
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of uniform OCL-OEG block cooligomers†
Philipp
Bohn
a,
Valerian
Hirschberg
b,
Simon
Buchheiser
c,
Dafni
Moatsou
 d,
Hermann
Nirschl
c and
Michael A. R.
Meier
*ad
d,
Hermann
Nirschl
c and
Michael A. R.
Meier
*ad
aLaboratory of Applied Chemistry, Institute of Organic Chemistry (IOC), Karlsruhe Institute of Technology (KIT), Straße am Forum 7, 76131 Karlsruhe, Germany. E-mail: m.a.r.meier@kit.edu; Web: https://www.meier-michael.com
bInstitute of Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), Engesserstraße 18, 76131 Karlsruhe, Germany
cInstitute for Mechanical Process Engineering and Mechanics (MVM), Karlsruhe Institute of Technology (KIT), Straße am Forum 8, 76131 Karlsruhe, Germany
dLaboratory of Applied Chemistry, Institute of Biological and Chemical Systems – Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology (KIT), Eggenstein-Leopoldshafen, 76344 Germany
First published on 5th October 2023
Abstract
Block copolymers are an interesting class of materials, offering the opportunity to form nanostructured morphologies, making them suitable for a broad range of applications in nanotechnology, medicine, or biotechnology. Especially for the pharmaceutical sector, a uniform structure and a distinct structure–property relationship is desirable to manufacture highly reproducible and tailor-made materials. Herein, we report the synthesis and characterization of uniform (Đ = 1.01) oligo(ε-caprolactone)-oligo(ethylene glycol) (OCL-OEG) block co-oligomers (BCOs). Three different BCOs, varying in the length of the hydrophobic caprolactone segment, were obtained via Steglich esterification of the corresponding homo oligomers. A clear dispersity and composition dependent structure–property relationship based on the thermal properties is observed, compared to identical structures similar in Mn and dispersities of Đ = 1.06, obtained via ring-opening polymerization (ROP) of ε-caprolactone. In addition, increased long-range-order distances L0 with increasing dispersity of the BCOs are found for the formed morphologies after solvent vapor annealing (SVA). These results highlight the importance of uniform structures for a better understanding of the structure–property relationship of block copolymers.
Introduction
Inspired by the highly defined structures of biomacromolecules (e.g. DNA, RNA or proteins), which exhibit architectures of synthetically unreached complexity and thus enable complex natural processes, current research in polymer chemistry focuses on the synthesis of uniform and sequence-defined macromolecules. Especially block copolymers (BCP)s are of interest due to their self-assembly (SA) capability,1,2 which is the basis for several applications including drug delivery,3 organic optoelectronics,4 or as supramolecular materials for membranes.5 The experimental SA of copolymers, supported by theoretical studies such as the Flory–Huggins theory6 or the self-consistent mean field theory (SCMFT),7 has been extensively investigated since the 1960s for the SA in bulk2,8 and in solution.9 In bulk, the SA of a diblock copolymers (AnBm) depends on the Flory–Huggins Parameter, χAB, describing the energy of the thermodynamic immiscibility normalized to kbT of the two homoblocks A and B, the degree of polymerization, N, and the volume fractions of block A (ΦA) and block B (ΦB). More recently, the tuneability and predictability of the formed morphologies, depending on the dispersity10,11,12–15 and the shape of the molecular weight distribution,16–18 have gained interest in polymer chemistry and nanotechnology.However, since the field of polymer chemistry has always been the science of molecular weight distribution and structural dispersity, the synthesis of perfectly defined structures, as present in some biopolymers in nature, remained an unreached goal for a long time.19 In the last decade, progress in the preparation of tailor-made materials was described by controlling the monomer sequence and molecular weight distribution down to uniformity18,20 and insights into the distinct structure–property relationship of macromolecules became accessible. Thus, these structures made a significant contribution to material and life science.21,22
Pioneering work for the synthesis of uniform and sequence-defined structures has been set by Robert B. Merrifield with the development of the Solid-Phase-Peptide-Synthesis (SPPS) in 1963,23 for which he was awarded the 1984 Nobel Prize.24 The concept was transferred to other classes of polymers, such as peptoids,25 glycopeptides,26 or oligonucleotides,27 and has set the foundation for an enormous variety of synthetic sequence-defined macromolecules.22,28 Furthermore, different approaches, including solid-29 and liquid phase,30 or solid supported synthesis,31 single unit monomer insertion (SUMI),32 or template assisted synthesis were investigated.33 In this context, the iterative exponential growth (IEG) strategy was developed, allowing a fast build-up of uniform macromolecules.34
The first studies using an iterative synthesis protocol towards uniform block copolymers was reported by Meijer et al. in 2016.12 The obtained dimethylsiloxane (DMS)-lactic acid BCOs up to the dononacontamer showed distinct order–disorder transitions and were compared to narrowly distributed analogues. An increase in domain spacing and surprising stability of the morphology (increase of TODT), and a decrease of the overall degree of ordering with increasing dispersity was observed.13 The same group has shown the temperature-controlled formation of 1D and 2D nanostructures of hydrazone-oDMS di- and triblockcooligomers.35 The mixing of these discrete BCOs, the tuneability of the phase behavior for blends with Đ ≤ 1.10, as well as the difference to a disperse sample was recently investigated by Fors and Meijer. Furthermore, microphase segregation was observed for disperse oligomer mixtures containing more than 50 mol% of the discrete compound similar in length and volume fraction, underlining the differences of discrete and disperse BCOs.36 In 2020, the group of Hawker demonstrated an efficient and rapid preparation of discrete diblock copolymer libraries using automated chromatographic fractionation of a single narrowly distributed poly(dodecyl acrylate)-block-poly(lactide) (PDDA-b-PLA).14 This technique simplifies the investigations of the phase behavior, and allows insights into new morphologies of novel BCPs. Using the same strategy, oligo(3-hexylthiophene), which showed a distinct crystallization behavior and optical properties, dependent on chain length,37 were described. Moreover, lipid monodisperse PEG derivatives showed a reduced anti-PEG antibody recognition compared to molecular weight disperse analogues.38 The same group demonstrated the application of this method to triblock terpolymers. The purified samples showed an increase in long-range order compared to the parent polymers.39 The stereochemical effect in uniform PLA-PEG BCPs were reported by Kim et al. in 2021. Irregular nanostructures were observed for complementary configured monodisperse poly(lactic acid), whereas triangular and vascular nanostructures were formed for mismatched or stereochemical sequences.40
Here, we report the synthesis of uniform (Đ = 1.01) as well as narrowly distributed (Đ = 1.06) oligo(ε-caprolactone)-oligo(ethylene glycol) (OCL-OEG) block co-oligomers (BCOs), obtained via an iterative exponential approach or ring-opening polymerization, respectively. The PEG-PCL system is of special interest due to its biocompatibility, biodegradability, and application in the pharmaceutical sector as drug delivery systems, where a distinct structure–property relationship and the control of it via chemical reactions is crucial. We aimed for three comparable BCO pairs, varying in the length of the hydrophobic caprolactone block (mOEGn-b-OCLm-TBDMS; n = 16, m = 16, 32, 64; ΦOCL = 0.71–0.92) to investigate their dispersity and constitution dependent structure–property relationship. Comparison of the thermal properties and shifts in the long-range-order distances highlight the difference of uniform and narrowly distributed (Đ = 1.06), often incorrectly labeled as monodisperse, macromolecules.
Results and discussion
OEG-OCL block cooligomers were synthesized via coupling of the monoprotected homooligomers (Scheme 1), which were obtained using an iterative exponential growth strategy. The caprolactone oligomer was prepared according to the procedure of Hawker et al. performing first a base-catalyzed nucleophilic ring opening of ε-caprolactone to obtain the starting monomer, 6-hydroxycaproic acid, HO(CL)1CO2H.41 Half of the sample was protected using a tert-butyldimethylsilyl (TBDMS) ether as protecting group of the alcohol and the other half using a benzyl ester for the carboxylic acid. The orthogonally protected building units TBDMS(CL)1CO2H and HO(CL)1Bn were coupled via a Steglich esterification using N,N′-dicyclohexylcarbodiimide (DCC) and 4-(dimethylamino)pyridine (DMAP) up to the tetramer, TBDMS(CL)4Bn and 4-(dimethylamino)pyridinium p-toluene sulfonate (DPTS) from the octamer on to suppress the reported side reaction towards the unreactive N-acylurea. Afterwards, the sample of the doubly protected tetramer was split, and the protecting groups were cleaved orthogonally. The benzyl ester was deprotected via reductive hydrogenation, affording the carboxylic acid, and the silyl ether was deprotected by treatment with tetra-n-butyl ammonium fluoride (TBAF) under acidic conditions to yield the desired alcohol. By repetition of the iterative reaction cycle (Scheme 1), consisting of a coupling step and subsequent separate deprotection, an oligo caprolactone containing 64 repeating units was obtained in 20 steps in an overall yield of 33.1%, always considering the lowest yield of the divergent step. The general reaction scheme for the synthesis of uniform polycaprolactone is shown in Scheme 1. | ||
| Scheme 1 Reaction scheme for the synthesis of uniform OCL-OEG BCOs (i-TBDMS(CL)n-(EG)16OMe (n = 16, 32, 64)) from the corresponding OCL and OEG homooligomers, which were obtained via an iterative exponential growth strategy; green iteration cycle:41 I: hydrogenation of benzyl ester with H2, Pd/C, in EA at rt. o.n.; II: TBDMS deprotection with TBAF, AcOH, in THF at 50 °C o.n.; III: Steglich esterification with DCC, DMAP in DCM at rt o.n.; purple iteration cycle: IV: hydrogenation of benzyl ether with H2, Pd/C, in EA at reflux o.n.; V: THP deprotection: p-TsOH in MeOH at rt, 36 h; tosylation: NaOH, p-TsCl in H2O/THF at rt, 15 h; VI: ether coupling: KOtBu (potassium tert-butoxide) in THF, 0 – rt, o.n.; THP deprotection: p-TsOH in MeOH at rt, 36 h; methylation: NaH (sodium hydride), MeI in THF, 0 – rt, o.n.; hydrogenation of benzyl ether with H2, Pd/C, in ethanol at rt o.n.; i: prepared via an iterative approac. | ||
Each of the individual products was characterized by 1H, 13C, and IR spectroscopy, as well as MS (Mass Spectrometry) and SEC to confirm their high purity (≥99% SEC purity). The analytical data are provided in the ESI.† Further, the scale, yield, dispersity, and purity (determined by SEC) are summarized in ESI Table 13.† In accordance to the description of Hawker et al.,41 minor impurities that match to the precursor molecules in terms of respective retention times were observed from the synthesis of the hexadecamer on. Therefore, we focused on the purification of the respective compounds via fractionating column chromatography. As an example, a 15.5 mmol (31.8 g) batch of TBDMS(CL)16Bn was applied on a silica column and eluted as slowly as possible to remove impurities with almost similar retention times. Three of these fractionating isolation steps were performed successively, and in total 46 fractions were collected, 31 of which contained the product in purities ranging from 50.2 to >99% (see ESI† for SEC chromatograms and the evaluated data sets). Only the samples with a SEC-purity of >99% were selected for further synthesis (ESI Tables 7–9,† highlighted in green). It is worthwhile to note that the molecular weight distributions of the unpurified and the purified TBDMS(CL)16Bn are almost indistinguishable (ESI Fig. 63†), yet the purification was necessary to claim uniformity, as the separated byproducts observed by SEC (ESI Fig. 60–62†) clearly demonstrate.
After extensive comparison of different synthesis protocols to achieve uniform OEGs,42 a hexadeca(ethylene glycol) HO(EG)16OMe was prepared (see Scheme 1) according to the procedure of Baker et al.43 and Bruce et al.44 The starting unit, tetra(ethylene glycol), was commercially obtained in a purity of 99%. A benzyl (Bn) ether and a tetrahydropyranyl (THP) ether turned out to be the most efficient orthogonal protecting groups. The THP protection was performed according to a procedure of Baker et al.43 and the product THP(EG)4OH was obtained in a yield of 74%, whereas the benzyl group was introduced as described by Bruce et al. leading to product Bn(EG)4OH (83%).44 Having installed the orthogonal protecting groups, the next step was the activation of the hydroxyl group of Bn(EG)4OH into a tosylate Bn(EG)4Ts, thus opening the possibility for an ether coupling of THP(EG)4OH and Bn(EG)4Ts, and thus a chain doubling. Using NaH as a base, 68% yield of Bn(EG)8THP was obtained, whereas KOtBu resulted in a yield of 48% after purification via silica column chromatography (ESI Tables 2 and 3†). A detailed MS study of the occurring side products was described in a previous report and summarized in the ESI Table 1.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 42 Separate deprotection of the orthogonal protecting groups generated the respective unsymmetric alcohols Bn(EG)8OH and THP(EG)8OH in yields >98%. The THP group was cleaved by treatment with p-toluenesulfonic acid in methanol, and the benzyl ether was removed by reductive hydrogenation. By repetition of the tosylation of the benzyl protected species Bn(EG)8OH, and a coupling reaction with THP(EG)8OH, the double protected hexadecamer Bn(EG)16THP was obtained. In order to obtain an identical structure for the uniform and the non-uniform BCOs, the protecting group had to be exchanged. Therefore, the THP protected alcohol was cleaved under acidic conditions, resulting in Bn(EG)16OH in a quantitative yield. Subsequently, a methylation was conducted using methyl iodide in ten-fold excess and NaH as base. Unfortunately, only a decreased yield of 30% of pure product Bn(EG)16OMe and 70% with a purity of 93–98% were obtained after purification via silica column chromatography. In the last step, the benzyl group was deprotected according to a procedure of Haag et al.45 Replacing ethyl acetate with ethanol and conducting the reaction at room temperature instead of reflux, the side reaction was avoided and product HO(EG)16OMe was obtained in a yield of 98% (overall yield of 3% in 11 steps) and was used without further purification. Full characterization by nuclear magnetic resonance (NMR) and infrared (IR) spectroscopy as well as high resolution-electrospray ionization mass spectrometry (HR-ESI MS) of all oligo(ethylene glycol)s THP(EG)4OH – HO(EG)16OMe and a summary of the SEC chromatograms are provided in the ESI.†
42 Separate deprotection of the orthogonal protecting groups generated the respective unsymmetric alcohols Bn(EG)8OH and THP(EG)8OH in yields >98%. The THP group was cleaved by treatment with p-toluenesulfonic acid in methanol, and the benzyl ether was removed by reductive hydrogenation. By repetition of the tosylation of the benzyl protected species Bn(EG)8OH, and a coupling reaction with THP(EG)8OH, the double protected hexadecamer Bn(EG)16THP was obtained. In order to obtain an identical structure for the uniform and the non-uniform BCOs, the protecting group had to be exchanged. Therefore, the THP protected alcohol was cleaved under acidic conditions, resulting in Bn(EG)16OH in a quantitative yield. Subsequently, a methylation was conducted using methyl iodide in ten-fold excess and NaH as base. Unfortunately, only a decreased yield of 30% of pure product Bn(EG)16OMe and 70% with a purity of 93–98% were obtained after purification via silica column chromatography. In the last step, the benzyl group was deprotected according to a procedure of Haag et al.45 Replacing ethyl acetate with ethanol and conducting the reaction at room temperature instead of reflux, the side reaction was avoided and product HO(EG)16OMe was obtained in a yield of 98% (overall yield of 3% in 11 steps) and was used without further purification. Full characterization by nuclear magnetic resonance (NMR) and infrared (IR) spectroscopy as well as high resolution-electrospray ionization mass spectrometry (HR-ESI MS) of all oligo(ethylene glycol)s THP(EG)4OH – HO(EG)16OMe and a summary of the SEC chromatograms are provided in the ESI.†
To obtain the desired block cooligomers i-TBDMS(CL)n-(EG)16OMe (n = 16, 32, 64), the oligocaprolactones TBDMS(CL)nCO2H (n = 16, 32, 64) were coupled via a Steglich esterification with HO(EG)16OMe (Scheme 1). Quantitative conversions were achieved by using equimolar amounts of DPTS and 6.00 equiv. of DCC. The formation of the individual products was first confirmed via SEC, with the appearance of a new signal at retention times of 15.9 min (i-TBDMS(CL)16-(EG)16OMe), 14.2 min (i-TBDMS(CL)32-(EG)16OMe), and 13.2 min (i-TBDMS(CL)64-(EG)16OMe), respectively. For each reaction, separate peaks at earlier retention times indicated the formation of side products of larger hydrodynamic volumes, which were not analyzed further. Similar to the homooligomers, the purification of the three BCOs was challenging via silica column chromatography and the products were obtained in yields of 37% (i-TBDMS(CL)16-(EG)16OMe), 43% (i-TBDMS(CL)32-(EG)16OMe) and 18% (i-TBDMS(CL)64-(EG)16OMe), respectively. Purities of >99% and dispersities of Đ = 1.01 were determined via SEC, respectively. 1H and 13C NMR spectroscopy proved to be helpful to follow the formation of the BCOs. The CH2 signal next to the terminal hydroxyl moiety is shifted upfield from 3.59 to 4.22 ppm in the proton spectrum after esterification (Fig. 1). All other signals remain at the same chemical shift.
 | ||
| Fig. 1 Comparison of 400 MHz 1H NMR spectra of the monomethyl hexadeca(ethylene glycol) HO(EG)16OMe, the carboxyl-terminated hexadeca(ε-caprolactone) TBDMS(CL)16CO2H and the corresponding product i-TBDMS(CL)16-(EG)16OMe. Signal 3 of the mOEG16 is shifted upfield from 3.59 to 4.22 ppm due to the esterification. All other signals remain at the same chemical shift. These spectra are representative for the synthesis of all three BCP. A full characterization for each of them is provided in the ESI.† | ||
COSY and HMBC as well as a comparison of the 13C NMR spectra are provided in the ESI (ESI Fig. 85†) to clarify the peak assignment. DOSY NMR further confirmed the successful coupling reaction (ESI Fig. 86, 97 and 103†). Additionally, HR ESI-MS analysis was performed. The found mass and the experimental isotopic pattern of the single or double charged sodium adducts matched with the calculated m/z values (i-TBDMS(CL)16-(EG)16OMe [M + Na]+m/z calc. 2698.6106, detected 2698.6143; i-TBDMS(CL)32-(EG)16OMe [M + Na]+m/z calc. 4523.6999, detected 4523.6934; i-TBDMS(CL)64-(EG)16OMe [M + 2Na]2+m/z calc. 4100.9427, detected 4100.9558). Unfortunately, impurities of mOEGn-b-OCLm−1 species were observed for i-TBDMS(CL)32-(EG)16OMe (relative intensity, Irel.(mOEG16-b-OCL31[M + 2Na]2+) = 33), and i-TBDMS(CL)64-(EG)16OMe (relative intensity Irel.(mOEG16-b-OCL63[M + 3Na]3+) = 17, see ESI Fig. 98 and 104†) in the ESI-MS spectra. Therefore, only i-TBDMS(CL)16-(EG)16OMe can be considered as truly uniform with respect to MS analysis, whereas i-TBDMS(CL)32-(EG)16OMe and i-TBDMS(CL)64-(EG)16OMe must be classified as non-uniform according to the definition of IUPAC.46
In order to investigate the structure–property relationship in terms of the self-assembly behavior of the amphiphilic block cooligomers, identical structures, similar in Mn with the uniform BCOs, but exhibiting a broader molecular weight distribution, were prepared. First, a base-catalyzed ring-opening polymerization of ε-caprolactone was conducted using a monomethyl oligo(ethylene glycol) (Mn = 750 Da) as macroinitiator. 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD) was utilized as organo-catalyst in 0.5 mol% relative to the monomer according to a procedure of Hedrick, Waymouth and coworkers.47 All chemicals were dried carefully prior to usage to suppress unwanted side products due to hydrolyzation of the ester groups or initiation by water. Furthermore, to prevent intramolecular backbiting or intermolecular chain transfer, increased monomer to initiator ([M]0/[I]0) values were used to reduce the reaction time and thus keeping the dispersity at a relatively low value. Since we aimed for BCOs similar in Mn compared to i-TBDMS(CL)n-(EG)16OMe (n = 16, 32, 64) and therefore the same retention time in SEC (assuming a symmetric peak shape), reaction monitoring via SEC was performed. Five different approaches, varying in their [M]0/[I]0 values, ranging from [M]0/[I]0 = 40–1226 were performed. A linear relationship of the molecular weight depending on the reaction time was observed for all reactions, whereas the dispersity remained constant (Đ < 1.08) up to a reaction time of three hours. Afterwards, a slight increase was recorded as caused by the already mentioned side reactions (see ESI Table 14 and Fig. 111†). To prevent post-polymerization transesterification, the catalyst was quenched with benzoic acid after the respective reaction time. The most promising results were achieved with [M]0/[I]0 = 167 and 52 min (p-TBDMS(CL)17-(EG)17OMe) as reference for i-TBDMS(CL)16-(EG)16OMe, [M]0/[I]0 = 335 and 109 min (p-TBDMS(CL)34-(EG)17OMe) for i-TBDMS(CL)32-(EG)16OMe, and [M]0/[I]0 = 1226 and 225 min (p-TBDMS(CL)74-(EG)17OMe) for i-TBDMS(CL)64-(EG)16OMe. Unfortunately, the formation of a high molecular weight shoulder, as well as a tailing towards higher retention times via mentioned side reactions for p-TBDMS(CL)74-(EG)17OMe could not be suppressed completely or separated via silica column chromatography. In order to have similar structures to the uniform BCOs, the alcohol end-group was subsequently converted into the TBDMS ether. An excess of TBDMS-Cl was used to guarantee quantitative protection, which was confirmed by NMR spectroscopy and SEC (see ESI†). The resulting products were purified via precipitation, washing and afterwards twice by column chromatography, yielding narrow distributed block cooligomers (p-TBDMS(CL)n-(EG)17OMe, n = 17, 34, 74), varying in the domain size of the OCL block. Similar retention times and thus Mn values were obtained from SEC analysis for the three BCO pairs (see Fig. 2), which is important for the following comparison.
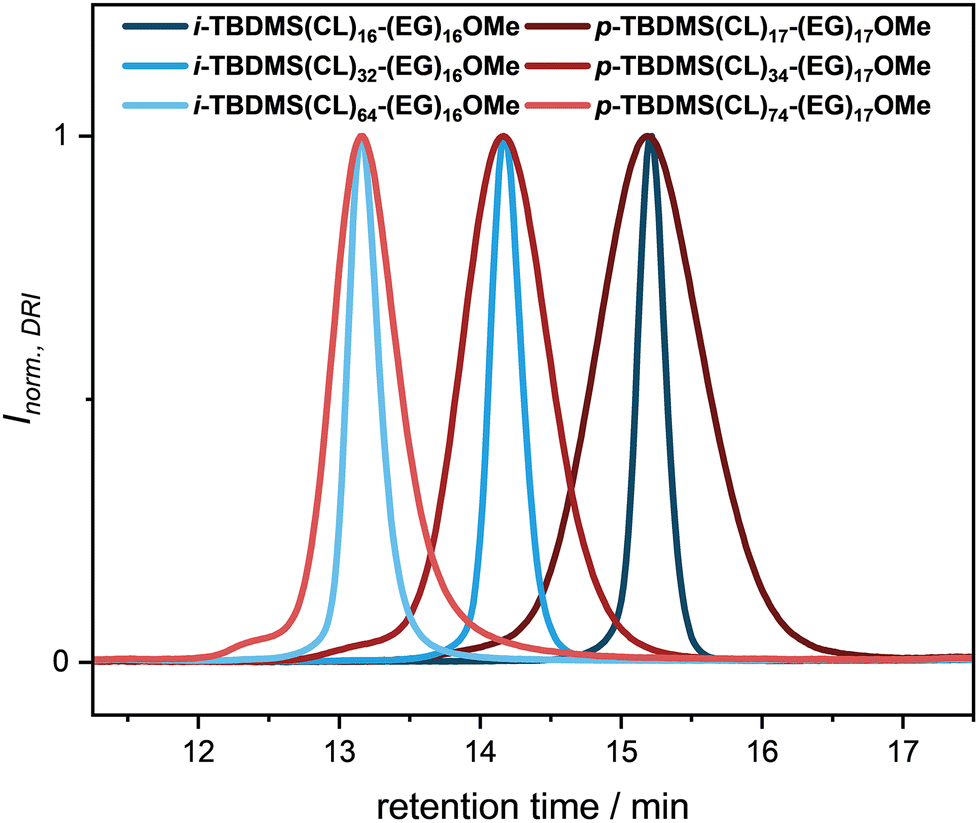 | ||
| Fig. 2 Comparison of the SEC traces of the three uniform BCOs (i-TBDMS(CL)n-(EG)16OMe, n = 16, 32, 64, blue) prepared via an iterative synthesis strategy and the corresponding disperse (p-TBDMS(CL)n-(EG)17OMe, n = 17, 34, 74, red) BCOs obtained from ROP of ε-caprolactone. | ||
To investigate the dispersity-dependent structure–property-relationship in terms of the self-assembly behavior of the presented BCOs, differential scanning calorimetry (DSC) and small-angle X-ray scattering (SAXS) analyses were conducted. The corresponding DSC graphs of the BCO pairs of similar size (uniform (red traces) and non-uniform (blue traces)) are individually compared in Fig. 3a–c. Thermal properties and potential differences in thermal transitions were analyzed using the following heating program with two identical cycles: heating from −15 °C to 70 °C at 20 °C min−1 and subsequent cooling from 70 °C to −15 °C at −10 °C min−1. A general trend of an increase in both the melting temperature, Tm, and the crystallization temperature, Tc, with increasing degree of polymerization, N, of the OCL block was observed, which are summarized in Table 1.
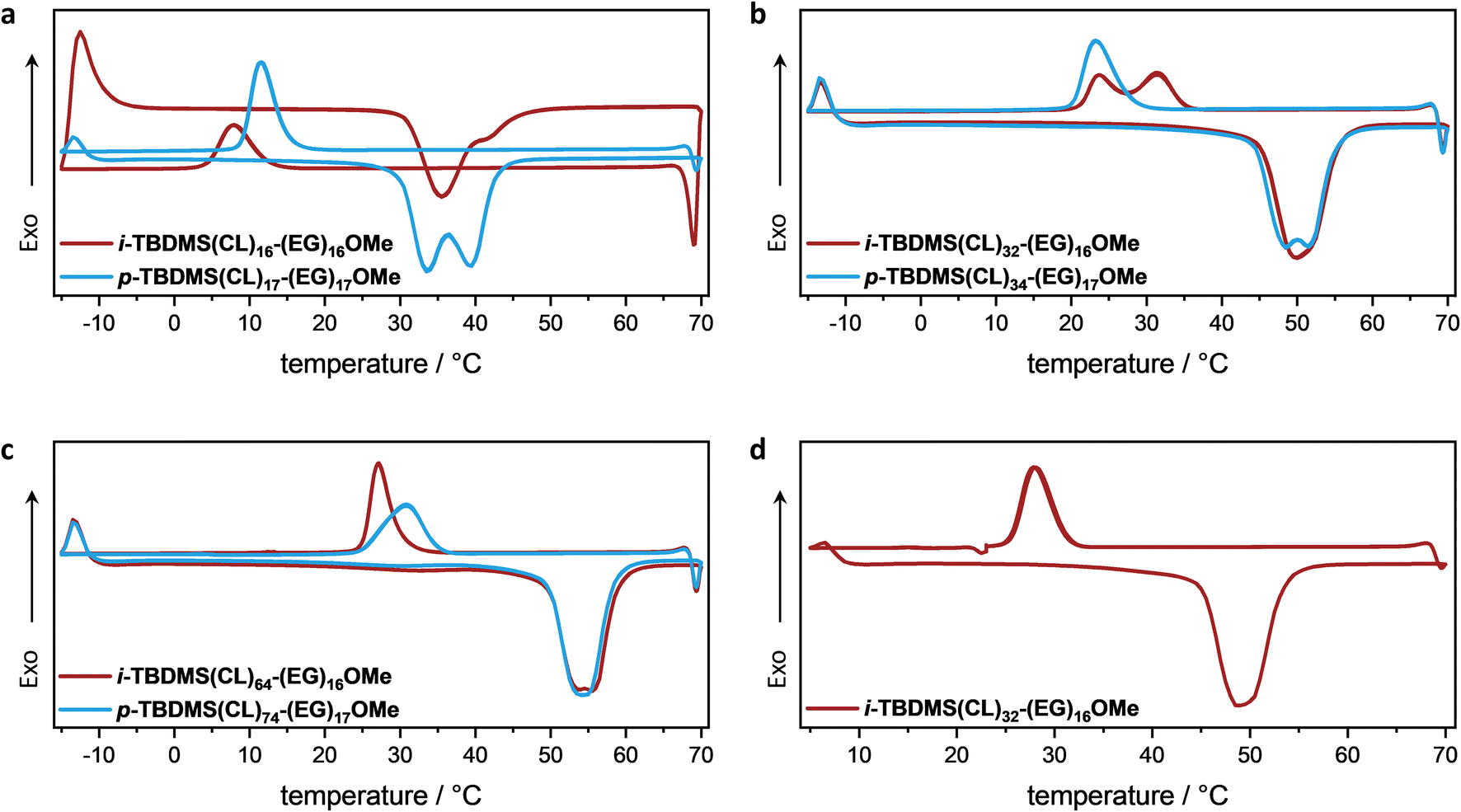 | ||
| Fig. 3 DSC traces of the individual uniform (red) and non-uniform (blue) BCO pairs (a–c) using a heating program with two identical cycles: heating from −15 °C to 70 °C at 20 °C min−1 and subsequent cooling from 70 °C to −15 °C at −10 °C min−1. (d) DSC trace of i-TBDMS(CL)32-(EG)16OMe using a heating program as follows: 5 °C to 70 °C at 20 °C min−1 and subsequent cooling from 70 °C to 23 °C at −10 °C min−1, keeping that temperature for 5 min; cooling from 23 °C to 5 °C at −10 °C min−1. | ||
| Sample | m/mg | T onsetc/°C | T peakc/°C | ΔHc /J g−1 | T onsetm/°C | T peakm/°C | ΔHm/J g−1 |
|---|---|---|---|---|---|---|---|
| i-TBDMS(CL) 16 -(EG) 16 OMe | 5.3 | 13 | 8 | 36 | 31 | 35 | −38 |
| p-TBDMS(CL) 17 -(EG) 17 OMe | 5.3 | 15 | 12 | 56 | 30 | 34/40 | −58 |
| i-TBDMS(CL) 32 -(EG) 16 OMe | 6.5 | 28 | 24/32 | 60 | 44 | 48 | −63 |
| p-TBDMS(CL) 34 -(EG) 17 OMe | 6.5 | 30 | 24 | 60 | 45 | 49/52 | −63 |
| i-TBDMS(CL) 64 -(EG) 16 OMe | 5.5 | 30 | 28 | 58 | 49 | 55 | −57 |
| p-TBDMS(CL) 74 -(EG) 17 OMe | 5.5 | 35 | 31 | 57 | 49 | 54 | −55 |
For the samples i-TBDMS(CL)16-(EG)16OMe and p-TBDMS(CL)17-(EG)17OMe, two melting points were observed for both oligomer samples, with the non-uniform showing two distinct Tms, whereas for the uniform sample a shoulder towards higher temperatures was observed. A clear trend of a decrease in ΔTm of the two melting points with increasing NOCL was observed for the non-uniform samples. For p-TBDMS(CL)17-(EG)17OMe, a difference of ΔTm = 6 °C and 3 °C for p-TBDMS(CL)34-(EG)17OMe were observed, whereas p-TBDMS(CL)74-(EG)17OMe showed only one melting temperature at 54 °C. In comparison, for the uniform BCOs with NOCL = 32 and NOCL = 64, only one melting transition at 48 °C and 55 °C was observed, respectively. For the crystallization transition, a shift towards lower temperatures was observed with decreasing temperature for the samples i-TBDMS(CL)16-(EG)16OMe and i-TBDMS(CL)64-(EG)16OMe compared to the corresponding non-uniform BCOs (p-TBDMS(CL)17-(EG)17OMe and p-TBDMS(CL)74-(EG)17OMe). This behavior could be explained by larger macromolecules only present within the non-uniform samples, which could act as crystallization nuclei. Interestingly, the uniform BCP with NOCL = 32 (i-TBDMS(CL)32-(EG)16OMe) showed two crystallization transitions at Tc = 24 °C and Tc = 32 °C, whereas only one transition at Tc = 24 °C was observed for the corresponding non-uniform sample (p-TBDMS(CL)34-(EG)17OMe). To slow down the crystallization process, the temperature program was adjusted by inserting an isotherm for 5 min at 23 °C (Fig. 3d). Thus, only a single crystallization transition was observed for i-TBDMS(CL)32-(EG)16OMe, indicating the crystallization being slower than the initial cooling rate. Furthermore, no noticeable difference in the comparison of the melting enthalpy, ΔHm, of the uniform and non-uniform samples with NOCL = 32 and NOCL = 64 (see Table 1, please note: for i-TBDMS(CL)16-(EG)16OMe an exothermic phase transition was observed at −13 °C in the heating cycle. Therefore, these results were not considered in the comparison) were observed.
For investigations of the long-range-order distance and to observe first insights into the morphology of the BCOs, SAXS analysis was performed. In this context, the uniform and non-uniform BCOs (i-TBDMS(CL)n-(EG)16OMe, n = 16, 32, 64 and p-TBDMS(CL)n-(EG)17OMe, n = 17, 34, 74) were self-assembled directly on Kapton® foil via either thermal or solvent vapor annealing (SVA) with acetone. For thermal annealing, the sample was heated to 70 °C (21–40 °C above Tonsetm) under vacuum, kept at that temperature for three hours and was subsequently cooled to room temperature overnight. To evaluate the long-range-order distance (domain size, L0 = 2π/q0) in the uniform and non-uniform BCOs, SAXS was performed at room temperature. The 1D SAXS patterns for the thermally annealed samples are shown in Fig. 4a–c. In general, the samples show SAXS reflections at 1q0 and 3q0 consistent with a symmetric lamellar morphology, except for i-TBDMS(CL)16-(EG)16OMetherm. (Fig. 4a, red trace), which had an exact degree of polymerization of N = 16 for both the OEG and OCL block, that lacked the higher-order peak. Compared to the corresponding non-uniform BCO (p-TBDMS(CL)17-(EG)17OMetherm., Fig. 4a, blue trace), a single broad peak at q0 = 0.42 Å−1 was observed indicating a less ordered structure. These observations are inconsistent with the findings of Meijer and Palmans et al. as well as with Hawker, Bates and coworkers, who describe the opposite effect for uniform and non-uniform oligo(DMS-b-LA) BCPs13 and oligo(DMS-b-MMA) BCPs,15 respectively. With a decrease in dispersity, an increase in the (long-range) order was described.13,15 On the other hand, note that the reported difference in dispersity of the compared BCPs is twice as large (ΔĐ ∼ 0.13)15 as for the BCOs described herein (ΔĐ = 0.05). i-TBDMS(CL)16-(EG)16OMe and p-TBDMS(CL)17-(EG)17OMe are similar in their volume fraction of OCL (ΦOCL = 0.71), but the latter BCO contains longer-chain oligomers, which might form an ordered state due to a higher χN value and could thus be decisive for the effect in segregation.
 | ||
| Fig. 4 : SAXS data for the uniform (i-TBDMS(CL)n-(EG)16OMe, n = 16, 32, 64, red traces) and non-uniform BCP (p-TBDMS(CL)n-(EG)17OMe, n = 17, 34, 74, blue traces). Self-assembly via thermal (a–c) or solvent vapor (d–f) annealing on Kapton® foil. | ||
Similar results for the degree of order, as described for the thermal annealing of i-TBDMS(CL)16-(EG)16OMe and p-TBDMS(CL)17-(EG)17OMe, were observed for the self-assembly via SVA in acetone (Fig. 4d). Due to the less pronounced phase separation, a broader peak was observed at q0 = 0.59 Å−1 (Fig. 4a), corresponding to a decrease in the long-range-order distance of ΔL0 = 3.1 nm (L0(i-TBDMS(CL)16-(EG)16OMeSVA) = 10.5 nm) compared to the non-uniform p-TBDMS(CL)17-(EG)17OMe (L0(p-TBDMS(CL)17-(EG)17OMeSVA) = 13.6 nm). A similar trend is observed for i-TBDMS(CL)32-(EG)16OMeSVA (L0 = 13.6 nm), and p-TBDMS(CL)34-(EG)17OMeSVA (L0 = 13.8 nm) (ΔL0 = 0.2 nm, Fig. 4e) as well as i-TBDMS(CL)64-(EG)16OMeSVA (L0 = 12.6 nm) and p-TBDMS(CL)74-(EG)17OMeSVA (L0 = 13.0 nm) with a difference of L0 = 0.4 nm (Fig. 4f).
Thus, even small differences of ΔĐ = 0.05 in relation to the uniform BCOs affected the expansion of the lamellar period for the SVA, resulting in an increase of the primary Bragg reflection q0, which is in agreement with the results of previous experimental reports and predictions by self-consistent field theory (SCFT).10,13,15
However, a contradictory trend was obtained for the samples self-assembled via thermal annealing (Fig. 4a–c). A decrease in the dispersity resulted in an increase of the L0 up to 4% (0.5 nm for N = 32) compared to the non-uniform BCPs. Since the scattering vector, q0, is proportional to the radius of gyration, Rg, which in turn is proportional to N2/3 (assuming lamellar morphologies), shorter chains in a non-uniform oligomer have greater impact on Rg. Thus, smaller values for q0 are expected for a symmetrical widening of the molecular weight distribution, resulting in larger L0 (Table 2, thermal). Similar results were reported by Fors et al. for truly dispersed polymers showing a positively and negatively skewed molecular weight distribution.16 Furthermore, an increase of ΦOCL resulted in an expected increase in L0 for the thermal annealing (excluding the less ordered i-TBDMS(CL)16-(EG)16OMetherm), where an inverted trend was observed for the SVA of the samples in acetone.
| Sample | N OEG | N CL | Đ |
Φ
OCLc |
L
0d/nm |
L
0e/nm |
|---|---|---|---|---|---|---|
| a Determined via SEC (system III). b Determined via1H NMR. c OCL volume fraction using densities of 1.094 g mL−1 for mOEG (Mn = 750 Da) and 1.146 g mL−1 for PCL (average Mw ∼ 14k; average Mn ∼ 10k by SEC). d Long-range order distance calculated via L0 = 2π/q0 (thermal annealing). e Long-range order distance calculated via L0 = 2π/q0 (solvent vapor annealing). | ||||||
| i-TBDMS(CL) 16 -(EG) 16 OMe | 16 | 16 | 1.01 | 0.71 | 14.8 | 10.5 |
| i-TBDMS(CL) 32 -(EG) 16 OMe | 16 | 32 | 1.01 | 0.83 | 14.6 | 13.6 |
| i-TBDMS(CL) 64 -(EG) 16 OMe | 16 | 64 | 1.01 | 0.91 | 16.1 | 12.6 |
| p-TBDMS(CL) 17 -(EG) 17 OMe | 17a | 17b | 1.06 | 0.71 | 14.1 | 13.6 |
| p-TBDMS(CL) 34 -(EG) 17 OMe | 17a | 34b | 1.06 | 0.83 | 14.1 | 13.8 |
| p-TBDMS(CL) 74 -(EG) 17 OMe | 17a | 74b | 1.06 | 0.92 | 15.8 | 13.0 |
In summary, a clear difference in the self-assembly behavior of the uniform and non-uniform OCL-OEG BCOs with ΦOCL = 0.71 was demonstrated for thermal as well as solvent vapor annealing. Furthermore, an increase of the long-range-order distance L0 with increasing dispersity was obtained for all BCOs via SVA, which is in accordance with the literature. Contradictory results were obtained for both annealing processes and no expected narrowing of the signals was observed. However, differences in solvent and thermal annealing are a literature-known phenomenon.48
Conclusion
We describe the synthesis of uniform OCL-OEG block cooligomers via a Steglich esterification of the corresponding OEG and OCL homooligomers, which were prepared via an iterative exponential growth strategy. A OCL containing 64 repeating units was obtained in 20 steps in an overall yield of 33% according to the optimized reaction protocol of Hawker et al.41 TBDMS ether and benzyl ester were employed as orthogonal protecting groups. Consistently high yields for both the deprotection (>95%) and the coupling steps (>83%) were achieved. For the synthesis of uniform OEGs, THP and benzyl ether as protecting groups and KOtBu as base for the etherification showed the most promising results and a uniform mOEG16 was obtained in 11 steps in an overall yield of 3%. Three uniform OCL-OEG BCOs (Đ = 1.01), varying in the length of the OCL domain, were synthesized. Identical structures, similar in Mn with the uniform BCO, but exhibiting a slightly broader molecular weight distribution (Đ = 1.06) were prepared via ROP of ε-caprolactone and the influence of the dispersity on thermal properties and morphologies obtained via self-assembly through thermal and solvent-vapor annealing was investigated by DSC and SAXS analysis, respectively. Significantly increased crystallization temperatures, Tc, were observed for p-TBDMS-(CL)17-(EG)17OMe and p-TBDMS-(CL)34-(EG)17OMe in comparison with their uniform analogues. Furthermore, SAXS analysis revealed an increase in the long-range order distance, L0, and a less pronounced phase separation for i-TBDMS(CL)16-(EG)16OMe, with a decrease in dispersity of the BCOs.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
Parts of this work was supported by the DFG within the framework of the collaborative research center 1176 (SFB 1176, project C3). The authors would like to thank Prof. Dr. Manfred Wilhelm for helpful discussions and the analytical team from KIT for analytical support.References
- (a) Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC; (b) S. B. Darling, Prog. Polym. Sci., 2007, 32, 1152 CrossRef CAS; (c) A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267 CrossRef CAS PubMed.
- P. Alexandridis and B. Lindman, Amphiphilic block copolymers. Self-assembly and applications, Elsevier, Amsterdam, 1st edn, 2000 Search PubMed.
- A. Rösler, G. W. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270 CrossRef.
- R. A. Segalman, B. McCulloch, S. Kirmayer and J. J. Urban, Macromolecules, 2009, 42, 9205 CrossRef CAS.
- H.-A. Klok and S. Lecommandoux, Adv. Mater., 2001, 13, 1217 CrossRef CAS.
- (a) M. L. Huggins, J. Chem. Phys., 1941, 9, 440 CrossRef CAS; (b) P. J. Flory, J. Chem. Phys., 1942, 10, 51 CrossRef CAS.
- L. Leibler, Macromolecules, 1980, 13, 1602 CrossRef CAS.
- (a) F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32 CrossRef CAS; (b) S. Förster and T. Plantenberg, Angew. Chem., Int. Ed., 2002, 41, 688 CrossRef; (c) J. K. Kim, S. Y. Yang, Y. Lee and Y. Kim, Prog. Polym. Sci., 2010, 35, 1325 CrossRef CAS; (d) M. C. Orilall and U. Wiesner, Chem. Soc. Rev., 2011, 40, 520 RSC.
- (a) J. C. van Hest, D. A. Delnoye, M. W. Baars, M. H. van Genderen and E. W. Meijer, Science, 1995, 268, 1592 CrossRef CAS PubMed; (b) L. Zhang, K. Yu and A. Eisenberg, Science, 1996, 272, 1777 CrossRef CAS PubMed; (c) L. Zhang and A. Eisenberg, Science, 1995, 268, 1728 CrossRef CAS PubMed.
- N. A. Lynd and M. A. Hillmyer, Macromolecules, 2005, 38, 8803 CrossRef CAS.
- (a) K. E. B. Doncom, L. D. Blackman, D. B. Wright, M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2017, 46, 4119 RSC; (b) O. Terreau, L. Luo and A. Eisenberg, Langmuir, 2003, 19, 5601 CrossRef CAS; (c) S. R. George, R. Champagne-Hartley, G. A. Deeter, J. D. Campbell, B. Reck, D. Urban and M. F. Cunningham, Macromolecules, 2017, 50, 315 CrossRef CAS; (d) Y. Hirai, T. Terashima, M. Takenaka and M. Sawamoto, Macromolecules, 2016, 49, 5084 CrossRef CAS; (e) A. Buckinx, M. Rubens, N. R. Cameron, C. Bakkali-Hassani, A. Sokolova and T. Junkers, Polym. Chem., 2022, 13, 3444 RSC; (f) E. Konishcheva, D. Häussinger, S. Lörcher and W. Meier, Eur. Polym. J., 2016, 83, 300 CrossRef CAS; (g) Y. Jiang, M. R. Golder, H. V.-T. Nguyen, Y. Wang, M. Zhong, J. C. Barnes, D. J. C. Ehrlich and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 9369 CrossRef CAS PubMed; (h) M. R. Golder, Y. Jiang, P. E. Teichen, H. V.-T. Nguyen, W. Wang, N. Milos, S. A. Freedman, A. P. Willard and J. A. Johnson, J. Am. Chem. Soc., 2018, 140, 1596 CrossRef CAS.
- B. van Genabeek, B. F. M. de Waal, M. M. J. Gosens, L. M. Pitet, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 4210 CrossRef CAS.
- B. van Genabeek, B. F. M. de Waal, B. Ligt, A. R. A. Palmans and E. W. Meijer, ACS Macro Lett., 2017, 6, 674 CrossRef CAS PubMed.
- C. Zhang, M. W. Bates, Z. Geng, A. E. Levi, D. Vigil, S. M. Barbon, T. Loman, K. T. Delaney, G. H. Fredrickson, C. M. Bates, A. K. Whittaker and C. J. Hawker, J. Am. Chem. Soc., 2020, 142, 9843 CAS.
- B. Oschmann, J. Lawrence, M. W. Schulze, J. M. Ren, A. Anastasaki, Y. Luo, M. D. Nothling, C. W. Pester, K. T. Delaney, L. A. Connal, A. J. McGrath, P. G. Clark, C. M. Bates and C. J. Hawker, ACS Macro Lett., 2017, 6, 668 CrossRef CAS PubMed.
- D. T. Gentekos, J. Jia, E. S. Tirado, K. P. Barteau, D.-M. Smilgies, R. A. DiStasio and B. P. Fors, J. Am. Chem. Soc., 2018, 140, 4639 CrossRef CAS PubMed.
- D. T. Gentekos and B. P. Fors, ACS Macro Lett., 2018, 7, 677 CrossRef CAS PubMed.
- D. T. Gentekos, R. J. Sifri and B. P. Fors, Nat. Rev. Mater., 2019, 4, 761 CrossRef.
- J.-F. Lutz, Polym. Chem., 2010, 1, 55 RSC.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- (a) P. Nanjan and M. Porel, Polym. Chem., 2019, 10, 5406 RSC; (b) M. A. R. Meier and C. Barner-Kowollik, Adv. Mater., 2019, 31, e1806027 CrossRef PubMed; (c) S. A. Hill, C. Gerke and L. Hartmann, Chem. – Asian J., 2018, 13, 3611 CrossRef CAS PubMed; (d) R. Aksakal, C. Mertens, M. Soete, N. Badi and F. Du Prez, Adv. Sci., 2021, 8, 2004038 CrossRef CAS PubMed; (e) A. S. Knight, E. Y. Zhou, M. B. Francis and R. N. Zuckermann, Adv. Mater., 2015, 27, 5665 CrossRef CAS PubMed; (f) Z. Deng, Q. Shi, J. Tan, J. Hu and S. Liu, ACS Mater. Lett., 2021, 3, 1339 CrossRef CAS.
- S. C. Solleder, R. V. Schneider, K. S. Wetzel, A. C. Boukis and M. A. R. Meier, Macromol. Rapid Commun., 2017, 38, 1600711 CrossRef PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149 CrossRef CAS.
- R. B. Merrifield, Angew. Chem., Int. Ed. Engl., 1985, 24, 799 CrossRef.
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg and C. K. Marlowe, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367 CrossRef CAS PubMed.
- H. Herzner, T. Reipen, M. Schultz and H. Kunz, Chem. Rev., 2000, 100, 4495 CrossRef CAS PubMed.
- S. L. Beaucage and M. H. Caruthers, Tetrahedron Lett., 1981, 22, 1859 CrossRef CAS.
- B. Genabeek, B. A. G. Lamers, C. J. Hawker, E. W. Meijer, W. R. Gutekunst and B. V. K. J. Schmidt, J. Polym. Sci., 2021, 59, 373 CrossRef.
- (a) J. W. Grate, K.-F. Mo and M. D. Daily, Angew. Chem., 2016, 128, 3993 CrossRef; (b) A. Al Ouahabi, M. Kotera, L. Charles and J.-F. Lutz, ACS Macro Lett., 2015, 4, 1077 CrossRef CAS PubMed; (c) S. Martens, J. van den Begin, A. Madder, F. E. Du Prez and P. Espeel, J. Am. Chem. Soc., 2016, 138, 14182 CrossRef CAS PubMed.
- (a) W. Konrad, C. Fengler, S. Putwa and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2019, 58, 7133 CrossRef CAS PubMed; (b) J. C. Barnes, D. J. C. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7, 810 CrossRef CAS PubMed.
- X. Tong, B. Guo and Y. Huang, ChemComm, 2011, 47, 1455 RSC.
- J. J. Haven, J. A. de Neve and T. Junkers, ACS Macro Lett., 2017, 6, 743 CrossRef CAS PubMed.
- (a) S. Ida, T. Terashima, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2009, 131, 10808 CrossRef CAS PubMed; (b) S. Ida, M. Ouchi and M. Sawamoto, Macromol. Rapid Commun., 2011, 32, 209 CrossRef CAS PubMed.
- (a) O. I. Paynter, D. J. Simmonds and M. C. Whiting, J. Chem. Soc., Chem. Commun., 1982, 1165 RSC; (b) G. M. Brooke, S. Burnett, S. Mohammed, D. Proctor and M. C. Whiting, J. Chem. Soc., Perkin Trans. 1, 1996, 1635 RSC.
- B. A. G. Lamers, R. Graf, B. F. M. de Waal, G. Vantomme, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 15456 CrossRef CAS PubMed.
- B. A. G. Lamers, B. P. Fors and E. W. Meijer, J. Polym. Sci., 2022, 61, 929 CrossRef.
- Y. Kim, H. Park, A. Abdilla, H. Yun, J. Han, G. E. Stein, C. J. Hawker and B. J. Kim, Chem. Mater., 2020, 32, 3597 CrossRef CAS.
- J. Chen, A. Rizvi, J. P. Patterson and C. J. Hawker, J. Am. Chem. Soc., 2022, 144, 19466 CrossRef CAS PubMed.
- E. A. Murphy, Y.-Q. Chen, K. Albanese, J. R. Blankenship, A. Abdilla, M. W. Bates, C. Zhang, C. M. Bates and C. J. Hawker, Macromolecules, 2022, 55, 8875 CrossRef CAS.
- Y. Kwon and K. T. Kim, Macromolecules, 2021, 54, 10487 CrossRef CAS.
- K. Takizawa, C. Tang and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 1718 CrossRef CAS PubMed.
- P. Bohn and M. A. R. Meier, Polym. J., 2020, 52, 165 CrossRef CAS.
- Q. Zhang, H. Ren and G. L. Baker, Tetrahedron Lett., 2014, 55, 3384 CrossRef CAS.
- K. Maranski, Y. G. Andreev and P. G. Bruce, Angew. Chem., Int. Ed., 2014, 53, 6411 CrossRef CAS PubMed.
- M. Shan, A. Bujotzek, F. Abendroth, A. Wellner, R. Gust, O. Seitz, M. Weber and R. Haag, ChemBioChem, 2011, 12, 2587 CrossRef CAS PubMed.
- V. Gold, in The IUPAC Compendium of Chemical Terminology, ed. V. Gold, International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC, 2019. https://goldbook.iupac.org/terms/view/U06558, (accessed June 2023) Search PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574 CrossRef CAS.
- X. Gu, I. Gunkel, A. Hexemer and T. P. Russell, Macromolecules, 2016, 49, 3373 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01052j |
| This journal is © The Royal Society of Chemistry 2023 |