
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of poly(methyl methacrylate)-b-polyethylene (PMMA-b-PE) block copolymers via conventional emulsion polymerization†
L.
Sinniger
 a,
O.
Boyron
a,
P. Y.
Dugas
a,
G.
Patias
b,
D.
Lester
b,
D. M.
Haddleton
b,
V.
Monteil
a,
M.
Lansalot
*a and
F.
D'Agosto
*a
a,
O.
Boyron
a,
P. Y.
Dugas
a,
G.
Patias
b,
D.
Lester
b,
D. M.
Haddleton
b,
V.
Monteil
a,
M.
Lansalot
*a and
F.
D'Agosto
*a
aUniversité de Lyon, Université Lyon 1, CPE Lyon, CNRS UMR 5128, Laboratoire CP2M, Equipe PCM, Villeurbanne, 69616, France. E-mail: franck.dagosto@univ-lyon1.fr; muriel.lansalot@univ-lyon1.fr
bUniversity of Warwick, Department of Chemistry, Gibbet Hill, Coventry, CV4 7AL, UK
First published on 18th September 2023
Abstract
Poly(methyl methacrylate)-b-polyethylene (PMMA-b-PE) block copolymers were recently obtained simply by involving ω-unsaturated PMMA oligomers prepared by CCTP in the radical polymerization of ethylene performed in dimethylcarbonate (DMC) solution (F. Baffie, G. Patias, A. Shegiwal, F. Brunel, V. Monteil, L. Verrieux, L. Perrin, D. M. Haddleton and F. D’Agosto, Angew. Chem., Int. Ed., 2021, 60, 25356–25364). However, ω-unsaturated PMMA oligomers are more effectively obtained as a latex by emulsion CCTP. In the present paper, these resulting latexes were used as seeds in the radical emulsion polymerization of ethylene in water. The effects of the methacrylic oligomer molar mass and the initial solids content of the seeds were investigated. Cryo-TEM analysis enabled demonstration of the formation of stable latexes composed of faceted spherical particles. The increase of molar mass correlated to the consumption of unsaturated oligomers with the polymerization time indicated the formation of block copolymers. The present study successfully transposed the chemistry performed in DMC solution and PMMA-b-PE block copolymers were obtained by a simple post-treatment of a PMMA latex.
Introduction
Due to its low production costs and excellent mechanical properties, polyethylene (PE) has long been the most widely produced polymer and indeed chemical globally. Its fully hydrocarbon-based backbone ensures excellent chemical and mechanical properties and stability. The production of macromolecular architectures containing ethylene and polar monomers is thus particularly relevant as such polymers will combine the properties of PE with the functionality of the polar units. However, the synthesis of such structures e.g., by copolymerization of ethylene with polar monomers, is not trivial,1,2 and only few industrial solutions are available even though the potential applications are many and a great deal of research has been carried out over many decades. Reversible-deactivation radical polymerization (RDRP) represents today one of the most versatile tools to result in complex architectures combining polar and apolar monomers. Recently, ethylene-based copolymers were developed by our group through the use of reversible addition–fragmentation chain transfer (RAFT),3–5 organotellurium mediated radical polymerization (TERP)6 and iodine transfer polymerization (ITP)7,8 under mild conditions (≤200 bar, ≤80 °C) in dimethylcarbonate (DMC), but also in dispersed media or via polymerization-induced self-assembly (PISA).9,10 These techniques, however, require the use of controlling agents, which is not always compatible with the cost of the final structures that has to remain very low, particularly when considering ethylene-based products.Thus, using much more conventional radical polymerization techniques, complex architectures could be obtained simply by involving ω-unsaturated methacrylic oligomers in the radical polymerization of a wide range of monomers. These types of oligomers can be obtained by catalytic chain transfer polymerization (CCTP) as discovered by Smirnov and Marchenko.11,12 CCTP involves the use of [Co]II or [Co]III catalysts such as cobaloximes as catalytic chain transfer agents (CCTA) in the radical polymerization of methacrylates.13–19 CCTP has been carried out successfully in all processes used for free radical polymerization including solution, suspension and bulk as well as commercial processes using emulsion polymerization and indeed very recently to photopolymerization to 3D networks.20 The use of CCTP in dispersed media is not just used efficiently commercially but has raised a lot of interest in academia.21–26 The transition of carrying out CCTP in solution to dispersed media is however not trivial. Indeed, the compartmentalization effects associated to emulsion polymerization27 affect the efficiency of the CCTA, which needs to partition between the aqueous phase and the polymer particles. In most cases, lower catalytic activity and thus higher molar mass are observed when CCTP is conducted in dispersed media due to mass transport limitations, partitioning and potential deactivation of catalysts in aqueous phase. It is also important for the polymerizations to be at less than 100% instantaneous conversion so the glass transition of the swollen polymer particle allows the catalyst to move in and out of the aqueous phase.11,21,23,28–31
Low molar mass ω-unsaturated methacrylic oligomers have been used in radical polymerization for the synthesis of branched copolymers, amphiphilic dispersants or multi-block copolymers.32–36 In most of the cases, the propagating radical is added on the unsaturated chain end leading to the formation of an intermediate radical. Depending on the monomer used for the radical polymerization, and thus the corresponding propagation rate, two types of architectures can be obtained. For mostly styrene, acrylate, acrylonitrile the propagation rate is relatively high and propagation step occurs leading to grafted structures.37,38 However, in the case of acrylates, depending on the nature of the acrylate and its molecular weight, block copolymer structures can be obtained after termination steps of the intermediate radical.32,39–41 When methacrylates are used, the rate of propagation is lower than the rate of β-scission leading to an addition–fragmentation chain transfer mechanism and to the formation of block copolymers.34,42–45
The use of ω-unsaturated oligomers synthesized by CCTP in emulsion has also been established. For instance, block copolymers with a poly(n-butyl methacrylate) block have been produced in emulsion polymerization from PMMA macromonomers.34,42,44,45 They can also be used as in situ stabilizer in order to conduct a surfactant-free emulsion polymerization of n-butyl acrylate (BA).42,46,47
We recently showed that PMMA produced by CCTP (PMMAn) can serve as efficient degradative chain transfer agent in the radical polymerization of ethylene conducted in DMC under relatively mild conditions (80 bar and 70 °C).48 Detailed characterizations of the obtained products combined with DFT calculations showed that the propagating polyethylenyl radicals produced upon initiation of ethylene polymerization rapidly react with the ω-chain end of the PMMAn to form an intermediate radical (Fig. 1). The fate of this intermediate radical was demonstrated to be termination by H-abstraction to lead to PMMAn-b-PE for n > 2. Upon consumption of the double bond of PMMAn, the molar mass of the PE segment increased consistently with a polymerization mechanism implying a degradative chain transfer reaction. Depending on the amount of ω-unsaturated oligomers, the molar mass of the PE block could be adjusted.
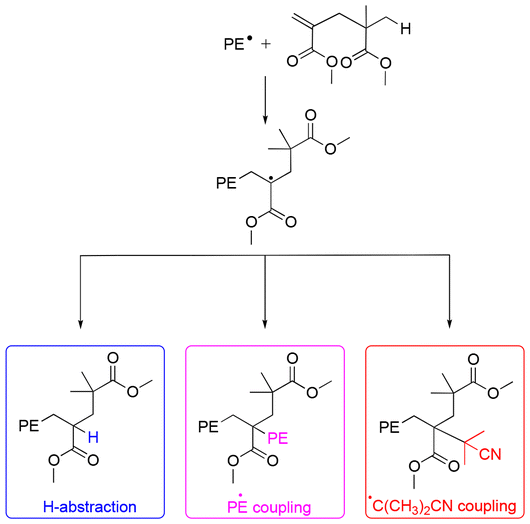 | ||
| Fig. 1 Possible fate of the propagating radicals in ethylene polymerization in the presence of PMMA2. Reproduced from ref. 48. | ||
As mentioned above, CCTP of MMA can easily be carried out in emulsion using sodium dodecylsulfate (SDS) as surfactant to produce a stable latex incorporating ω-vinyl functional PMMA.34,42,44,45 Building on our results obtained in DMC, we now report application in aqueous dispersed media by the direct use of PMMAn from CCTP latexes as seeds in ethylene polymerization. Indeed, this approach would be a powerful way to produce PMMA-b-PE copolymers directly in water, simply by post-treating the seed latex with initiator under ethylene pressure. The study explores the effect of the molar mass of PMMA oligomers and of the initial solids content of the seed latex on the polymer architecture. Morphologies of the produced particles are also investigated.
Materials and methods
Materials
Methyl methacrylate (MMA, Aldrich, ≥99%), 4,4′-azobis(4-cyanovaleric acid) (ACVA, Aldrich, ≥98%), sodium dodecylsulfate (SDS, MP Biomedicals, LLC), ammonium persulfate (APS, Aldrich, ≥98%) and sodium bicarbonate (NaHCO3, Aldrich, Bioreagent) were used as received. Water was deionized before use (Purelab Classic UV, Elga LabWater). The catalyst bis[(difluoroboryl)dimethylglyoximato]cobalt(II) (CoBF) was synthesised as previously described.32,49 Deuterated solvents were supplied by Eurisotop.Analytical techniques
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v:v) using a Bruker Avance 400 spectrometer. Proton NMR spectra were recorded with a 5 mm BBFO + probe with a z-gradient coil. Chemical shifts are given in parts per million (ppm) with the benzene deuterated solvent used as internal standard.
570000–580 g mol−1. Mn and Đ values were determined by conventional calibration using Agilent GPC/SEC software.
:1 v:v) using a Bruker Avance 400 spectrometer. Proton NMR spectra were recorded with a 5 mm BBFO + probe with a z-gradient coil. Chemical shifts are given in parts per million (ppm) with the benzene deuterated solvent used as internal standard.
570000–580 g mol−1. Mn and Đ values were determined by conventional calibration using Agilent GPC/SEC software.
where ni is the number of particles with diameter Di.
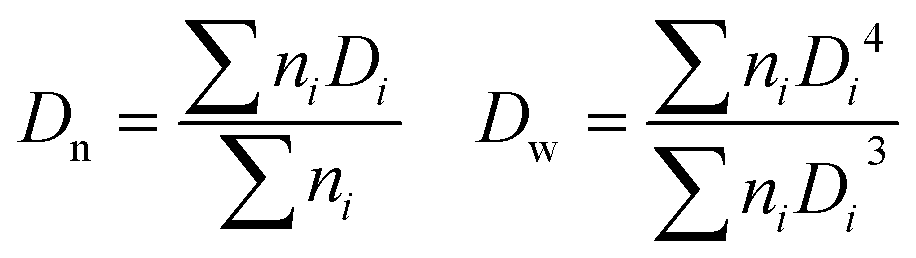
Synthetic procedures
Results and discussion
Our previous study demonstrated that PMMA-b-PE copolymers can be formed by combining CCTP and radical polymerization of ethylene in DMC.48 In that work, PMMAn oligomers obtained by CCTP were solubilized in DMC in the presence of AIBN. PMMAn chain-end double bonds were quickly consumed, and the molar mass of the PE block increased with the reaction time. Herein, latexes of PMMAn particles are used as seeds in the radical emulsion polymerization of ethylene to transpose the chemistry performed in DMC to aqueous media and thus produce particles of PMMAn-b-PE block copolymers.Seeded emulsion polymerization of ethylene with PMMA16
Using SDS as surfactant and ACVA as initiator, two batches of latex were produced with two polymerization degrees (namely PMMA16 and PMMA36) adjusted by the amount of CoBF involved in the recipe (Table 1). Both latexes were stable with particle size of 147 and 101 nm, respectively. The two significantly different Tg values (34 °C for PMMA16 and 93 °C for PMMA36) are directly related to the difference of molar masses.| PMMAn | τ s (%) | Z ave (nm)/PDIb | M n,NMR (g mol−1) | M n, SEC (g mol−1)/Đd | T g (°C) |
|---|---|---|---|---|---|
| a Determined by gravimetric analysis. b Determined by DLS. c Calculated by assuming that there is one vinyl per chain. d Determined by SEC-THF using conventional PMMA calibration. e Determined by DSC at 10 °C min−1. | |||||
| PMMA16 | 16.9 | 147 | 1 470 | 1200 | 34 |
| 0.07 | 1.8 | ||||
| PMMA36 | 19.9 | 101 | 3 630 | 3000 | 93 |
| 0.09 | 2.2 | ||||
Seeded radical emulsion polymerization of ethylene was first carried out using the PMMA16 latex. The seed latex was diluted to an initial solids content of 5 wt% and introduced into the reactor. Subsequently, an aqueous phase containing APS was injected in the reactor with NaHCO3 used as buffer. APS was chosen as initiator in our system although persulfate initiators could react with the remaining CoBF31 in the PMMA16 seed latex. To counterbalance this effect, the APS concentration was increased compared to the reference concentration used in more classical emulsion polymerization of ethylene previously performed in our group.50
After 4 h of reaction under 100 bar of ethylene at 80 °C (run 1, Table 2), a stable latex was obtained. The particle size did not vary significantly (147 nm for the seed PMMA16 and 155 nm for the obtained latex). The 2.4 wt% increase in solids content correlated with the appearance of an intense signal in the 1H NMR spectrum of the dry extract at 1.3 ppm (Fig. 2), corresponding to the methylene protons of PE, are consistent with the formation of PE during the reaction. The almost total disappearance of the characteristic signals of the vinylic protons of PMMA16, in the 5.6 to 6.5 ppm region, indicated that they were almost quantitatively consumed (99% conversion after 4 h). The polymerization degree of PE (DPPE) was measured by NMR (eqn (1)) assuming that the obtained product is a PMMA-b-PE block copolymer. The integral of the methoxy signal from PMMA at 3.5 ppm (Ic′) was thus fixed at a value of 48.
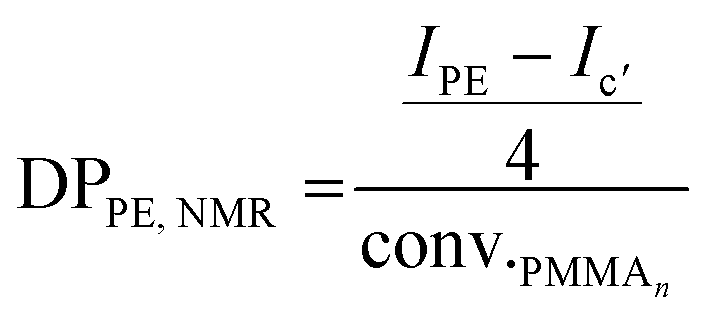 | (1) |
| Run | PMMAn | T (h) |
τ
s,i
(%) |
τ
s,f
(%) |
Z
avec (nm) |
PDIc | PMMA conv.d (%) | DPPEe |
M
n,SECf (g mol−1) |
Đ |
T
gg (°C) |
T
cg (°C) |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a APS (0.54 mmol), NaHCO3 (pH = 8) at 80 °C and 100 bar in water (Vtot = 50 mL). b The initial and final solids content determined by gravimetric analysis. c Determined by DLS. d Calculated by comparing the 1H NMR signals intensities of the vinylic protons of PMMA16 and residual vinylic protons at the end of the reaction. e Calculated by assuming that there is one methacrylic oligomer per PE chain, DPPE is the degree of polymerization and is calculated according to the eqn (1) using NMR. f Determined by HT-SEC with a conventional calibration based on PS standards. Two separate entries per row corresponds to a bipopulated molar mass distribution. g Determined by DSC (“—” is used when glass transition or crystallization was not visible). h This experiment can also be considered as a run 3-t5. i Experiment conducted without ethylene pressure. | |||||||||||||
| 1h | PMMA16 | 4 | 5.0 | 7.4 | 155 | 0.06 | 99 | 26 | 4400 | 6.1 | — | 42 | 56 |
| 505200 |
1.4 | ||||||||||||
| 2i | PMMA16 | 4 | 5.0 | 5.4 | 146 | 0.05 | 27 | 0 | 1500 | 2.0 | n.d. | n.d. | |
| 3-t1 | PMMA16 | 0.5 | 5.0 | 5.4 | 142 | 0.04 | 49 | 3 | 1500 | 1.9 | 55 | — | |
| 3-t2 | PMMA16 | 1 | 5.0 | 4.9 | 151 | 0.02 | 72 | 2 | 2300 | 2.2 | 68 | — | |
| 3-t3 | PMMA16 | 2 | 5.0 | 5.5 | 154 | 0.05 | 93 | 5 | 2900 | 4.7 | 72 | 42 | |
| 3-t4 | PMMA16 | 3 | 5.0 | 6.1 | 159 | 0.04 | 99 | 13 | 4000 | 5.9 | 86 | 45 | |
| 419100 |
1.6 | ||||||||||||
| 4-t1 | PMMA36 | 0.5 | 10.0 | 9.4 | 113 | 0.09 | 72 | 3 | 3000 | 2.2 | 87 | — | |
| 4-t2 | PMMA36 | 1 | 10.0 | 9.8 | 100 | 0.07 | 96 | 11 | 8700 | 4.1 | 88 | 42 | |
| 4-t3 | PMMA36 | 2 | 10.0 | 12.1 | 110 | 0.02 | 95 | 48 | 6700 | 4.8 | 97 | 42 | |
| 410300 |
5.7 | ||||||||||||
| 4-t4 | PMMA36 | 4 | 10.0 | 14.6 | 120 | 0.07 | 92 | 105 | 5900 | 5.7 | — | 42 | 56 |
| 837000 |
1.5 | ||||||||||||
| 4-t5 | PMMA36 | 6 | 10.0 | 15.6 | 117 | 0.03 | 92 | 123 | 7 300 | 5.2 | — | 42 | 56 |
| 1016000 |
1.5 | ||||||||||||
| 5 | PMMA16 | 4 | 10.0 | 11.6 | 155 | 0.03 | 98 | 3 | 5200 | 3.9 | 80 | — | |
| 6 | PMMA-FRP | 4 | 5.0 | 14.8 | 68 | 0.03 | n.d. | n.d. | 7200 | 72.2 | — | 55 | |
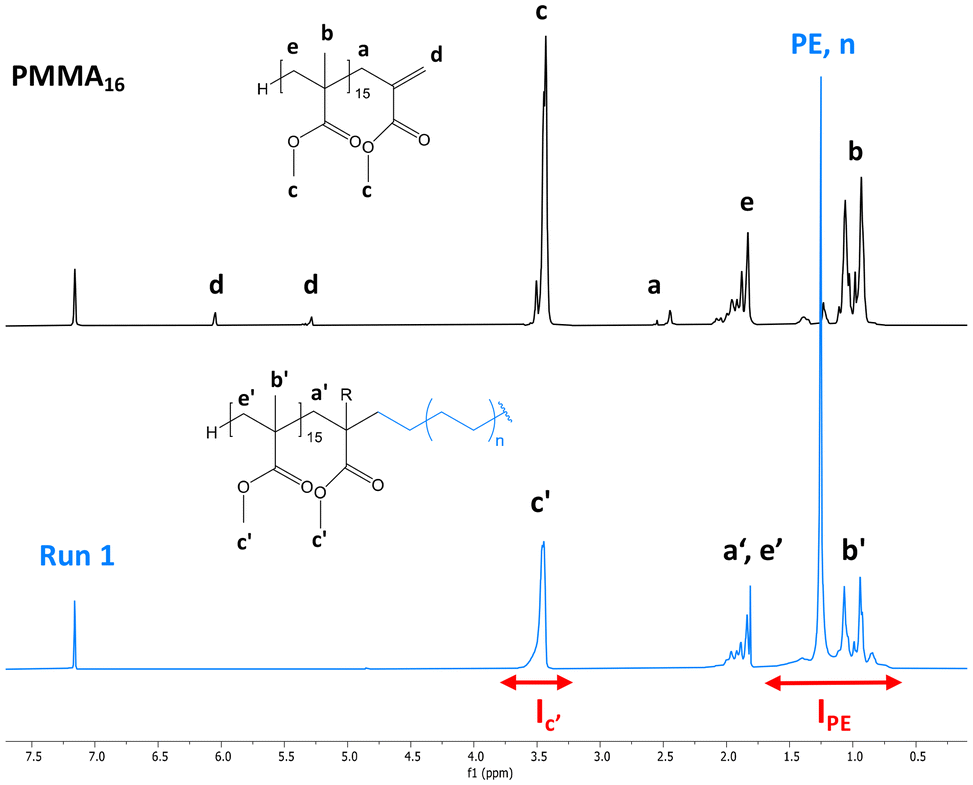 | ||
| Fig. 2 1H NMR spectra (TCE/C6D6 at 90 °C) of the dry extract (a) from the PMMA16 seed latex and (b) from the latex obtained by ethylene emulsion polymerization in the presence of PMMA16 (run 1, Table 2). | ||
HT-SEC chromatograms of the starting PMMA16 and the product obtained in run 1 are compared in Fig. 3. It shows the consumption of the starting PMMA16 and the formation of higher molar mass product at lower elution time. In addition, a second minor population at even lower elution time (tel = 12.4 min), i.e., higher molar masses, appeared. A similar behavior was observed in our previous study conducted in DMC, in which the high molar mass population was assigned to the formation of PE homopolymer taking place when all the PMMAn chain transfer agents have been consumed.48
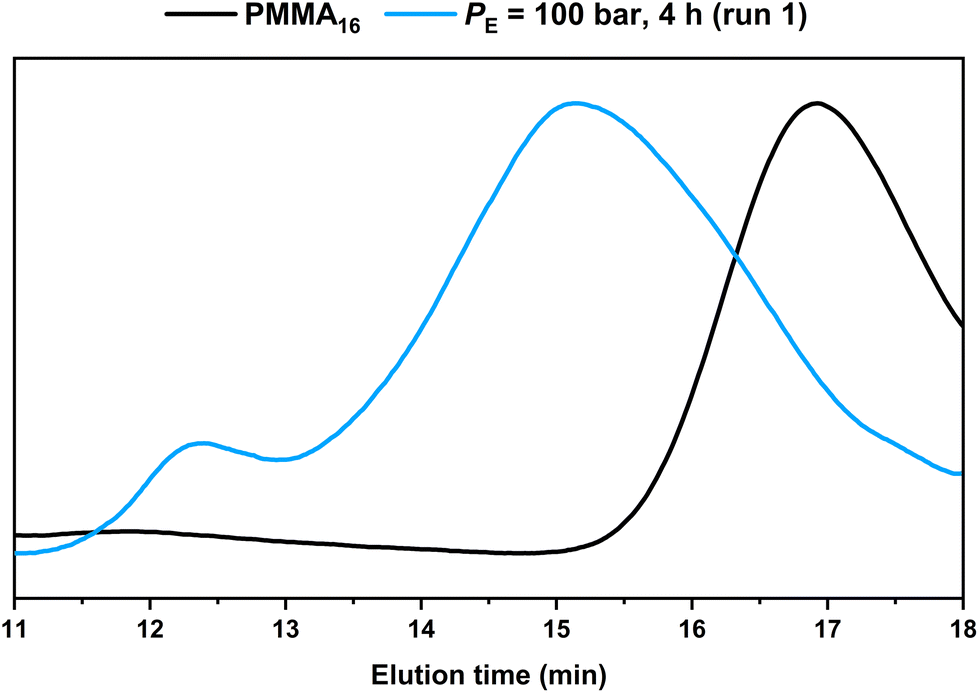 | ||
| Fig. 3 HT-SEC chromatograms of the dry extract from the PMMA16 seed and from the latex obtained by ethylene emulsion polymerization in the presence of PMMA16 (run 1, Table 2). | ||
However, under the conditions employed here, one might ask whether the consumption of the PMMA16 unsaturated chain-ends is indeed only related to the reaction of polyethylenyl radicals formed in water (this is made possible by the solubility of ethylene in water from 0.3 g L−1 at 4.5 bars to 4.6 g L−1 at 110 bar at 75 °C51) and entering the particles to react with PMMA16. An additional experiment was thus carried out under the exact same conditions but without ethylene pressure (run 2, Table 2) to further investigate this point. After 2 h of reaction at 80 °C, a plateau of 27% was reached for the consumption of the PMMA16 vinyl end groups. Consumption of vinyl end groups of PMMAn was not observed when similar experiments were carried out in DMC in the presence of AIBN. The nature of the primary radicals generated (carbon- or oxygen-centered for DMC or emulsion polymerization, respectively) might be put forward to explain this different behavior. It is worth mentioning here that in emulsion polymerization of MMA initiated with APS, depending on the initial conditions (notably the initiator and surfactant concentration), PMMA oligomers will form in water with a degree of polymerization lower than 10 (the DP for surface activity, z, is close to 5, and the oligomers remain water-soluble up to a DP of 10 (jcrit)52). The formation of our PMMA seeds is based on CCTP for which the majority of the chains are not initiated by a charged group. The corresponding critical DPs should then be much lower and the amount of PMMA oligomers remaining in water small. The isolation of the aqueous phase of PMMA16 latex by centrifugation confirmed nevertheless the presence of some oligomers that quantitatively reacted after 2 h at 80 °C in the presence of APS (Fig. S1†). However, a reaction between the water-soluble radicals generated from APS with these water-soluble species could reasonably not account for the 27% of double bond consumption mentioned above. SEC analyses of the dry extract recovered from run 2 (Table 2) indeed revealed a significant conversion of the lowest molar mass species into higher molar mass ones shifted in the molar mass main distribution (Fig. 4). The fraction of double bonds consumed in run 2 is thus probably explained also by the reaction of part of the PMMA16 chains with entering radicals after initiation with APS as explained above. The shift of molar mass distribution observed in Fig. 3 when ethylene is polymerized is thus very likely related to the polyethylenyl radicals entering the particles and reacting with PMMA16 as expected. The following investigations aim at demonstrating that PMMA-b-PE are effectively formed during this process.
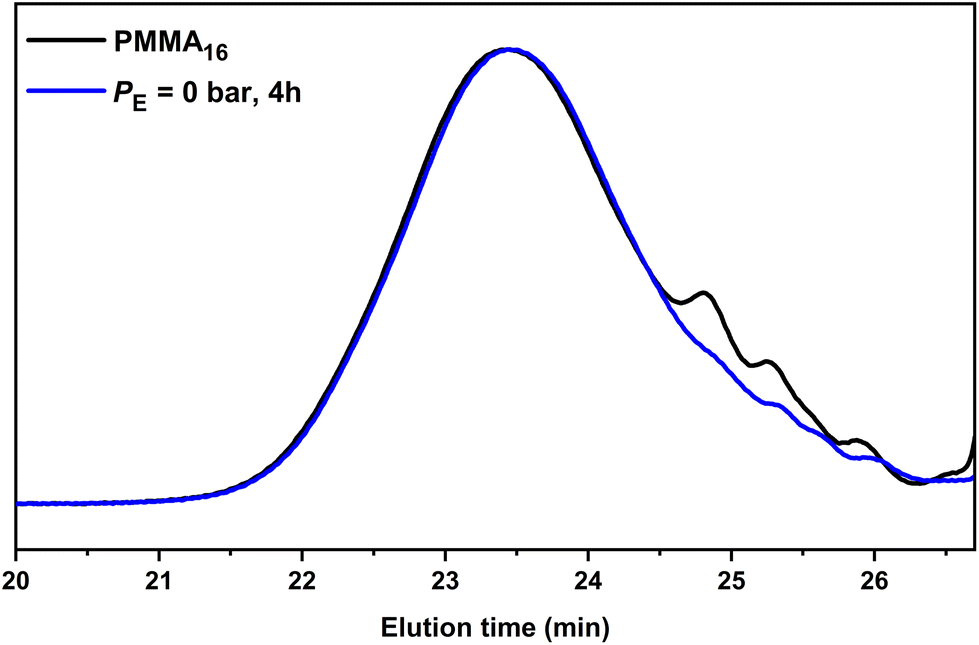 | ||
| Fig. 4 SEC-THF chromatograms of the seed PMMA16 and the polymer obtained after 4 h of reaction without ethylene pressure (run 2, Table 2). | ||
Kinetic study
The influence of the molar mass of the PMMAn latexes (i.e., the value of n) on the polymerization kinetics was first investigated. A series of polymerizations were conducted at 80 °C and 100 bar of ethylene with both the PMMA16 and PMMA36 latexes, but with two different solids content (5 wt% for PMMA16 and 10 wt% for PMMA36) in order to keep the same amount of vinyl functions in both systems. Other parameters were kept identical to those described above with PMMA16. As the reactions were conducted in a pressurized autoclave, the withdrawal of aliquots during the experiment was not possible. Therefore, each point of the kinetics (Table 2) represents an experiment. The trends observed through the data obtained (Fig. 5) are indeed showing the good consistency of the different experiments.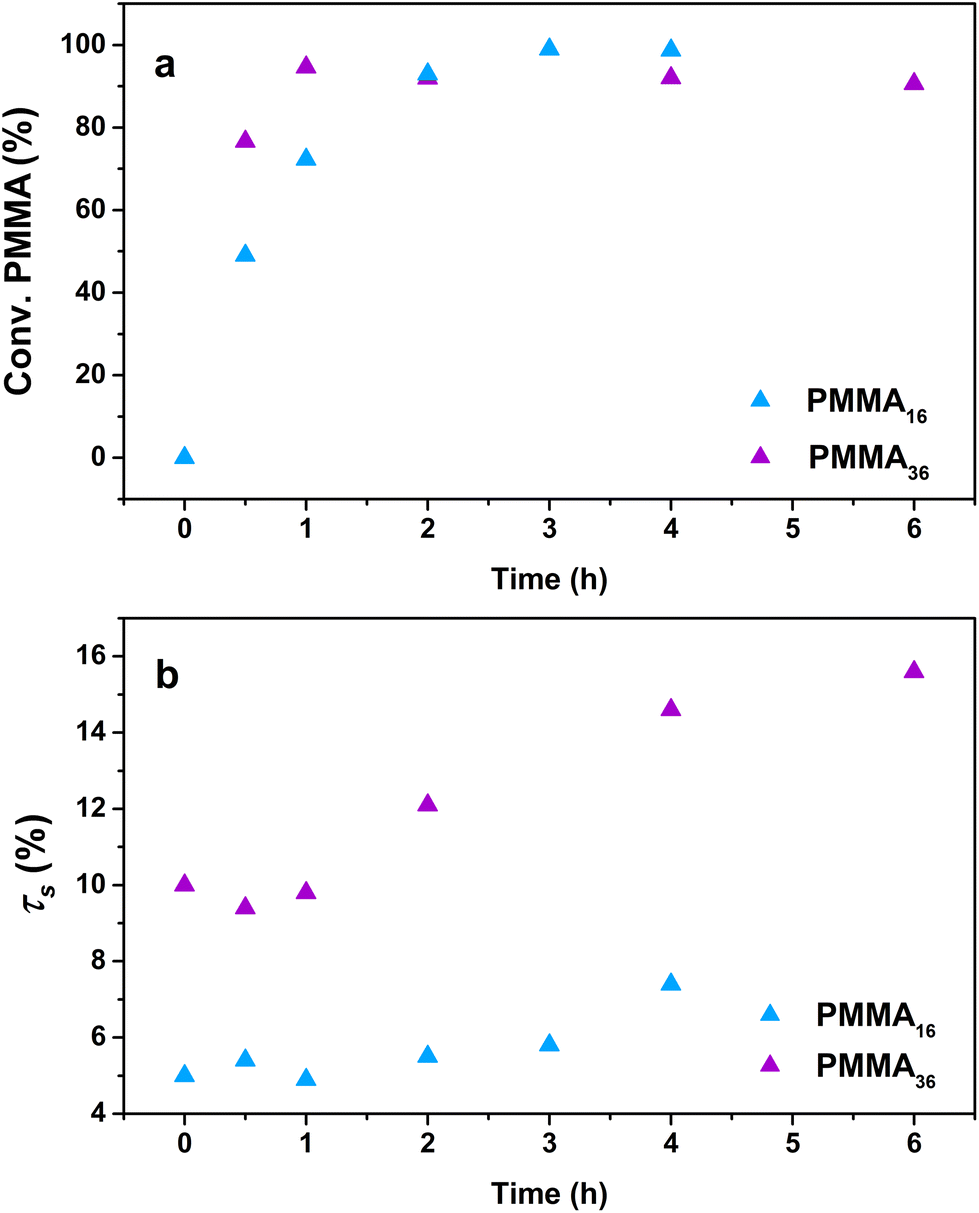 | ||
| Fig. 5 Evolution of (a) PMMAn conversion and (b) solids content with polymerization time at 80 °C and 100 bar of ethylene pressure (respectively runs 3 and 4, Table 2). | ||
For both the PMMA16 and PMMA36 seeds, the double bonds on the methacrylic oligomers are quickly consumed at the beginning of the reaction (> 90% after 2 h) until almost being entirely consumed. Again, DPPE was calculated with eqn (1) by assuming that the obtained product is a PMMA-b-PE block copolymer. The integral of the methoxy signal from PMMA at 3.5 ppm (Ic′) was fixed at a value of 48 for PMMA16 and 108 for PMMA36 (Fig. 6). As shown in Fig. 5b, the solids content starts to strongly increase only when most of PMMAn is consumed. This might be showing rapid termination of growing polyethylenyl radicals with PMMAn and formation of PE homopolymer when all PMMAn has been consumed as previously observed in DMC.48 After 4 h, an increase of both solids content and DPPE was observed for the two PMMAn (after 4 h: τs,f = 7.4 wt% and DPPE = 26 and 16.6 wt% and 123 for PMMA16 and PMMA36, respectively).
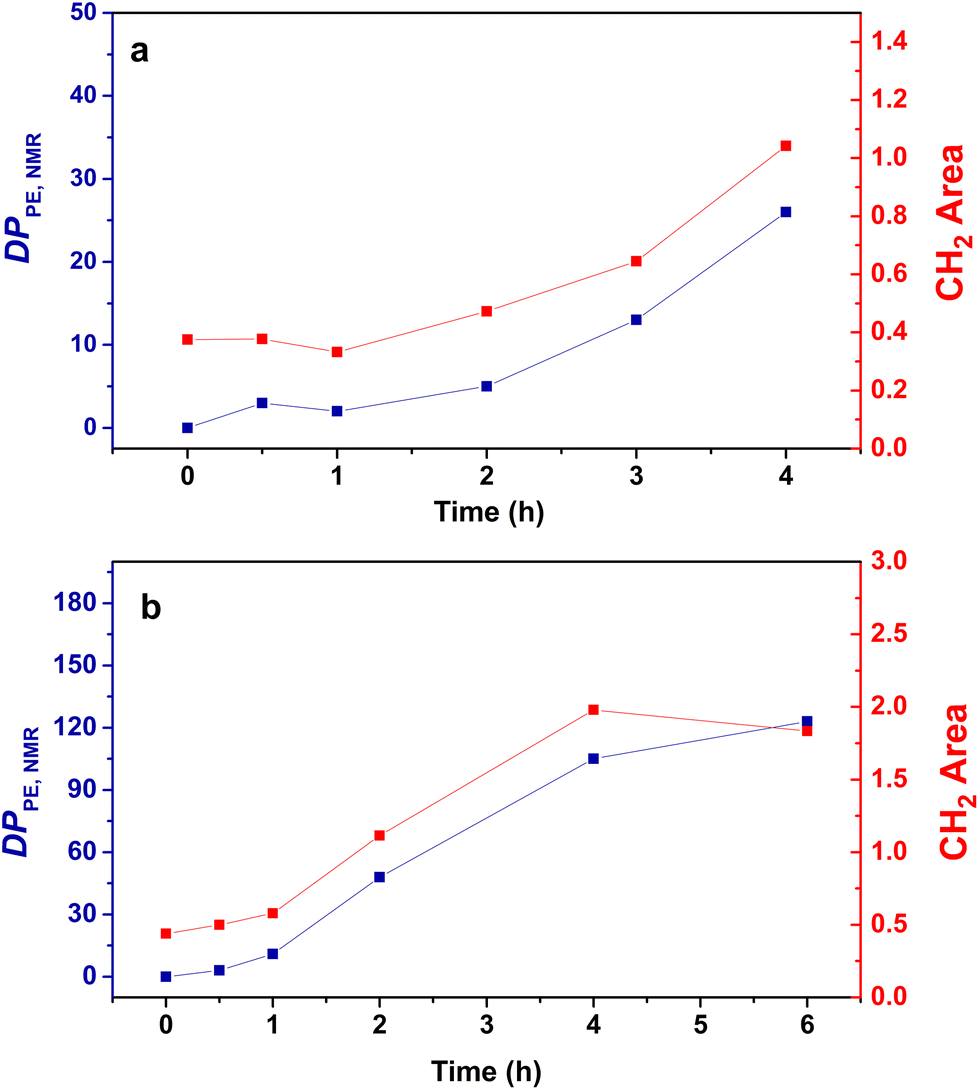 | ||
| Fig. 6 Evolution of the DPPE determined by NMR (blue) and the area of the absorption band of CH2 determined by FT-IR (red) with the polymerization time for the copolymers obtained with (a) PMMA16 and (b) PMMA36 (respectively runs 3 and 4, Table 2). | ||
This is indeed consistent with HT-SEC analyses (Fig. 7) that shows the shift of the chromatograms towards higher molar masses with polymerization times in both cases and as long as PMMAn remains. Once PMMAn has been consumed, a second population of much higher molar mass appears. As observed in DMC, block copolymers seem to be formed early during the polymerization, and PMMAn seems to be similarly acting as degradative chain transfer agent by terminating the growing PE chains entering the particles. When the reaction time increases, the amount of unsaturated oligomer decreases and the probability for a polyethylenyl radical to react on the double bound of the methacrylic oligomer is lower, so the PE block is longer. When all the vinyl functions are consumed, homopolymerization of ethylene might then take place.
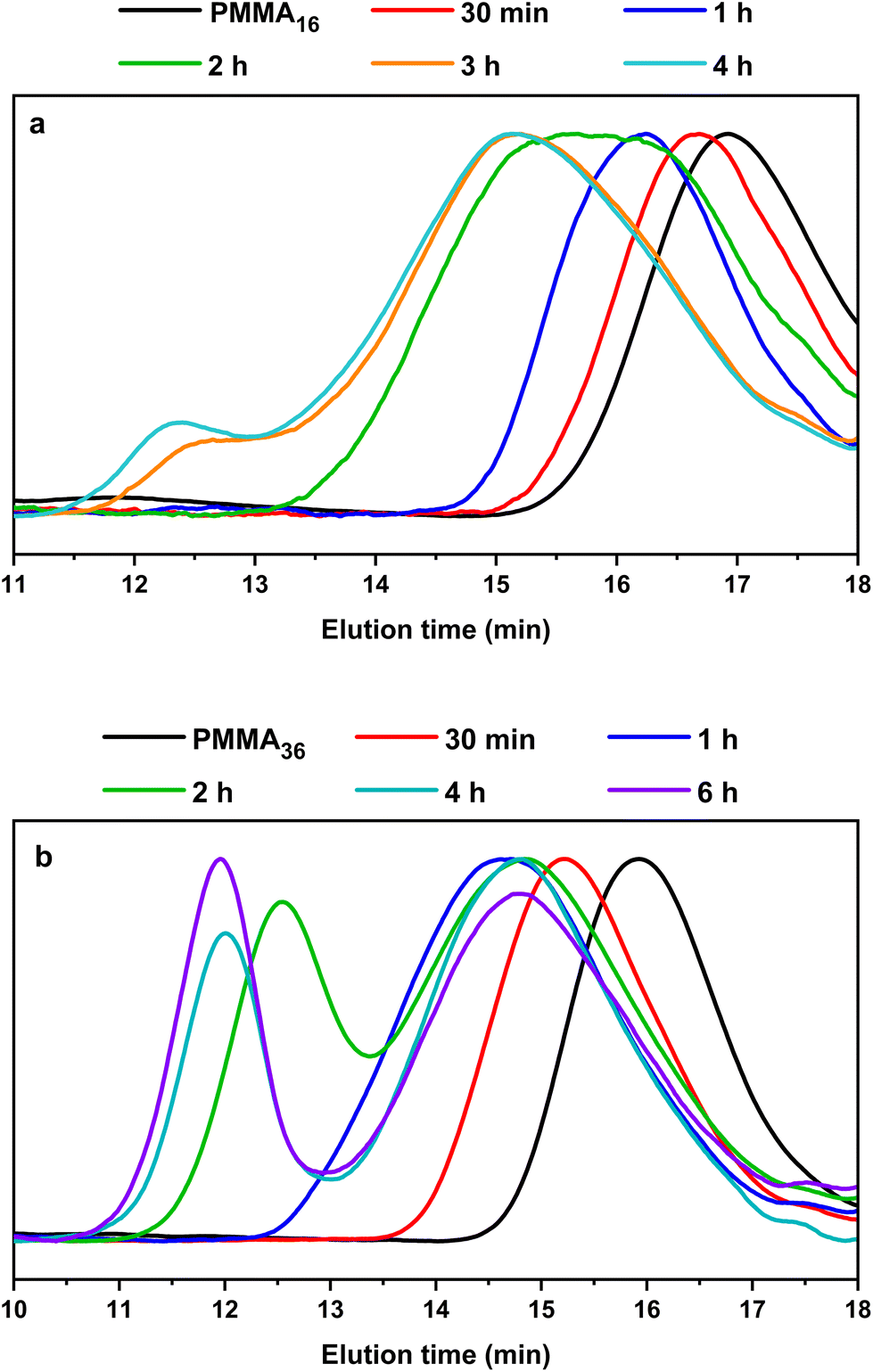 | ||
| Fig. 7 HT-SEC chromatograms of the polymers obtained during seeded emulsion polymerization of ethylene with (a) PMMA16 and (b) PMMA36, at 80 °C and 100 bar of ethylene pressure after the indicated polymerization time (respectively runs 3 and 4, Table 2). | ||
No control experiment of ethylene polymerization that could be performed without PMMAn in the exact same conditions can be compared to these experiments to show that this high molar mass population represents PE chains. Indeed, a typical radical emulsion polymerization of ethylene would not be of relevance here since it would not represent the seeded emulsion polymerization systems used. To confirm the nature of this second population, the final products were analyzed by FT-IR spectroscopy. The evolution of the absorption band area of the CH2 stretching from ethylene units (3000–2800 cm−1) was measured by normalizing the area of the absorption band of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from MMA units (1735 cm−1) (see Fig. S3†). If we compare the evolution of DPPE,NMR and the CH2 area with the polymerization time (Fig. 6), we can observe two different regimes of PE production. A first slow regime (up to one hour) where methacrylic oligomers are still available in the medium, and a second regime for which the production rate of PE is higher, when there are no methacrylic oligomers remaining. The transition to this second regime is also correlated with the appearance of a second high molar mass population in HT-SEC observed for elution times between 11 and 13 min (Fig. 7). This transition is observed after 2 h of polymerization for PMMA16 and after 1 h for PMMA36. These additional results show that a high molar mass PE homopolymer is formed when all the methacrylic oligomers have been consumed. The molar masses measured by high-temperature SEC in PS equivalent are particularly high, exceeding several hundred thousand grams per mol.53
O from MMA units (1735 cm−1) (see Fig. S3†). If we compare the evolution of DPPE,NMR and the CH2 area with the polymerization time (Fig. 6), we can observe two different regimes of PE production. A first slow regime (up to one hour) where methacrylic oligomers are still available in the medium, and a second regime for which the production rate of PE is higher, when there are no methacrylic oligomers remaining. The transition to this second regime is also correlated with the appearance of a second high molar mass population in HT-SEC observed for elution times between 11 and 13 min (Fig. 7). This transition is observed after 2 h of polymerization for PMMA16 and after 1 h for PMMA36. These additional results show that a high molar mass PE homopolymer is formed when all the methacrylic oligomers have been consumed. The molar masses measured by high-temperature SEC in PS equivalent are particularly high, exceeding several hundred thousand grams per mol.53
Thermal analysis was conducted on the copolymer dried extracts. The cooling curves plotted in Fig. 8 show that for short polymerization times (up to 1 h), an increase of Tg of the PMMA block is clearly observed (Table 2). In our case, the addition of ethylene units to a low molar mass PMMA extends the polymer chains leading to chain stiffening and hence an increase of the Tg value. This behavior can be interpreted by Fox and Flory54 with the use of the following equation:
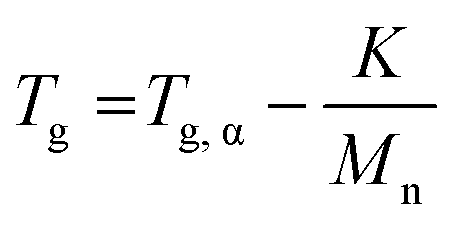 | (2) |
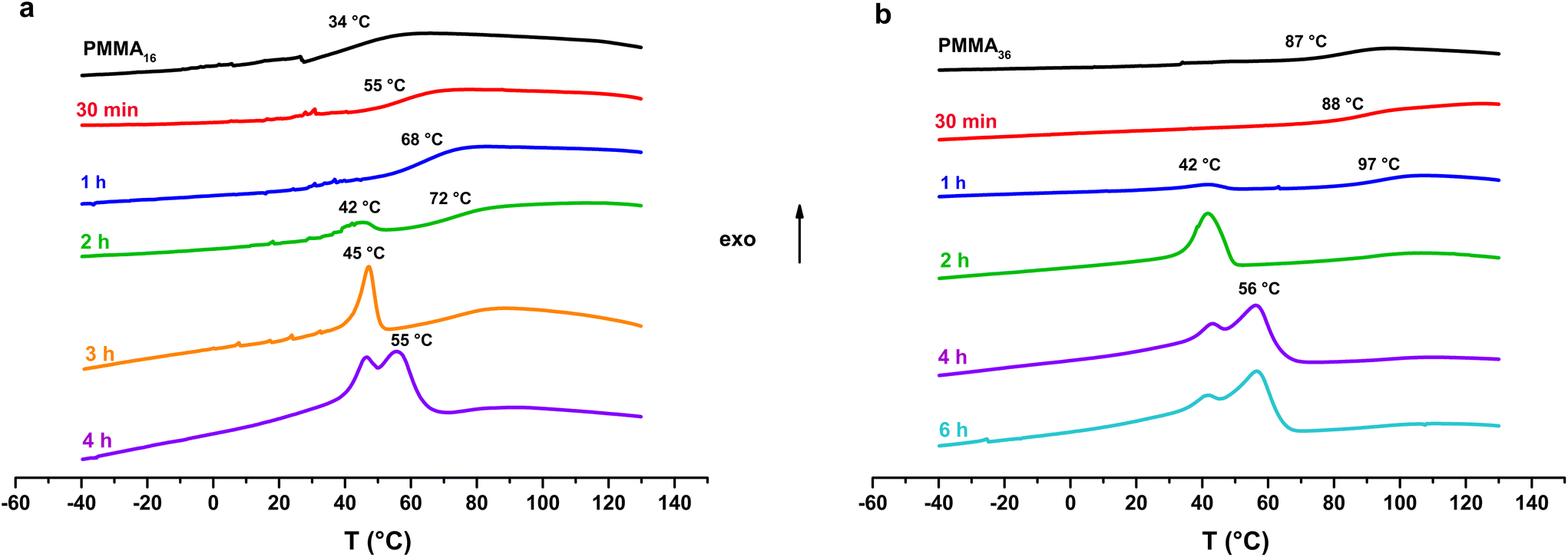 | ||
| Fig. 8 DSC analysis (cooling) of the samples obtained with (a) PMMA16 and (b) PMMA36 (respectively runs 3 and 4, Table 2). | ||
Fox and Flory's relation also explains the lower increase of Tg value of our copolymers in the case of PMMA36 since its molar mass is higher and so its Tg value is closer to the Tg,α. For both PMMA16 and PMMA36, a first population of chains crystallizing at 42 °C is visible at 2 h for PMMA16 and at 1 h for PMMA36, followed by a second at higher temperature (55–56 °C) for longer reaction times (4 h for the two PMMAn). The population of chains crystallizing at 55–56 °C may correspond to PE chains since they appear at the same time a second population of high molar mass chains is observed in HT-SEC (Fig. 7). The other population could mostly correspond to PMMA-b-PE chains, where DPPE are smaller and thus crystallize at lower temperature. The thermal behavior of these copolymers was compared to the behavior an ethylene homopolymer obtained by radical emulsion polymerization with the same initiating and stabilization system (see ESI for details on its synthesis†) and to a physical blend of PMMA16 and the same PE homopolymer. Firstly, the PE sample (dark) and the PMMA16/PE physical blend (blue) behave similarly when subjected to the same temperature profile (Fig. 9). A single crystallization peak is observed at close values, at 61 °C for the blend and at 64 °C for the PE homopolymer. The thermal behaviour of the copolymers synthesized with PMMA16 is different. The crystallization of the sample obtained after 3 h of polymerization (orange) occurs at a significantly lower temperature than that of the blend (45 °C vs. 61 °C for the blend), showing that PMMA interferes with crystallization of the PE segments and proving that it is indeed bound to PE. By comparing the thermograms of PE and of the copolymer obtained after 4 h of reaction (green), the second crystallization is most likely caused by PE chains formed when all the PMMA16 has been consumed.
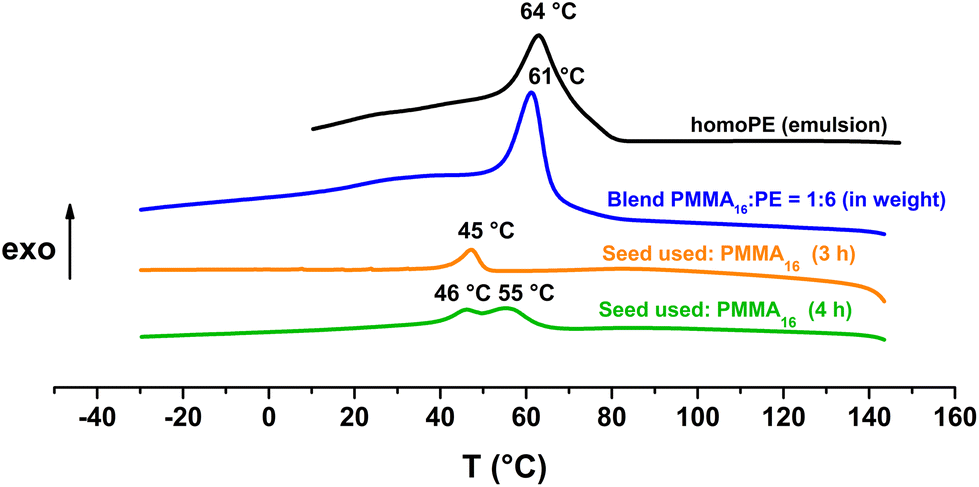 | ||
| Fig. 9 DSC analyses (cooling) of the polymers obtained after polymerization with PMMA16 as a seed (run 3-t4 and run 1 in Table 2), a blend of PMMA16 and PE (1:6 in weight), and homoPE obtained by radical emulsion polymerization in water. | ||
Finally, the colloidal features of the obtained dispersions, stable in each case, were investigated by DLS and TEM analyses.
The particle sizes determined by DLS (Fig. 10) do not significantly increase during the polymerization for the reaction with PMMA16 even after a long polymerization time (142 nm at the beginning and 155 nm after 4 h), contrary to PMMA36 where the particle size slightly increases (101 nm at the beginning and 117 nm after 6 h). Surprisingly, the polydispersity index decreases with the polymerization time. The reaction starts with a quite polydisperse latex, but during the reaction the size distribution seems to become narrower around higher particle sizes.
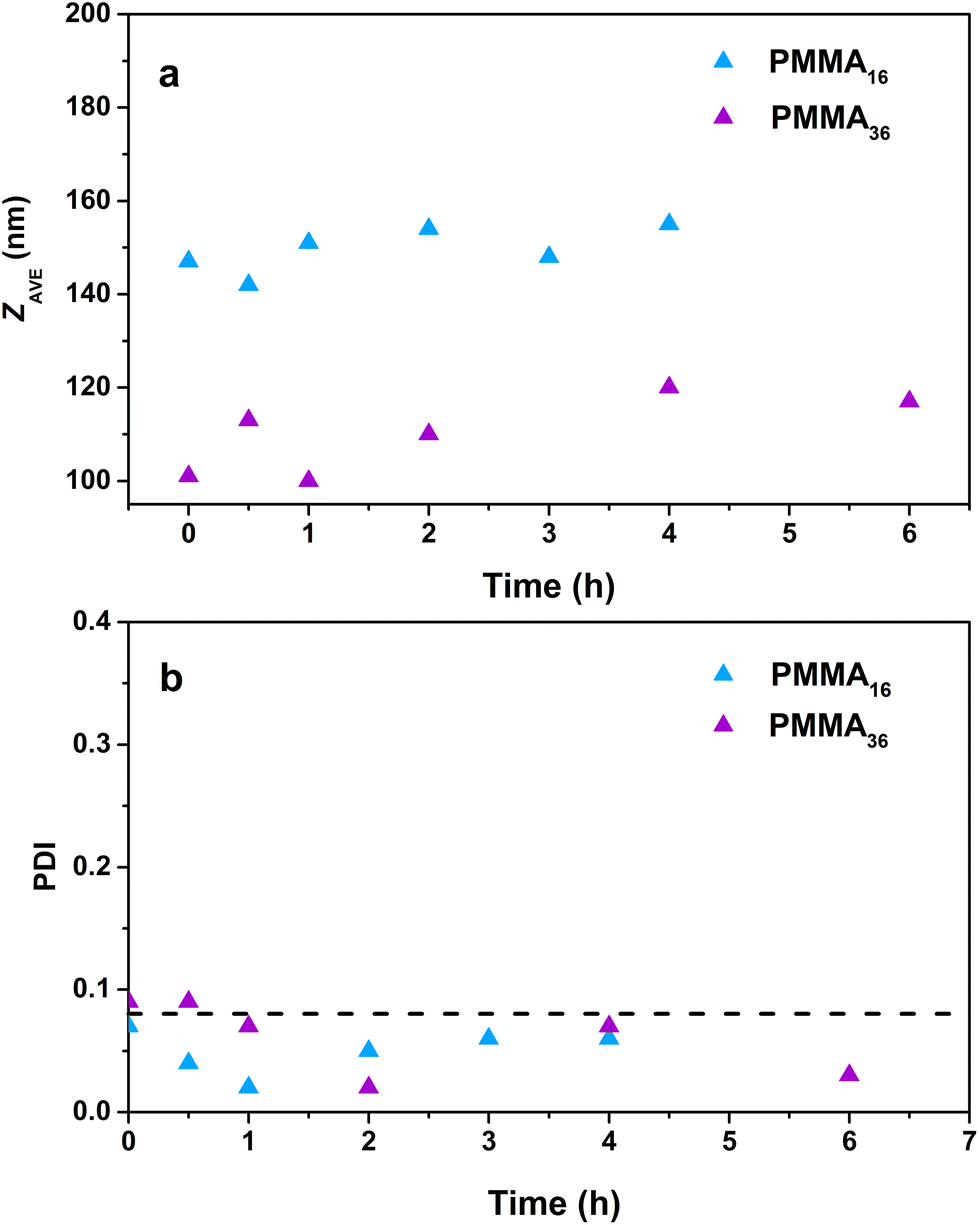 | ||
| Fig. 10 Evolution of the (a) particle size and (b) polydispersity index with polymerization time for polymer latexes obtained during seeded emulsion polymerization of ethylene with (a) PMMA16 and (b) PMMA36, at 80 °C and 100 bar of ethylene pressure (respectively runs 3 and 4, Table 2). | ||
Cryo-TEM analysis of the different latexes revealed that both the PMMA16 and PMMA36 particles were spherical and mostly polydisperse (Fig. 11a and Fig. 12a, respectively), explaining the discrepancies between the particle diameters determined by DLS and TEM. As they are initially polydisperse, it is difficult to conclude on any secondary nucleation that could occur during ethylene polymerization, especially when PE homopolymer starts to form. However, the particle surface seems to change when PE is formed, being more heterogeneous with some facets (Fig. 11b and 12b). This type of surface was already observed for PE particles by TEM in our group,50,55 when PE lamellas prohibit the formation of perfect spheres. However, we do not observe any phase separation with PE excrescences as already observed when PE was polymerized from PS seeds.56
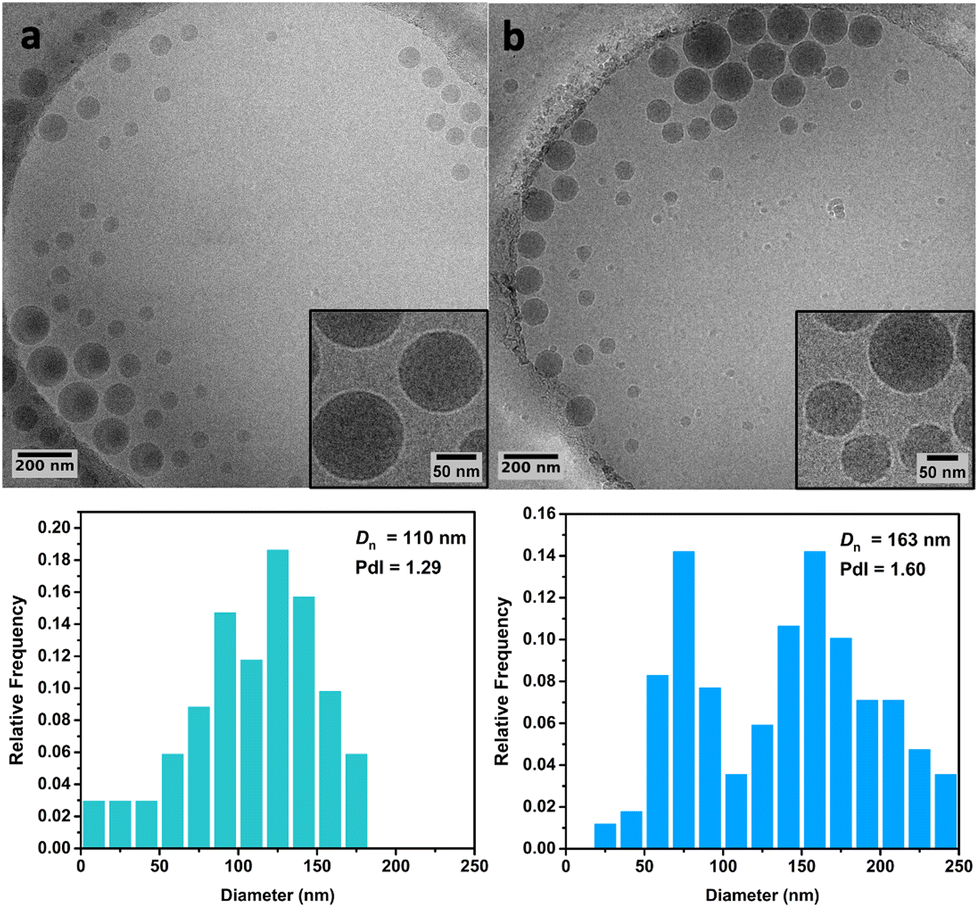 | ||
| Fig. 11 Cryo-TEM images and particle size distributions of (a) the PMMA16 seed particles and (b) of the particles obtained after 3 h of polymerization at 80 °C under 100 bar of ethylene pressure with PMMA16 seed (run 3-t4, Table 2). | ||
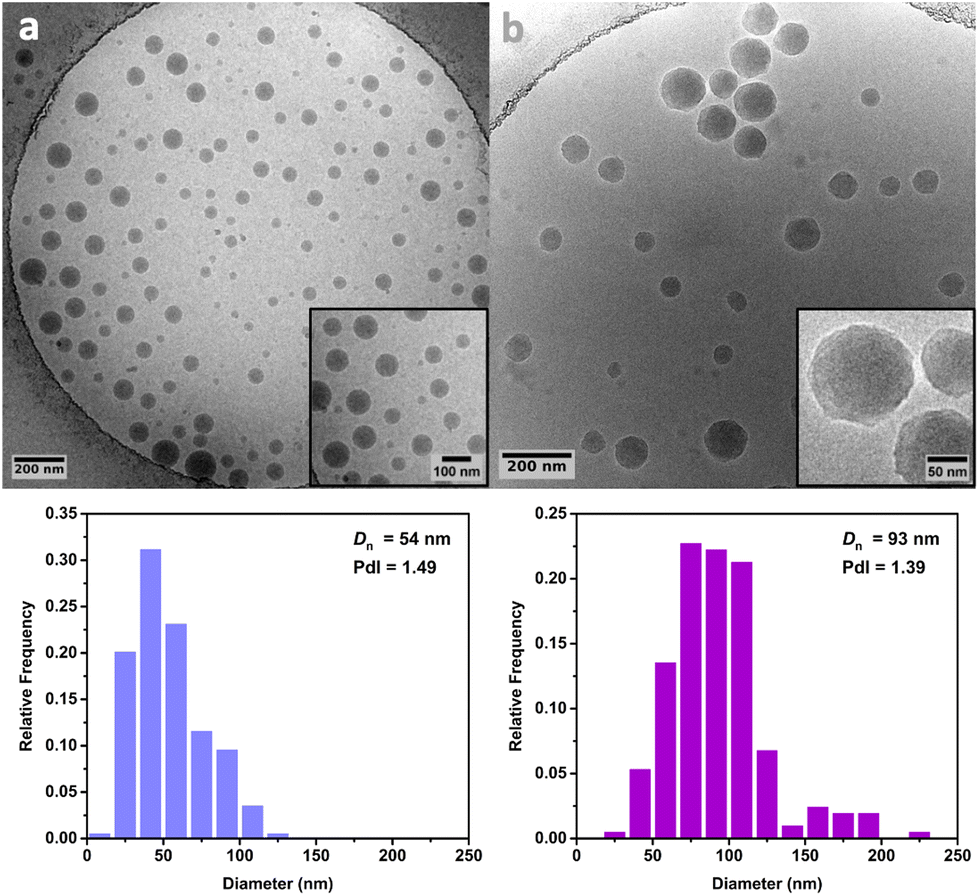 | ||
| Fig. 12 Cryo-TEM images and particle size distributions of (a) the PMMA36 seed particles and (b) the particles obtained after 4 h of polymerization at 80 °C under 100 bar of ethylene pressure with PMMA36 seed (run 4-t4, Table 2). | ||
Solids content effect
In our previous study polymerizations were performed in DMC,48 and it was shown that the amount of methacrylic oligomers added greatly affected the yield and thus the molar mass of the PE block formed. Thus, this part aims to study the behavior of our system when we increase the initial solids content (τs,i) of the PMMA16 seeds from 5 to 10 wt% while keeping all the other parameters constant (run 5, Table 2).Increasing τs,i to 10 wt% still led to the formation of a stable latex, and neither the particle size nor the polydispersity index are affected by this change. But the polymerization degree of PE and thus the molar mass are. Indeed, after 4 h of polymerization DPPE decreases from 26 down to 3. In DMC, when polymerizations were carried out with a molar ratio of [PMMA11]:[AIBN]:[ethylene] = 3:1:1900 instead of 1:1:1900, a drastic drop in yield but also in DPPE was observed (after 6 h, DPPE of 221 down to 44 for [PMMA11]:[AIBN] ratios increasing from 1 to 3).48 Thus, our results seem to be consistent with the results obtained in DMC and a degradative chain transfer role of PMMAn in ethylene emulsion polymerization. Indeed, by increasing the solids content, although the overall amount of vinyl functions is higher, the concentration of double bonds per particle, where the polymerization takes place, remains the same. For the same concentration of initiator, the time needed to consume the double bonds increases. Thus, after 4 hours, homopolymerization of ethylene has consumed more ethylene monomer at 5 wt% solids and the chains will then be longer in that case. In brief, when increasing the solids content, shorter PE chains are produced before being terminated reflecting in a decrease of DPPE.
Polymerization of ethylene in the presence of a PMMA seed synthesized by aqueous radical emulsion polymerization
With the aim of comparing the behavior of the PMMA seeds obtained by CCTP to a PMMA seed obtained by free radical emulsion polymerization in water, a PMMA latex was synthesized under conventional emulsion polymerization conditions, using SDS as surfactant and KPS as thermal initiator (PMMA-FRP, Table S1†). The conversion of MMA was almost complete (96%) and a stable latex was obtained.This latex was then diluted to set the initial solids content at 5 wt% and used as a seed in ethylene polymerization conducted at 80 °C, under 100 bar of ethylene for 4 h (run 6, Table 2). All of the parameters were kept constant between this reaction and run 1 (Table 2).
The increase of solids content with PMMA-FRP seed is significantly higher than with the PMMA16 CCTP seed (9.8 wt% with PMMA-FRP against 2.4 wt% with PMMA16). The latex remained stable and the particle size increased from 42 nm for the seeds to 68 nm for the final particles. It is worth mentioning that, during a MMA free radical polymerization, disproportionation reactions occur leading to around half of the chains carrying an unsaturated chain ends.57 Thus, this seed cannot be strictly speaking considered as a real “blank” experiment without double bonds. Furthermore, the molar mass of the chains for PMMA-FRP seed is much higher than the one for the PMMA16 seed (Mn = 138000 g mol−1 and Mn = 1600 g mol−1, respectively). So, for a fixed initial solids content, there are fewer double bonds in the PMMA-FRP seed. Accordingly, the probability/efficiency for an ethylene oligomer to terminate on a double bound is probably lower with these FRP seeds than with the PMMA16 explaining the above obtained results.
This experiment was mainly realized to compare the morphology of the obtained particles to those obtained from PMMA seeds produced by CCTP. Fig. 13 shows the cryoTEM images of the PMMA-FRP seed and of the particles obtained after 4 h of ethylene polymerization. The morphology of the latter looks elongated as made of two incompatible phases that would separate. This morphology is in sharp contrast with that of the particles obtained when using PMMA-CCTP seeds (Fig. 11 and 12) and for which the chemical link between PMMA and PE blocks seems to prevent phase separation.
 | ||
| Fig. 13 Cryo-TEM images of (a) PMMA-FRP particles (Table S2†) and (b) the particles obtained after 4 h of polymerization at 80 °C under 100 bar with PMMA-FRP seed (run 6, Table 2). | ||
Conclusions
The seeded radical emulsion polymerization of ethylene was performed using ω-unsaturated PMMA oligomer latexes synthesized by CCTP. In each case, stable latexes were produced. Thus, this chemistry previously performed in organic DMC solution was successfully transposed to a polymerization in aqueous dispersed media and yielding stable latexes. As in DMC, the chain end double bonds carried by PMMAn synthesized by CCTP are quickly consumed while ethylene is polymerized. For short reaction times, PMMA-b-PE block copolymers were produced directly in the particles, while for longer polymerization times PE homopolymer was additionally formed. Depending on the solids content of the seed latex, the molar mass of the PE block could be adjusted. Cryo-TEM analyses of the final latex confirmed that the chains in the particles do not phase separate. The synthesis of PMMA-b-PE copolymers in emulsion polymerization, a widely used industrial process, is thus now easily accessible by simply post-polymerization treatment of ω-unsaturated PMMA oligomer latexes synthesized industrially by CCTP for many years. Such copolymers may be useful for compatibilization. The strategy should be easily transposable to other kind of ω-unsaturated oligomers synthesized by CCTP, and thus enable the formation of new PE-based block copolymers in emulsion. To the best of our knowledge, the synthesis of such aqueous copolymer latexes is unprecedented.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
L. S. acknowledges funding from the Ministère de l'Enseignement Supérieur, de la Recherche et de l'Innovation. We also thank the Research Technology Platforms (RTP) of the University of Warwick and EPSRC for equipment funded in part by EPSRC EP/V036211/1 and EP/V007688/1.References
- Z. Chen and M. Brookhart, Acc. Chem. Res., 2018, 51, 1831–1839 CrossRef CAS PubMed
.
- C. J. Kay, P. D. Goring, C. A. Burnett, B. Hornby, K. Lewtas, S. Morris, C. Morton, T. McNally, G. W. Theaker, C. Waterson, P. M. Wright and P. Scott, J. Am. Chem. Soc., 2018, 140, 13921–13934 CrossRef CAS PubMed
- C. Dommanget, F. D'Agosto and V. Monteil, Angew. Chem., Int. Ed., 2014, 53, 6683–6686 CrossRef CAS PubMed
- A. Wolpers, C. Bergerbit, B. Ebeling, F. D'Agosto and V. Monteil, Angew. Chem., Int. Ed., 2019, 58, 14295–14302 CrossRef CAS PubMed
- A. Wolpers, F. Baffie, V. Monteil and F. D'Agosto, Macromol. Rapid. Commun., 2021, 42, 2100270 CrossRef CAS PubMed
- Y. Nakamura, B. Ebeling, A. Wolpers, V. Monteil, F. D'Agosto and S. Yamago, Angew. Chem., Int. Ed., 2018, 57, 305–309 CrossRef CAS PubMed
- A. Wolpers, F. Baffie, L. Verrieux, L. Perrin, V. Monteil and F. D'Agosto, Angew. Chem., Int. Ed., 2020, 59, 19304–19310 CrossRef CAS PubMed
- F. Baffie, M. Lansalot, V. Monteil and F. D'Agosto, Polym. Chem., 2022, 13, 2469–2476 RSC
- C. Bergerbit, F. Baffie, A. Wolpers, P.-Y. Dugas, O. Boyron, M. Taam, M. Lansalot, V. Monteil and F. D'Agosto, Angew. Chem., 2020, 132, 10471–10476 CrossRef
- J. Delorme, O. Boyron, P.-Y. Dugas, P.-E. Dufils, D. J. Wilson, V. Monteil, F. D'Agosto and M. Lansalot, Polym. Chem., 2020, 11, 7410–7420 RSC
- A. A. Gridnev and S. D. Ittel, Chem. Rev., 2001, 101, 3611–3660 CrossRef CAS PubMed
-
S. Slavin, K. McEwan and D. M. Haddleton, in Polymer Science: A Comprehensive Reference, Elsevier, 2012, pp. 249–275 Search PubMed
- D. M. Haddleton, E. Depaquis, E. J. Kelly, D. Kukulj, S. R. Morsley, S. A. F. Bon, M. D. Eason and A. G. Steward, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2378–2384 CrossRef CAS
- D. M. Haddleton, D. R. Maloney, K. G. Suddaby Adam Clarke and S. N. Richards, Polymer, 1997, 38, 6207–6217 CrossRef CAS
-
W. H. Moser, G. D. Joly, L. R. Krepski, B. N. Gaddam, A. S. Abuelyaman, B. D. Craig, T. D. Dunbar, C. Cao, J. D. Oxman, A. Falsafi and H. T. Bui, United States, US20150238389A1, 2015
-
G. D. Joly, A. S. Abuelyaman, R. S. Davidson, T. D. Jones, B. N. Gaddam and S. J. Moench, World Intellectual Property Organization, WO2014116461A1, 2014
-
W. H. Moser, A. R. Fornof, G. D. Joly, S. Yurt, L. R. Krepski, A. S. Abuelyaman, A. Falsafi, B. D. Craig, J. D. Oxman and B. N. Gaddam, World Intellectual Property Organization,WO2016133668A1, 2016
-
W. H. Moser, A. R. Fornof, G. D. Joly, S. Yurt, L. R. Krepski, A. S. Abuelyaman, A. Falsafi, B. D. Craig, J. D. Oxman and B. N. Gaddam, United States, US20200031961A1, 2020
-
G. D. Joly, L. R. Krepski, A. R. Fornof, S. Yurt, B. N. Gaddam and A. S. Abuelyaman, United States, US20150203658, 2015
- N. R. Bagnall, M. H. Jones, G. C. Jernigan, C. Routt, L. C. Dar and B. T. Worrell, J. Am. Chem. Soc., 2023, 145, 14202–14207 CrossRef CAS PubMed
- D. Kukulj, T. P. Davis, K. G. Suddaby, D. M. Haddleton and R. G. Gilbert, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 859–878 CrossRef CAS
- D. Kukulj, T. P. Davis and R. G. Gilbert, Macromolecules, 1997, 30, 7661–7666 CrossRef CAS
- K. G. Suddaby, D. M. Haddleton, J. J. Hastings, S. N. Richards and J. P. O'Donnell, Macromolecules, 1996, 29, 8083–8091 CrossRef CAS
- D. M. Haddleton, D. R. Morsley, J. P. O'Donnell and S. N. Richards, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3549–3557 CrossRef CAS
- S. A. F. Bon, D. R. Morsley, J. Waterson, D. M. Haddleton, M. R. Lees and T. Horne, Macromol. Symp., 2001, 165, 29–42 CrossRef CAS
- S. C. J. Pierik, B. Smeets and A. M. van Herk, Macromolecules, 2003, 36, 9271–9274 CrossRef CAS
- P. B. Zetterlund and D. R. D′hooge, Macromolecules, 2019, 52, 7963–7976 CrossRef CAS
- N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk, M. F. Cunningham and A. M. van Herk, Macromolecules, 2009, 42, 7332–7341 CrossRef CAS
- N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk, M. F. Cunningham and A. M. van Herk, Macromolecules, 2009, 42, 6422–6428 CrossRef CAS
- N. M. B. Smeets, J. P. A. Heuts, J. Meuldijk and A. M. van Herk, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5839–5849 CrossRef CAS
- A. A. Gridnev, Polym. J., 1992, 24, 613–623 CrossRef CAS
- G. Patias, A. M. Wemyss, S. Efstathiou, J. S. Town, C. J. Atkins, A. Shegiwal, R. Whitfield and D. M. Haddleton, Polym. Chem., 2019, 10, 6447–6455 RSC
- D. J. Krasznai, T. F. L. McKenna, M. F. Cunningham, P. Champagne and N. M. B. Smeets, Polym. Chem., 2012, 3, 992–1001 RSC
- N. G. Engelis, A. Anastasaki, G. Nurumbetov, N. P. Truong, V. Nikolaou, A. Shegiwal, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Nat. Chem., 2017, 9, 171–178 CrossRef CAS PubMed
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, ChemComm, 2009, 2727–2729 RSC
- C. J. Atkins, G. Patias, J. S. Town, A. M. Wemyss, A. M. Eissa, A. Shegiwal and D. M. Haddleton, Polym. Chem., 2019, 10, 646–655 RSC
- P. Cacioli, D. G. Hawthorne, R. L. Laslett, E. Rizzardo and D. H. Solomon, J. Macromol. Sci., Part A: Pure Appl.Chem., 1986, 23, 839–852 CrossRef
- L. M. Muratore, K. Steinhoff and T. P. Davis, J. Mater. Chem., 1999, 9, 1687–1691 RSC
- B. Yamada, P. B. Zetterlund and E. Sato, Prog. Polym. Sci., 2006, 31, 835–877 CrossRef CAS
- B. Yamada and S. Kobatake, Prog. Polym. Sci., 1994, 19, 1089–1131 CrossRef CAS
- B. Yamada, F. Oku and T. Harada, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 645–654 CrossRef CAS
- G. Nurumbetov, N. Engelis, J. Godfrey, R. Hand, A. Anastasaki, A. Simula, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2017, 8, 1084–1094 RSC
- L. Hutson, J. Krstina, C. L. Moad, G. Moad, G. R. Morrow, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2004, 37, 4441–4452 CrossRef CAS
- J. Krstina, G. Moad, E. Rizzardo, C. L. Winzor, C. T. Berge and M. Fryd, Macromolecules, 1995, 28, 5381–5385 CrossRef CAS
- A. Shegiwal, A. M. Wemyss, E. Liarou, J. Town, G. Patias, C. J. Atkins, A. Marathianos, D. W. Lester, S. Efstathiou and D. M. Haddleton, Eur. Polym. J., 2020, 125, 109491 CrossRef CAS
- I. Schreur-Piet and J. P. A. Heuts, Polym. Chem., 2017, 8, 6654–6664 RSC
- L. Chen, L. Yan, Q. Li, C. Wang and S. Chen, Langmuir, 2010, 26, 1724–1733 CrossRef CAS PubMed
- F. Baffie, G. Patias, A. Shegiwal, F. Brunel, V. Monteil, L. Verrieux, L. Perrin, D. M. Haddleton and F. D'Agosto, Angew. Chem., Int. Ed., 2021, 60, 25356–25364 CrossRef CAS PubMed
- A. Shegiwal, A. M. Wemyss, M. A. J. Schellekens, J. de Bont, J. Town, E. Liarou, G. Patias, C. J. Atkins and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, E1–E9 CrossRef
- G. Billuart, E. Bourgeat-Lami, M. Lansalot and V. Monteil, Macromolecules, 2014, 47, 6591–6600 CrossRef CAS
- E. J. Bradbury, D. McNulty, R. I. Savage and E. E. McSweeney, Ind. Eng. Chem., 1952, 44, 211–212 CrossRef CAS
- I. A. Maxwell, B. R. Morrison, D. H. Napper and R. G. Gilbert, Macromolecules, 1991, 24, 1629–1640 CrossRef CAS
- T. Williams and I. M. Ward, J. Polym. Sci., Part B: Polym. Lett., 1968, 6, 621–624 CrossRef CAS
- T. G. Fox and P. J. Flory, J. Polym. Sci., 1954, 14, 315–319 CrossRef CAS
- E. Grau, P.-Y. Dugas, J.-P. Broyer, C. Boisson, R. Spitz and V. Monteil, Angew. Chem., Int. Ed., 2010, 49, 6810–6812 CrossRef CAS PubMed
-
G. Billuart, PhD thesis, Université de Lyon, 2015
- G. Ayrey and C. G. Moore, J. Polym. Sci., 1959, 36, 41–53 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00897e |
| This journal is © The Royal Society of Chemistry 2023 |