
Pt(IV) complex selectively oxidizes alpha-synuclein methionine as disclosed by NMR†
Bin-Bin
Pan
,
Hui-Zhong
Liu
,
Wei-Han
Meng
and
Xun-Cheng
Su
 *
*
State Key Laboratory of Elemento-organic Chemistry, Tianjin Key Laboratory of Biosensing and Molecular Recognition, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: xunchengsu@nankai.edu.cn
First published on 9th November 2022
Abstract
Alpha-synuclein (αS) is an intrinsically disordered neuronal protein, and the oxidative modification of αS promotes its oligomerization and accumulation in Parkinson's disease. By combining high-resolution NMR spectroscopy and mass spectrometry, we investigated the interaction of a platinum(IV) complex with αS at the residual specific resolution. The data indicate that the Pt(IV) complex was able to oxidize the methionine sidechains in a distinct manner under physiological conditions. The C-terminal M116 and M127 were first oxidized in a mutually exclusive manner followed by the oxidation of M5 with a large excess of Pt(IV), whereas M1 could not be oxidized. The oxidized methionines were unable to bind to Pt(II) and the Pt(II)-coordinated methionine could not be oxidized by H2O2. These results show the distinct oxidation preferences of αS methionines exhibited by the Pt(IV) complex at the residual specific resolution and imply the presence of long-range spatial contacts for the segments containing methionines in this intrinsically disordered protein.
Introduction
Oxidative stress has been implicated in various neurodegenerative diseases including Parkinson's disease (PD).1–3 αS is an intrinsically disordered protein.4 The oxidative modification of αS affects protein aggregation5,6 and has great impact on the physiological functions of αS including membrane binding,7 vesicular trafficking, and vesicle fusion.8 Recent findings suggest that αS is also a modulator of mRNA stability in gene expression.9 The four methionine residues of αS (M1, M5, M116, and M127) can be oxidized, in vivo and in vitro.6 Methionine oxidation is the most common oxidative modification of αS, which is especially mediated by dopamine or its oxidation products and H2O2.10,11 The oxidative modification of methionine leads αS to aggregate into soluble oligomers rather than fibrils and display different degrees of cytotoxicity.12αS binds many transition metal ions, which generally promote αS aggregation.13–15 The Cu(I)-coordinated M1 and M5 are rapidly oxidized under air-exposure conditions.16,17 The N-terminally acetylated αS-bound Cu(I) activates O2, resulting in an intermolecular Y39–Y39 cross-linking within the fibrillar core and intramolecular cross-linking.18,19 Similarly, dityrosine (diTyr) cross-linked αS was produced in the presence of Cu(II) and H2O2, preventing αS aggregation.20,21
The kinetic properties of platinum complexes differ greatly from those of Fe(III) and Cu(II)/Cu(I) complexes, which are generally kinetically labile. In contrast to most transition metal ions, cisplatin interacts with αS by coordinating the side-chains of methionine and histidine in the formation of an intramolecular complex.22 The formation of this kinetically inert complex αS-Pt(II) maintains an overall random coil structure and prevents αS fibrillation, which differs significantly from other metal ions that accelerate fibril formation. Pt(IV) complexes, the next generation of platinum drugs, have been developed to address the issues of classical Pt(II) drugs including the onset of severe side effects, intrinsic resistance and general toxicity.23–28 Pt(IV) complexes are thought to be activated by intracellular sulfur-containing small molecules, ascorbic acid (VC), and cysteine-containing proteins,24,29–32 while methionine and cysteine,33–35 dGMP,36 serum albumin,37 and flavoproteins38 have been reported to reduce platinum(IV) complexes. Recent studies have showed that some platinum(IV) complexes are resistant to glutathione (GSH) but are rapidly reduced in cancer cells or cell lysates, and high molecular weight species (MW > 3000 Dalton) were therefore proposed for activating Pt(IV).39–44 Satraplatin is reduced to a reactive Pt(II) complex (JM118) by cytochrome c and liver microsomes with the help of NADH.45,46 Although DNA binding is the main reason for the antitumor activity of platinum, only 1–5% of platinum binds to DNA. In contrast, most platinum complexes are coordinated by thiols and methionines to the intracellular components of the cytoplasm.47,48 However, most of these interactions between Pt(IV) and proteins are poorly understood due to the lack of high-resolution details.
Following the recent discovery via high-resolution NMR of cisplatin coordinating to αS in the formation of a stable complex and thus inhibiting αS fibrillation,22 we aimed to understand how Pt(IV) interacts with αS under physiological conditions. To gain a better understanding of the interaction between αS and Pt(IV) compounds, we selected cis,cis,trans-[Pt(NH3)2Cl2Br2], referred to as Pt(IV) hereafter, which is active for human ovarian cells.49 We used the Pt(IV) complex to investigate the interaction with αS in vitro mainly by means of high-resolution NMR and mass spectrometry. The results indicated that the Pt(IV) complex is able to oxidize the methionine side-chains of αS, but in a distinct manner. M116 and M127 can be readily oxidized by the Pt(IV) complex, but in a mutually exclusive manner, whereas M5 can only be oxidized with a large excess of Pt(IV) while M1 remains unchanged under these conditions. Partially oxidized αS is able to coordinate to the reduced Pt(II) complex with the remaining unoxidized methionines as well as H50. Interestingly, the Pt(II)-coordinated methionines are difficult to oxidize using H2O2 and the oxidized methionines are neither able to coordinate to Pt(II) nor interact with the Pt(IV) complex.
Results and discussion
αS is oxidized by cis,cis,trans-[Pt(NH3)2Cl2Br2]
The interaction of αS with Pt(IV) was first characterized for a reaction mixture of αS and one equivalent of Pt(IV) at 25 °C by ESI-Q-TOF spectrometry. In addition to the peak of free αS, three major peaks corresponding to the mass of free αS plus 16.6 Dalton, free αS plus 193.2 Dalton, and free αS plus the sum of 16.2 and 193.2 Dalton were observed in the mass spectrum (Fig. 2A–B and Fig. S3†), in which 16 Dalton is the mass of one oxygen and 193.2 Dalton is the molecular mass of one platinum. The result suggested that Pt(IV) does have an impact on the molecular mass of αS, which is probably due to the oxidation and metal binding. These three additional peaks increased with the incubation time, whereas that of free αS decreased concomitantly (Fig. S3†), suggesting that the oxidation process proceeds slowly. Finally, 33% of free αS was oxidized and 63% of αS was converted into one platinum-bound state after the incubation of one equivalent of Pt(IV) over 100 h (Fig. S3 and S4†) in 20 mM 2-(N-morpholino) ethanesulfonic acid (MES) buffer at pH 6.4. In contrast, the simultaneous incubation of αS with six equivalents of VC and two equivalents of Pt(IV) resulted in no αS oxidation but in a Pt-bound αS complex as determined by mass spectrometry (Fig. S5†). These results could be attributed to the faster reduction rate of Pt(IV) to Pt(II) by VC than the oxidation of αS by the Pt(IV) complex. | ||
| Fig. 1 (A) The primary structure of αS is composed of three regions: the N-terminus, NAC region, and C-terminus are highlighted with white, gray, and orange backgrounds, respectively. (B) The chemical structure of cisplatin and cis,cis,trans-[Pt(NH3)2Cl2Br2] (Pt(IV)). | ||
 | ||
| Fig. 2 Pt(IV) is able to oxidize αS. (A) Mass spectrum recorded for the sample of 0.1 mM αS. (B) Mass spectrum recorded for the solution of 0.1 mM αS after incubation with one equivalent of Pt(IV) at 25 °C for 24 h; the major species are labeled with their molecular mass. The labels in parentheses represent the corresponding atoms added to αS. (C) An overlay of 2D 1H–15N NMR spectra recorded for the sample of 0.1 mM αS before (black) and after incubation with 0.1 mM Pt(IV) at 25 °C for 28 h (red). (D) The plot of the cross-peak intensity ratio between αS and the mixture of αS and Pt(IV) with the function of amino acid sequence, of which the 1H–15N NMR spectra were recorded for 0.1 mM αS incubation with 0.1 mM Pt(IV) at varied times. The segments containing residues 1–21, 39–60, and 115–136 significantly attenuated are indicated in dark gray. I0 and I are the cross-peak intensities recorded in the absence and presence of Pt(IV), respectively. (E) An overlay of 1H–15N HSQC spectra recorded for the sample of 0.15 mM 15N-methionine-labeled αS (black) and after being incubated with 0.15 mM Pt(IV) at 298 K for 17 h (red) in 20 mM MES at pH 6.4. The cross-peaks produced due to the amino scrambling in the protein biosynthesis are marked with aa. (F) Mass spectra recorded for the sample of the αS M1G/M5I/M116I/M127I (4M) mutant (black) and αS 4M after incubation with one equivalent of Pt(IV) for 48 h (red). | ||
To acquire the residual specific information of the interaction between αS and the Pt(IV) complex, high resolution NMR spectroscopy was used to map the oxidation sites in αS. 15N-enriched αS was overexpressed in E. coli and purified using an FPLC column. The interaction of 15N-αS upon treatment with one equivalent of Pt(IV) complex was monitored by 1H–15N HSQC spectroscopy with the incubation time varying from 0 to 50 h. As shown in Fig. 2C and D, the structural segments containing residues 1–20, 39–62, and 115–136 experienced significant line-broadening effects after incubation with Pt(IV). The cross-peak attenuation pattern as shown in Fig. 2D is rather similar to that of the interaction between cisplatin and αS,22 in which only line-broadening was determined for the residues containing M1, M5, M116, M127 and H50 and no new cross-peaks were observed. These data suggest that the oxidation of αS by Pt(IV) is accompanied by the generation of Pt(II) and the consequent coordination to αS. The formed complex exhibits the conformation exchange at a slow to intermediate time-scale in the NMR spectrum.
αS has four methionine residues (M1, M5, M116 and M127) and four tyrosine residues, and the former are more susceptible to oxidation under mild oxidation conditions. To evaluate the role of methionine residues in the interaction between αS and Pt(IV), we selectively used 15N-Met-enriched αS to simplify the 15N-HSQC spectrum and resolve the overlapped signals. The amide signals of M5, M116 and M127 were well-resolved and could be assigned unambiguously50 (Fig. 2E and Fig. S2A†). It was noted that a small portion of acetylated αS was produced during the overexpression in E. coli,51 which was marked as Ac-M1 in the 15N-HSQC spectra (Fig. 2E and Fig. S2A†). Incubation of αS with one equivalent of Pt(IV) for about one hour produced a few new cross-peaks in the 15N-HSQC spectrum (Fig. S6†). The four αS methionines were fully oxidized to sulfoxides by 4% H2O2,5,50 corresponding to an increase of 63.8 Dalton in the molecular mass, which was verified by NMR and ESI-Q-TOF (Fig. S2†). Comparison of the 15N-HSQC spectra of αS oxidized by H2O2 and the mixture of αS with Pt(IV), revealed two new well-resolved cross-peaks with chemical shifts (8.28, 120.40; 8.29, 120.69 ppm) and (8.45, 122.26; 8.44, 122.52 ppm), which were assigned to the sulfoxides of the C-terminal M116 and M127, respectively. The results are consistent with the reported data that both oxidized methionines have two diastereoisomers, MetO(R) and MetO(S).50 However, another new cross-peak (labeled as M5*) with a chemical shift (8.15, 122.37 ppm) as shown in Fig. 2E is absent in the methionines fully oxidized by H2O2 (Fig. S2A†), and this does not match those of any fully oxidized αS methionine residues. It is very likely that the new cross-peak labeled M5* is due to the oxidation of M116 or M127 or both M116 and M127 in the C-terminal region, which changes the chemical shift of M5 because of long-range contacts between these residues. The intramolecular coordination of αS methionines with Pt(II) suggests that a state containing these spatially close residues exists in the flexible polypeptide chain,22 since the transient contacts between the N- and C-domains are present in αS.52–54
To confirm that the interaction of Pt(IV) with αS is generally dominated by methionine, we constructed a mutant αS by mutating all methionines, M1G/M5I/M116I/M127I (4M), and analyzed its interaction with Pt(IV). Treatment of the 4M mutant with one equivalent of Pt(IV) for 48 h produced no changes in the molecular mass of αS (Fig. 2F). This result further indicates that the methionine was oxidized but not tyrosine. Based on the previous report that Pt(IV) prodrugs are generally converted into Pt(II) analogs by reduction24–28,39 and from the residual-specific profile of αS as shown in Fig. 2D, we conclude that the interaction of αS with Pt(IV) is mainly due to the oxidation of methionine sidechains by Pt(IV), which is reduced to Pt(II). Similar to cisplatin, the reduced metal product, Pt(II), is able to coordinate with the remaining free methionines as well as H50.
Pt(IV) preferentially oxidizes the C-terminal methionines of αS
To unambiguously distinguish the methionine oxidation preferences in αS by Pt(IV), we used M116I/M127I and M1G/M5I mutants to monitor the oxidation reaction with one equivalent of Pt(IV) by mass spectrometry (Fig. 3A–B and Fig. S7–S8†). As for the M1G/M5I mutant, the ESI-Q-TOF result showed that three peaks with molecular mass increments of 16.1, 193.0, and 209.8 Dalton (Fig. S7†), respectively, were present in addition to that of the free protein in the reaction mixture. In contrast, no molecular mass changes were determined for the mixture of M116I/M127I mutants with the Pt(IV) complex even after incubation for over 20 h (Fig. S8†). 15N-enriched single point mutant M1G was constructed and its interaction with Pt(IV) was characterized similar to the wild type αS (Fig. S9†). In the M1G mutant, M116 and M127 were oxidized by mixing them with one equivalent of Pt(IV). In addition, the species M5* (8.12, 122.57 ppm) was determined, which has slight chemical shift differences from those of the αS product. Taken together, it is conclusive that M116 and M127 are preferably oxidized by one equivalent of Pt(IV), whereas M1 and M5 are resistant to oxidation under such conditions (Fig. 3D).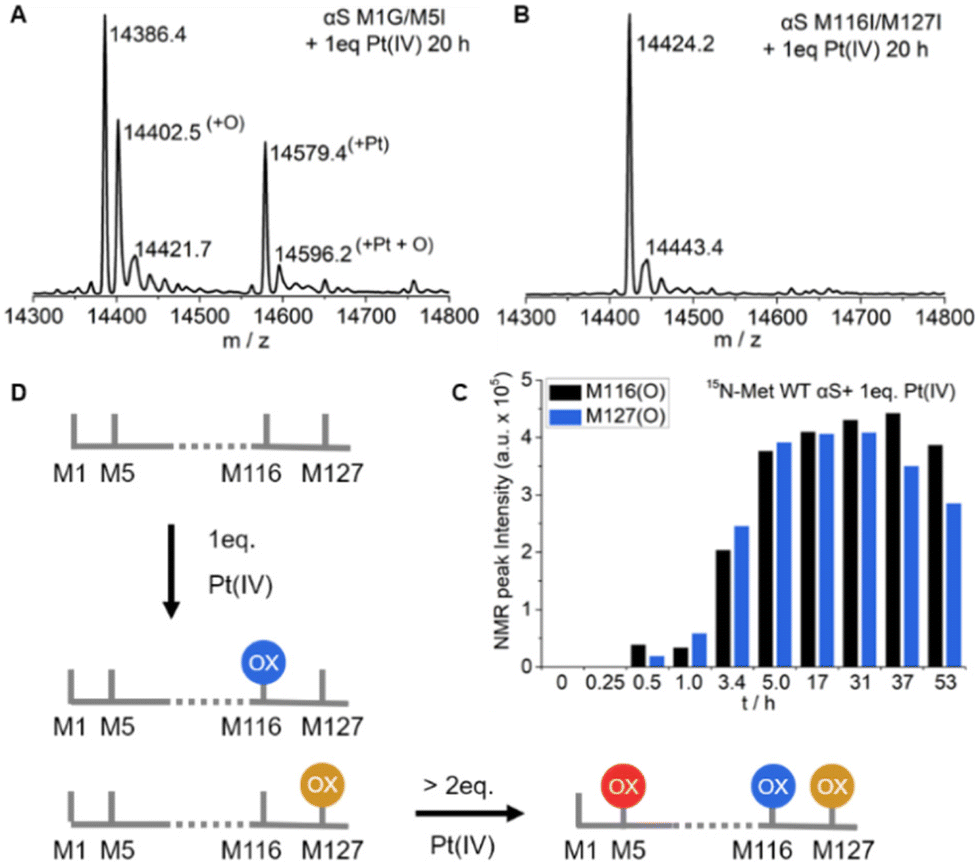 | ||
| Fig. 3 Pt(IV) preferentially oxidizes the C-terminal methionines of αS. (A) The reaction mixture of 0.1 mM αS M1G/M5I mutant incubated with 0.1 mM Pt(IV) for 20 h was characterized by ESI-Q-TOF spectrometry. (B) The ESI-Q-TOF spectrum of 0.1 mM αS M116I/M127I mutant incubated with 0.1 mM Pt(IV) for 20 h. (C) Real-time NMR profiles of the cross-peak intensities of methionine sulfoxide species, M116(O) and M127(O), produced by αS and one equivalent of Pt(IV), with increasing incubation time. (D) Schematic representation of Pt(IV) oxidizing αS methionine in a distinct manner. Ox denotes methionine sulfoxide. | ||
Pt(IV) oxidizes C-terminal methionines of αS in a mutually exclusive manner
In the reaction mixture recorded for the solution of αS with one equivalent of Pt(IV), the fractions of M116(O) and M127(O) signals were approximately in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry in the 1H,15N-HSQC NMR spectrum (Fig. 3C and Fig. S6†). A slight decrease in the intensity of M116(O) and M127(O) was observed after incubation for over 31 h, which was perhaps due to the formation of oligomer species.5 However, only one peak with an additional mass of 16 Dalton was observed and no peak with an additional mass of 32 Dalton was found in the ESI-Q-TOF spectra (Fig. 1B and Fig. S3–S4†). Similar results were determined for the M1G/M5I mutant treated by one equivalent of Pt(IV) (Fig. 3A and Fig. S7†). An increased mass of 32 Dalton was produced for the mixture of αS with two equivalents of Pt(IV), but the fraction corresponding to the mass of one oxidized methionine is dominant over that of two oxidized methionines (Fig. S4B, S12 and S13†), in which only traces of M5 were oxidized when incubated under such conditions (Fig. S18†). Overall, these data indicated that the C-terminal M116 and M127 were selectively oxidized but in a mutually exclusive manner upon interaction with one equivalent of Pt(IV), that is, either M116 or M127 is oxidized for each αS, but the two are not simultaneously oxidized.
:1 stoichiometry in the 1H,15N-HSQC NMR spectrum (Fig. 3C and Fig. S6†). A slight decrease in the intensity of M116(O) and M127(O) was observed after incubation for over 31 h, which was perhaps due to the formation of oligomer species.5 However, only one peak with an additional mass of 16 Dalton was observed and no peak with an additional mass of 32 Dalton was found in the ESI-Q-TOF spectra (Fig. 1B and Fig. S3–S4†). Similar results were determined for the M1G/M5I mutant treated by one equivalent of Pt(IV) (Fig. 3A and Fig. S7†). An increased mass of 32 Dalton was produced for the mixture of αS with two equivalents of Pt(IV), but the fraction corresponding to the mass of one oxidized methionine is dominant over that of two oxidized methionines (Fig. S4B, S12 and S13†), in which only traces of M5 were oxidized when incubated under such conditions (Fig. S18†). Overall, these data indicated that the C-terminal M116 and M127 were selectively oxidized but in a mutually exclusive manner upon interaction with one equivalent of Pt(IV), that is, either M116 or M127 is oxidized for each αS, but the two are not simultaneously oxidized.
Stoichiometric oxidation of αS by Pt(IV) and H2O2 reveals long-range contacts between methionines
The four methionines of αS can be fully oxidized to sulfoxides by 300 mM H2O2.54 Our results demonstrate that one equivalent of Pt(IV) only oxidizes the C-terminal methionine M116 or M127. To address whether higher concentrations of Pt(IV) could oxidize all the methionines, we next proceeded to evaluate the impact of the stoichiometry between Pt(IV) and αS on the oxidation products. The reaction of αS with 2–20 fold Pt(IV) at 25 °C in 20 mM MES buffer at pH 6.5 was characterized by NMR and mass spectrometry (Fig. 4 and Fig. S12–S20†). NMR and MS showed a significant increase in the oxidation fractions, and a significant amount of oxidized M5 was observed in the NMR spectrum. However, 8 or 20 equivalents of Pt(IV) resulted in only a molecular mass increase of 48.1 Dalton and no mass increment of 64 Dalton was observed (Fig. 4A–B, and Fig. S16–S17†). In addition, no mass increment was observed for the M5I/M116I/M127I mutant treated with one to two equivalents of Pt(IV) (Fig. S10–S11, Table S1†). The results suggested that M5, M116, and M127 but not M1 in αS can be oxidized by an excess of Pt(IV). The species with the molecular mass of the Pt(II) coordinated protein was detected in the ESI-Q-TOF (Fig. 4A and B). On combining the NMR data (Fig. 4C–D and S19–S20†), we concluded that M1 cannot be oxidized by a large excess of Pt(IV) and it rather coordinates to Pt(II) together with H50 and other acidic amino acids such as Asp and Glu in such processes.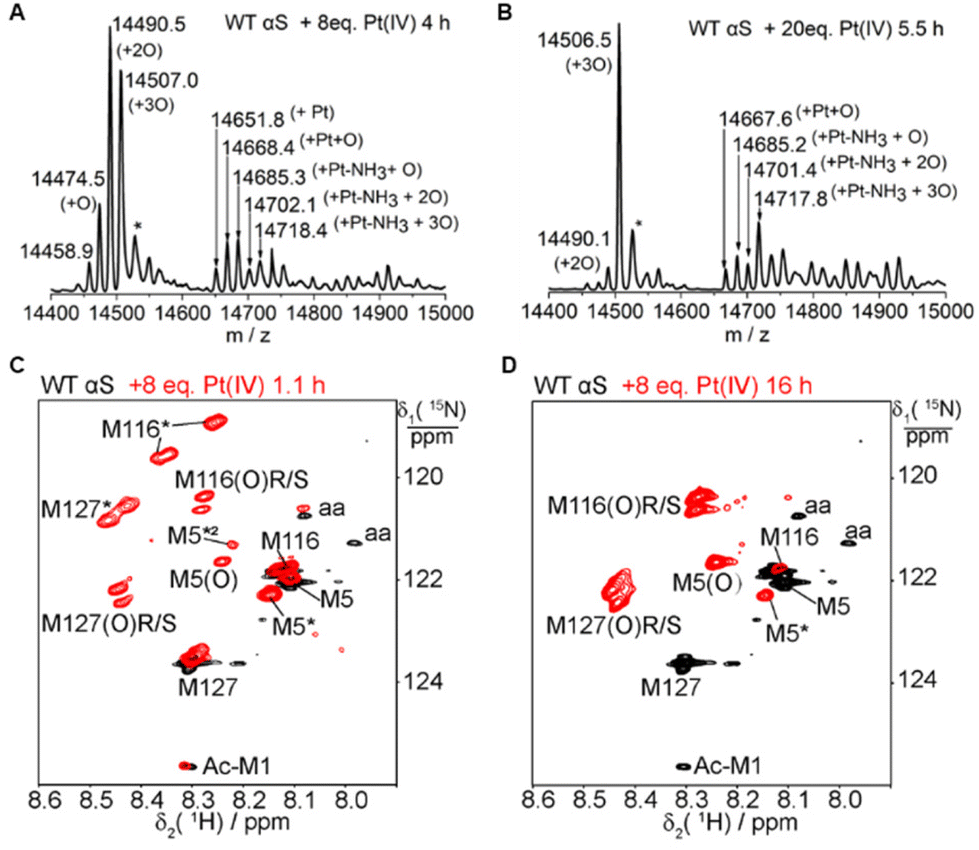 | ||
| Fig. 4 The effect of stoichiometry on αS oxidation by Pt(IV) assessed by mass spectrometry and 2D NMR. (A) and (B) The reaction mixture of 0.1 mM αS after incubation with 8- and 20-fold amounts of Pt(IV) for 4–6 h was characterized by ESI-Q-TOF spectrometry. The peak value plus one Na+ ion is marked with an asterisk (*). (C) and (D) Overlay of 1H–15N HSQC spectra recorded for the sample of 0.15 mM 15N-methionine-labeled αS (black) and 0.15 mM 15N-methionine-labeled αS incubated with 8-fold amounts of Pt(IV) for 1.1 h and 16 h at 298 K (red), respectively. The Ac-M1 denotes the N-acetylated M1, and the aa represents cross-peaks produced by amino scrambling from 15N Met in the protein biosynthesis. | ||
A notable observation is that two new cross-peaks labeled as M116* and M127* in the NMR spectra are produced close to those of M116(O) and M127(O) upon the addition of four- or eight-fold amounts of Pt(IV) (Fig. 4C). These two new species increase transitorily with time and then decay gradually (Fig. S19 and S20†). Similar observations were also produced for the mixture of αS incubated with two equivalents of Pt(IV), but the two species were at much lower intensities. In general, during the oxidation process of αS by Pt(IV), the cross-peak intensities of M5*(8.15 ppm, 122.37 ppm) and M5*2 (8.22 ppm, 121.30 ppm) first increased and then vanished with time, whereas that of M5(O) increased gradually during the entire reaction (Fig. 4C–D and Fig. S18–S20†). These observations of the intermediate species determined from the NMR spectra during the oxidation process indicate the presence of transient long-range contacts for the structural segments containing methionines in the intrinsically disordered protein.
For a better comparison, we set out to understand whether H2O2 oxidation can produce similar observations as seen for Pt(IV). We characterized oxidation products from incubating 0.3 mM αS with H2O2 in 20 mM MES buffer at pH 6.4 and 298K (Fig. 5 and Fig. S21–S22†). 1H–15N HSQC results showed that only 6 mM H2O2 oxidizes the C-terminal M116 and M127. In addition, two resonances labeled as M5* and M5*2 close to the cross-peaks of M5 and M116 were produced (Fig. 5A). The NMR signals of the C-terminal M116(O) and M127(O) increased with the H2O2 concentration, whereas the cross-peaks of M5* and M5*2 increased with the H2O2 concentration to a certain degree and then decayed gradually. Meanwhile, the oxidation state of M5(O) increases with the H2O2 concentration (Fig. 5B).
 | ||
| Fig. 5 Residual specific assay of αS oxidation by H2O2. (A) An overlay of 1H–15N HSQC spectra recorded for the sample of 0.3 mM 15N-methionine-enriched αS (black) and 0.3 mM 15N-methionine-enriched αS incubated with 6 mM and 160 mM (red) for 0.3 h, respectively. (B) The plot of the cross-peak peak intensity as a function of H2O2 concentration, in which M(O) represents the oxidation product and M5*(1 + 2) is the sum of intermediate species (M5* and M5*2) due to the partial oxidation of αS. | ||
The oxidation reaction of αS with H2O2 is significantly faster than that with Pt(IV) but the oxidation preferences are similar, with M116 and M127 being preferably first oxidized rather than the N-terminal M1 and M5. The intermediate species marked as M5* and M5*2 in the partial oxidation state of αS suggest the difference in the chemical environment for the residues containing N-terminal residues between the free αS and partially oxidized C-terminal M116 or M127. The difference is that Pt(IV) oxidation is accompanied by the coordination of Pt(II) binding to αS, resulting in cross-peak attenuations for the residues close to the metal binding sites as demonstrated by high-resolution NMR spectroscopy (Fig. 2).
It is noted that the αS oxidation preference exhibited by Pt(IV) and H2O2 in this work differs greatly from the previous mass spectrometry report that showed M5 is more prone to be oxidized by H2O2.55 We argue that the inconsistency might be due to the different concentrations of H2O2, since concentration-dependent oxidation was not carried out in the previous study as it was in the work presented herein (Fig. 5 and Fig. S21–S22†). In addition, high-resolution NMR-powered isotope labeling shows a more detailed and residual specific level of protein change upon undergoing the oxidation process.
The methionine sulfoxide is unable to coordinate with the Pt(II) complex
To obtain a more detailed description of Pt(IV) or Pt(II) interactions with αS, we have characterized the reaction between H2O2 oxidized αS (αS_Met(O)) with Pt(IV) or cisplatin by means of mass spectrometry and NMR. It is notable that no changes in the cross-peak intensity in NMR spectra were determined. Similarly, no molecular mass changes of αS_Met(O) were observed in the mass spectra (Fig. 6A and B). These data clearly demonstrated that oxidized methionine neither interacts with Pt(IV) nor binds with Pt(II).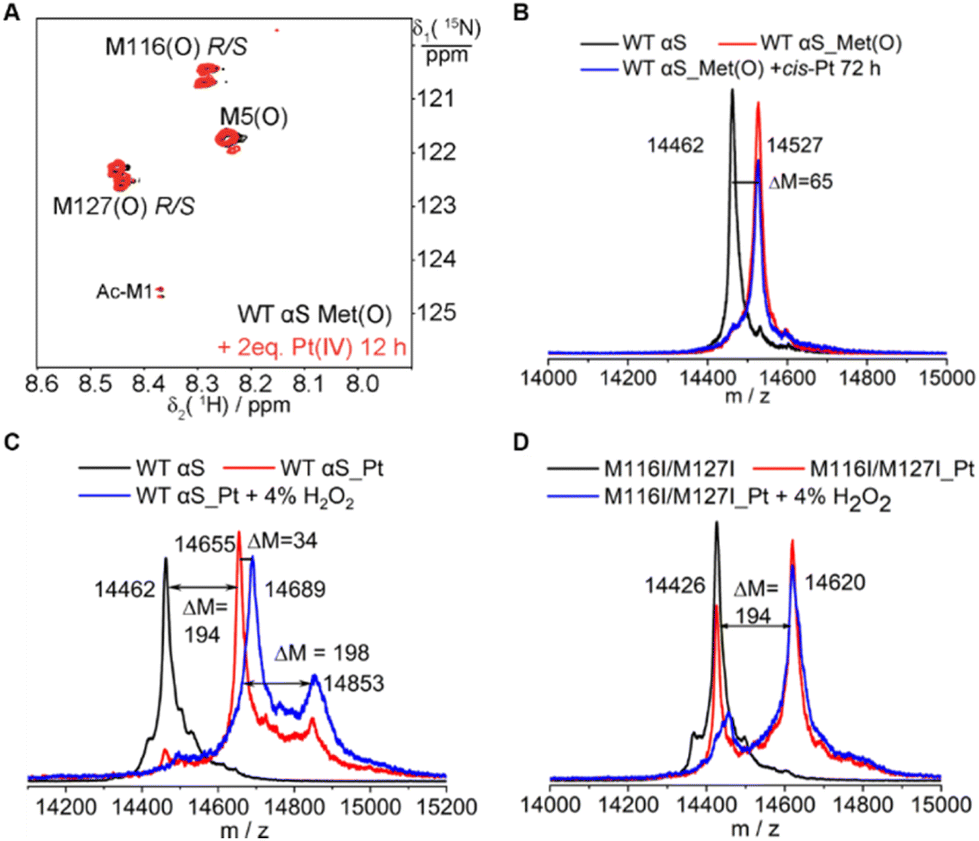 | ||
| Fig. 6 αS methionine sulfoxide is unable to coordinate with the Pt complex and the coordinated methionine is difficult to oxidize. (A) 1H–15N HSQC spectra recorded for the sample of 0.1 mM H2O2-oxidized 15N-methionine-enriched αS, αS_Met(O) (black) and after incubation with 0.2 mM Pt(IV) for 12 h (red). (B) MALDI-TOF mass spectra of αS (black), αS_Met(O) (red) and αS_Met(O) incubated with one equivalent of cisplatin for 72 h (blue). (C) MALDI-TOF mass spectra recorded for the sample of αS (black), αS_Pt complex (red) and αS_Pt treated with 4% H2O2 (blue). (D) MALDI-TOF mass spectra of the αS M116I/M127I mutant (black), αS M116I/M127I_Pt complex (red) and αS M116I/M127I_Pt treated with 4% H2O2 (blue). | ||
Interestingly, it is difficult to oxidize the cisplatin-bound αS. Incubation of cisplatin Pt-bound αS (αS-Pt) with 4% H2O2 for 0.5 h produced a molecular mass increase of 34 Dalton (Fig. 6C), suggesting that two methionines were oxidized. In the Pt-bound αS, the fractions of M116- and M127-coordinated Pt are lower than M1 and M5 in addition to H50,22 and it was proposed that M116 and M127 are the least spatial residues in coordination with cisplatin. As expected, the M116/M127 mutant-bound cisplatin remained inert after treatment with H2O2 (Fig. 6D). Hence, the Pt(II)-coordinated methionine prevents oxidation by H2O2.
Conclusions
In this work, we characterized the interaction of Pt(IV) with αS by a combination of high-resolution NMR, mass spectrometry (Table S1†), and isotope labeling. The oxidation preferences of Pt(IV) and H2O2 show a similar trend for αS methionines. The results indicated that Pt(IV) oxidizes the αS methionines but shows large deviations in the stoichiometry and oxidation preference. M116 and M127 are preferably oxidized by Pt(IV), but this proceeds in a mutually exclusive manner, and while M5 can be oxidized by an excess of Pt(IV), M1 remains unchanged in the oxidation process of Pt(IV) (Fig. 4). This is because oxidation of αS methionines by Pt(IV) generates the Pt(II) species that is readily coordinated to M1 and H50.22 The M1 coordinated with Pt(II) both by sulfur and N-terminal amino groups is difficult to oxidize with Pt(IV). In addition, the oxidation process of αS discloses the presence of long-range contacts between the structural segment containing M5 and that containing either M116 or M127 or both M116 and M127 in this intrinsically disordered protein.52–54 Finally, we measured the inhibitory efficiency of Pt(IV) for αS fibrillation using a thioflavin T (ThT) fluorescence assay (Fig. S23†), which suggests that the Pt(IV) prevents fibrillation aggregation. The impact of the local environment effect on the redox potential might be essentially disclosed by a similar oxidation trend of Pt(IV) and H2O2 in αS. The above results are different from the previous reports on methionine oxidation in αS. In particular, M5 is more prone to oxidation by H2O2;55 Cu(II)/Cu(I) and the peroxidized lipids selectively oxidize the N-terminal M1 and M5.16,17,56 M127 in the 125YEMPS129 motif showed a relatively slow kinetics for oxidative modification in a recent report.57 Our results show that the C-terminus M116 and M127 are more susceptible to oxidation in agreement with previous reports that M116 and M127 were partially oxidized to the sulfoxide in the αS isolated from oxidatively stressed neuron PC12 cells.58 It is indeed important to compare the oxidation potential of Pt(IV)/Pt(II) and the reduction potential of methionines to gain insight into oxidation preferences for the interaction with proteins; however, the local effect and transient association on the redox potential might vary greatly between proteins59 as well as the different positions of residues in in a protein.60To summarize, αS can be oxidized by Pt(IV) under physiological conditions and the methionines in αS show varied oxidation preferences. The C-terminal M116 and M127 were firstly oxidized in a mutually exclusive manner, followed by the oxidation of M5, however, M1 cannot be oxidized by Pt(IV). The oxidation process is accompanied by a resultant Pt(II) coordination to the protein. αS is also highly expressed in several non-neuronal tissues, including blood and kidneys,9,61 and cisplatin and oxaliplatin are generally neurotoxic and would accumulate in the brain after chronic treatment.62 Increasing evidence indicates that the methionine residue of proteins is an important antioxidant;63 how Pt(IV) interacts with αS in cells remains unclear. It is of importance to understand the intracellular interactions between Pt(IV)/Pt(II) complexes with target proteins at an atomic resolution in live cells.
Experimental
Detailed experimental procedures and supplementary figures can be found in the ESI.† The ESI† contains details of the protein expression and purification procedure; cis,cis,trans-[Pt(NH3)2Cl2Br2] synthesis; ESI-Q-TOF measurements; NMR measurements; MALDI-TOF measurements; the fluorescence assay; and supplementary figures.Author contributions
X. C. Su conceived the project; B. B. Pan, H. Z. Liu and W. H. Meng performed the experiments; H. Z. Liu synthesized the Pt(IV) complex; B. B. Pan and W. H. Meng analyzed the data; B. B. Pan and X. C. Su wrote the paper. All authors made contributions to the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the National Natural Science Foundation of China (22161142018, 22174074 and 21991081).References
- B. I. Giasson, J. E. Duda, I. V. Murray, Q. Chen, J. M. Souza, H. I. Hurtig, H. Ischiropoulos, J. Q. Trojanowski and V. M. Lee, Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions, Science, 2000, 290, 985–989 CrossRef CAS.
- K. A. Conway, J.-C. Rochet, R. M. Bieganski and P. T. Lansbury Jr., Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct, Science, 2001, 294, 1346–1349 CrossRef CAS.
- K. J. Barnham, C. L. Masters and A. I. Bush, Neurodegenerative diseases and oxidative stress, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS.
- F.-X. Theillet, A. Binolfi, T. Frembgen-Kesner, K. Hingorani, M. Sarkar, C. Kyne, C. Li, P. B. Crowley, L. Gierasch and G. J. Pielak, Physicochemical properties of cells and their effects on intrinsically disordered proteins (IDPs), Chem. Rev., 2014, 114, 6661–6714 CrossRef CAS PubMed.
- C. B. Glaser, G. Yamin, V. N. Uversky and A. L. Fink, Methionine oxidation, α-synuclein and Parkinson's disease, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1703, 157–169 CrossRef CAS PubMed.
- S. Schildknecht, H. R. Gerding, C. Karreman, M. Drescher, H. A. Lashuel, T. F. Outeiro, D. A. Di Monte and M. Leist, Oxidative and nitrative alpha-synuclein modifications and proteostatic stress: implications for disease mechanisms and interventions in synucleinopathies, J. Neurochem., 2013, 125, 491–511 CrossRef CAS.
- P. K. Auluck, G. Caraveo and S. Lindquist, α-Synuclein: membrane interactions and toxicity in Parkinson's disease, Annu. Rev. Cell Dev. Biol., 2010, 26, 211–233 CrossRef CAS PubMed.
- B. H. Hunn, S. J. Cragg, J. P. Bolam, M.-G. Spillantini and R. Wade-Martins, Impaired intracellular trafficking defines early Parkinson's disease, Trends Neurosci., 2015, 38, 178–188 CrossRef CAS.
- E. Hallacli, C. Kayatekin, S. Nazeen, X. H. Wang, Z. Sheinkopf, S. Sathyakumar, S. Sarkar, X. Jiang, X. Dong, R. Di Maio, W. Wang, M. T. Keeney, D. Felsky, J. Sandoe, A. Vahdatshoar, N. D. Udeshi, D. R. Mani, S. A. Carr, S. Lindquist, P. L. De Jager, D. P. Bartel, C. L. Myers, J. T. Greenamyre, M. B. Feany, S. R. Sunyaev, C. Y. Chung and V. Khurana, The Parkinson's disease protein alpha-synuclein is a modulator of processing bodies and mRNA stability, Cell, 2022, 185, 2035–2056 CrossRef CAS PubMed.
- S. L. Leong, R. Cappai, K. J. Barnham and C. L. L. Pham, Modulation of alpha-synuclein aggregation by dopamine: a review, Neurochem. Res., 2009, 34, 1838–1846 CrossRef CAS.
- E. Coelho-Cerqueira, C. de Araújo Correia Campos and C. Follmer, Formation of large oligomers of DOPAL-modified α-synuclein is modulated by the oxidation of methionine residues located at C-terminal domain, Biochem. Biophys. Res. Commun., 2019, 509, 367–372 CrossRef CAS PubMed.
- B. Winner, R. Jappelli, S. K. Maji, P. A. Desplats, L. Boyer, S. Aigner, C. Hetzer, T. Loher, M. Vilar and S. Campioni, In vivo demonstration that α-synuclein oligomers are toxic, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4194–4199 CrossRef CAS.
- G. Yamin, C. B. Glaser, V. N. Uversky and A. L. Fink, Certain metals trigger fibrillation of methionine-oxidized α-synuclein, J. Biol. Chem., 2003, 278, 27630–27635 CrossRef CAS.
- K. J. Barnham and A. I. Bush, Biological metals and metal-targeting compounds in major neurodegenerative diseases, Chem. Soc. Rev., 2014, 43, 6727–6749 RSC.
- N. González, T. Arcos-López, A. König, L. Quintanar, M. M. Márquez, T. F. Outeiro and C. O. Fernández, Effects of alpha-synuclein post-translational modifications on metal binding, J. Neurochem., 2019, 150, 507–521 CrossRef PubMed.
- M. C. Miotto, E. E. Rodriguez, A. A. Valiente-Gabioud, V. Torres-Monserrat, A. Binolfi, L. Quintanar, M. Zweckstetter, C. Griesinger and C. O. Fernandez, Site-specific copper-catalyzed oxidation of α-synuclein: Tightening the link between metal binding and protein oxidative damage in Parkinson's disease, Inorg. Chem., 2014, 53, 4350–4358 CrossRef CAS.
- A. Binolfi, A. A. Valiente-Gabioud, R. Duran, M. Zweckstetter, C. Griesinger and C. O. Fernandez, Exploring the structural details of Cu(I) binding to α-synuclein by NMR spectroscopy, J. Am. Chem. Soc., 2011, 133, 194–196 CrossRef CAS PubMed.
- D. L. Abeyawardhane, R. D. Fernández, D. R. Heitger, M. K. Crozier, J. C. Wolver and H. R. Lucas, Copper induced radical dimerization of α-synuclein requires histidine, J. Am. Chem. Soc., 2018, 140, 17086–17094 CrossRef CAS.
- H. R. Lucas, S. DeBeer, M.-S. Hong and J. C. Lee, Evidence for copper-dioxygen reactivity during α-synuclein fibril formation, J. Am. Chem. Soc., 2010, 132, 6636–6637 CrossRef CAS PubMed.
- S. R. Paik, H.-J. Shin and J.-H. Lee, Metal-catalyzed oxidation of α-synuclein in the presence of copper(II) and hydrogen peroxide, Arch. Biochem. Biophys., 2000, 378, 269–277 CrossRef CAS.
- C. Sahin, E. C. Østerlund, N. Österlund, J. Costeira-Paulo, J. N. Pedersen, G. Christiansen, J. Nielsen, A. L. Grønnemose, S. K. Amstrup, M. K. Tiwari, R. S. P. Rao, M. J. Bjerrum, L. L. Ilag, M. J. Davies, E. G. Marklund, J. S. Pedersen, M. Landreh, I. M. Møller, T. J. D. Jørgensen and D. E. Otzen, Structural basis for dityrosine-mediated inhibition of α-synuclein fibrillization, J. Am. Chem. Soc., 2022, 144, 11949–11954 CrossRef CAS PubMed.
- B. B. Pan, Y. Yang, H. Z. Liu, Y. H. Li and X. C. Su, Coordination of platinum to α-synuclein inhibits filamentous aggregation in solution, ChemBioChem, 2019, 20, 1953–1958 CrossRef CAS.
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Platinum compounds: a new class of potent antitumour agents, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- E. Wexselblatt and D. Gibson, What do we know about the reduction of Pt(IV) pro-drugs?, J. Inorg. Biochem., 2012, 117, 220–229 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, The next generation of platinum drugs: targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- V. Venkatesh and P. J. Sadler, Platinum(IV) prodrugs, Met. Ions Life Sci., 2018, 18, 69–108 CAS.
- X. Wang, X. Wang, S. Jin, N. Muhammad and Z. Guo, Stimuli-responsive therapeutic metallodrugs, Chem. Rev., 2018, 119, 1138–1192 CrossRef PubMed.
- Z. Xu, Z. Wang, Z. Deng and G. Zhu, Recent advances in the synthesis, stability, and activation of platinum(IV) anticancer prodrugs, Coord. Chem. Rev., 2021, 442, 213991 CrossRef CAS.
- N. Graf and S. J. Lippard, Redox activation of metal-based prodrugs as a strategy for drug delivery, Adv. Drug Delivery Rev., 2012, 64, 993–1004 CrossRef CAS PubMed.
- M. Sinisi, F. P. Intini and G. Natile, Dependence of the reduction products of platinum(IV) prodrugs upon the configuration of the substrate, bulk of the carrier ligands, and nature of the reducing agent, Inorg. Chem., 2012, 51, 9694–9704 CrossRef CAS.
- S. Choi, C. Filotto, M. Bisanzo, S. Delaney, D. Lagasee, J. L. Whitworth, A. Jusko, C. Li, N. A. Wood and J. Willingham, Reduction and anticancer activity of platinum(IV) complexes, Inorg. Chem., 1998, 37, 2500–2504 CrossRef CAS.
- M. D. Hall and T. W. Hambley, Platinum(IV) antitumour compounds: their bioinorganic chemistry, Coord. Chem. Rev., 2002, 232, 49–67 CrossRef CAS.
- Y. Liu, H. Tian, L. Xu, L. Zhou, J. Wang, B. Xu, C. Liu, L. I. Elding and T. Shi, Investigations of the kinetics and mechanism of reduction of a carboplatin Pt(IV) prodrug by the major small-molecule reductants in human plasma, Int. J. Mol. Sci., 2019, 20, 5660 CrossRef CAS PubMed.
- S. Jovanović, B. Petrović, Ž. D. Bugarčić and R. van Eldik, Reduction of some Pt(IV) complexes with biologically important sulfur-donor ligands, Dalton Trans., 2013, 42, 8890–8896 RSC.
- T. Niu, W. Wan, X. Li, D. Su, S. Huo and S. Shen, Reduction of platinum(IV) prodrug model complex trans-[PtCl2(CN)4]2− by a peptide containing cysteine and methionine groups: HPLC and MS studies, J. Mol. Liq., 2018, 252, 24–29 CrossRef CAS.
- A. Ariafard, N. M. Ghohe, K. K. Abbasi, A. J. Canty and B. F. Yates, Theoretical Investigation into the Mechanism of 3′-dGMP Oxidation by [PtIVCl4(dach)], Inorg. Chem., 2013, 52, 707–717 CrossRef CAS PubMed.
- N. A. Kratochwil, A. I. Ivanov, M. Patriarca, J. A. Parkinson, A. M. Gouldsworthy, P. del Socorro Murdoch and P. J. Sadler, Surprising reactions of iodo Pt(IV) and Pt(II) complexes with human albumin: detection of Cys34 sulfenic acid, J. Am. Chem. Soc., 1999, 121, 8193–8203 CrossRef CAS.
- S. Alonso-de Castro, A. L. Cortajarena, F. López-Gallego and L. Salassa, Bioorthogonal catalytic activation of platinum and ruthenium anticancer complexes by FAD and flavoproteins, Angew. Chem., Int. Ed., 2018, 57, 3143–3147 CrossRef CAS PubMed.
- C. K. Chen, J. Z. Zhang, J. B. Aitken and T. W. Hambley, Influence of equatorial and axial carboxylato ligands on the kinetic inertness of platinum(IV) complexes in the presence of ascorbate and cysteine and within DLD-1 cancer cells, J. Med. Chem., 2013, 56, 8757–8764 CrossRef CAS PubMed.
- A. Nemirovski, Y. Kasherman, Y. Tzaraf and D. Gibson, Reduction of cis,trans,cis-[PtCl2(OCOCH3)2(NH3)2] by aqueous extracts of cancer cells, J. Med. Chem., 2007, 50, 5554–5556 CrossRef CAS PubMed.
- M. D. Hall, G. J. Foran, M. Zhang, P. J. Beale and T. W. Hambley, XANES determination of the platinum oxidation state distribution in cancer cells treated with platinum(IV) anticancer agents, J. Am. Chem. Soc., 2003, 125, 7524–7525 CrossRef CAS PubMed.
- C. K. Chen, P. Kappen, D. Gibson and T. W. Hambley, trans-Platinum(IV) pro-drugs that exhibit unusual resistance to reduction by endogenous reductants and blood serum but are rapidly activated inside cells: 1H NMR and XANES spectroscopy study, Dalton Trans., 2020, 49, 7722–7736 RSC.
- S. Yuan, Y. Zhu, Y. Dai, Y. Wang, D. Jin, M. Liu, L. Tang, F. Arnesano, G. Natile and Y. Liu, 19F NMR allows the investigation of the fate of Platinum(IV) prodrugs in physiological conditions, Angew. Chem., Int. Ed., 2022, 61, e202114250 CAS.
- H. Yao and G. Zhu, A platinum-based fluorescent “turn on” sensor to decipher the reduction of platinum(IV) prodrugs, Dalton Trans., 2022, 51, 5394–5398 RSC.
- J. L. Carr, M. D. Tingle and M. J. McKeage, Satraplatin activation by haemoglobin, cytochrome C and liver microsomes in vitro, Cancer Chemother. Pharmacol., 2006, 57, 483–490 CrossRef CAS.
- A. Lasorsa, O. Stuchlíková, V. Brabec, G. Natile and F. Arnesano, Activation of platinum(IV) prodrugs by cytochrome c and characterization of the protein binding sites, Mol. Pharm., 2016, 13, 3216–3223 CrossRef CAS.
- F. Arnesano, M. Losacco and G. Natile, An updated view of cisplatin transport, Eur. J. Inorg. Chem., 2013, 2701–2711 CrossRef CAS.
- S. Göschl, E. Schreiber-Brynzak, V. Pichler, K. Cseh, P. Heffeter, U. Jungwirth, M. A. Jakupec, W. Berger and B. K. Keppler, Comparative studies of oxaliplatin-based platinum(IV) complexes in different in vitro and in vivo tumor models, Metallomics, 2017, 9, 309–322 CrossRef.
- Z. Xu, Z. Wang, S. M. Yiu and G. Zhu, Mono- and di-bromo platinum(IV) prodrugs via oxidative bromination: synthesis, characterization, and cytotoxicity, Dalton Trans., 2015, 44, 19918–19926 RSC.
- A. Binolfi, A. Limatola, S. Verzini, J. Kosten, F.-X. Theillet, H. M. Rose, B. Bekei, M. Stuiver, M. Van Rossum and P. Selenko, Intracellular repair of oxidation-damaged α-synuclein fails to target C-terminal modification sites, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- C. S. Hwang, A. Shemorry and A. Varshavsky, N-terminal acetylation of cellular proteins creates specific degradation signals, Science, 2010, 327, 973–977 CrossRef CAS.
- C. W. Bertoncini, Y.-S. Jung, C. O. Fernandez, W. Hoyer, C. Griesinger, T. M. Jovin and M. Zweckstetter, Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1430–1435 CrossRef CAS.
- M. M. Dedmon, K. Lindorff-Larsen, J. Christodoulou, M. Vendruscolo and C. M. Dobson, Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations, J. Am. Chem. Soc., 2005, 127, 476–477 CrossRef CAS.
- P. Bernadó, C. W. Bertoncini, C. Griesinger, M. Zweckstetter and M. Blackledge, Defining long-range order and local disorder in native α-synuclein using residual dipolar couplings, J. Am. Chem. Soc., 2005, 127, 17968–17969 CrossRef.
- W. Zhou, C. Long, S. H. Reaney, D. A. Di Monte, A. L. Fink and V. N. Uversky, Methionine oxidation stabilizes non-toxic oligomers of α-synuclein through strengthening the auto-inhibitory intra-molecular long-range interactions, Biochim. Biophys. Acta, Mol. Basis Dis., 2010, 1802, 322–330 CrossRef CAS PubMed.
- A. S. Maltsev, J. Chen, R. L. Levine and A. Bax, Site-specific interaction between α-synuclein and membranes probed by NMR-observed methionine oxidation rates, J. Am. Chem. Soc., 2013, 135, 2943–2946 CrossRef CAS PubMed.
- E. Ponzini, A. De Palma, L. Cerboni, A. Natalello, R. Rossi, R. Moons, A. Konijnenberg, J. Narkiewicz, G. Legname and F. Sobott, Methionine oxidation in α-synuclein inhibits its propensity for ordered secondary structure, J. Biol. Chem., 2019, 294, 5657–5665 CrossRef CAS PubMed.
- H. Mirzaei, J. L. Schieler, J.-C. Rochet and F. Regnier, Identification of rotenone-induced modifications in α-synuclein using affinity pull-down and tandem mass spectrometry, Anal. Chem., 2006, 78, 2422–2431 CrossRef CAS PubMed.
- M. C. McCormick, K. Keijzer, A. Polavarapu, F. A. Schultz and M. H. Baik, Understanding intrinsically irreversible, non-Nernstian, two-electron redox processes: a combined experimental and computational study of the electrochemical activation of platinum(IV) antitumor prodrugs, J. Am. Chem. Soc., 2014, 136, 8992–9000 CrossRef CAS.
- A. S. Mackay, R. J. Payne and L. R. Malins, Electrochemistry for the Chemoselective Modification of Peptides and Proteins, J. Am. Chem. Soc., 2022, 144, 23–41 CrossRef CAS PubMed.
- M. Bozic, M. Caus, R. R. Rodrigues-Diez, N. Pedraza, M. Ruiz-Ortega, E. Garí, P. Gallel, M. J. Panadés, A. Martinez and E. Fernández, Protective role of renal proximal tubular alpha-synuclein in the pathogenesis of kidney fibrosis, Nat. Commun., 2020, 11, 1–16 CrossRef.
- E. Gamelin, L. Gamelin, L. Bossi and S. Quasthoff, Clinical aspects and molecular basis of oxaliplatin neurotoxicity: current management and development of preventive measures, Semin. Oncol., 2002, 29, 21–33 CrossRef CAS PubMed.
- G. Kim, S. J. Weiss and R. L. Levine, Methionine oxidation and reduction in proteins, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 901–905 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and supplementary figures. See DOI: https://doi.org/10.1039/d2qi01705a |
| This journal is © the Partner Organisations 2023 |