
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ABA block copolymers comprising a water soluble poly(N-hydroxyethyl acrylamide) B block form self-assemblies of varied morphologies in an aqueous environment†
Alshaimaa
Almehmady
,
Robert
Cavanagh
,
Giuseppe
Mantovani
* and
Snow
Stolnik
 *
*
School of Pharmacy, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: giuseppe.mantovani@nottingham.ac.uk; snow.stolnik@nottingham.ac.uk
First published on 3rd August 2023
Abstract
In this study, we apply a combination of SET-LRP and photoinduced LRP techniques to synthesize novel ABA block copolymers comprising poly(N-hydroxyethyl acrylamide) as a water soluble middle B block, flanked by poly(n-butyl acrylate) A blocks: (n-BA)m-b-(N-HEA)n-b-(n-BA)m. NMR analysis of the copolymers in the ‘best performing’ common solvents indicates the presence of partially solvated poly(n-butyl acrylate) blocks and a well-solvated poly(N-hydroxyethyl acrylamide) block in a DMSO-d6/acetone-d6 mixture, whilst the inverse occurs in MeOH-d4. This partial solvation of the constitutive blocks in the best common solvents may be a contributing factor for the formation of self-assembled structures with varied morphologies and broad particle size distribution on the introduction of a solvent (water) selective for the central poly(N-hydroxyethyl acrylamide) block. With the view of potential biomedical applications, toxicity studies show that the copolymers do not cause appreciable damage to the cell membrane leading to undesirable necrotic cell death, often observed for amphipathic polymeric surfactants, even at relatively high concentrations with 24-hour exposure. We envisage that the middle poly(N-hydroxyethyl acrylamide) block offers the potential to further introduce different surface functionalities, including targeting ligands.
Introduction
Block copolymers self-assemble in a selective solvent and these self-assemblies can assume interesting supramolecular morphologies, including spheres, worms or vesicles. Self-assembly of ABA triblock copolymers in solvents predominantly dissolving their middle B block can bring the A blocks into a core surrounded by the B block forming a loop – a micellar architecture referred to as flower-like micelles in the literature.1,2 Alternatively, the entropic penalty of such an arrangement of the middle B block may be overcome by the formation of a ‘network’ structure with ‘nodes’ of poorly solvated A blocks connected with solvated middle B blocks, leading to the formation of gel-type systems, particularly in a higher concentration regime. AB and ABA block copolymers have been studied for their potential applications in different fields, including biomedical purposes and drug delivery.3,4 The biomedical applications dictate the nature of the B block to be hydrophilic and water soluble, due to the aqueous nature of biological environments. Consequently, most explored ABA copolymers in this field include poly(ε-caprolactone)-b-poly(ethylene glycol)-b-poly(ε-caprolactone), poly(N-isopropylacrylamide)-b-poly(ethylene glycol)-b-poly(N-isopropylacrylamide) or poly(L-lactic acid)-b-poly(ethylene glycol)-b-poly(L-lactic acid), whereby the water soluble middle block is typically based on poly(ethylene glycol), with poly(acrylic acid) or poly(isopropylacrylamide) explored in some studies.5–7 In an interesting study, ABA copolymers were synthesized from cationic amphiphilic polycarbonates, which in a water environment present the cationic B block at the surface of approximately 200–300 nm sized micelles.8Here, we employed poly(N-hydroxyethyl acrylamide) as the water soluble B block, flanked by poly(n-butyl acrylate) as the water insoluble A blocks. To the best of our knowledge, this ABA copolymer structure has not yet been studied. We based the synthesis of (n-BA)m-b-(N-HEA)n-b-(n-BA)m copolymers on a combination of single electron transfer living radical polymerisation (SET-LRP) and photoinduced living radical polymerisation (FT-LRP) as synthetic methods.9–11 This approach exploits the principal advantage of SET-LRP to produce a polymer in a fast manner while retaining chain fidelity, if the optimal balance is maintained between initiation and termination, while the synthesis of poly(n-butyl acrylate) blocks utilizes previously described conditions for photoinduced living radical polymerisation (FT-LRP).12
Common methods for preparing self-assembled systems from block copolymers when these do not directly disperse in a relevant selective solvent, as is the case in the current study, in principle involve mixing of a copolymer solution in a common solvent with a miscible solvent which is selective for one block. In the present study, water represents a selective solvent that preferentially dissolves the poly(N-hydroxyethyl acrylamide) middle A block. We screened a number of solvents and solvent mixtures that are miscible with water to be used as a potential common solvent for (n-BA)m-b-(N-HEA)n-b-(n-BA)m copolymers, with the data for a DMSO/acetone mixture and methanol as the ‘best performers’ upon visual inspection presented here. The cytotoxicity of the copolymers was tested in epithelial cells with the view of their potential application at mucosal surfaces. In vitro testing focuses on the potential mechanism of toxicity to assess if, due to their amphiphilic architecture similar to amphiphilic surfactants, the copolymers are causing cell membrane damage which normally leads to non-desirable necrotic cell death.
The (n-BA)m-b-(N-HEA)n-b-(n-BA)m copolymers offer a potential advantage, relative to ABA copolymers with the PEG middle block, of possible further chemistry on the poly(N-hydroxyethyl acrylamide) block by applying e.g. orthogonal covalent attachment (‘click chemistry’) to introduce targeting surface functionalities.
Experimental
Materials
Diethylene glycol bis(2-bromoisobutyrate) (2f-DEBiB), tris[2-(dimethylamino)ethyl]amine (Me6TREN), propargyl folate and triethylene glycol methacrylate-N3 (TEGMA-N3) were synthesised with >98% purity, as described below.2,2-Bipyridine (bpy, 99%), N,N-dimethylformamide (DMF, anhydrous, 99%), copper(I) bromide (Cu(I)Br, 99%), N-hydroxyethylacrylamide (N-HEA, 97%), dimethylsulfoxide (DMSO, 99%), deuterated solvents like dimethylsulfoxide (DMSO-d6, 99%), chloroform (CDCl3, 99%), and acetone (acetone-d6, 99%) and ethylenediaminetetraacetic acid (EDTA, 99%) were purchased from Sigma-Aldrich. Diethyl ether (Et2O, 99%) and methanol (MeOH, 99%) were purchased from Fisher Scientific. Copper(II) bromide (Cu(II)Br2, 99%), copper(I) chloride (Cu(I)Cl, 99%) and n-butyl acrylate (n-BA, 99%) were purchased from Alfa Aesar. Triethylamine (Et3N, 99%), formaldehyde aqueous solution (37%), tetrahydrofuran (THF, anhydrous, 99%), tris(2-aminoethyl)amine (99%), 2-bromoisobutyryl bromide (98%), diethylene glycol (99%), folic acid (97%) and propargylamine (98%) were purchased from Sigma-Aldrich. Methacryloyl chloride (97%) was purchased from Alfa Aesar. Petroleum ether (pet. ether, 99%), ethyl acetate (EtOAc, 99%), toluene (99%), dichloromethane (DCM, 99%), acetone (99%), and MgSO4 were purchased from VWR International.
Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, 2 mL). The mixture was stirred until complete dissolution (typically 5 min); then the solution was bubbled with Ar for 10 min and finally cannulated into the Schlenk tube containing Me6TREN/Cu0–CuII, previously cooled to 0 °C (t0). The polymerisation was allowed to proceed at 0 °C under stirring. Purification of the resulting polymer reaction mixture was performed by dialysis, using a wetted dried snake dialysis membrane (cut-off 3.5 kDa) for 5 days against 5 L of water, changing the water every 24 hours. To remove copper from the mixture, EDTA was added to the reaction mixture before dialysis, and the dialysis cycles were carried out vs. aqueous EDTA until the disappearance of the green colour from the dialysis bag. For the subsequent cycles, dialysis was performed against DI water. After dialysis, the dialysis tube content was freeze-dried (yield = 200 mg, 79%). Ratio of [2f-DEBiB]:[N-HEA]:[2-CEA]:[Cu(I)Br]:[Me6TREN] = [1]:[36]:[4]:[0.80]:[0.40], target conversion >99%. Quantities used are as follows: 2f-DEBiB (113 mg, 0.28 mmol), Cu(I)Br (32.13 mg, 0.22 mmol), Me6TREN (25.80 mg, 0.11 mmol) and DMSO:H2O (4:1, 2 mL).
:1 v/v, 2 mL). The mixture was stirred until complete dissolution (typically 5 min); then the solution was bubbled with Ar for 10 min and finally cannulated into the Schlenk tube containing Me6TREN/Cu0–CuII, previously cooled to 0 °C (t0). The polymerisation was allowed to proceed at 0 °C under stirring. Purification of the resulting polymer reaction mixture was performed by dialysis, using a wetted dried snake dialysis membrane (cut-off 3.5 kDa) for 5 days against 5 L of water, changing the water every 24 hours. To remove copper from the mixture, EDTA was added to the reaction mixture before dialysis, and the dialysis cycles were carried out vs. aqueous EDTA until the disappearance of the green colour from the dialysis bag. For the subsequent cycles, dialysis was performed against DI water. After dialysis, the dialysis tube content was freeze-dried (yield = 200 mg, 79%). Ratio of [2f-DEBiB]:[N-HEA]:[2-CEA]:[Cu(I)Br]:[Me6TREN] = [1]:[36]:[4]:[0.80]:[0.40], target conversion >99%. Quantities used are as follows: 2f-DEBiB (113 mg, 0.28 mmol), Cu(I)Br (32.13 mg, 0.22 mmol), Me6TREN (25.80 mg, 0.11 mmol) and DMSO:H2O (4:1, 2 mL).
In the further purification step, a copolymer present in DMSO was additionally dialysed against L-cysteine solution for a week, with daily changes of the dialysis medium, with subsequent dialysis against DI water. This was undertaken due to cell toxicity observed in the initial screening of copolymers dialysed against EDTA to remove copper.
:[Me6TREN] = [0.06]:[0.12] w/w, [n-BA]:[DMSO] = 1:3 v/v. Typical quantities used to target 24 units of n-BA (target DP) of (N-HEA)40 (MI) are as follows: MI (256 mg, 0.05 mmol), n-BA (164.08 mg, 1.28 mmol), Cu(II)Br2 (1.25 mg, 0.005 mmol), and Me6TREN (7.741 mg, 0.03 mmol). Depending on the target DP of n-BA, the quantities of n-BA and DMSO were changed accordingly.
The UV-irradiation source used during photoinduced LRP was a commercially available nail gel curing device (λmax ∼ 360 nm) equipped with four 36 W bulbs.
136000 g mol−1). Data were elaborated using Cirrus GPC/SEC 3.0 software.
:1 mixture or methanol, and placed in a wetted snakeskin dialysis tube (cut-off 3.5 kDa). The contents of the dialysis tube were dialyzed against 1 L of deionized water for 3 days, with changes of deionized water every 24 hours.
Statistical analysis
The cell response curves for the MTS assays were fitted using a nonlinear normalized response equation through computer-assisted curve fitting with GraphPad Prism software 7.0. This software was also used to carry out statistical analysis. A comparison between groups was performed using one-way ANOVA followed by Tukey's multiple comparison post hoc test (N = 3, n = 3).Results and discussion
The synthesis of the water soluble B block i.e. (N-HEA)n macromolecular initiator was conducted by applying the Single Electron Transfer Living Radical Polymerisation (SET-LRP) technique. This process is based on the phenomenon that in the presence of an aqueous medium, or a binary mixture of water and polar organic solvents (e.g. DMSO, THF and alcohols),15 Cu(I) species tend to disproportionate to produce Cu(0), which then acts as an activator in the SET reaction, and Cu(II)Br2 which, by reacting with radical species, acts as the radical deactivator (Fig. 1). Typically, polydentate ligands, e.g. Me6TREN, are required to stabilize the copper species involved in the catalytic cycle.10 SET-LRP was adopted in this study instead of ‘conventional’ ARTP, due to its higher ability to maintain the halide chain-end fidelity,16–20 essential to be able to, in the next stage, extend the hydrophilic B block and generate the desired ABA triblock copolymer structure. This technique has been shown to work more efficiently with monomers such as vinyl chloride,21 acrylates22–24 and acrylamides,25,26 compared to those characterized by lower propagation rates, such as methacrylates. A degree of polymerisation (DP) of 40 was targeted for the (N-HEA)n macroinitiator with the view to potentially allow the middle block to form a water-solvated loop when the copolymers are present in a water environment.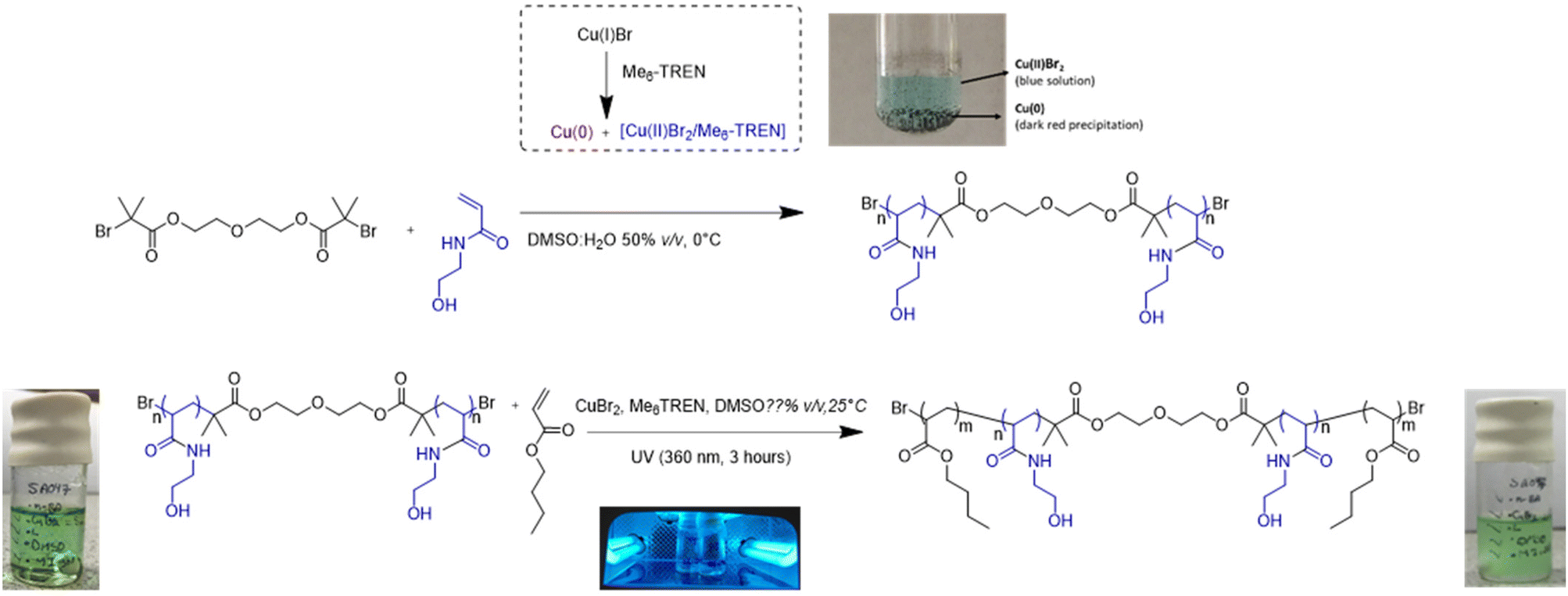 | ||
| Fig. 1 Schematic of synthesis of ABA (n-BA)m-b-(N-HEA)n-b-(n-BA)m copolymers. Synthesis of the (N-HEA)n B block using the macromolecular initiator by single electron transfer-living radical polymerisation (SET-LRP). The growth of (n-BA)m A blocks by photoinduced LRP using Cu(II)Br2 and Me6TREN as catalyst complexes. | ||
Data in Fig. 2 show that a near complete (N-HEA)n conversion was achieved within approximately 30 minutes. Very high (n-BA)m conversion was also accomplished in all chain extension reactions, yielding polymers with low molecular weight distributions, although it should be noted that DMF was used as an elution solvent in SEC.
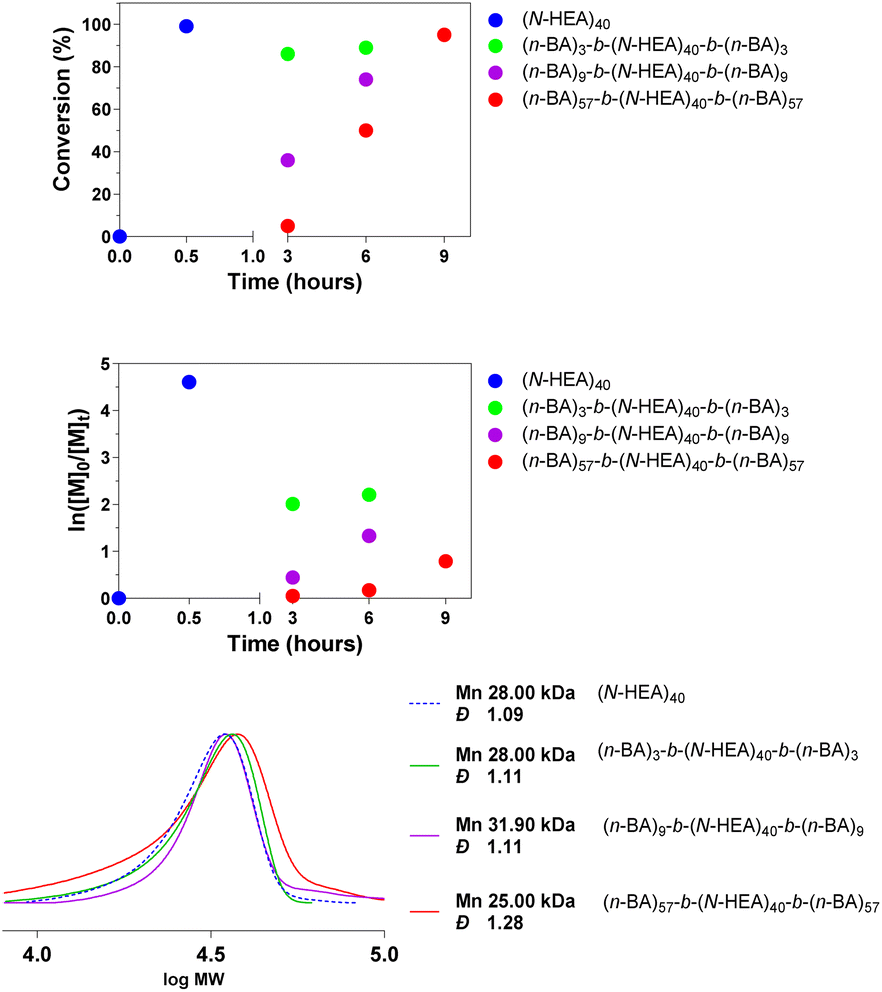 | ||
| Fig. 2 Conversion to (n-BA)m-b-(N-HEA)40-b-(n-BA)m copolymers of the precursor (N-HEA)40 (top). SEC trace of the synthesized copolymers recorded in DMF (with LiBr 1%) (bottom). | ||
Attributes of the synthesized copolymers are summarized in Table 1. FTIR analysis was applied to confirm the presence of the main functional groups and block composition of the synthesized copolymers27 (Fig. 3). More specifically, for (n-BA)m-b-(N-HEA)40-b-(n-BA)m copolymers, functional groups are: amide (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1680–1630 cm−1, stretching, and N–H at 1640–1550 cm−1, bending), ester (CO at 1750–1730 cm−1, stretching), and alkane (3000–2850 cm−1, stretching). The analysis of FTIR spectra revealed the increased relative intensity of alkane (2875 cm−1, stretching) and ester (1730 cm−1 stretching) signals with the increasing length of the poly(n-butyl acrylate) hydrophobic blocks i.e., in order of (n-BA)57-b-(N-HEA)40-b-(n-BA)57 > (n-BA)9-b-(N-HEA)40-b-(n-BA)9 > (n-BA)3-b-(N-HEA)40-b-(n-BA)3.
O at 1680–1630 cm−1, stretching, and N–H at 1640–1550 cm−1, bending), ester (CO at 1750–1730 cm−1, stretching), and alkane (3000–2850 cm−1, stretching). The analysis of FTIR spectra revealed the increased relative intensity of alkane (2875 cm−1, stretching) and ester (1730 cm−1 stretching) signals with the increasing length of the poly(n-butyl acrylate) hydrophobic blocks i.e., in order of (n-BA)57-b-(N-HEA)40-b-(n-BA)57 > (n-BA)9-b-(N-HEA)40-b-(n-BA)9 > (n-BA)3-b-(N-HEA)40-b-(n-BA)3.
 | ||
| Fig. 3 FTIR spectra of the synthesized (n-BA)m-b-(N-HEA)40-b-(n-BA)m copolymers and starting (N-HEA)40 macroinitiator. The copolymers demonstrated similar transmittance, apart from (n-BA)57-b-(N-HEA)40-b-(n-BA)57, (Mn,theo 19.60 kDa), which shows stronger transmittance in the ester region (CO at 1750–1730 cm−1, stretching). | ||
| Compositiona | t.DPb [M] | Convc (%) |
M
n(theo)d (kDa) |
(BA)m:(N-HEA)x:(BA)m ratio |
M n (1H NMR)f (kDa) | M n (GPC)e (kDa) | Đ |
|---|---|---|---|---|---|---|---|
| a Number of units in each block of the ABA copolymer was calculated (t.DP × fractional conversion calculated by 1H NMR) assuming 100% initiator efficiency.28. b t.DP is the target degree of polymerisation relative to initiator [2f-DBiB] i.e., targeted number of monomeric units (M). c Conv (%) is the percent of the monomer converted to the polymer, calculated by 1H NMR. d M n theo is the theoretical number-average molecular weight calculated as follows = [((t.DP of M × Mwt of monomer) × fractional conversion] + 404.10 g mol−1. e Determined by SEC using DMF as the eluent (+1% LiBr). The system was calibrated using a PMMA standard. f Calculated from 1H NMR of the (co)polymer in MeOH-d4. g Calculated from 1H NMR of the (co)polymer in a mixture of DMSO-d6/CDCl3. | |||||||
| (N-HEA)40 initiator | 40 | >99 | 5.00 | 1.0f | 10.00 | 28.40 | 1.09 |
| (n-BA)3-b-(N-HEA)40-b-(n-BA)3 | 10 | 89 | 6.50 | 0.11:1.0:0.11f |
12.40 | 28.20 | 1.11 |
| (n-BA)9-b-(N-HEA)40-b-(n-BA)9 | 24 | 76 | 8.40 | 0.5:1.0:0.5g |
73.00 | 31.80 | 1.11 |
| (n-BA)57-b-(N-HEA)40-b-(n-BA)57 | 120 | 95 | 19.60 | 1.57:1.0:1.57f |
51.00 | 25.20 | 1.28 |
Initially, 1H NMR spectra of (n-BA)m-b-(N-HEA)40-b-(n-BA)m copolymers were obtained in a DMSO-d6/acetone-d6 5:1 mixture (Fig. 4). The addition of a less polar solvent, acetone-d6, to DMSO-d6 was aimed to promote solvation of more hydrophobic (n-BA)m blocks (ENT 0.356 and 0.460 for acetone and DMSO, respectively).29 This was based on the observation during copolymer synthesis that the starting macroinitiator, (N-HEA)40, dissolves initially in DMSO as a clear solution, but the reaction mixture becomes gradually turbid, i.e., indicating decreased solubility, with the growth of (n-BA)m blocks (as illustrated by the image on the right in Fig. 1). As shown in Fig. 4, the distinguished signals for (N-HEA)40 are –NH and –OH (labelled a and b), appearing as broad peaks at ∼7.75–7.35 ppm and ∼5.13–4.70 ppm, respectively. For the (n-BA)m moiety, prominent signals observed are –COOCH2 and –CH2CH3 as broad peaks at ∼4.20–3.75 ppm and ∼0.95–0.70 ppm, respectively. Regarding copolymers, (n-BA)3-b-(N-HEA)40-b-(n-BA)3 shows a small –CH2CH3 peak with the absence of an ester peak at 3.75 ppm, which might be due to the relatively short (n-BA)3 block length. The (n-BA)9-b-(N-HEA)40-b-(n-BA)9 spectrum, on the other hand, reveals both –OH peaks at ∼5.13–4.70 ppm of (N-HEA)40, as well as a broad peak at ∼3.75 ppm which resonates for (n-BA)m ester (–COOCH2), with a high broad peak at ∼0.70 ppm for the (n-BA)m methyl group (–CH2CH3). For the (n-BA)57-b-(N-HEA)40-b-(n-BA)57 copolymer, smaller peaks were observed for (n-BA)m ester (–COOCH2) and methyl (–CH2CH3) groups, presumably due to the vicinity of a large peak of water which might be present due to the hygroscopicity of DMSO.
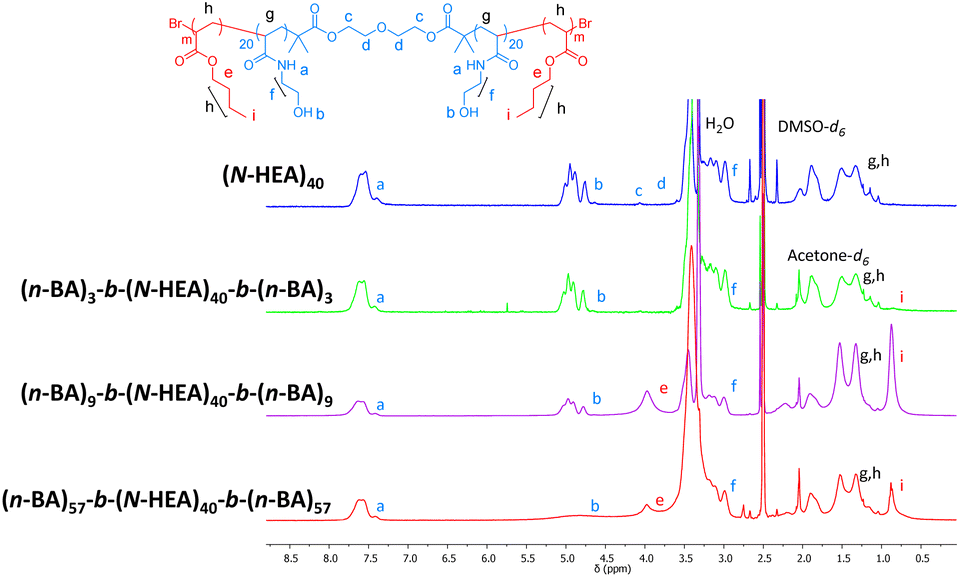 | ||
| Fig. 4
1H NMR spectra of the synthesized (n-BA)m-b-(N-HEA)40-b-(n-BA)m copolymers in the DMSO-d6/acetone-d6 mixture. Spectrum of the (N-HEA)40 macroinitiator recorded in DMSO-d6 and spectra of the copolymers in the DMSO-d6/acetone-d6 mixture at a ratio of 5:1 v/v. | ||
Fig. 4 further shows the differences in the NMR spectra for (n-BA)9-b-(N-HEA)40-b-(n-BA)9 and (n-BA)57-b-(N-HEA)40-b-(n-BA)57 copolymers when present in a relatively polar DMSO-d6/acetone-d6 solvent mixture. For (n-BA)9-b-(N-HEA)40-b-(n-BA)9, peaks for both structural components, –(N-HEA) and (n-BA), are distinguishable in the spectrum, indicating that they are, to some extent, solvated and, if the copolymer forms some supramolecular structures, both structural components are fairly solvated.
In the case of the (n-BA)57-b-(N-HEA)40-b-(n-BA)57 copolymer, an (N-HEA) relevant p-NH peak at ∼7.75–7.35 ppm is present in the spectrum, while peaks at ∼5.13–4.70 ppm for –OH and (n-BA)m relevant ester (–COOCH2) peak at ∼4.20–3.75 ppm both appear obstructed. An (n-BA)m relevant –CH2CH3 peak at ∼0.95–0.70 ppm is significantly reduced, relative to (n-BA)9-b-(N-HEA)40-b-(n-BA)9, despite substantially larger (n-BA) blocks. The latter would indicate that, with an increase in their size, the (n-BA)m blocks are less solvated in the DMSO-d6/acetone-d6 mixture pointing to the presence of the copolymer structure where (n-BA)57 end-chains are forming a less solvated regions in the presence of more solvated (N-HEA)40 moieties.
1H NMR spectra were also recorded in MeOH-d4 (Fig. 5). In these spectra, the amide peak (–NH, δ 7.90 ppm) of the hydrophilic (N-HEA)40 macroinitiator appears smaller than expected. Moreover, hydroxyl –OH groups are not visible in the spectrum, likely attributable to deuterium exchange with MeOH-d4.
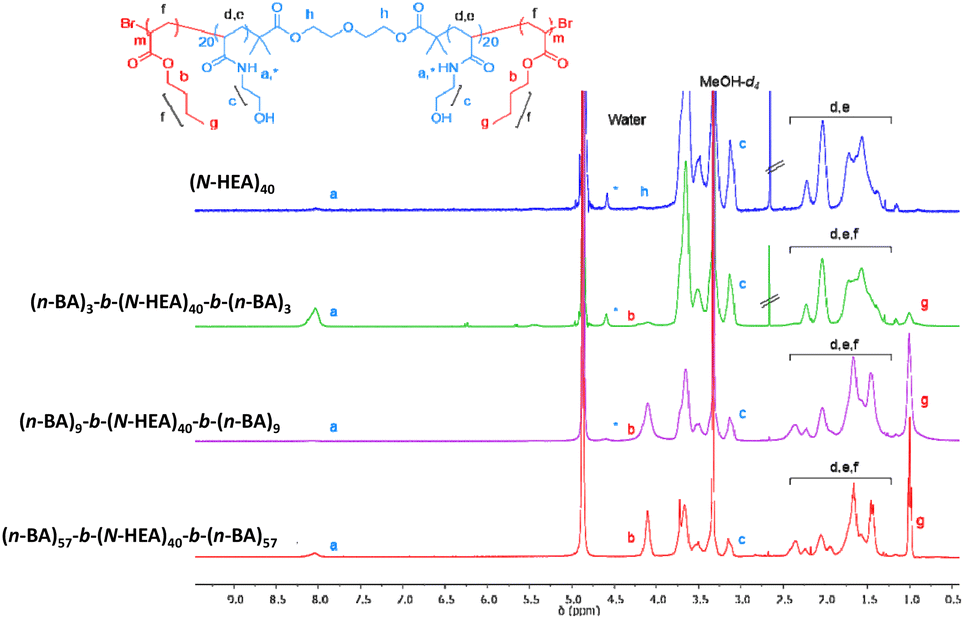 | ||
| Fig. 5 1H NMR spectra of the synthesized (n-BA)m-b-(N-HEA)40-b-(n-BA)m copolymers and starting (N-HEA)40 macroinitiator recorded in MeOH-d4. Concentration 3 mg/0.6 ml w/v. | ||
An interesting peak was observed at ∼4.50 ppm, which might be referred to as either another shift of the amide peak (–NH) of (N-HEA)40, or resonance of hydrogen bonding between solvent molecules and amide.28–30 This small peak is present in (n-BA)3-b-(N-HEA)40-b-(n-BA)3 and (n-BA)9-b-(N-HEA)40-b-(n-BA)9, but not in (n-BA)57-b-(N-HEA)40-b-(n-BA)57. This corroborates with the observation of increased intensity in the ester peak (–COOCH2, δ ∼ 4.00 ppm) and methyl group (–CH2CH3, δ 0.90 ppm) as (n-BA)m blocks increase in length. This would indicate that in MeOH-d4, a solvent preferential for (n-BA)m, these solvated blocks might be masking (N-HEA)40 from interaction with the MeOH-d4 solvent. In other words, with an increase in n-BA blocks, the structures formed in MeOH-d4 may comprise well-solvated flank (n-BA)m blocks and partially solvated (N-HEA)40 areas, the structure inverse to the situation in the DMSO-d6/acetone-d6 mixture.
As 1H NMR analyses in both solvent media used (DMSO-d6/acetone-d6 mixture in Fig. 4 and MeOH-d4 in Fig. 5) reveal the evidence of incomplete solvation of (N-HEA)40 or (n-BA)m copolymer blocks, the actual degree of polymerisation for each block was calculated from monomer conversion (Table 1).
The analysis of the glass transition temperature, Tg, of the copolymers is summarized in Fig. 6. A Tg value of 123.23 °C was determined for the starting macroinitiator, (N-HEA)40, and indicates that, at approximately room temperatures, the (N-HEA)40 chain motion is restricted, i.e., the material is glassy. Conversely, Tg values for poly(n-butyl acrylate) homopolymers, (n-BA)12 and (n-BA)60, were determined to be −52.89 °C and −50.36 °C, respectively, which is consistent with previous reports.31 These Tg values indicate that under experimental conditions close to room temperature, (n-BA)m chains are mobile and the material is rubber in nature.
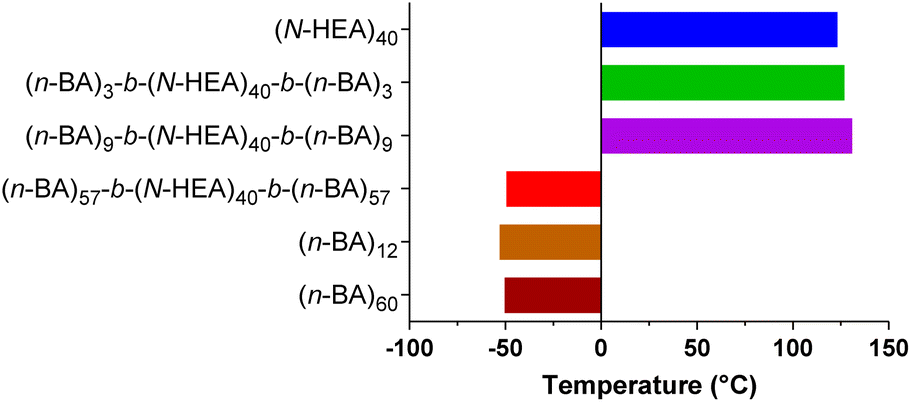 | ||
| Fig. 6 Glass transition temperature (Tg) of the synthesized (n-BA)m-b-(N-HEA)40-b-(n-BA)m copolymers. Comparisons were made with poly(hydroxy acrylamide) and poly(butyl acrylate) homopolymers. | ||
Regarding ABA copolymers, Tg for (n-BA)3-b-(N-HEA)40-b-(n-BA)3 and (n-BA)9-b-(N-HEA)40-b-(n-BA)9 are at 126.9 °C and 131.0 °C, respectively, values close to Tg at 123.23 °C for the starting (N-HEA)40 macroinitiator. This would indicate that, in a dry state, the chain motion of these copolymers is restricted and that their behaviour is predominantly influenced by the (N-HEA)40 block. Inversely, (n-BA)57-b-(N-HEA)40-b-(n-BA)57, the copolymer with the (n-BA)m blocks larger than (N-HEA)40 (Mn,theo 14.6 kDa vs. 5.0 kDa, respectively), has a Tg of −49.38 °C, which is closer to that of (n-BA)60 at −50.36 °C. This might indicate that the copolymer chains are relatively mobile at room temperature, i.e., copolymer behaviour is dominated by the (n-BA)57 blocks, important information for storage conditions of potential drug delivery systems based on these copolymers.
It should be noticed that only one glass transition peak was observed in DSC thermograms for the (n-BA)m-b-(N-HEA)n-b-(n-BA)m copolymers, including (n-BA)57-b-(N-HEA)40-b-(n-BA)57 (ESI, Fig. S1†). This would indicate that the poly(n-BA) and poly(N-HEA) moieties in these copolymers are miscible in the dry state. A similar observation was observed for (n-BA)m-g-polystyrene copolymer series where with an increase in the number of grafted polystyrene chains, the Tg of (n-BA)m was increased.32 By comparing the Tg values of (n-BA)3-b-(N-HEA)40-b-(n-BA)3, (n-BA)9-b-(N-HEA)40-b-(n-BA)9 and (n-BA)57-b-(N-HEA)40-b-(n-BA)57 at 123.23, 126.96 and −49.38 °C, respectively, a clear relationship emerges between the length of the poly(n-BA) blocks and Tg, whereby as the (n-BA)m block length is increased, the glass-transition temperature shifts towards its glass-transition temperature indicating that (n-BA)57-b-(N-HEA)40-b-(n-BA)57 would show soft and elastic behaviour at room temperature.
Fig. 7 shows hydrodynamic particle size distribution profiles for (n-BA)m-b-(N-HEA)40-b-(n-BA)m copolymer assemblies obtained by different protocols; addition of a solvent selective for the B block (water) into the copolymer present in methanol solvent, or dialysis of copolymers from either methanol or the DMSO:acetone mixture against the solvent selective for the B block (water). The profiles, in general, show the average particle size in the 250–450 nm range, with relatively broad particle size distributions and high polydispersities. Inspecting the distribution profiles obtained, it is difficult to distinguish possible trends and impacts on the particle size distribution of either a formulation protocol of selective solvent introduction, common solvent quality, i.e., the use of methanol or the DMSO/acetone mixture, or different copolymer compositions. It, however, appears that the (n-BA)57-b-(N-HEA)40-b-(n-BA)57 copolymer forms smaller self-assemblies, relative to other copolymers tested. Referring here to 1H NMR spectra in MeOH-d4 (Fig. 5), where the structures formed comprise well-solvated (n-BA)m blocks and partially solvated (N-HEA)40 blocks, the assembly reorganization would need to occur on the addition of a solvent (water) selective for (N-HEA) blocks, which differs from the scenario where copolymers in the DMSO-d6/acetone-d6 mixture form a less solvated core of (n-BA)m blocks in the presence of more solvated (N-HEA)40 blocks; the latter structure would not need to undergo reorganization on water addition.
 | ||
| Fig. 7 Hydrodynamic particle size distribution profiles for the self-assembled structure formed from (n-BA)3-b-(N-HEA)40-b-(n-BA)3, (n-BA)9-b-(N-HEA)40-b-(n-BA)9 and (n-BA)57-b-(N-HEA)40-b-(n-BA)57 copolymers by applying different protocols. Protocols are addition of the (N-HEA)40 selective solvent (water) to the copolymer in methanol, followed by evaporation (graphs in the left column); dialysis of the copolymer in methanol against water as the (N-HEA)40 selective solvent (middle column), and dialysis of the copolymer in a DMSO/acetone mixture against the (N-HEA)40 selective solvent (right column). Data show particle size distribution (intensity) profiles from dynamic light scattering measurements for one batch per protocol. PDI is the polydispersity index. Concentration of polymers in MetOH and the DMSO/acetone 5:1 mixture at 0.5 mg mL−1. | ||
The morphologies of (n-BA)m-b-(N-HEA)40-b-(n-BA)m self-assembled structures assessed by transmission electron microscopy (TEM) are presented in Fig. 8. The images depict different morphologies and point to their dependence on both, the copolymer composition and assembly protocol. For example, for the (n-BA)3-b-(N-HEA)40-b-(n-BA)3 copolymer present in methanol, the addition of water as the solvent selective for the central (N-HEA)40 block resulted in clusters of discrete but irregular (somewhat angular) species around 40–50 nm in size, while its dialysis from the DMSO/acetone mixture against water produced discrete, spherical species of approximately 50–200 nm in size with interesting ‘hairy protrusions’. The (n-BA)9-b-(N-HEA)40-b-(n-BA)9 copolymer produced a significant population of elongated self-assemblies around 30–40 nm in size, not observed for other copolymers; the (n-BA)57-b-(N-HEA)40-b-(n-BA)57 copolymer produced around 40–50 nm sized assemblies regardless of the protocol, but in the case of dialysis from the DMSO/acetone protocol, these appear to be connected with ‘thick bridges’ between individual species. Considering the latter observation, TEM images were taken on ‘dried samples’ prepared at room temperature. Taking into account the DSC data (Fig. 6), in their dry state, the copolymers will possess different chain mobilities, with (n-BA)57-b-(N-HEA)40-b-(n-BA)57 copolymer behaviour dominated by relatively mobile chains of (n-BA)57 blocks (Tg −49.38 °C). This may result in a formation of the observed ‘thick bridges’ (Fig. 8) during TEM sample preparation, however, it does not provide an explanation for differences in the morphologies of (n-BA)57-b-(N-HEA)40-b-(n-BA)57 self-assemblies formed by two different protocols. In contrast, one would not expect changes in the morphology of (n-BA)3-b-(N-HEA)40-b-(n-BA)3 and (n-BA)9-b-(N-HEA)40-b-(n-BA)9 assemblies during TEM sample preparation due to a dominant effect of high Tg of the (N-HEA)40 block in these copolymers (Fig. 6).
 | ||
| Fig. 8 Transmission electron microscopy (TEM) images of self-assembled structures formed from (n-BA)3-b-(N-HEA)40-b-(n-BA)3, (n-BA)9-b-(N-HEA)40-b-(n-BA)9 and (n-BA)57-b-(N-HEA)40-b-(n-BA)57 copolymers by applying different protocols. Protocols are addition of water as the (N-HEA)40 selective solvent to copolymers in methanol followed by evaporation (images in left two columns), and dialysis of copolymers in a DMSO/acetone 5:1 mixture against the (N-HEA)40 selective solvent (images in right two columns). Images at different magnifications (as indicated in the scale bar) and different fields of view are shown. | ||
We observed profound discrepancies in particle size analysis obtained from dynamic light scattering (Fig. 7) and particle sizes seen in TEM microscopy images (Fig. 8). At this stage, it is not clear what may be the reason(s) for this. For some cases, one may suggest that this might be due to the presence of aggregated or ‘bridged’ polymeric self-assemblies, e.g. for (n-BA)3-b-(N-HEA)40-b-(n-BA)3 formed from methanol on the addition of the selective solvent, or for (n-BA)57-b-(N-HEA)40-b-(n-BA)57 formed by dialysis against the selective solvent; however, this does not provide an appropriate explanation for all the studied systems.
Observations in NMR spectra (Fig. 4 and 5) may contribute to the discussion on morphologies and particle size of (n-BA)m-b-(N-HEA)40-b-(n-BA)m self-assemblies. The NMR spectra were recorded in the same common solvents as used in the self-assembly formation, and indicate that in the two common solvent systems used (DMSO/acetone and methanol; selected as the ‘best performing’ in visual observations of solubility) ‘pre-assembled’ structures exist even prior to the addition of a solvent selective for the (N-HEA)40 middle B block. The structures of these ‘pre-assemblies’ could remain relatively unchanged during the introduction in the selective solvent in the case when DMSO/acetone is used as a common solvent in which copolymer chains form well-solvated (N-HEA)40 and poorly solvated (n-BA)m regions. Alternatively, they need to undergo structural reorganisation on the introduction of the selective solvent for (N-HEA)40 to copolymers initially present in methanol where solvated (n-BA)m and poorly solvated (N-HEA)40 regions are present.33 Furthermore, one needs to consider the tendency of block copolymers to become kinetically trapped in non-equilibrium states, meaning that the structures they adopt depend on the processing route taken,34 as it would be the case in this study (Fig. 8).
It should be noted that the addition of a solvent preferential for the (N-HEA)40 B-block into copolymers present in a common solvent in all cases resulted in the formation of particulate self-assemblies, rather than hydrophobic blocks forming interconnected domains separated by a hydrophilic B block that could result in the formation of a gel structure. This would indicate that for (n-BA)m-b-(N-HEA)40-b-(n-BA)m ABA copolymers, the concentrations used in their self-assembly process are relatively low, resulting in isolated particles, where the two hydrophobic A-blocks preferentially reside in the core, and the mid B-block will create a hydrophilic surface,33 as would also be indicated from the observed colloidal stability of their suspensions to aggregation.
In further analysis of the synthesized materials, the cellular toxicity of (n-BA)m-b-(N-HEA)40-b-(n-BA)m copolymers was initially assessed by applying LDH release and MTS assays to evaluate cell membrane integrity and cellular metabolic activity, respectively. The data in Fig. 9 illustrate that on 24 hour exposure, the applied copolymers did not cause statistically significant damage to the cell plasma membrane that would lead to the release of intracellular LDH enzymes. The metabolic MTS assay demonstrates that the exposure to the copolymers results in molecular structure-dependent cellular toxicity, with relatively high calculated 50% lethal dose (LD50) values (shown as a bar chart in the inset) for (n-BA)3-b-(N-HEA)40-b-(n-BA)3, (n-BA)9-b-(N-HEA)40-b-(n-BA)9 and (n-BA)57-b-(N-HEA)40-b-(n-BA)57 of 163.7 ± 6.9, 236.9 ± 8.0 and 625.2 ± 25.5 μg mL−1, respectively, all of which differ significantly from each other (p > 0.0001), and indicate relatively low toxicity of the copolymers to epithelial cells.
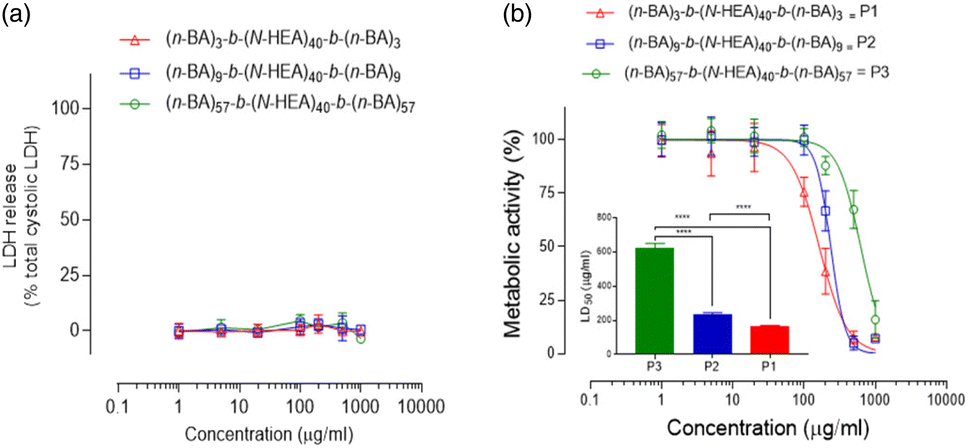 | ||
| Fig. 9 Toxicity profiles of the synthesized (n-BA)m-b-(N-HEA)40-b-(n-BA)m copolymers. (a) Plasma membrane integrity of Calu-3 cells following 24 hour exposure to compounds determined by the LDH release assay. Data presented as mean ± SD, and represent triplicates from three independent experiments. (b) Metabolic activity of Calu-3 cells following 24 hour exposure to copolymers determined by the MTS assay. Data presented as mean ± SD and represent triplicate samples from three independent experiments. Statistical significance was determined by one-way ANOVA followed by Tukey's post hoc test (****, p < 0.0001). | ||
The data hence indicate that a metabolic decline occurs in the absence of cell membrane damage. This suggests that the copolymers do not cause perturbations of the cell plasma membrane, as typically seen for amphipathic surfactants which damage the plasma membrane by the insertion of their hydrophobic moiety.35 Thus, an alternative toxicity mechanism may have occurred.
We hence investigated a potential mechanism(s) of cell death induced by the copolymers by assessing cellular ROS generation, effector caspase-3/7 activation, and nuclear morphology and permeability (Fig. 10). The application of copolymers at concentrations above determined LD50 (from Fig. 9) resulted in the significant generation of ROS, with all copolymers inducing ROS levels 2-fold greater than the untreated control (Fig. 10a). Moreover, at these concentrations, the copolymers generated significant levels of activated caspases-3/7, between 2 to 3-fold greater than untreated cells (Fig. 10b). Fluorescence microscopy imaging of Hoechst 33342 and PI-stained cells, presented in Fig. 10c, demonstrates that application of the copolymers above their LD50 concentrations induces changes in nucleus morphology, observed via Hoechst staining, and results in increases in nuclear permeability, as indicated by positive PI staining. The nuclei of the treated cells exhibit signs of chromatid condensation, nuclear fragmentation, and the presence of apoptotic bodies, which are well-known pro-apoptotic features, and observed following exposure to all copolymers and the apoptotic control (staurosporine treatment). Furthermore, it was noted that positive PI nuclear staining was not observed in all cells treated with the copolymers or staurosporine, but predominately in those showing pro-apoptotic nuclear morphologies. Together, these data suggest that cells, under the conditions tested, underwent late apoptosis.
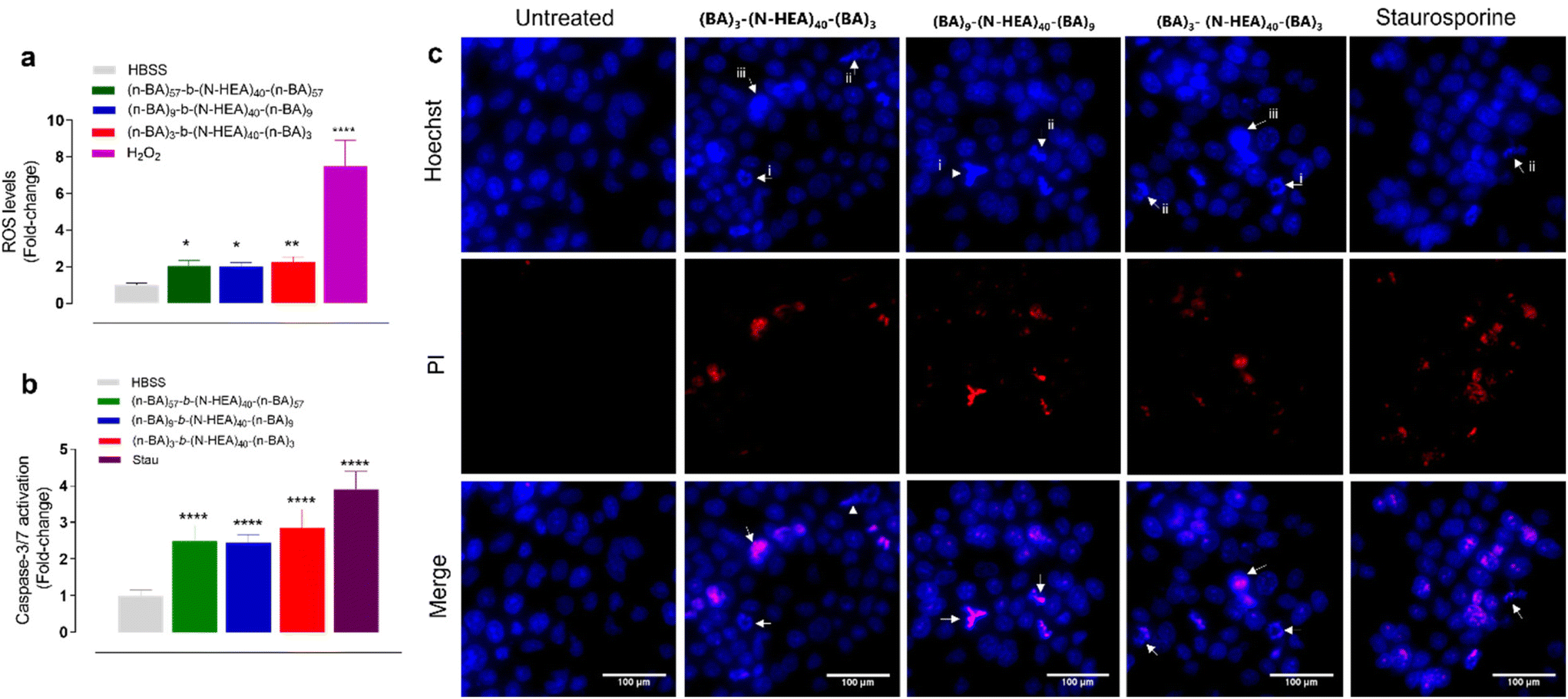 | ||
| Fig. 10 Characteristic features of the synthesized (n-BA)m-b-(N-HEA)40-b-(n-BA)m toxicity on Calu-3 cells. (a) Intracellular ROS levels were assessed using the CM-H2DCFDA probe. H2O2 was used as the positive ROS-inducing control and applied at 1 mM. (b) Activation of pro-apoptotic caspase-3/7 was measured using the CellEvent probe. Staurosporine (Stau) was used as the positive apoptotic control and applied at 10 μM. (c) Images of Hoechst/propidium iodide (PI) staining of nuclei. White arrows indicate the presence of apoptotic features including (i) nuclear fragmentation, (ii) apoptotic bodies, and (iii) chromatin condensation. The scale bar is 100 μm. (a–c) Copolymers were incubated for 24 hours in a concentration with their LD50 values. (a and b) Data presented as mean ± S.D (n = 3). Statistical significance was determined by one-way ANOVA followed by Tukey's post hoc test (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001). | ||
The toxicity at higher concentrations thus appears to arise from the initiation of cellular apoptosis, i.e., by decreasing mitochondrial activity. In comparison, Pluronics®, poly(ethylene oxide)-co-poly(propylene oxide)-co-poly(ethylene oxide), a linear triblock copolymer, appears to exert its cytotoxic activity by interrupting cell membrane integrity and mitochondrial activity.36,37 Considering the differences in chain composition and properties between (n-BA)m-b-(N-HEA)40-b-(n-BA)m and Pluronics®, including their ‘opposite’ block sequences, these all may contribute to the observed differences in the toxicity mechanism.
Conclusions
This study applies a combination of SET-LRP and photoinduced LRP techniques to synthesize block copolymers – (n-BA)m-b-(N-HEA)n-b-(n-BA)m – containing poly(N-hydroxyethyl acrylamide) as a water soluble middle A block, the structure that, to the best of our knowledge, has not been previously studied. NMR analysis of the copolymers in their common solvents (DMSO-d6/acetone-d6 mixture and MeOH-d4) indicates the presence of partially solvated poly(n-butyl acrylate) blocks and a well-solvated poly(N-hydroxyethyl acrylamide) block in a DMSO-d6/acetone-d6 mixture, whilst in MeOH the presence of well-solvated (n-BA)m B blocks is seen with partially solvated (N-HEA)40 areas, inverse to the solvation in a DMSO-d6/acetone-d6 mixture.This partial solvation of the constitutive blocks in the best common solvents, together with differences in the tested copolymer composition, i.e., length of the B blocks, may contribute to the formation of self-assembled structures with varied morphologies and broad size distribution on the introduction of a miscible solvent (water) selective for the poly(N-hydroxyethyl acrylamide) block. It is interesting to note that despite their amphiphilic architecture, the copolymers do not cause appreciable damage to the cell membrane, typical of amphipathic materials, even on 24 hour exposure, and possess relatively high LD50 in the range of 160–625 μg mL−1. The toxicity mechanism observed upon cellular exposure to the copolymers at LD50 concentrations involves apoptosis-mediated cell death. We envisage that the poly(N-hydroxyethyl acrylamide) block as the middle block in ABA copolymers can be further explored to, via e.g. ‘click-chemistry’, introduce different surface functionalities to the assemblies formed.
Conflicts of interest
There are no conflicts to declare.References
- N. P. Balsara, M. Tirrell and T. P. Lodge, Micelle Formation of BAB Triblock Copolymers in Solvents That Preferentially Dissolve the A Block, Macromolecules, 1991, 24(8), 1975–1986, DOI:10.1021/ma00008a040.
- H. Wada, Y. Kitazawa, S. Kuroki, Y. Tezuka and T. Yamamoto, NMR Relaxometry for the Thermal Stability and Phase Transition Mechanism of Flower-like Micelles from Linear and Cyclic Amphiphilic Block Copolymers, Langmuir, 2015, 31(32), 8739–8744, DOI:10.1021/acs.langmuir.5b01902.
- A. J. De Graaf, I. I. Azevedo Próspero Dos Santos, E. H. E. Pieters, D. T. S. Rijkers, C. F. Van Nostrum, T. Vermonden, R. J. Kok, W. E. Hennink and E. Mastrobattista, A Micelle-Shedding Thermosensitive Hydrogel as Sustained Release Formulation, J. Controlled Release, 2012, 162(3), 582–590, DOI:10.1016/j.jconrel.2012.08.010.
- M. A. Moretton, C. Hocht, C. Taira and A. Sosnik, Rifampicin-Loaded ‘Flower-like’ Polymeric Micelles for Enhanced Oral Bioavailability in an Extemporaneous Liquid Fixed-Dose Combination with Isoniazid, Nanomedicine, 2014, 9(11), 1635–1650, DOI:10.2217/nnm.13.154.
- C. Bruce, I. Javakhishvili, L. Fogelström, A. Carlmark, S. Hvilsted and E. Malmström, Well-Defined ABA- and BAB-Type Block Copolymers of PDMAEMA and PCL, RSC Adv., 2014, 4(49), 25809–25818, 10.1039/C4RA04325A.
- G. Bonacucina, M. Cespi, G. Mencarelli, G. Giorgioni and G. F. Palmieri, Thermosensitive Self-Assembling Block Copolymers as Drug Delivery Systems, Polymer, 2011, 3(2), 779–811, DOI:10.3390/polym3020779.
- M. Najafi, N. Kordalivand, M. A. Moradi, J. Van Den Dikkenberg, R. Fokkink, H. Friedrich, N. A. J. M. Sommerdijk, M. Hembury and T. Vermonden, Native Chemical Ligation for Cross-Linking of Flower-Like Micelles, Biomacromolecules, 2018, 19(9), 3766–3775, DOI:10.1021/acs.biomac.8b00908.
- F. Nederberg, Y. Zhang, J. P. K. Tan, K. Xu, H. Wang, C. Yang, S. Gao, X. D. Guo, K. Fukushima, L. Li, J. L. Hedrick and Y.-Y. Yang, Biodegradable Nanostructures with Selective Lysis of Microbial Membranes, Nat. Chem., 2011, 3(5), 409–414, DOI:10.1038/nchem.1012.
- A. Simula, G. Nurumbetov, A. Anastasaki, P. Wilson and D. M. Haddleton, Synthesis and Reactivity of Homotelechelic Polymers by Cu(0)-Mediated Living Radical Polymerisation, Eur. Polym. J., 2015, 62, 294–303, DOI:10.1016/j.eurpolymj.2014.07.014.
- Q. Zhang, P. Wilson, Z. Li, R. Mchale, J. Godfrey, A. Anastasaki, C. Waldron and D. M. Haddleton, Aqueous Copper-Mediated Living Polymerisation: Exploiting Rapid Disproportionation of CuBr with Me 6 TREN, J. Am. Chem. Soc., 2013, 135(19), 7355–7363 CrossRef CAS PubMed https://pubmed.ncbi.nlm.nih.gov/23597244/ .
- S. R. Samanta, H. Sun, A. Anastasaki, M. Haddleton and V. Percec, Self-activation and activation of Cu(0) wire for SET-LRP mediated by fluorinated alcohols, Polym. Chem., 2014, 5, 89–95 RSC https://pubs.rsc.org/en/content/articlelanding/2014/py/c3py01007d .
- A. Anastasaki, V. Nikolaou, A. Simula, J. Godfrey, M. Li, G. Nurumbetov, P. Wilson and D. M. Haddleton, Expanding the Scope of the Photoinduced Living Radical Polymerisation of Acrylates in the Presence of CuBr2 and Me6-Tren, Macromolecules, 2014, 47(12), 3852–3859, DOI:10.1021/ma500787d.
- A. Simula, V. Nikolaou, A. Anastasaki, F. Alsubaie, G. Nurumbetov, P. Wilson, K. Kempe and D. M. Haddleton, Synthesis of Well-Defined αalfa,omega-Telechelic Multiblock Copolymers in Aqueous Medium: In Situ Generation of α-Diols, Polym. Chem., 2015, 6(12), 2226–2233, 10.1039/C4PY01802H.
- C. MacDonald, W. Lyzenga, D. Shao and R. U. Agu, Water-Soluble Organic Solubilizers for in Vitro Drug Delivery Studies with Respiratory Epithelial Cells: Selection Based on Various Toxicity Indicators, Drug Delivery, 2010, 17(6), 434–442, DOI:10.3109/10717541003777548.
- G. Lligadas, B. M. Rosen, M. J. Monteiro and V. Percec, Solvent Choice Differentiates SET-LRP and Cu-Mediated Radical Polymerisation with Non-First-Order Kinetics, Macromolecules, 2008, 41(22), 8360–8364, DOI:10.1021/ma801774d.
- S. R. Samanta, H.-J. Sun, A. Anastasaki, D. M. Haddleton and V. Percec, Self-activation and activation of Cu(0) wire for SET-LRP mediated by fluorinated alcohols, Polym. Chem., 2014, 5, 89–95 RSC https://pubs.rsc.org/en/content/articlelanding/2014/py/c3py01007d .
- C. Boyer, A. H. Soeriyadi, B. Zetterlund and M. R. Whittaker, Synthesis of Complex Multiblock Copolymers via a Simple Iterative Cu(0)-Mediated Radical Polymerisation Approach, Macromolecules, 2011, 44, 8028–8033, DOI:10.1021/ma201529j.
- F. Nyström, A. H. Soeriyadi, C. Boyer, P. B. Zetterlund and M. R. Whittaker, End-Group Fidelity of Copper(0)-Meditated Radical Polymerisation at High Monomer Conversion: An ESI-MS Investigation, J. Polym. Sci., Part A: Polym. Chem., 2011, 49(24), 5313–5321, DOI:10.1002/pola.25010.
- M. E. Levere, N. H. Nguyen and V. Percec, No Reduction of CuBr2 during Cu(0)-Catalyzed Living Radical Polymerisation of Methyl Acrylate in DMSO at 25 °C, Macromolecules, 2012, 45, 8267–8274, DOI:10.1021/ma301547n.
- G. Lligadas and V. Percec, Synthesis of Perfectly Bifunctional Polyacrylates by Single-Electron-Transfer Living Radical Polymerisation, J. Polym. Sci., Part A: Polym. Chem., 2007, 45(20), 4684–4695, DOI:10.1002/pola.22307.
- T. Hatano, B. M. Rosen and V. Percec, SET-LRP of Vinyl Chloride Initiated with CHBr3 and Catalyzed by Cu(0)-Wire/TREN in DMSO at 25 °C, J. Polym. Sci., Part A: Polym. Chem., 2010, 48(1), 164–172, DOI:10.1002/pola.23774.
- X. Leng, N. H. Nguyen, B. van Beusekom, D. A. Wilson, V. Percec, M. E. Levere, D. A. Wilson, M. J. Monteiro and V. Percec, SET-LRP of 2-Hydroxyethyl Acrylate in Protic and Dipolar Aprotic Solvents, Polym. Chem., 2013, 4(10), 2995, 10.1039/c3py00048f.
- G. Lligadas and V. Percec, Ultrafast SET-LRP of Methyl Acrylate at 25 °C in Alcohols, J. Polym. Sci., Part A: Polym. Chem., 2008, 46(8), 2745–2754, DOI:10.1002/pola.22607.
- N. H. Nguyen, J. Kulis, H.-J. Sun, Z. Jia, B. van Beusekom, M. E. Levere, D. A. Wilson, M. J. Monteiro, V. Percec, J. A. Yoon, S. S. Sheiko, S. Z. D. Cheng, A. Zhang, G. Ungar, D. J. P. Yeardley, B. M. Rosen, A. O. Argintaru, M. J. Sienkowska, K. Rissanen, S. Nummelin and J. Ropponen, A Comparative Study of the SET-LRP of Oligo(Ethylene Oxide) Methyl Ether Acrylate in DMSO and in H2O, Polym. Chem., 2013, 4(1), 144–155, 10.1039/C2PY20782F.
- N. H. Nguyen, B. M. Rosen and V. Percec, SET-LRP of N,N-Dimethylacrylamide and of n-Isopropylacrylamide at 25 C in Protic and in Dipolar Aprotic Solvents, J. Polym. Sci., Part A: Polym. Chem., 2010, 48(8), 1752–1763, DOI:10.1002/pola.23940.
- Y. Deng, J. Z. Zhang, Y. Li, J. Hu, D. Yang and X. Huang, Thermoresponsive Graphene Oxide-PNIPAM Nanocomposites with Controllable Grafting Polymer Chains via Moderate in Situ SET-LRP, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(21), 4451–4458, DOI:10.1002/pola.26259.
- Introduction to Spectroscopy, ed. D. L. Pavia, G. M. Lampman, G. S. Kriz and J. R. Vyvyan, Sengage Learning, 2015 Search PubMed.
- D. Neugebauer, B. S. Sumerlin, K. Matyjaszewski, B. Goodhart and S. S. Sheiko, How Dense Are Cylindrical Brushes Grafted from a Multifunctional Macroinitiator?, Polymer, 2004, 45(24), 8173–8179, DOI:10.1016/j.polymer.2004.09.069.
- C. Reichardt, Solvatochromic Dyes as Solvent Polarity Indicators, Chem. Rev., 1994, 94(8), 2319–2358 CrossRef CAS.
- J. E. Del Bene, S. A. Perera and R. J. Bartlett, Hydrogen Bond Types, Binding Energies, and 1H NMR Chemical Shifts, J. Phys. Chem. A, 1999, 103(40), 8121–8124, DOI:10.1021/jp9920444.
- D. Fioretto, A. Livi, P. A. Rolla, G. Socino and L. Verdini, The Dynamics of Poly(n-Butyl Acrylate) above the Glass Transition, J. Phys.: Condens. Matter, 1994, 6(28), 5295, DOI:10.1088/0953-8984/6/28/007.
- J. L. de la Fuente, M. Fernández-García and E. López Madruga, Characterization and Thermal Properties of Poly(n-Butyl Acrylate-g-Styrene) Graft Copolymers, J. Appl. Polym. Sci., 2001, 80(5), 783–789, DOI:10.1002/1097-4628(20010502)80:5<783::AID-APP1155>3.0.CO;2-5.
- C. Lang, J. A. LaNasa, N. Utomo, Y. Xu, M. J. Nelson, W. Song, M. A. Hickner, R. H. Colby, M. Kumar and R. J. Hickey, Solvent-Non-Solvent Rapid-Injection for Preparing Nanostructured Materials from Micelles to Hydrogels, Nat. Commun., 2019, 10(1), 3855, DOI:10.1038/s41467-019-11804-7.
- R. C. Hayward and D. J. Pochan, Tailored Assemblies of Block Copolymers in Solution: It Is All about the Process, Macromolecules, 2010, 43(8), 3577–3584, DOI:10.1021/ma9026806.
- R. J. Cavanagh, P. A. Smith and S. Stolnik, Exposure to a Nonionic Surfactant Induces a Response Akin to Heat-Shock Apoptosis in Intestinal Epithelial Cells: Implications for Excipients Safety, Mol. Pharm., 2019, 16(2), 618–631, DOI:10.1021/acs.molpharmaceut.8b00934.
- M. Redhead, G. Mantovani, S. Nawaz, P. Carbone, D. C. Gorecki, C. Alexander and C. Bosquillon, Relationship between the Affinity of PEO-PPO-PEO Block Copolymers for Biological Membranes and Their Cellular Effects, Pharm. Res., 2012, 29(7), 1908–1918, DOI:10.1007/s11095-012-0716-6.
- G. Pembouong, N. Morellet, T. Kral, M. Hof, D. Scherman, M. F. Bureau and N. Mignet, A Comprehensive Study in Triblock Copolymer Membrane Interaction, J. Controlled Release, 2011, 151(1), 57–64, DOI:10.1016/j.jconrel.2011.01.007.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00615h |
| This journal is © The Royal Society of Chemistry 2023 |