
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of original polymeric hydroperoxides as innovative oxidizing agents for self-cure dental materials†
Paul
Morandi
a,
Yohann
Catel
b,
Jörg
Angermann
b,
Pascal
Fässler
b,
Jean-Jacques
Robin
a and
Sophie
Monge
 *a
*a
aICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: sophie.monge-darcos@umontpellier.fr
bIvoclar Vivadent AG, Schaan, FL-9494, Liechyenstein
First published on 1st August 2023
Abstract
Redox initiator systems based on cumene hydroperoxide are currently widely used for the curing of self-cure (SC) and dual-cure (DC) dental materials. Unfortunately, cumene hydroperoxide exhibits a strong odor, which can be unpleasant if high amounts of materials are required for a restoration. In order to reduce smell, innovative cumyl hydroperoxide containing oligomers were prepared and tested as oxidizing agents in current self-curing dental formulations. In a first step, a methacrylate monomer containing a cumyl group was synthesized, namely the (4-isopropylbenzoate) 2-ethyl methacrylate (IBEMA). Then, homopolymer from IBEMA (PIBEMA) and copolymers from IBEMA and methyl methacrylate (MMA) (P(MMA-st-IBEMA)) were successfully synthesized using telomerization in the presence of 2-mercaptoethanol with two different low molecular weights and two monomer ratios for the copolymers. The chain-end sulfide groups of all produced oligomers were quantitatively oxidized to sulfonyl groups. Finally, hydroperoxide groups were obtained on the IBEMA units using the oxidation of isopropyl groups, thus leading to poly(4-(2-hydroxyperoxypropylbenzoate) 2-ethyl methacrylate) (PHPPBEMA) homopolymer and poly(methyl methacrylate-st-(4-(2-hydroxyperoxypropyl)benzoate) 2-ethyl methacrylate) (P(MMA-st-HPPBEMA)) copolymers. Self-cure composites based on the latter were formulated and the working time as well as the mechanical properties (flexural strength and modulus) of the cured materials were assessed. The two-component composites prepared with the oligomers containing the highest amounts of hydroperoxide groups provided high flexural strength and modulus values. To the best of our knowledge, we describe here the first synthesis of hydroperoxide-based oligomeric materials that can be used for well identified application. Indeed, such compounds appeared to be a promising alternative to cumene hydroperoxide for the formulation of odorless SC and DC dental materials.
1. Introduction
Self-cure (SC) and dual-cure (DC) materials are nowadays commonly used in dentistry. Indeed, such two-component materials are employed for the cementation of indirect restorations (luting composites, self-adhesives cements) as well as for the placement of direct (DC dental composites) or temporary (DC temporary filling material) restorations. SC materials are cured without light via a redox polymerization whereas DC materials are additionally light-cured (redox polymerization + photopolymerization). The SC reaction is essential for luting composites and self-adhesive cements. Indeed, the amount of light that can reach the cement through the restoration is most of the time insufficient for an efficient photocuring (due to the opacity, shade or thickness of the indirect restoration). The main advantage of DC composites in comparison to light-cure filing materials is their unlimited depth of cure, as the redox polymerization enables a curing which is independent of the light penetration depth.SC and DC two-component dental materials are made up of an organic matrix (mixture of (di)methacrylates, initiator system and additives) and inorganic fillers (e.g. silanized glass fillers, ytterbium fluoride, spherical mixed oxides, etc.). The most commonly used dimethacrylates are 2,2-bis[4-(2-hydroxy-3-methacryloyloxypropoxy)-phenyl]propane (Bis GMA), 1,6-bis-(methacryloyloxy-2-ethoxycarbonylamino)-2,4,4-trimethylhexane (UDMA), ethoxylated bisphenol A dimethacrylate (BisEMA), triethylene glycol dimethacrylate (TEGDMA), and 1,10-decanediol dimethacrylate,1–3 which polymerize during the restoration process.4 The first component of SC/DC dental materials contains an oxidant whereas the second component typically contains a reducing agent combined with a metal catalyst. Upon mixing of both components, radicals are generated via a redox reaction, yielding radicals that initiate the polymerization. DC materials additionally contain a photoinitiator system that enables a curing on demand. The benzoyl peroxide (BPO)/tertiary aromatic amine system has been widely used to initiate free-radical polymerization of two-component dental materials.5–7 The main disadvantage of this system lies in the thermal instability of BPO. Indeed, dental materials containing this peroxide are not stable at room temperature and must therefore be stored in the refrigerator. Consequently, the dental industry developed alternative redox initiator systems for the formulation of room temperature stable SC/DC dental materials. Nowadays, hydroperoxides (e.g. cumene hydroperoxide (CHP), amyl hydroperoxide, etc.) are mainly used as oxidizing agents in SC/DC dental materials, as they present a significantly improved thermal stability and can be stored at room temperature. However, the reaction between hydroperoxides and tertiary aromatic amines is rather inefficient for redox polymerization. Therefore, alternative reducing agents are required. Reducing agents such as (thio)barbituric, sulfinic, and ascorbic acid derivatives have been proposed and evaluated in dental materials.8–16 Another efficient redox initiator system that is widely used in two-component dental materials involves (acyl)thioureas.17–21 Such reducing agents are typically combined with a hydroperoxide as the oxidizing agent, and a metal salt (e.g. copper(II) acetylacetonate (Cu(acac)2)). Although SC/DC dental materials based on this system are storage stable, they also present some drawbacks. First of all, (acyl)thiourea compounds are bitter and can be responsible for an unpleasant taste in the patient's mouth in case of insufficient curing or of a leaching of the reducing agent. To solve this problem, polymerizable thioureas have been developed.22 Another disadvantage is the strong odor of the currently used hydroperoxides. To date, no satisfactory solution was found to overcome this problem. In that context, the synthesis of high molecular weight molecules bearing hydroperoxide would be an interesting approach to avoid the high vapor pressure and, as a consequence, the unpleasant smell in the dental practice.
In the present contribution, we report, to our knowledge, the first example of the synthesis of oligomers bearing cumyl hydroperoxide groups. Indeed, the main objective of the reported work was to prepare innovative CHP-based homopolymers and statistical copolymers showing different characteristics (different monomer ratios and as a consequence different amounts of hydroperoxide groups after post-polymerization modification) to determine the most interesting chemical structure for the targeted application. Telomerization23,24 was chosen to yield low molecular weight polymers (≤5000 g mol−1) to favor the solubility of these oxidizing agents in methacrylate mixtures commonly used in dental products. Then, obtained polymers were judiciously functionalized. Finally, preliminary results concerning the development of new self-cure materials were reported using the hydroxyperoxide-based oligomer/acetylthiourea/Cu(acac)2 redox initiator systems. The influence of parameters such as the amount of cumyl hydroperoxide groups or the quantity of added oligomer on the mechanical properties (flexural strength and modulus) and the working time was studied.
2. Experimental
2.1. Materials and methods
4-Isopropylbenzoic acid (IBA), 4-dimethylaminopyridine (DMAP), triethylamine (TEA), 2-hydroxyethyl methacrylate (HEMA), methyl methacrylate (MMA), 2-mercaptoethanol (2-ME), 2-(methylthio)ethanol (MTE), magnesium monoperoxyphthalate (MMPP), anhydrous sodium sulfate (Na2SO4), hydrochloric acid (HCl), dichloromethane (DCM), toluene, ethanol (EtOH) and acetonitrile (MeCN) were purchased from Sigma Aldrich and used without further purification. N-(3-Dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC, HCl) from Fluorochem and N-hydroxyphthalimide (NHPI) from Alfa Aesar were used without further purification. 2,2-Azobis(2-methylpropionitrile) (AIBN) was purchased from Sigma Aldrich and recrystallized twice in methanol before use.1H NMR and 13C NMR spectra were recorded using a Bruker Advance DRX 400 (400 MHz) and Bruker Advance DRX 600 (600 MHz) with deuterated chloroform (CDCl3) as the solvent. For 1H NMR, chemical shifts were normalized with respect to the peak of residual non-deuterated chloroform (7.26 ppm). Size Exclusion Chromatography (SEC) measurements were performed on a Varian 390-LC apparatus with a refractive index detector from Agilent Technologies (USA), equipped with two columns PLGel 5 μm mixed-D, using THF + toluene (0.05 wt%) as the eluent at 30 °C with a flow rate of 1 mL min−1. Linear poly(methyl methacrylate)s (PMMA) were used as standards.
2.2. Synthetic procedures
1H NMR (CDCl3, 400 MHz) δ [ppm]: 7.96–7.29 (d, d, 4H, aromatic CH), 6.14–5.58 (m, 2H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 4.54–4.49 (m, 4H, CH2–O), 2.95 (m, 1H, CH), 1.95 (m, 3H, CH3), 1.26 (d, 6H, isopropyl (CH3)2).
CH2), 4.54–4.49 (m, 4H, CH2–O), 2.95 (m, 1H, CH), 1.95 (m, 3H, CH3), 1.26 (d, 6H, isopropyl (CH3)2).
1H NMR (CDCl3, 400 MHz) δ [ppm]: 8–7.2 (4H, aromatic CH), 4.5–4.1 (4H, CH2–O), 3.6–3.5 (CH2–OH), 3–2.8 (1H, CH), 2.7–2.4 (2H, CH2–S), 2.1–1.5 (2H, CH2–C), 1.3–1.15 (6H, (CH3)2), 1.15–0.8 (3H, CH3–C).
Regarding the different copolymer compositions, four different copolymers with molecular weights equal to 5000 and 2500 g mol−1 were synthesized varying [MMA]/[IBEMA] ratios (50/50 or 25/75, mol%/mol%) (Table 1). Details for the synthesis of other copolymers are reported in ESI.† MMA/IBEMA/ME/AIBN molar ratios were equal to 50/50/5.54/1 for P(MMA50-st-IBEMA50)5000, 25/75/6.84/1 for P(MMA25-st-IBEMA75)5000, 50/50/11.26/1 for P(MMA50-st-IBEMA50)2500, and 25/75/13.90/1 for 25/75 P(MMA25-st-IBEMA75)2500.
| Oligomer name | Conv.a (%) | [MMA]/[IBEMA] targ. ratiob | [MMA]/[IBEMA] exp. ratioa |
M
n targ.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (g mol−1) c (g mol−1) |
M
n exp.d (g mol−1) |
Đ |
|---|---|---|---|---|---|---|
|
a Determined by 1H NMR in deuterated chloroform.
b Initial [MMA]:[IBEMA] molar ratio.
c
M
n targ. = Mw telogen + n.Mw MMA + m.Mw IBEMA, with n and m, number of MMA and IBEMA units, respectively.
d Determined by size exclusion chromatography in THF (+0.05 wt% toluene) using PMMA standards.
|
||||||
| PIBEMA2500 | 99 | — | — | 2500 | 2800 | 1.35 |
| P(MMA50-st-IBEMA50)2500 | MMA: 98 | 50/50 | 49.5/50.5 | 2500 | 2600 | 1.57 |
| IBEMA: 99 | ||||||
| P(MMA25-st-IBEMA75)2500 | MMA: 99 | 25/75 | 26/74 | 2500 | 2400 | 1.47 |
| IBEMA: 99 | ||||||
| P(MMA50-st-IBEMA50)5000 | MMA: 96 | 50/50 | 50/50 | 5000 | 6000 | 1.43 |
| IBEMA: 97 | ||||||
| P(MMA25-st-IBEMA75)5000 | MMA: 96 | 25/75 | 26/74 | 5000 | 4900 | 1.45 |
| IBEMA: 96 | ||||||
1H NMR (CDCl3, 400 MHz) δ [ppm]: 7.95–7.27 (4H, aromatic CH), 4.6–4.1 (4H, CH2–O), 3.7–3.4 (3H, CH3–O and CH2–OH), 3.1–2.9 (1H, CH), 2.7–2.4 (CH2–S), 2.1–1.7 (4H, CH2–C), 1.3–1.2 (6H, (CH3)2), 1.2–0.7 (6H, CH3–C).
1H NMR (CDCl3, 400 MHz) δ [ppm]: 8–7.2 (4H, aromatic CH), 4.5–4.1 (4H, CH2–O), 4–3.9 (CH2–OH), 3.1–3 (CH2–SO2), 3–2.8 (1H, CH), 2.1–1.5 (2H, CH2–C), 1.3–1.15 (6H, (CH3)2), 1.15–0.8 (3H, CH3–C).
1H NMR (CDCl3, 400 MHz) δ [ppm]: 7.95–7.27 (4H, aromatic CH), 4.6–4.1 (4H, CH2–O), 4.1–3.9 (CH2–OH), 3.7–3.3 (3H, CH3–O), 3.2–3 (CH2–SO2), 3–2.8 (1H, CH), 2.1–1.7 (4H, CH2–C), 1.3–1.2 (6H, (CH3)2), 1.2–0.7 (6H, CH3–C).
Regarding the different copolymer compositions, four different copolymers were prepared, from the P(MMA-st-IBEMA) oligomers previously synthesized: oxidized P(MMA50-st-IBEMA50)5000, oxidized P(MMA25-st-IBEMA75)5000, oxidized P(MMA50-st-IBEMA50)2500, and oxidized P(MMA25-st-IBEMA75)2500. Details for the synthesis of all oxidized P(MMA-st-IBEMA) oligomers are reported in the ESI.†
1H NMR (CDCl3, 400 MHz) δ [ppm]: 8.1–7.4 (4H, aromatic CH), 4.6–4 (4H, CH2–O), 4–3.9 (CH2–OH), 3.1–3 (CH2–SO2), 3–2.9 (remaining CH), 2.1–1.5 (2H, CH2–C), 1.6–1.5 (6H, hydroperoxypropyl (CH3)2), 1.3–1.15 (remaining ((CH3)2 from isopropyl group), 1.1–0.7 (3H, CH3–C).
1H NMR (CDCl3, 400 MHz) δ [ppm]: 8.1–7.4 (4H, aromatic CH), 4.6–4.1 (4H, CH2–O), 4.1–4 (2H, CH2–OH), 3.7–3.3 (3H, CH3–O), 3.2–3 (2H, CH2–SO2), 3–2.9 (1H, remaining CH), 2.1–1.5 (4H, CH2–C), 1.7–1.5 (6H, hydroperoxipropyl (CH3)2), 1.3–1.2 (6H, remaining (CH3)2 from isopropyl group), 1.2–0.6 (6H, CH3–C).
Regarding the different copolymer compositions, four different copolymers were prepared, respectively, from the oxidized P(MMA-st-IBEMA) oligomers previously synthesized: P(MMA50-st-HPPBEMA50)5000, P(MMA25-st-HPPBEMA75)5000, P(MMA50-st-HPPBEMA50)2500, P(MMA25-st-HPPBEMA75)2500. Details for the synthesis of all P(MMA-st-HPPBEMA) oligomers are reported in the ESI.†
1H NMR (CDCl3, 400 MHz) δ [ppm]: 7.98–7.28 (d, d, 4H, aromatic CH), 4.48 (t, 2H, CH2–O), 2.96 (m, 1H, CH), 2.86 (t, 2H, CH2–S), 2.21 (s, 3H, CH3–S), 1.26 (d, 6H, (CH3)2).
1H NMR (CDCl3, 400 MHz) δ [ppm]: 7.96–7.30 (d, d, 4H, aromatic CH), 4.78 (t, 2H, CH2–O), 3.45 (t, 2H, CH2–SO2), 3.03 (s, 3H, CH3–SO2), 2.96 (m, 1H, CH), 1.27 (d, 6H, (CH3)2.
1H NMR (CDCl3, 400 MHz) δ [ppm]: 8.03–7.56 (d, d, 4H, aromatic CH), 7.44 (s, 1H, O–OH), 4.79 (t, 2H, CH2–O), 3.46 (t, 2H, CH2–SO2), 3.02 (s, 3H, CH3–SO2), 1.62 (s, 6H, (CH3)2).
3. Results and discussion
Synthesis of valuable hydroperoxide containing oligomers was achieved in three steps from an original monomer, namely (4-isopropylbenzoate) 2-ethyl methacrylate (IBEMA) (Scheme 1). Telomerization was chosen as it allowed the synthesis of low molecular weight oligomers, thus favoring solubility in dental formulations. The use of methyl methacrylate (MMA) for the synthesis of statistical copolymers also favored solubility, and allowed us determining the influence of hydroperoxide content on mechanical properties and working time of self-cure materials formulations. Thus, poly((4-isopropylbenzoate) 2-ethyl methacrylate (PIBEMA) homopolymer and poly(methyl methacrylate-st-(4-isopropylbenzoate) 2-ethyl methacrylate) (PMMA-st-IBEMA) copolymers were prepared. Then, oxidation of the sulfur atom to sulfide group was achieved, and finally hydroperoxidation reaction was carried out, thus leading to cumyl hydroperoxide moieties along the macromolecular chain. The different (co)polymers prepared were accurately characterized at each step of the synthesis in order to correlate their chemical structure with performances for targeted dental application.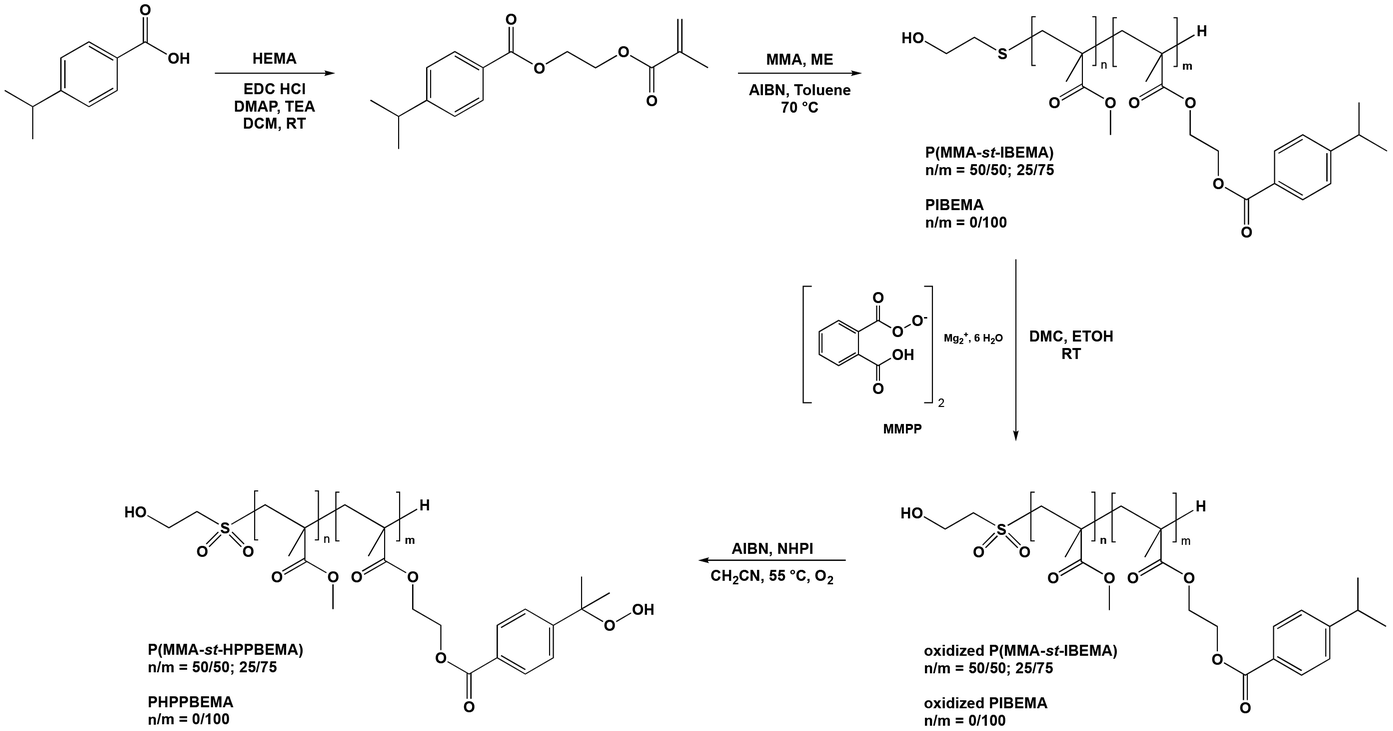 | ||
| Scheme 1 Synthetic pathway for the synthesis of PHPPBEMA and P(MMA-st-HPPBEMA) oligomers, bearing cumyl hydroperoxide moieties. | ||
3.1. Synthesis of poly((4-isopropylbenzoate) 2-ethyl methacrylate) (PIBEMA) and poly(methyl methacrylate-st-(4-isopropylbenzoate) 2-ethyl methacrylate) (PMMA-st-PIBEMA) oligomers
A cumyl containing monomer, namely (4-isopropylbenzoate) 2-ethyl methacrylate (IBEMA), was first prepared to obtain in a second step oligomers, which contain multiple cumyl groups. IBEMA was synthesized by Steglich esterification of 4-isopropylbenzoic acid with hydroxyethyl methacrylate (HEMA) in the presence of 4-dimethylaminopyridine (DMAP) and N-ethyl-N-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, HCl). IBEMA, isolated in a 65% yield, was fully characterized (Fig. S3 and S4†). It was then both homopolymerized or copolymerized with methyl methacrylate (MMA) in order to lead to poly((4-isopropylbenzoate) 2-ethyl methacrylate) (PIBEMA) homopolymer and statistical poly(methyl methacrylate-st-(4-isopropylbenzoate) 2-ethyl methacrylate) (P(MMA-st-IBEMA)) copolymers, respectively. The latter were prepared by telomerization, using 2-mercaptoethanol (2-ME) as telogen agent. The transfer constant (CT) used was equal to 0.67, which was the value usually employed for the polymerization of methacrylate monomers when aliphatic thiols are used as telogen, as mentioned in the literature (ESI†).30–32 Two different proportions of cumyl based monomer ([MMA]/[IBEMA] = 50/50 and 25/75, mol%/mol%) were considered as we wanted to study the influence of the number of hydroperoxide groups on the formulation of self-cured materials for dental applications. All polymerizations were carried out in toluene at 70 °C with 2,2′-azobis(2-methylpropionitrile) (AIBN) as initiator. At the end of the reaction, oligomers were purified by precipitation in methanol. One molecular weight was targeted for PIBEMA homopolymers (2500 g mol−1) whereas P(MMA-st-IBEMA) oligomers statistical copolymers were prepared with molar masses equal to 2500 and 5000 g mol−1 (Table 1). All (co)polymers were fully characterized by 1H NMR (Fig. 1 for P(MMA50-st-IBEMA50)5000, and Fig. S5, S9, S12 and S15† for other oligomers), and 13C NMR (Fig. S6, S8, S10, S13 and S16†). Conversions were determined from 1H NMR spectra, comparing integrations of the methacrylate double bond (6.18, 5.63 ppm, and 6.14, 5.58 ppm for MMA, and IBEMA, respectively) of the monomers and the methylene in the α position of the ester oxygen atom (3.5 ppm) for MMA units and the proton corresponding to the methine of the isopropyl group (2.96 ppm) for IBEMA in the monomers and the resulting oligomers. Final monomers ratios for P(MMA-st-IBEMA) copolymers were determined from 1H NMR spectra of the purified products, comparing integrations of the methylene in the α position of the ester oxygen atom of MMA units (3.5 ppm), and methine proton of the isopropyl group of IBEMA units (2.96 ppm) (Fig. 1 for P(MMA50-st-IBEMA50)5000 and Fig. S5, S9, S12 and S15† for other oligomers). For all copolymers, the final monomers ratios were very close from theoretical ones, ([MMA]/[IBEMA] = 50/50, 25/75, mol%/mol%). Additionally, it is valuable to notice that reactivity ratios for MMA and IBEMA were determined in toluene using Jaacks method.33 For such purpose, two free radical polymerization of MMA and IBEMA were carried out, each one with an excess of one monomer (ESI†). The same order of magnitude of reactivity ratios was found (rMMA = 0.9; rIBEMA = 1), thus confirming the ability of both methacrylate monomers to copolymerize in a statistical way (Fig. S1 and S2†). Whatever the molecular weight targeted, oligomers were prepared with a good control over the molecular weight and low dispersities (Đ) (Fig. S7, S11, S14 and S17†). In particular, in the case of P(MMA50-st-IBEMA50)5000 oligomer, chromatogram showed monomodal peak (Fig. 2) with an experimental molecular weight equal to 6000 g mol−1 and a dispersity of 1.43.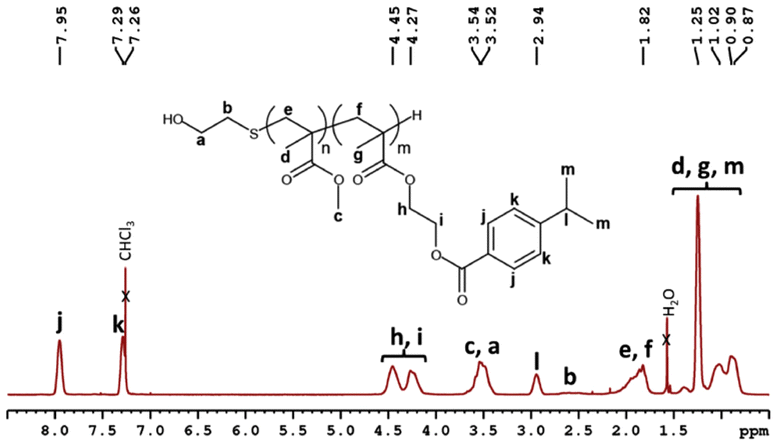 | ||
| Fig. 1 1H NMR (400 MHz, CDCl3) spectrum, and proton assignment for P(MMA50-st-IBEMA50)5000 oligomer. | ||
 | ||
| Fig. 2 Size exclusion chromatogram in the case of P(MMA50-st-IBEMA50)5000 statistical copolymer (eluent: THF, PMMA standards). | ||
Other steps of the synthesis dealt with the modification of produced (co)polymers in order to introduce the hydroperoxide moieties on the cumyl groups.
3.2. Synthesis of poly(4-(2-hydroxyperoxypropylbenzoate) 2-ethyl methacrylate) (PHPPBEMA) and poly(methyl methacrylate-st-(4-(2-hydroxyperoxypropyl)benzoate) 2-ethyl methacrylate) (P(MMA-st-HPPBEMA)) oligomers
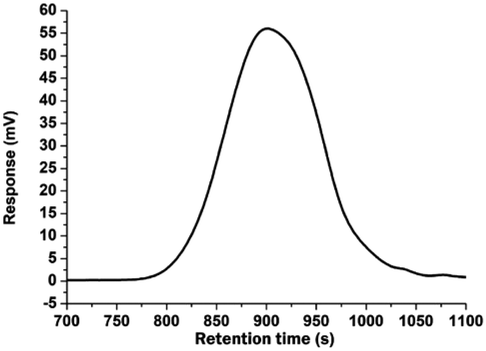
Hydroperoxidation of the cumyl groups was first achieved on poly((4-isopropylbenzoate) 2-ethyl methacrylate) (PIBEMA) homopolymers. Reaction was carried out in the presence of N-hydroxyphthalimide (NHPI) and 2,2′-azobis(2-methylpropionitrile) (AIBN) at 55 °C under air-flow bubbling through the solution.28,29 Unfortunately, hydroperoxidation did not occur properly. We have assumed that the presence of the thioether group could inhibit or lead to side reactions. Indeed, it has been reported in the literature that hydroperoxide compounds could be used for the oxidation of sulfide to sulfoxide34–36 or to sulfonyl group in the presence of a catalyst.37,38 As a result, we thought that it would be better to oxidize thioether group before achieving the hydroperoxidation reaction in order to avoid any side reaction. To check that the presence of thioether was problematic, we decided to first synthesize a model molecule, namely the (2-methylthioethyl) 4-isopropylbenzoate (MTEPB), bearing both cumyl group and sulfur atom, and to try carrying out hydroperoxidation reaction on it (Scheme 2). MTEBP was prepared by Steglich esterification25 from 2-(methylthio)ethanol (MTE) and 4-isopropylbenzoic acid (IBA) in the presence of 4-dimethylaminopyridine (DMAP) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC, HCl) in anhydrous dichloromethane (DCM). Resulting product was purified by gel chromatography on silica gel with petroleum ether/ethyl acetate (50/50, v/v). 1H NMR spectrum allowed characterizing the synthesized MTEBP, notably with signals at 2.21 ppm and 1.26 ppm, corresponding to the methylene in the α position of the sulfur atom and to the methyls of the isopropyl group, respectively (Fig. S36†). Hydroperoxidation of MTEBP was carried out using the same experimental conditions than in the case of hydroperoxidation of PIBEMA oligomer. Once again, reaction was unsuccessful. As a result, we decided to convert thioether to sulfide group. (2-Methylsulfonylethyl) 4-isopropylbenzoate (MSEPB) was prepared by oxidation reaction using magnesium monoperoxyphthalate (MMPP) in a mixture of dichloromethane/ethanol (2/1, v/v). Characterization by 1H NMR (Fig. S37†) confirmed the oxidation of the sulfur atom, notably with the chemical shift of the methyl group in the α of the sulfur going from 1.26 to 3.03 ppm. Finally, hydroperoxidation of MSEPB was carried out with NHPI and AIBN in acetonitrile at 55 °C under air-flow bubbling through the solution. Reaction was this time successful, and confirmed with the appearance on the 1H NMR spectrum of the signal corresponding to the proton of the hydroperoxide moiety at 7.44 ppm (Fig. 3). | ||
| Scheme 2 Synthetic pathway for the study of hydroperoxidation reaction on both (2-methylthioethyl) 4-isopropylbenzoate (MTEPB), and (2-methylsulfonylethyl) 4-isopropylbenzoate (MSEPB) model molecules. | ||
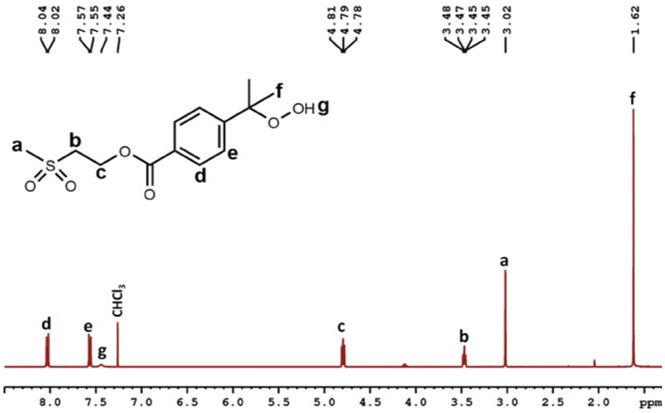 | ||
| Fig. 3 1H NMR (400 MHz, CDCl3) spectrum, and proton assignment for (2-methylsulfonylethyl) 4-hydroxyperoxipropylbenzoate (MSEHPB). | ||
From these experimental results, it was clear that sulfur atom needed to be first oxidized into sulfonyl group to then allow successful hydroperoxidation reaction. The reason remained not fully clear, as we did not find any valuable explanation in the literature indicating why hydroperoxidation reaction of cumyl groups could be inefficient in the presence of thioether function. Synthetic pathway for the synthesis of hydroperoxide-based oligomers was based on results obtained on model molecules. So, oxidation of thioether groups on PIBEMA and P(MMA-st-IBEMA) oligomers was achieved following the same experimental procedures than for the synthesis of MSEHPB from MTEBP. The amount of needed reactant (MMPP) for the chain end oxidation depended on the molecular weight (Mn) of the oligomers. As the Mn was determined by size exclusion chromatography using a calibration based on PMMA standards, it was only approximate and, as a consequence, MMPP was used in large excess (1.4 molar equivalent in relation with oligomer) to oxidize sulfur to sulfide group. After 18 hours, oxidized oligomers were purified by precipitation into methanol. Modification was characterized by 1H NMR spectroscopy (Fig. 4 for P(MMA50-st-IBEMA50)5000, and Fig. S18, S21, S23 and S25† for other oligomers), following protons borne by methylene in the α and β positions of the sulfur atom (coming from telogen agent), shifting from 2.7–2.4 to 3.2–3 ppm (Fig. 4, Hb) and 3.7–3.4 to 4.1–3.9 ppm (Fig. 4, Ha), from thioether to sulfonyl groups, respectively.
 | ||
| Fig. 4 1H NMR (400 MHz, CDCl3) spectrum, and proton assignment for oxidized P(MMA50-st-IBEMA50)5000 oligomer. | ||
1H NMR spectra logically showed no significant changes concerning the monomer ratios, by comparing integrations of the methylene in the α position of the oxygen atom of MMA blocks (3.5 ppm), and proton of the isopropyl group of IBEMA blocks (2.9 ppm). For instance, in the case of P(MMA50-st-IBEMA50)5000, the [MMA]/[IBEMA] ratio remained equal to 1 after chain end oxidation. 1H–13C NMR HSQC (Fig. 5) also confirmed that the oxidation reaction was successful, demonstrating an important chemical shift for the carbon in the α position of the sulfur atom, going from 36.5 to 57.5 ppm when oxidation occurred. Finally, the size exclusion chromatography analysis of oxidized oligomers confirmed that no degradation of the macromolecular chain took place, since traces exhibited similar profiles without any additional shoulder after oxidation reaction (Fig. 6 for P(MMA50-st-IBEMA50)5000, and Fig. S19, S20, S22, S24 and S26† for other oligomers).
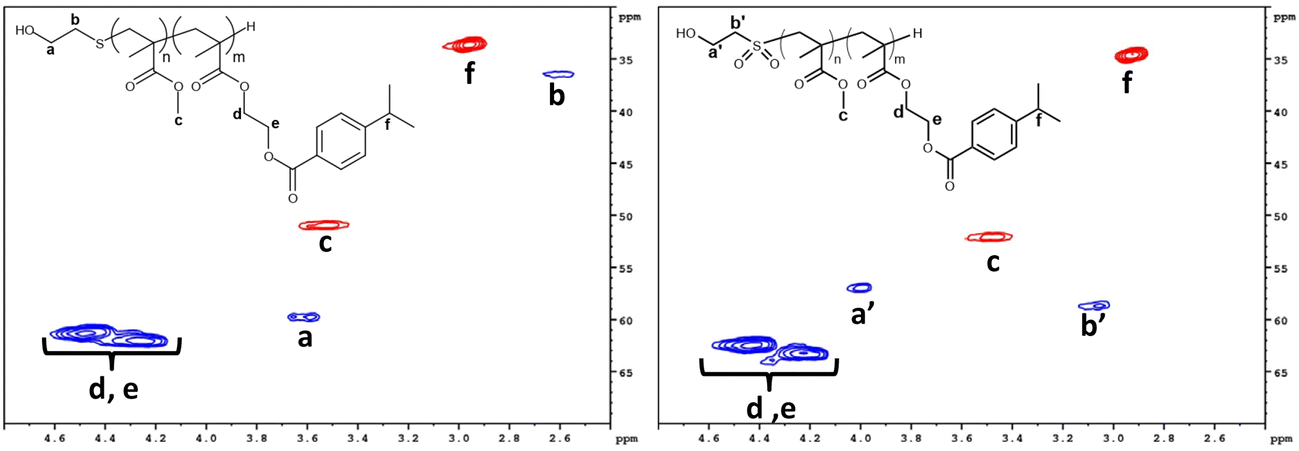 | ||
| Fig. 5 Zoom on 1H–13C HSQC NMR (600 MHz, CDCl3) spectra and proton/carbon assignments for P(MMA50-st-IBEMA50)5000 oligomers before (left) and after (right) chain end thioether group oxidation into sulfonyl group. Red spots are corresponding to CH/CH3 groups; blue spots are corresponding to CH2 groups. | ||
 | ||
| Fig. 6 Size exclusion chromatogram of P(MMA50-st-IBEMA50)5000 oligomer before (in black) and after (in red) chain end sulfide group oxidation into sulfonyl group. | ||
Last step of the synthesis dealt with the hydroperoxidation reaction on all oxidized oligomers. As for model molecule, reaction was carried out using N-hydroxyphthalimide (NHPI) and AIBN in acetonitrile at 55 °C under air-flow bubbling through the solution, and was followed by 1H NMR using the decrease of the signal corresponding to the CH of the isopropyl group at 2.94 ppm. The reactants were added in catalytic amount ([oligomer]/[NHPI]/[AIBN] was equal to 1/0.05/0.06). Reaction was stopped when a maximum of conversion of cumyl group was reached (around 90%). Then, resulting poly(4-(2-hydroxyperoxypropylbenzoate) 2-ethyl methacrylate) (PHPPBEMA) and poly(methyl methacrylate-st-(4-(2-hydroxyperoxypropyl)benzoate) 2-ethyl methacrylate) (P(MMA-st-HPPBEMA)) oligomers were purified by precipitation in ultra-pure water. Obtained yields were in the range of 95%. Experimental characteristics of the prepared oligomers are detailed in Table 2. In all cases, the maximum of conversion was reached after 120 hours of reaction. The final conversion of cumyl to cumyl hydroperoxide groups was determined by 1H NMR of the purified products (Fig. 7, and Fig. S27, S30, S32 and S34† for other oligomers), using the integration of the signal corresponding to the CH of the isopropyl group of non-converted chains residues at 3–2.9 ppm.
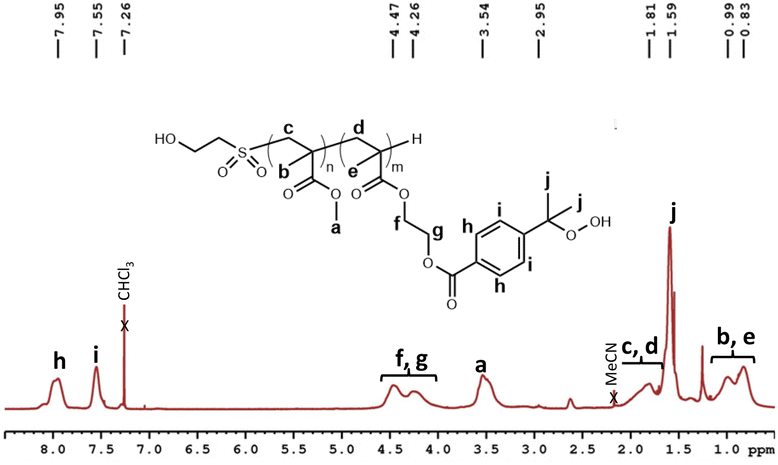 | ||
| Fig. 7 1H NMR (400 MHz, CDCl3) spectrum, and proton assignment for oxidized P(MMA50-st-HPPBEMA50)5000 oligomer. | ||
| Oligomer name | Average number of cumyl groupsa | Conv.b (%) | Average number of hydroperoxide groupsc |
|---|---|---|---|
|
a Calculated from Mn exp of PIBEMA or P(MMA-st-IBEMA) oligomers and [MMA]:[IBEMA] experimental ratios.
b Determined by 1H NMR in deuterated chloroform in the case of P(MMA-st-HPPBEMA) copolymers.
c Determined by 1H NMR in deuterated chloroform using integration of the CH of the remaining cumyl group.
|
|||
| PHPPBEMA2500 | 10 | 78 | 8 |
| P(MMA50-st-HPPBEMA50)2500 | 7 | 88 | 6 |
| P(MMA25-st-HPPBEMA75)2500 | 7.5 | 85 | 6 |
| P(MMA50-st-HPPBEMA50)5000 | 16 | 86 | 13.5 |
| P(MMA25-st-HPPBEMA75)5000 | 15.5 | 84 | 13 |
The hydroperoxidation reaction did not change the oligomer chemical structures, as the [MMA]/[HPPBEMA] ratios remained the same whatever the oligomer considered before and after the hydroperoxidation reaction. Final produced homopolymers and copolymers were also characterized by 13C NMR, which confirmed their chemical structures (Fig. S28, S29, S31, S33 and S35†). Odorless compounds were obtained, which was a key-parameter for considering these materials for the targeted application, i.e. their use as innovative oxidizing agents in self-cure dental materials. 1H NMR and SEC also demonstrated that no additional molecules/impurities were found in the synthesized PHPPBEMA-based telomers. Finally, it is important to mention that the cotelomers were stable with time as same 1H NMR and SEC results were obtained six months after the synthesis, still without bad smell.
To conclude, we managed to prepare original cumyl hydroperoxide-based oligomers with control over the molecular weight, low dispersities, and control of the [MMA]/[HPPBMA] ratios. This represents, to the best of our knowledge, the first example in the literature of macromolecular hydroperoxides prepared by telomerization reaction.
3.3. Formulation and evaluation of self-cure composites containing hydroperoxide oligomers
In order to evaluate the potential of hydroperoxide containing oligomers as oxidizing agents in two components dental materials, self-cure composites based thereon were formulated. A bisphenol A–glycidyl methacrylate/urethane dimethacrylate/triethylene glycol dimethacrylate (BisGMA/UDMA/TEGDMA) (3/4/3, wt/wt/wt) monomer mixture was chosen for the organic matrix. First of all, the solubility of the hydroperoxide oligomers in the selected monomer mixture (5 and 10 wt% of oxidizing agent) was checked. Unfortunately, both oligomers having a molecular weight of 5000 g mol−1 (Table 2) were not fully soluble and were not further considered. On the other hand, all of the 2500 g mol−1 oligomers were fully soluble in the dimethacrylate solution. This result showed that, as expected, the molecular weight of the hydroperoxide oligomers plays a significant role on the solubility properties, and fully justify the choice of telomerization process as it allowed the synthesis of well-controlled low molecular weight oligomers.To prepare SC materials based on the new hydroperoxide oligomers, composites CA1–5 and CB1 were formulated (Table 3). The CA composites contained a hydroperoxide oligomer, PHPPBEMA2500, P(MMA50-st-HPPBEMA50)2500, or P(MMA25-st-HPPBEMA75)2500 (CA1–5) as oxidizing agents, while the CB1 composite contained an acylthiourea derivative as reducing agent and Cu(acac)2 as metal catalyst. P(MMA50-st-HPPBEMA50)2500 and P(MMA25-st-HPPBEMA75)2500 were added in two different concentrations (1.75 wt% and 3.5 wt%) whereas a single composite was formulated based on PHPPBEMA2500 (CA5, 3.5 wt% of oligomer). Additionally, a reference composite CA6 containing cumyl hydroperoxide (CHP) as oxidizing agent was prepared (Table 3). 65 wt% of a barium–aluminum–borosilicate glass filler (GM27884 d50 = 0.7 μm, Schott) was added to each composite as an inorganic filler. monomethylether of hydroquinone (MEHQ) and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) were used as stabilizers.
| Component | CA1 (wt%) | CA2 (wt%) | CA3 (wt%) | CA4 (wt%) | CA5 (wt%) | CA6 (wt%) | CB1 (wt%) |
|---|---|---|---|---|---|---|---|
| Bis-GMA | 9.970 | 9.445 | 9.970 | 9.445 | 9.445 | 9.970 | 10.324 |
| UDMA | 13.292 | 12.592 | 13.292 | 12.592 | 12.592 | 13.292 | 13.765 |
| TEGDMA | 9.970 | 9.445 | 9.970 | 9.445 | 9.445 | 9.970 | 10.324 |
| P(MMA50-st-HPPBEMA50)2500 | 1.750 | 3.500 | — | — | — | — | — |
| P(MMA25-st-HPPBEMA75)2500 | — | — | 1.750 | 3.500 | — | — | — |
| PHPPBEMA2500 | — | — | 3.500 | — | — | ||
| CHP | — | — | — | 1.750 | — | ||
| Cu(acac)2 | — | — | — | — | — | — | 0.007 |
| Acetylthiourea | — | — | — | — | — | — | 0.555 |
| MEHQ | 0.018 | 0.018 | 0.018 | 0.018 | 0.018 | 0.018 | 0.018 |
| TEMPO | — | — | — | — | — | — | 0.007 |
| Glass filler GM27884 0.7 μm sil 6% | 65.00 | 65.00 | 65.00 | 65.00 | 65.00 | 65.00 | 65.00 |
The self-cure composites SCC1–6 were two-component materials resulting from the following composite formulations: CA1/CB1, CA2/CB1, CA3/CB1, CA4/CB1, CA5/CB1, and CA6/CB1 (1/1, wt/wt). The mechanical properties of SCC1–6 were measured (Fig. S39–S44†). The results are gathered in Table 4. First, the reference composite based on CHP (SCC6) provided the best mechanical properties. However, the working time was too short and the amount of CHP would need to be significantly reduced for a clinical application. In a general manner, obtained results with the hydroperoxide oligomers clearly showed that the latter were less reactive than CHP. Additionally, the higher the amount of hydroperoxide oligomer was, the higher the flexural strength and the flexural modulus were. Indeed, SCC2 and SCC4 led to significantly better mechanical properties than SCC1 and SCC3. Moreover, shorter working times were obtained with the self-cure composites containing the higher contents of hydroperoxide oligomer (SCC2, 4, and 5). This result was expected, as a higher amount of oxidizing agent would accelerate the formation of radicals, leading to faster polymerization rates. P(MMA25-st-HPPBEMA75)2500 was found to be a more efficient oxidizing agent than P(MMA50-st-HPPBEMA50)2500. Indeed, SCC4 provided higher flexural strength and modulus values than SCC1 and SCC2, respectively. Additionally, the working times of SCC3 and SCC4 were slightly shorter than for the corresponding SCC1 and SCC2 materials. This result could be explained by the higher amount of hydroperoxide groups present in the P(MMA25-st-HPPBEMA75)2500 oligomer in comparison with P(MMA50-st-HPPBEMA50)2500.
| Cement | HP samplea | Wt% in composites CA1–6a | Flexural strength 24 h, H2O, 37 °Cb (MPa) | Flexural modulus 24 h, H2O, 37 °Cb (MPa) | Working timeb (s) |
|---|---|---|---|---|---|
| a Wt% of corresponding HP sample in the formulation. b Number in brackets indicated the margin of error. | |||||
| SCC1 | P(MMA50-st-HPPBEMA50)2500 | 1.75 | 65.9 (6.8) | 2959 (76) | 266 (19) |
| SCC2 | P(MMA50-st-HPPBEMA50)2500 | 3.5 | 89.5 (8.2) | 6289 (327) | 157 (11) |
| SCC3 | P(MMA25-st-HPPBEMA75)2500 | 1.75 | 70.0 (7.6) | 4057 (141) | 226 (5) |
| SCC4 | P(MMA25-st-HPPBEMA75)2500 | 3.5 | 100.2 (11.2) | 6847 (144) | 113 (2) |
| SCC5 | PHPPBEMA2500 | 3.5 | 87.8 (7.8) | 6229 (610) | 149 (2) |
| SCC6 | Cumene hydroperoxide (CHP) | 1.75 | 118.1 (8.9) | 7906 (298) | 71 (4) |
Surprisingly, the SCC5 composite containing the PHPPBEMA2500 oligomer exhibited lower mechanical properties than the corresponding SCC4 material based on the P(MMA25-st-HPPBEMA75)2500 copolymer although the homopolymer contained slightly more cumyl hydroperoxide groups than the copolymer (Table 2, entries 1 & 3). One explanation could be that the hydroperoxide groups were less accessible in the homopolymer, due to steric hindrance. Indeed, in the case of the copolymer, adding MMA led to a statistical distribution of the hydroperoxide moieties along the macromolecular chains, thus probably improving their accessibility and their reactivity. Additionally, it is worthwhile mentioning that determination of the number of cumyl hydroperoxide groups was achieved by 1H NMR spectrum considering that all telomers bore 2-mercaptoethanol-based chain end. Unfortunately, functionalization of all macromolecular chains by telogen agent is unlikely as already reported in the literature concerning telomerization.32 As a result, the amount of hydroperoxide groups could be in reality slightly higher in the P(MMA25-st-HPPBEMA75)2500 copolymer than in PHPPBEMA2500 homopolymer. Amongst the tested composites, SCC4 exhibited the highest flexural strength and modulus values. The measured mechanical properties were similar to those obtained with commercially available two-component dental composites. To conclude, tailored hydroperoxide oligomers proved to be suitable oxidizing agents for an application in dental materials. Valuable advantage of using such polymeric materials is that they were odorless, thus improving patient comfort.
4. Conclusions
Innovative oligomers containing cumyl hydroperoxide groups were synthesized following a four-steps synthesis strategy, leading to five oligomers with increasing amounts of cumyl hydroperoxide groups. For such purpose, one poly((4-isopropyl benzoate) 2-ethyl methacrylate) (PIBEMA) homopolymer and four poly(methyl methacrylate-st-(4-isopropyl benzoate) 2-ethyl methacrylate) (P(MMA-st-IBEMA)) statistical copolymers were successfully prepared by telomerization reaction in the presence of 2-mercaptoethanol as telogen agent. Then, they were judiciously functionalized in two steps. First, thioether function located at the chain end of the oligomers was oxidized to sulfonyl group, thus avoiding any side reaction during further hydroperoxidation reaction. Then, the cumyl groups of the IBEMA moieties were converted into cumyl hydroperoxide groups leading to poly(4-(2-hydroxyperoxypropylbenzoate) 2-ethyl methacrylate) (PHPPBEMA) homopolymer and poly(methyl methacrylate-st-(4-(2-hydroxyperoxypropyl) benzoate) 2-ethyl methacrylate) (P(MMA-st-HPPBEMA)) statistical copolymers. Two molecular weights were targeted: 2500 and 5000 g mol−1 to ensure solubility of the produced materials in dental formulations. Experimentally, only lowest molecular weight oligomers (2500 g mol−1) were fully soluble in employed formulations. Concerning statistical copolymers, two different proportions of [MMA]/[IBEMA] monomeric units were considered: 50/50, and 25/75 (mol%/mol%). Telomerization allowed a good control over the molecular weight, low dispersities, and control of copolymer monomeric content.All prepared homo- and copolymers were then evaluated as oxidizing agents in self-cure dental composites in order to determine their ability to initiate the polymerization of methacrylate based resins, using acetylthiourea as co-initiator of the redox system and copper acetylacetonate as the catalyst. Due to their polymeric structure, the hydroperoxide oligomers were odorless. These cumyl hydroperoxide-containing oligomers successfully initiated the polymerization of methacrylate-based two-component composites. Different mechanical properties were obtained, depending on the structure of the used oligomers. P(MMA25-st-HPPBEMA75)2500 was found to be the most promising oligomer, as the corresponding SC composite exhibited the highest flexural strength and modulus values. The results also clearly showed that the replacement of CHP with a hydroperoxide oligomer led to a reduction of the polymerization rate.
To conclude, the obtained results demonstrated the ability of cumyl hydroperoxide-based oligomers to be used as valuable oxidizing agents in redox initiator systems. In a more general manner, the hydroperoxides-based telomers could be used to initiate polymerization in macromolecular chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Ivoclar Vivadent AG for mechanical tests, and more generally for financial support.References
- E. Asmussen and A. Peutzfeldt, Influence of UEDMA, BisGMA and TEGDMA on selected mechanical properties of experimental resin composites, Dent. Mater., 1998, 14(1), 51–56 CrossRef CAS PubMed.
- A. Peutzfeldt, Resin composites in dentistry: The monomer systems, Eur. J. Oral Sci., 1997, 105(2), 97–116 CrossRef CAS PubMed.
- K. L. Van Landuyt, J. Snauwaert, J. De Munck, M. Peurnans, Y. Yoshida, A. Poitevin, E. Coutinho, K. Suzuki, P. Lambrechtsa and B. Van Meerbeek, Systematic review of the chemical composition of contemporary dental adhesives, Biomaterials, 2007, 28(26), 3757–3785 CrossRef CAS PubMed.
- R. V. Monteiro, C. M. C. Taguchi, R. G. Machado, S. B. Silva, J. K. Bernardon and S. Monteiro, Bulk-Fill Composite Restorations Step-by-Step Description of Clinical Restorative Techniques Case Reports, Odovtos, 2019, 21(2), 23–31 Search PubMed.
- D. S. Achilias and I. D. Sideridou, Kinetics of the benzoyl peroxide/amine initiated free-radical polymerization of dental dimethacrylate monomers: experimental studies and mathematical modeling for TEGDMA and Bis-EMA, Macromolecules, 2004, 37, 4254–4265 CrossRef CAS.
- I. D. Sideridou, D. S. Achilias and O. Karava, Reactivity of benzoyl peroxide/amine system as an initiator for the free radical polymerization of dental and orthopaedic dimethacrylate monomers: effect of the amine and monomer chemical structure, Macromolecules, 2006, 39, 2072–2080 CrossRef CAS.
- A. Zoller, D. Gigmes and Y. Guillaneuf, Simulation of radical polymerization of methyl methacrylate at room temperature using a tertiary amine/BPO initiating system, Polym. Chem., 2015, 6, 5719–5727 RSC.
- I. Yuji, K. Yoshinori and K. Toru, Polymerization initiator composition controlling polymerization at interface and curable composition containing same, EP0480785A2, 1991.
- R. Hecht and M. Ludsteck, Initiator system for acid dental formulations, US6953535B2, 2002.
- R. Hecht, M. Ludsteck, A. Stippschild, G. Raia and R. Guggenberger, Two-component self-adhesive dental composition, process of production and use thereof, WO2016/007453A1, 2016.
- A. Falsafi, R. S. Kalgutkar and J. D. Oxman, Dental compositions and methods with arylsulfinate salts, US7465758B2, 2007.
- M. Kawashima and I. Omura, Polymerizable composition, EP0408357B1, 1990.
- R. A. Guggenberger, R. Hecht, M. Ludsteck, G. Raia and A. Stippschild-Boxler, Twocomponent self-adhesive dental composition, process of production and use thereof, US10932994B2, 2015.
- M. N. Nyunt and Y. Imai, Adhesion to dentin with resin using sulfinic acid initiator system, Dent. Mater. J., 1996, 15(2), 175–182 CrossRef CAS PubMed.
- J. M. Antonucci, C. L. Grams and D. J. Termini, New initiator systems for dental resins based on ascorbic acid, J. Dent. Res., 1979, 58, 1887–1899 CrossRef CAS PubMed.
- T.-Y. Kwon, R. Bagheri, Y. K. Kim, K.-H. Kim and M. F. Burrow, Cure mechanisms in materials for use in esthetic dentistry, J. Invest. Clin. Dent., 2012, 3(1), 3–16 CrossRef PubMed.
- G. S. Misra, U. D. N. Bajpai and J. Trekoval, Role of thiourea Oin redox polymerization, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 1984, C24(3), 335–353 CrossRef CAS.
- H. Heike and A. Utterodt, Two-component initiator system (amine- free) with very good storage stability and particular suitability for acid systems, EP1754465B1, 2006.
- P. Burstscher, A. Gianasmidis and N. Moszner, Photopolymerizable and dual-curing dental materials based on thiourea derivatives, US10195121B2, 2015.
- A. Falsafi and S. B. Mitra, Composition containing a polymerizable reducuing agent, kit, and method, US7541393B2, 2002.
- I. Lamparth, P. Fässler, T. Schnur, E. Thetiot, J. Lalevée and Y. Catel, Polymerizable thioureas as innovative reducing agents for self-cured and dual-cured dental materials, Dent. Mater., 2022, 38, 1108–1116 CrossRef CAS PubMed.
- A. R. Mukherjee, R. P. Mitra, A. M. Biswas and S. Maiti, Redox-initiated vinyl polymerization with thiourea as reductant, J. Polym. Sci., Part A: Polym. Chem., 1967, 5, 135–149 CrossRef CAS.
- C. Boyer, G. Boutevin, J. J. Robin and B. Boutevin, Study of the telomerization of dimethylaminoethyl methacryalte (DMAEMA) with mercaptoethanol. Application to the synthesis of a new macromonomer, Polymer, 2004, 45(23), 7863–7876 CrossRef CAS.
- B. Boutevin, G. David and C. Boyer, Telechelic oligomers and macromonomers by radical techniques, Adv. Polym. Sci., 2007, 206, 31–135 CrossRef CAS.
- B. Neises and W. Steglich, Simple Method for the Esterification of Carboxylic Acids, Angew. Chem., Int. Ed. Engl., 1978, 17(7), 522–524 CrossRef.
- M. H. Ali and G. J. Bohnert, Chemoselective oxidation of sulfides to sulfones with magnesium monoperoxyphthalate (MMPP) on silica gel support in methylene chloride solvent, Synth. Commun., 1998, 28(16), 2983–2998 CrossRef CAS.
- M. De Vleeschauwer and J. Y. Gauthier, Remarkably mild and simple preparations of sulfinates, sulfonyl chlorides and sulfonamides from thioanisoles, Synlett, 1997,(4), 375–377 CrossRef CAS.
- K. Sugamoto, Y. Matsushita, T. Yamamoto and T. Matsui, Regioselective hydroperoxygenation of aralkanes and alpha,beta-unsaturated carbonyl compounds catalyzed by N-hydroxyphthalimide and 2,2′-azobis (4-methoxy-2,4-dimethylvaleronitrile), Synth. Commun., 2005, 35(14), 1865–1874 CrossRef CAS.
- B. Orlinska and J. Zawadiak, Aerobic oxidation of isopropylaromatic hydrocarbons to hydroperoxides catalyzed by N-hydroxyphthalimide, React. Kinet., Mech. Catal., 2013, 110(1), 15–30 CrossRef CAS.
- J. L. De La Fuente and E. L. Madruga, Homopolymerization of methyl methacrylate and styrene: determination of the chain-transfer constant from the Mayo equation and the number distributuin for n-dodecanethiol, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 170–178 CrossRef CAS.
- J. L. O'Brien and F. Gornick, Chain transfer in the polymerization of methyl methacrylate. I. Transfer with monomer and thiols. The mechanism of the termination reaction at 60 °C, J. Am. Chem. Soc., 1955, 77, 4757–4763 CrossRef.
- C. Loubat and B. Boutevin, Etude de la télomérisation du méthacrylate de méthyle par l'acide thioglycolique. Application à la détermination des probabilités de structures des télomères synthétisés, Macromol. Chem. Phys., 2000, 201, 2853–2860 CrossRef CAS.
- V. Jaacks, A novel method of determination of reactivity ratios in binary and ternary copolymerizations, Makromol. Chem., 1972, 161, 161–172 CrossRef CAS.
- A. A. Linden, M. Johansson, N. Hermanns and J. E. Backvall, Efficient and selective sulfoxidation by hydrogen peroxide, using a recyclable flavin- BMIm PF6 catalytic system, J. Org. Chem., 2006, 71(10), 3849–3853 CrossRef CAS PubMed.
- E. Dumitriu, C. Guimon, A. Cordoneanu, S. Casenave, T. Hulea, C. Chelaru, H. Martinez and V. Hulea, Heterogeneous sulfoxidation of thioethers by hydrogen peroxide over layered double hydroxides as catalysts, Catal. Today, 2001, 66(2–4), 529–534 CrossRef CAS.
- K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Oxidation of sulfides to sulfoxides. Part 2: Oxidation by hydrogen peroxide, Tetrahedron, 2005, 61(35), 8315–8327 CrossRef CAS.
- E. Baciocchi, M. F. Gerini and A. Lapi, Synthesis of sulfoxides by the hydrogen peroxide induced oxidation of sulfides catalyzed by iron tetrakis(pentafluorophenyl)porphyrin: Scope and chemoselectivity, J. Org. Chem., 2004, 69(10), 3586–3589 CrossRef CAS PubMed.
- D. J. Robinson, L. Davies, N. McGuire, D. F. Lee, P. McMorn, D. J. Willock, G. W. Watson, P. C. B. Page, D. Bethell and G. J. Hutchings, Oxidation of thioethers and sulfoxides with hydrogen peroxide using TS-1 as catalyst, Phys. Chem. Chem. Phys., 2000, 2(7), 1523–1529 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterizations. See DOI: https://doi.org/10.1039/d3py00639e |
| This journal is © The Royal Society of Chemistry 2023 |