
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inverse vulcanisation of self-activating amine and alkyne crosslinkers†
Liam James
Dodd
 *a,
William
Sandy
a,
Romy A.
Dop
a,
Bowen
Zhang
a,
Amy
Lunt
a,
Daniel R.
Neill
bc and
Tom
Hasell
*a
*a,
William
Sandy
a,
Romy A.
Dop
a,
Bowen
Zhang
a,
Amy
Lunt
a,
Daniel R.
Neill
bc and
Tom
Hasell
*a
aUniversity of Liverpool, School of Physical Sciences, Department of Chemistry, Crown Street, Liverpool, L697ZD, Merseyside, UK. E-mail: sgldodd@liverpool.ac.uk; t0m@liverpool.ac.uk
bDepartment of Clinical Infection, Microbiology and Immunology, University of Liverpool, Liverpool L69 7BE, UK
cDivision of Molecular Microbiology, School of Life Sciences, University of Dundee, Dundee, DD1 5EH, UK
First published on 8th August 2023
Abstract
Inverse Vulcanisation is a versatile route to the synthesis of high sulfur content polymers. Developments to the field include expanding the variety of organic crosslinker molecules that can be used in the reaction, and the application of catalysis, which lowers the reaction time and the required temperature, as well as improving the yield and properties of the resultant polymers. However, concerns remain that the polymers may have a residual metal content from the catalysts, which is undesirable when considering environmental ramifications. There is also the question of whether the catalyst should be extracted from the polymer, which adds another processing step. Presented here is a study on crosslinkers that contain a non-metallic activating moiety built into their structure, thereby eliminating the aforementioned concerns whilst still providing several benefits. Also explored is the relatively untouched field of using alkynes, rather than alkenes, as crosslinkers, which have the potential to provide much higher crosslink densities in the resultant polymers, which may give favourable properties. The work presented here demonstrates the capability of the self-activating crosslinkers to be used as the sole crosslinker, where they can polymerise below the melting point of sulfur, or as a secondary crosslinker in another reaction, bringing the aforementioned benefits of catalysis.
Introduction
The field of inverse vulcanisation has expanded and diversified rapidly since its conception in 2013, when Pyun et al. discovered that small organic molecules containing more than one alkene bond can be co-polymerised with radical-capped chains of sulfur atoms, which are produced by heating molten sulfur.1 The result of this reaction is a polymeric material of organic units connected by chains of sulfur atoms, which can be stable to depolymerisation, unlike sulfur homopolymers.1 This class of intriguing polymers has uniquely high sulfur contents, ranging up to ninety percent by mass in some cases, which gives them a complement of attractive properties that makes them amenable to a wide variety of applications.2 Inverse vulcanisation has also been found to accept a wide variety of different co-monomers and crosslinkers, some of which can be sourced renewably in line with the principles of green chemistry, and since sulfur is produced in excess of sixty million tons a year as an industrial by-product, the polymers can be remarkably low cost and sourced sustainably.1,3–6Expanding the library of known crosslinkers and observing the effects of crosslinker structure upon the polymers has highlighted the versatility of inverse vulcanisation with regard to what crosslinkers the reaction can accept: vegetable oils like canola oil, rigid polycyclics like dicyclopentadiene, flexible aliphatics like squalene (which contains more than two alkene bonds), alkenes conjugated to aromatics such as divinylbenzene, and many other types of crosslinker are amenable to inverse vulcanisation.8–10 Different crosslinkers provide inverse vulcanised polymers with different properties, which in turn steers those polymers to a wide array of different applications. Example applications include cheap but effective cathode materials in lithium–sulfur batteries, antimicrobial coatings, and mercury sorbents for water remediation, amongst many others that currently exist, as well as those yet to be discovered.3,8,11–13
Another way inverse vulcanisation is adaptable, is by modification of the synthetic process to achieve different outcomes with the same inverse vulcanised polymer. Some examples of this are salt templating the polymers, or foaming the polymers with supercritical carbon dioxide to achieve porosity.8,11 Fillers can also be added to the polymers to modify their mechanical properties, or alternatively, blends of two different crosslinkers in different proportions can be used to achieve different outcomes.7,14,15 Another important example of modifying the synthetic route is the inclusion of a catalyst in the reaction, though this term is used tentatively since regeneration of the catalytic molecule has yet to be proven.16–18 Catalysis has been shown to give numerous benefits to inverse vulcanisation, including lowering the minimum reaction temperature, decreasing the reaction time, permitting the use of otherwise unreactive crosslinkers, decreasing the production of the toxic hydrogen sulfide by-product, improving the yield of the reaction, and increasing the glass transition temperatures (Tg's) of the resultant polymers.17
Whilst catalysis is versatile in the crosslinkers it positively affects, there are some remaining concerns regarding its application.17 First is whether the catalyst can or should be extracted from the polymer, and what consequences such a post synthetic step might have on the polymers and their production cost. This concern could be eliminated if the catalytic molecule was intentionally incorporated into the polymer structure. Though it would no longer be a true catalyst due to its consumption in the reaction, the ‘catalyst’ molecule would then contribute to the polymer's properties as a crosslinker, rather than an impurity. Thus the ‘catalyst’ would be considered an activator, and this term will be used henceforth when referring to this work. The second concern is that many established catalysts are derived from metal dialkyldithiocarbamates, leaving a trace metal residue in the polymers, raising environmental concerns.17 If the metallic catalyst could be replaced by a fully organic molecule, which both Hasell et al. and Pyun et al. have demonstrated with amines, then this issue would also be circumvented.17,18 It has previously been reported that DCPD can be used as a second organic comonomer in an inverse vulcanisation reaction that mainly consists of some other organic comonomer, and it was found that this was beneficial to the properties and allowed tuning of the Tg. Thus, it is not difficult to imagine an amine containing crosslinker taking up a similar role as DCPD in this regard.14
In line with these ideas, this work focused upon the inverse vulcanisation of self-activating amine crosslinkers: molecules that contain both an activating amine moiety and crosslinker moieties. In doing so, alkyne crosslinkers, which have thus far seen limited research attention in inverse vulcanisation, were also explored as a natural branch of this research avenue.19 Alkynes have the potential to react with sulfur twice, forming twice as many crosslinks as an alkene would, and this could lead to a higher crosslink density (Fig. 1). As such, it was thought that alkynes may permit a smaller load of crosslinker to be capable of stabilising more sulfur, leading to even higher sulfur contents in the resultant polymers, which could be valuable to the electrochemical applications, and the antimicrobial applications of inverse vulcanised polymers. That being said, recent developments in the field of inverse vulcanisation show that there could be complications to this statement. Pyun et al. recently found evidence that polymers of 1,3-diisopropenylbenzene do not react as was initially thought; rather than radical addition across the double bond to create two new C–S bonds, a more complex mechanism dominates, resulting primarily in a linear polymer unit where each double bond produces one new C–S bond.20 Whether reactions with alkynes could be subject to the same sort of mechanism is unknown, but because Pyun's new findings centre around the formation of thiocumyl moieties and there seems there is no intuitive way such a moiety could form from an inverse vulcanised polymer of an alkyne, it will be initially assumed that radical addition as traditionally expected of inverse vulcanisation occurs here.20 Furthermore, it cannot be that all co-monomers in inverse vulcanisation are subject to this new mechanism proposed by Pyun, because if that were the case, styrene would not be able to form an inverse vulcanised polymer, but it is documented that it can.21
 | ||
| Fig. 1 Names, abbreviations, molecular structures, and potential for crosslinking (κ) values, for the crosslinkers used in this study, as well as schematics illustrating their potential to crosslink chains of sulfur. For comparison, divinylbenzene, a common crosslinker, has κ = 15.4. | ||
Several self-activating crosslinkers to be used in inverse vulcanisation reactions were identified (Fig. 1). Tripropargylamine (TPA) is an example of an alkyne and an amine containing crosslinker, which is comparable to the already published alkene analogue triallylamine (TAA) which was used here as a direct comparison to TPA to examine the differences between alkyne and alkene co-monomers.22 1,8-Nonadiyne (NON), although not directly comparable to TPA in terms of its structure, was used as the best available comparison to TPA in terms of an alkyne crosslinker without a self-activating amine moiety. Additionally, diallylamine (DAA) and monopropargylamine (MPA) were also successfully polymerised, which is particularly exciting since they can be polymerised at below the melting temperature of sulfur, and MPA has only a single alkyne moiety. Finally, TPA and TAA were shown to successfully blend with DCPD and linseed oil in inverse vulcanisations, showing their value as trace presence activators.
Results and discussion
Method
To begin this study, the aforementioned crosslinkers were used as the sole co-monomer in inverse vulcanisation reactions, as this provided a simpler starting point compared to reactions where they would be used in conjunction with another crosslinker. Initial attempts to synthesize inverse vulcanised polymers of TPA, TAA, and NON through bulk polymerisation were unsuccessful. NON reacted to form an unacceptably inhomogeneous polymer, whereas TPA and TAA violently underwent an exothermic auto-acceleration reaction, the Trommsdorff–Norrish effect, perhaps not surprising given that in a 50% by mass sulfur reaction, the remaining 50% of the mass is all activator.Bulk polymerisations are prone to the Trommsdorff–Norrish effect due to their poor heat dissipation, however the addition of a solvent to the polymerisation can significantly decrease the risk by diluting the heat production over a larger volume. As such a dispersion polymerisation method was employed, which after optimisation, came to the following general method (full details can be found in the ESI, section II.A.†). In brief, mass X of sulfur and mass Y of a chosen crosslinker (X plus Y always equalled 10 g in every reaction) were reacted overnight in refluxing xylene with stirring. This afforded two products: one that was insoluble in the xylene, and one that remained soluble in the xylene. The two were separated by filtration and the xylene-soluble product was evaporated to dryness, and then cured overnight in an oven at 140 °C. The resultant xylene-soluble product after curing was termed, the Sol product, of which it is important to note, may no longer be soluble in xylene and other solvents due to reacting and crosslinking further upon curing. That is, the Sol product is the material given from reaction that was soluble in xylene immediately after the reaction, but may no longer be soluble due to being cured overnight to form a more crosslinked structure. On the other hand, the xylene-insoluble material obtained from the reaction was purified by Soxhlet extraction on toluene overnight, after which it was cured in an oven at 140 °C to give what was termed, the Insol product. In summary, the Sol product is the material that, after purification and curing, was obtained from the xylene solution of the reaction. It is important to remember that although the Sol product originated from a soluble species, it may no longer be soluble after the processing. Meanwhile, the Insol product was obtained by purifying and curing the material that naturally precipitated out of the xylene reaction solution. The products of these reactions will be referred to by the following naming convention: NAMEα-Sβ-X, where NAME is the abbreviation of the crosslinker in use, α is the feed ratio of that crosslinker in the reaction, β is the feed ratio of sulfur in that reaction, and X is replaced with either Sol, to refer to the Sol product, or Insol, to refer to the Insol product.
The method for polymerising MPA and DAA had to be modified, as detailed in the ESI, section II.B.† because these crosslinkers boil below the melting temperature of sulfur (83 °C for MPA and 111 °C for DAA). To account for this, MPA and DAA were reacted as above, but at the initial temperatures of 70 °C and 100 °C, respectively. After 24 hours, a pre-polymer had formed, and so the temperature was then increased to reflux, and left to react overnight. Other than this modification, the method remained the same as the general method. What is interesting is that MPA and DAA were able to react with sulfur despite the sulfur not having sufficient thermal energy to melt or undergo ring-opening, which is the generally accepted route for initiation of the polymerisation. This suggests that amine containing crosslinkers may offer an alternative route for initiation, perhaps by direct nucleophilic attack of the nitrogen upon the sulfur rings. This would cleave open the sulfur rings to yield reactive sulfide anions, which would open up an anionic polymerisation pathway. Such anionic polymerisations with an electron rich alkene might be unexpected, especially because it yields a carbanion, which is generally accepted to be unreactive, however, the mechanism proposed in the ESI, section III,† avoids this intermediate through a concerted transition state. This conclusion of an amine initiated anionic pathway is supported by the fact that when 1,7-octadiene (boiling point 114 to 121 °C, initial reaction temperature 100 °C), which was thought to be a good comparison to NON, was used in such a reaction, no changes were observed. 1,7-Octadiene has no amine moiety, and since it was exposed to temperatures insufficient to ring open sulfur, there were no sulfur radicals to initiate the polymerisation, so without an alternate source of initiation, no reaction occurred. The lack of reaction was confirmed by the fact that after the reaction period, yellow sulfur powder in a clear and colourless solvent was observed, exactly the appearance of the reagents. 1H NMR of the xylene solvent revealed unreacted 1,7-octadiene was the only impurity.
Yields
Fig. 2 shows the yields of the Sol and Insol products of dispersion polymerisation inverse vulcanisations of TPA, TAA, NON, DAA, and MPA. As the mass loading of TPA was increased, the yield of the TPA Insol product also increased, up to a maximum at 50% mass loading, after which it began to decrease. An explanation for this is that as more crosslinker is added, more sulfur is able to be incorporated into the polymer, giving a greater yield, until it reaches the point where there is so much crosslinker that there is then not enough sulfur to react with all the crosslinker, resulting in wastage of crosslinker and thus a lower yield. | ||
| Fig. 2 The yields of the Sol and Insol products at different weight percentages of crosslinker in the reaction feed for (a) TPA and MPA, (b) TAA and DAA, (c) NON. | ||
The yield of the Sol product remained relatively constant for different mass loadings of TPA, with a significant dip at 50% mass loading. An explanation for this is that as more crosslinker is added, there is the potential to form more products overall, both Sol and Insol. However, because there is a limited supply of reagents, the Sol and Insol products are in competition with one another. Higher crosslinker loads would favour the more highly crosslinked Insol product. This would explain why the yield of the Sol remains relatively constant, even though there is more crosslinker available to form products, and it would also explain the dip in yield for the sol product when the Insol product was at its highest yield; where the Insol product was taking up the majority of the reagents, leaving few to form the Sol product. At high crosslinker loadings of TPA (70%) the yield of the Sol product went up whilst the yield of the Insol went down. This could be due to an insufficient supply of sulfur to fully connect together all of the crosslinker molecules into a network, instead yielding more of the less crosslinked Sol product.
In comparison to TPA, MPA shows a relatively similar behaviour, but with some minor differences that likely arise due to the difference in potential for crosslinking between them. Overall, the Insol yields for MPA were lower than those for TPA, which is reasonable because MPA contains fewer double bonds and so has less capacity to crosslink chains of sulfur. Thus, it would be expected that MPA would not be able to stabilise as much sulfur as TPA. It is an interesting result that MPA, which contains only a single alkyne unit, was able to produce polymers sufficiently crosslinked that they became insoluble in the reaction solution, which provides evidence that both double bonds of the alkyne react. Conversely, at high loadings of sulfur, MPA gave greater yields of the Sol product than TPA, again explained as MPA having less crosslinking capacity, and so being more prone to forming lower molecular weight, less crosslinked products. Just as for TPA, at high loadings of MPA, the yield of the Insol product increased at the expense of the Sol product, explained with the same reasoning as for TPA.
The trends observed for TPA are also observed in the yield trends of NON, suggesting that these two alkyne crosslinkers behave relatively similarly, despite the presence or absence of an activating amine moiety. That being said, the absolute values for the yields of NON are higher for its Sol product and lower for its Insol product. This could be due to the fact that NON has one fewer alkyne groups to react and form a polymer, and thus NON has less crosslinking capacity per molecular weight unit, which would make it favour the less crosslinked Sol product over the more crosslinked Insol product.
TAA shows different trends in its yield data than TPA or NON, however TAA's yield trends could still be explained by the same principles. In this case the Sol products’ yields were higher than that of the Insol products. This could be because since TAA is an alkene, it can only react once per alkene moiety, whereas its analogue, TPA which is an alkyne could potentially react twice. As a result, TAA would only be able to react half as many times, and would therefore favour the less crosslinked Sol product over the more crosslinked Insol product. In this case TAA favours the Sol product sufficiently that the Sol product has a higher yield than the Insol product. This yield of Sol product rises as the loading of TAA in the reaction increases. This could be explained by a greater incorporation of sulfur into the polymer with more crosslinker being available to react with it. Otherwise the TAA Insol products follow the same rise and fall in yield as the TAA loading is increased, likely for the same reasoning as TPA and NON's Insol products.
Surprisingly, DAA does not behave similarly to TAA, and instead DAA's yields much more closely match those trends seen in TPA and NON. This difference in behaviour between TAA and DAA is ascribed to the fact that TAA is a tertiary amine, whereas DAA is a secondary amine, and so has an N–H bond, which might show unexpected chemistry. Another surprising difference is that DAA overall shows higher absolute yields than TAA, despite having one fewer double bond to react with sulfur, which is suggested to be the effect of unexpected chemistry involving the N–H bond.
It can be seen in several cases in the yield data, that the Sol and Insol yields for a particular polymer at a particular feed percentage of crosslinker, sums to less than 100%. This could be due to the loss of hydrogen sulfide gas. Another likely loss to the yields was the formation of a volatile fraction, as it was noted that solvent condensed during the rotary evaporation stage of the reaction processing, was frequently coloured yellow. Since this solvent was discarded, any volatised oligomers of polymer contained therein would not contribute to the yield.
Solubility studies
Fig. 3 shows the soluble fractions of the Sol and Insol products of dispersion polymerisation inverse vulcanisations of TPA, TAA, NON, MPA, and DAA. It is important to remember that during the post-reaction processing, the Sol and Insol products are made by curing their precursors after extracting them from the reaction. This curing leads to increased crosslinking, and so the Sol and Insol products will have decreased in solubility during this curing. As such, just because the Sol product originated from the xylene soluble product of the reaction, it may not be soluble after this processing. For each solubility study, approximately 50 mg (within 2 mg) of polymer was left in 2 cm3 of chloroform for 24 hours. After this time the chloroform was filtered off into a pre-weighed vial. The chloroform was left for 3 days to fully evaporate, after which time the vial was reweighed to determine the mass of the soluble fraction, which was then taken as a percentage of the initial 50 mg of polymer. | ||
| Fig. 3 The soluble fractions of the Sol and Insol products at different weight percentages of crosslinker in the reaction feed for (a) TPA and MPA, (b) TAA and DAA, (c) NON. | ||
The Insol products of all the different crosslinkers in all cases showed very low to no soluble fraction, indicating a highly crosslinked and therefore insoluble structure for the Insol products. In line with the conclusions from the yield data, the TAA Sol products all showed very high soluble fractions, indicating low crosslink density and low molecular weight in the TAA Sol products, similarly observed for DAA. The Sol products of NON and TPA show a similar trend in that they show a high soluble fraction for crosslinker loadings of 30% or lower, and a lower soluble fraction for crosslinker loadings of 50% and higher. This could be because at such high crosslinker loadings for the alkynes, there is sufficient crosslinking upon curing, that even the Sol product is quite crosslinked and of high molecular weight.
It is worth noting that polymers of NON do not contain a nitrogen atom, whereas polymers of TPA, MPA, TAA, and DAA do, and it cannot be known from this study, whether the nitrogen atom has an effect on the solubility of the polymers in chloroform, or whether the soluble fraction is solely influenced by the crosslink density and molecular weight. Both the solubility study and the yield data suggest that polymers of NON and TPA, which are both alkyne crosslinkers, behave similarly to each other and differently to polymers of TAA, which is an alkyne crosslinker. This is observed in spite of the fact that NON does not contain a nitrogen atom, and so is not self-activating, but TPA does contain a nitrogen atom and is self-activating. This does not mean that TPA's self-activation is not beneficial, as polymers of TPA typically showed yields higher than those of NON, but instead points to the conclusion that the alkyne moiety's capacity to react multiple times is the dominant factor in terms of the crosslink density. Why the Sol products of MPA become increasingly soluble with rising crosslinker loading cannot be explained at this time.
Differential scanning calorimetry
Note that for Fig. 4, the I bars do not show the error in the measurement but instead show the onset and end temperatures of the Tg. Observing Fig. 4, it can be seen that the Tg's obtained from the second heating cycle of DSC for TAA follow an expected trend. For both the Sol and Insol products, as the mass loading of TAA was increased, the Tg increased too, explained by the increasing amount of TAA giving more crosslinking in the product polymers. The Tg's of the Sol products of TAA inverse vulcanisations were always lower that the corresponding Insol products, which provides evidence to the conclusion that the Insol products are more crosslinked than the Sol products. The Sol products for DAA show a similar trend to those of TAA, however for the DAA Insol products, unexpectedly the Tg decreases as the loading of crosslinker increases (accepting that the Tg for DAA50-S50-Insol could not be identified). A possible explanation is that as the quantity of crosslinker is increased, there becomes insufficient sulfur to form a well crosslinked network, leaving more and more double bonds unreacted at higher crosslinker loadings. This would give the polymer more linear character, resulting in a lower Tg. However, if this was the case then a raise in the solubility of DAA Insol polymers with increasing crosslinker loading would be expected, which is not observed, and furthermore, polymers of TPA and TAA would be expected to show the same trend, as they have even more double bonds to react, and so should suffer from this effect more so than DAA, which is not the case. As such, the best explanation that can be postulated here is that the unexpected behaviour of DAA is due to its N–H bond which could introduce unexpected chemistry, whereas TAA and TPA are both tertiary amines and contain no N–H bonds. | ||
| Fig. 4 The Tg's, obtained from the second heating cycle of DSC, of the Sol and Insol products at different weight percentages of crosslinker in the reaction feed for (a) TPA and MPA, (b) TAA and DAA, (c) NON. Note that the error bars indicate the onset and end temperatures of the Tg. | ||
Sol polymers of NON show a similar trend to that of TAA, however the Insol products of NON, MPA, and TPA showed an interesting result in that the Insol polymers often did not show a Tg at all, except at very low crosslinker loadings. TGA analysis gave no meaningful trends between the polymers’ decomposition temperatures, and it was not accurate to determine the mass losses as the steps were drawn out. Additionally, TGA experiments were particularly detrimental to the instrument, and so only a few selected samples were run, with the protocol chosen to minimise the maintenance load on the TGA. The TGA analysis did show that the polymers’ decomposition temperatures are all above 190 °C (see the ESI, section IV†), yet they show no Tg below this temperature. This suggests that the alkyne crosslinkers have such a high crosslink density, that their Tg is actually higher than their decomposition temperature, such that the polymer will decompose before becoming a melt or flexible material, which cannot be said of any other inverse vulcanised polymer at this time. To confirm that this was indeed the case, the Insol polymers of TPA and NON were heated upon a hotplate to 170 °C and then agitated with spatula, revealing that they were still glass-like and brittle. Note that an explanation for the change in Tg of TPA and MPA's Sol products with increasing mass loading of TPA cannot be given at this time, though they appear to have a similar trend, with the exception that MPA is staggered to the left. Additionally, MPA Insol products showed an endothermic peak in their heating cycles, at −48 °C, and a corresponding exothermic peak in their cooling cycles at −52 °C. These peaks were weaker in samples with less sulfur, implying that they are a melt transition linked to either free sulfur species, or the sulfurous component of the polymer. In some cases, particularly for the Insol products, the onset to end range of the polymers’ Tg's are quite broad, which may indicate a microscopically inhomogeneous polymer with many different environments for the polymer chains, each with different degrees of immobilisation, and this could result in a more drawn out Tg. see the ESI, section V† for representative DSC thermograms.
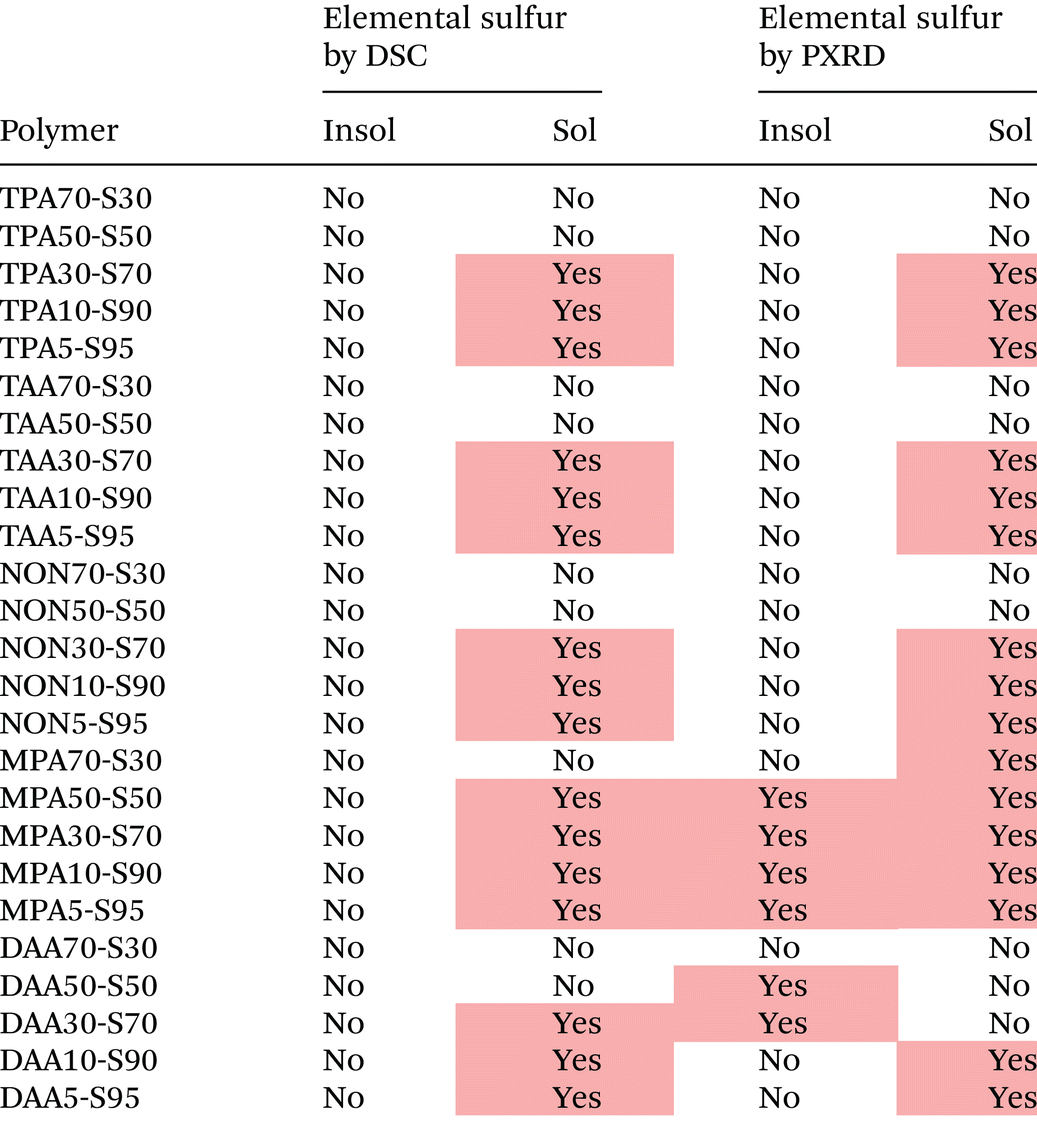
DSC was able to detect the presence of crystalline elemental sulfur in the polymers (endothermic peaks in the heating cycles, corresponding to the melt transitions of elemental sulfur at 110 to 120 °C), with a corroborating technique being PXRD, which can detect the crystalline scattering peaks amongst the polymers’ amorphous scattering signal. In general, as seen in Table 1, there was good agreement between DSC and PXRD in terms of detecting elemental sulfur in the polymers. DSC and PXRD found that Sol products of 30% or less crosslinker feed ratio, regardless of crosslinker identity, contained crystalline elemental sulfur. This suggests that these polymers do not have sufficient crosslinking to stabilise all of their sulfur, and either some left over sulfur reactant remains as an impurity, or some polymerised sulfur is depolymerising to give the crystalline elemental sulfur contaminant. DSC suggested that none of the Insol products contained elemental sulfur, which for TPA, TAA, and NON, PXRD agreed with. This suggests that these products are sufficiently crosslinked to stabilise all their sulfur, at the various sulfur loadings used here, though it is worth remembering that the Soxhlet extraction used to purify the Insol products, may also have purified the Insol products of elemental sulfur too. In the cases of MPA50-S50-insol, MPA30-S70-insol, MPA10-S90-insol, MPA5-S95-insol, DAA50-S50-insol, and DAA30-S70-insol, PXRD detected crystalline elemental sulfur where DSC did not. For MPA insol products, it is perhaps not surprising that they contain some elemental sulfur, as MPA contains only two double bonds by which it can crosslink, in the form of an alkyne, and so it lacks the crosslinking potential of the other crosslinkers. As such it seems that the PXRD results are trustworthy, even though they are in contradiction with the DSC; it may be that DSC was not sufficiently sensitive to detect the crystalline elemental sulfur. For the DAA Insols that contain elemental sulfur by PXRD, it is hard to explain the results, given that polymers with a greater loading of sulfur reactant, did not display elemental sulfur presence by PXRD or DSC. One potential explanation for the unexpected results could be the time difference between taking the DSC and PXRD measurements. It has been shown that over time, elemental sulfur can crystallise from inverse vulcanised polymers. As such, the DSC measurements, taken shortly after the polymers were synthesized may not show elemental sulfur signals, whereas the PXRD measurements, which were taken as soon as the instrument was available sometime after the polymer synthesis, do show elemental sulfur signals. To help corroborate this, the DSC experiments were repeated after the PXRD experiments, and in this case, DSC was able to detect some elemental sulfur in agreement with the PXRD results. It is hard to know exactly when a polymer will begin to display elemental sulfur signals due to ageing, and it may be that given long enough, all polymers synthesized here will begin to develop sulfur crystals that can be detected by DSC and PXRD. Raman spectroscopy was also attempted for the detection of elemental sulfur, but was unsuccessful due to the intense fluorescence of the dark coloured samples.23 TLC was also unsuccessful at detecting elemental sulfur where PXRD and DSC could not, perhaps because there was no elemental sulfur, or perhaps because the samples were sufficiently crosslinked that elemental sulfur remained entrapped and immobilised in the polymer structure, preventing its extraction into the TLC eluent.
|
Fourier transform infrared spectroscopy
Because of the high IR transparency of the polymers, FT-IR was of limited use in characterisation. There was little difference in the spectra of the same polymer with different sulfur loadings. Because of this, even though IR spectra were obtained for all products, only the spectra of the neat crosslinkers, and the Sol and Insol products at 50% crosslinker loading are shown in Fig. 5, though all spectra are available in the ESI, section VI.† To assist with the assignment of the spectra, density functional theory was used to predict the IR spectra of abbreviated models of the polymers. The spectral data for these polymer models as well as the computational method can be found in the ESI, sections VII and I† respectively. Calculations were performed according to pre-established literature methods found to be successful for inverse vulcanised polymers.23 | ||
| Fig. 5 FTIR spectra of (a) Neat TPA, TPA50-S50-Insol, and TPA50-S50-Sol; (b) Neat MPA, MPA50-S50-Insol, and MPA50-S50-Sol; (c) Neat TAA, TAA50-S50-Insol, and TAA50-S50-Sol; (d) Neat DAA, DAA50-S50-Insol, and DAA50-S50-Sol; (e) Neat NON, NON50-S50-Insol, and NON50-S50-Sol. | ||
Regarding TPA, MPA, and NON, the alkyne C–H vibration at approximately 3300 cm−1 in the neat crosslinker spectra, does not appear in the spectra of the Sol or Insol products, indicating that the alkyne bond has been consumed in the inverse vulcanisation reaction as expected. Unfortunately, the carbon–carbon triple bond vibration was too weak in the neat spectra to be diagnostic in the polymer spectra, which were always of high raw transmittance. Regarding TAA and DAA, the IR spectra suggest a nearly complete reaction. The alkene C–H mode at about 3100 cm−1 in the neat crosslinker spectra is absent in the spectra of the Sol and Insol products. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C mode at about 1650 cm−1 in the neat crosslinker spectra is plausibly present in the spectra of the sol and insol products of TAA and DAA, as there is a peak in the polymer spectra that could be assigned to the CC bond, but it seems too broad and too intense to be solely the result of the CC mode. The CC peak at 990 cm−1 is also plausibly absent in the spectra of the Sol and Insol products versus the neat crosslinker. The CC peak at approximately 910 cm−1 is much more diagnostic however as it is significantly more intense in the neat crosslinker spectra. TAA50-S50-sol, TAA50-S50-insol, and DAA50-S50-sol all show a weak peak that can be attributed to the CC mode at 910 cm−1. DAA50-S50-insol does not show any evidence of this peak, nor does it show evidence of the 1650 cm−1 peak, suggesting that of the alkene crosslinkers, DAA sees the greatest consumption of its double bonds, which is reasonable, because DAA starts with the fewest double bonds, and the Insol product is expected to be the most thoroughly reacted. Even so, all the aforementioned conclusions drawn from the FT-IR data are somewhat ambiguous due to the weak signal strength of the polymers which makes it hard to assign peaks with certainty.
C mode at about 1650 cm−1 in the neat crosslinker spectra is plausibly present in the spectra of the sol and insol products of TAA and DAA, as there is a peak in the polymer spectra that could be assigned to the CC bond, but it seems too broad and too intense to be solely the result of the CC mode. The CC peak at 990 cm−1 is also plausibly absent in the spectra of the Sol and Insol products versus the neat crosslinker. The CC peak at approximately 910 cm−1 is much more diagnostic however as it is significantly more intense in the neat crosslinker spectra. TAA50-S50-sol, TAA50-S50-insol, and DAA50-S50-sol all show a weak peak that can be attributed to the CC mode at 910 cm−1. DAA50-S50-insol does not show any evidence of this peak, nor does it show evidence of the 1650 cm−1 peak, suggesting that of the alkene crosslinkers, DAA sees the greatest consumption of its double bonds, which is reasonable, because DAA starts with the fewest double bonds, and the Insol product is expected to be the most thoroughly reacted. Even so, all the aforementioned conclusions drawn from the FT-IR data are somewhat ambiguous due to the weak signal strength of the polymers which makes it hard to assign peaks with certainty.
Where the alkyne crosslinkers are concerned, the FT-IR spectra may be indicative of a facet of the mechanism, as at about 1670 cm−1 there are weak peaks in the spectra of TPA50-S50-sol, TPA50-S50-insol, and NON50-S50-insol, which could be assigned to trisubstituted CC bonds. This would suggest that the inverse vulcanisation of alkynes occurs progressively, from C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C to CC to C–C. There is a peak at 1530 cm−1 in the sol and Insol spectra of TPA, which the computational data suggest could also be assigned to the presence of a CC mode, and similarly the Sol and Insol spectra of NON show a peak at 1030 cm−1 and 1220 cm−1 both of which the computational chemistry suggests could result from a CC bond. The NON polymer spectra also show a weak peak at 815 cm−1, whilst TPA polymers show a peak at 790 cm−1, both of which could be tentatively assigned to trisubstituted CC modes, which provides a little more evidence to the conclusion of a progressive reaction. CC bond modes are not observed in the spectra of MPA polymers, which could be explained by the fact that MPA has fewer double bonds per molecule, and so sees greater consumption of its double bonds in comparison to TPA and NON, which have enough double bonds that some are left over in the product polymers as CC. As a final note, the FT-IR spectra indicate that the organic backbone of the crosslinkers is left intact upon polymerisation, which is supported by the results of the computationally predicted IR data.
C to CC to C–C. There is a peak at 1530 cm−1 in the sol and Insol spectra of TPA, which the computational data suggest could also be assigned to the presence of a CC mode, and similarly the Sol and Insol spectra of NON show a peak at 1030 cm−1 and 1220 cm−1 both of which the computational chemistry suggests could result from a CC bond. The NON polymer spectra also show a weak peak at 815 cm−1, whilst TPA polymers show a peak at 790 cm−1, both of which could be tentatively assigned to trisubstituted CC modes, which provides a little more evidence to the conclusion of a progressive reaction. CC bond modes are not observed in the spectra of MPA polymers, which could be explained by the fact that MPA has fewer double bonds per molecule, and so sees greater consumption of its double bonds in comparison to TPA and NON, which have enough double bonds that some are left over in the product polymers as CC. As a final note, the FT-IR spectra indicate that the organic backbone of the crosslinkers is left intact upon polymerisation, which is supported by the results of the computationally predicted IR data.
Sulfur content
Since each dispersion polymerisation produced two products simultaneously, the Sol and Insol, it was not possible to predict how much sulfur was incorporated into each product. Combustion microanalysis was attempted, but gave unreliable results, especially since the total of all elements present added up to greater than 100% total mass. Additionally, analysis of the polymers in combustion microanalysis was unacceptably detrimental to the instrument. As such, an alternative method to determine the sulfur content was sought out. X-Ray Fluorescence (XRF) was identified as a potential route to the analysis of the elemental sulfur content, which has not been applied in the field of inverse vulcanisation thus far. Unfortunately, although XRF could give estimates of the sulfur content, these estimates were subject to doubt due to the fact that a stable baseline could not be obtained even after method optimisation. One interesting result was that in all cases, repeated measurements upon the same sample showed that the sulfur content increased incrementally with each progressive measurement, which was attributed to destruction of the organic component of the polymer by exposure to the X-rays. This indicates that inverse vulcanised polymers must be analysed as rapidly as possible, with the minimum energy directed on them as possible, in order to minimise sample degradation, and if repeat measurements are desired, one must analyse a different sample of the same polymer rather than analysing the same sample multiple times. This could also have ramifications for other mainstream analysis techniques of inverse vulcanised polymers that use X-rays, for example PXRD. Regardless, even though XRF was unsuccessful in analysing the polymers, further research attention may be warranted as it presents a rapid and convenient technique to analyse the sulfur content of the polymers. Although the data is unreliable, CHNS and XRF data is provided in the ESI, section VIII.†NMR characterisation
In order to better understand the structure of these polymer, an NMR characterisation using the method developed by Pyun et al. was attempted.20 In short, this method involves breaking down the polymer with LiAlH4 and analysing the degradation products, which in Pyun et al.'s case, were monomeric units of the polymer (see the ESI, section IX† for the full method). Here this analysis method seems not to have worked so effectively. After overnight reaction with 1 M LiAlH4 in THF, the polymers had not fully broken down, and there remained insoluble polymeric material (note that this analysis was only carried out upon the Sol and Insol products of TPA50-S50, TAA50-S50, NON50-S50, MPA50-S50, and DAA50-S50). When the reaction mixtures were quenched with water, the reaction mixture fizzed violently, indicating that there was still plentiful LIAlH4 remaining, and this suggests that the polymers were largely chemically impervious to LiAlH4. This may be because these polymers are crosslinked, whereas the polymer analysed by Pyun et al. was largely linear with few branching units. Thus, it may be that highly crosslinked polymers are less amenable to this analysis technique, which is supported by the fact that TPA50-S50-Insol and NON50-S50-Insol yielded no degradation product at all after the reaction with LiAlH4, and it is predicted that these would be the most crosslinked of the polymers. Nevertheless, the other eight polymers yielded degradation products which were analysed by 1H NMR, 13C NMR, 13C DEPT135 NMR, 1H 1H COSY NMR, and 1H 13C HSQC NMR. see the ESI, section IX† for the NMR spectra. In line with the idea that the LiAlH4 degradation was not going to completion, the NMR shown in the ESI, section IX† does not seem to show the spectra of a clean monomeric unit cut from the polymer, and instead seems to show the NMR of an oligomer, with broadened peaks that are characteristic of the many similar environments of an oligomer. Despite the challenge in analysing these spectra, they still provided useful information.The NMR spectra of the polymers indicated that the structure mostly consisted of alkyl groups, consistent with the expected structure, and where the crosslinker structure was more simple, the NMR spectra of the degradation products was more simple as well. Alkene region resonances were observed, concurrent with the results of the FT-IR analyses, suggesting that the reactions do leave some leftover alkene resonances and that the alkynes react progressively from alkyne to alkene to sp3 hybridised centres. The spectra showed similarities and consistencies between the degradation products suggesting that the reaction pathways are largely the same between the different crosslinkers, and thus yield related products. The Sol products gave spectra with a greater population of broad resonances whereas the Insol products generally gave less cluttered spectra with less broad peaks, suggesting that the Insol products are more well defined compared to the Sol products, and that the Sol products have a greater variety of different molecular substructures. Lesser amounts of aromatic byproduct fragment were detected in the spectra which are tentatively attributed to benzenes, thiophenols, and thiophenes, the latter of which have been shown in the literature to form from intramolecular reactions under inverse vulcanisation conditions when multiple alkynes are present.19 The polymer products of inverse vulcanisations with alkynes are the only ones where thiophenol and benzene resonances could be detected, suggesting that only alkynes can form these moieties. Additionally, NON50-S50-Sol has weaker aromatic resonances than the other amine containing polymers, suggesting that the reaction pathway to form the aromatic structures is kinetically assisted by an amine activated pathway. However, in all cases, the aromatic signals were much weaker than the alkyl signals, so it seems likely that the aromatic components are a small population substructural by-product.
Antimicrobial activity
The polymers TPA30-S70-Sol, TPA30-S70-Insol, TAA30-S70-Sol, TAA30-S70-Insol, NON30-S70-Sol, and NON30-S70-Insol were selected to be tested for antimicrobial activity. The S70 polymers were chosen because it is generally accepted that the sulfurous component of the polymers is responsible for antimicrobial activity, so it is sensible to choose polymers with higher sulfur content. Additionally, as reported by Dop et al., polymers that have a glass transition temperature close to the ambient temperature of their environment tend to have better antimicrobial activity, and the S70 polymers gave Sol products with glass transition temperatures close to room temperature, whilst the Insol products had glass transition temperatures too high to detect.25,26 Finally, NON provides an interesting comparison to TAA and TPA because the latter contain nitrogen which could have an effect on the antimicrobial activity, whilst NON lacks nitrogen. TPA is an alkyne crosslinker whereas TAA is an alkene crosslinker, and it was interesting to observe whether this had an effect. The polymers were ground to powder and tested for solution antibacterial activity because the Insol products were not amenable to moulding, and could not be shaped appropriately for other test methods. The full method of testing can be found in the ESI, section X,† while the results can be seen in Fig. 6. | ||
| Fig. 6 The log reductions, relative to untreated culture, in bacterial populations of S. aureus strain USA300 in solution when different inverse vulcanised polymers were present in those solutions. | ||
As can be seen in Fig. 6, all of the tested polymers were effective in reducing the bacterial populations of S. aureus strain USA300 in solution, far better than elemental sulfur alone. However, the differences in results between the different polymers was quite small. In general, the Sol products produced larger log reductions than their corresponding Insol counterparts. This agrees with the conclusions of Dop et al. in that polymers with glass transition temperatures close to the temperature of the study are more effective in inhibiting viable cells, whereas those that are at a temperature below their glass transition temperature are less effective.25,26 Interestingly there was little difference between NON and TPA polymers, suggesting that the presence of the nitrogen is not important to the antibacterial activity. The polymers of TAA were less effective than those of TPA and NON, suggesting that the alkyne crosslinkers gave polymers that had greater antibacterial activity, perhaps due to their greater ability to stabilise sulfur. It should be noted that in solution, the polymer samples sometimes aggregated, and this may have resulted in underestimation of the antibacterial activity of the polymers.
Phosphorus containing crosslinkers
An interesting comparison to the aforementioned amine crosslinkers, are analogues with a phosphorus atom. Trivalent phosphorus is widely considered to be a stronger nucleophile than trivalent nitrogen, as phosphorus has a lower electronegativity, and higher principal quantum number for its outermost electrons, making its lone pair more readily donated. The phosphorus containing crosslinkers used here are shown in Fig. 7(a). Triallylphosphine (TAPIN) is directly comparable to TAA, whereas triallylphosphite (TAPIT) and triallylphosphate (TAPAT) are an interesting comparison to TAPIN for the increasing number of oxygens bonded to the phosphorus. For TAPIT, it would be expected that the phosphorus would be less electron rich than TAPIN, and for TAPAT, the phosphorus no longer has a lone pair and instead has an oxygen atom. What effect these factors had on the catalytic properties, or whether indeed these molecules where catalytic at all was a point of interest. | ||
| Fig. 7 (a) Chemical structures of TAPIN, TAPIT, and TAPAT as well as a table indicating whether elemental sulfur could be detected in the polymers by DSC or PXRD; (b) the yields of the Sol and Insol products at different weight percentages of crosslinker in the reaction feed for TAPIN, TAPIT, and TAPAT; (c) the soluble fractions of the Sol and Insol products at different weight percentages of crosslinker in the reaction feed for TAPIN, TAPIT, and TAPAT; (d) the Tg's, obtained from the second heating cycle of DSC, of the Sol and Insol products at different weight percentages of crosslinker in the reaction feed for TAPIN, TAPIT, and TAPAT. Note that the error bars indicate the onset and end temperatures of the Tg. Where data points are missing, no Tg could be identified in the thermogram. | ||
Unfortunately, due to the cost of these crosslinkers, they could not all be tested in the full range of feed ratios as the aforementioned crosslinkers. This cost would likely prevent widespread use of such phosphorus containing crosslinkers, except in specialty applications, but they are still a useful comparison to the already tested nitrogen crosslinkers.
Interestingly, both TAPIT and TAPIN required week-long reaction times to reach completion, which in comparison to TAA, indicates that they might not possess any rate enhancing moieties within the crosslinker structure. Tonkin et al. recently reported the formation of phosphine sulfides from phosphines in an inverse vulcanisation reaction, so it seems sensible that an analogous reaction pathway could be occurring here, which would deactivate the phosphorus atoms’ lone pair, and replace it with an inactive sulfide, explaining the longer reaction time for TAPIN and TAPIT.24 The formation of these phosphine sulfides is hypothesized to be driven by the formation of the strong phosphorus – sulfur double bond. The analogous nitrogen – sulfur double bond is not strong and so is not exergonic to form, which would explain why amines are not subject to this reaction and do provide rate enhancements.
Observing Fig. 7(b) it can be seen that the yields of TAPIN, TAPIT, and TAPAT largely follow the same trends as that of TAA, excepting that TAPIT and TAPIN are missing the data points for 50 and 70% sulfur loading. As expected, the Insol products of TAPIN, TAPIT, and TAPAT all had negligible soluble fractions. Meanwhile, as expected, the Sol products showed decreasing solubility as the feed percentage of crosslinker was increased, indicating a more crosslinked Sol product. As before, DSC and PXRD indicated that the Insol products contained no crystalline elemental sulfur, whereas Sol products at higher feed percentages of sulfur did contain crystalline elemental sulfur, with explanations mirroring those for TPA, TAA, and NON. Observing Fig. 7(d), the Sol products show rising Tg values with increasing crosslinker loading, in a fashion similar to TPA, TAA, and NON. The Insol products are more difficult to explain with regards to their Tg's. From the results of TAA, it would be expected that Insol products of alkene crosslinkers should show Tg's, but this was not the case for TAPIT, which might suggest that TAPIT Insols are like the alkyne crosslinker Insol products, in that they are sufficiently crosslinked to not show a Tg, though this is a surprising result. It is also hard to explain why for TAPAT and TAPIN Insols, their Tg's decrease and then increase with rising crosslinker loading.
One interesting observation from Fig. 7, is that TAPIT Insol and TAPAT Insol show very similar results for their yields, soluble fractions and Tg's, which might imply that they are in fact, the same product. This seems plausible because if TAPIT were to be oxidised, the product would be TAPAT. Attempts to prove such a transformation by 31P NMR were unsuccessful, as even the most soluble polymers were not sufficiently soluble to provide a signal.
Crosslinker blends
One potential benefit of crosslinkers like TPA and TAA, is that they could be blended with other crosslinkers, to act as an activator to the inverse vulcanisation reaction. TPA and TAA as activators would be intentionally incorporated into the polymer structure, eliminating concerns about extracting the catalyst, whilst also raising no issues with heavy metal contamination as do catalysts like zinc dimethyldithiocarbamate.17 To demonstrate this, TPA and TAA were blended with dicyclopentadiene and linseed oil in bulk polymerisations as described in the ESI, section II.D.† Of note, blends were also attempted with divinylbenzene which is a more reactive crosslinker, but these proved to react too quickly, resulting the Trommsdorff–Norrish effect. This reinforces the conclusion that catalysis and activation should be applied with due care and consideration in these reactions, and should not be attempted where a reaction is already reasonably quick.Initial attempts to polymerise linseed oil by the method detail in the ESI, section II.D.† Resulted in the polymer product bubbling up the reaction vial in a manner that appeared similar to an auto-acceleration, which was unexpected for linseed oil. Linseed oil showed reaction times much longer than would be expected to produce the Trommsdorff–Norrish effect. Regardless, the proportion of linseed oil in the reactions was increased to seventy percent by mass, to slow the reaction down and mitigate auto—accelerations. For some of the catalysed reactions with seventy percent linseed, the reaction mixture bubbled up, still at reaction times too long for the Trommsdorff–Norrish effect to be expected. Closer inspection suggested this bubbling up of the reaction may not have been due to the Trommsdorff–Norrish effect. Such an auto-acceleration would likely occur when the reaction is still stirring, or most likely, very soon after the stirrer ceases to rotate, which is when heat transfer to the surroundings becomes poorest but there is still sufficient reactive material in the mixture for auto-acceleration to occur. In these linseed oil reactions, the bubbling up did not occur immediately after the stirring ceased, and occurred sometime during the overnight cure when the reaction was not being observed. This suggests that the bubbling up of the reaction might not be due to an auto-acceleration, but instead, a much slower release of a gaseous side-product during the curing step. This suggests that reactions with a fifty percent mass loading of linseed oil could be viable, but since the reactions with a seventy percent mass loading of linseed oil gave results that were convenient to measure, reactions with a fifty percent mass loading of linseed oil were not attempted.
The results shown in Fig. 8 indicate that both TAA and TPA are effective activators for both DCPD and linseed oil, as the vitrification times (the time between adding the crosslinker into the reaction, and the point where the reaction mixture was sufficiently viscous that the stirrer could no longer rotate) were decreased when TA and TPA were present. The yield of DCPD reactions were improved when TAA and TPA were included in activator quantities, which suggests that either hydrogen sulfide generation is suppressed by the activated pathway that outcompetes it, or more likely, that because the reaction takes less time to reach completion, there is less crosslinker evaporation. It should be noted that, as an improvement to the method used previously by Dodd et al., the method employed here used a sealed reaction vessel with an air balloon for pressure regulation, with the intention of minimising crosslinker evaporation.17 Reactions with DVB followed by CHNS analysis showed this method to be highly effective for this purpose, giving sulfur and carbon contents very close to the predicted values. There are no clear trends in the linseed oil reaction yields, which may be because the linseed oil reaction achieves such a high yield without activation that it is hard to enhance it further. It can be noted that TPA was a stronger activator than TAA in terms of the vitrification time. It was considered that because TPA is a lighter molecule than TAA, 0.1 g or 0.3 g (a 1% or 3% mass loading respectively) of TPA constitutes more moles of activator than TAA. However, 0.1 g of TAA is 0.72 mmol, and 0.1 g of TPA is 0.76 mmol which is a small difference. Thus, it is not surprising that when a reaction was performed with 0.76 mmol of TAA, the results were only marginally different to a 0.72 mmol loading. Both DCPD and linseed oil reactions benefitted from the presence of TAA and TPA in terms of the glass transition temperatures of the products, suggesting that these activators help to achieve a more crosslinked, fully reacted, final structure. Overall, there is not much difference between TAA and TPA in terms of their benefits as activators.
 | ||
| Fig. 8 (a) Vitrification times, (b) yields and (c) glass transition temperatures of DCPD and linseed oil polymers blended with either 0, 1 or 3% by mass TAA or TPA in the feed ratio. Note that the DCPD polymers were 50% by mass sulfur in the feed ratio, and the linseed oil polymers were 30% by mass sulfur. The error bars show the standard deviation, and are too small to be seen in (a). (d) The integration ratio of the norbornene region and the cyclopentene region from 1H NMR spectra taken at different reaction times of a DCPD reaction with 50% by mass sulfur and either 0% or 1% by mass TPA or TAA. | ||
For DCPD, the molecule has its two alkene bonds in different environments, resulting in different activation energies, and as described by Smith et al. at low temperatures, only one bond reacts in inverse vulcanisation, giving more linear character to the resultant polymer.2,14 It was theorised here that TAA and TPA might encourage both bonds to react by lowering the reaction energy, or producing sufficiently reactive sulfur species, capable of attacking both double bonds. To answer this, reactions of fifty percent by weight sulfur, and either 50% DCPD, 49% DCPD + 1% TPA, or 49% DCPD + 1% TAA were conducted as described in the ESI, section II.D.,† with aliquots of the reaction taken at 30, 60, 90 and 120 minutes reaction times. 1HNMR was performed on these aliquots to observe the changes in the integration ratios of the norbornene alkene hydrogens as compared to the cyclopentene alkene hydrogens, the results of which can be seen in Fig. 8(d).
The first note to make is that the 1HNMR signals were not straightforward to analyse as would be expected of pristine DCPD (ESI, section XI†). This is because of several reasons, including the different ways DCPD can react (as described by Smith et al., like retro Diels Alder before polymerisation) and the fact that when one alkene bond reacts, the other alkene bond is no longer in a molecule of DCPD, but is instead in a DCPD unit within a polymer, and therefore its chemical shift is no longer the same.2,14 These factors result in small complications to the signals meaning they were no longer well defined multiplets and were instead rough regions which corresponded to either the norbornene alkene hydrogens of the cyclopentene alkene hydrogens. Such complications may explain why the two environments for the two cyclopentene hydrogens did not always have equal integrations. Note that in all NMR spectra the regions that were integrated over were always kept consistently to 6.09 to 5.85 ppm for the norbornene region and 5.58 to 5.4 ppm for the cyclopentene region. Note that an aliquot of the activator free reaction could not be taken at 30 minutes due to the inhomogeneity of the reaction mixture. Integration ratios at longer reaction times may be less accurate to the decreased intensity of the signals, particularly for the activated reactions.
From Fig. 8(d) it can be observed that with no activator, the norbornene and cyclopentene alkenes react at similar rates in the early stage of the reaction, when there is plentiful unreacted high energy sulfur species. However, as the reaction progresses, the integration ratio begins to decrease, indicating that the norbornene alkene is being consumed more quickly than the cyclopentene alkene. For the reactions where TPA or TAA is present, this decrease in the integration ratio occurs much sooner and is more pronounced. Because Fig. 8(d) does not provide information on the overall rate of reaction, but only indicates whether the cyclopentene or norbornene alkene has a greater population at a given time, this observation could mean either of two things: that the addition of an activator accelerates consumption of the norbornene alkene more than the cyclopentene alkene, or that the addition of an activator accelerates consumption of both alkenes proportionally, thus the reaction reaches completion sooner, and the naturally higher rate of consumption of the norbornene alkene occurs sooner in time. Regardless, it appears that activation with TPA and TAA does not seem to favour promotion of reaction upon the less reactive cyclopentene.
It is worth noting that this nucleophilic activation by including an amine containing crosslinker is likely to be amenable to other co-monomers in inverse vulcanisation. Previous publications have already demonstrated that several co-monomers can benefit from amine activation, including divinylbenzene, 1,3-diisopropenylbenzene, styrene, 4-amino styrene, methyl methacrylate, and ethylene glycol dimethacrylate.17,18 As such it is expected that nucleophilic activation by amines should work on a broad scope of co-monomers in inverse vulcanisation, and that the extent of this scope warrants further investigation beyond this proof of concept example with dicyclopentadiene.
Conclusions
It has been shown here that previously unreported amine containing alkyne crosslinkers can be polymerised successfully in inverse vulcanisation, provided measures are in place to manage their high reactivity, yielding polymers with glass transition temperatures higher than their decomposition temperature. By using a dispersion polymerisation, highly reactive crosslinkers can reliably be polymerised whilst avoiding hazardous auto-accelerations, giving two products which can have different applications. Amine containing crosslinkers have also been polymerised by this method to harness the advantages of amine activation without concerns of metallic contamination of the polymers, nor the need to extract the catalyst from the polymer in post-synthesis. It has been found that amines provide an alternate route of activation for inverse vulcanisation polymerisations, and that this permits inverse vulcanisation of low boiling crosslinkers, at temperatures where sulfur is in the solid state. By this method, propargylamine was polymerised, which is of note because it contains only a single alkyne bond, and yet was still able to form a crosslinked polymer. FT-IR revealed that it is likely that alkynes react progressively in inverse vulcanisation, first converting to alkene, before reacting again to form a saturated system, and that these resultant polymers are reasonably effective at inhibiting antimicrobial growth. The phosphorus containing analogues of amine containing crosslinkers were found to have no activating effects which may be due to the formation of phosphorus-sulfur bonds, which deactivate the phosphorus. Finally, it was shown that amine containing crosslinkers can be used as a secondary crosslinker, present in catalytic quantities, in an inverse vulcanisation of a primary comonomer, and this brings several benefits, such as improved yield, decreased reaction time, and increased glass transition temperature.Author contributions
Experimental work was performed by Liam Dodd and William Sandy. Amy Lunt carried out PXRD. Romy Dop performed the antimicrobial activity studies, under the supervision of Daniel Neill. All work was performed under the supervision of Tom Hasell.Conflicts of interest
There are no conflicts to declare.References
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, A. Somogyi, P. Theato, M. E. Mackay, Y. E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- J. A. Smith, X. Wu, N. G. Berry and T. Hasell, J. Mater. Chem. A, 2018, 56, 1777–1781 CAS.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS PubMed.
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC.
- M. J. H. Worthington, R. L. Kucera, I. S. Albuquerque, C. T. Gibson, A. Sibley, A. D. Slattery, J. A. Campbell, S. F. K. Alboaiji, K. A. Muller, J. Young, N. Adamson, J. R. Gascooke, D. Jampaiah, Y. M. Sabri, S. K. Bhar-gava, S. J. Ippolito, D. A. Lewis, J. S. Quinton, A. V. Ellis, A. Johs, G. J. L. Bernardes and J. M. Chalker, Chem. – Eur. J., 2017, 23, 16219–16230 CrossRef CAS PubMed.
- R. J. Angelici, Acc. Chem. Res., 1988, 21, 387–394 CrossRef CAS.
- F. Stojcevskia, M. K. Stanfield, D. J. Hayne, M. Mann, N. A. Lundquist, J. M. Chalker and L. C. Henderson, Sustainable Mater. Technol., 2022, 32, e00400 CrossRef.
- D. J. Parker, H. A. Jones, S. Petcher, L. Cervini, J. M. Griffin, R. Akhtarb and T. Hasell, J. Mater. Chem. A, 2017, 5, 11682–11692 RSC.
- I. Gomez, D. Mecerreyes, J. A. Blazquez, Q. Leonet, H. Ben Youcef, C. Li, J. L. Gomez-Camer, O. Bondarchuk and L. Rodriguez-Martinez, J. Power Sources, 2016, 329, 72–78 CrossRef CAS.
- T. S. Sahu, S. Choi, P. Jaumaux, J. Zhang, C. Wang, D. Zhou and G. Wang, Polyhedron, 2019, 162, 147–154 CrossRef CAS.
- S. Petcher, D. J. Parker and T. Hasell, Environ. Sci.: Water Res. Technol., 2019, 5, 2142–2149 RSC.
- A. G. Simmonds, J. J. Griebel, J. Park, K. R. Kim, W. J. Chung, V. P. Oleshko, J. Kim, E. T. Kim, R. S. Glass, C. L. Soles, Y. Sung, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 229–232 CrossRef CAS PubMed.
- T. S. Kleine, T. Lee, K. J. Carothers, M. O. Hamilton, L. E. Anderson, L. R. Diaz, N. P. Lyons, K. R. Coasey, W. Q. Parker, L. Borghi, M. E. Mackay, K. Char, R. S. Glass, D. L. Lichtenberger, R. A. Norwood and J. Pyun, Angew. Chem., Int. Ed., 2019, 58, 17656–17660 CrossRef CAS.
- J. A. Smith, S. J. Green, S. Petcher, D. J. Parker, B. Zhang, M. J. H. Worthington, X. Wu, C. A. Kelly, T. Baker, C. T. Gibson, J. A. Campbell, D. A. Lewis, M. J. Jenkins, H. Willcock, J. M. Chalker and T. Hasell, Chem. – Eur. J., 2019, 25, 10433–10440 CrossRef CAS PubMed.
- V. Hanna, P. Yan, S. Petcher and T. Hasell, Polym. Chem., 2022, 13, 3930–3937 RSC.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 647 CrossRef PubMed.
- L. J. Dodd, O. Omar, X. Wu and T. Hasell, ACS Catal., 2021, 11, 4441–4455 CrossRef CAS.
- Y. Zhang, N. G. Pavlopoulos, T. S. Kleine, M. Karayilan, R. S. Glass, K. Char and J. Pyun, Polym. Chem., 2019, 57, 7–12 CrossRef CAS.
- P. T. Dirlam, A. G. Simmonds, T. S. Kleine, N. A. Nguyen, L. E. Anderson, A. O. Klever, A. Florian, P. J. Costanzo, P. Theato, M. E. Mackay, R. S. Glass, K. Char and J. Pyun, RSC Adv., 2015, 5, 24718–24722 RSC.
- J. Bao, K. P. Martin, E. Cho, K. Kang, R. S. Glass, V. Coropceanu, J. Bredas, W. O. Parker, J. T. Njardarson and J. Pyun, J. Am. Chem. Soc., 2023, 145, 12386–12397 CrossRef CAS PubMed.
- Y. Y. Zhang, J. J. Griebel, P. T. Dirlam, N. A. Nguyen, R. S. Glass, M. E. Mackay, K. Char and J. Pyun, Environ. Sci.: Water Res. Technol., 2017, 55, 107–116 CAS.
- G. Li, Y. Gao, X. He, Q. Huang, S. Chen, S. H. Kim and D. Wang, Nat. Commun., 2017, 8, 850 CrossRef PubMed.
- L. J. Dodd, C. Lima, D. Costa-Milan, A. R. Neale, B. Saunders, B. Zhang, A. Sarua, R. Goodacre, L. J. Hardwick, M. Kuball and T. Hasell, Polym. Chem., 2023, 14, 1369–1386 RSC.
- S. J. Tonkin, C. T. Gibson, J. A. Campbell, D. A. Lewis, A. Karton, T. Hasell and J. M. Chalker, Chem. Sci., 2020, 11, 5537–5546 RSC.
- R. A. Dop, D. R. Neill and T. Hasell, Biomacromolecules, 2021, 22, 5223–5233 CrossRef CAS PubMed.
- R. L. Upton, R. A. Dop, E. Sadler, A. M. Lunt, D. R. Neill, T. Hasell and C. R. Crick, J. Mater. Chem. B, 2022, 10, 4153–4162 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI document provides content on the following headings: I. General considerations, II. Reaction methods, III. Proposed mechanism of nucleophilic initiation, IV. Representative TGA data, V. Representative DSC thermograms, VI. FTIR spectra, VII. Computationally predicted IR data, VIII. CHNS and XRF, IX. NMR of the degradation products, X. Antimicrobial activity methods, XI. NMR kinetics. See DOI: https://doi.org/10.1039/d3py00757j |
| This journal is © The Royal Society of Chemistry 2023 |