
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Strands vs. crosslinks: topology-dependent degradation and regelation of polyacrylate networks synthesised by RAFT polymerisation†
Frances
Dawson
a,
Touseef
Kazmi
b,
Peter J.
Roth
 b and
Maciej
Kopeć
*a
b and
Maciej
Kopeć
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: mk2297@bath.ac.uk
bSchool of Chemistry and Chemical Engineering, University of Surrey, Guildford, Surrey GU2 7XH, UK
First published on 6th November 2023
Abstract
Degradable poly(n-butyl acrylate) networks were synthesised by reversible addition–fragmentation chain transfer (RAFT) polymerisation using a cleavable disulfide diacrylate crosslinker or a cleavable comonomer, dibenzo[c,e]oxepine-5(7H)-thione (DOT). Through the analysis of gelation kinetics, equilibrium swelling ratio and storage modulus, it was found that incorporation of the degradable units in both cases did not significantly impact the mechanical properties of the prepared gels compared to non-degradable controls. While both types of networks were found to readily degrade, either by thiol–disulfide exchange or aminolysis, they produced degradation fragments of different topology, namely monodisperse linear chains in the case of degradable crosslinkers and branched polydisperse fragments in the case of degradable strands. A simple oxidation of thiols to disulfide bonds in air at 30 °C was successfully used to repolymerise both types of degraded fragments back to solid networks through multiple degradation/regelation cycles. Interestingly, the branched, disperse fragments from degradation of the DOT-containing networks repolymerised more readily and produced networks with properties closer to the original material. This is in contrast to polyacrylate gels made by conventional free radical polymerisation (FRP) which do not degrade through cleavable crosslinks, only through cleavable strands. However, these networks cannot successfully reform after degradation. Furthermore, the apparent dependence of the regelation efficiency on topology of the degraded fragments can open a pathway to better understand reprocessing and recycling of crosslinked polymer networks.
Introduction
Crosslinked polymers such as thermosets, elastomers and polymer gels are indispensable engineering materials, with their covalently crosslinked structure responsible for exceptional mechanical and thermal properties. However, this crosslinked structure also makes recycling of these materials challenging.1 In order to afford degradable or reprocessable crosslinked polymers, labile bonds, e.g. based on covalent dynamic chemistry or noncovalent interactions, can be installed into the network structure, and cleaved by some external stimulus.2–5 Beyond recyclable thermosets, degradable and/or reversible crosslinks enable many advanced applications of polymer networks, such as drug delivery systems, or self-healing materials.6–9In addition to cleavable/reversible bond chemistry, their location within the network also significantly affects degradability. For example, degradable units can either be installed in the primary chains (strands) of the polymer network, or within the crosslinks. Indeed, using polydicyclopentadiene synthesised by ring opening metathesis polymerisation (ROMP) Johnson et al. have shown both theoretical and experimental evidence that networks with cleavable strands are more readily degradable than those with cleavable crosslinks.10
The influence of network topology becomes even more pronounced for gels synthesised by free radical polymerisation (FRP). In principle, using a crosslinker with a labile bond incorporated into it should be a simple method of making a degradable network. However, it is apparent that both polymerisation technique and monomer choice can affect the degradability of the resulting networks. For example, disulfide-containing crosslinkers have been used to impart degradability into poly(acrylamide)-based hydrogels synthesised by FRP.11,12 In contrast, there are also reports of crosslinked poly(N-isopropylacrylamide),13 poly(methyl methacrylate)14 and poly(hydroxyethyl acrylate)15 networks not degrading when synthesised by conventional FRP despite possessing cleavable crosslinkers. This is thought to be due to their clustered heterogeneous structure resulting from the kinetics of FRP which produces very high molecular weight, intermolecularly crosslinked chains right from the onset of the reaction.16,17
On the other hand, networks made by reversible deactivation radical polymerization (RDRP) techniques have a more homogenous structure due to the mechanism of controlled chain growth without formation of microgel clusters.18,19 This structure allows gels containing cleavable crosslinkers to degrade when made by RDRP.14,15,20 This difference was shown in work by Konkolewicz, Matyjaszewski et al., where polyacrylate gels containing disulfide crosslinker degraded when synthesised by RDRP techniques, but not when synthesised by FRP.15 The use of RDRP techniques has also allowed for preparation of vinyl-polymer-based vitrimers as demonstrated by Leibler and Sumerlin with respective co-workers.21,22
Another strategy to produce degradable vinyl polymers is to install cleavable units in the polymer backbone using radical ring-opening (co)polymerisation (rROP). Recently, the thionolactone dibenzo[c,e]oxepine-5(7H)-thione (DOT) has been used as a cleavable comonomer for a variety of monomers, including acrylates,23–26 acrylamides,24,27,28 maleimides29 and styrene.14,30 DOT has also been shown to be compatible with a variety RDRP techniques: reversible-addition fragmentation chain transfer (RAFT),24,26,31 atom-transfer radical polymerization (ATRP)29 and nitroxide-mediated polymerisation (NMP).31 The thioester linkage formed by the DOT copolymerisation can be cleaved by various nucleophiles such as primary amines or strong bases. In addition, DOT can be incorporated into crosslinked polymer networks as shown by our group23 and Guillaneuf et al. for polyacrylates32 as well as by Johnson and Guillaneuf and their respective co-workers for polystyrene.30,33
Recently, we have reported the synthesis and degradation of crosslinked poly(n-butyl acrylate-DOT) networks synthesised by FRP, with a critical DOT to crosslinker ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for complete degradation.23 This showed that the cleavable comonomer approach was more suited to impart degradability into gel networks made by FRP than a cleavable crosslinker approach.
:1 for complete degradation.23 This showed that the cleavable comonomer approach was more suited to impart degradability into gel networks made by FRP than a cleavable crosslinker approach.
Around the same time, Kiel et al. reported the use of DOT copolymerised with styrene or tert-butyl acrylate to make degradable linear polymers that could be repolymerised through thiol oxidation to reform the polymer at near identical molecular weight.30 Crosslinked networks containing DOT were also shown to successfully degrade under basic conditions, however these were not repolymerised.
Generally, all DOT-containing networks reported to date have been made by FRP, but the effect of DOT on the synthesis of crosslinked networks by RDRP has not been investigated. In this work, RAFT polymerisation was used to prepare model poly(n-butyl acrylate) (PBA) networks containing either cleavable crosslinks using a disulfide diacrylate (DSDA) crosslinker (Scheme 1) or cleavable strands using DOT (Scheme 2). The effect of each of these strategies on polymerisation kinetics, mechanical properties and degradability of the networks were investigated. Furthermore, these degradation methods produced thiol-functional fragments, allowing their reoxidation back to solid crosslinked networks. The comparison of the regelation efficiency of fragments with different topology provided valuable insights for the future design of recyclable networks.
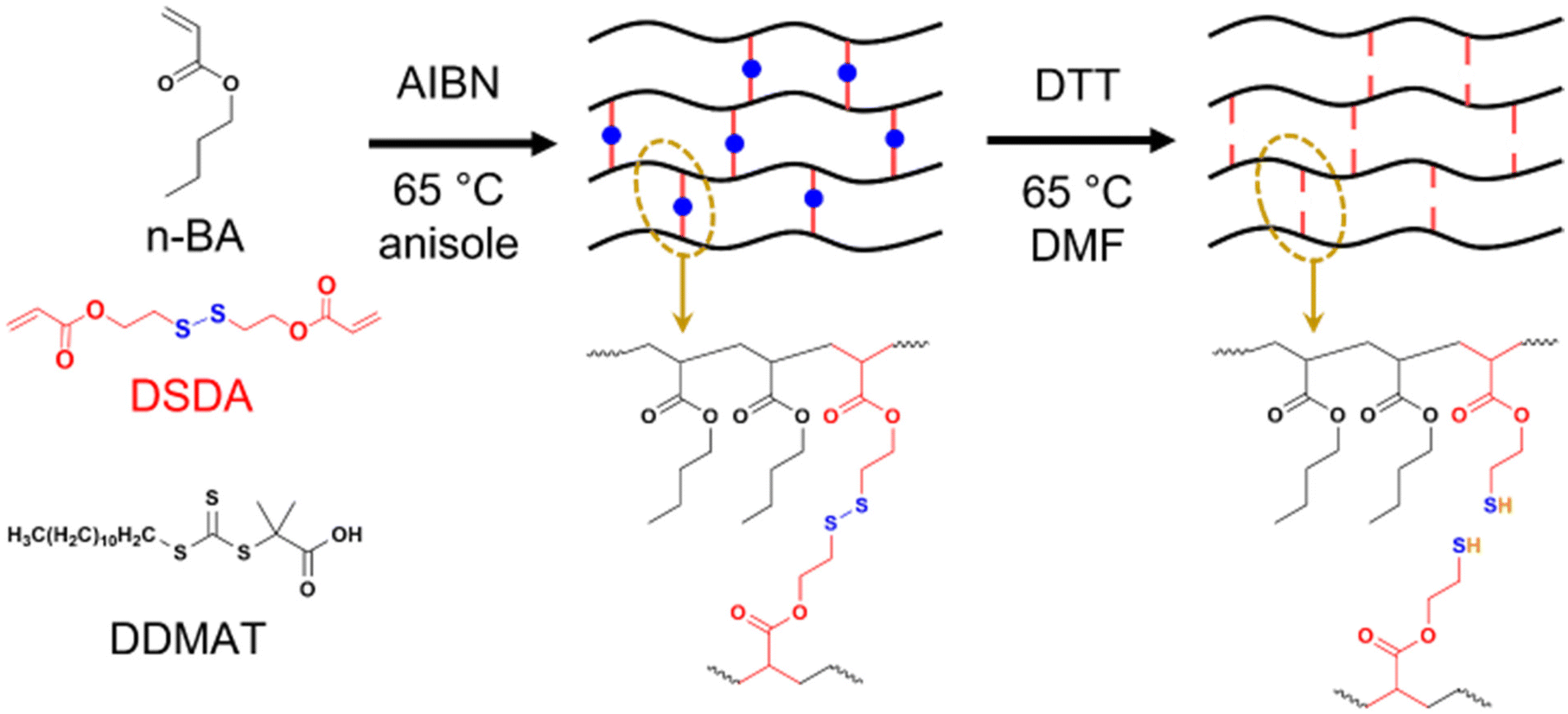 | ||
| Scheme 1 Synthesis of PBA networks containing disulfide crosslinker (DSDA) by RAFT polymerisation and their degradation by thiol–disulfide exchange with dithiothreitol (DTT). | ||
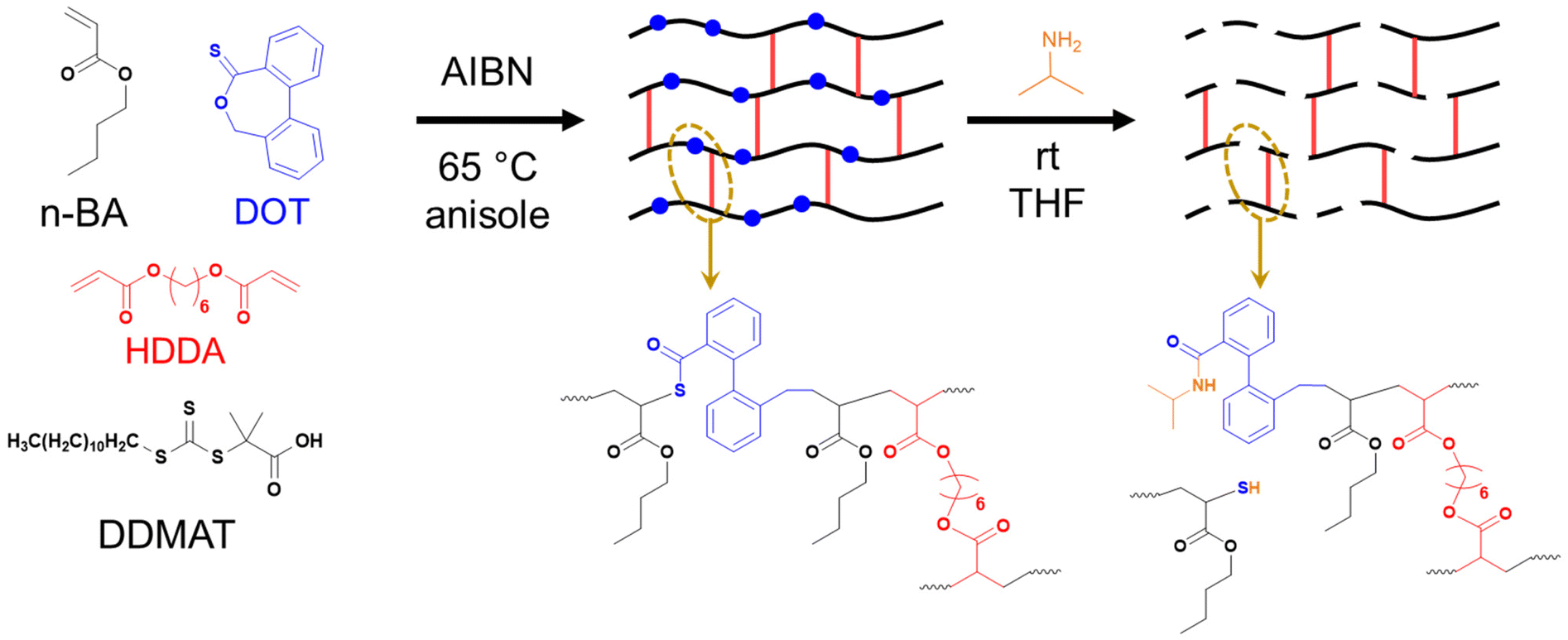 | ||
| Scheme 2 Synthesis of PBA-DOT networks by RAFT polymerisation and their degradation by aminolysis with isopropylamine. | ||
Results and discussion
Synthesis of PBA gels with disulfide crosslinker
To first assess how disulfide crosslinkers impart degradability to polyacrylate networks, PBA gels were synthesised with both a degradable (2,2′-dithiodiethanol diacrylate, DSDA) and non-degradable (1,6-hexanediol diacrylate, HDDA) crosslinker at two different crosslink densities, namely 2 mol% and 4 mol% vs. the monomer (Scheme 1). To compare the effect of using RDRP vs. conventional FRP on how the PBA gels degrade, samples of each crosslink density and type was synthesised using both RAFT polymerisation (Table 1) and FRP (Table S1†).| Sample | Mol% crosslinker | Gelation timea (min) | Estimated acrylate conversion at gel pointb | ESRc | Gel fractiond (%) |
|---|---|---|---|---|---|
| a Defined as the moment when the reaction mixture lost mobility upon inversion. b Acrylate conversion estimated from gelation time and previous conversion measurements prior to gelation. All reactions were conducted for 24 h. c Average value from triplicate measurements of gel equilibrium swelling ratio (ESR), determined gravimetrically by swelling in excess THF for 24 hours. ESR = mswollen/mdry. d Average value from triplicate measurements for different parts of a gel, determined by weighing a fully formed dried gel and solids extracted from the gel upon washing in THF (sol); % GF = mgel/(mgel + msol) × 100%. | |||||
| PBA-DSDA2 | 2% DSDA | 155 | 0.69 | 13.2 ± 0.2 | 65 ± 3.2 |
| PBA-HDDA2 | 2% HDDA | 110 | 0.76 | 13.7 ± 1.2 | 73 ± 1.3 |
| PBA-DSDA4 | 4% DSDA | 125 | 0.39 | 6.1 ± 0.5 | 86 ± 1.4 |
| PBA-HDDA4 | 4% HDDA | 73 | 0.48 | 6.3 ± 0.1 | 88 ± 2.1 |
For polymerisations with 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT) as the RAFT agent (DPtarget = 100), the reactions before the gel point were monitored by 1H NMR showing linear first order kinetics in all cases (Fig. 1). Despite thorough purification procedures applied both monomer and crosslinkers, an induction period was observed in the reactions with DSDA (both RAFT and FRP). However, after the induction time polymerisations with DSDA were virtually identical as those with HDDA. The time for gelation was recorded, and the monomer conversion past gel point was estimated by linear regression of the semilogarithmic kinetic plot. Although the gel times for the DSDA-containing gels were longer than for the HDDA analogues, the gel point conversions were similar, if slightly lower. This extended gelation time is due to an induction time in the PBA-DSDA reactions.
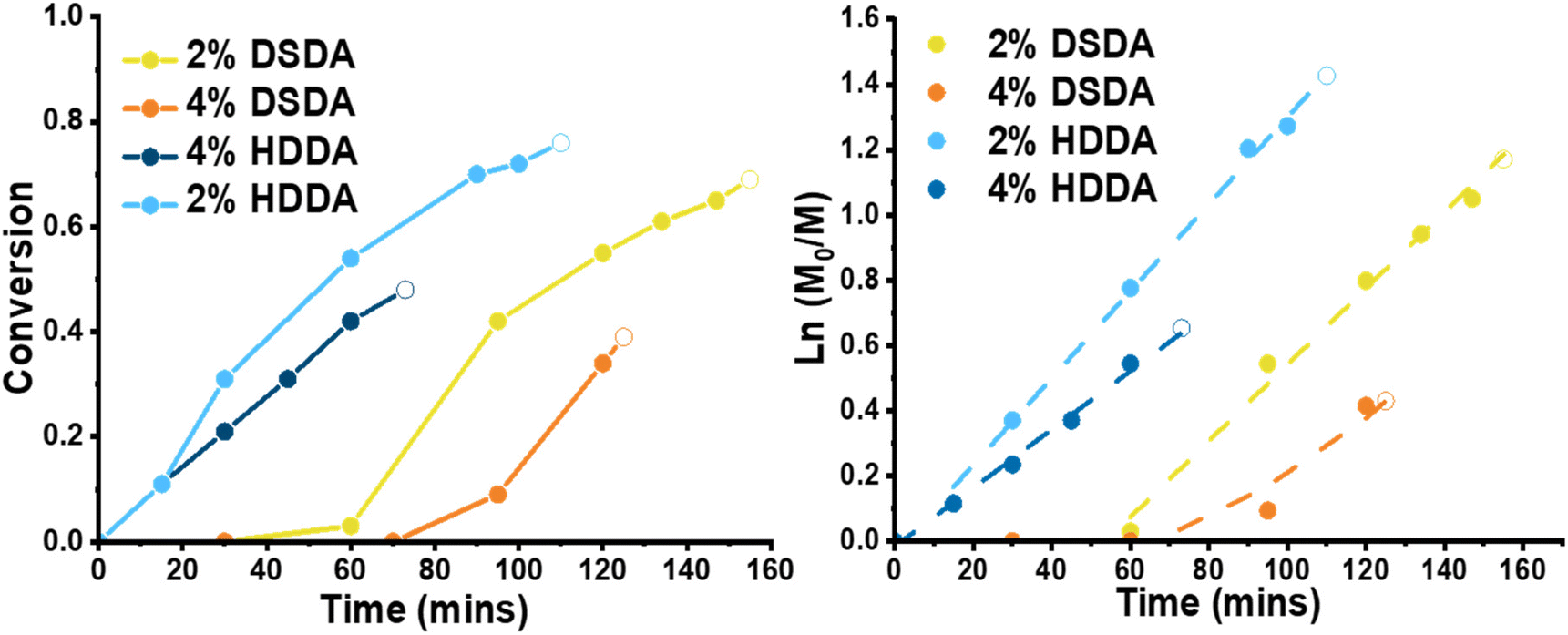 | ||
| Fig. 1 Conversion plots (left) and first-order semi-logarithmic kinetic plots (right) for RAFT polymerisations of PBA-DSDA and PBA-HDDA networks. The final point on each data set (open symbol) is the estimated acrylate conversion at the gel point from gel times and through linear regression of the kinetic plots. [BA]:[DDMAT]:[AIBN]:[X] = 100:1:0.2:2 or 4, X = DSDA or HDDA. BA:anisole 1:1 v/v; 65 °C, 24 h. | ||
When designing a degradable polymer network, the aim is to install degradable units into the network without compromising the material's mechanical properties. For gels synthesised by FRP, there was no difference in equilibrium swelling ratio (ESR) or rheological behaviour between the two types of crosslinker used (Table S1 and Fig. S1†). However, for gels synthesised by RAFT polymerisation, the shorter chain length and more homogeneous network structure allowed for the effect of the disulfide bonds on the mechanical properties of the networks to be visualised (Fig. 2). For 2 mol% crosslinker loading, the PBA-DSDA2 sample had a lower storage modulus (G′) value at 0.1 rad s−1 of 1.3 ± 0.4 kPa, compared to 3.5 ± 0.9 kPa of the corresponding PBA-HDDA2 sample. The gel fraction for the PBA-DSDA2 gel was relatively low, so a significant sol fraction was present in the sample which may have a plasticising effect. In addition, the DSDA-containing networks at both 2 and 4 mol% loadings showed a larger frequency dependence in their storage modulus than the corresponding HDDA gel. This is consistent with other networks containing reversible covalent bonds in environments where the mechanism of reversibility is accessible.34,35 Disulfides are known to show reversible covalent nature in basic conditions,36 and in our system the polymerisation/gelation solvent anisole could be acting as a mild base to facilitate the disulfide–thiol exchange, which would explain the observed frequency dependence. Namely, at low angular frequencies, the timescale probed was longer than the timescale of the disulfide/thiol exchange, so the network had time to restructure and exhibit more flow-like behaviour, thus reducing the elastic response.34 At shorter timescales i.e., at higher angular frequencies, the timescale of the kinetically labile crosslinks was too long to be relevant, and the networks had a similar G′ value as the networks with HDDA with the same average crosslink density. The increased fluid-like property of networks containing DSDA was also evident by their higher phase angle than the corresponding HDDA network (Fig. S2†). The difference in G′ and phase angle between the two types of networks was less prevalent at higher crosslink densities. In comparison, the ESRs are very similar between networks with DSDA and HDDA, made by both RAFT and FRP, as this measurement was at equilibrium with the average effective crosslink density being the same (Fig. 2 and Table S1†).
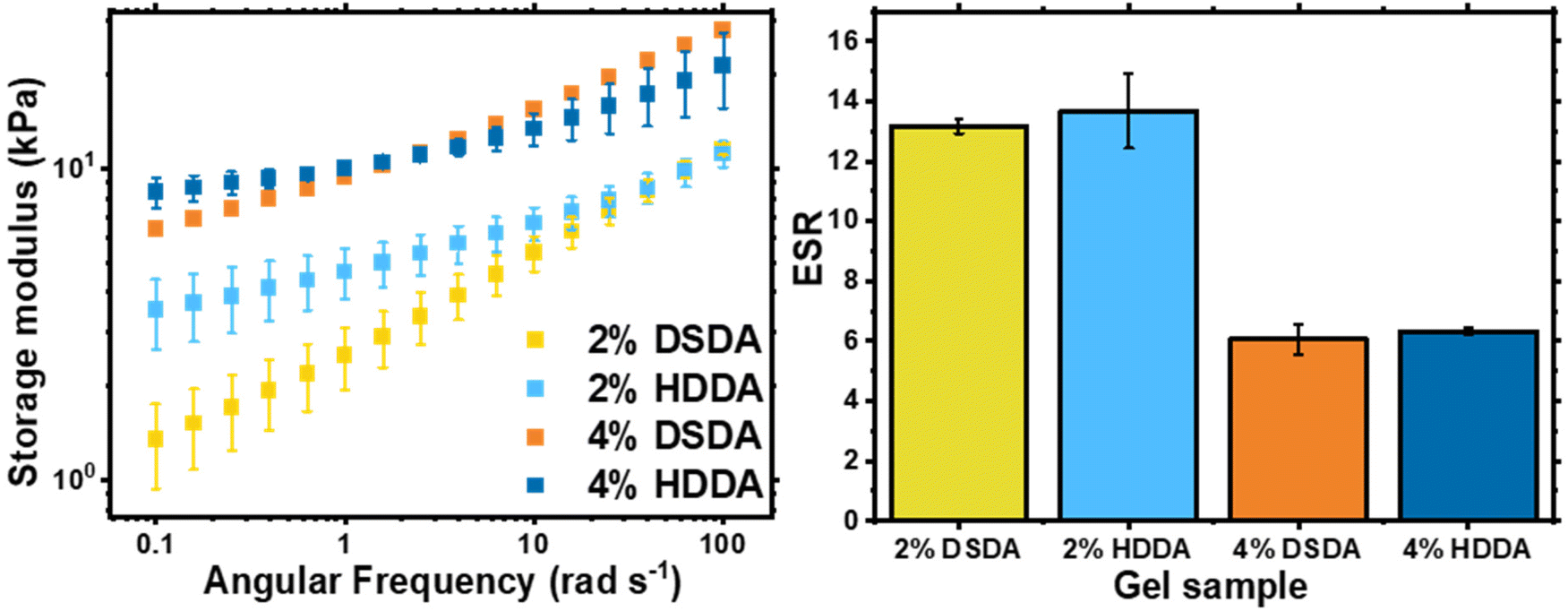 | ||
| Fig. 2 Characterisation of PBA-DSDA and PBA-HDDA networks: shear storage modulus, G′, of as-synthesised gel discs by oscillatory rheology (left) and ESR of gel samples in THF (right). | ||
Degradation of DSDA containing gels synthesised by FRP and RAFT
Degradation of the DSDA containing networks was carried out using a 25 mg mL−1 dithiothreitol (DTT) solution in DMF at 65 °C for 24 hours, according to previously published procedure.15 As expected, the two gels synthesised by RAFT fully degraded under these conditions, whereas the FRP-made gels did not and macroscopic gel fragments were still present in the vial after attempted degradation. This clearly shows that the homogeneous network structures achieved by the controlled growth of polymer chains in RAFT allowed the disulfide bond cleavage to fully degrade the network. The FRP samples did show partial degradation, as the 2% DSDA and 4% DSDA FRP-made gels lost 18% and 9% of their original mass, respectively (Table 2 and Fig. 3). This incomplete degradation could either be due to not enough of the disulfide units being cleaved or due to other structures such as entanglements holding the remaining network together. To evaluate the cleavage of the disulfide units, the ESR of the FRP networks was measured before and after the degradation procedure. The ESR increased from 6 to 21 for 2% DSDA and from 4 to 14 for 4% DSDA, indicating a considerable reduction in effective crosslink density of the remaining gel (Fig. S3†). This suggests that a significant number of the disulfide units were cleaved, but the entangled polymer strands in the clusters likely prevented the material from dissolving. However, it is also possible that dense microgel clusters present in the FRP networks prevented solvent/DTT penetration and therefore cleavage of some disulfide bonds.37 Gels made by both FRP and RAFT containing HDDA were also submitted to this procedure as a control, and they all showed <1% mass loss and no change in ESR after degradation.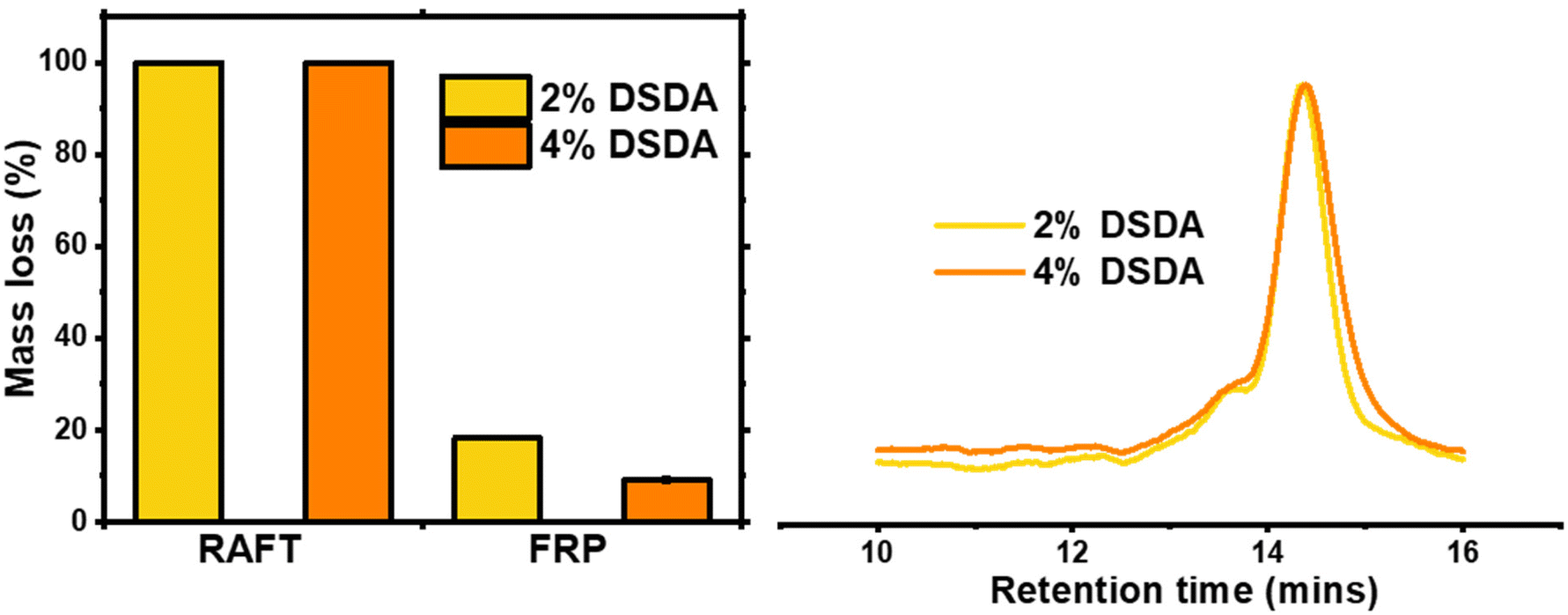 | ||
| Fig. 3 Degradation of PBA-DSDA gels by 25 mg mL−1 DTT solution in DMF. Left: Mass loss % referenced to original mass of dry gel sample before degradation. 100% mass loss corresponds to macroscopic degradation of gel to soluble fragments. Right: GPC traces of soluble fragments from fully degraded PBA-DSDA RAFT networks. | ||
| Polymerisation method | Mol% crosslinkera | Macroscopic degradationb | Mass lossc (%) |
M
nd (g mol−1) |
Đ |
|---|---|---|---|---|---|
| a PBA-HDDA networks showed <1% mass loss for all samples. b Determined visually-macroscopic degradation successful when no solids remained after degradation. c Measured gravimetrically by washing and drying remaining solids after degradation. % mass loss = (mdry − mdry,deg)/mdry × 100%. When full macroscopic degradation was observed, the mass loss was assumed to be 100%. d Measured by GPC; no GPC trace observed for partially degraded samples. | |||||
| RAFT | 2% DSDA | Yes | 100 | 14700 |
1.09 |
| 4% DSDA | Yes | 100 | 14000 |
1.10 | |
| FRP | 2% DSDA | No | 18 ± 0.4 | — | — |
| 4% DSDA | No | 9 ± 0.8 | — | — | |
As the gels synthesised by RAFT fully degraded at both crosslink densities, the solutions containing the degraded fragments were analysed by GPC. The fragments of both samples had very similar molecular weights and dispersity, around 14000 g mol−1 with Đ ≈ 1.10, which is consistent with a targeted DP of 100 (i.e, Mn,theo at full conversion = 13200 g mol−1). As the cleavable units were located in the crosslinks, the degraded fragments were structurally very similar to linear PBA chains synthesised by RAFT with no crosslinker present, which had a Mn of 13500 g mol−1 and a Đ = 1.14 (Fig. S4†). This confirms that the structure of networks synthesised by RAFT are highly uniform, in line with previously published results.15,20
Synthesis of PBA-DOT gels by RAFT
In order to prepare networks with degradable strands, a series of PBA gels were synthesised with a DOT content of 0–5 mol% vs. BA, with a targeted DP of 100 (Scheme 2). A constant crosslink density of 2 mol% was chosen to ensure consistent gelation under RAFT conditions. A parallel reaction with no crosslinker was used to monitor BA and DOT conversion, and the polymerisations were carried out for 24 h to maximise conversion.At all DOT loadings, the RAFT polymerisation was well controlled showing first order kinetics and similar measured Mn to theoretical values. The GPC traces of the linear polymers were all monomodal with low dispersities (1.14–1.25), with a high molecular weight shoulder appearing in the peaks at 24 hours reaction time likely due to some coupling occurring at near-complete monomer conversion (Table 3 and Fig. 4).
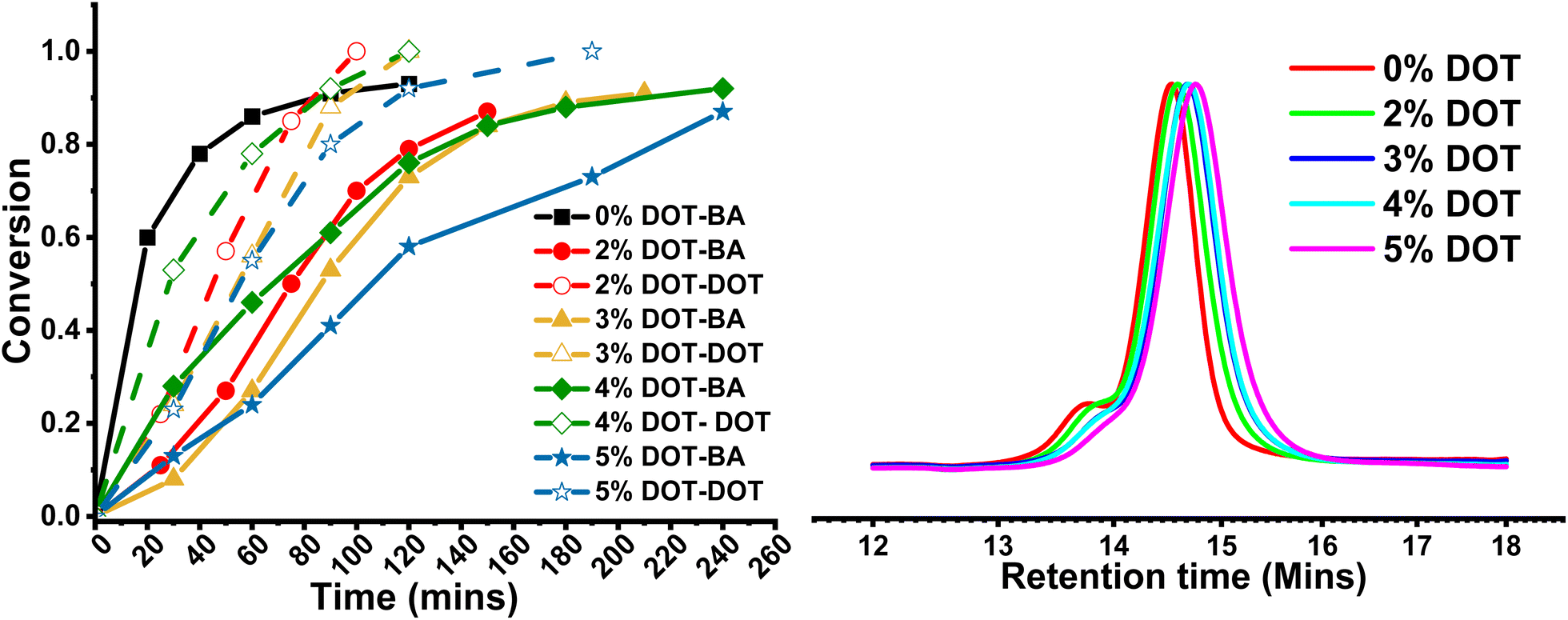 | ||
| Fig. 4 Left: Conversion plots for PBA-DOT-L linear polymers. Closed markers indicate BA conversion, open markers indicate DOT conversion. Right: GPC traces for PBA-DOT linear polymers after 24 h reaction time. [BA]:[DOT]:[DDMAT]:[AIBN] = 100:x:1:0.4, x = 0, 2, 3, 4, 5. BA:anisole 1:1 v/v, 65 °C. | ||
| Sample | DOT mol% | Acrylate conversion at 24 hoursa | Acrylate conversion at 100% DOT conversiona |
M
n,theob (g mol−1) |
M
n,GPCc (g mol−1) |
Đ |
|---|---|---|---|---|---|---|
| a Acrylate and DOT conversion measured using 1H NMR. b Theoretical Mn calculated using measured monomer conversions at 24 hours. c Measured by GPC. | ||||||
| PBA-L | 0 | 0.98 | — | 12900 |
13500 |
1.14 |
| PBA-DOT2-L | 2 | 0.88 | 0.70 | 12100 |
12000 |
1.17 |
| PBA-DOT3-L | 3 | 0.95 | 0.73 | 13200 |
11200 |
1.15 |
| PBA-DOT4-L | 4 | 0.93 | 0.76 | 13200 |
11100 |
1.17 |
| PBA-DOT5-L | 5 | 0.87 | 0.73 | 12600 |
9500 | 1.25 |
We previously reported the effect of DOT on the gelation of n-butyl acrylate (BA) in FRP.23 Increasing the DOT content increased the gelation time and significantly reduced the molecular weight of the polymer. This effect was rationalized through the slower propagation rate of the benzyl radical on the DOT unit compared to the BA unit, with the rate of termination remaining unchanged. The chains produced were shorter which delayed the point of gelation, and the overall polymerisation rate was slower which further increased the time taken to reach the gel point.
Herein, when the networks were produced by RAFT, the gelation time also increased with increasing DOT loading (Table 4). The rate of polymerisation in the presence of DOT was visibly slower than for pure BA, but all reactions reached between 87 and 95% BA conversion after 24 hours. Gel points were then estimated by comparing gelation times with rates recorded for corresponding linear reactions, and found to be similar, as was expected given the same crosslinker content. This suggests that even though gelation time increased with more DOT, the gel points stayed roughly the same.
| Sample | DOT mol% | Gel timea (min) | Acrylate conversion at gel pointb | ESRc | Gel fractiond (%) |
|---|---|---|---|---|---|
| a Defined as the moment when the reaction mixture lost mobility upon inversion. b Estimated from parallel linear PBA-DOT kinetic measurements. c Average value from triplicate measurements of gel equilibrium swelling ratio (ESR), determined gravimetrically by swelling in excess THF for 24 hours. ESR = mswollen/mdry. d Average value from triplicate measurements for different parts of a gel, determined by weighing a fully formed dried gel and solids extracted from the gel upon washing in THF (sol); % GF = mgel/(mgel + msol) × 100%. | |||||
| PBA-G | 0 | 60 | 0.86 | 10 ± 0.4 | 83 ± 1.7 |
| PBA-DOT2-G | 2 | 135 | 0.82 | 11 ± 2.5 | 78 ± 2.4 |
| PBA-DOT3-G | 3 | 165 | 0.86 | 10 ± 0.8 | 77 ± 2.9 |
| PBA-DOT4-G | 4 | 180 | 0.87 | 11 ± 0.5 | 76 ± 2.6 |
| PBA-DOT5-G | 5 | 220 | 0.77 | 13 ± 1.0 | 79 ± 1.0 |
Indeed, GPC analysis of the linear polymers made by RAFT showed only a minimal reduction in Mn with increasing DOT loading (Table 3 and Fig. 4). This is clearly the effect of reversible deactivation by the RAFT agent, which prevents termination despite the lower reactivity of the DOT radical. Thus, in RAFT the delayed gelation time is exclusively due to slower polymerisation rate and not termination and/or any side possible reactions (e.g., branching).
Furthermore, it is known that DOT reacts preferentially in the copolymerisation with acrylates, forming a slight gradient structure.24,28 For each DOT loading the conversion of butyl acrylate at the point where all the DOT has reacted was between 70–76%.
The physical properties of the networks were analysed by rheology and swelling (Fig. 5). As previously shown with RAFT-synthesised gel networks, the storage modulus is frequency dependent. The storage moduli at 0.1 rad s−1 for 0–4 mol% DOT loadings were fairly similar, however at 5 mol% DOT this reduced significantly but did converge on the other samples’ values at high frequencies. This is likely due to the lower Mn and longer gelation time which may result in a less-developed gel with more fluid-like behaviour. The ESR values for the networks were largely similar, with PBA-DOT5 sample having a slightly higher value. Overall, incorporation of DOT into the PBA network had little impact on the physical properties of the gels, at least up to 5 mol% DOT loading.
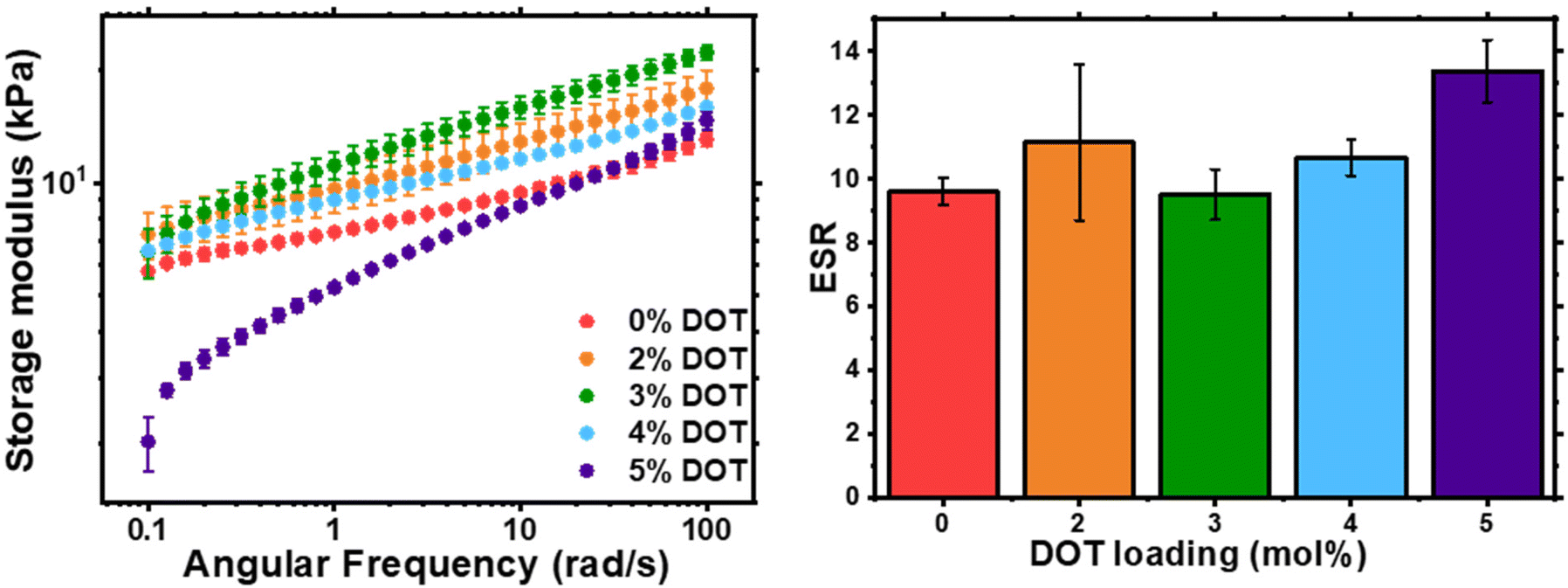 | ||
| Fig. 5 Characterisation of PBA-DOT-G networks: shear storage modulus, G′, of as-synthesised gel discs by oscillatory rheology (left) and ESR of gel samples in THF (right). | ||
Degradation of PBA-DOT RAFT gels
Degradation of the DOT-containing gels was carried out using 5.8 M isopropylamine in THF based on our previous work.23,27 After 24 hours in the solution at room temperature, the PBA-G and PBA-DOT2-G samples remained solid, whereas the samples with higher DOT loading fully degraded into solution (Table 5).| Sample | DOT mol% | Macroscopic degradationa | Mass loss upon degradationb (%) | M n of degraded fragmentsc (g mol−1) | Đ of degraded fragmentsc |
|---|---|---|---|---|---|
| a Determined visually; macroscopic degradation successful when no solids remain after degradation. b Measured gravimetrically by washing and drying remaining solids after degradation. When full macroscopic degradation was observed, the mass loss was assumed to be 100%. c Measured by GPC. | |||||
| PBA-G | 0 | No | 2.7 ± 0.6 | — | — |
| PBA-DOT2-G | 2 | No | 54 ± 1.7 | 5300 | 4.86 |
| PBA-DOT3-G | 3 | Yes | 100 | — | — |
| PBA-DOT4-G | 4 | Yes | 100 | 5500 | 7.26 |
| PBA-DOT5-G | 5 | Yes | 100 | 4600 | 5.26 |
The remaining gels were washed and dried, then weighed to determine the mass loss. As expected, the control PBA-G gel lost very little mass, below 3%-likely due to removal of any remaining sol. The PBA-DOT2-G sample lost 54 ± 1.7% of its original mass, and its ESR increased from 11 ± 2.5 to 28 ± 0.3, indicating a significant amount of the network was cleaved (Fig. 6 and S5†). PBA-DOT3-G, PBA-DOT4-G and PBA-DOT5-G samples all fully degraded; however, the fragments of PBA-DOT3-G could not be filtered through a 0.45 μm PTFE filter and therefore analysed by GPC. This suggests that PBA-DOT3-G is close to the critical DOT loading for degradation of this system.
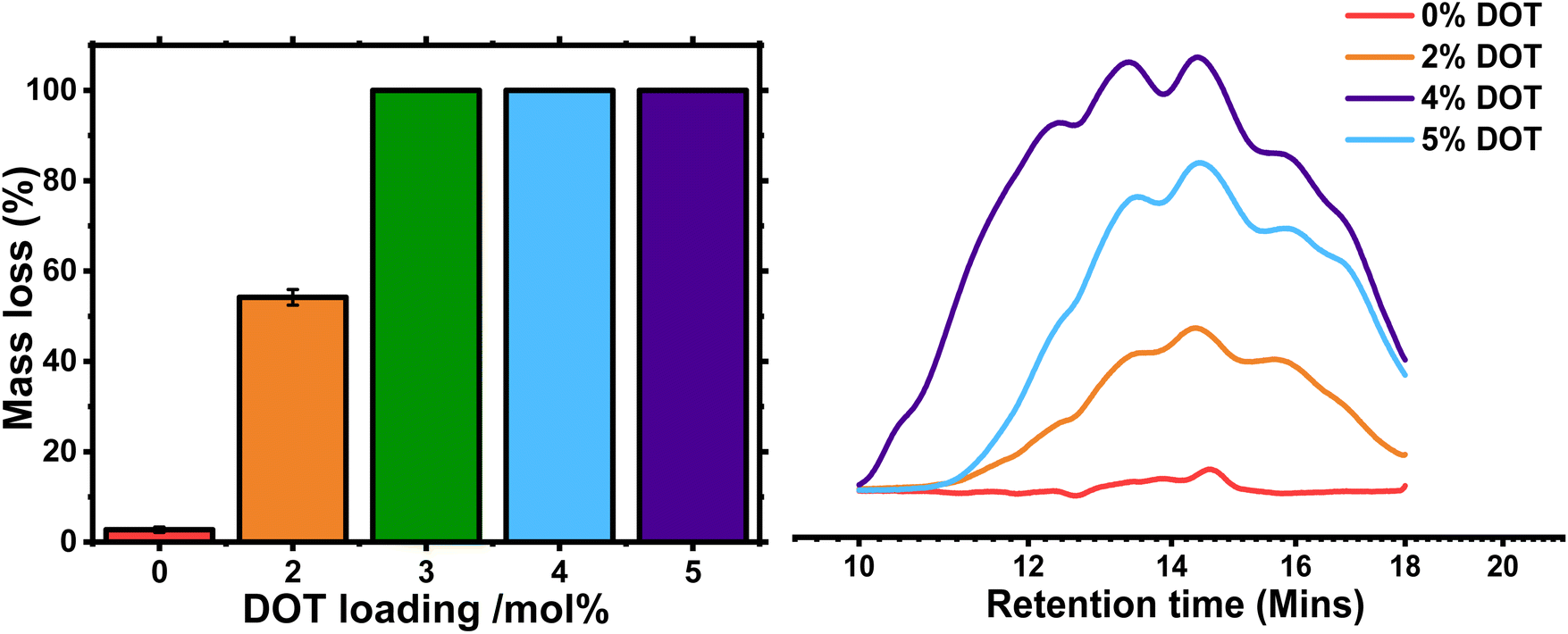 | ||
| Fig. 6 Degradation of PBA-DOT gels by isopropylamine/THF solution. Left: Mass loss % referenced to original mass of dry gel sample before degradation. 100% mass loss corresponds to macroscopic degradation of gel to soluble fragments. Right: GPC traces of soluble fragments from degraded PBA-DOTx networks. No trace was recorded for PBA-DOT3 degraded fragments as the solution could not be filtered for GPC analysis. | ||
Compared to the DOT:crosslinker ratio needed to degrade gels made by FRP of 4:1,23 the amount of DOT needed relative to crosslinker is much lower to degrade the RAFT gels, namely 3:2. Undoubtedly, this is another manifestation of a better homogeneity of polymer networks prepared by RDRP techniques. The degraded fragments were analysed by GPC, and the traces had a high Đ and broad, multimodal distribution, indicative of significant branching (Fig. 6). As the network is cleaved through the polymer backbone, the crosslinks remain intact and produce branched fragments. Indeed, when linear PBA-DOT copolymers were degraded, the fragments were more monomodal with the Mn of the degraded fragments are similar, namely 4600–5500 g mol−1 and dispersities tending towards the most probable distribution (Đ = 1.74–2.03, Fig. S6 and Table S2†), in line with previously reported DOT-containing polymers.24,28
It has been shown that DOT is incorporated preferentially into the PBA-DOT copolymer, and so the final section of the chain contains no cleavable units. Considering this, it is possible that increasing the degree of polymerisation of the network chains while retaining the same DOT:crosslinker ratio could lead to this final fragment becoming insoluble due to its high molecular weight. To investigate this, networks with [BA]:[DDMAT]:[DOT]:[HDDA] ratios of 200:1:4:2 and 500:1:4:2 were synthesised to give networks with a targeted DP (of the primary chains) of 200 and 500 respectively (Table S3 and Fig. S7†).
The two higher DP networks both degraded with isopropyl amine; however, the DPtarget = 500 sample's fragments could not be analysed by GPC due to their size. The GPC traces for degraded fragments of the DPtarget = 200 sample were also multimodal with large Đ, having fragments with roughly double the molecular weight of the DPtarget = 100 sample (Table S4 and Fig. S8†). As the higher DP networks have larger fragments and the DPtarget = 500 could not be filtered, it does suggest that as the DP increases higher, there may be a point where the networks no longer degrade. However, it should be pointed out that even though this sample contained only 0.8 mol% of DOT vs. the monomer, it still underwent full macroscopic degradation. Indeed, the corresponding linear polymers with higher DPtarget all degraded as efficiently as the DPtarget = 100 ones, despite the progressively lower DOT content (Table S5 and Fig. S9†). This indicates that the DOT loading as an additive can be minimised significantly by increasing the length of the polymer chain while retaining similar degradation properties.
Regelation studies
As shown so far, incorporating degradability into poly(n-butyl acrylate) networks synthesised by RAFT polymerisation can be achieved by using a disulfide crosslinker or DOT as a cleavable comonomer. However, the fragments produced from the degradation of the comonomer or crosslinker are structurally very different, therefore next we were interested if that difference in fragments’ topology may affect their ability to repolymerise back to a solid network.Thiol-functional fragments offer a route to reforming the network through oxidation to disulfide linkages (Fig. 7 and 8).11,12,34 The fragments produced by degrading PBA-DSDA gels through dithiol addition already contain multiple thiol functionalities per chain, and the reforming of these disulfide linkages has been shown in hydrogels for medical applications.11,12 In contrast, the degradation of PBA-DOT gels with isopropyl amine produces only one thiol end group per DOT repeat unit. However, using a thiol-functional amine can install a thiol functionality on each end of the degraded fragment, which can also be oxidised to form disulfide linkages. This has been recently shown by Kiel et al. where DOT was copolymerised with styrene to made degradable linear polymers, which were degraded with cysteamine and repolymerised through oxidation with I2/pyridine.30 In the case of our PBA-DOT networks, I2 was not a suitable oxidising agent due to the viscosity of the network fragments and precipitation of I2 at highly concentrated polymer solution. Instead, oxidation in air promoted by base has been shown to successfully oxidise thiols to disulfides, and this method was chosen for further studies.38
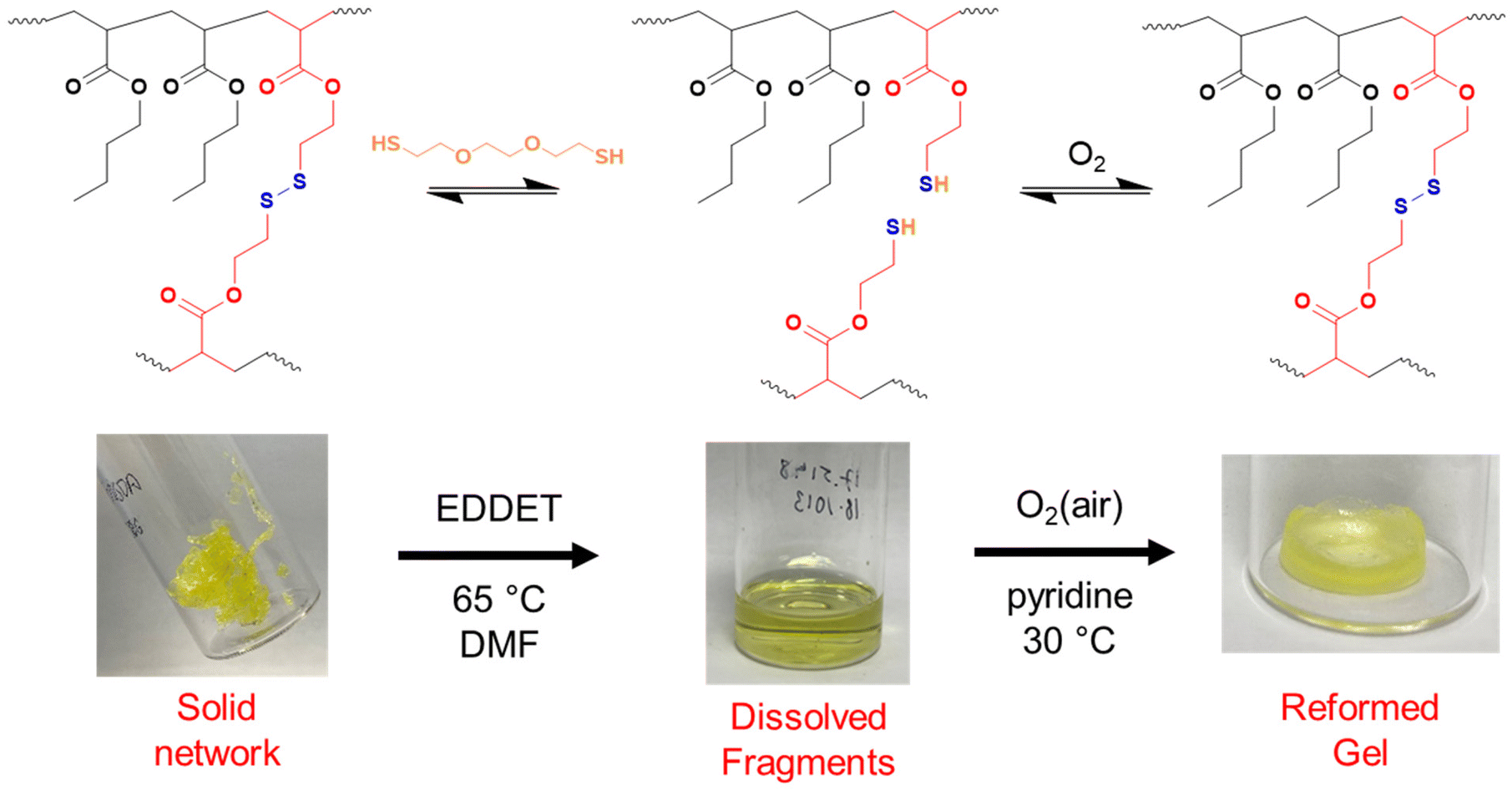 | ||
| Fig. 7 Degradation and regelation scheme for PBA-DSDA networks. Images of PBA-DSDA5 sample. | ||
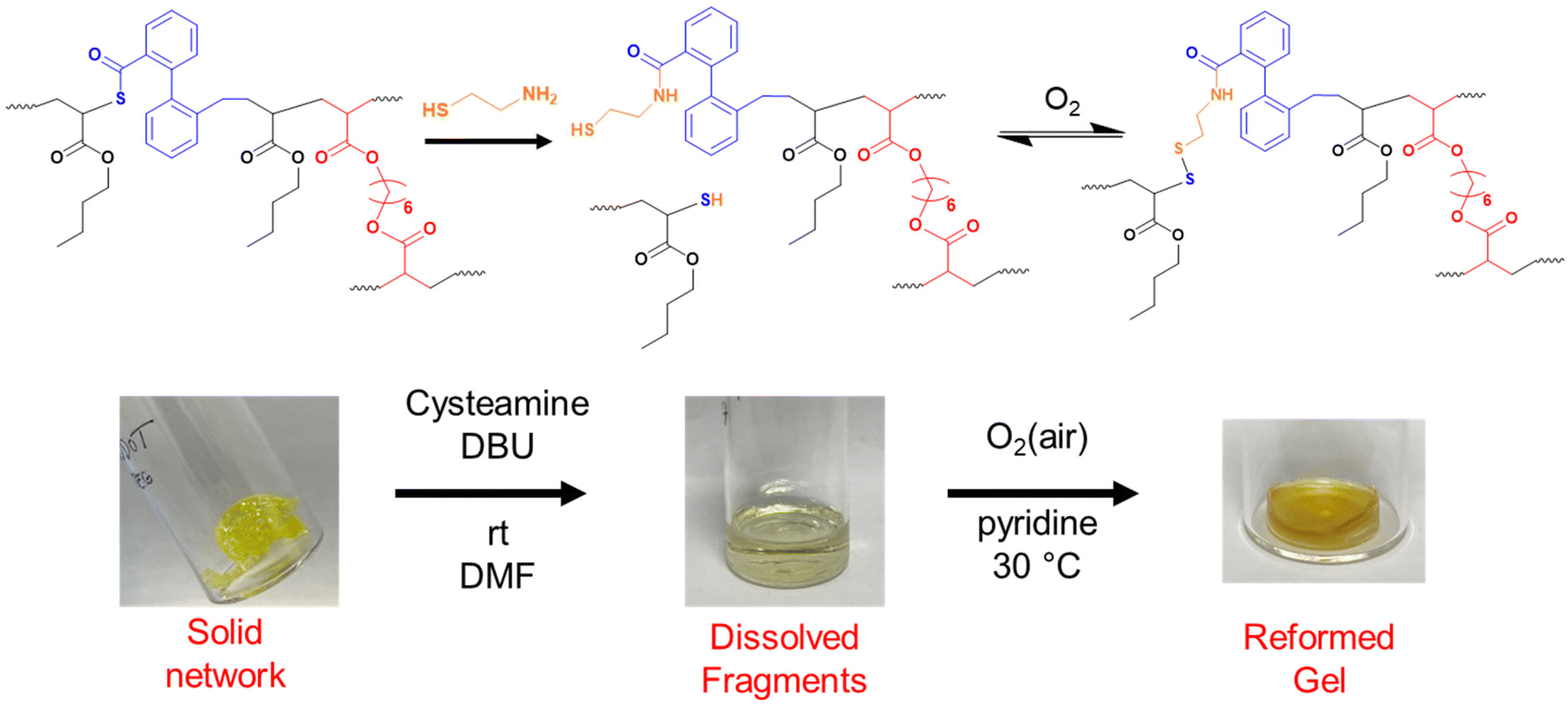 | ||
| Fig. 8 Degradation and regelation scheme for PBA-DOT networks. Images of PBA-DOT3 sample. | ||
First, the dry networks were degraded with 1.5 equivalents (vs. the degradable bonds) of either 2,2′-(ethylenedioxy)diethanethiol (EDDET) or cysteamine for DSDA or DOT-containing networks, respectively, in DMF. The fragments were then quenched with AcOH to preserve the thiol functionality and precipitated from cold methanol/water (90:10 v/v) to remove excess degradation agent and/or side products. For regelation, pyridine was added as a base to promote disulfide formation, and the fragment solutions were heated in air at 30 °C, to evaporate the residual solvent off and to simultaneously oxidise the thiols to disulfide linkages.
PBA-DSDAx gel networks with 2–5 mol% DSDA were degraded using EDDET (instead of DTT used previously), precipitated from methanol/water, then heated at 30 °C in air with pyridine to attempt regelation. The regelation was successful for samples with 4 and 5 mol% DSDA, whereas gels with lower crosslink loadings produced viscous liquids rather than solid gels. The ESRs of the successfully regelled networks were measured to be higher than pre-degradation, indicating a loss in overall crosslink density (Table S6† and Fig. 9). This implies that not all the disulfide linkages are reformed under these conditions, preventing networks with a lower crosslink density (PBA-DSDA2) from reaching the critical number of crosslinks for gelation when they are repolymerised (on average one crosslink per chain as dictated by the Flory-Stockmeyer theory).39,40 A second degradation/regelation process was successfully performed for PBA-DSDA4 and PBA-DSDA5 networks, with the latter retaining its ESR, however this was still higher than before first degradation. Additionally, the PBA-DSDA2 network was successfully reformed when the regelation was attempted at a higher temperature (65 °C), for 48 hours and with double the amount of pyridine. This suggests that with the optimisation of reaction conditions, materials with a range of crosslink densities could be reformed after degradation.
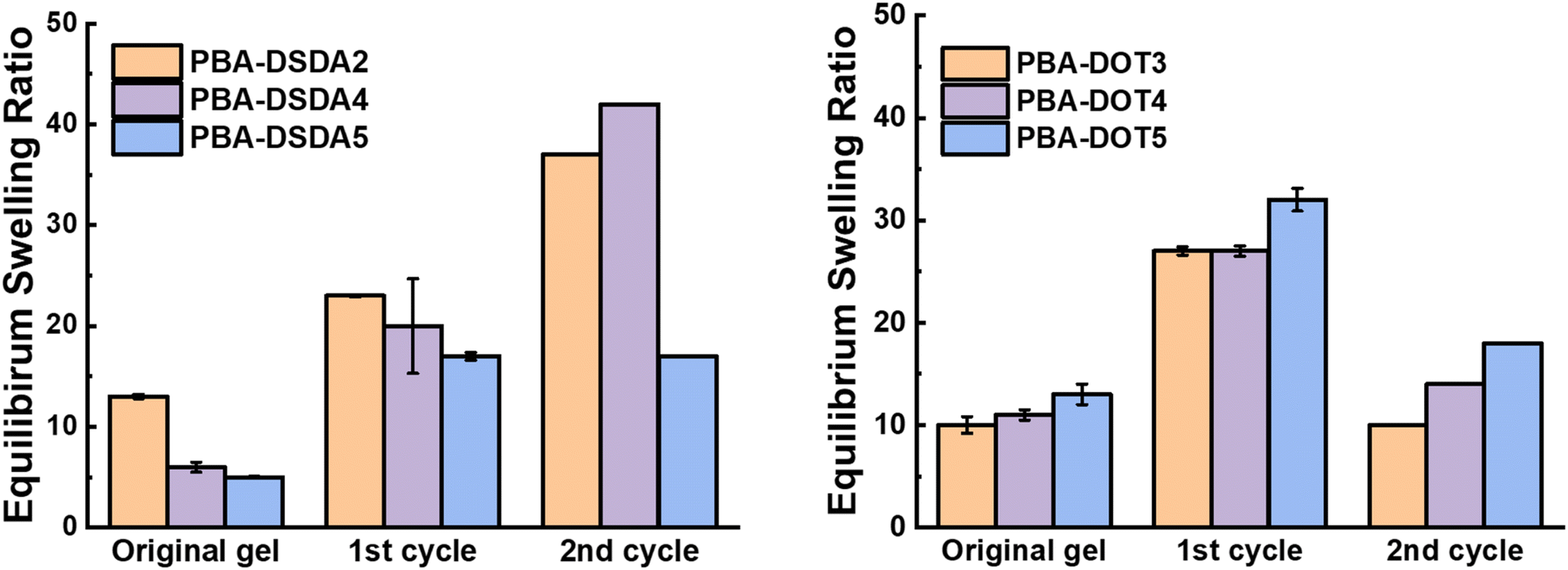 | ||
| Fig. 9 ESRs before and after two degradation/regelation cycles of PBA-DSDA networks (left) and PBA-DOT networks (right). | ||
PBA-DOTx gels at 3, 4 and 5 mol% loading were degraded using cysteamine and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) catalyst, before heating in air at 30 °C with pyridine to form disulfide linkages. All three samples successfully regelled into a solid disc, and their ESRs were measured. The reformed PBA-DOT samples swelled with an ESRs of 27–32, compared to 10–13 prior to the degradation/regelation process (Table S6† and Fig. 9). After regelation, the PBA-DOT networks contain disulfide linkages instead of the original thioesters (see Fig. 8); the second degradation was then performed using EDDET and all three samples successfully regelled after this 2nd degradation. Interestingly, the ESRs of the networks after the second cycle returned to values close to the ESRs prior to any degradation (ESRs of 10–18), suggesting retention of a similar network connectivity and crosslinking density, despite being prepared by a different polymerisation mechanism (RAFT vs. step-growth for regelation). This resembles the remarkable ‘molecular weight memory’ effect demonstrated by Kiel et al. by repolymerising DOT-containing linear polymers.30
Noteworthy, while regelation of the PBA-DOT networks was easily achieved when left in air at 30 °C, some samples were found to spontaneously regel when left in the methanol/water mixture in the fridge overnight, although they were generally not as developed as those made at elevated temperature (Fig. S10†).
In addition, a sample containing 4 mol% DOT and 1 mol% HDDA was synthesised using conventional FRP, which was then successfully degraded using the cysteamine/DBU method described above. These fragments could not regel using either the standard conditions of 30 °C or at an elevated temperature of 65 °C, both with the addition of 1.2 equivalents of pyridine. Likely, the more homogenous network structure in RAFT-made gels is critical to impart efficient reversibility.
The apparent easier regelation of the PBA-DOT networks can be explained by analysing the topology and dispersity of the degraded fragments. Degraded PBA-DSDA networks produce essentially linear chains with low Đ, which require on average more disulfide bonds to be reformed to reach the critical number of crosslinks and consequently the gel point.40–42 Thus, if the total number of reversible bonds is small, as in PBA-DSDA2, the imperfect efficiency of thiol oxidation and some intramolecular cyclisation will prevent gelation. In contrast, highly branched, polydisperse degradation fragments obtained by cleaving strands in PBA-DOT networks require fewer bonds to be reformed to reach the gel point, and therefore facilitate network reversibility as well as produce gels with properties more similar to the original materials.
Interestingly, samples with lower DOT loadings (i.e., PBA-DOT3) produce reformed gels with ESRs closer to their original values compared to the higher DOT loading (PBA-DOT5) sample. Using a minimum amount of DOT to achieve degradation is both economical with respect to the amount of comonomer needed and beneficial for targeted regelation of the network. In contrast, networks with the minimum amount of DSDA were the least effective at regelling, and higher DSDA loadings are required to produce consistent network reformation.
Conclusions
In summary, reversible poly(n-butyl acylate) networks were synthesised by RAFT polymerisation using either disulfide crosslinker or degradable comonomer. Whereas disulfide crosslinkers are ineffective at making networks fully degradable when synthesised by FRP, the use of RAFT polymerisation affords degradability networks at a range of crosslinking densities. The fragments produced by degradation of the crosslinks are similar structurally to linear poly(n-butyl acrylate) chains synthesised by RAFT, with relatively high Mn and low dispersity. However, due to this low dispersity, regelation of PBA-DSDA networks proved less efficient, as a significant number of crosslinks need to reform to hit the critical crosslink density.We have previously shown the use of DOT comonomer to make gels synthesised by FRP degrade, and the current work shows that when synthesising networks by RAFT polymerisation, much less DOT is needed to make the networks fully degrade (namely, DOT:crosslinker ratio = 3:2) due to their more homogeneous network structures. The degraded fragments have lower molecular weight and higher Đ than the disulfide-containing network fragments and are significantly branched. This makes reforming the network far easier as the original crosslinks remain intact after degradation and the fragments have a higher Đ, resulting in more facile regelation. Indeed, the DOT-containing networks returned to very similar equilibrium swelling ratios after two degradation/regelation cycles showing restoration of the original crosslinking density. This work underscores the importance of network topology, polymerisation method, location of degradable units and the structure of the degradation products when designing recyclable thermosets or reversibly crosslinked polymers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EPSRC New Investigator Award grant no. EP/W034778/1. T. K. thanks the Commonwealth Scholarship Commission for funding. Analytical facilities were provided through the Material and Chemical Characterization Facility (MC2) at the University of Bath. The authors thank Dr Gavin Irvine for assisting with the column chromatography.References
- W. Post, A. Susa, R. Blaauw, K. Molenveld and R. J. I. Knoop, Polym. Rev., 2020, 60, 359–388 CrossRef CAS.
- S. Ma and D. C. Webster, Prog. Polym. Sci., 2018, 76, 65–110 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159 CrossRef CAS.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- P. Shieh, M. R. Hill, W. Zhang, S. L. Kristufek and J. A. Johnson, Chem. Rev., 2021, 121, 7059–7121 CrossRef CAS PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed.
- S. Talebian, M. Mehrali, N. Taebnia, C. P. Pennisi, F. B. Kadumudi, J. Foroughi, M. Hasany, M. Nikkhah, M. Akbari, G. Orive and A. Dolatshahi-Pirouz, Adv. Sci., 2019, 6, 1801664 CrossRef PubMed.
- K. S. Anseth, A. T. Metters, S. J. Bryant, P. J. Martens, J. H. Elisseeff and C. N. Bowman, J. Controlled Release, 2002, 78, 199–209 CrossRef CAS PubMed.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, Nature, 2020, 583, 542–547 CrossRef CAS PubMed.
- N. Hisano, N. Morikawa, H. Iwata and Y. Ikada, J. Biomed. Mater. Res., 1998, 40, 115–123 CrossRef CAS PubMed.
- H. A. Aliyar, P. D. Hamilton and N. Ravi, Biomacromolecules, 2005, 6, 204–211 CrossRef CAS PubMed.
- H. Lee and T. G. Park, Polym. J., 1998, 30, 976–980 CrossRef CAS.
- J. K. Oh, C. Tang, H. Gao, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 5578–5584 CrossRef CAS PubMed.
- J. Cuthbert, S. V. Wanasinghe, K. Matyjaszewski and D. Konkolewicz, Macromolecules, 2021, 54, 8331–8340 CrossRef CAS.
- S. Seiffert, Prog. Polym. Sci., 2017, 66, 1–21 CrossRef CAS.
- S. Seiffert, Polym. Chem., 2017, 8, 4472–4487 RSC.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- A. Bagheri, C. M. Fellows and C. Boyer, Adv. Sci., 2021, 8, 2003701 CrossRef CAS PubMed.
- S. V. Wanasinghe, M. Sun, K. Yehl, J. Cuthbert, K. Matyjaszewski and D. Konkolewicz, ACS Macro Lett., 2022, 1156–1161 CrossRef CAS PubMed.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- H. Elliss, F. Dawson, Q. un Nisa, N. M. Bingham, P. J. Roth and M. Kopeć, Macromolecules, 2022, 55, 6695–6702 CrossRef CAS.
- N. M. Bingham and P. J. Roth, Chem. Commun., 2018, 55, 55–58 RSC.
- P. Galanopoulo, N. Gil, D. Gigmes, C. Lefay, Y. Guillaneuf, M. Lages, J. Nicolas, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2022, 61, e202117498 CrossRef CAS PubMed.
- P. Galanopoulo, N. Gil, D. Gigmes, C. Lefay, Y. Guillaneuf, M. Lages, J. Nicolas, F. D'Agosto and M. Lansalot, Angew. Chem., Int. Ed., 2023, 62, e202302093 CrossRef CAS PubMed.
- M. P. Spick, N. M. Bingham, Y. Li, J. De Jesus, C. Costa, M. J. Bailey and P. J. Roth, Macromolecules, 2020, 53, 539–547 CrossRef CAS.
- R. A. Smith, G. Fu, O. McAteer, M. Xu and W. R. Gutekunst, J. Am. Chem. Soc., 2019, 141, 1446–1451 CrossRef CAS PubMed.
- Q. Un Nisa, W. Theobald, K. S. Hepburn, I. Riddlestone, N. M. Bingham, M. Kopeć and P. J. Roth, Macromolecules, 2022, 55, 7392–7400 CrossRef CAS.
- G. R. Kiel, D. J. Lundberg, E. Prince, K. E. L. Husted, A. M. Johnson, V. Lensch, S. Li, P. Shieh and J. A. Johnson, J. Am. Chem. Soc., 2022, 144, 12979–12988 CrossRef CAS PubMed.
- M. Lages, T. Pesenti, C. Zhu, D. Le, J. Mougin, Y. Guillaneuf and J. Nicolas, Chem. Sci., 2023, 14, 3311–3325 RSC.
- N. Gil, C. Thomas, R. Mhanna, J. Mauriello, R. Maury, B. Leuschel, J.-P. Malval, J.-L. Clément, D. Gigmes, C. Lefay, O. Soppera and Y. Guillaneuf, Angew. Chem., Int. Ed., 2022, 61, e202117700 CrossRef CAS PubMed.
- N. Gil, B. Caron, D. Siri, J. Roche, S. Hadiouch, D. Khedaioui, S. Ranque, C. Cassagne, D. Montarnal, D. Gigmes, C. Lefay and Y. Guillaneuf, Macromolecules, 2022, 55, 6680–6694 CrossRef CAS.
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jiang and Y. Chen, ACS Macro Lett., 2012, 1, 275–279 CrossRef CAS PubMed.
- M. C. Roberts, M. C. Hanson, A. P. Massey, E. A. Karren and P. F. Kiser, Adv. Mater., 2007, 19, 2503–2507 CrossRef CAS.
- Z. Rodriguez-Docampo and S. Otto, Chem. Commun., 2008, 5301–5303 RSC.
- J. Bastide and L. Leibler, Macromolecules, 1988, 21, 2647–2649 CrossRef CAS.
- J. L. G. Ruano, A. Parra and J. Alemán, Green Chem., 2008, 10, 706–711 RSC.
- Y. Gao, D. Zhou, J. Lyu, S. A, Q. Xu, B. Newland, K. Matyjaszewski, H. Tai and W. Wang, Nat. Rev. Chem., 2020, 4, 194–212 CrossRef CAS PubMed.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, 1953 Search PubMed.
- F. Dawson, H. Jafari, V. Rimkevicius and M. Kopeć, Macromolecules, 2023, 56, 2009–2016 CrossRef CAS PubMed.
- W. Li, H. Gao and K. Matyjaszewski, Macromolecules, 2009, 42, 927–932 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01008b |
| This journal is © The Royal Society of Chemistry 2023 |