
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmphiphilic oligo(2-ethyl-2-oxazoline)s via straightforward synthesis and their self-assembly behaviour†
James
Lefley
a,
Zivani
Varanaraja
a,
Ben
Drain
a,
Steven
Huband
b,
James
Beament
c and
C. Remzi
Becer
 *a
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: remzi.becer@warwick.ac.uk
bX-ray Diffraction RTP, Department of Physics, University of Warwick, Coventry, CV4 7AL, UK
cInfineum UK Ltd., Milton Hill Business & Technology Centre, Abingdon, Oxfordshire OX13 6BB, UK
First published on 25th September 2023
Abstract
The synthesis of poly(2-oxazoline)s offers an unparalleled degree of functionalization when fabricating smart, functional polymers for biomedical uses. The termination of 2-oxazoline polymerisations by direct endcapping can be exploited to introduce a wide variety of end groups and could potentially offer an easier synthetic route to amphiphilic polymers that usually block copolymer synthesis. Herein, we report a facile one-pot synthesis and preparation of dodecyl-end capped oligo(2-ethyl-2-oxazoline)s via direct endcapping and thiol–yne click chemistry. A small set of propargyl tosylate initiated PEtOx oligomers were synthesised and subsequently functionalized with varying equivalents of dodecanethiol. GPC, NMR and MALDI-ToF were utilised for molecular weight analysis and determination of end-group fidelity. Film rehydration was employed to prepare self-assembled nanoparticles due the inexpensive set-up and practical simplicity of the technique. DLS, SAXS and TEM revealed that mono- and di-functionalized PEtOx self-assembled into micelles around 10 nm in diameter whereas tri-functionalized PEtOx was too hydrophobic and precipitated in aqueous solution. All oligomers were screened for their ability to encapsulate a model hydrophobic drug, curcumin, and UV-Vis spectrometry was utilized to determine the encapsulation efficiencies and drug loading capacities. Di-functionalized PEtOx provided the greatest drug loading capacity (8 wt%) of this study.
Introduction
Amphiphilic copolymers have gained significant attention in the field of polymer science due to their unique ability to form self-assembled nanostructures in a selective solvent such as water.1–3 By exploiting this phenomenon, researchers have been able to synthesise amphiphilic copolymers for a variety of biomedical applications such as the encapsulation and delivery of therapeutic drugs, complexation and delivery of genes, as well as the synthesis of artificial cells and nanoreactors.4–9 Given the potential significance of amphiphilic copolymers in future biomedical studies, it is therefore important to explore new strategies to synthesise amphiphilic macromolecules that exhibit self-assembly behaviour.Poly(2-oxazoline)s (POxs) are a class of polymeric materials that have been extensively studied for a diverse range of potential applications with a heavy focus in the biomedical field.10 Due to the living nature of the ω-chain end, POxs can be terminated with a variety of nucleophiles such as carboxylic acids,11,12 amines13,14 and thio-compounds,15,16 thus offering a facile way to introduce functionality to the chain end. The ability to further functionalize the α-chain end and the pendent side chain offers an incomparable level of functionalization and control over the chemical and physical properties of POx-based materials.17,18 Particularly within the past decade, POxs have been suggested as a viable alternative to PEG in biomedical applications due to their comparable stealth properties19–21 and the emergence of antiPEG-antibodies recently discovered in humans.22 As a result, many amphiphilic POx-based drug delivery systems have been reported for the encapsulation of a number of hydrophobic drugs.23–29 In one of the most recent examples, Hoogenboom et al. investigated the micellization and drug loading (DL) behaviours of block and gradient analogues of MeOx-PhOx copolymers of varying overall degrees of polymerization (DP).30 They found very little difference between the DL capacities between gradient copolymers and their block copolymer equivalents and achieved a maximum curcumin DL capacity of 16 wt%. So far, the gold standard of POx-based micelles for drug encapsulation has been reported by Luxenhofer et al.31 The use of doubly amphiphilic POx/POz-based copolymers (such as MeOx-PrOz-MeOx) allows for the encapsulation and co-formulation of a library of hydrophobic drugs and, in some cases, achieving DL values >50 wt%. This is considerably higher than DL values that are usually reported in the literature.
Evidently, copolymer systems such as the examples mentioned above can offer great drug solubilization properties. However, in most cases, their use of commercially unavailable and specialist reagents could potentially hinder their success if such a copolymer system needed to be synthesised on a larger scale.
Other than the simplest 2-alkyl-2-oxazolines (C1–C4) and 2-phenyl-2-oxazoline, most other 2-oxazoline monomers are not commercially available. Furthermore poly(2-oxazines), which are a relatively new class of poly(2-oxazoline) derivatives, are not commercially available either and require a multi-step synthesis and purification strategy to yield the monomer.32 Therefore, we propose whether drug loading capacities, similar to the values currently reported in the literature, can be obtained from synthetically simple endcapped-POx amphiphiles. Only a few examples exist in the literature and have been reported by Amiel and co-workers.33–35 In these studies, homopolymers of 2-methyl-2-oxazoline were synthesised via the use of a dodecyl-iodide initiator to form simple micelles. Compared to tosylates, iodide-initiated CROP reactions suffer from slower propagation rates and poorer initiation efficiencies.36 Recently, we have reported that thiol-endcapping of POxs is a very efficient termination reaction that results in complete functionalization of the ω-chain end, thus allowing access to the synthesis of multi block copolymers.37 Furthermore, a wide variety of thiols can be purchased from commercial suppliers in cost-effective, large quantities. The use of alkyl thiols to end-functionalize POxs would also circumvent the use of alkyl iodide initiators with poor initiation efficiencies. Inspired by this, we have demonstrated the use of thiol-endcapping for the preparation pseudo-block copolymers.
In this investigation, we report a very easy, one pot synthesis of the direct endcapping of dodecanethiol onto PEtOx. Furthermore, we employed the use of a functional and clickable initiator, propargyl tosylate, that allowed us to vary the degree of dodecyl functionalization onto PEtOx via a combination of direct-endcapping and thiol–yne click chemistry. Thus, we were able to synthesise a small set of oligomers with varying ratios of hydrophilic/hydrophobic mass fractions. From this, we investigated how the thermal, self-assembly and drug encapsulation behaviours of these polymers were affected by varying the extent of dodecyl-functionalization of PEtOx.
Results and discussion
Synthesis of PEtOx with a monomer-to-initiator ratio [M]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I] = 10 was carried out at a lower than optimal temperature of 110 °C to facilitate immediate endcapping after polymerization. The optimal conditions (140 °C in acetonitrile) reported by Hoogenboom were used as the basis for the modified procedure.38 Propargyl tosylate (PropTos) was selected as the initiator to introduce alkyne functionality at the α-end of the chain for post-polymerization modification using thiol–yne click chemistry. In a recent study from our group,39 we found that using PropTos resulted in the complete and efficient initiation of EtOx at a lower temperature of 100 °C with no sign of non-linear first order kinetics which is indicative of slow initiation. The synthetic procedures of all oligomers is outlined in Fig. 1. Despite the lower reaction temperature, the polymerization reactions all achieved full conversion within 30 minutes. TF0 and TF2 were terminated with an excess of piperidine at the ω-end and TF0 was purified. A thiol–yne reaction of the TF2 intermediate was carried out with 5 equivalents of dodecanethiol at the α-end to yield the di-substituted product, TF2. For TF1 and TF3, both were terminated with an excess of dodecanethiol thiol at the ω-end and TF1 was purified. For TF3, a solution of AIBN/acetonitrile was added to the mono-substituted TF3 intermediate to further functionalize the α-end with the remaining equivalents of dodecanethiol in the reaction vessel. The thiol–yne reaction yielded the tri-substituted product, TF3.
:[I] = 10 was carried out at a lower than optimal temperature of 110 °C to facilitate immediate endcapping after polymerization. The optimal conditions (140 °C in acetonitrile) reported by Hoogenboom were used as the basis for the modified procedure.38 Propargyl tosylate (PropTos) was selected as the initiator to introduce alkyne functionality at the α-end of the chain for post-polymerization modification using thiol–yne click chemistry. In a recent study from our group,39 we found that using PropTos resulted in the complete and efficient initiation of EtOx at a lower temperature of 100 °C with no sign of non-linear first order kinetics which is indicative of slow initiation. The synthetic procedures of all oligomers is outlined in Fig. 1. Despite the lower reaction temperature, the polymerization reactions all achieved full conversion within 30 minutes. TF0 and TF2 were terminated with an excess of piperidine at the ω-end and TF0 was purified. A thiol–yne reaction of the TF2 intermediate was carried out with 5 equivalents of dodecanethiol at the α-end to yield the di-substituted product, TF2. For TF1 and TF3, both were terminated with an excess of dodecanethiol thiol at the ω-end and TF1 was purified. For TF3, a solution of AIBN/acetonitrile was added to the mono-substituted TF3 intermediate to further functionalize the α-end with the remaining equivalents of dodecanethiol in the reaction vessel. The thiol–yne reaction yielded the tri-substituted product, TF3.
 | ||
| Fig. 1 Synthesis of amphiphilic oligo(2-ethyl-2-oxazoline)s via cationic ring opening polymerization (CROP). TF0 and TF1 are obtained by direct endcapping after CROP with piperidine or dodecanethiol respectively. Radical thiol–yne reactions of the propargyl group on TF0 and TF1 with dodecanethiol yields TF2 and TF3 respectively. | ||
1H spectroscopic analysis of TF1–TF3 revealed the successful functionalization of PEtOx with dodecanethiol after purification due to the appearance of peaks at 1.3 ppm and 0.9 ppm corresponding to –CH2– and –CH3 of the alkyl chains respectively (Fig. S1–S4†). The functionalization efficiencies were calculated by comparing the ratio of the proton integrals between the –CH3 groups of dodecanethiol (0.9 ppm) and PEtOx (1.10–1.20 ppm). For TF0 and TF1, complete functionalization (>99%) was achieved whereas TF2 and TF3 achieved functionalization efficiencies of 61% and 72% respectively which suggests the thiol–yne reaction was not 100% efficient and that a mixture of products are obtained. However, further end-group analysis revealed by MALDI-ToF MS suggests the absence of partially functionalized products (Fig. 2). The MALDI spectra of TF0–TF3 each show a single distribution that corresponds to the sodium adduct of DP10 PEtOx assuming complete functionalization of end groups.
 | ||
| Fig. 2 Full spectrum and magnified inset (dashed box) of MALDI-ToF for piperidine and/or dodecyl-functionalized PEtOx (A) TF0, (B) TF1, (C) TF2 and (D) TF3. All major distributions correspond to the sodium adduct of the completely functionalized product. | ||
Although TF2 shows a minor secondary distribution, this can be attributed to the hydrogen adduct of the fully functionalized product. A difference of 22 Da separates the major distribution (Na adduct) and the minor distribution (H adduct) of TF2. Within each spectrum, the peaks of the major distribution are separated by 99 Da which corresponds to the mass of the EtOx repeat unit. Verification of the end-groups was conducted by comparing the calculated and found masses of an individual n-mer. Taking TF2 as an example, the theoretical mass of the sodium adduct of the difunctionalized 11-mer endcapped with piperidine is 1640.432 Da. The observed mass for the 11-mer is 1640.422 Da. A difference of 0.01 Da. End-group verification for all oligomers can be found in the ESI (Table S3†). GPC-SEC traces of TF0–TF3 showed that oligomers had monomodal molecular weight distributions with narrow dispersity values ranging from 1.08–1.13 (Fig. 3). A clear shift to successive higher molecular weights can clearly be seen as the number of dodecyl moieties per PEtOx chain increases. Upon comparing the physical appearances of the purified products, it was noted that TF0–TF3 exhibited interesting physical properties. Whereby TF0 and TF2 were both dry powders and TF1 and TF3 were tacky, amorphous solids. DSC and TGA analysis was employed to elucidate how the increasing number dodecyl moieties affected the thermal properties of the oligomers (Fig. S5†). Firstly, it should be noted that the Tg value of TF0 (Tg = 50 °C) is lower than the reported Tg value of PEtOx homopolymers of higher DPs (Tg = 60–62 °C).40,41 This is due to the very low molecular weight of the oligomer. Short chain polymers have more free volume than long chain polymers which gives a lower Tg as stated by the Flory-Fox equation. In the case of TF0, TF1 and TF3, the greater extent of dodecyl functionalization resulted in an expected reduction in glass transition temperature (Tg) (50 °C, 18 °C and 10 °C respectively). Interestingly in the case of TF2, the Tg increased to 33 °C. To verify if this was an anomalous result or not, a second batch of TF2 was synthesized using the same procedure as before (characterization data found in ESI Fig. S7†). The second batch of TF2 provided a not too dissimilar Tg value of 30 °C. The increase in Tg going from TF1 to TF2 was unexpected and could potentially be attributed to the differences in the packing arrangement of the chains. TGA measurements showed that the onset of thermal degradation for all dodecyl endcapped PEtOx oligomers starts at 250 °C onwards (Fig. S6†).
 | ||
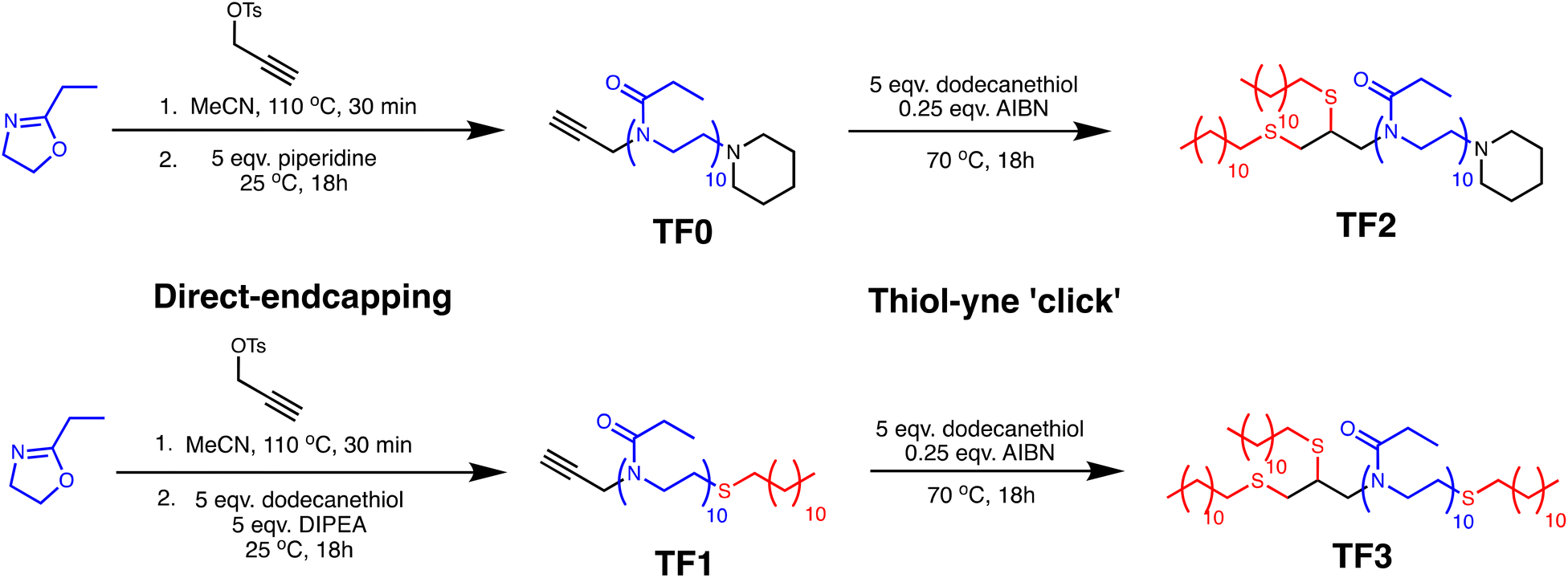
| Fig. 3 GPC-SEC traces of the dodecyl-functionalized PEtOx polymers TF0–TF3. Measurements performed using THF (2% TEA and 0.01% BHT) as the eluent. PMMA standards were used for the calibration. | ||
Next, for each oligomer, the self-assembly behaviour in aqueous solution was studied. In theory, all dodecyl functionalized PEtOx oligomers have some degree of amphiphilicity with all having a hydrophilic mass fraction between f = 61% and 89% and should exhibit some sort of self-assembly behaviour. Thin film hydration was chosen as the nanoparticle preparation method and the procedure used was adapted from literature. This technique is simple, very easy and requires no specialist equipment to perform. As expected, TF0 did not show any self-assembly due to the lack of a hydrophobic moiety. Surprisingly, TF3, functionalized with three dodecyl moieties, did not self-assemble and instead macroprecipitated shortly after rehydration. Self-assembly was confirmed for TF1 and TF2, the unloaded (UL) and curcumin-loaded (L) micelles were studied using dynamic light scattering (DLS), small angle X-ray scattering (SAXS) and transmission electron microscopy (TEM).
Compared to other polyoxazoline-based amphiphiles found in literature, the self-assembled structures of TF1-UL and TF2-UL were limited to very small micelles with number-averaged diameter sizes 5.5 and 5.4 nm respectively obtained by DLS (Fig. S10–S12 and Table S4†). Given the very short length of PEtOx and dodecyl chains, this was expected. The diameters of TF1-L (6.8 nm) and TF2-L (7.7 nm) were found to be slightly larger than the unloaded micelles. Small angle X-ray scattering (SAXS) was employed to gain further insight into the overall size of the nanoparticles and the internal structures (Fig. 4A–D). Seen as TF0 and TF3 did not self-assemble, SAXS measurements of these samples were not conducted. The obtained SAXS responses for TF1-UL, TF1-L and TF2-UL micelles were fitted with SASview using a core–shell sphere model. TF2-L showed signs of agglomeration with an increase in scattering at low q values. Therefore, a power law slope was integrated into the core–shell fit to model the agglomeration. Comparing the measured diameters obtained from DLS and SAXS for TF1 (6.82 nm and 7.67 nm respectively) and TF2 (7.65 nm and 8.95 nm respectively), the values obtained show good agreement with each other between each of the analytical techniques. It can also be seen that TF2 micelles are slightly larger, albeit very similar, in diameter than TF1. Values of the mean core radius and mean shell thickness for TF1 and TF2 unloaded and loaded micelles can be found in the ESI (Table S2†).
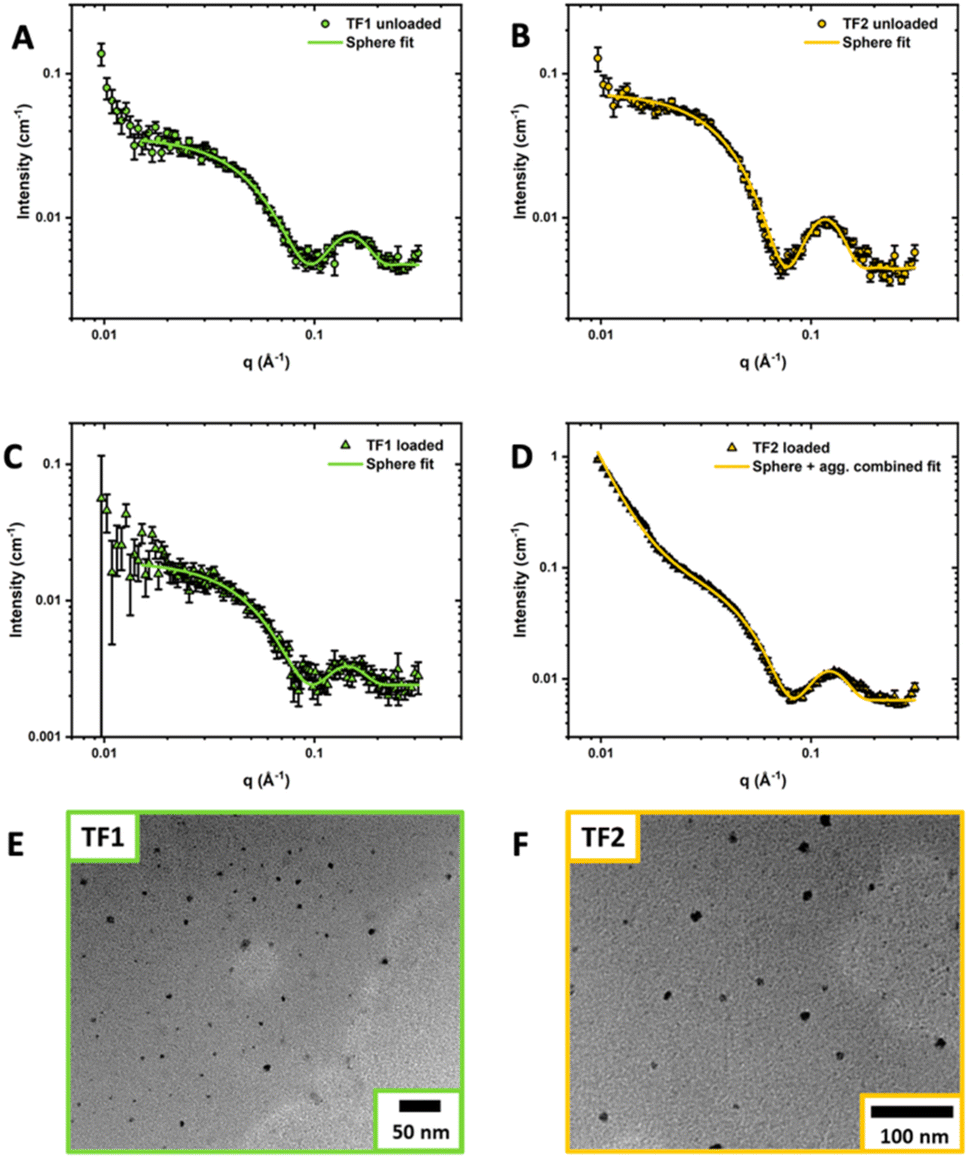 | ||
| Fig. 4 SAXS patterns and data fits of TF1-UL (A), TF1-L (B), TF2-UL (C) and TF2-L (D) nanoparticle solutions. Representative TEM images taken of TF1-L (E) and TF2-L (F). Oligomer and curcumin feed concentrations used were Coligo = 10 mg mL−1 and CCUR = 2 mg mL−1. | ||
The thermodynamic stability of micelles is important to quantify as micellar drug formulations need to be stable whilst circulating the body. Higher thermodynamic stability can reduce leaching of the therapeutic drug and prevent disassembly of the micelles upon large dilutions.42 A critical micelle concentration (CMC) is defined as the minimum concentration of a surfactant or polymer required to form micellar aggregates and is used to evaluate the thermodynamic stability of polymeric micelles. CMCs were determined for unloaded and loaded TF1 and TF2 micelles via pyrene fluorescence spectroscopy and ranged from 2–30 mg L−1 (Fig. S8†) which corroborates nicely with other CUR-loaded POx-based nanoformulations.26,30,31
Thermoresponsivity of the oligomeric micelles was studied to observe if there was any temperature-sensitive behaviour.
For P(EtOx) homopolymers, LCST-behaviour is usually observed at high DPs (>100).43 However, given the very short length of the chain and the very hydrophobic end groups, we hypothesized that there could be some potential thermoresponsive behaviour of TF1 and TF2. Turbidimetry studies of the unloaded and loaded micelles of TF1 and TF2 were conducted and cloud point temperatures (Tcp) were determined at 50% transmittance (Fig. S9†). LCST-type behaviour for both TF1-UL and TF1-L was observed whilst TF2 showed no LCST-type behaviour. It was found that drug loading of the micelles results in a substantial reduction of Tcp. TF1-UL gave a cloud point of Tcp = 45 °C whilst the drug loaded micelles TF1-L gave a much lower cloud point of Tcp = 32 °C. The decrease in Tcp is mostly likely caused by the formation of oligomer-drug complexes resulting in the increased hydrophobization of the system. Interestingly though, TF2 which has two dodecyl substituents as opposed to TF1 with one substituent, showed no LCST-type behaviour despite its greater hydrophobicity. This result deviates from the expected outcome and warrants further investigation to determine the underlying cause of this unexpected result.
The ability of the prepared oligomers to solubilize a model hydrophobic drug was studied including both TF0 and TF3. Curcumin was chosen as a model hydrophobic compound to encapsulate due to its recent interest among research groups as a potential therapeutic compound. However, curcumin suffers from extremely poor water solubility (<8 μg mL−1), bioavailability and rapid metabolism by the body thus making it a promising candidate to study.44 Thin film hydration was selected for the preparation method for this study as it has been employed in previous studies for the preparation of POx-based curcumin nanoformulations.24,26,30,31 Aliquots of pre-made stock solutions of the oligomers (30 mg mL−1) and curcumin (5 mg mL−1) in ethanol were mixed in pre-determined ratios. The ethanol was removed via gentle heating under a stream of nitrogen and the films were dried in a vacuum oven overnight.
Upon rehydration, the desired oligomer (10 mg mL−1) and curcumin (1, 2 and 4 mg mL−1) concentrations were achieved and stirred for 24 hours. The nanoformulations were centrifuged and filtered to remove any curcumin that had not been incorporated in the micelles. A 50 μL aliquot of the drug-loaded micelle solution was diluted up to 1 mL with ethanol. UV-Vis spectroscopy was then used to determine the amount of curcumin solubilized by comparing the absorbance values against a calibration curve of known concentrations of curcumin (Fig. 5A). From these values, the encapsulation efficiencies and drug loading capacities were calculated (see ESI eqn (1) and (2)†). TF2 with a 2 mg ml−1 curcumin feed provided a nanoformulation with the highest drug loading capacity of 8 wt% (Table S5†). Furthermore, TF2 exhibited the best loading efficiency (LE) and drug loading (DL) values at all concentrations of the curcumin feeds compared to all other oligomers. In the case of the nanoformulations using 1 mg mL−1 and 2 mg mL−1 curcumin feeds, TF2 managed to solubilize nearly 3 times the amount of curcumin than TF1. This is most likely due to the greater hydrophobicity of the core, with TF2 having double the number of dodecyl-chains than TF1. TF0 and TF3 encapsulated a negligible amount of curcumin at all concentration feeds as expected. Nanoformulations made using a 4 mg mL−1 curcumin feed resulted in a significant decrease in the amount of drug loading. Higher concentrations of curcumin lead to the formation of insoluble oligomer-drug aggregates and results in the increased hydrophobicity of the entire system. This phenomenon is consistent with similar investigations in literature.28Fig. 5B shows the loss of curcumin 6 months after preparation of the nanoformulations of TF1 and TF2 using a 2 mg mL−1 curcumin feed. A significant loss of the encapsulated payload is recorded for TF1 after 6 months whilst TF2 shows a much smaller loss (73% and 13% respectively). The substantial leakiness of TF1 compared to TF2 can be attributed to the lower thermodynamic stability of TF1. TF2-L has a CMC of 2 mg L−1 compared to TF1-L with a CMC of 30 mg L−1. A lower CMC corresponds to more thermodynamically stable micelles which may result in slower leaching of encapsulated drugs.42
 | ||
| Fig. 5 (A) Solubilization data of dodecyl functionalized PEtOx showing the encapsulation efficiencies and drug loading capacities of each sample prepared via thin film hydration. (B) Nanoformulation stability of TF1-L and TF2-L with 2 mg mL−1 CUR feed recorded at the time of preparation (blue) and 6 months later (pink). Oligomer and curcumin feed concentrations used were Coligo = 10 mg mL−1 and CCUR = 1, 2 and 4 mg mL−1. Values expressed as a mean from three repeats of each experiment. Error expressed as 1 SD from the mean (n = 3). | ||
Conclusions
Functionalization of PEtOx via direct endcapping and thiol–yne click chemistry has shown to produce simple, amphiphilic, amphiphilic oligomers that are able to self-assemble into micelles sub 10 nm in diameter. Mono- and di-functionalized P(EtOx) oligomers were capable of solubilizing a hydrophobic drug, curcumin, and achieved a maximum DL capacity of 8 wt%. Indeed, POx-based block and gradient copolymers have been shown to solubilize greater amounts of curcumin and should be utilized to achieve greater DL capacities. However, to our knowledge, this is the first reported example of simple alkyl functionalized PEtOx showing DL capacities this high. Future work may include screening a variety of hydrophobic alkyl and aryl endcapping reagents to further study the effect on self-assembly and drug encapsulation.Author contributions
JL – synthesis, self-assembly, drug encapsulation, NMR, GPC, DLS, UV-Vis and TEM analysis. ZV – DSC and TGA. BD – MALDI-ToF. SH – SAXS. JB – supervision and manuscript proof reading. CRB – supervision and manuscript proof reading.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JL thanks the Warwick Polymer Characterisation RTP for the use of their DLS, DSC and TGA instruments. We also thank the Warwick X-ray Diffraction RTP and Electron Microscopy RTP for the use of their SAXS and TEM facilities respectively. This research was sponsored by Infineum UK and we thank them for their financial support.References
- G. Riess, Micellization of block copolymers, Prog. Polym. Sci., 2003, 28(7), 1107–1170, DOI:10.1016/S0079-6700(03)00015-7.
- S. Hornig, T. Heinze, C. R. Becer and U. S. Schubert, Synthetic polymeric nanoparticles by nanoprecipitation, J. Mater. Chem., 2009, 19(23), 3838–3840, 10.1039/B906556N.
- M. Karayianni and S. Pispas, Self-Assembly of Amphiphilic Block Copolymers in Selective Solvents, in Fluorescence Studies of Polymer Containing Systems, ed. K. Procházka, Springer International Publishing, Cham, 2016, pp. 27–63 Search PubMed.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Block Copolymer Micelles in Nanomedicine Applications, Chem. Rev., 2018, 118(14), 6844–6892, DOI:10.1021/acs.chemrev.8b00199.
- J. P. K. Armstrong, M. N. Holme and M. M. Stevens, Re-Engineering Extracellular Vesicles as Smart Nanoscale Therapeutics, ACS Nano, 2017, 11(1), 69–83, DOI:10.1021/acsnano.6b07607.
- A. K. Blakney, R. Liu, G. Yilmaz, Y. Abdouni, P. F. McKay, C. R. Bouton, R. J. Shattock and C. R. Becer, Precisely targeted gene delivery in human skin using supramolecular cationic glycopolymers, Polym. Chem., 2020, 11(22), 3768–3774, 10.1039/D0PY00449A.
- A. K. Blakney, G. Yilmaz, P. F. McKay, C. R. Becer and R. J. Shattock, One Size Does Not Fit All: The Effect of Chain Length and Charge Density of Poly(ethylene imine) Based Copolymers on Delivery of pDNA, mRNA, and RepRNA Polyplexes, Biomacromolecules, 2018, 19(7), 2870–2879, DOI:10.1021/acs.biomac.8b00429.
- R. J. R. W. Peters, I. Louzao and J. C. M. van Hest, From polymeric nanoreactors to artificial organelles, Chem. Sci., 2012, 3(2), 335–342, 10.1039/C2SC00803C.
- B. C. Buddingh’ and J. C. M. van Hest, Artificial Cells: Synthetic Compartments with Life-like Functionality and Adaptivity, Acc. Chem. Res., 2017, 50(4), 769–777, DOI:10.1021/acs.accounts.6b00512.
- S. Nemati Mahand, S. Aliakbarzadeh, A. Moghaddam, A. Salehi Moghaddam, B. Kruppke, M. Nasrollahzadeh and H. A. Khonakdar, Polyoxazoline: A review article from polymerization to smart behaviors and biomedical applications, Eur. Polym. J., 2022, 178, 111484, DOI:10.1016/j.eurpolymj.2022.111484.
- A. Krieg, C. Weber, R. Hoogenboom, C. R. Becer and U. S. Schubert, Block Copolymers of Poly(2-oxazoline)s and Poly(meth)acrylates: A Crossover between Cationic Ring-Opening Polymerization (CROP) and Reversible Addition–Fragmentation Chain Transfer (RAFT), ACS Macro Lett., 2012, 1(6), 776–779, DOI:10.1021/mz300128p.
- L. M. Stafast, C. Weber, M. T. Kuchenbrod, S. Hoeppener, M. Behnke, S. Schubert, K. Mehmetaj, A. T. Press, M. Bauer and U. S. Schubert, Poly(2-oxazoline) Homopolymers and Diblock Copolymers Containing Retinoate ω-End Groups, ACS Appl. Polym. Mater., 2022, 4(5), 3417–3425, DOI:10.1021/acsapm.2c00037.
- J. Stadermann, H. Komber, M. Erber, F. Däbritz, H. Ritter and B. Voit, Diblock Copolymer Formation via Self-Assembly of Cyclodextrin and Adamantyl End-Functionalized Polymers, Macromolecules, 2011, 44(9), 3250–3259, DOI:10.1021/ma200048a.
- L. Tauhardt, M. Frant, D. Pretzel, M. Hartlieb, C. Bücher, G. Hildebrand, B. Schröter, C. Weber, K. Kempe, M. Gottschaldt, K. Liefeith and U. S. Schubert, Amine end-functionalized poly(2-ethyl-2-oxazoline) as promising coating material for antifouling applications, J. Mater. Chem. B, 2014, 2(30), 4883–4893, 10.1039/C4TB00193A.
- V. R. de la Rosa, Z. Zhang, B. G. De Geest and R. Hoogenboom, Colorimetric Logic Gates Based on Poly(2-alkyl-2-oxazoline)-Coated Gold Nanoparticles, Adv. Funct. Mater., 2015, 25(17), 2511–2519, DOI:10.1002/adfm.201404560.
- A.-K. Trützschler, M. N. Leiske, M. Strumpf, J. C. Brendel and U. S. Schubert, One-Pot Synthesis of Block Copolymers by a Combination of Living Cationic and Controlled Radical Polymerization, Macromol. Rapid Commun., 2019, 40(1), 1800398, DOI:10.1002/marc.201800398.
- E. Rossegger, V. Schenk and F. Wiesbrock, Design Strategies for Functionalized Poly(2-oxazoline)s and Derived Materials, Polymers, 2013, 5(3), 956–1011 CrossRef.
- M. Glassner, M. Vergaelen and R. Hoogenboom, Poly(2-oxazoline)s: A comprehensive overview of polymer structures and their physical properties, Polym. Int., 2018, 67(1), 32–45, DOI:10.1002/pi.5457.
- I. Muljajew, S. Huschke, A. Ramoji, Z. Cseresnyés, S. Hoeppener, I. Nischang, W. Foo, J. Popp, M. T. Figge, C. Weber, M. Bauer, U. S. Schubert and A. T. Press, Stealth Effect of Short Polyoxazolines in Graft Copolymers: Minor Changes of Backbone End Group Determine Liver Cell-Type Specificity, ACS Nano, 2021, 15(7), 12298–12313, DOI:10.1021/acsnano.1c04213.
- M. N. Leiske, M. Lai, T. Amarasena, T. P. Davis, K. J. Thurecht, S. J. Kent and K. Kempe, Interactions of core cross-linked poly(2-oxazoline) and poly(2-oxazine) micelles with immune cells in human blood, Biomaterials, 2021, 274, 120843, DOI:10.1016/j.biomaterials.2021.120843.
- J. R. Finnegan, E. H. Pilkington, K. Alt, M. A. Rahim, S. J. Kent, T. P. Davis and K. Kempe, Stealth nanorods via aqueous living crystallization-driven self-assembly of poly(2-oxazoline)s, Chem. Sci., 2021, 12, 7350–7360, 10.1039/D1SC00938A.
- Q. Yang, T. M. Jacobs, J. D. McCallen, D. T. Moore, J. T. Huckaby, J. N. Edelstein and S. K. Lai, Analysis of Pre-existing IgG and IgM Antibodies against Polyethylene Glycol (PEG) in the General Population, Anal. Chem., 2016, 88(23), 11804–11812, DOI:10.1021/acs.analchem.6b03437.
- R. Luxenhofer, A. Schulz, C. Roques, S. Li, T. K. Bronich, E. V. Batrakova, R. Jordan and A. V. Kabanov, Doubly amphiphilic poly(2-oxazoline)s as high-capacity delivery systems for hydrophobic drugs, Biomaterials, 2010, 31(18), 4972–4979, DOI:10.1016/j.biomaterials.2010.02.057.
- M. M. Lübtow, L. Hahn, M. S. Haider, R. Luxenhofer and D. Specificity, Synergy and Antagonism in Ultrahigh Capacity Poly(2-oxazoline)/Poly(2-oxazine) based Formulations, J. Am. Chem. Soc., 2017, 139(32), 10980–10983, DOI:10.1021/jacs.7b05376.
- S. Datta, A. Jutková, P. Šrámková, L. Lenkavská, V. Huntošová, D. Chorvát, P. Miškovský, D. Jancura and J. Kronek, Unravelling the Excellent Chemical Stability and Bioavailability of Solvent Responsive Curcumin-Loaded 2-Ethyl-2-oxazoline-grad-2-(4-dodecyloxyphenyl)-2-oxazoline Copolymer Nanoparticles for Drug Delivery, Biomacromolecules, 2018, 19(7), 2459–2471, DOI:10.1021/acs.biomac.8b00057.
- M. M. Lübtow, M. S. Haider, M. Kirsch, S. Klisch and R. Luxenhofer, Like Dissolves Like? A Comprehensive Evaluation of Partial Solubility Parameters to Predict Polymer–Drug Compatibility in Ultrahigh Drug-Loaded Polymer Micelles, Biomacromolecules, 2019, 20(8), 3041–3056, DOI:10.1021/acs.biomac.9b00618.
- L. Loukotová, P. Švec, O. Groborz, T. Heizer, H. Beneš, H. Raabová, T. Bělinová, V. Herynek and M. Hrubý, Direct Comparison of Analogous Amphiphilic Gradient and Block Polyoxazolines, Macromolecules, 2021, 54(17), 8182–8194, DOI:10.1021/acs.macromol.0c02674.
- D. Babuka, K. Kolouchova, L. Loukotova, O. Sedlacek, O. Groborz, A. Skarkova, A. Zhigunov, E. Pavlova, R. Hoogenboom, M. Hruby and P. Stepanek, Self-Assembly, Drug Encapsulation, and Cellular Uptake of Block and Gradient Copolymers of 2-Methyl-2-oxazine and 2-n-Propyl/butyl-2-oxazoline, Macromolecules, 2021, 54(23), 10667–10681, DOI:10.1021/acs.macromol.1c01794.
- S. Datta, V. Huntošová, A. Jutková, R. Seliga, J. Kronek, A. Tomkova, L. Lenkavská, M. Máčajová, B. Bilčík, B. Kundeková, I. Čavarga, E. Pavlova, M. Šlouf, P. Miškovský and D. Jancura, Influence of Hydrophobic Side-Chain Length in Amphiphilic Gradient Copoly(2-oxazoline)s on the Therapeutics Loading, Stability, Cellular Uptake and Pharmacokinetics of Nano-Formulation with Curcumin, Pharmaceutics, 2022, 14(12), 2576 CrossRef CAS PubMed.
- O. Sedlacek, V. Bardoula, E. Vuorimaa-Laukkanen, L. Gedda, K. Edwards, A. Radulescu, G. A. Mun, Y. Guo, J. Zhou, H. Zhang, V. Nardello-Rataj, S. Filippov and R. Hoogenboom, Influence of Chain Length of Gradient and Block Copoly(2-oxazoline)s on Self-Assembly and Drug Encapsulation, Small, 2022, 18(17), 2106251, DOI:10.1002/smll.202106251.
- M. M. Lübtow, L. C. Nelke, J. Seifert, J. Kühnemundt, G. Sahay, G. Dandekar, S. L. Nietzer and R. Luxenhofer, Drug induced micellization into ultra-high capacity and stable curcumin nanoformulations: Physico-chemical characterization and evaluation in 2D and 3D in vitro models, J. Controlled Release, 2019, 303, 162–180, DOI:10.1016/j.jconrel.2019.04.014.
- Z. Varanaraja, J. Kim and C. R. Becer, Poly(2-oxazine)s: A comprehensive overview of the polymer structures, physical properties and applications, Eur. Polym. J., 2021, 147, 110299, DOI:10.1016/j.eurpolymj.2021.110299.
- G. Volet, V. Chanthavong, V. Wintgens and C. Amiel, Synthesis of Monoalkyl End-Capped Poly(2-methyl-2-oxazoline) and Its Micelle Formation in Aqueous Solution, Macromolecules, 2005, 38(12), 5190–5197, DOI:10.1021/ma050407u.
- G. Volet, L. Auvray and C. Amiel, Monoalkyl Poly(2-methyl-2-oxazoline) Micelles. A Small-Angle Neutron Scattering Study, J. Phys. Chem. B, 2009, 113(41), 13536–13544, DOI:10.1021/jp9029634.
- G. Volet, A.-C. L. Deschamps and C. Amiel, Association of hydrophobically α,ω-end-capped poly(2-methyl-2-oxazoline) in water, J. Polym. Sci., Part A: Polym. Chem., 2010, 48(11), 2477–2485, DOI:10.1002/pola.24019.
- R. Hoogenboom, M. W. M. Fijten and U. S. Schubert, Parallel kinetic investigation of 2-oxazoline polymerizations with different initiators as basis for designed copolymer synthesis, J. Polym. Sci., Part A: Polym. Chem., 2004, 42(8), 1830–1840, DOI:10.1002/pola.20024.
- T. Zhao, B. Drain, G. Yilmaz and C. R. Becer, One-pot synthesis of amphiphilic multiblock poly(2-oxazoline)s via para-fluoro-thiol click reactions, Polym. Chem., 2021, 12(44), 6392–6403, 10.1039/D1PY00944C.
- F. Wiesbrock, R. Hoogenboom, M. A. M. Leenen, M. A. R. Meier and U. S. Schubert, Investigation of the Living Cationic Ring-Opening Polymerization of 2-Methyl-, 2-Ethyl-, 2-Nonyl-, and 2-Phenyl-2-oxazoline in a Single-Mode Microwave Reactor, Macromolecules, 2005, 38(12), 5025–5034, DOI:10.1021/ma0474170.
- Z. Varanaraja, N. Hollingsworth, R. Green and C. R. Becer, Poly(2-alkyl-2-oxazoline)-Based Copolymer Library with a Thermoresponsive Behaviour in Dodecane, ACS Appl. Polym. Mater., 2023, 5(7), 5158–5168, DOI:10.1021/acsapm.3c00625.
- E. F.-J. Rettler, J. M. Kranenburg, H. M. L. Lambermont-Thijs, R. Hoogenboom and U. S. Schubert, Thermal, Mechanical, and Surface Properties of Poly(2-N-alkyl-2-oxazoline)s, Macromol. Chem. Phys., 2010, 211, 2443–2448, DOI:10.1002/macp.201000338.
- M. Everaerts, A. Tigrine, V. R. de la Rosa, R. Hoogenboom, P. Adriaensens, C. Clasen and G. V. den Mooter, Unravelling the Miscibility of Poly(2-oxazoline)s: A Novel Polymer Class for the Formulation of Amorphous Solid Dispersions, Molecules, 2020, 25(16), 3587, DOI:10.3390/molecules25163587.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Block Copolymer Micelles in Nanomedicine Applications, Chem. Rev., 2018, 118(14), 6844–6892, DOI:10.1021/acs.chemrev.8b00199.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Tuning the LCST of poly(2-oxazoline)s by varying composition and molecular weight: alternatives to poly(N-isopropylacrylamide)?, Chem. Commun., 2008, 5758–5760, 10.1039/B813140F.
- K. Suresh and A. Nangia, Curcumin: pharmaceutical solids as a platform to improve solubility and bioavailability, CrystEngComm, 2018, 20(24), 3277–3296, 10.1039/C8CE00469B.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR, GPC, MALDI ToF, DSC. See DOI: https://doi.org/10.1039/d3py00809f |
| This journal is © The Royal Society of Chemistry 2023 |