
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring telogen chemical structure and reactivity when forming highly branched polyesters derived from ethylene glycol dimethacrylate homopolymerisation under transfer-dominated branching radical telomerisation (TBRT) conditions†
Sean
Flynn
ab,
Bethany
Linthwaite
ab,
Oliver B.
Penrhyn-Lowe
ab,
Samuel
Mckeating
ab,
Stephen
Wright
ab,
Savannah R.
Cassin
ab,
Pierre
Chambon
ab and
Steve P.
Rannard
 *ab
*ab
aDepartment of Chemistry, University of Liverpool, Crown Street, L69 7ZD, UK. E-mail: srannard@liv.ac.uk
bMaterials Innovation Factory, University of Liverpool, Crown Street, L69 7ZD, UK
First published on 10th November 2023
Abstract
Transfer-dominated Branching Radical Telomerisation (TBRT) is a free radical telomerisation reaction that allows the homopolymerisation of multi-vinyl substrates. When applied to dimethacrylates, highly branched polyesters are formed with essentially complete consumption of vinyl functional groups and without gel formation. To date, no reports of TBRT have investigated a diverse series of thiol telogens with considerable variation in chain transfer coefficients (CT) when using the same multi-vinyl taxogen (MVT). Here, the impact of CT on reaction conditions and structural telogen sub-units is studied for the homopolymerisation of ethylene glycol dimethacrylate (EGDMA). Kinetic studies explore the different rates of vinyl group consumption, and model linear telomer studies using a monovinyl taxogen investigate the distribution of sub-units that are formed when using the different telogens. The formation of a library of branched polyesters with varying telogen-derived pendant groups with weight average molecular weights up to 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 930000 g mol−1 has allowed a detailed comparison of glass transition temperatures and the calculation of Tg∞ values using classical or modified Flory–Fox relationships.
930000 g mol−1 has allowed a detailed comparison of glass transition temperatures and the calculation of Tg∞ values using classical or modified Flory–Fox relationships.
Introduction
Relative to linear polymers, the complex macromolecular architecture of branched polymers has led to properties such as low solution viscosities and high functionality and established their importance in the fields of viscosity modification,1–3 drug delivery applications4,5 and degradable materials.6–8 The pursuit of innovative synthetic strategies to enable the formation of high molecular weight, highly branched macromolecules, whilst maintaining control over their physical and chemical properties, has challenged synthetic polymer chemists for decades. Classical approaches to hyperbranched polymers employed step-growth polymerisation of multifunctional monomers, including but not limited to ABx and ABx + Bx systems.9 These approaches generate terminal, dendritic, linear and focal point units within the branched macromolecular architecture whilst avoiding the possibility of gelation. Significant efforts have also been made to prepare branched polymers via step-growth polymerisation of A2 + B3 monomers although this approach is inherently susceptible to chemical cross-linking.10In recent years, free radical (co)polymerisation strategies using multifunctional vinyl monomers have emerged as efficient alternative routes.11,12 These often require manipulation of the prevalence of competing reactions which occur during chain-growth polymerisation. For example, Wang and co-workers utilised deactivation enhanced atom transfer radical polymerisation to control the radical growth boundaries during homopolymerisations of divinyl monomers.13,14 This influenced the relative number of propagation and deactivation steps and provided greater control over the formation of hyper-branched and highly cyclised polymer architectures.15–17 Gao and co-workers employed a similar strategy to increase the degree of branching of hyperbranched polymers formed by self-condensing vinyl polymerisation of alkyl halides containing inimers in emulsion conditions. This approach utilised activators generated by electron transfer atom transfer radical polymerisation to increase the number of activation/reactivation cycles per propagation step and enhance branch point formation via higher levels of alkyl halide activation. Most recently, Matyjazewski and co-workers reported an elegant approach to activate branching units in situ using visible-light-mediated controlled radical branching polymerisation of “inibramers”.18 This approach capitalised on polymerisation-induced changes in the bond dissociation energy of alkyl bromide bonds within α-bromo acrylates to enable branch point activation within growing polymer chains.
Control over the relative ratio of different chain-growth polymerisation steps has proven equally important in the preparation of (hyper)branched (co)polymers via conventional free radical polymerisation (FRP). Sherrington and co-workers pioneered the preparation of statistical copolymers by chain transfer mediated FRP.19–21 This strategy, known as the ‘Strathclyde approach’, relies on competing propagation and chain transfer reactions to limit the statistical incorporation of divinyl monomers into growing polymer chains formed from monovinyl monomer propagation, hence avoiding network formation.
In contrast, Jiang et al. used a chain transfer monomer approach to generate hyperbranched polymers by FRP in emulsion.22 This approach placed chain initiating groups, namely thiols and peroxides, within the pendant group of (meth)acrylic monomers,23–26 enabling in situ branch point formation through chain transfer reactions with propagating radicals. Interestingly, the degree of branching achieved was highly dependent upon the relative magnitudes of the propagation and chain transfer rate constants, Kp and Ktr respectively; i.e. the chain transfer coefficient; CT = Ktr/Kp.27 Chain transfer monomers exhibiting CT values ∼1 produced highly branched polymers due to high competition between transfer and propagation reactions. In contrast, chain transfer monomers which exhibited CT values ≫1 or ≪1 produced polymers with predominantly linear topologies, due to reactions dominated by thiol–ene mono-addition and or vinyl group propagation mechanisms respectively.28
Previously, we introduced Transfer-dominated Branching Radical Telomerisation (TBRT) as a new approach for the preparation of hyperbranched polymers via a facile and commercially scalable one pot synthesis.29 TBRT utilises thiol-mediated radical telomerisation of multivinyl taxogens (MVT), such as dimethacrylates, to generate polymers containing ester units within the polymer backbone (Fig. 1).
 | ||
| Fig. 1 Schematic representation of the formation of branched polyesters via TBRT of ethylene glycol dimethacrylate (EGDMA) using 1-dodecanethiol as the thiol-based telogen. The figure includes a chemical description of the polyester backbone, the presence of telomers with degree of polymerisation (DP) of 1 and 2 units, and the nominal repeat structure for the final polymer. | ||
Under TBRT conditions, thiols act as telogens and are incorporated as side-chains, in contrast to becoming chain ends when used as a conventional chain transfer agents. The success of TBRT depends on limitation of the number-average degree of polymerisation (DPn) of telomer chains, ideally to <2 units, to prevent three-dimensional network formation via crosslinking. We recently identified several critical factors that influence branched polymer formation, either through telomer formation or the nature of vinyl group consumption, during TBRT. These include the initial molar ratio of MVT to telogen ([MVT]0/[Tel]0),30 MVT chain dimensions,31 polymerisation temperature32 and reaction concentration.33 Optimisation of reaction conditions can therefore enable the preparation of hyperbranched, high molecular weight polyesters with control over the chemical functionality of both MVT and telogen residues within polymer repeat units.
Here, we more closely explore the role of the telogen chain transfer coefficient (CT) in the formation of branched polymers during TBRT. We aim to utilise thiol structure, and thus reactivity, to control the relative propensities of chain transfer and propagation reactions to influence the extent of branching reactions and thus polymer molecular weight. Telogen CT has been selected as the only chemical variable within the studies of ethylene glycol dimethacrylate (EGDMA) TBRT with four structurally varied thiol-based telogens. All other chemical and experimental parameters such as MVT selection, reaction solvent, radical source and concentration, and polymerisation temperature have been normalised. Although high molecular weight branched polymers were obtained using each of the telogens studied, telogen structure led to significant contrast in the [MVT]0/[Tel]0 required to generate high molecular weight branched polymers. Detailed kinetic and model linear telomerisation reactions revealed considerable differences in telomer formation and explain variations in the extent of branching reactions observed. As previously shown, variation in the structure of telogen residues also significantly impacts the thermal properties of the resulting high molecular weight branched polymers.
Results and discussion
Thiol chemistry impact on branched polymer synthesis using TBRT
Having recently demonstrated the preparation of mixed-telogen copolymers using thiols with comparable CT values, a series of TBRT reactions using EGDMA were conducted using four thiol-based telogens with highly variable CT values. Cyclohexanethiol (CHT), phenyl ethyl mercaptan (PhEM), benzyl mercaptan (BzM) and 4-tert-benzyl mercaptan (tBBM) were selected with previously reported CT values of 0.14, 0.23, 0.35, 0.42 respectively during the FRP of methyl methacrylate (MMA).34 The structural variation within these telogens was also expected to generate a series of p(EGDMA)-based branched polyesters containing different pendent group functionality (Scheme 1). | ||
| Scheme 1 Synthesis of four branched polyesters using the TBRT of EGDMA and (A) cyclohexane thiol, CHT, (B) phenyl ethyl mercaptan, PhEM, (C) 4-tert-benzyl mercaptan, tBBM, and (D) benzyl mercaptan, BzM as the telogen species. Each polymer is shown as the nominal repeat unit with telogen residues as pendant group functionality. | ||
TBRTs were conducted using a nominal fixed molar ratio of EGDMA to telogen ([EGDMA]0/[Tel]0) at which all reactions resulted in the formation of soluble branched polymers and avoided the formation of insoluble crosslinked samples ([EGDMA]0/[Tel]0 = 0.45); accurate values of [EGDMA]0/[Tel]0 were determined by 1H nuclear magnetic resonance (NMR) spectroscopy of the reaction mixture prior to addition of 2,2′-azobis(2-methylpropionitrile), (AIBN) as the radical source, at 1.5 mole percent (mol%) equivalent with respect to EGDMA vinyl groups (ESI Fig. S1–4, and Table S1†). To ensure comparable reaction conditions, all TBRTs were conducted using a fixed MVT concentration of 4.2 mol dm−3 in ethyl acetate (EtOAc) and were allowed to proceed for 24 hours. Regardless of the telogen employed, the absence of vinylic protons in 1H NMR spectra of crude reactions mixtures indicated that all TBRTs had achieved high vinyl conversion (>99%, ESI Fig. S5–S8†).
Interestingly, analysis of the purified polymer products by triple detection size exclusion chromatography (TD-SEC) showed vastly contrasting molecular weight distributions for the branched polyesters generated using the different telogens at a this fixed [EGDMA]0/[Tel]0 ratio (Fig. 2); weight-average molecular weight (Mw) values of approximately 329000 g mol−1, 52000 g mol−1, 14800 g mol−1 and 9000 g mol−1 were observed for p(CHT-EGDMA), p(PhEM-EGDMA), p(BzM-EGDMA) and p(tBBM-EGDMA) respectively (ESI Table S1†). Corresponding respective number average molecular weight (Mn) values were approximately 14600 g mol−1, 5500 g mol−1, 4100 g mol−1 and 1350 g mol−1, and all purified polymers showed [EGDMA]F/[Tel]F values close to unity, indicating low or no meaningful cyclisation under these conditions.33 The vast contrast in Mw values indicates significant variation in the extent of intermolecular branching occurring in each of these reactions and showed an inverse correlation with the reported CT; values decreased with increasing telogen CT.
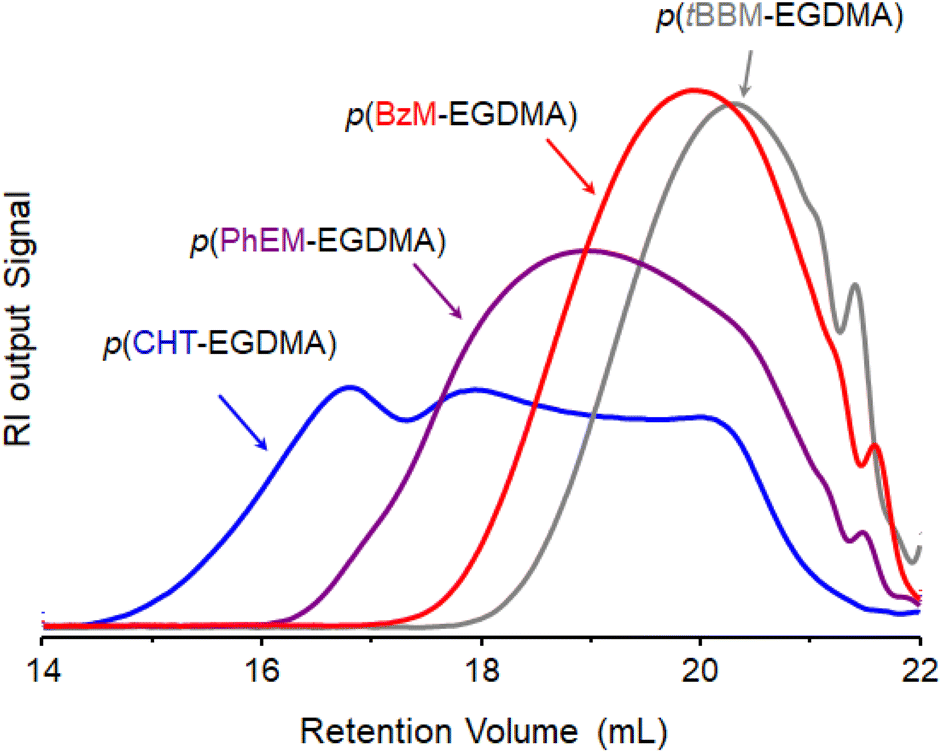 | ||
| Fig. 2 Variation in molecular weight distributions of branched polyesters formed by TBRT using telogens with differing chain transfer coefficients. Overlaid refractive index (RI) chromatograms obtained from the triple-detection size exclusion chromatography of p(CHT-EGDMA) (blue), p(PhEM-EGDMA) (purple), p(BzM-EGDMA) (red), and p(tBBM-EGDMA) (grey). | ||
The higher Mw values obtained for polymers generated using telogens with lower CT values implies chain transfer kinetics playing an active role in branched polyester formation by TBRT. This is analogous to our recent description of the impact of temperature on the TBRT of a single MVT/telogen combination.32 At low CT, enhanced propagation relative to chain transfer would suggest longer telomer subunit formation relative to reactions utilising telogens with higher CT values. Furthermore, the low Mw (and Mn) obtained for p(PhEM-EGDMA), p(BzM-EGDMA) and p(tBBM-EGDMA) indicated that TBRTs conducted using these telogens would require significant increases in the [EGDMA]0/[Tel]0 ratio to generate branched polyesters with molecular weights comparable to those obtained for p(CHT-EGDMA).
Structural analysis of p(EGDMA) with varying pendant groups
Several of the branched polyesters formed here, are new macromolecular structures that have not been previously described. As such detailed 1H and 13C NMR studies were conducted on purified materials to compare the effects of the different telogens on the resonances within the polymers, with the potential to derive structural insight. As mentioned above, the telogen acts as a pendant group within TBRT polymers and the EGDMA homopolymerisations will generate identical polyester backbone structures but with considerable variation in the pendant group structure, analogous to polymerisation of monovinyl monomers with varying side chain chemistries.1H NMR spectroscopy of the four polymers showed very negligible impact of telogen structure on the polyester backbone methylene groups (H1) as may be expected, with a broad resonance between δ = 4.0–4.4 ppm in all spectra, Fig. 3.
 | ||
| Fig. 3 Pendant group variation in branched polyesters generated by TBRT. Overlaid 1H NMR spectra obtained for (A) p(CHT-EGDMA), (B) p(PhEM-EGDMA), (C) p(BzM-EGDMA) and (D) p(tBBM EGDMA). | ||
No clear modification of the 13C resonances for this region of the polymers (δ ∼ 62.0 ppm) was observed either (ESI Fig. S32–35†). The methyl protons pendant to the polyester (H2) also appeared unaffected in the 1H NMR study, but as described previously, these groups are represented by two resonances at δ = 1.22 and 1.14 ppm, Fig. 3. This has been attributed to different environments associated with either a DP1 telomer sub-unit, where the telogen has added to just one vinyl group and created a tertiary carbon alpha to the backbone carbonyl group, Fig. 1, or the limited propagation of the telomer subunits at DP ≥ 2, where quaternary carbons result. Importantly, each telomer with a DP ≥ 2 contains a tertiary carbon, also as a result of chain transfer and capping of the telomer chain with a proton (ESI Fig. S36†).
The aromatic protons of the telogen pendant groups within p(BzM-EGDMA), p(tBBM-EGDMA) and p(PhEM-EGDMA) are all clearly visible, Fig. 3B–D. The benzyl methylene protons in p(BzM-EGDMA) and p(tBBM-EGDMA) are observed at δ = 3.69 ppm, Fig. 3C & D, and absent for p(PhEM-EGDMA) which shows resonances at δ = 2.84 and 2.76 ppm, Fig. 3B. The 1H NMR analysis of the non-aromatic p(CHT-EGDMA), showed ring resonances at δ = 2.84, 1.94, 1.74 and 1.25 ppm, Fig. 3A.
In contrast, telogen residue structure had a tangible impact on the resonances observed within the 13C NMR spectra of the polymers, specifically within the proximity of the pendant group (ESI Fig. S32–35†). The methylene carbon within the EGDMA residue, and formed by addition of the thiyl radical to the vinyl group of EGDMA, exhibits variation in its chemical shift that is telogen dependent. For the benzyl-containing polymers p(tBBM-EGDMA) and p(BzM-EGDMA), this methylene group resonance was seen at δ = 34.6 ppm. For p(CHT-EGDMA) and p(PhEM-EGDMA) the methylene was observed at δ = 33.5 and 34.3 ppm respectively. A 13C attached proton test (APT) NMR analysis was used to identify the terminal carbons from each telomer sub-unit chain, formed after chain-transfer, and two resonances were observed for each polymer; this has previously been attributed to the formation of DP1 units and the terminal vinyl group addition in telomers with DP ≥ 2.30,31 The values observed for resonances from the DP1 units of p(CHT-EGDMA), p(PhEM-EGDMA), p(BzM-EGDMA) and p(tBBM-EGDMA) were δ = 40.6, 40.3, 39.9 and 40.0 ppm respectively. Resonances associated with tertiary carbons situated at the ω termini of telomer sub-units with DP ≥ 2 were practically identical in their chemical shifts with values obtained for p(CHT-EGDMA), p(PhEM-EGDMA), p(tBBM-EGDMA) and p(BzM-EGDMA) being δ = 36.0 36.0 35.9 35.9 ppm respectively. This was somewhat expected given the distance from the telogen residue.
Kinetic analysis of TBRTs conducted at fixed [MVT]0/[Tel]0
To understand the extent that chain transfer kinetics impacts telomer formation and branching during TBRT, kinetic studies were conducted on each of the EGDMA/telogen reactions using the conditions described earlier.During a conventional linear vinyl polymerisation, the conversion of monomer to polymer is intrinsically linked to the consumption of vinyl functional groups. Under TBRT conditions, vinyl consumption does not correlate directly to polymer formation as the large number of pendant vinyl groups may react via intermolecular branching, or through DP1 formation, without any further addition of unreacted MVT to the growing branched polymer structures.
Vinyl consumption was monitored using 1H NMR spectroscopy, and reactions were spiked with anisole for use as an internal standard to determine conversions of EGDMA and telogen by gas chromatography (GC; Fig. 4; ESI Fig. S9†). In all cases, TBRTs achieved high vinyl group consumption (>95%) after two hours, with no indication of remaining vinyl groups after 180 minutes. 1H NMR analysis indicated that TBRTs showed slight variations in induction time following heating, with PhEM, BzM, tBBM and CHT showing induction times of approximately 3, 3, 9 and 9 minutes respectively (ESI Tables S2–5†). Taking differences in induction time into account, EGDMA TBRT in the presence of PhEM, CHT and BzM proceeded at highly comparable rates of vinyl group consumption (ESI Fig. S10†) with, for example, 72%, 75% and 78% vinyl consumption respectively after approximately 50 minutes. In contrast, the TBRT of EGDMA with tBBM proceeded at a lower rate of vinyl consumption, showing just 60% vinyl conversion after 51 minutes.
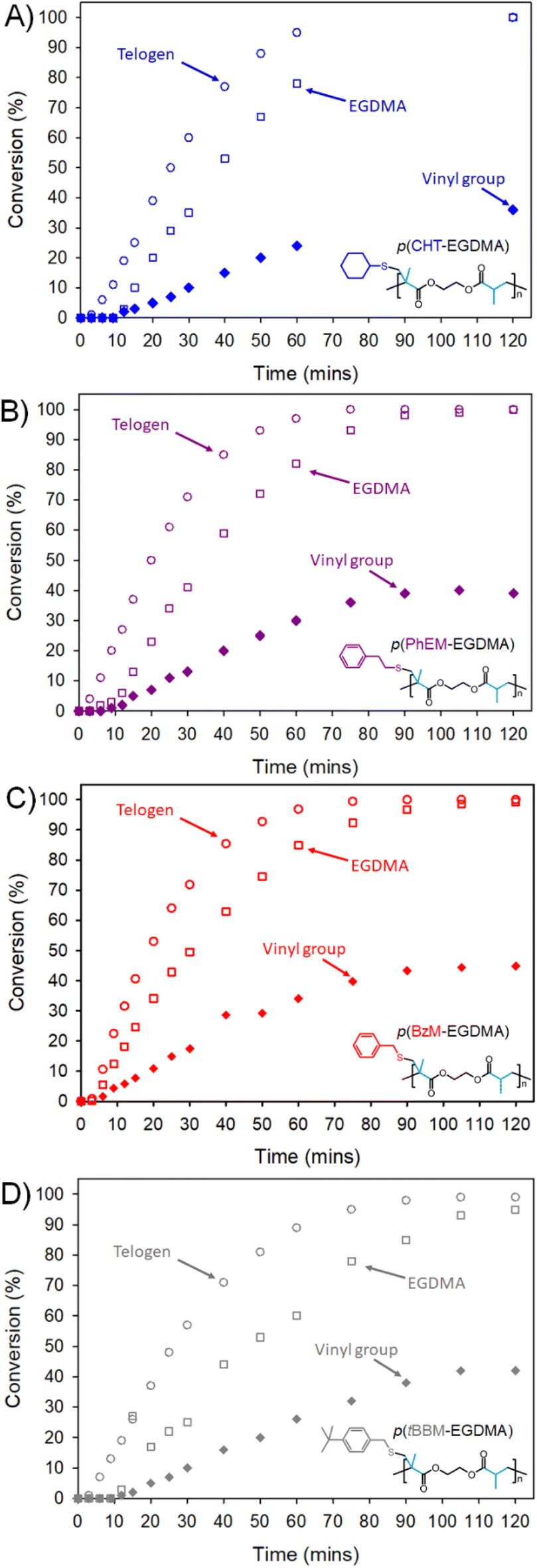 | ||
| Fig. 4 Kinetic analysis of TBRTs of EGDMA with (A) CHT, (B) PhEM, (C) BzM and (D) tBBM ([EGDMA]0/[Tel]0 = 0.45). GC monitoring of telogen (filled diamonds), and EGDMA (hollow squares) conversion, overlaid with 1H NMR monitoring of vinyl group (hollow circles) conversion over 120 minutes. | ||
This observation is important as it distinguishes the role of chain transfer reactions in TBRT, i.e. thiols acting as telogens, being different to conventional chain transfer within a linear polymerisation. Due to the high concentration of telogen used to ensure telomer formation, a transfer-dominated reaction is seen, with transfer of the thiyl radical to thiol being a critical reaction, Scheme 2.
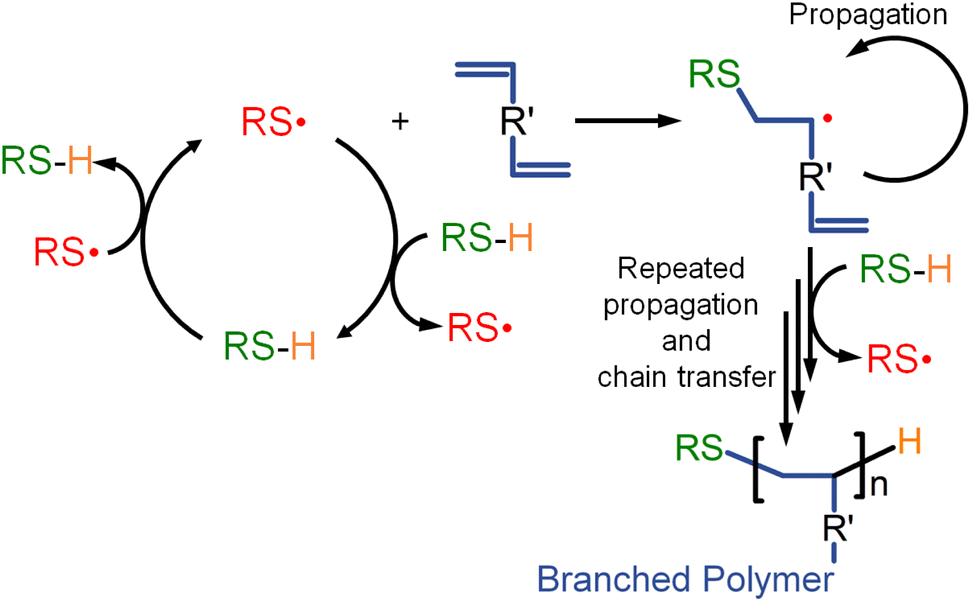 | ||
| Scheme 2 TBRT reaction mechanism showing chain transfer to telogen (thiol) as the dominant reaction step. | ||
As tBBM has the highest CT value, established through MMA polymerisation studies, it is probable that it also has the highest thiyl radical-to-thiol telogen chain transfer rate of the telogens studied here. The slower vinyl group consumption within the EGDMA/tBBM TBRT reaction can therefore be explained by the combined dual effects of thiyl radicals being engaged in rapid chain transfer to telogen and rapid chain transfer of propagating carbon-centred radicals to telogen.
In all cases, plotting vinyl consumption using semi-logarithmic co-ordinates showed an ln([M]0/[M]) trend considerably deviating from linearity during the latter stages of TBRT (ESI Fig. S11†). The observed deviations from steady-state kinetics, commonly associated with free-radical polymerisation, are highly indicative of an increase in radical concentration; this likely occurs through suppression of radical termination via extensive chain transfer reactions, which is a key characteristic of TBRT.
GC monitoring of EGDMA consumption across all reactions showed >99% within 120 minutes. Telogen consumption, however, varied depending on the thiol used. Final consumptions of 36%, 39%, 46% and 47% were achieved during TBRTs using CHT, PhEM BzM and tBBM respectively. Given that all TBRTs were conducted using a fixed molar ratio of EGDMA to telogen ([EGDMA]0/[Tel]0 = 0.45), these results indicated theoretical crude branched polymer compositions [EGDMA]C/[Tel]C(Theory) = 1.25, 1.15, 0.98 and 0.96 for p(CHT-EGDMA), p(PhEM-EGDMA), p(BzM-EGDMA) and p(tBBM-EGDMA) respectively. GC analysis therefore clearly demonstrated that telogen compositions within crude branched polymers increased with CT (Fig. 4). Furthermore, these values indicate that theoretical telomer subunit DPn = 2.50, 2.30, 1.96 and 1.92 were achieved during TBRTs of EGDMA with CHT, PhEM, BzM and tBBM respectively. This calculation has been previously described but, in summary, an ideal TBRT polymer will contain n MVT residues, 2n vinyl group residues, and n + 1 telogen residues allowing the number average kinetic chain length of the telomer subunits (or DPn) to be simply calculated as the ratio of vinyl group residues per telogen.29 The telomer subunit DPn values provide strong evidence that telogen CT impacts the extent of branching during TBRT via influence over telomer chain length. The Mw values are also consistent with the increase in [EGDMA]C/[Tel]C(Theory) and the expected higher number of taxogens in the final branched polymer structure, based on theoretical calculations adhering to the Flory–Stockmayer limits of gelation in step-growth polymerisations (ESI Fig. S12†).35–39
The different induction periods and rates of vinyl group consumption made direct comparison of the chain transfer kinetics complex. Plots of the consumption of vinyl groups vs. telogen were therefore constructed from kinetic data to investigate the impact of thiol structure (Fig. 5a) within these reactions. Vinyl group consumption per telogen appears to increase with decreasing thiol CT under these TBRT conditions; for example, at 20% telogen conversion, 1H NMR analysis showed that TBRTs with CHT, PhEM, BzM and tBBM had achieved approximately 67%, 59%, 56% and 51% vinyl conversion respectively. Furthermore, this analysis demonstrates that vinyl group consumption per telogen followed the order of tBBM < BzM < PhEM < CHT at all stages of the telomerisation.
 | ||
| Fig. 5 Monitoring chain transfer kinetics during TBRTs of EGDMA. Analysis of kinetic data to show: (A) relative vinyl consumption with telogen conversion (CHT (blue squares), PhEM (purple hexagons), BzM (red diamonds) and tBBM (grey circles)). (b) Determination of chain transfer coefficient of CHT (hollow blue squares), PhEM (hollow purple hexagons), BzM (hollow red diamonds) and tBBM (hollow grey circles). | ||
An analysis was also carried out to determine CT vales for each telogen during TBRT. Our previous reports of TBRT have shown that the combination of relatively small vinyl-functional structures lead to molecular weight increase following a process analogous to step-growth polymerisation.29
Methods for determination of CT which rely on molecular weight data, for example Mayo's method, are therefore unsuitable for determination of telogen CT values in TBRT, and molecular weight independent methods are more appropriate. Smith's method offers an elegant alternative to obtain CT values simply by measuring changes in the relative concentrations of thiol and vinyl groups (eqn (1)),40 and has been used in thiol chain transfer agent-mediated FRP of styrenes and methacrylates.41,42
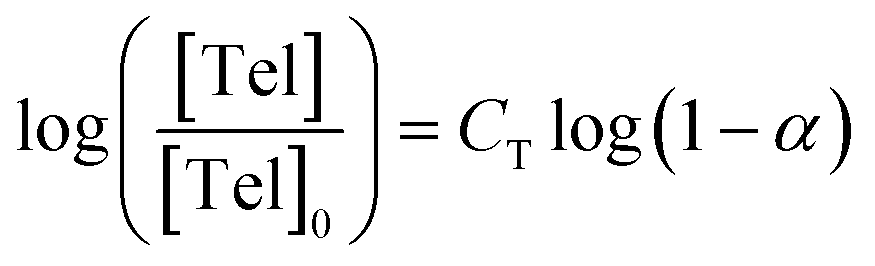 | (1) |
Application of Smith's method to TBRT involves generating logarithmic plots of telogen ([Tel]/[Tel]0) and vinyl consumption (α) from the kinetic measurements. In all cases studied here, the Smith plots showed a linear increase in −log([Tel]/[Tel]0) vs. −log(1 − α) during the early stages of TBRT (Fig. 5b), allowing determination of CT values for the four thiol-based telogens used in this study. CT values of 0.18, 0.21, 0.25, and 0.40 were obtained for CHT, PhEM, BzM, and tBBM respectively. These are in close accordance with values reported from Mayo analysis of conventional linear polymerisations, as discussed earlier.29 Significant deviations from linearity occurred within each of these plots at higher monomer conversion (ESI Fig. S13†), presumably due to significant deviations in the molar ratio of EGDMA to telogen, from [EGDMA]0/[Tel]0 in the latter stages of TBRT.43
Model linear telomerisations derived from MMA
Thiol-based telogens with higher CT values are more effective at moderating the growth of p(EGDMA) during TBRT, apparently due to the control of telomer subunit length. To add evidence to this conclusion, a series of model linear telomerisations were conducted using MMA as a surrogate measure of the propensity of each thiol to limit telomer growth within TBRT reactions.Linear MMA telomerisations were conducted in EtOAc under identical conditions using fixed concentrations of MMA (6.41 mol dm−3) and the AIBN (and 0.1 mol dm−3). A fixed nominal molar ratio of MMA (taxogen) to thiol-based telogen ([MMA]0/[Tel]0 = 5) was also utilised, ensuring identical telogen concentrations (1.29 mol dm−3) when investigating the four study thiol telogens. Again, accurate [MMA]0/[Tel]0 ratios were determined by 1H NMR analysis of telomerisation mixtures prior to thermal initiation (Table 1, and ESI Fig. S14–17†).
The model linear telomerisations were conducted over 24 hours at 70 °C, following which 1H NMR analysis of crude reaction mixtures indicated near-quantitative vinyl conversion of the MMA taxogen (≥99%) (ESI Fig. S18–21†). Following solvent removal in vacuo, the crude reaction mixtures were analysed using MALDI-TOF mass spectrometry without further purification (Table 1, ESI Fig. S22–25†).
Detailed analysis of the crude spectra revealed the presence of species matching the exact theoretical molecular weights of linear telomers containing up to 27 MMA repeat units. The length of the telogen chains identified by MALDI-TOF appears to be impacted by the different telogens, assuming no significant differences in the ability of each sample to charging during the analysis. For example, the maximum DP species identified within tBBM-p(MMA) and CHT-p(MMA) (telogen CT = 0.42 and 0.14) molecular weight distributions contained 23 and 27 MMA repeat units respectively. The spectra were used to calculate estimates of telomer Mn and peak molecular weight (Mp) values (Fig. 6Ai–iv, and Table 1), leading to estimated DPn and peak degree of polymerisation (DPp). Simplified distributions were therefore generated to enable a direct comparison of the linear MMA telomer distributions formed using each telogen, Fig. 6.
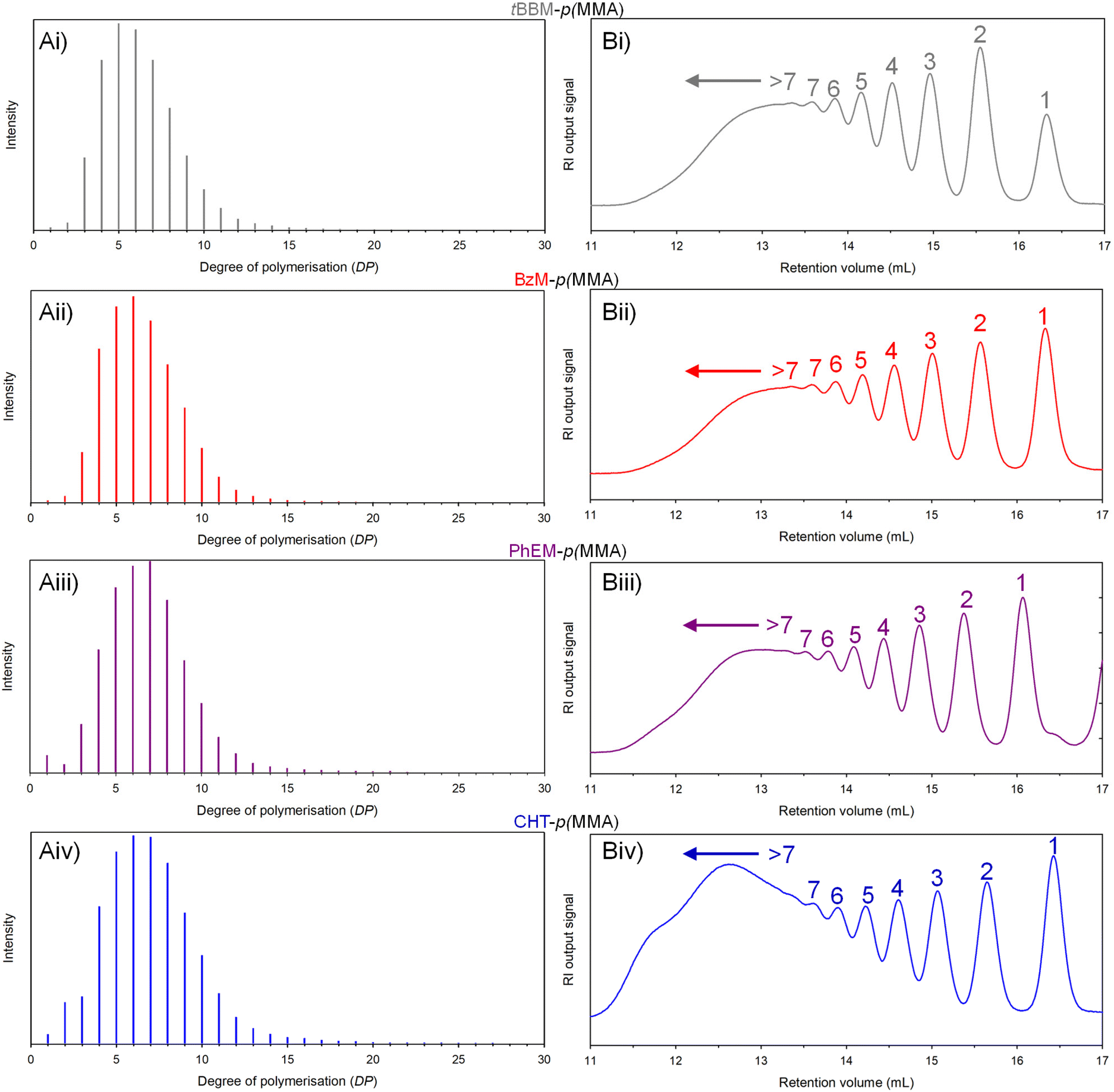 | ||
| Fig. 6 The impact of thiol structure on linear telomer molecular weight distribution. Overlaid (A) MALDI-TOF mass spectra and (B) oligomer-SEC RI chromatograms obtained for (i) tBBM-p(MMA), (ii) BzM-p(MMA), (iii) PhEM-p(MMA) and (iv) CHT-p(MMA). | ||
Using this analysis, telomers prepared using tBBM were shown to have a DPp of approximately 5 monomer units. In contrast, telomer distributions prepared using PhEM and CHT had higher DPp values of approximately 7 units. DPn calculations led to very similar values across the four model telomerisations when using MALDI-TOF data.
To further analyse the model linear telomerisations, single detection oligomer SEC analysis was conducted on the crude reaction mixtures (Fig. 6Bi-iv). Telomers containing DP values up to 7 MMA units were resolved at higher retention volumes and the presence of higher molecular weight telomers were also detected at retention volumes ≤13 minutes. The refractive index (RI) chromatograms of the PhEM-p(MMA) and CHT-p(MMA) telomers showed significantly higher signal intensities at lower elution volumes than their tBBM- and BzM-derived equivalents (ESI Fig. S26†), suggesting the presence of much larger telomers. This supports the detection of higher DP telomers within the CHT-p(MMA) molecular weight distribution observed during MALDI-TOF analysis.
Understanding the limiting [EGDMA]0/[Tel]0 gel point values for EGDMA TBRT using telogens with highly variable CT values
Telomer subunit length varies the intermolecular branching and final molecular weights obtainable when varying the [EGDMA]0/[Tel]0 ratio with the TBRT feedstock. The large differences in CT across the four telogens was therefore expected to highly influence the [EGDMA]0/[Tel]0 ratios at which TBRT results in the formation of an insoluble gel network, previously defined as the limiting [EGDMA]0/[Tel]0 gel point values.31Each telogen was therefore used to generate a series of TBRTs with systematically increasing [EGDMA]0/[Tel]0 values, using the same conditions to those outlined above. All reactions achieved high vinyl group consumption with no indication of gelation. The resulting branched polyesters were purified via precipitation in methanol, dried in vacuo and characterised using TD-SEC and both 1H and 13C NMR spectroscopy, Table 2.
| Telogen | 1H NMR | TD-SEC (THF)e | DSC | |||||
|---|---|---|---|---|---|---|---|---|
| [MVT]0/[Tel]0a |
Conv.b (%) | [MVT]F/[Tel]Fc |
M w (g mol−1) | M n (g mol−1) | Đ | α | T g (°C) | |
| a Calculated by 1H NMR analysis prior to thermal initiation. b Calculated by 1H NMR analysis of crude reaction mixtures after 24 hours. c Calculated by 1H NMR analysis of the purified polymers. d Determined by visual inspection of the reaction mixture after 24 hours and subject to polymers passing through a 0.200 μm filter. Molecular weight values determined using a p(MMA) conventional calibration. e Calculated as (Mn(SEC) − Mr(telogen)/Mr(MMA)). | ||||||||
| CHT | 0.35 | >99 | 1.03 | 59940 |
7550 | 7.89 | 0.356 | 11 |
| 0.40 | >99 | 1.06 | 98530 |
10145 |
9.71 | 0.349 | 26 | |
| 0.43 | >99 | 1.06 | 320020 |
13305 |
24.05 | 0.342 | 31 | |
| 0.45 | >99 | 1.07 | 521120 |
18185 |
28.65 | 0.352 | 33 | |
| 0.48 | >99 | 1.07 | 4927000 |
351770 |
14.01 | 0.528 | 38 | |
| 0.53 | — d | — d | — d | — d | — d | — d | — d | |
| PhEM | 0.50 | >99 | 1.04 | 42538 |
5950 | 7.15 | 0.346 | −8 |
| 0.55 | >99 | 1.00 | 86299 |
4960 | 17.41 | 0.356 | −13 | |
| 0.58 | >99 | 1.08 | 135892 |
9105 | 14.92 | 0.442 | 5 | |
| 0.60 | >99 | 1.06 | 280461 |
11425 |
24.55 | 0.336 | 7 | |
| 0.63 | >99 | 1.06 | 528656 |
17795 |
29.71 | 0.340 | 9 | |
| 0.65 | >99 | 1.06 | 2100000 |
35345 |
59.43 | 0.360 | 14 | |
| 0.67 | — d | — d | — d | — d | — d | — d | — d | |
| BzM | 0.65 | >99 | 1.06 | 116150 |
7020 | 16.55 | 0.319 | 2 |
| 0.68 | >99 | 1.06 | 262405 |
9675 | 27.12 | 0.337 | 11 | |
| 0.70 | >99 | 1.09 | 315530 |
8765 | 36.00 | 0.374 | 6 | |
| 0.73 | >99 | 1.09 | 642015 |
12690 |
50.59 | 0.312 | 14 | |
| 0.75 | >99 | 1.08 | 2,093,000 | 31295 |
66.89 | 0.366 | 16 | |
| 0.78 | — d | — d | —d | —d | — d | — d | — d | |
| tBBM | 0.70 | >99 | 1.02 | 102870 |
6205 | 16.58 | 0.362 | 15 |
| 0.75 | >99 | 1.08 | 314035 |
8200 | 38.31 | 0.409 | 27 | |
| 0.77 | >99 | 1.06 | 638155 |
10230 |
62.37 | 0.327 | 32 | |
| 0.80 | >99 | 1.08 | 774325 |
20590 |
37.61 | 0.339 | 36 | |
| 0.82 | >99 | 1.07 | 1,543,000 | 25795 |
59.81 | 0.455 | 37 | |
| 0.85 | —d | — d | — d | — d | — d | — d | — d | |
As expected, a considerable variation in the limiting [EGDMA]0/[Tel]0 gel point ratios was seen, with values of 0.53, 0.67, 0.78 and 0.85 when using CHT, PhEM, BzM and tBBM respectively, Table 2. Our previous studies of 1-dodecane thiol (DDT, CT = 0.45) as telogen have reported a limiting gel point [EGDMA]0/[Tel]0 ratio of 0.88.30,34
Again, the clear implication of these values is that telogens with low CT values are unable to provide the same level of telomer subunit growth control to limit intermolecular branching. Importantly, this does not prevent molecular weight manipulation overall and [EGDMA]0/[Tel]0 ratios that yield low and very high Mw values are clearly available for all telogens studied. Low CT does, however, compress the range of [EGDMA]0/[Tel]0 values available experimentally. Surprisingly, a plot of limiting gel point [EGDMA]0/[Tel]0 ratio vs. telogen CT showed a very strong linear correlation, Fig. 7, including a literature value for DDT added for additional comparison.
 | ||
| Fig. 7 Relationship between telogen CT and limiting [EGDMA]0/[Tel]0 gel point ratio. CHT (blue square), PhEM (purple hexagon), BzM (red diamond), tBBM (grey circle) and DDT (green triangle). | ||
In agreement with our previous studies, increases in both the Mw and Mn values of each of the branched polyesters was seen with increasing [EGDMA]0/[Tel]0 ratio, regardless of the telogen used, and these rose exponentially as the ratio approached the limiting gel point value (Fig. 8).30,44,45 A considerable shift to lower [EGDMA]0/[Tel]0 values for the onset of the exponential rise of molecular weight is seen, in line with the ordering of telogen CT values, as may be expected. Additionally, the Mark–Houwink–Sakurada α values of the purified polymers spanned a range from 0.319 to 0.528 across the samples produced, confirming the compact nature of the polymers.
 | ||
| Fig. 8 Variation in (A) Mw and (B) Mn in the TBRT of EGDMA at varied [EGDMA]0/[Tel]0 using CHT (blue squares), PhEM (purple hexagons), BzM (red diamonds) and tBBM (grey circles). | ||
1H NMR spectroscopy was also used to determine the final molar ratios of telogen and MVT residues, [MVT]f/[Tel]f, within the purified polymers (ESI Fig. S28–S31†), with a variation from 1.02–1.09. As mentioned earlier, a value of 1.00 would indicate ideal branching and deviation above unity is highly indicative of cyclisation; here we see comparable values across the samples, suggesting a low level of telogen-independent cyclisation (2–8 mol%) is present using previously described equations.31
P(EGDMA) polyester homopolymer thermal properties when synthesised using TBRT and varying telogens
As a pendant group, the telogen structure can impact the thermal properties in a TBRT polymer in a manner that is unique to this chemistry. Previous reports have shown the tuning of thermal properties within a new range of TBRT copolymers, using chemical approaches that are not readily available by other means.34 Three of the polyesters here have not been previously studied, namely p(CHT-EGDMA), p(PhEM-EGDMA), and p(BzM-EGDMA), therefore differential scanning calorimetry (DSC) was used to characterise their glass transition temperature (Tg) behaviour and enable a comparison with previous TBRT examples (Fig. 9; and Table 2). | ||
| Fig. 9 Overlaid differential scanning calorimetry (DSC) thermograms obtained from the highest molecular weight TBRT p(telogen-EGDMA) homopolymers generated using each telogen. | ||
The polymer samples shown in Table 2 allowed a study of the relationship of Tg with increasing molecular weight for each of the four polymer structures. There has been much debate about the mechanism of Tg within branched polymers for many years, with dendrimers initially established as having a peripheral group effect that dominated thermal behaviour, leading to a modification of the Flory–Fox equation (eqn (2)),46 but this has been questioned through comparison of a range of dendrimers showing that a modification of the classical equation was unnecessary.47
Additionally, further suggestions have been made that highly disperse samples should factor in the presence of the high molecular weight fraction within the broad distribution, leading to a modification that includes a square root of the product of Mn and Mw (eqn (3)). This modification approximates very closely to the classical equation when studying monodisperse samples as Mw approaches Mn.
 | (2) |
 | (3) |
Across the range of samples studied the Tg increased with polymer Mn as would be expected. For example, an increase in the Mn of p(tBBM-EGDMA) from 6204 to 20590 g mol−1 led to a Tg increase from 15 to 36 °C. Each of the polymers seemed to follow a classical curve with a clear indication of the onset of a plateau region, although the range of available Mn values was not as wide as would be required to fully establish a Tg∞ value experimentally (Fig. 10).
 | ||
| Fig. 10 Relationship between Mnvs. Tg for (A) p(tBBM-EGDMA), (B) p(CHT-EGDMA), (C) p(PhEM-EGDMA) and (D) p(BzM-EGDMA). Dotted lines show calculated Tg∞ values using either the conventional Flory–Fox equation or modified Flory–Fox calculations that take polydispersity into consideration. | ||
When comparing the highest molecular weight samples formed from each telogen, at [EGDMA]0/[Tel]0 ratios just before the limiting gel values, a clear influence of telogen residue structure on the thermal properties of the polymers was observed (Fig. 9). Tg values of 38 °C, 37 °C, 16 °C, and 14 °C were obtained for p(CHT-EGDMA), p(tBBM-EGDMA), p(BzM-EGDMA) and p(PhEM-EGDMA) respectively. To add comparative data from previous reports, DSC analysis was also conducted on a comparable p(DDT-EGDMA) sample (Mn = 19400 g mol−1), which exhibited a Tg = −48 °C.34
The highly comparable Tg behaviour obtained for p(PhEM-EGDMA) and p(BzM-EGDMA) is somewhat surprising as the additional spacing and flexibility within the PhEM telogen residue may be expected to lead to a slight reduction in any restriction to segmental motion. Although samples with identical Mn and Mw are not available, Tg values of 7 °C and 14 °C for p(PhEM-EGDMA) with an Mn = 11425 g mol−1 (Mw = 280460 g mol−1) and p(BzM-EGDMA) with a comparable Mn = 12690 g mol−1 (Mw = 642015 g mol−1) respectively, do suggest that the extra methylene group does indeed offer less hindrance to the polyester backbone.
Placing a t-butyl substituent on the aromatic ring of the telogen, using the tBBM telogen, has a considerable impact on the thermal behaviour of the EGDMA polyester. The best comparator within the samples shown in Table 2 is a p(tBBM-EGDMA) with an Mn value of 10230 g mol−1 (Mw = 638155 g mol−1) which exhibited a Tg = 32 °C. This a clear 18 °C higher than the comparable p(BzM-EGDMA) and a 25 °C increase over the p(PhEM-EGDMA) sample. The analogy with chain-growth polymers may be drawn through comparison of literature Tg values for poly(styrene) and poly(t-butylstyrene) (Tg = 100 and 127 °C respectively).
The presence of the flexible linear pendant group of p(DDT-EGDMA) has been shown previously to lead to very low Tg values, but somewhat surprisingly, the presence of the cyclic aliphatic telogen-residue within p(CHT-EGDMA) led to a Tg value for the comparable sample (Mn = 13305 g mol−1, Mw = 320020 g mol−1) of 31 °C, which is not dissimilar to the value obtained for the p(tBBM-EGDMA) presumably due to the proximity of the pendant group to the polyester backbone, derived from a secondary thiol, and the contorted nature of the cyclohexyl ring.
As mentioned above the debate about whether the classical Flory–Fox relationship is relevant to highly branched polymers is important to investigate for new materials, thereby adding more experimental data to the discussion. As a number of polymer samples was available with varying molecular weights, calculations of the plateau Tg∞ value were conducted for each branched p(telogen-EGDMA) polyester series. Both the original and modified Flory–Fox relationships, eqn (2) & (3), were utilised. When considering the classical relationship, (ESI Table S6, and Fig. S37†), Tg∞ values of 45 °C, 42 °C, 21 °C and 19 °C were determined for p(tBBM-EGDMA), p(CHT-EGDMA), p(BzM-EGDMA) and p(PhEM-EGDMA) respectively. The values are somewhat higher than those experimentally determined within the series synthesised here, however, a relatively limited range of Mn values is available within the polymers produced. That said, it is unlikely that the samples with the highest Mn values across the four polymer structures are considerably below the critical entanglement molecular weight.38 Importantly, the fit of the linear regression when using the classical Flory–Fox relationship varied considerably from 0.76–0.97 which may indicate the reliability of the outcome of this analysis (ESI Fig. S37†).
The dispersity and the high values of Mw present within TBRT polymers suggests that the modified Flory–Fox relationship may be more reliable, therefore a similar analysis was conducted, but relacing Mn with (Mn·Mw)½, as in eqn (3) (ESI Table S6, and Fig. S38†). The fit of the data to a linear regression was considerably more robust using this approach. As an example, when considering p(BzM-EGDMA) the r2 value improved from 0.76 (Flory–Fox) to 0.91 (modified Flory–Fox). Three of the four polymer series showed an increase in the r2 value, but a decrease was observed for p(PhEM-EGDMA). Analysis of the predicted Tg∞ values using this approach showed consistently lower values than those generated using the classical relationship; Tg∞ values of 40 °C, 39 °C, 18 °C and 15 °C were determined for p(tBBM-EGDMA), p(CHT-EGDMA), p(BzM-EGDMA) and p(PhEM-EGDMA) respectively. These values are much closer to those for the highest molecular weight samples generated during this study, lying within only 1–2 °C of the experimentally derived data for three of the four polymers.
Conclusions
Accessing structural variation within branched step-growth polymers is not trivial. Given conventional branched polymerisation techniques, the synthesis of a range of new ABn monomers is often required to create diverse libraries of materials with varying properties. TBRT offers a relatively facile and scalable synthetic route for the production of a new classes of high molecular weight branched polyesters, with structural variation derived either through bespoke or commercially available MVTs, or simply through telogen selection.When choosing to vary telogen, it is clear from the work presented here that the nature of the pendant group has a considerable impact on physical properties of the resulting branched polymers, but there are several chemical considerations that must be taken into account when planning polymer synthesis. Along with our previous reports of reaction concentration, temperature, [MVT]0/[Tel]0 ratio, and the propensity of the MVT to cyclise, telogen CT values clearly impact both the global polymer molecular weight distribution and the internal telomer sub-unit length distribution within the polymer architecture. Success from TBRT polymerisations therefore has many variables to use to optimise outcomes. For example, a very low CT may be enhanced by increasing reaction temperature, and a low Tg may be improved, if required, by careful selection of telogen.
Glass transition temperature, and its relevance within highly branched polymer architectures, has been subject to much debate, but polymers with very high dispersity and high architectural variation have not been readily available to evaluate different mathematical approaches to Tg determination. At least for the high molecular weight, high disperse, branched materials produced here, the inclusion of Mw into the Flory–Fox relationship appeared to produce a better model of glass transition behaviour. Further studies of this nature are required to establish the generic capability of the modified Flory–Fox relationship to accurately reflect the complex transitions within materials of this class.
TBRT is a new polymerisation tool, offering significant opportunities for manipulation and control over polymer physical and chemical properties. As the study of materials generated from TBRT grows, it is expected to become a valuable asset in the creation of new materials for future applications.
Author contributions
SF contributed to conceptualisation of the study and conducted the bulk of the experimental investigation, data analysis and production of the draft manuscript. BL, OBP-L, SM, and SW conducted aspects of the experimental investigation and data analysis. SRC contributed to methodologies utilised within the study. PC contributed to methodologies, supervision and manuscript review. SPR was responsible for conceptualisation, progress review, supervision, resources, and manuscript review and editing. SF and SPR were responsible for data and concept visualisation.Conflicts of interest
SRC, PC and SPR are co-inventors on patents that protect the TBRT chemistry; these patents have been licensed to Scott Bader and form the basis of Polymer Mimetics Ltd (Company number 12598928).Acknowledgements
The Engineering & Physical Sciences Research Council (EPSRC) are grateful acknowledged for funding through grant EP/R010544/1. OBPL is grateful for studentship funding from EPSRC and the University of Liverpool. SM is grateful for studentship funding from EPSRC and Unilever. BL is grateful for studentship funding from Scott Bader. The authors would like to thank the Materials Innovation Factory (University of Liverpool) for access to analytical facilities and are especially grateful to Stephen Moss for assistance with the collection and processing of gas chromatography data.References
- J. W. Robinson, Y. Zhou, J. Qu, J. T. Bays and L. Cosimbescu, React. Funct. Polym., 2016, 109, 52–55 CrossRef CAS.
- Y. Zheng, K. J. Thurecht and W. Wang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 629–637 CrossRef CAS.
- Z. Ye, F. AlObaidi and S. Zhu, Macromol. Chem. Phys., 2004, 205, 897–906 CrossRef CAS.
- J. D. Simpson, G. R. Ediriweera, C. B. Howard, N. L. Fletcher, C. A. Bell and K. J. Thurecht, Biomater. Sci., 2019, 7, 4661–4674 RSC.
- D. J. Coles, B. E. Rolfe, N. R. B. Boase, R. N. Veedu and K. J. Thurecht, Chem. Commun., 2013, 49, 3836–3838 RSC.
- M. R. Martinez and M. Matyjaszewski, CCS Chem., 2022, 4, 2176–2211 CrossRef CAS.
- H. Sun, C. P. Kabb and B. S. Sumerlin, Chem. Sci., 2014, 5, 4646–4655 RSC.
- H. E. Rogers, P. Chambon, S. Flynn, F. Y. Hern, A. Owen and S. P. Rannard, Nanoscale Adv., 2020, 2, 5468–5477 RSC.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS PubMed.
- H. Chen and J. Kong, Polym. Chem., 2016, 7, 3643–3663 RSC.
- Z. Li, H. Yong, K. Wang, Y.-N. Zhou, J. Lyu, L. Liang and D. Zhou, Chem. Commun., 2023, 59, 4142–4157 RSC.
- Y. Gao, D. Zhou, J. Lyu, S. A. Q. Xu, B. Newland, K. Matyjaszewski, H. Tai and W. Wang, Nat. Rev. Chem., 2020, 4, 194–212 CrossRef CAS PubMed.
- T. Zhao, Y. Zheng, J. Poly and W. Wang, Nat. Commun., 2013, 4, 1873–1881 CrossRef PubMed.
- Y. Gao, B. Newland, D. Zhou, K. Matyjaszewski and W. Wang, Angew. Chem., Int. Ed., 2017, 56, 450–460 CrossRef CAS PubMed.
- Y. Zheng, H. Cao, B. Newland, Y. Dong, A. Pandit and W. Wang, J. Am. Chem. Soc., 2011, 133, 13130–13137 CrossRef CAS PubMed.
- A. Aied, B. Glynn, H. Cao, Y. Zheng, H. Tai, A. Pandit and W. Wang, Polym. Chem., 2012, 3, 332–334 RSC.
- H. Cao, C. Chen, D. Xie, X. Chen, P. Wang, Y. Wang, H. Song and W. Wang, Polym. Chem., 2018, 9, 169–177 RSC.
- K. Kapil, G. Szczepaniak, M. R. Martinez, H. Murata, A. M. Jazani, J. Jeong, S. R. Das and K. Matyjaszewski, Angew. Chem., Int. Ed., 2023, 62, e202217658 CrossRef CAS PubMed.
- N. O'Brien, A. McKee, D. C. Sherrington, A. T. Slark and A. Titterton, Polymer, 2000, 41, 6027–6031 CrossRef.
- F. Isaure, P. A. G. Cormack and D. C. Sherrington, J. Mater. Chem., 2003, 13, 2701–2710 RSC.
- G. Saunders, P. A. G. Cormack, S. Graham and D. C. Sherrington, Macromolecules, 2005, 38, 6418–6422 CrossRef CAS.
- L. Jiang, W. Huang, X. Xue, H. Yang, B. Jiang, D. Zhang, J. Fang, J. Chen, Y. Yang, G. Zhai, L. Kong and S. Wang, Macromolecules, 2012, 45, 4092–4100 CrossRef CAS.
- Q. Jiang, J. Li, W. Huang, D. Zhang, J. Chen, H. Yang, X. Xue and B. Jiang, Polym. Chem., 2017, 8, 4428–4439 RSC.
- W. Huang, W. Gu, H. Yang, X. Xue, B. Jiang, D. Zhang, J. Fang, J. Chen, Y. Yang and J. Guo, Polymers, 2017, 9 Search PubMed.
- W. Huang, J. Li, H. Wu, Z. Ren, S. Wang, Q. Jiang, X. Xue, H. Yang, B. Jiang and J. Chen, Mater. Res. Innovations, 2018, 22, 379–384 CAS.
- W. Huang, Y. Xia, T. Hou, J. Chen, J. Fang, X. Xue, H. Yang, Y. Yang, L. Kong and B. Jiang, Mater. Res. Innovations, 2015, 19, 380–384 CrossRef CAS.
- J. Liu, Y. Wang, Q. Fu, X. Zhu and W. Shi, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1449–1459 CrossRef CAS.
- C. Li, R. Huang, J. Wang and Z. Liu, Chin. J. Polym. Sci., 2013, 31, 1404–1414 CrossRef CAS.
- S. R. Cassin, P. Chambon and S. P. Rannard, Polym. Chem., 2020, 11, 7637–7649 RSC.
- S. R. Cassin, S. Flynn, P. Chambon and S. P. Rannard, RSC Adv., 2021, 11, 24374–24380 RSC.
- O. B. Penrhyn-Lowe, S. Flynn, S. R. Cassin, S. Mckeating, S. Lomas, S. Wright, P. Chambon and S. P. Rannard, Polym. Chem., 2021, 12, 6472–6483 RSC.
- S. Flynn, O. B. Penrhyn-Lowe, S. Mckeating, S. Wright, S. Lomas, S. R. Cassin, P. Chambon and S. P. Rannard, RSC Adv., 2022, 12, 31424–31431 RSC.
- S. R. Cassin, S. Wright, S. Mckeating, O. B. Penrhyn-Lowe, S. Flynn, S. Lomas, P. Chambon and S. P. Rannard, Polym. Chem., 2023, 14, 1905–1914 RSC.
- S. R. Cassin, S. Flynn, P. Chambon and S. P. Rannard, Polym. Chem., 2022, 13, 2295–2306 RSC.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3083–3090 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3091–3096 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3096–3100 CrossRef CAS.
- W. H. Stockmayer, J. Chem. Phys., 1943, 11, 45–55 CrossRef CAS.
- W. H. Stockmayer, J. Chem. Phys., 1944, 12, 125–131 CrossRef CAS.
- W. V. Smith, J. Am. Chem. Soc., 1946, 68, 2059–2064 CrossRef CAS PubMed.
- R. A. Gregg, D. M. Alderman and F. R. Mayo, J. Am. Chem. Soc., 1948, 70, 3740–3743 CrossRef CAS PubMed.
- J. L. O'Brien and F. Gornick, J. Am. Chem. Soc., 1955, 77, 4757–4763 CrossRef.
- M. K. Donald and S. A. F. Bon, Polym. Chem., 2020, 11, 4281–4289 RSC.
- S. Flynn, A. B. Dwyer, P. Chambon and S. Rannard, Polym. Chem., 2019, 10, 5103–5115 RSC.
- S. Flynn, S. D. Dale, A. B. Dwyer, P. Chambon and S. P. Rannard, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3963–3967 CrossRef CAS.
- K. L. Wooley, C. J. Hawker, J. M. Pochan and J. M. J. Frechet, Macromolecules, 1993, 26, 1514–1519 CrossRef CAS.
- A. Stoddart, W. J. Feast and S. P. Rannard, Soft Matter, 2012, 8, 1096–1108 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, full experimental details and characterisation. See DOI: https://doi.org/10.1039/d3py01208e |
| This journal is © The Royal Society of Chemistry 2023 |