
Electrocatalytic production of hydrogen peroxide enabled by post-synthetic modification of a self-assembled porphyrin cube†
Matthew R.
Crawley
 ,
Daoyang
Zhang
and
Timothy R.
Cook
*
,
Daoyang
Zhang
and
Timothy R.
Cook
*
Department of Chemistry, University at Buffalo, The State University of New York, Buffalo, New York 14260, USA. E-mail: trcook@buffalo.edu
First published on 25th November 2022
Abstract
Self-assembled metallacyles and cages formed via coordination chemistry have been used as catalysts to enforce 4H+/4e− reduction of oxygen to water with an emphasis on attenuating the formation of hydrogen peroxide. That said, the kinetically favored 2H+/2e− reduction to H2O2 is critically important to industry. In this work we report the synthesis, characterization, and electrochemical benchmarking of a hexa-porphyrin cube which catalyses the electrochemical reduction of molecular oxygen to hydrogen peroxide. An established sub-component self-assembly approach was used to synthesize the cubic free-base porphryin topologies from 2-pyridinecarboxaldehyde, tetra-4-aminophenylporphryin (TAPP), and Fe(OTf)2 (OTf− = trifluoromethansulfonate). Then, a tandem metalation/transmetallation was used to introduce Co(II) into the porphyrin faces of the cube, and exchange with the Fe(II) cations at the vertices, furnishing a tetrakaideca cobalt cage. Electron paramagnetic resonance studies on a Cu(II)/Fe(II) analogue probed radical interactions which inform on electronic structure. The efficacy and selectivity of the CoCo-cube as a catalyst for hydrogen peroxide generation was investigated using hydrodynamic voltammetry, revealing a higher selectivity than that of a mononuclear Co(II) porphyrin (83% versus ∼50%) with orders of magnitude enhancement in standard rate constant (ks = 2.2 × 102 M−1 s−1versus ks = 3 × 100 M−1 s−1). This work expands the use of coordination-driven self-assembly beyond ORR to water by exploiting post-synthetic modification and structural control that is associated with this synthetic method.
Introduction
Meeting ever-growing global energy demand is one of the greatest challenges of the 21st century.1 Harvesting energy from chemical changes of small molecules is a major thrust in the field of sustainable energy production.2 Of particular interest is the oxygen reduction reaction (ORR). Molecular oxygen (O2) possesses several reductive pathways, the 4-proton, 4-electron (4H+/4e−) pathway to water (H2O), and the 2H+/2e− pathway to hydrogen peroxide (H2O2). The former is well known as the cathodic reaction in a hydrogen fuel cell. Due to the undesirable effects of H2O2 on fuel cell materials, many research efforts aim to quell this kinetically more accessible reduction pathway; yet H2O2 production is a crucial industrial process and a valuable product, making selective peroxide catalysts important.Currently, H2O2 production exceeds 4 million metric tons annually, and is critical to the paper-and-pulp industry, as well as water treatment, cosmetics, and rocket propulsion.3,4 Although industrialized as early as 1818 via a BaO2/BaSO4 route, current production of H2O2 uses the anthraquinone process (or AO process, see Fig. 1), in which an alkyl anthraquinone serves as a catalyst in the conversion of H2(g) and O2(g) to H2O2(aq), forming aqueous peroxide solutions, typically between 15–40% (w/w). Initially, the solution of anthraquinone is reduced by H2 on a Pd or RANEY® Ni catalyst to form the corresponding hydroquinone. The hydroquinone is then treated with molecular oxygen, which is reduced to H2O2, and the anthraquinone is reformed. A major drawback to the AO process is the large energetic demand. The hydrogenation of anthraquinone uses H2 gas feedstock, which is industrially produced from endergonically intensive steam methane reforming.5 In addition to energetic hurtles, during the hydrogenation step it is possible to over-hydrogenate the anthraquinone at the aromatic rings which deactivates the catalyst. Electrocatalytic H2O2 production provides an attractive alternate synthetic route using O2 and H+ as the feedstocks. Additionally, electrocatalysts are more scalable to meet small to large demand and when driven by renewable energy sources environmental impact is minimized. Of particular interest are catalysts derived from earth abundant elements. In terms of metal-containing molecular catalysts, Co-based complexes have attracted the greatest attention of all the non-noble metals for ORR to H2O2.6 Of these catalysts, almost all are mononuclear porphyrin or macrocycle complexes.7–9 Homogeneous catalysis using monomeric Co complexes are often highly selective (>90%) for the kinetically favored 2H+/2e− reduction to H2O2. Examples include Co complexes of tetra-(N-methyl-4-pyridium)porphyrin (CoTMPyP),10 cyclam,7 and chlorin ligands.11 Notably, when these monomeric species are immobilized in films, drastic changes in selectivities are observed. For example, in dilute solution CoTPP shows a near 100% selectivity for H2O2 production;12 however when immobilized in a film we found ∼60% selectivity, and Chang ∼50%.13 Upon aggregation, bimolecular pathways become relevant that may favor 4H+/4e− chemistry. Other examples of molecular ORR to H2O2 catalysts include dinuclear Cu-based catalysts bearing N/O-donor ligands that almost exclusively form H2O2.14 Along with CoTMPyP, MnTMPyP was found to reduce O2 to H2O2 with a selectivity of 76%, comparable with the cubic assembly reported here; however, this was reported in dilute solution where bimolecular pathways are avoided.10 There are numerous examples of peroxide-selective heterogeneous ORR catalysts reviewed elsewhere.6 Due to the industrial importance of H2O2, furthered understanding of electrocatalytic ORR, and the factors governing the selectivity of catalysts is imperative for future designs.
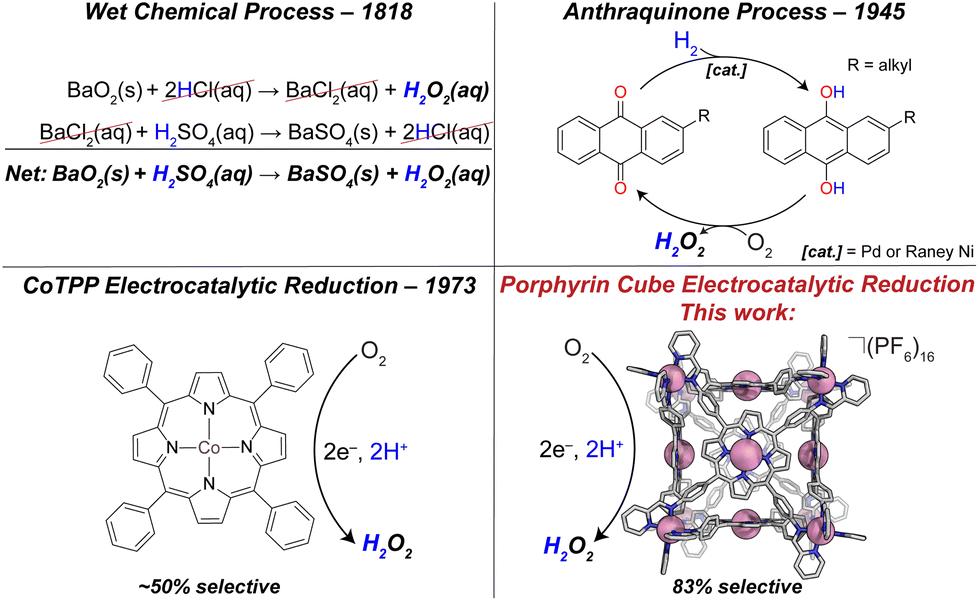 | ||
| Fig. 1 (Top left) Early hydrogen peroxide production method using BaO2 in acidic media; (top right) anthraquinone process for H2O2 production; (bottom right) mononuclear cobalt tetraphenylporphyrin (CoTPP) is a model catalyst for oxygen reduction studies; (bottom left) Co(II)-metalated porphyrin cube reported here. | ||
Our lab and others have been interested in pre-organizing multiple metal centers using coordination-driven self-assembly chemistry,15–18 inspired by several covalently tethered architectures that are among the most selective molecular ORR catalysts known.19–21 The broader motivation behind these designs are the active sites of metalloenzymes such as cytochrome c oxidase,22 which allow for cooperative interactions with substrate. This is traditionally thought of as an effective way to promote the thermodynamically favored 4H+/4e− pathway to water. We hypothesized that if, on the other hand, catalysts are purposely designed to prevent such cooperativity (i.e., large metal–metal separations are designed into molecular structure), then the kinetically favored 2H+/2e− pathway should become operative. This is supported by examples of cofacial porphyrin-corrole dyads which catalyze the reduction O2 to H2O2 when divergent organic linkers are used.23
Previously, we have used Lewis-basic porphyrin-based building blocks to populate a library of self-assembled cofacial prisms which have demonstrated remarkable catalytic activity and selectivity for the ORR to water.24–27 Literature reports of covalently synthesized cubic multinuclear catalysts applied to ORR and other small molecule activations encouraged our efforts to develop self-assembled catalysts with different topologies.13,28,29 We have used a subcomponent self-assembly route to synthesize porphyrin-based cubes with tris chelated vertices reported by Nitschke (see Fig. 2).30
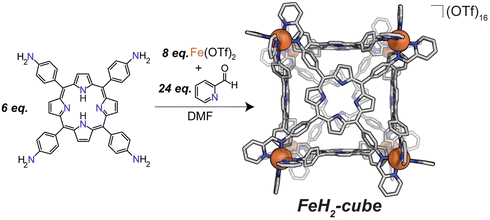 | ||
| Fig. 2 Synthesis of cubic porphyrin self-assemblies by sub-component coordination-driven self-assembly from tetra-4-aminophenylporphryin (TAPP), 2-pyridinecarboxaldehyde, and Fe(OTf)2 by Nitschke (2011). | ||
The goal of this work is to further understand how geometric factors influence catalytic performance for ORR. These studies are enabled by our self-assembly approach with allows for relatively straightforward changes to a number of parameters that would be difficult using traditional covalent and step-wise synthetic methods. In previous work, we have shown that metal–metal separation is not the sole parameter governing selectivity.24 Exploiting the modularity of self-assembly and adapting our studies of polynuclear catalysts to a sub-component assembly method16,31 we have increased nuclearity, expanded metal–metal separation (ca. 15 Å), and significantly altered the relative orientations of the porphyrin faces. Our cubic architecture joins a small library of similar structures formed by covalent and self-assembly routes.13 These compounds all share an important advantage over monomers, namely that they minimize aggregation due to their rigid and porous structures so that the local environment around each porphyrin center is generally consistent throughout the bulk, even when immobilized in films. When monomers aggregate and adopt certain intermolecular metal–metal separations, new pathways become available and non-selective catalysis results.13
We also discovered an unanticipated feature of our self-assembly and post-assembly metalation strategy wherein transmetallation of the vertices metals from Fe to Co occurred quantitatively. This furnished the “all Co(II) metallated cube” or CoCo-cube, (Note: the naming scheme is MM′-cube, where M is the structural metal ion, i.e. the metal at the vertices of the cube, and M′ is in the porphyrin). Under catalytically relevant conditions, CoCo-cube selectively catalyzed the 2H+/2e− reduction to peroxide with a selectivity of 83%. This is the first example of a H2O2 selective coordination-driven self-assembled ORR catalyst.
Experimental
All transformations were performed under inert conditions unless otherwise stated, using standard Schlenk techniques. Reagents were purchased and used without subsequent purification. Solvents were degassed prior to use. NMR spectra were acquired on a Bruker AVANCE NEO 500 operating at 500 MHz. High resolution mass spectra were acquired on a Bruker Solarix 12T ESI-FT-ICR. EPR spectra were acquired on a Bruker EMX X-band spectrometer. Fe(OTf)2 was purchased from Strem Chemicals, tetra-4-aminophenylporphryin (TAPP) was purchased from Frontier Scientific, and 2-pyridinecarboxaldehyde from Beantown Chemical. CuTAPP was synthesized as outlined in the literature.32FeH2-cube was synthesized following a literature procedure.30
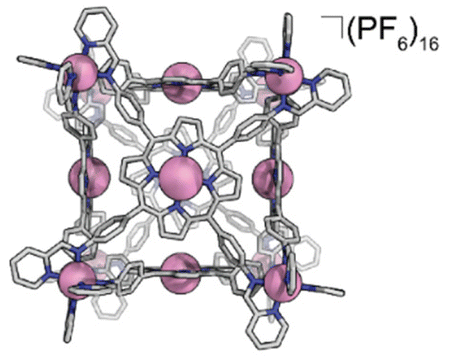
Synthesis of CoCo-cube
Under a nitrogen atmosphere, 13.01 mg of FeH2-cube (1.443 μmol) was dissolved in 10 mL of dry, degassed acetonitrile. To this solution was added 11.85 mg of Co(OAc)2·4H2O (47.58 μmol, 33 eq.). The reaction mixture was heated to 60 °C for 24 hours. The reaction mixture transitioned from a deep purple to a deep red/orange. The reaction was then cooled to 25 °C and stirred for an additional 18 hours. Half of the solvent was removed by rotary evaporation; no efforts were made to exclude air or water from this point forward. In a 20 mL vial, 26.50 mg of KPF6 (144.0 μmol, 100 eq.) was dissolved in 10 mL of DI H2O. The reduced-volume reaction mixture was then pipetted into the KPF6 solution. A brownish orange precipitate immediately formed. The suspension was then filtered through a fine glass frit. The solid on the frit was washed with 3 × 15 mL DI H2O and 2 × 15 mL diethyl ether. The remaining residue was then dissolved and passed through a frit using 15 mL of acetonitrile. The solvent was removed from the deep red/orange filtrate in vacuo to give CoCo-cube as a red/orange solid. Yield: 11.83 mg (1.269 μmol, 87.94%). Paramagnetic 1H NMR (500 MHz, CD3CN, 298 K): δ 246.64, 90.35, 75.97, 53.40, 18.08, 15.55, 7.82, 5.38, 3.22, −3.73, −62.90. ESI-FT-ICR (CH3CN, M = (C408H264Co14N72)(PF6)16, m/z): [M − 6(PF6)−]6+ = 1408.1814, [M − 7(PF6)−]7+ = 1186.3047, [M − 8(PF6)−]8+ = 1019.8938, [M − 9(PF6)−]9+ = 890.4639, [M − 10(PF6)−]10+ = 786.9205, [M − 11(PF6)−]11+ = 702.2025. Soret band: 427 nm (ε = 1.31 × 106 M−1 cm−1), Q bands: 546 nm (ε = 1.56 × 105 M−1 cm−1), 583 nm (ε = 9.54 × 104 M−1 cm−1). FT-IR (ATR, cm−1): 3636, 3387, 3095, 1629, 1598, 1499, 1351, 1206, 1005, 836, 773, 642, 556.
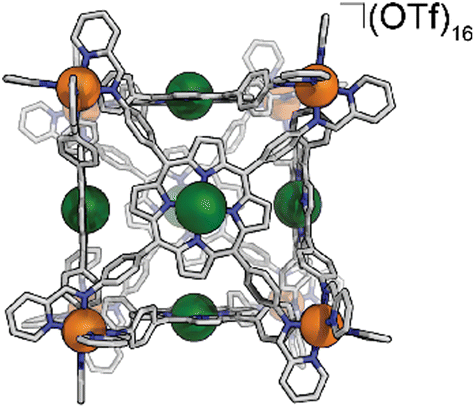
Synthesis of FeCu-cube
In a 2 dram vial, 32.2 mg Fe(OTf)2 (91.0 μmol, 8 eq.), 51.1 mg CuTAPP (69.4 μmol, 6 eq.), 26.1 μL 2-pyridinecarboxyaldehyde (274 μmol, 24 eq.), and 2.0 mL of DMF were combined. The vial was sealed with a rubber septum and three evacuation/N2 cycles were performed to exclude oxygen. The reaction mixture was heated to 70 °C with stirring for 24 hours. The reaction mixture was then cooled to 25 °C and added to 18 mL diethyl ether to precipitate the FeCu-cube as a purple solid. The solid was collected by centrifugation, washed with diethyl ether, and dried in vacuo. Yield: 81.4 mg (8.67 μmol, 76.2%). ESI-FT-ICR (CH3CN, M = (C408H264Cu6Fe8N72)(OTf)16, m/z): [M − 6(OTf)−]6+ = 1415.6466, [M − 7(OTf)− + Cl−]6+ = 1396.6495, [M − 7(OTf)−]7+ = 1192.1328, [M − 8(OTf)− + Cl−]7+ = 1175.8494, [M − 8(OTf)−]8+ = 1024.3723. UV-Vis (CH3CN) Soret band: 409 nm (ε = 2.67 × 106 M−1 cm−1), Q bands: 539 nm (ε = 2.35 × 105 M−1 cm−1), 576 nm (ε = 1.22 × 105 M−1 cm−1). FT-IR (ATR, cm−1): 3448, 3081, 3035, 2932, 1648, 1497, 1387, 1249, 1154, 1028, 998, 799, 635, 515.Electrochemical experiments
All electrochemical experiments were performed using a BioLogic SP-300 bipotentiostat. Homogeneous non-aqueous electrochemical experiments were performed in acetonitrile dried and degassed with a Pure Process Technology solvent purification system. Tetrabutylammonium hexafluorophosphate (TBAPF6) was used as the supporting electrolyte and was recrystallized three times from absolute ethanol before use. A glassy carbon button was used at the working electrode, a platinum wire as the counter electrode, and referenced to a Ag/AgNO3 nonaqueous reference electrode, which was then referenced to the Fc+/0 couple. The working electrode was polished before and after each experiment. Aqueous experiments were performed in 0.5 M H2SO4. The working and counter electrodes remained the same as in non-aqueous experiments; however, the reference electrode was changed to Ag/AgCl (3 M KCl). For hydrodynamic voltammetry experiments, a Pine MSR rotator was used along with a glassy carbon disk/Pt ring, rotating ring-disk electrode (RRDE). Scan rates were 100 mV s−1 for all CV experiments, and 20 mV s−1 for all linear sweep hydrodynamic voltammetry experiments. Catalyst ink films were formed as outlined previously.24Results and discussion
We assembled two related cages for study. The cubes containing Co(II)-metallated porphyrins were assembled to enable investigations of electrochemical ORR, and the Cu(II)-based assemblies provided the basis for straightforward EPR experiments. The FeCu-cube was accessed in a similar route to Nitschke's free-base Fe cube assembly, with the only difference being that we used TAPP metalated with Cu in the sub-component assembly reaction illustrated in Fig. 2. However, when we attempted to extend this strategy to install Co(II) in the faces of the cube, the use of CoTAPP resulted in intractable and insoluble products. Several different solvents, reaction times, and temperatures were used yet in all cases a dark purple/grey insoluble material was the primary product. We spectulate that coordination of the TAPP amine nitrogen atoms to the Co(II) centres is a likely culprit for the insolubility, due to the formation of oligomers/coordination polymers. To circumvent this, we hypothesized that a post-synthetic metalation could be used with a free-base porphyrin cube. This has been demonstrated on trimeric porphyrin barrels synthesized by Lipke et al.33 The free-base porphyrin cube, FeH2-cube, was synthesized according to literature protocol (see Fig. 2).30 It was then treated with an excess of Co(OAc)2·4H2O (33 equivalents) (see Fig. 3). During the reaction, a distinct colour change occurred, the solution transitioned from a deep purple to a deep red/orange. Mass spectral analysis of the reaction mixture suggested that although the porphyrin metalation had indeed occurred as intended, the pyridylimine sites also underwent chemistry. The structural metal Fe(II) cations in the vertices of the cube exchanged, resulting in CoCo-cube rather than the expected FeCo-cube. Although there have been reports of metal cation exchange in self-assembled bis-chelated helicate structures,34 and others with lanthanides,35 there are no previous reports of such a phenomenon in assemblies constructed with tris-chelated transition metal nodes. This straightforward approach furnished an assembly that was otherwise inaccessible without post-assembly modification. Even more intriguingly, vertex exchange provides some insight into the solution-state dynamics of self-assembled architectures. To exchange a tris-chelated vertex, some degree of disassembly must occur under the metalation conditions. The generality of this phenomenon is point of ongoing research.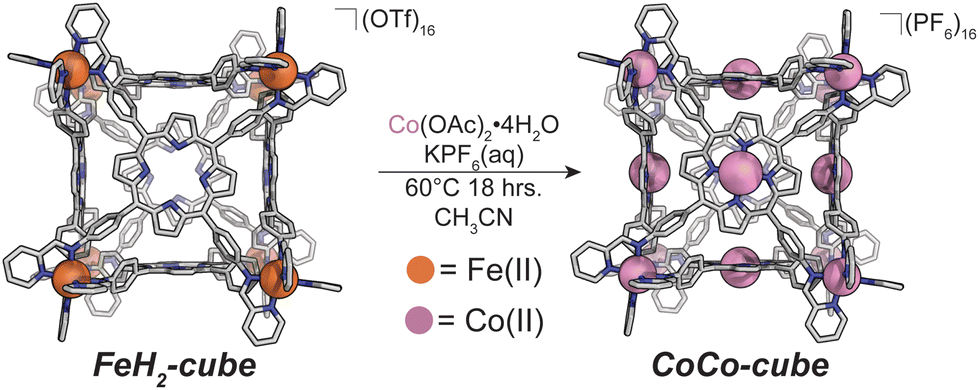 | ||
| Fig. 3 Post-synthetic metalation/transmetallation of FeH2-cube to form CoCo-cube. | ||
UV-Vis spectroscopy was used to verify the metalation FeH2-cube to CoCo-cube by the diagnostic collapse of the four Q-bands of free-base to the two Q-bands of the metalated prisms. FeCu-cube analogously shows the expected two Q-bands. For all porphyrin-based assemblies their Soret bands dominated the spectra (see Fig. S5 and S6†).
High-resolution mass spectrometry was used to verify the stoichiometry and elemental composition of the self-assembled cubic structures. The high-resolution mass spectrum of the CoCo-cube supports the full exchange of all Fe(II) centres of the vertices for Co(II) centres. The dominant peaks correspond to the intact CoCo-cube core ionized by loss of PF6− counterions (ranging from a loss of 5 to 12 counterions, see Fig. 4). Closer examination of the 9+ parent ion peak shows excellent agreement between the experimental and simulated isotopic distribution of [CoCo-cube − 9PF6−]9+. A similar mass spectrum was obtained for FeCu-cube; however, each peak corresponding to the intact prisms was flanked a second peak that corresponded to the exchange of a OTf− for a Cl−. For both cubes, high-resolution mass spectrometry strongly supports the assigned stoichiometries and formulations of the porphyrin cube assemblies.
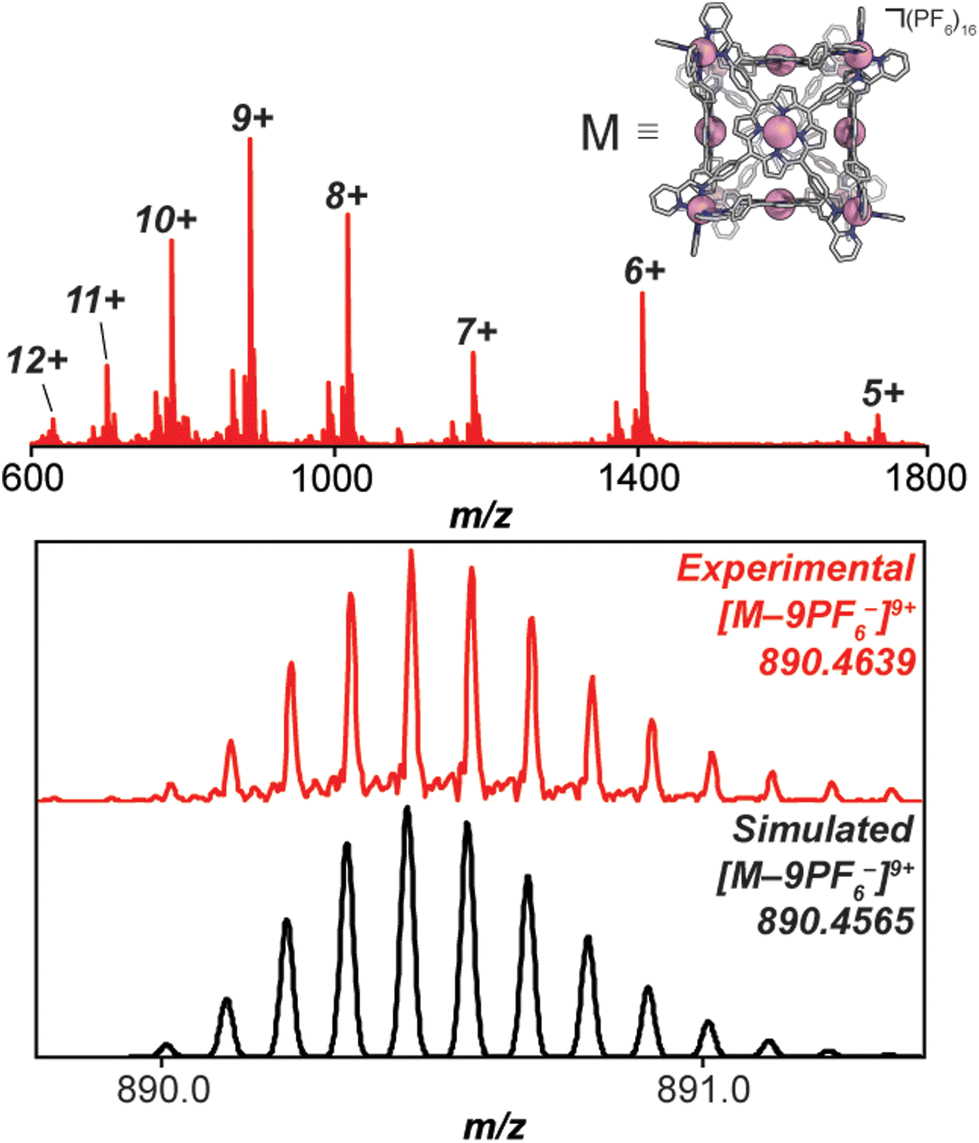 | ||
| Fig. 4 High resolution ESI-FT-ICR mass spectrum of CoCo-cube acquired in acetonitrile. (upper) Full mass spectrum, labelled peaks correspond to the intact cube ionized by loss of outer sphere PF6− (i.e. [M − nPF6−]n+). (lower) Experimental (red) and simulated (black) mass spectrum of the 9+ peak corresponding to the intact cube less 9 PF6− counterions. | ||
NMR spectroscopy was used to further interrogate the molecular structure, as well as probe spin-state. The paramagnetic 1H NMR spectrum of CoCo-cube was consistent the presence of high-spin Co(II) metal centres (see Fig. S1†). The most downfield resonance was found at 247 ppm, while the most upfield resonance occurred at −63 ppm. These resonances are consistent with related high-spin Co(II) assemblies and further supports the exchange of the Fe(II) centres with Co(II) at the vertices.16,36 Closer inspection of the 0 to 20 ppm range reveals broad resonances characteristics of square planar Co(II) porphyrin with a single unpaired electron per porphyrin-ligated metal centre.33
Electron paramagnetic resonance (EPR) informed on the spin states and local structure of paramagnetic centres. The 77 K X-band EPR of CuTAPP starting material shows an axial signal for an S = ½ spin system (see Fig. 5), with clear hyperfine coupling to the 63/65Cu nucleus (I = 3/2) and clear superhyperfine coupling to the 14N nuclei (I = 1) of the porphyrin ligand. The hyper- and superhyperfine interactions support that CuTAPP possesses a d9 Cu(II) centre with the unpaired electron residing in a molecular orbital of Cu dx2−y2 parentage. After assembly, the 77 K EPR spectrum of FeCu-cube showed similar axial spectral features of doublet spin system. The g∥ was found to be 2.19, in good agreement with 2.18 for CuTAPP. Similar g values support that no major geometric, or electronic perturbation occurs at the metal centres upon assembly. The hyperfine coupling of the radical to the 63/65Cu nuclei was still observed; however, the superhyperfine coupling was less observable. In the assembled cubic structure, there may be slight variations, from porphyrin to porphyrin, in the local coordination environment, and as such the resonance conditions may vary, leading to line broadening. Notably, there is no evidence of coupling of the unpaired electrons from two Cu-centres to form a triplet spin system after assembly.37 No measurable half-field transition was observed, and the line shape is consistent with a doublet spin state. Therefore, each Cu(II) centre can be thought of as an isolated radical, well separated from other metal centres. Finally, the 77 K EPR spectrum of CoCo-cube shows an asymmetric resonance at a g of ca. 2.00 (see Fig. 6). This transition is attributed to a porphyrin-ligated Co(II)-based radical. Due to spin–orbit coupling, the high-spin pseudo-octahedral Co(II) centres of the vertices are not expected to be observable at X-band under the experimental conditions. The observed transition is consistent with a doublet spin state, suggesting that Co(II) centres are electronically isolated from one another, as with the FeCu-cube. The asymmetric shape of the transition is similar to those observed by others for related O2 bound Co(II) complexes.38–43
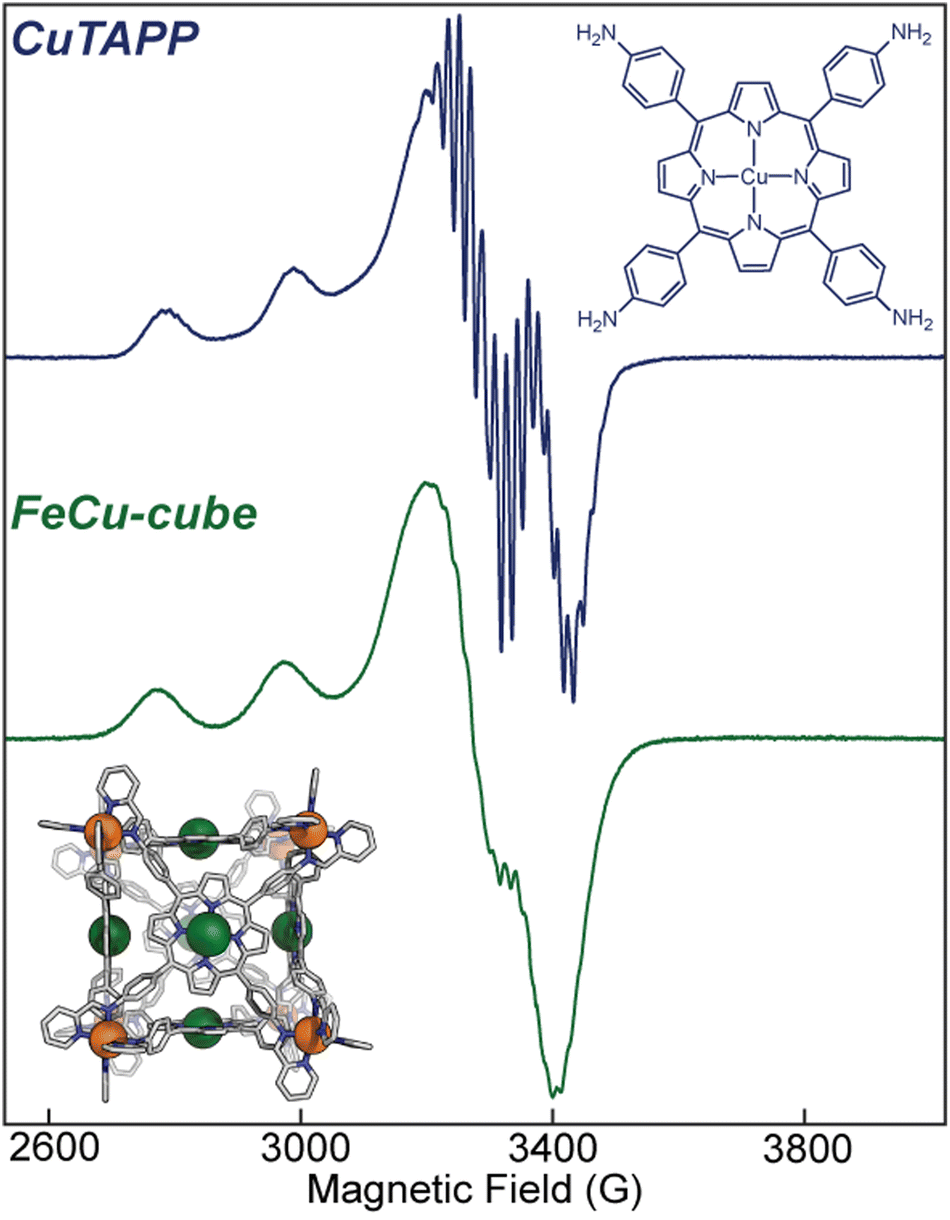 | ||
Fig. 5 (top) X-band EPR of CuTAPP at 9.445 GHz frozen in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 CH3CN:toluene glass at 77 K. (bottom) X-band EPR of FeCu-cube at 9.442 GHz frozen in a 50:50 CH3CN:toluene glass at 77 K. :50 CH3CN:toluene glass at 77 K. (bottom) X-band EPR of FeCu-cube at 9.442 GHz frozen in a 50:50 CH3CN:toluene glass at 77 K. | ||
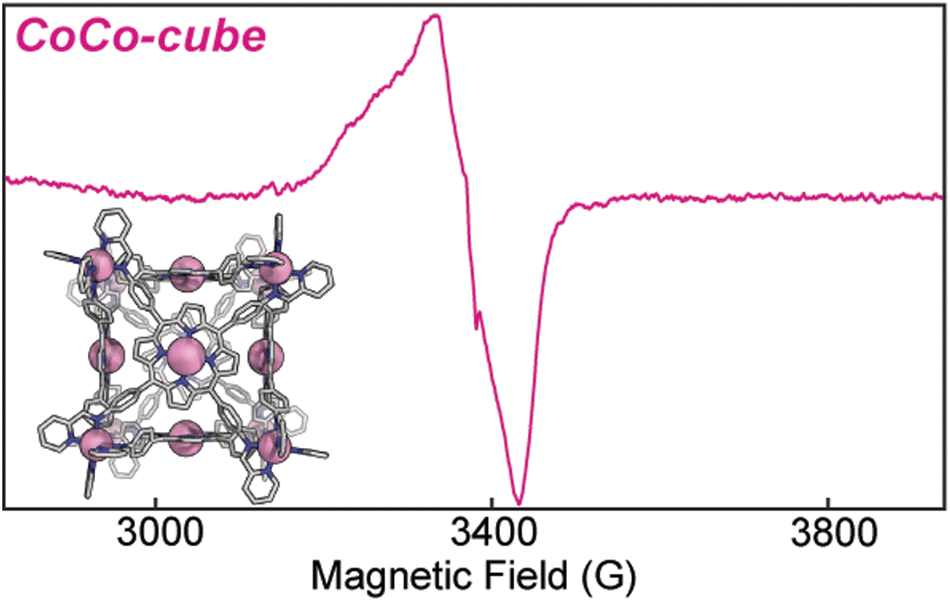 | ||
| Fig. 6 X-band EPR of CoCo-cube at 9.439 GHz frozen in a 50:50 CH3CN:toluene glass at 77 K. | ||
Under inert conditions, the cyclic voltammogram (CV) of CuTAPP possesses two quasi-reversible reduction events at −1.74 V and −2.22 V vs. Fc+/0 (Fig. S8†). Previous literature reports on related Cu(II) porphyrins attribute these to the π-centred reductions of the macrocycle.44 Three oxidation events were also observed at 0.33, 0.49, and 0.88 V vs. Fc+/0. The CVs of both FeCu- and CoCo-cubes suggest they tenaciously form films on the working electrode surface, this was evident by significant changes in the voltammogram from cycle to cycle when reducing potentials were scanned (see Fig. S11 and S15†). Film formation persisted even at high scan rate and lower concentrations of analyte (Fig. S16†). When oxidizing potentials were isolated, little change in current responses were observed during cycling. For both cubes, if reducing potentials were scanned first, large oxidation events were observed that were absent if the electrode was first polarized towards oxidizing potentials (Fig. S9 and S12†). Comparing the current response of FeCu-cube to CoCo-cube, an additional feature found at −0.97 V vs. Fc+/0 was observed (Fig. S12†). This is assigned to the Co(II/I) couple. If isolated, this event is electrochemically quasi-reversible. In previous studies on cofacial porphyrins two sequential Co(II/I) redox were observed; however, in this case there is a singular event, further supporting that each Co-centre is electrochemically isolated.24 To support that observed couple results from Co(II)'s in the faces of the cube UV-Vis spectroelectrochemistry was performed. When the working electrode was held at −1.2 V vs. Fc+/0, two new bands grew in at 364 and 520 nm (Fig. S14†), in excellent agreement with other Co(I) porphyrin species.45
To screen CoCo-cube for catalytic activity, cyclic voltammograms were acquired under catalytically relevant conditions. When the nitrogen atmosphere was replaced with oxygen, no major current response was observed until the formation of superoxide by the glassy carbon working electrode (brown trace in Fig. 7). Upon the addition of a proton source, in this case trifluoroacetic acid (TFA), there was a significant catalytic current response (green trace in Fig. 7). When CV were acquired after sparging with N2 in the presence of TFA, a reductive current response was observed beyond −1.5 V vs. Fc+/0 (Fig. S17†), this is well beyond the onset of catalytic current when sparged with O2. These results support that CoCo-cube is an effective ORR catalyst and the current response is not due to proton reduction.
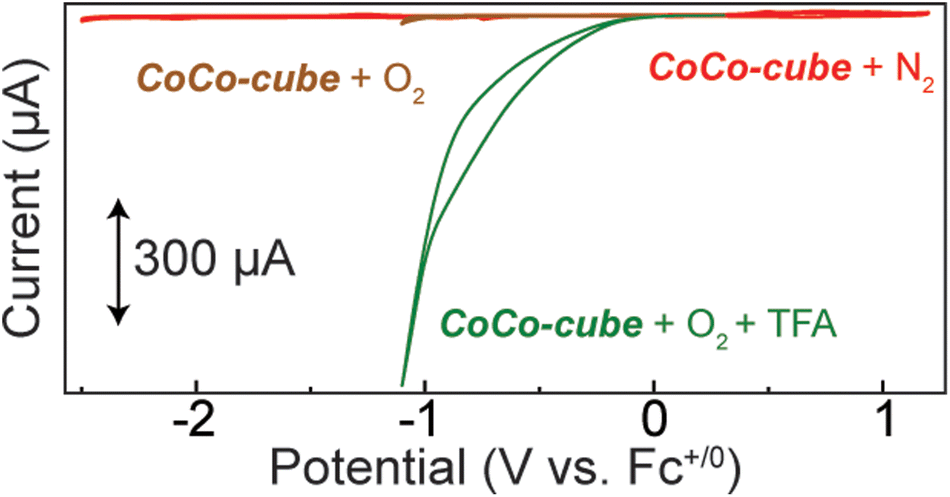 | ||
| Fig. 7 Cyclic voltammogram of CoCo-cube (0.05 mM) in CH3CN with 100 mM TBAPF6 under a nitrogen atmosphere (red), oxygen atmosphere (brown), and oxygen atmosphere with the addition of 100 mM trifluoroacetic acid (green). | ||
Hydrodynamic rotating ring disk voltammetry was then used to determine the selectivity and kinetics of ORR catalysed by CoCo-cube. A catalyst ink was cast onto the surface of a glassy carbon electrode, and immersed in 0.5 M H2SO4, the CV under N2 was featureless, ruling out any contribution from proton reduction in the observed current response (see Fig. S18†). UV-Vis studies were used to confirm the stability of CoCo-cube under the catalytic conditions (Fig. S19†). Under O2, the catalyst film produced a catalytic current response, consistent with homogeneous experiments. Rotating ring disk studies allowed for an assessment of selectivity. Analogous to the heterogeneous CVs, a catalyst film was drop cast on the glassy carbon disk of a RRDE. The film was then swept to reducing potentials while the platinum ring electrode was maintained at 1.0 V, therefore, any oxygen reduced to hydrogen peroxide was then re-oxidized back to oxygen. A substantial current response was measured at the ring electrode, implying considerable peroxide production. Using eqn (S4)† and the ring and disk currents (see Fig. 8), the %H2O2 was found to be 82.7%. This is significantly higher than most monomeric porphyrin complexes, which typically show near 50/50 product distributions.46 Monomeric CoTPP and CoTPyP27 films prepared identically to the films of CoCo-cube showed a selectivity of 63% and 61% respectively towards H2O2 (Fig. S21†). To rule out catalytic activity from the Co(II) vertices, tris(N-phenyl-pyridinaldimine)cobalt(II) hexafluorophosphate was synthesized according to literature protocol.47 No catalytic active was observed (Fig. S20†). Using the hydrodynamic linear sweep voltammograms (LSV, top of Fig. 8) Koutecký–Levich analysis (Fig. S22 and S23†) was used to determine the standard rate constant (ks).24,27,48 The observed standard rate constant was 2.2 ± 0.7 × 102 M−1 s−1. This is a significant enhancement when compared to CoTPyP performed under identical conditions by our lab previously.27 The catalyst ink used to determine ks was normalized in Co concentration to previously reported CoTPyP to allow for a direct comparison at the same effective Co-site concentration. Furthermore, this value assumes that every Co-centre in the film is catalytically active, meaning that the reported rate constant is the lower limit, and the actual kinetics may be even faster. It should be noted that in other supramolecular ORR catalysts there is evidence in the LSV of multiple features that were attributed to alternate, competing pathways. In our RRDE LSV a single, smooth current response was observed, suggesting that these competing pathways are not operative in the self-assembled catalyst.13
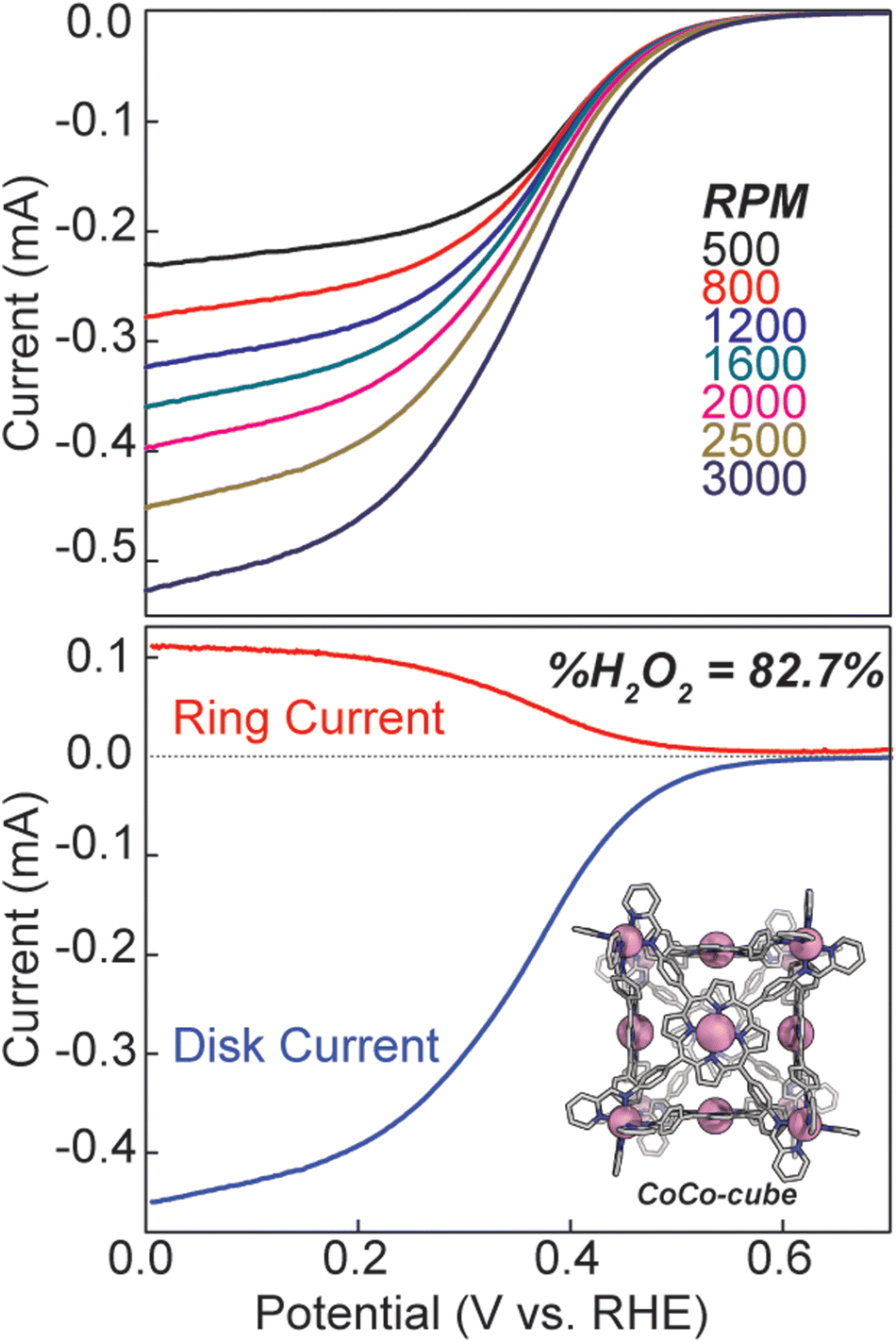 | ||
| Fig. 8 (top) Linear sweep voltammograms (LSV) of CoCo-cube in heterogeneous films on RRDE at differing rotation rates. (bottom) Plot of ring and disk current responses using RRDE at 2500 rpm. Electrolyte: 0.5 M H2SO4, pH = 0.29. | ||
In a catalyst ink, the intermolecular attractive forces such as van der Waal's forces, π–π interactions, and C–H—π interactions, between mononuclear porphyrin rings will promote aggregation, and lead to randomly distributed domains where multiple metal centres may approach one another, while others are isolated. This random distribution gives rise to the mixed product selectivity due to the range metal–metal separation which allows access to both reduction pathways at different domains within a single sample. Through the formation of supramolecular assemblies, greater metal–metal separation is built into the molecular structure of the catalyst. The singular Co(II/I) couple observed in the CV of CoCo-cube, and the doublet spin state observed in the EPR spectrum of FeCu-cube both support this claim. Through assembly, the “mononuclearity” of each porphyrin site is enhanced, and by isolating each metal centre, the more redox demanding 4e−/4H+ pathway is shutdown. These results are consistent with other hydrogen peroxide producing supramolecular catalysts.13 On the other hand, this specific cubic design also shuts down the thermodynamically favoured pathway to water because the metal nodes are not arranged in the correct configuration to promote O–O bond cleavage. A final point which supports these explanations comes from %H2O2 values reported for CoTPP. In a 1999 report by Anson et al. CoTPP was found to produced ca. 100% H2O2 based on the slopes of Koutecký–Levich plots.49 In their report, the electrode was dipped into a CHCl3 solution of porphyrin to form the film. In a more recent report from Chang et al. a catalyst is of CoTPP was reported to produced ca. 50% H2O2 based on RRDE data using eqn (S4)†, and in our hands 63%.13 It is tempting to account for the disparity with the fact that two different methods were used (K–L plot vs. RRDE); however, Anson reports that comparable results were obtained with a RRDE. Therefore, it is likely that the large range in reported H2O2 selectivity is a result of different film morphologies caused by differences in film preparation. The edge plane graphite electrode formed a film which minimized Co–Co interactions, while the drop cast catalyst ink allowed for a greater degree of aggregation, and thus greater H2O formation. This is a cautionary tale of the importance of consistency in film preparation, as well as the difficulties of comparing faradaic efficiencies from one film to another.
Conclusions
A supramolecular ORR catalyst which selectively produces H2O2, CoCo-cube, was synthesized in two steps from commercially available building blocks in overall yields >73%. This report is the first example of a tandem post-synthetic metalation/transmetallation of a self-assembled porphyrin cube. The exchange of the tris-chelated vertex metal cations suggests that in solution, the assembled cubes are dynamic and may undergo at least partial disassembly and reassembly. CoCo- and FeCu-cube were synthesized and fully characterized using a variety of spectroscopic techniques. High resolution mass spectrometry was used to confirm the metalation/transmetallation. EPR studies of FeCu-cube were used to probe metal–metal separation and verify that each metal-centre is spatially, and electronically isolated. Electrochemical studies using both standard CV and hydrodynamic voltammetric studies were used to study ORR catalysed by CoCo-cube. By designing larger molecular architectures, which enforce the isolation of each metal centre, we have formed an electrocatalytic hydrogen peroxide generating catalyst with a selectivity of 83%. This work demonstrates how structural tuning can in turn optimize the selectivity of ORR catalysts between two important pathways, water24–27 and now hydrogen peroxide. This work is an important next step in our efforts to expand our library of polynuclear catalysts to new architectures and reactivities, further establishing coordination-driven self-assembly as an important tool in small molecule activation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSF CAREER Award #1847950 (T. R. C.). Characterization work was performed in part in the Chemistry Instrument Center (CIC), University at Buffalo, SUNY, Buffalo, NY. This work used the 12T Bruker SolariXR 12 Hybrid FTMS purchased with NIH S10 RR029517. Bruker AVANCE NEO 500 MHz NMR purchased with NSF CHE-2018160, a part of the UB Department of Chemistry Magnetic Resonance Center.References
- N. S. Lewis and D. G. Nocera, Powering the planet: chemical challenges in solar energy utilization, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Recent Advances in Electrocatalysts for Oxygen Reduction Reaction, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS.
- M. Melchionna, P. Fornasiero and M. Prato, The Rise of Hydrogen Peroxide as the Main Product by Metal-Free Catalysis in Oxygen Reductions, Adv. Mater., 2019, 31, 1802920 CrossRef PubMed.
- A. T. Murray, S. Voskian, M. Schreier, T. A. Hatton and Y. Surendranath, Electrosynthesis of Hydrogen Peroxide by Phase-Transfer Catalysis, Joule, 2019, 3, 2942–2954 CrossRef CAS.
- V. Subramani, P. Sharma, L. Zhang and K. Liu, in Hydrogen and Syngas Production and Purification Technologies, 2009, pp. 14–126, DOI:10.1002/9780470561256.ch2.
- Y. Wang, G. I. N. Waterhouse, L. Shang and T. Zhang, Electrocatalytic Oxygen Reduction to Hydrogen Peroxide: From Homogeneous to Heterogeneous Electrocatalysis, Adv. Energy Mater., 2021, 11, 2003323 CrossRef CAS.
- T. Geiger and F. C. Anson, Homogeneous catalysis of the electrochemical reduction of dioxygen by a macrocyclic cobalt(III) complex, J. Am. Chem. Soc., 1981, 103, 7489–7496 CrossRef CAS.
- R. J. H. Chan, Y. O. Su and T. Kuwana, Electrocatalysis of oxygen reduction. 5. Oxygen to hydrogen peroxide conversion by cobalt(II) tetrakis(N-methyl-4-pyridyl)porphyrin, Inorg. Chem., 1985, 24, 3777–3784 CrossRef CAS.
- Y.-H. Wang, M. L. Pegis, J. M. Mayer and S. S. Stahl, Molecular Cobalt Catalysts for O2 Reduction: Low-Overpotential Production of H2O2 and Comparison with Iron-Based Catalysts, J. Am. Chem. Soc., 2017, 139, 16458–16461 CrossRef CAS PubMed.
- Q. He, T. Mugadza, G. S. Hwang and T. Nyokong, Mechanisms of electrocatalysis of oxygen reduction by metal porphyrins in trifluoromethane sulfonic acid solution, Int. J. Electrochem. Sci., 2012, 7, 7045–7064 CAS.
- K. Mase, K. Ohkubo and S. Fukuzumi, Efficient Two-Electron Reduction of Dioxygen to Hydrogen Peroxide with One-Electron Reductants with a Small Overpotential Catalyzed by a Cobalt Chlorin Complex, J. Am. Chem. Soc., 2013, 135, 2800–2808 CrossRef CAS PubMed.
- S. Fukuzumi, S. Mochizuki and T. Tanaka, Efficient reduction of dioxygen with ferrocene derivatives, catalyzed by metalloporphyrins in the presence of perchloric acid, Inorg. Chem., 1989, 28, 2459–2465 CrossRef CAS.
- P. T. Smith, Y. Kim, B. P. Benke, K. Kim and C. J. Chang, Supramolecular Tuning Enables Selective Oxygen Reduction Catalyzed by Cobalt Porphyrins for Direct Electrosynthesis of Hydrogen Peroxide, Angew. Chem., Int. Ed., 2020, 59, 4902–4907 CrossRef CAS PubMed.
- S. Fukuzumi, L. Tahsini, Y.-M. Lee, K. Ohkubo, W. Nam and K. D. Karlin, Factors That Control Catalytic Two- versus Four-Electron Reduction of Dioxygen by Copper Complexes, J. Am. Chem. Soc., 2012, 134, 7025–7035 CrossRef CAS.
- T. R. Cook and P. J. Stang, Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- E. G. Percástegui, J. Mosquera, T. K. Ronson, A. J. Plajer, M. Kieffer and J. R. Nitschke, Waterproof architectures through subcomponent self-assembly, Chem. Sci., 2019, 10, 2006–2018 RSC.
- N. W. Wu, L. J. Chen, C. Wang, Y. Y. Ren, X. Li, L. Xu and H. B. Yang, Hierarchical self-assembly of a discrete hexagonal metallacycle into the ordered nanofibers and stimuli-responsive supramolecular gels, Chem. Commun., 2014, 50, 4231–4233 RSC.
- M. Fujita, Metal-directed self-assembly of two- and three-dimensional synthetic receptors, Chem. Soc. Rev., 1998, 27, 417–425 RSC.
- C. J. Chang, C.-Y. Yeh and D. G. Nocera, Porphyrin Architectures Bearing Functionalized Xanthene Spacers, J. Org. Chem., 2002, 67, 1403–1406 CrossRef CAS PubMed.
- J. P. Collman, M. Marrocco, P. Denisevich, C. Koval and F. C. Anson, Potent catalysis of the electroreduction of oxygen to water by dicobalt porphyrin dimers adsorbed on graphite electrodes, J. Electroanal. Chem. Interfacial Electrochem., 1979, 101, 117–122 CrossRef CAS.
- J. P. Collman, C. M. Elliott, T. R. Halbert and B. S. Tovrog, Synthesis and characterization of “face-to-face” porphyrins, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 18–22 CrossRef CAS.
- K. Muramoto, K. Hirata, K. Shinzawa-Itoh, S. Yoko-o, E. Yamashita, H. Aoyama, T. Tsukihara and S. Yoshikawa, A histidine residue acting as a controlling site for dioxygen reduction and proton pumping by cytochrome c oxidase, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7881–7886 CrossRef CAS.
- K. M. Kadish, L. Frémond, J. Shen, P. Chen, K. Ohkubo, S. Fukuzumi, M. El Ojaimi, C. P. Gros, J.-M. Barbe and R. Guilard, Catalytic Activity of Biscobalt Porphyrin-Corrole Dyads Toward the Reduction of Dioxygen, Inorg. Chem., 2009, 48, 2571–2582 CrossRef CAS.
- M. R. Crawley, D. Zhang, A. N. Oldacre, C. M. Beavers, A. E. Friedman and T. R. Cook, Tuning the Reactivity of Cofacial Porphyrin Prisms for Oxygen Reduction Using Modular Building Blocks, J. Am. Chem. Soc., 2021, 143, 1098–1106 CrossRef CAS PubMed.
- A. N. Oldacre, M. R. Crawley, A. E. Friedman and T. R. Cook, Tuning the Activity of Heterogeneous Cofacial Cobalt Porphyrins for Oxygen Reduction Electrocatalysis through Self-Assembly, Chem. – Eur. J., 2018, 24, 10984–10987 CrossRef CAS.
- A. N. Oldacre, A. E. Friedman and T. R. Cook, A Self-Assembled Cofacial Cobalt Porphyrin Prism for Oxygen Reduction Catalysis, J. Am. Chem. Soc., 2017, 139, 1424–1427 CrossRef CAS.
- D. Zhang, M. R. Crawley, A. N. Oldacre, L. J. Kyle, S. N. MacMillan and T. R. Cook, Lowering the Symmetry of Cofacial Porphyrin Prisms for Selective Oxygen Reduction Electrocatalysis, Inorg. Chem., 2022 DOI:10.1021/acs.inorgchem.2c01109.
- P. T. Smith, B. P. Benke, L. An, Y. Kim, K. Kim and C. J. Chang, A Supramolecular Porous Organic Cage Platform Promotes Electrochemical Hydrogen Evolution from Water Catalyzed by Cobalt Porphyrins, ChemElectroChem, 2021, 8, 1653–1657 CrossRef CAS.
- P. T. Smith, B. P. Benke, Z. Cao, Y. Kim, E. M. Nichols, K. Kim and C. J. Chang, Iron Porphyrins Embedded into a Supramolecular Porous Organic Cage for Electrochemical CO2 Reduction in Water, Angew. Chem., Int. Ed., 2018, 57, 9684–9688 CrossRef CAS PubMed.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, A Self-Assembled M8L6 Cubic Cage that Selectively Encapsulates Large Aromatic Guests, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS PubMed.
- P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, An Unlockable–Relockable Iron Cage by Subcomponent Self-Assembly, Angew. Chem., Int. Ed., 2008, 47, 8297–8301 CrossRef CAS PubMed.
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J.-L. Wang, D.-Q. Yuan and Y.-Q. Lan, Rational Design of Crystalline Covalent Organic Frameworks for Efficient CO2 Photoreduction with H2O, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS.
- P. T. Blackburn, I. F. Mansoor, K. G. Dutton, A. M. Tyryshkin and M. C. Lipke, Accessing three oxidation states of cobalt in M6L3 nanoprisms with cobalt-porphyrin walls, Chem. Commun., 2021, 57, 11342–11345 RSC.
- J. Dömer, J. C. Slootweg, F. Hupka, K. Lammertsma and F. E. Hahn, Subcomponent Assembly and Transmetalation of Dinuclear Helicates, Angew. Chem., Int. Ed., 2010, 49, 6430–6433 CrossRef.
- A. M. Johnson, M. C. Young, X. Zhang, R. R. Julian and R. J. Hooley, Cooperative Thermodynamic Control of Selectivity in the Self-Assembly of Rare Earth Metal–Ligand Helices, J. Am. Chem. Soc., 2013, 135, 17723–17726 CrossRef CAS PubMed.
- T. K. Ronson, C. Giri, N. Kodiah Beyeh, A. Minkkinen, F. Topić, J. J. Holstein, K. Rissanen and J. R. Nitschke, Size-Selective Encapsulation of Hydrophobic Guests by Self-Assembled M4L6 Cobalt and Nickel Cages, Chem. – Eur. J., 2013, 19, 3374–3382 CrossRef CAS.
- S. S. Eaton, G. R. Eaton and C. K. Chang, Synthesis and geometry determination of cofacial diporphyrins. EPR spectroscopy of dicopper diporphyrins in frozen solution, J. Am. Chem. Soc., 1985, 107, 3177–3184 CrossRef CAS.
- R. S. Drago and B. B. Corden, Spin-pairing model of dioxygen binding and its application to various transition-metal systems as well as hemoglobin cooperativity, Acc. Chem. Res., 1980, 13, 353–360 CrossRef CAS.
- B. S. Tovrog, D. J. Kitko and R. S. Drago, Nature of the bound oxygen in a series of cobalt dioxygen adducts, J. Am. Chem. Soc., 1976, 98, 5144–5153 CrossRef CAS.
- J. P. Collman, C. S. Bencosme, C. E. Barnes and B. D. Miller, Two new members of the dimeric .beta.-linked face-to-face porphyrin family: FTF4* and FTF3, J. Am. Chem. Soc., 1983, 105, 2704–2710 CrossRef CAS.
- U. Keirei, T. Kiyoshi, S. Yoichi and I. Taira, Synthesis of Box-shaped Pentaporphyrin with Cobalt(II) Porphyrin in the Center; Reversible O2 Binding in the Box, Chem. Lett., 2005, 34, 474–475 CrossRef.
- J. P. Collman, J. I. Brauman, K. M. Doxsee, T. R. Halbert, S. E. Hayes and K. S. Suslick, Oxygen binding to cobalt porphyrins, J. Am. Chem. Soc., 1978, 100, 2761–2766 CrossRef CAS.
- B. B. Wayland, J. V. Minkiewicz and M. E. Abd-Elmageed, Spectroscopic studies for tetraphenylporphyrincobalt(II) complexes of carbon monoxide, nitrogen oxide, molecular oxygen, methylisonitrile, and trimethyl phosphite, and a bonding model for complexes of carbon monoxide, nitrogen oxide, and molecular oxygen with cobalt(II) and iron(II) porphrins, J. Am. Chem. Soc., 1974, 96, 2795–2801 CrossRef CAS.
- Y. Fang, M. O. Senge, E. Van Caemelbecke, K. M. Smith, C. J. Medforth, M. Zhang and K. M. Kadish, Impact of Substituents and Nonplanarity on Nickel and Copper Porphyrin Electrochemistry: First Observation of a CuII/CuIII Reaction in Nonaqueous Media, Inorg. Chem., 2014, 53, 10772–10778 CrossRef CAS.
- P. T. Blackburn, I. F. Mansoor, K. G. Dutton, A. M. Tyryshkin and M. C. Lipke, Accessing three oxidation states of cobalt in M6L3 nanoprisms with cobalt–porphyrin walls, Chem. Commun., 2021, 57, 11342–11345 RSC.
- Y. Li, N. Wang, H. Lei, X. Li, H. Zheng, H. Wang, W. Zhang and R. Cao, Bioinspired N4-metallomacrocycles for electrocatalytic oxygen reduction reaction, Coord. Chem. Rev., 2021, 442, 213996 CrossRef CAS.
- I. A. Riddell, M. M. J. Smulders, J. K. Clegg, Y. R. Hristova, B. Breiner, J. D. Thoburn and J. R. Nitschke, Anion-induced reconstitution of a self-assembling system to express a chloride-binding Co10L15 pentagonal prism, Nat. Chem., 2012, 4, 751–756 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2000 Search PubMed.
- E. Song, C. Shi and F. C. Anson, Comparison of the Behavior of Several Cobalt Porphyrins as Electrocatalysts for the Reduction of O2 at Graphite Electrodes, Langmuir, 1998, 14, 4315–4321 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qi02050e |
| This journal is © the Partner Organisations 2023 |