
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface mannosylation of dispersion polymerisation derived nanoparticles by copper mediated click chemistry†
Daniela V.
Tomasino
a,
Ashfaq
Ahmad
b,
Tauseef
Ahmad
 c,
Golestan
Salimbeigi
c,
Jennifer
Dowling
c,
Mark
Lemoine
cd,
Ruth M.
Ferrando
e,
Alan
Hibbitts
cd,
Ruairí P.
Branningan
f,
Mathew I.
Gibson
bg,
Luigi
Lay
*e and
Andreas
Heise
*adh
c,
Golestan
Salimbeigi
c,
Jennifer
Dowling
c,
Mark
Lemoine
cd,
Ruth M.
Ferrando
e,
Alan
Hibbitts
cd,
Ruairí P.
Branningan
f,
Mathew I.
Gibson
bg,
Luigi
Lay
*e and
Andreas
Heise
*adh
aDepartment of Chemistry, RCSI University of Medicine and Health Sciences, 123 St Stephen's Green, Dublin 2, Ireland. E-mail: andreasheise@rcsi.ie
bDepartment of Chemistry, University of Warwick, Gibbet Hill Road, CV4 7AL, Coventry, UK
cTissue Engineering Research Group, Dept. of Anatomy and Regenerative Medicine, RCSI University of Medicine and Health Sciences, 123 St. Stephen's Green, Dublin 2, Ireland
dAMBER, The SFI Advanced Materials and Bioengineering Research Centre, RCSI, Dublin D02, Ireland
eDepartment of Chemistry and CRC Polymeric Materials (LaMPo), University of Milan, via Golgi 19, 20133 Milan, Italy. E-mail: luigi.lay@unimi.it
fSchool of Chemical Sciences, Dublin City University, Glasnevin, Dublin 9, Ireland
gDivision of Biomedical Sciences, Warwick Medical School, University of Warwick, Gibbet Hill Road, CV4 7AL, Coventry, UK
hScience Foundation Ireland (SFI) Centre for Research in Medical Devices (CURAM), RCSI, Dublin 2, Ireland
First published on 9th February 2024
Abstract
The synthesis of spherical polymeric nanoparticles containing alkyne surface functionalities for post polymerisation glycosylation is described. The nanoparticles were obtained by a polymerisation induced self-assembly (PISA) inspired methodology in dispersed media by Cu(0) mediated polymerisation. A water soluble poly(ethylene glycol methacrylate-stat-propargyl methacrylate), poly(PEGMA18-stat-PgMA5), macroinitiator was first synthesised and chain extended with 2-hydroxypropyl methacrylate (HPMA) in water using a copper wire catalyst. It was found that irrespective of the macroinitiator to HPMA ratio and the reaction time the desired spherical morphologies (<100 nm) were obtained while the absence other morphologies suggest a deviation from the classical PISA process due to chain termination in the nanoparticle's core. The obtained nanoparticles contained alkyne functionalities in the shell, which were successfully reacted by copper mediated click chemistry with fluoresceine azide and mannosides with hydrophobic and hydrophilic spacers of different lengths. The obtained mannosylated nanoparticles displayed no significant cytotoxicity against human alveolar basal epithelial adenocarcinomic (A549) cells at any dose <0.5 mg mL−1. Preliminary binding studies confirm the ability of the mannosylated nanoparticles to bind to human lectin dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN). The methodology reported here is a convenient route to well-defined spherical and shell-functionalisable nanoparticles to create libraries of bio-active nanomaterials.
Introduction
Nanoparticles (NP) have become important materials in medical applications ranging from therapeutic delivery to diagnostics.1,2 While it is long understood that the NP interface defines their biological interaction, controlling this interfacial interaction remains a significant challenge in NP design and their clinical translation. When NP come in contact with complex media, such as cell culture media or blood plasma, the surface forms a strong and nearly irreversible interaction with biomolecules, the so-called corona.3,4 This corona layer consists of proteins, glycoproteins, and lipids and imparts a new, recognisable biological identity to the nanomaterial, which can modulate its interaction with cells. The corona can mask and bury complex targeting moieties that are grafted onto the NP surface, leading to the loss, or attenuation, of targeting properties. A strategy to minimize non-specific interactions and prevent corona formation is through modification of the NP surface, for example with poly(ethylene glycol) (PEG).5,6 Glycopolymers, on the other hand, are frequently proposed to facilitate targeted delivery,7,8 whereby the multivalent presentation of glycans enhances their interaction with cell surface lectin receptors via the cluster glycoside effect.9,10 Nanomaterials engineered with glycans and PEG thus offer multiple possibilities in drug delivery when arranged on a nanomaterial surface by restricting non-specific interactions with proteins as well as promoting specific receptor binding.11,12 Our long-term goal is the design of polymeric drug delivery NP, which favours specific over non-specific binding by facile surface modification with PEG and glycans. Here we report on the development of a synthetic strategy towards that goal by the combination of dispersion polymerisation and copper mediated click chemistry.Several examples of glycosylated polymer NP have been reported in the literature, mostly micelles or vesicles readily obtained by self-assembly of amphiphilic block copolymers.11,13–16 These are typically synthesised by either controlled polymerisation of glycosylated monomers or by post polymerisation glycosylation. Direct NP formation by emulsion and dispersion polymerisation techniques is an attractive alternative as they are carried out in an aqueous medium omitting or minimising the use of organic solvents. For instance, we used glycosylated amphiphilic block copolymers as surfactants in mini emulsion polymerisations, where the glycosylated hydrophilic block formed the NP shell.17–19 Similarly, dispersion polymerisation techniques such as polymerisation-induced self-assembly (PISA) are suitable for the in situ preparation of block copolymer nano-objects of controlled size, morphology, and surface chemistry in aqueous media.20–28 Moreover, the post PISA modification of the NP shell, including bioconjugation, utilising different coupling chemistries has been demonstrated.29–36
Here we devise a PISA inspired methodology by which a hydrophilic statistical copolymer comprising poly(ethylene glycol) methyl ether methacrylate (PEGMA) and propargyl methacrylate (PgMA) is first synthesised. This copolymer acts as macroinitiator for the polymerisation of 2-hydroxypropyl methacrylate (HPMA) in water thereby triggering block copolymer self-assembly due to the water insolubility of the polyHPMA block. This gives rise to NP with PEG side chains as well as reactive sites for mannose conjugation by copper mediated click reaction. A single-electron transfer living-radical polymerization (SET-LRP) approach was selected for the dispersion polymerisation as it enables low catalyst loading by using copper wire as a catalyst thus facilitating biocompatibility.37–40 While metal mediated approaches are still underutilised in PISA, low catalyst loading atom transfer radical type polymerisations (ATRP) such as initiator for continuous activator regeneration (ICAR ATRP) or activator regenerated by electron transfer (ARGET ATRP), have been successfully employed.41–44 To date, SET-LRP PISA was only disclosed by Neufeld et al. for spherical particles with diameters of 35–350 nm applying a partially depolymerized alginate macroinitiator and poly(methyl methacrylate) (PMMA) as the core block in a water/methanol mixture.45
Experimentals
Materials and methods
All chemicals and solvents were purchased from Sigma Aldrich and used as received unless otherwise noted. For Cu(0) mediated PISA, the copper wire was washed with acetone and its surface was scratched with sandpaper prior the polymerization to remove any presence of Cu2O on the surface. Propargyl methacrylate (98%) (PgMA) was purchased from Alfa Aesar and used without further purification. Tris [2-(dimethylamino)ethyl]amine (Me6TREN) (98+%), ethyl α-bromoisobutyrrate (>98%) (EBiB), 2-hydroxypropyl methacrylate (97%) (HPMA), 3-chloro-1-propanol (>98%), ethylene glycol mono-2-chloroethyl ether (>96%) 6-chloro-1-hexanol (>98%), ethylene glycol mono-2-chloroethyl ether (>98%) and 2-[2-(2-chloroethoxy)ethoxy]ethanol (>96%) were purchased from Tokyo Chemical Industry (TCI). Monomers were passed through a basic alumina column prior to use to remove the inhibitor. Copper(I) chloride (≥99.995%) and copper(I) bromide (99.999%) were activated in glacial acetic acid followed by washing with ethanol and diethyl ether. Tetrabutyl ammonium bromide (99%) (TBAB) was purchased from FluoroChem. Syntheses and characterizations of mannosides M1–M5 are reported in the ESI.† Reactions were monitored by thin layer chromatography (TLC) using Merck precoated 60 F254 glass plates (0.25 mm). Compounds were visualised by use of H2SO4 acidic solution in methanol or in Pankaldi solution (0.5 eq. ammonium molybdate salt and 1 eq. cerium ammonium molybdate dissolved in 6% H2SO4 aqueous solution).1H-NMR spectra were recorded on a Bruker Avance 400 (400 MHz) in DMSO-d6 or CDCl3 as solvents. 13C NMR spectra were measured at 100 MHz and 298 K with a Bruker Avance III spectrometer; δC values are reported in ppm relative to the signal of CDCl3 (δC = 77.0, CDCl3). NMR signals were assigned by homonuclear and heteronuclear 2-dimensional correlation spectroscopy (COSY, HSQC). All chemical shifts are reported as δ in parts per million (ppm) and are relative to sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS, δ = 0 ppm) or residual solvent peaks (CDCl3δ = 7.26 ppm, DMSO δ = 2.50 ppm). Mass of mannosides were measured by Electron Spray Ionization (ESI) or Matrix-Assisted Laser Ionization (MALDI) mass spectrometry. Dynamic light scattering (DLS) was done at 20 °C using a Malvern Zetasizer Nano ZSP instrument (Malvern Instruments, Malvern UK) with a detection angle of 173° and a 3 mW He–Ne laser operating at a wavelength of 633 nm. Gel permeation chromatography (GPC) was measured using a PSS SECurity GPC system equipped with a SDV 7 μm 8 × 50 mm pre-column, a SDV 100 Å, 7 μm 8 × 300 mm and a SDV 1000 Å, 7 μm 8 × 300 mm column in series and a differential refractive index (RI) detector at a flow rate of 1.0 mL × min−1 (THF). The system was calibrated against Agilent Easi-Vial linear poly(methyl methacrylate) (PMMA) standards and analysed by PSS winGPCUniChrom. All GPC samples were prepared using a concentration of 2 mg × mL−1, and were filtered through a 0.2 μm millipore filter prior to injection. Dry-state TEM imaging of the NPs was performed on a Hitachi H-7650 instrument at 150–50 K magnification. The samples (5 μL) were dropped on a Cu grid coated with SiO and Formvar and were wiped off after 10 minutes and let dry overnight. ImageJ software was used for particles size analysis. Attenuated total reflection (ATR) FTIR spectra were recorded using PerkinElmer Spectrum 100 in the region of 4000–650 cm−1. Eight scans were completed with a resolution of 2 cm−1. Mannose modification reactions were monitored by thin-layer chromatography (TLC) on Silica Gel 60 F254 (Sigma Aldrich) or with high performance thin-layer chromatography (HPTLC); compounds were visualised by heating with 10% (v/v) ethanolic H2SO4. Column chromatography was performed using Silica Gel 200–400 mesh or Biotage SNAP Ultra.
The safety profile of starting and mannosylated NP was assessed using Human alveolar basal epithelial adenocarcinomic (A549) cells. Experimental design was similar to prior studies.46,47 Briefly, 24 hours prior to treatment, A549 cells were seeded in 96 well plates at a density of 1.25 × 104 cells per well and incubated overnight at 37 °C 5% CO2. Cells were then treated with synthesized NPs at a range of concentrations and returned to the incubator. Cells were then assessed for changes in metabolic activity using the WST-1 cell viability reagent (Merck, Ireland). Cytotoxicity was assessed using the CyQUANT™ LDH Cytotoxicity Assay (Thermo Fisher Scientific, Ireland). Both kits were used according to manufacturer's instructions. In both cases, treated cells were compared against those of untreated controls and cells treated with media spiked with 0.1% Triton-X detergent (Merck, Ireland) as a positive control and background adjusted against cell medial only wells. Studies were undertaken with samples in triplicate and repeated three independent times.
Bio-layer Interferometry (BLI) was performed on an Octet® RED96 Bio-Layer Interferometry system (Forte Bio, USA) with Octet® Streptavidin (SA) used for lectin binding studies at 30 °C with shaking at 1000 rpm. For SA biosensors, the lectin DC-SIGN was first biotinylated using the EZ-Link™ Sulfo-NHS-LC-Biotin kit as per the manufacturer's instruction, in the assay buffer (10 mM HEPES, 100 mM NaCl, 10 mM CaCl2, pH 7.4). The biosensors were pre-hydrated in 200 μL of BLI assay buffer for at least 10 min in the biosensor's plate to remove the protective sucrose coating. Flat bottom black 96-Well microplates were used and loaded with 200 μL of liquid per well. The assay plate was prepared as follows: column 1 (assay buffer), column 2 (200 μg mL−1 of the DC-SIGN in assay buffer), column 3 (assay buffer), column 4 (nanomaterials at concentration NP-M1 = 2.7 mg mL−1, NP-M2 = 3.1 mg mL−1, NP-M3 = 6.4 mg mL−1, NP-M4 = 5.2 mg mL−1, NP-M5 = 3 mg mL−1 in assay buffer), and column 5 (assay buffer). Furthermore, the BLI assay was carried out as follow, Baseline 1 in column 1 (Equilibration), loading in column 2 (immobilization of the lectin on the biosensor), baseline 2 in column 3 (wash off loosely bound lectins), binding/association in column 4 (immobilized lectin binding to nanomaterials in solution), and finally dissociation in column 5 (wash off loosely bound complexes).
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I]:[L]:[NaAsc]:[TBAB] = [150]:[1]:[20]:[1.5]:[5]. The feed ratios are summarized in Table S1.† In a Schlenk-Tube, Me6TREN (138.6 μL, 0.5 mmol) was dissolved in deionised H2O and bubbled with nitrogen for 20 min before copper wire (10 cm) rolled onto a stirring bar was placed into the purged solution. Nitrogen was bubbled through the solution for a further 20 min under moderate stirring. Once the deoxygenation was completed, the tube was placed in an ice bath, ready for the subsequent step. In a separate flask, poly(PEGMA18-stat-PgMA5) (0.15 g, 0.025 mmol), sodium ascorbic acid (7.9 mg, 0.04 mmol) and TBAB salt (40 mg, 0.12 mmol) were dissolved in deionised water, before being placed in an ice-bath. HPMA was then added to the cooled macroinititator solution and the resulting solution was degassed via 3 freeze–pump–thaw cycles.
:[I]:[L]:[NaAsc]:[TBAB] = [150]:[1]:[20]:[1.5]:[5]. The feed ratios are summarized in Table S1.† In a Schlenk-Tube, Me6TREN (138.6 μL, 0.5 mmol) was dissolved in deionised H2O and bubbled with nitrogen for 20 min before copper wire (10 cm) rolled onto a stirring bar was placed into the purged solution. Nitrogen was bubbled through the solution for a further 20 min under moderate stirring. Once the deoxygenation was completed, the tube was placed in an ice bath, ready for the subsequent step. In a separate flask, poly(PEGMA18-stat-PgMA5) (0.15 g, 0.025 mmol), sodium ascorbic acid (7.9 mg, 0.04 mmol) and TBAB salt (40 mg, 0.12 mmol) were dissolved in deionised water, before being placed in an ice-bath. HPMA was then added to the cooled macroinititator solution and the resulting solution was degassed via 3 freeze–pump–thaw cycles.
The monomer/macroinitiator solution was added to the Schlenk tube containing the catalytic copper catalyst via cannula transfer and the reaction was allowed to proceed for 40 min. Under a nitrogen flux, aliquot of the reaction mixture was withdrawn via degassed syringe at 10 min intervals and monitored by DLS analysis (10 μL dissolved in 1 ml of H2O) and 1H NMR spectroscopy (in DMSO-d6 with a DSS internal standard). The final solution was diluted with water, purged with air and dialysed against water for 72 h. Monomer consumption was determined by comparing the integrals at time t (At) the integral at time t = 0 (A0) of the R-CH2 HPMA signal at 6.03–6.01 ppm, following the equation:
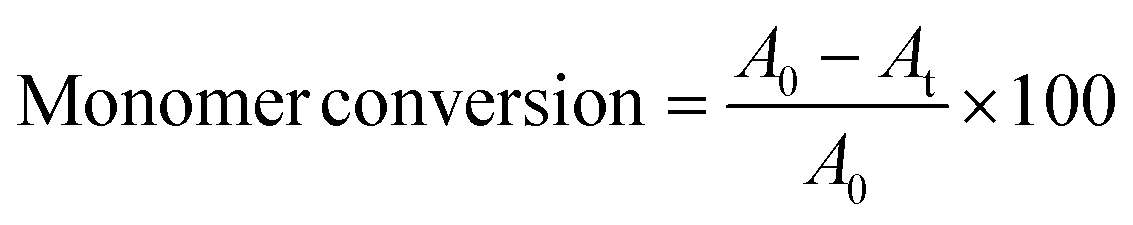
Results and discussion
Optimisation of alkyne functional nanoparticle synthesis
In the first step, a water soluble macroinitiator was synthesised via statistical copolymerisation of PEGMA (Mn = 300 g × mol−1) and PgMA at a 4:1 ratio by conventional ATRP (Fig. 1A, reaction (a)). 1H NMR analysis confirmed the copolymer structure and a monomer conversion of 97% and 92% for PgMA and PEGMA, respectively, corresponding to a degree of polymerisation (DP) of 23 at a ratio of 18 units PEGMA and 5 units of PgMA (Fig. S1 and S2†). The presence of the alkyne groups in the copolymer was further confirmed by FTIR (3657 cm−1, Fig. S3†). A dispersity (Đ) of around 1.3 was obtained from Gel Permeation Chromatography (GPC) (Fig. S4†). NP formation was achieved by chain extension of the water miscible macroinitiator (MI) with HPMA via Cu(0) using a copper wire under aqueous dispersion conditions at 10 wt% solid content (Fig. 1A, reaction (b)). Three theoretical DPs were targeted through the ratios of MI to HPMA, i.e. MI/HPMA = 150, 300 and 450. For the following discussion these samples are denoted as DP = 150, DP = 300 and DP = 450. The molar feed ratio of [NaAsc]:[TBAB]:[Me6TREN]:[poly(PEGMA18-stat-PgMA5)] was maintained constant at [1.2]:[5]:[20]:[1] for all reactions. While the addition of sodium ascorbate (NaAsc) is commonly not required for Cu(0) mediated polymerisation, we observed an improved stability of the system supressing gelation in its presence.
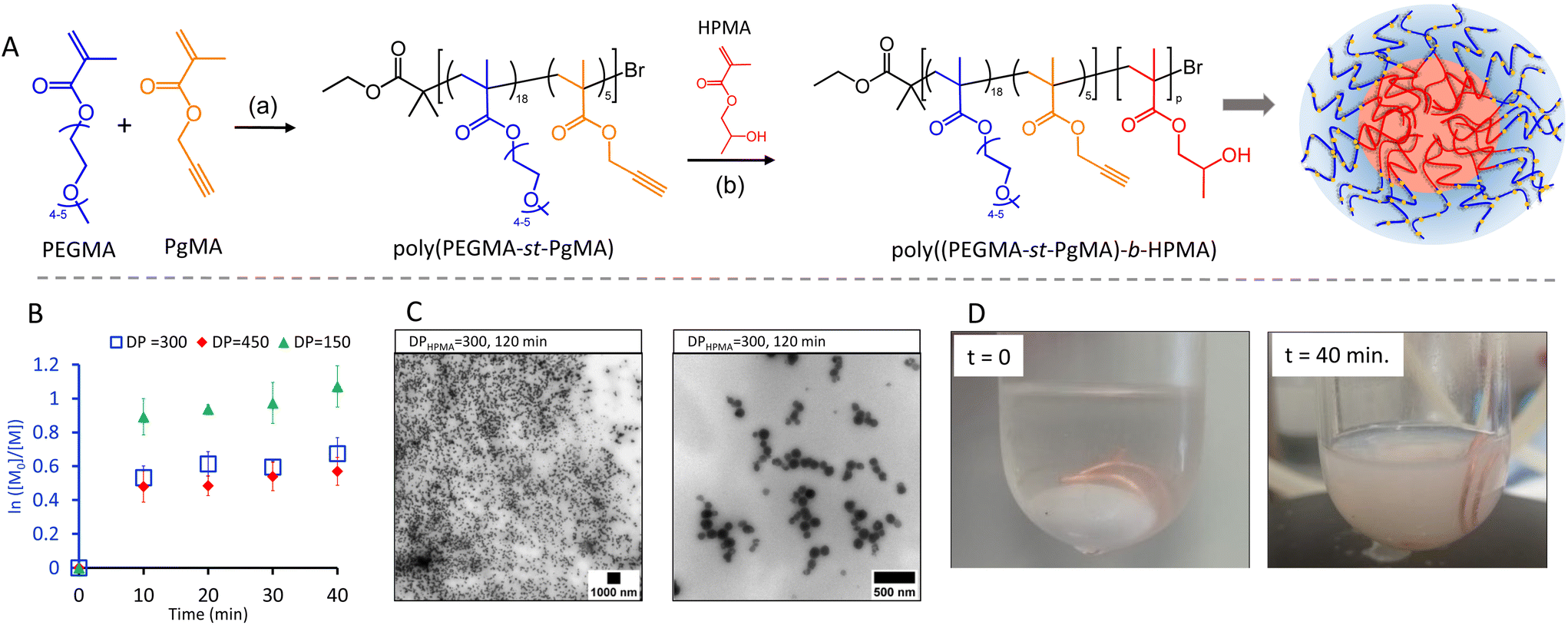 | ||
| Fig. 1 (A): Synthesis of nanoparticles by dispersion polymerisation; (a) Cu(I)/CuBr/PMDETA, bromoisobutyrate (EBIB) (1 eq.) PhOME, 60 °C; (b) Cu(0) wire (10 cm, Æ 0.5 mm), Me6TREN (20 equiv.), NaAsc (1.5 equiv.), TBAB (5 equiv.), 30 C. (B): Semilogarithmic plot of HPMA consumption over time for DPHPMA = 150, DPHPMA = 300 and DPHPMA = 450. Error bars represent standard deviation (SD; n = 3). (C): TEM images of nano particles obtained from DPHPMA = 300 after 120 min. (D): Image of the nanoparticle synthesis at the start of the reaction (t = 0 min) and at t = 40 min. | ||
For each targeted ratio, the polymerisation was repeated in triplicate and samples withdrawn every 10 min for 40 min. The initially transparent reaction solutions turned increasingly opaque with reaction time indicating the formation of polymer particles (Fig. S6†). Optical inspection revealed the highest colloidal stability at a DP = 300. We hypothesize that at a ratio of DP = 150 the HPMA block is too short to sufficiently stabilise the nanoparticles, while at DP = 450 gelation around the copper wire catalyst was observed (Fig. S12†). This is a common limitation of aqueous SET-LRP and is due to the ability of amphiphilic growing chains to compete with the ligand in complexation to the hydrophobic catalyst surface.48,491H NMR analysis confirmed the presence of HMPA signals in the final product as well as signals assigned to the macroinitiator (Fig. S5†). However, regardless of the MI:HPMA ratio, only spherical morphologies were observed in TEM images (Fig. S11†) with no morphological transition even at longer reaction times. Morphological transitions from spheres to worms or vesicles are a typical characteristic of PISA due to the continued polymer chain extension within the NP.21,50 The absence of it suggests a deviation from the classical PISA mechanism. To obtain more insight, HPMA conversions were calculated from 1H NMR analysis for samples withdrawn at time intervals. It was found that HPMA conversions reached a plateau between 46 and 60% at the first timepoint at 10 min with longer reaction times not resulting in any increase (Fig. 1B and Fig. S7–S9†). This suggests that the disproportionation equilibrium of the polymerisation is disrupted due to the slow rate at which Cu/ligand complexes diffuse into the particle core or by the presence of NaAsc producing additional Cu(I) activator. Ultimately both effects compromise reaction control leading to bimolecular termination. GPC analysis corroborates these results displaying broad dispersities and identical elution times for all samples (Fig. S10†). Therefore, the process does not strictly follow a PISA mechanism. It is, however, suitable to produce the sought-after spherical NP evident from the TEM images of the lead sample from DP = 300 (Fig. 1C). Software-based size analysis revealed a mean size of 90 nm (standard deviation 20 nm, n = 54).
One motivation for using Cu(0) mediated polymerisation was the low copper concentration rendering the materials cyto-compatible. Indeed, when exposing human alveolar basal epithelial adenocarcinomic A549 cells to the purified NP from DP = 300 there were no significant decreases in cell metabolic activity as measured by two assays. The WST-1 assay monitors the conversion of water-soluble tetrazolium salt (WST) by metabolically active cells into a formazan dye. As seen in Fig. 2a, their metabolic activity remained high at any of the doses examined compared to untreated cells. The Lactate dehydrogenase (LDH) assay confirmed these results. LDH is released by damaged cells and a convenient marker to determine the level of cell death. The low LDH levels as well as the high WST readings, as compared to the positive control samples (Triton-X), confirm that the NP were very well tolerated, which is in keeping with classical PEGylated NP highlighting the potential of these NP for safe in vitro/in vivo application.51–53
 | ||
| Fig. 2 Human alveolar basal epithelial adenocarcinomic A549 cells were treated with the synthesised nanoparticles from DP = 300 at a range of concentrations and examined for changes in (a) cell metabolic activity and (b) cytotoxicity at 24 hours (n = 3, ±SD). | ||
Mannosylation of the nanoparticle shell by click chemistry
For all following click reactions, lead NP with DP = 300 were selected. The availability of pendant propargylic moieties in the NP shell for Huisgen 1,3-dipolar cycloaddition was first demonstrated using fluorescein azide (Fig. 3, reaction 1). After mixing of the NP suspension with fluorescein azide in the presence of a copper catalyst for 24 h, the nanoparticles were dialysed to remove any unreacted fluorescein azide evident from a colourless dialysis solution and the absence of bands around 2080 cm−1 in the FTIR spectrum attributed to the stretching vibration of –N3 (Fig. S14†). Visual confirmation for the successful functionalisation was obtained from the green fluorescence of the NP suspension (Fig. 4a). DLS traces remained monomodal indicating a slight increase in DH from 148 (PDI = 0.18) to 177 nm (PDI = 0.26) upon nanoparticle shell functionalisation (Fig. 4b and Fig. S15†). Next, a set of azide containing mannosides with varying spacers in the anomeric position was synthesised. The spacer chains were varied in lengths and in composition, including hydrophobic (Fig. 3, M2 and M3) as well as hydrophilic spacers (M4 and M5). Literature reports suggest a dependence of lectin binding activity of glycopolymers on the spacer length.54 An overview of the synthetic routes is shown in Scheme S1.† Briefly, mannose derivative M1 (1-azido-α-D-mannopyranose), which presents the azido directly at the anomeric carbon was exclusively obtained as the α-anomer in high yield (95%), as confirmed by NMR spectroscopy (Fig. S17†).55 For the synthesis of M2–M5 a chloro derivative of the alcoholic spacer was used as a glycosyl acceptor and BF3OEt2 as a promoter for the O-glycosylation of penta-O-acetyl-α-D-mannopyranose.56–60 After column chromatography purification, the chloro intermediates were reacted with sodium azide to obtain the azide bearing products followed by O-deacetylation to yield M2–M5. For each mannose intermediate, the structure was confirmed by 1H-NMR and 13C-NMR spectroscopy and the final anomeric configuration was assessed via HSQC NMR techniques and COSY NMR (Fig. S16–S68†). | ||
| Fig. 3 Functionalisation of the NP surface by fluoresceine and mannose azides with different spacer lengths. | ||
 | ||
| Fig. 4 (a) Photograph of a NP suspension functionalised with fluorescein azide (DPHPMA = 300); (b) Comparison of the hydrodynamic diameter before and after the shell functionalization of the NP with fluorescein (correlograms see Fig. S15†). | ||
The mannose derivatives were co-clicked with fluorescein azide to the NP shell at a ratio of mannose to fluoresceine of 3:2, resulting in five sets of mannosylated fluorescein-conjugated NP (NP-M1 to NP-M5). While the total number of clickable sites per hydrophilic chain is about 5 (PgMA repeating units), the exact number of PgMA units per NP is unknown. Consequently, no stoichiometric ratio of the clickable moieties (R–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) to azide compounds could be calculated and a mass ratio of 1:0.6 mg of NPs to azide was used. After extensive dialysis to remove unreacted mannosides and fluoresceine, the five distinct NP solutions exhibit fluorescence when exposed to UV light, whilst appearing yellow under visible light (Fig. 5A). DLS measurements revealed a reproducible hydrodynamic diameter increase of the NPs from 183 nm (PDI = 0.13) to around 200 nm (PDI < 0.2) and a change in zeta potential from −8 mV to around −14 mV upon shell modification (Fig. 5B). TEM images confirmed similar spherical morphologies (Fig. 5C) for all samples although some aggregation was observed after extended storage time of >2 weeks.
CH) to azide compounds could be calculated and a mass ratio of 1:0.6 mg of NPs to azide was used. After extensive dialysis to remove unreacted mannosides and fluoresceine, the five distinct NP solutions exhibit fluorescence when exposed to UV light, whilst appearing yellow under visible light (Fig. 5A). DLS measurements revealed a reproducible hydrodynamic diameter increase of the NPs from 183 nm (PDI = 0.13) to around 200 nm (PDI < 0.2) and a change in zeta potential from −8 mV to around −14 mV upon shell modification (Fig. 5B). TEM images confirmed similar spherical morphologies (Fig. 5C) for all samples although some aggregation was observed after extended storage time of >2 weeks.
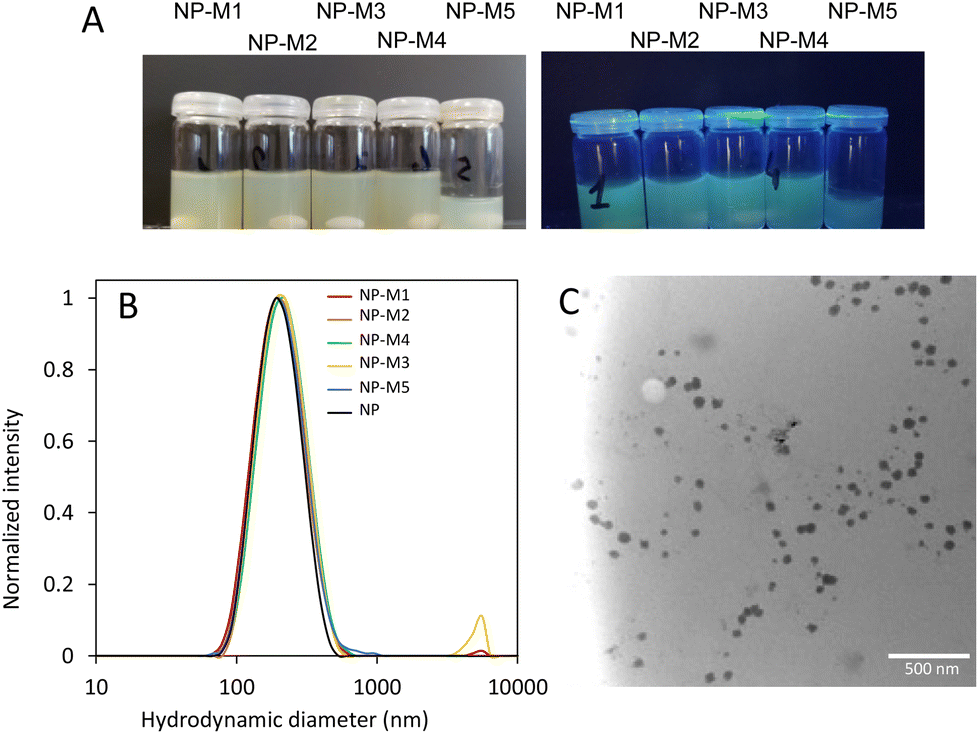 | ||
| Fig. 5 (A): Images of the solutions of (Fluor)-(Man)-NPs under visible light (left) and under UV light (right; λ = 254/365 nm). (B): Dynamic light scattering (DLS) plots of mannose/fluorescein conjugated NPs after dialysis against water for 72 h. (C): TEM image of mannosylated NP-M2. | ||
1H NMR spectroscopy performed on the dispersed NP provided further qualitative evidence for the successful shell modification for all samples (Fig. S69†). Characteristic triazole peaks are present around 8.2 ppm and 4.8 ppm as well as fluorescein peaks in the region of 6.6 ppm and at 9.5 ppm while the signals from 5 to 4 ppm are attributed to the mannose ring. FTIR spectroscopy corroborated the NMR results (Fig. S70†). A band at 3657 cm−1 corresponding to the –CC–H stretching vibration is clearly detectable in the alkyne-functional NP but is no longer present after co-clicking with fluorescein azide and the mannoside azides of M1, M3, M4 and M5 suggesting a near quantitative reaction of the alkyne groups. In case of M2, the band corresponding to the alkyne group was still visible hence a second click reaction was attempted. Although the band appeared to have reduced in intensity it was still detectable. The reason for the incomplete functionalisation of this sample is currently unclear. The change in surface polarity of the NP caused by the shell functionalisation was further demonstrated by solvent tests. Unfunctionalised NP and NP functionalised with fluorescein only resulted in a clear solution in tetrahydrofurane (THF) while they precipitate in hexafluoroisopropanol (HFiP). Functionalisation with fluorescein and mannosides inversed solvent dispersibility in these solvents attributed to the presence of OH groups from the mannose thus providing further evidence of the altered shell properties.
Cytotoxicity and lectin binding
The cytotoxicity profile of the mannosylated NP was assessed using the same experimental design as for the unfunctionalised NP on human alveolar basal epithelial adenocarcinomic A549 cells (vide supra). As can be seen in Fig. 6, a high initial dose of 1 mg mL−1 was deliberately chosen to ensure a cellular response. At 1 mg mL−1 there were significant decreases in A549 metabolic activity in NP that were mannosylated using M1, M2, and M4. The most impacted group was NP-M4 which demonstrated metabolic activity levels that were 46% (±15%) of untreated cells (STD-NP). Significant decreases at 0.5 mg mL−1 were only evident again for NP-M4 and 79% (±22%). Beyond this, metabolic activity was consistently above 95% of untreated cells at all samples and doses tested. LDH release at the highest dose indicated a significant difference between conjugated and unconjugated controls. However, it is possible that residual Cu2+ is distorting the reading at high doses. Specifically, since Cu2+ is known to bind and sequester LDH thereby reducing its absorbance intensity.61 This was also in contrast to the uniform behaviour observed in the WST-1 assay. Moreover, at lower doses where the effect of any residual copper is diluted out, LDH assays demonstrated minimal changes in LDH release. These were consistently within 20% (and often near or below 10%) of untreated cells from doses of 0.25 mg ml−1 and below. Overall, this indicates that there was minimal evidence of cytotoxicity following mannosylation regardless of final conformation. | ||
| Fig. 6 Human alveolar basal epithelial adenocarcinomic (A549 cells) treated with the mannosylated nanoparticles at a range of concentrations and examined for changes in (a) cell metabolic activity and (b) cytotoxicity at 24 hours (n = 3, ± SEM). STD-NP refers to NP not mannosylated. | ||
To demonstrate that the NP mannose shell residues were available for binding, preliminary biolayer interferometry (BLI) studies were carried out. DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin), a mannose-binding human lectin, was used at concentration of 0.2 μg mL−1 and immobilised to the surface of the BLI sensor, and nanoparticles were tested at concentrations of 2.7–6.4 mg mL−1. As illustrated in Fig. 7, a strong binding signal was observed for most of the nanoparticles used in the study, NP-M1, NP-M2, NP-M4, and NP-M5 qualitatively confirming binding availability of the mannose with the DC-SIGN. No signal was observed for the negative control, buffer alone, which validated the positive binding signal obtained from the mannosylated nanoparticles. No signal was observed for the NP-M3, which might suggest a linker effect in the binding availability of mannose, but it would require further in-depth studies to conclusively validate this.
 | ||
| Fig. 7 Lectin binding studies by biolayer interferometry (BLI) using DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin); NP concentrations: NP-M1 = 2.7 mg mL−1, NP-M2 = 3.1 mg mL−1, NP-M3 = 6.4 mg mL−1, NP-M4 = 5.2 mg mL−1, NP-M5 = 3 mg mL−1. | ||
Conclusions
The synthetic methodology presented here offers a convenient route to well-defined nanoparticles with surface functionality for further bio-conjugation. The Cu(0) approach under PISA dispersion conditions is particularly appealing due to its convenient use of copper wire as a catalyst. Compared to the more frequently used Reversible Addition–Fragmentation Chain-Transfer (RAFT) PISA process it lacks the ability to produce nanoparticles of different morphologies but proved robust if spherical nanoparticles are targeted. Moreover, the introduction of alkyne groups in the nanoparticle's shell allowed for the design of a set of mannosylated nanoparticles using various spacer chemistry. Overall, the developed synthetic protocol seems suitable to produce conjugated nanoparticle libraries for biological studies.Author contributions
D. V. T.: data curation, investigation, methodology, writing original draft; A. A.: investigation; T. A.: investigation; G. S.: investigation; J. D.: investigation; M. L.: investigation; R. M. F.: investigation; A. Hibbits: formal analysis, writing review and editing; R. P. B.: conceptualisation, writing review and editing; M. I. G.: formal analysis, writing review and editing; L. L.: supervision, writing review and editing; A. Heise: conceptualisation, methodology, supervision, writing review and editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 814236. Mass spectrometry analyses were performed at the Mass Spectrometry facility of the Unitech COSPECT at the University of Milan (Italy).References
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101 CrossRef CAS PubMed.
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826 CrossRef CAS PubMed.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779 CrossRef CAS PubMed.
- W. Richtering, I. Alberg and R. Zentel, Small, 2020, 2002162 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, J. Am. Chem. Soc., 2012, 134, 2139 CrossRef CAS PubMed.
- K. Partikel, R. Korte, N. C. Stein, D. Mulac, F. C. Herrmann, H.-U. Humpf and K. Langer, Eur. J. Pharm. Biopharm., 2019, 141, 70 CrossRef CAS PubMed.
- M. H. Stenzel, Macromolecules, 2022, 55, 4867 CrossRef CAS.
- C. R. Becer, Macromol. Rapid Commun., 2012, 33, 742 CrossRef CAS PubMed.
- Y. Miura, Y. Hoshino and H. Seto, Chem. Rev., 2016, 116, 1673 CrossRef CAS PubMed.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555 CrossRef CAS PubMed.
- P. Gurnani, A. M. Lunn and S. Perrier, Polymer, 2016, 106, 229 CrossRef CAS.
- B. Ka, P. Okwieka, S. Schöttler, S. Winzen, J. Langhanki, K. Mohr, T. Opatz, V. Mailänder, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2015, 54, 7436 CrossRef PubMed.
- J. Huang, C. Bonduelle, J. Thevenot, S. Lecommandoux and A. Heise, J. Am. Chem. Soc., 2012, 134, 119 CrossRef CAS PubMed.
- X. Lia and G. Chen, Polym. Chem., 2015, 6, 1417 RSC.
- A. Adharis, T. Ketelaar, A. G. Komarudin and K. Loos, Biomacromolecules, 2019, 20, 1325 CrossRef CAS PubMed.
- C. Cao, J. Zhao, F. Chen, M. Lu, Y. Y. Khine, A. Macmillan, C. J. Garvey and M. H. Stenzel, Chem. Mater., 2018, 30, 5227 CrossRef CAS.
- J. Jacobs, N. Gathergood, J. P. A. Heuts and A. Heise, Polym. Chem., 2015, 6, 4634 RSC.
- J. Jacobs, A. Byrne, N. Gathergood, T. E. Keyes, J. P. A. Heuts and A. Heise, Macromolecules, 2014, 47, 7303 CrossRef CAS.
- J. Jacobs, D. Pavlovic, H. Prydderch, M.-A. Moradi, E. Ibarboure, J. P. A. Heuts, S. Lecommandoux and A. Heise, J. Am. Chem. Soc., 2019, 141, 12522 CrossRef CAS PubMed.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1800325 CrossRef PubMed.
- C. György and S. P. Armes, Angew. Chem., Int. Ed., 2023, 62, e202308372 CrossRef PubMed.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef PubMed.
- S. L. Canning, G. N. Smith and S. P. A. Armes, Macromolecules, 2016, 49, 1985 CrossRef CAS PubMed.
- D. Ikkene, J.-L. Six and K. Ferji, Eur. Polym. J., 2023, 111848 CrossRef CAS.
- M. Lansalot and J. Rieger, Macromol. Rapid Commun., 2019, 40, e1800885 CrossRef PubMed.
- S. Varlas, J. C. Foster and R. K. O'Reilly, Chem. Commun., 2019, 55, 9066 RSC.
- E. J. Cornel, J. Jiang, S. Chen and J. Du, CCS Chem., 2021, 3, 2104 CrossRef CAS.
- L. E. B. Karagoz, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350 RSC.
- D. Le, D. Keller and G. Delaittre, Macromol. Rapid Commun., 2019, 40, 1800551 CrossRef PubMed.
- Z. An, W. Tang, M. Wu, Z. Jiao and G. D. Stucky, Chem. Commun., 2008, 6501 RSC.
- L. P. D. Ratcliffe, K. J. Bentley, R. Wehr, N. J. Warren, B. R. Saunders and S. P. Armes, Polym. Chem., 2017, 8, 5962 RSC.
- D. Keller, A. Beloqui, M. Martínez-Martínez, M. Ferrer and G. Delaittre, Biomacromolecules, 2017, 18, 2777 CrossRef CAS PubMed.
- H. Asem, W. Zheng, F. Nilsson, Y. Zhang, M. S. Hedenqvist, M. Hassan and E. Malmström, ACS Appl. Bio Mater., 2020, 4, 1045 CrossRef.
- B. Maiti, K. Bauri, M. Nandi and P. De, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 263 CrossRef CAS.
- D. Le, D. Keller and G. Delaittre, Macromol. Rapid Commun., 2018, 1800551 Search PubMed.
- Z. Jia, V. A. Bobrin, N. P. Truong, M. Gillard and M. J. Monteiro, J. Am. Chem. Soc., 2014, 136, 5824 CrossRef CAS PubMed.
- N. H. Nguyen, B. M. Rosen, G. Lligadas and V. Percec, Macromolecules, 2009, 42, 2379 CrossRef CAS.
- V. Percec, A. V. Popov, E. Ramirez-Castillo and O. Weichold, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3283 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 123, 14156 CrossRef PubMed.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- J. Wang, Z. Wu, G. Wang and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, e1800332 CrossRef PubMed.
- G. Wang, M. Schmitt, Z. Wang, B. Lee, X. Pan, L. Fu, J. Yan, S. Li, G. Xie, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2016, 49, 8605 CrossRef CAS.
- B. Shi, H. Zhang, Y. Liu, J. Wang, P. Zhou, M. Cao and G. Wang, Macromol. Rapid Commun., 2019, 40, 1900547 CrossRef CAS PubMed.
- K. Wang, Y. Wang and W. Zhang, Polym. Chem., 2017, 8, 6407 RSC.
- V. Kapishon, R. A. Whitney, P. Champagne, M. F. Cunningham and R. J. Neufeld, Biomacromolecules, 2015, 16, 2040 CrossRef CAS PubMed.
- A. Hibbitts, A. Lucía, I. Serrano-Sevilla, L. De Matteis, M. McArthur, J. M. de la Fuente, J. A. Aínsa and F. Navarro, PLoS One, 2019, 14, e0220684 CrossRef CAS PubMed.
- A. J. Hibbitts, Z. Kočí, S. Kneafsey, A. Matsiko, L. Žilić, A. Dervan, P. Hinton, G. Chen, B. Cavanagh, J. K. Dowling, C. E. McCoy, C. T. Buckley, S. J. Archibald and F. J. O'Brien, Matrix Biol., 2022, 106, 34 CrossRef CAS PubMed.
- Y. Li, N. Busatto and P. J. Roth, Macromolecules, 2021, 54, 3101 CrossRef CAS.
- S. R. Samanta, V. Nikolau, S. Keller, M. J. Monteiro, D. A. Wilson, D. M. Haddleton and V. Percec, Polym. Chem., 2015, 6, 2084 RSC.
- J. Y. Rho, G. M. Scheutz, S. Häkkinen, J. B. Garrison, Q. Song, J. Yang, R. Richardson, S. Perrier and B. S. Sumerlin, Polym. Chem., 2021, 12, 3947 RSC.
- A. Hibbitts, N. Lieggi, O. McCabe, W. Thomas, J. Barlow, F. O'Brien and S.-A. Cryan, Ther. Delivery, 2011, 2, 987 CrossRef CAS PubMed.
- A. Hibbitts, A. M. O'Connor, J. McCarthy, E. B. Forde, G. Hessman, C. M. O'Driscoll, S.-A. Cryan and M. Devocelle, ACS Omega, 2019, 4, 10078 CrossRef CAS PubMed.
- A. J. Hibbitts, J. M. Ramsey, J. Barlow, R. MacLoughlin and S. A. Cryan, Nanomaterials, 2020, 10, 1248 CrossRef CAS PubMed.
- G. Yilmaz, V. Uzunova, M. Hartweg, V. Beyer, R. Napier and C. R. Becer, Polym. Chem., 2018, 9, 611 RSC.
- V. Percec, P. Leowanawat, H. J. Sun, O. Kulikov, C. D. Nusbaum, T. M. Tran, A. Bertin, D. A. Wilson, M. Peterca, S. Zhang, N. P. Kamat, K. Vargo, D. Moock, E. D. Johnston, D. A. Hammer, D. J. Pochan, Y. Chen, Y. M. Chabre, T. C. Shiao, M. Bergeron-Brlek, A. Andre, R. Roy, H. J. Gobius and P. A. Heiney, J. Chem. Soc., 2013, 135, 9055–9077 CrossRef CAS PubMed.
- L. Gu, P. G. Luo, H. Wang, M. J. Meziani, Y. Lin, L. M. Veca, L. Cao, L. Fushen, X. Wang, R. A. Quinn, W. Wang, P. Zhang, S. Lacher and Y. P. Sun, Biomacromolecules, 2008, 9, 2408 CrossRef CAS PubMed.
- J. Moratz, F. Klepel and B. J. Ravoo, Org. Biomol. Chem., 2017, 15, 5089–5094 RSC.
- I. Kalograiaki, M. Abellan-Flos, L. A. Fernandez, M. Menedez, S. P. Vincent and D. Solis, Anal. Chem., 2018, 90, 12314 CrossRef CAS PubMed.
- W. C. Li, K. W. Han, B. Ghosh, P. H. Li, H. Y. Lin, P. H. Chan, C. Lin, Y. C. Chen and K. K. T. Mong, Chem. – Asian J., 2019, 9, 1786 CrossRef PubMed.
- M. Li, W. Ye, K. Zhang, C. Shi, Y. Huang, W. Chen, J. Hu, Z. Jiang and W. Zha, Eur. J. Med. Chem., 2020, 202, 11509 Search PubMed.
- X. Han, R. Gelein, N. Corson, P. Wade-Mercer, J. Jiang, P. Biswas, J. N. Finkelstein, A. Elder and G. Oberdörster, Toxicology, 2011, 287, 99 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterisation of nanoparticles, synthesis and characterisation of mannosides, additional characterisation of functional nanoparticles. See DOI: https://doi.org/10.1039/d3py01361h |
| This journal is © The Royal Society of Chemistry 2024 |