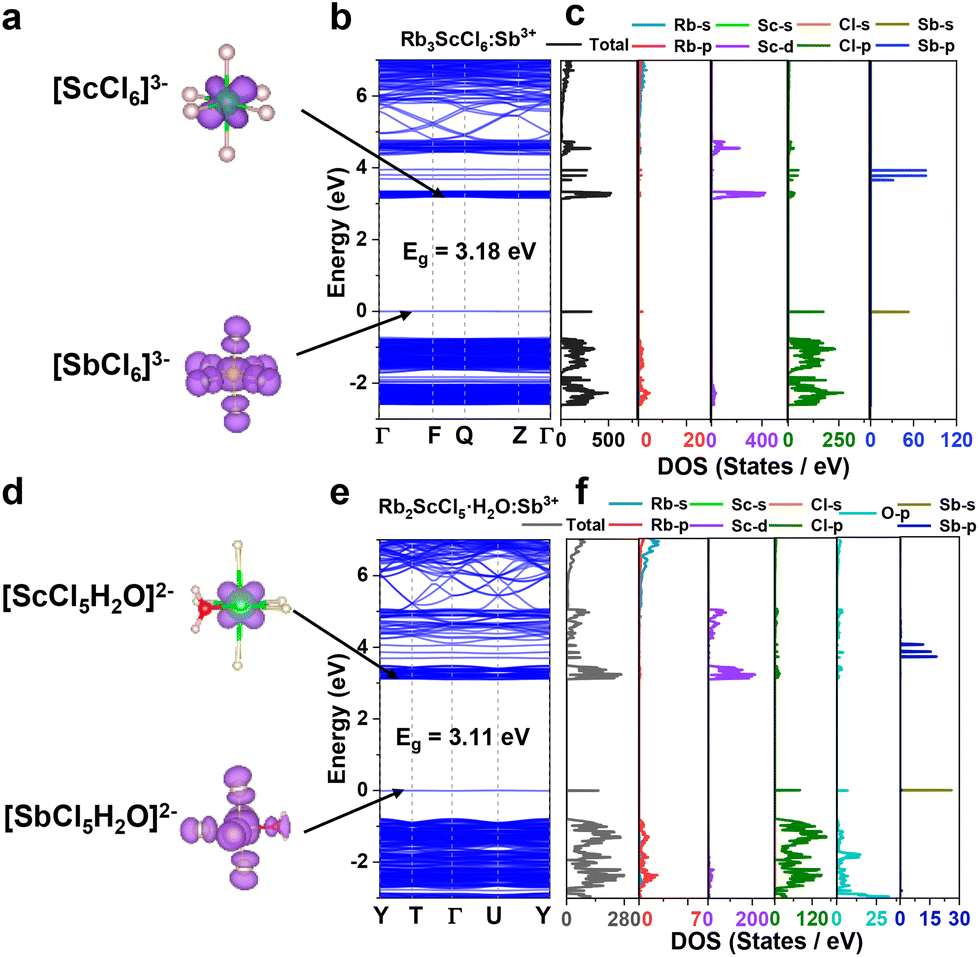 | ||
| Fig. 6 (a and d) The valence band maximum (VBM) and the conduction band minimum (CBM) associated charge densities of the (a) [SbCl6]3−, [ScCl6]3− and (d) [SbCl5(H2O)]2−, [ScCl5(H2O)]2−centers in Rb3ScCl6:Sb3+ and Rb2ScCl5·H2O:Sb3+ crystals, respectively. (b and e) Calculated electronic band structure and (c and f) density of states of (a–c) Rb3ScCl6:Sb3+ and (d–f) Rb2ScCl5·H2O:Sb3+ crystals. | ||
The conduction band minimum (CBM) states of both the Rb3ScCl6:Sb3+ and Rb2ScCl5·H2O:Sb3+ crystals mainly come from the Sc-d orbitals rather than the Sb-p orbitals (Fig. 6, S14 and S15†). The charges in the CBM are concentrated at the center ions of Sc3+ and those in the VBM are distributed uniformly in the whole [SbCl6]3− octahedron, which signifies that the electrons occupied in the Sb-s orbitals are readily transferred to the Sc-d orbitals upon UV excitation. A higher energy is necessary to promote the Sb-s electrons up to the Sb-p orbitals and non-radiative transition to the Sc-d orbitals would occur subsequently. Thus, these results demonstrate that the dominant contribution for the PL of Sb3+-doped Sc-based perovskite crystals originates from the intrinsic excitons in the host lattice rather than in [SbCl6]3−.44 The electrons in the ground state are mainly distributed in the [SbCl6]3− octahedron, while the electrons in the excited state migrate to the [ScCl6]3− octahedron, which is accompanied by the strong electron–phonon coupling caused by the distortion of the octahedron units after doping.45 The electrons in the Sb-s orbitals in the ground state can transfer to the Sc-d orbitals by creating trapping energy states upon the excitation of UV light and subsequently transfer back to the ground states. Through non-radiative relaxation and excited-state structural reorganization process, visible light with a large Stokes shift and broadband feature will be produced by the radiative transition process after the recombination of carriers.43 As presented in the electronic band structure of Rb2ScCl5·H2O:Sb3+ (Fig. 6e and f), the minimum energy for promoting the electrons from the VB to the CB is 3.11 eV (399 nm) and the Sb-p orbitals are located approximately at 4.06 eV (305 nm). There is no excitation band before 320 nm (Fig. S2†), which further indicates that the PL of Rb2ScCl5·H2O:Sb3+ does not originate from the s–p transition of Sb3+ but from STEs induced by Jahn–Teller distortion.
Fig. 7 reveals the charge density distributions and local structures of the [SbCl6]3− octahedron in Rb3ScCl6:Sb3+ and the [SbCl5(H2O)]2− octahedron in Rb2ScCl5·H2O:Sb3+. The charge is uniformly distributed around the six Cl− ions in [SbCl6]3− because of its symmetrical octahedral structure (Fig. 7a). The introduction of H2O breaks the symmetrical structure of the octahedron and changes the charge density distribution in the octahedral structure by forming an O–Sb bond in a distorted octahedral structure of [SbCl5(H2O)]2−. Moreover, most of the charge is observed to be dragged toward H2O in the [SbCl5(H2O)]2− octahedron (Fig. 7b). The local octahedral structures of the [SbCl6]3− octahedron in Rb3ScCl6:Sb3+ indicate that [SbCl6]3− possesses the perfect structure, and the Sb–Cl bond length is about 2.65 Å, while the Cl–Sb–Cl angle is 87.6° (Fig. 7c). Meanwhile, the Sb–O bond length reduces to 2.439 Å, and the Cl–Sb–O angle reduces to 87.4° for [SbCl5(H2O)]2− in Rb2ScCl5·H2O:Sb3+, which demonstrates the further evident distortion of the lattice (Fig. 7d). The orbitals between the neighboring Cl-s and Sb-p show few overlaps and the charge carriers from electron transport can be negligible according to the calculated charge density of the best charge density isosurface plane of [SbCl6]3−. Moreover, it is more difficult for the electrons in [SbCl5(H2O)]2− to transfer because of the slighter overlap of orbitals from Cl-s, Sb-p, and O-p.
 | ||
| Fig. 7 (a and b) Charge density distribution and (c and d) local structures of (a and c) [SbCl6]3− in Rb3ScCl6:Sb3+ and (b and d) [SbCl5(H2O)]2− in Rb2ScCl5·H2O:Sb3+ crystals calculated by DFT. | ||
Consequently, combining the theoretical calculation and the experimental characterization results of the optical properties, the luminescence mechanism of Rb2ScCl5·H2O:Sb3+ and Rb3ScCl6:Sb3+ is illustrated in Fig. S16.† The electrons in the ground state are promoted to a higher excited state (ES1) and then trapped in [SbCl6]3− under excitation at 335 nm for Rb3ScCl6:Sb3+, which subsequently induced the STE1 emission of Rb3ScCl6:Sb3+ centered at 500 nm. In the same way, upon 340 nm excitation, the low energy will lead to the transfer of electrons to a lower excited state (ES2) and then trapped in [SbCl5(H2O)]2− in Rb2ScCl5·H2O:Sb3+. The electrons follow a self-trapped process to form STE2, and the recombination of electrons from the STE2 state to the ground state yields a wide orange emission at 600 nm with a large Stokes shift.
4 Conclusions
In summary, we have developed a new strategy to probe water content based on the orange/cyan emissive sensors of Sb3+ during the reversible phase transformation between the two novel rare-earth-based perovskite crystals Rb2ScCl5·H2O and Rb3ScCl6 with high PLQYs and sensitivity. The in-depth analysis of the calculated energy of the bottom of the CBM suggests that the luminescence of Sb3+ in Sc-based perovskite crystals originates from STEs rather than an s–p transition of Sb3+ because the bottom of the CBM mainly consists of the 3d states of Sc3+ and the 5p states of Sb3+ which are far higher than the bottom of the CBM. After a hydration process through absorbing water in air, the [ScCl6]3− octahedron is converted into a distorted [ScCl5(H2O)]2− octahedron accompanied by the bond length becoming shorter and the charge deviating preferentially toward the H2O ligand, which causes the emission wavelength of Sb3+ to return from the cyan to orange region. This work provides a new water sensor by a switch of the PL color and sheds more light on the importance of the bandgap and charge distribution in determining the luminescence mechanism of Sb3+ in low dimensional perovskites.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is financially supported by the National Natural Science Foundation (NSF) of China (52172152, 51932009, 52172166), the Key Projects of NSF of Zhejiang Province (LZ20E020003, LQ22B010003), the Wenzhou Major Scientific and Technological Innovation Project (ZG2020025), and the Applied Research Programs of Guangdong-Hong Kong-Macao Innovation Center sponsored by Guangzhou Development District.References
- X. Y. Yu, L. Z. Wu, D. Yang, M. H. Cao, X. Fan, H. P. Lin, Q. X. Zhong, Y. Xu and Q. Zhang, Hydrochromic CsPbBr3 Nanocrystals for Anti-Counterfeiting, Angew. Chem., Int. Ed., 2020, 59, 14527–14532 CrossRef CAS PubMed.
- J. B. Luo, J. H. Wei, Z. Z. Zhang and D. B. Kuang, Water-Molecule-Induced Emission Transformation of Zero-Dimension Antimony-Based Metal Halide, Inorg. Chem., 2022, 61, 338–345 CrossRef CAS PubMed.
- X. L. Li, C. Peng, Y. H. Xiao, D. F. Xue, B. B. Luo and X. C. Huang, Guest-Induced Reversible Phase Transformation of Organic-Inorganic Phenylpiperazinium Antimony(III) Chlorides with Solvatochromic Photoluminescence, J. Phys. Chem. C, 2021, 125, 25112–25118 CrossRef CAS.
- L. Zhou, J. F. Liao, Z. G. Huang, J. H. Wei, X. D. Wang, W. G. Li, H. Y. Chen, D. B. Kuang and C. Y. Su, A Highly Red-Emissive Lead-Free Indium-Based Perovskite Single Crystal for Sensitive Water Detection, Angew. Chem., Int. Ed., 2019, 58, 5277–5281 CrossRef CAS PubMed.
- J. D. Majher, M. B. Gray, T. Y. Liu, N. P. Holzapfel and P. M. Woodward, Rb3InCl6: A Monoclinic Double Perovskite Derivative with Bright Sb3+-Activated Photoluminescence, Inorg. Chem., 2020, 59, 14478–14485 CrossRef CAS PubMed.
- P. G. Han, W. Zhou, D. Y. Zheng, X. R. Zhang, C. Li, Q. K. Kong, S. Q. Yan, R. F. Lu and K. L. Han, Lead-Free All Inorganic Indium Chloride Perovskite Variant Nanocrystals For Efficient Luminescence, Adv. Opt. Mater., 2022, 10, 2101344 CrossRef CAS.
- J. C. Jin, Y. H. Peng, Y. T. Xu, K. Han, A. R. Zhang, X. B. Yang and Z. G. Xia, Bright Green Emission from Self-Trapped Excitons Triggered by Sb3+ Doping in Rb4CdCl6, Chem. Mater., 2022, 34, 5717–5725 CrossRef CAS.
- X. F. Meng, Q. L. Wei, W. C. Lin, T. Huang, S. G. Ge, Z. M. Yu and B. S. Zou, Efficient Yellow Self-Trapped Exciton Emission in Sb3+-Doped RbCdCl3 Metal Halides, Inorg. Chem., 2022, 61, 7143–7152 CrossRef CAS PubMed.
- R. S. Zeng, L. L. Zhang, Y. Xue, B. Ke, Z. Zhao, D. Huang, Q. L. Wei, W. C. Zhou and B. S. Zou, Highly Efficient Blue Emission from Self-Trapped Excitons in Stable Sb3+-Doped Cs2NaInCl6 Double Perovskites, J. Phys. Chem. Lett., 2020, 11, 2053–2061 CrossRef CAS PubMed.
- Y. Y. Jing, Y. Liu, M. Z. Li and Z. G. Xia, Photoluminescence of Singlet/Triplet Self-Trapped Excitons in Sb3+-Based Metal Halides, Adv. Opt. Mater., 2021, 9, 2002213 CrossRef CAS.
- J. W. Lin, K. J. Liu, H. Ruan, N. Sun, X. Chen, J. Zhao, Z. N. Guo, Q. L. Liu and W. X. Yuan, Zero-Dimensional Lead-Free Halide with Indirect Optical Gap and Enhanced Photoluminescence by Sb Doping, J. Phys. Chem. Lett., 2022, 13, 198–207 CrossRef CAS.
- Y. Y. Jing, Y. Liu, X. X. Jiang, M. S. Molokeev, Z. S. Lin and Z. G. Xia, Sb3+ Dopant and Halogen Substitution Triggered Highly Efficient and Tunable Emission in Lead-Free Metal Halide Single Crystals, Chem. Mater., 2020, 32, 5327–5334 CrossRef CAS.
- Z. L. Gong, W. Zheng, P. Huang, X. W. Cheng, W. Zhang, M. R. Zhang, S. Y. Han and X. Y. Chen, Highly Efficient Sb3+ Emitters in 0D Cesium Indium Chloride Nanocrystals with Switchable Photoluminescence through Water-Triggered Structural Transformation, Nano Today, 2022, 44, 101460 CrossRef CAS.
- A. Noculak, V. Morad, K. M. McCall, S. Yakunin, Y. Shynkarenko, M. Wörle and M. V. Kovalenko, Bright Blue and Green Luminescence of Sb(III) in Double Perovskite Cs2MInCl6 (M = Na, K) Matrices, Chem. Mater., 2020, 32, 5118–5124 CrossRef CAS PubMed.
- J. L. Huang, T. Chang, R. S. Zeng, J. Yan, Q. L. Wei, W. C. Zhou, S. Cao and B. S. Zou, Controlled Structural Transformation in Sb-Doped Indium Halides A3InCl6 and A2InCl5·H2O Yields Reversible Green-to-Yellow Emission Switch, Adv. Opt. Mater., 2021, 9, 2002267 CrossRef CAS.
- B. Yang and K. L. Han, Ultrafast Dynamics of Self-Trapped Excitons in Lead-Free Perovskite Nanocrystals, J. Phys. Chem. Lett., 2021, 12, 8256–8262 CrossRef CAS PubMed.
- S. R. Li, J. J. Luo, J. Liu and J. Tang, Self-Trapped Excitons in All-Inorganic Halide Perovskites: Fundamentals, Status, and Potential Applications, J. Phys. Chem. Lett., 2019, 10, 1999–2007 CrossRef CAS PubMed.
- R. X. Liu, W. J. Zhang, G. J. Li and W. J. Liu, An Ultraviolet Excitation Anti-Counterfeiting Material of Sb3+ Doped Cs2ZrCl6 Vacancy-Ordered Double Perovskite, Inorg. Chem. Front., 2021, 8, 4035–4043 RSC.
- H. Arfin and A. Nag, Origin of Luminescence in Sb3+- and Bi3+-Doped Cs2SnCl6 Perovskites: Excited State Relaxation and Spin-Orbit Coupling, J. Phys. Chem. Lett., 2021, 12(41), 10002–10008 CrossRef CAS PubMed.
- C. G. Zhang, M. R. Zhang, W. Zheng, J. J. Wei, S. T. Wang, P. Huang, X. W. Cheng, T. Dao, Z. Chen and X. Y. Chen, A New Class of Luminescent Nanoprobes Based on Main-Group Sb3+ emitters, Nano Res., 2022, 15(1), 179–185 CrossRef CAS.
- A. S. Kshirsagar, H. Arfin, S. Banerjee, B. Mondal and A. Nag, Colloidal Sb3+-Doped Cs2InCl5·H2O Perovskite Nanocrystals with Temperature-Dependent Luminescence, J. Phys. Chem. C, 2021, 125, 27671–27677 CrossRef CAS.
- X. Y. Liu, X. Xu, B. Li, Y. Q. Liang, Q. Li, H. Jiang and D. S. Xu, Antimony-Doping Induced Highly Efficient Warm-White Emission in Indium-Based Zero-Dimensional Perovskites, CCS Chem., 2020, 2, 216–224 CrossRef CAS.
- J. C. Zhou, C. Shi, X. S. Li, Z. M. Sun, Y. J. Ji, J. Deng and B. Wang, A Heterovalent Doping Strategy Induced Efficient Cyan Emission in Sb3+-Doped CsCdCl3 Perovskite Microcrystal for Solid State Lighting, Ceram. Int., 2022, 48, 28327–28333 CrossRef CAS.
- R. L. Zhang, X. Xu, X. Mao, Z. Y. Wang, P. Y. Wang, Y. Yang, J. S. Chen, R. F. Lu, W. Q. Deng and K. L. Han, Excitation-Dependent Emission in All-Inorganic Lead-Free Cs2ScCl5·H2O Perovskite Crystals, Laser Photonics Rev., 2022, 2100689 CrossRef CAS.
- M. D. Smith and H. I. Karunadasa, White-Light Emission from Layered Halide Perovskites, Acc. Chem. Res., 2018, 51, 619–627 CrossRef CAS PubMed.
- X. Y. Chen, X. Wang, C. H. An, J. W. Liu and Y. T. Qian, Synthesis of Sb2O3 Nanorods under Hydrothermal Conditions, Mater. Res. Bull., 2005, 40, 469–474 CrossRef CAS.
- M. F. Hamza, Y. Z. Wei and E. Guibal, Quaternization of Algal/PEI Beads (A New Sorbent): Characterization and Application to Scandium Sorption from Aqueous Solutions, Chem. Eng. J., 2020, 383, 123210 CrossRef CAS.
- A. Elizabeth, S. K. Sahoo, H. Phirke, T. Kodalle, T. D. Kühne, J. N. Audinot, T. Wirtz, A. Redinger, C. A. Kaufmann, H. Mirhosseini and H. Mönig, Surface Passivation and Detrimental Heat-Induced Diffusion Effects in RbF-Treated Cu(In,Ga)Se2 Solar Cell Absorbers, ACS Appl. Mater. Interfaces, 2022, 14(29), 34101–34112 CrossRef PubMed.
- J. Y. Zhong, W. D. Zhuang, X. R. Xing, L. G. Wang, Y. F. Li, Y. L. Zheng, R. H. Liu, Y. H. Liu and Y. S. Hu, Blue Shift of Spectrum and Enhanced Luminescent Properties of YAG:Ce3+ Phosphor Induced by Small Amount of La3+ Incorporation, J. Alloys Compd., 2016, 674, 93–97 CrossRef CAS.
- C. J. Liu, X. M. Zhu and Z. F. Zhou, Effects of Composition Modulation on The Structural and Luminescence Properties of Mn2+ Doped Na2Mg1−xCaxSiO4 Green-Emitting Phosphors, Optik, 2019, 179, 875–882 CrossRef CAS.
- M. Z. Liu, C. K. Duan, P. A. Tanner, C. G. Ma and M. Yin, Rationalizing the Photoluminescence of Bi3+ and Sb3+ in Double Perovskite Halide Crystals, J. Phys. Chem. C, 2021, 125, 26670–26678 CrossRef CAS.
- Y. Y. Jing, Y. Liu, J. Zhao and Z. G. Xia, Sb3+ Doping-Induced Triplet Self-Trapped Excitons Emission in Lead-Free Cs2SnCl6 Nanocrystals, J. Phys. Chem. Lett., 2019, 10, 7439–7444 CrossRef CAS PubMed.
- S. L. Jin, R. F. Li, H. Huang, N. Z. Jiang, J. D. Lin, S. X. Wang, Y. H. Zheng, X. Y. Chen and D. Q. Chen, Compact Ultrabroadband Light-Emitting Diodes Based on Lanthanide-Doped Lead-Free Double Perovskites, Light: Sci. Appl., 2022, 11, 52 CrossRef CAS PubMed.
- C. Y. Peng, Q. L. Wei, S. F. Yao, X. F. Meng, Z. M. Yu, H. Peng, X. C. Zhong and B. S. Zou, H2O-NH4+-Induced Emission Modulation in Sb3+-Doped (NH4)2InCl5·H2O, Inorg. Chem., 2022, 61, 12406–12414 CrossRef CAS PubMed.
- J. H. Wei, J. B. Luo, J. F. Liao, W. T. Ou and D. B. Kuang, Te4+-Doped Cs2InCl5·H2O Single Crystals for Remote Optical Thermometry, Sci. China Mater., 2022, 65(3), 764–772 CrossRef CAS.
- J. H. Han, T. Samanta, Y. M. Park, H. B. Cho, J. W. Min, S. J. Hwang, S. W. Jang and W. B. Im, Effect of Self-Trapped Excitons in The Optical Properties of Manganese-Alloyed Hexagonal-Phased Metal Halide Perovskite, Chem. Eng. J., 2022, 450, 138325 CrossRef CAS.
- C. K. Zhou, H. R. Lin, Y. Tian, Z. Yuan, R. Clark, B. H. Chen, L. J. Burgt, J. C. Wang, Y. Zhou, K. Hanson, Q. J. Meisner, J. Neu, T. Besara, T. Siegrist, E. Lambers, P. Djurovich and B. W. Ma, Luminescent Zero-Dimensional Organic Metal Halide Hybrids with Near-Unity Quantum Efficiency, Chem. Sci., 2018, 9, 586–593 RSC.
- P. G. Han, C. Luo, S. Q. Yang, Y. Yang, W. Q. Deng and K. L. Han, All-Inorganic Lead-Free 0D Perovskites by a Doping Strategy to Achieve a PLQY Boost from <2% to 90%, Angew. Chem., Int. Ed., 2020, 59, 12709–12713 CrossRef CAS PubMed.
- X. W. Cheng, R. F. Li, W. Zheng, D. T. Tu, X. Y. Shang, Z. L. Gong, J. Xu, S. Y. Han and X. Y. Chen, Tailoring the Broadband Emission in All-Inorganic Lead-Free 0D In-Based Halides through Sb3+ Doping, Adv. Opt. Mater., 2021, 2100434 CrossRef CAS.
- J. Zhou, X. Y. Yun, R. Y. Wang, D. H. Xu and X. Li, Self-Trapped Exciton to Dopant Energy Transfer in Sb3+-Doped Cs2ZrCl6 Perovskite Variants, Mater. Chem. Front., 2021, 5, 6133 RSC.
- B. B. Su, M. Z. Li, E. H. Song and Z. G. Xia, Sb3+-Doping in Cesium Zinc Halides Single Crystals Enabling High-Efficiency Near-Infrared Emission, Adv. Funct. Mater., 2021, 31, 2105316 CrossRef CAS.
- Q. L. Wei, X. F. Meng, W. C. Lin, S. G. Ge, X. X. Han, L. Chen, R. S. Zeng and B. S. Zou, Green Triplet Self-Trapped Exciton Emission in Layered Rb3Cd2Cl7:Sb3+ Perovskite: Comparison with RbCdCl3:Sb3+, J. Phys. Chem. Lett., 2022, 13, 8436–8446 CrossRef CAS PubMed.
- B. Chen, Y. Guo, Y. Wang, Z. Liu, Q. Wei, S. X. Wang, A. L. Rogach, G. C. Xing, P. Shi and F. Wang, J. Am. Chem. Soc., 2021, 143, 17599–17606 CrossRef CAS.
- Y. R. Dai, Q. L. Wei, T. Chang, J. L. Zhao, S. Cao, B. S. Zou and R. S. Zeng, Efficient Self-Trapped Exciton Emission in Ruddlesden-Popper Sb-Doped Cs3Cd2Cl7 Perovskites, J. Phys. Chem. C, 2022, 126, 11238–11245 CrossRef CAS.
- B. Zhou, Z. X. Liu, S. F. Fang, H. Z. Zhong, B. B. Tian, Y. Wang, H. N. Li, H. L. Hu and Y. M. Shi, Efficient White Photoluminescence from Self-Trapped Excitons in Sb3+/Bi3+-Codoped Cs2NaInCl6 Double Perovskites with Tunable Dual-Emission, ACS Energy Lett., 2021, 6, 3343–3351 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qi02221d |
| This journal is © the Partner Organisations 2023 |